 Atsunobu Takeda
Atsunobu Takeda Ryoji Yanai
Ryoji Yanai Yusuke Murakami1
Yusuke Murakami1 Koh-Hei Sonoda
Koh-Hei Sonoda
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 03 July 2020
Sec. Autoimmune and Autoinflammatory Disorders
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01431
This article is part of the Research Topic Retinal Immunobiology and Retinopathy View all 13 articles
In the twentieth century, a conspicuous lack of effective treatment strategies existed for managing several retinal disorders, including age-related macular degeneration; diabetic retinopathy (DR); retinopathy of prematurity (ROP); retinitis pigmentosa (RP); uveitis, including Behçet's disease; and vitreoretinal lymphoma (VRL). However, in the first decade of this century, advances in biomedicine have provided new treatment strategies in the field of ophthalmology, particularly biologics that target vascular endothelial growth factor or tumor necrosis factor (TNF)-α. Furthermore, clinical trials on gene therapy specifically for patients with autosomal recessive or X-linked RP have commenced. The overall survival rates of patients with VRL have improved, owing to earlier diagnoses and better treatment strategies. However, some unresolved problems remain such as primary or secondary non-response to biologics or chemotherapy, and the lack of adequate strategies for treating most RP patients. In this review, we provide an overview of the immunological mechanisms of the eye under normal conditions and in several retinal disorders, including uveitis, DR, ROP, RP, and VRL. In addition, we discuss recent studies that describe the inflammatory responses that occur during the course of these retinal disorders to provide new insights into their diagnosis and treatment.
In the last 2 decades, advances in the interdisciplinary collaboration of the fields of molecular biology, biochemistry, genetics, and biomedicine have resulted in tremendous breakthroughs in the treatment of refractory ocular disorders. Infliximab (IFX), a chimeric antibody of the tumor necrosis factor (TNF)-α, is a biologics that is used for treating ocular symptoms of Behçet's disease that have not been adequately controlled (1). Anti-vascular endothelial factor (VEGF) agents such as ranibizumab and aflibercept are used as the first-line therapy in the management of intractable retinal disorders such as neovascular age-related macular degeneration and diabetic macular edema (DME). These agents can also maintain remission in such cases (2, 3). For a long time, laser photocoagulation alone has been used for the primary treatment of etinopathy of prematurity (ROP). However, in 2018, ranibizumab was also validated for the treatment of ROP in Japan. Gene therapy clinical trials targeting the treatment of autosomal recessive or X-linked retinitis pigmentosa (RP), which is an incurable genetic retinal disorder, have been initiated (4). Furthermore, the overall survival of patients with vitreoretinal lymphoma (VRL), which is a fetal retinal malignancy, has increased because of improved treatment strategies that involve intense systemic chemotherapy and/or radiotherapy (5–7). However, unmet needs remain in the management of these retinal disorders because of primary and secondary treatment failure or non-response to the biologics, or most cases of RP untargeted by gene therapy.
The eye, just like the brain and the testes, is an immune-privilege site (8). Ocular immune privilege is an active process in which the regulatory molecules and cells of the eye modulate the induction and the expression of inflammation (8–10). As long as the ocular immune-privilege system is working, a harmful immune response and degenerative eye diseases can be prevented. By limiting intraocular inflammation, immune privilege preserves the integrity of the visual axis and thereby prevents blindness (11). However, the mechanism of immune privilege can be compromised genetically and/or by environmental stimuli such as damage-associated molecular patterns, infection, and a chronic immune response, and thereby give rise to various retinal disorders (11).
Immunological responses to various environmental stimuli have been associated with the pathogenesis of uveitis and retinal vascular diseases such as diabetic retinopathy (DR) and ROP (11–13). Retinitis pigmentosa is a genetic disorder, although inflammatory responses to microenvironmental changes, such as rod cell death, which occur after the primary onset of the disease, may cause subsequent loss of cone cells (11). In VRL, oncogenic mutations of VRL cells and the evasion of immune surveillance because of the immunosuppressive ocular microenvironment may contribute to tumor growth.
In this review, we focus on the roles of immunological responses in a normal conditions and in several major retinal disorders including non-infectious uveitis (NIU), DR, ROP, RP, and VRL. In addition, we contemplate new approaches for the diagnosis and treatment of these intractable retinal disorders from an immunological point of view.
A properly elicited and regulated immune response by the human body is necessary for eliminating threats due to infectious microbes and tissue trauma to avoid irreversible tissue damage (11). Acute inflammation should be self-limiting and normally attenuated after the elimination of deleterious stimuli to enable physiological recovery. Chronic inflammation causes degenerative diseases with consequent loss of organ function.
Ocular immune privilege is believed to elicit self-limiting immune responses (14). Several soluble and cell-bound inhibitory factors are involved in the mechanism of ocular immune privilege to create an intraocular immunosuppressive microenvironment, which prevents excessive immune activation and subsequent tissue damage. These factors include transforming growth factor-beta (TGF-β)2 (15), retinoic acid (16), and multiple immunosuppressive factors in ocular fluids (17), and the constitutive expression of the Fas ligand (18), programmed death-ligand 1 (PD-L1) (19), galectins (20), membrane glycoprotein CD200 receptor 1 (21), cytotoxic T lymphocyte-associated protein (CTLA)-2a (22), B7 (23, 24) on the surface of ocular cells. For the maintenance of retinal homeostasis, these immunosuppressive molecules in the eye actively regulate the induction and the expression of inflammation to prevent excessive activation and subsequent tissue damage.
In addition to the abovementioned local immunosuppression, ocular immune privilege is associated with the development of a type of an antigen-specific systemic immune regulation, called “anterior chamber-associated immune deviation” (“ACAID”) (8, 25). ACAID is induced by intrinsic intraocular bone marrow-derived antigen-presenting cells (APCs) that trap an antigen within the anterior chamber and migrates to the spleen via circulating blood (8, 26). Antigens from the anterior chamber can be transported to the regional lymph nodes (27). Tolerogenic CD11c+ dendritic cells (DCs) also transport antigens to the thymus (27). APCs bearing the eye-derived antigen can elicit the development regulatory T (Treg) cells in these lymphoid organs. In the eye, innate immune cells such as microglial cells, neutrophils, monocyte-macrophages, natural killer (NK) cells, natural killer T (NKT) cells, and γδT cells can exhibit a broad range of antigen recognition and confer a widely distributed form of immunity, which constitutes the first line of defense against various invading pathogens (28).
The anterior chamber and the vitreous cavity or the posterior chamber of the eye have the capacity to induce systemic immune deviation (29). This phenomenon is called “vitreous cavity-associated systemic immune deviation” (“VCAID”). Research demonstrates that F4/80+ hyalocytes are distributed over the retinal surface (30) and that murine hyalocytes are bone marrow-derived and turned over in 4 months (31). Hyalocytes have strong scavenger activity (30); therefore, we postulated that ocular hyalocytes capture an antigen on the retinal surface and carry it via the blood to the spleen, which induces antigen-specific Tregs (30).
Non-infectious uveitis (NIU) is a sight-threatening disorder associated with systemic autoimmune diseases such as Behcet's disease, sarcoidosis, and Vogt–Koyanagi–Harada disease (32). NIU is often recurrent and causes tissue destruction and scarring, especially in the retina and uvea, which results in permanent loss of vision. Early studies showed that T-helper (Th) 1 and Th17 cells are the major effector cells and are crucial for development of uveitis (33). In many cases, the administration of several immunosuppressive drugs for an extended period is necessary to control inflammation in eyes with uveitis (34). These agents include corticosteroids, tacrolimus, and cyclosporine, which have a strong T cell-suppressive effect and have serious side effects, such as diabetes, hypertension, and nephrotoxicity (35, 36). A new generation of biological compounds that inhibit T cell activation such as monoclonal antibodies and recombinant forms of natural inhibitory molecules have emerged (36, 37). For example, IFX is validated for the treatment of refractory ocular Behçet's disease (1, 38, 39). Adalimumab (ADA), which is a recombinant human immunoglobulin G1 monoclonal antibody to TNF-α, has also been validated for the treatment of patients with NIU in the United States and in Japan. ADA has been proven as an effective corticosteroid-sparing agent that reduces adverse effects associated with long-term usage of corticosteroids (34). Immunosuppressive therapeutic agents for treating non-infections uveitis (based on the guidelines of an international expert steering committee consisting of uveitis specialists are summarized in Table 1) (40). However, several studies report that IFX can cause the development of autoimmune diseases, primarily cutaneous vasculitis, lupus-like syndrome, and malignant lymphoma (41). In recent reports, some patients had a decreased response to IFX or ADA during the course of the treatment, owing to the development of antidrug antibodies against IFX or ADA (42–44). Therefore, new effective therapeutic targets for uveitis with less severe adverse effects need to be identified.
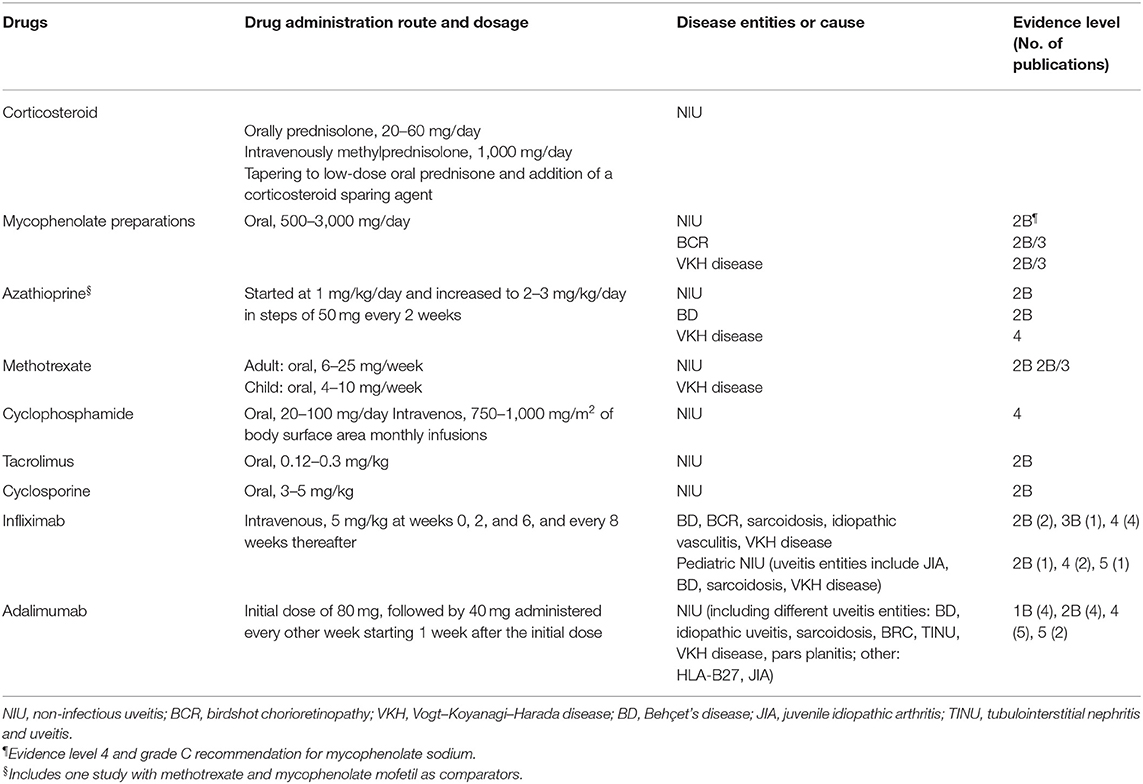
Table 1. Systemic corticosteroid and immunomodulatory therapeutic agents for non-infectious uveitis.
Experimental autoimmune uveitis (EAU) is an animal disease model of a T cell–mediated autoimmune disease that mimics many of the pathological features of human uveitis (45, 46). EAU is induced by injecting animals with purified retinal antigens such as S-Ag, a fragment (residues 1–20, GPTHLFQPSLVLDMAKVLLD) of the human interphotoreceptor retinoid-binding protein (hIRBP); rhodopsin (or opsin); phosducin, or recoverin. The disease is mediated by Th1 and Th17 cells (47, 48), which are generated from naive T cells in response to their exposure to proinflammatory cytokines and to the foreign antigens presented by APCs—including DCs, macrophages, NK cells, and B lymphocytes—in secondary lymphoid organs. Infiltration of inflammatory cells such as granulocytes and macrophages and other non-specific lymphocytes that can destroy ocular tissue also contributes to the development of EAU (48, 49). In the next section, we will discuss recent studies of EAU model mice and review studies on microglia, NK cells, NKT cells, DCs, the P2X7 receptor, Notch signaling, and the transcription factor Foxp3 (forkhead box P3), which have been identified as targets for translational medicine. Such research has provided insight into the mechanisms underlying disease pathogenesis and the basis for the development of new preventive or therapeutic approaches to human uveitis.
The important role of microglia in the regulation of inflammatory cell infiltration into the retina associated with the initiation of retinal autoimmune uveitis has recently been demonstrated (50). During the early phase of EAU (days 7–10), microglia have a direct effect on the increase in the number of various adherent vascular leukocytes, including T cells, major histocompatibility complex (MHC) class II+ cells, and CD11b+ cells. This effect was specific to microglia, given that it was not reproduced by other immune cell types such as monocytes-macrophages. Therefore, immunomodulatory therapies targeted at microglia are a primary focus in the development of new treatments for patients with uveitis.
We previously explained that NKT cells suppress the induction of Th17 cells and the ocular infiltration of hIRBP-specific T cells in EAU. However, in contrast to the ameliorating effects of NKT cell activation that is apparent during the initiation phase of EAU, the activation during the effector phase exacerbates disease pathology. This finding suggests that NKT cells have a dual role in EAU, depending on the phase of the disease (51).
CD83+CCR7+ NK cells induced by interleukin (IL)-18 released from DCs promote EAU (52). NK cells are negatively regulated by a soluble form of CD83 in EAU (53). In addition, DC–NK cell interactions that underlie the regulation of Th1 responses modulate the adaptive Th17 response and limit tissue-specific autoimmunity through the innate interferon (IFN)-γ-IL-27 axis in this model (54).
Treg cells are necessary for the resolution of EAU and the prevention of relapse (55, 56). A recent study demonstrated that PD-1+CD25+CD4+ Treg cells require programmed cell death 1 (PD-1) stimulation through a melanocortin-adenosine pathway to suppress EAU. These Treg cells did not induce suppressor activity in APCs through the PD-1 pathway (57).
Many studies have demonstrated that monocytes-macrophages have a role in the development of EAU (58). Activated bone marrow–derived macrophages are required during the effector phase of EAU (45). However, in addition to their proinflammatory function, macrophages have suppressive effects on ocular inflammation, especially in the chronic phase. Suppressor of cytokine signaling 3 (SOCS3) in macrophages was recently found to be important in the suppressing the inflammation caused by these cells in EAU (59).
Extracellular adenosine triphosphate (ATP) is a key chemotactic signal for the recruitment of innate immune cells to sites of brain injury (60). ATP is actively released via exocytosis or transporters during the early phase of apoptotic cell death, whereas it is passively released from necrotic cells after the rupture of the plasma membrane. Extracellular ATP acts at P2X and P2Y purinergic receptors and induces the formation of inflammasomes, which are large intracellular multiprotein complexes that are key players in host defense during the innate immune response (61). The P2X receptor family consists of ligand-gated cation channels that open in response to the binding of extracellular ATP. Among the seven mammalian P2X receptors, P2RX7 shows the highest affinity for ATP and is highly expressed in immune cells such as monocytes and T lymphocytes (62, 63). Research has demonstrated that the genetic ablation of P2RX7 or the administration of the P2RX7 antagonist BBG in mice suppresses EAU clinically and histopathologically by attenuating hIRBP-dependent induction of interferon (IFN)-γ and IL-17 (64).
DCs are highly efficient APCs and have the unique ability to prime and activate naive T lymphocytes (65, 66). They are divided into three types, based on their function: immature DCs, mature DCs, and regulatory DCs. Under physiological conditions, DCs are widely distributed among tissues and organs (67) where they are in an immature state and contribute to immune surveillance. In the eyes of mice and rats, these cells are at the peripheral margin and in juxtapapillary areas of the retina (68, 69). The functions of DCs in the quiescent retina include promoting the generation of Foxp3+ Treg cells and inhibiting the activation of naive T cells induced by splenic DCs and antigens (69).
DCs have an essential role in innate immunity. They also link the innate and adaptive immune systems and are key for the induction of late immune responses. Cell-based therapy involving the ex vivo manipulation of mature or regulatory DCs has been adopted as a means to induce tolerance in autoimmune disease (70–72). Studies of the mechanisms of DC function in uveitis are thus warranted to identify new therapeutic targets for this condition. Mature DCs pulsed with uveitogenic antigens induce the development of EAU (69). Treatment with fixed immature DCs, but not with fixed mature DCs, has also been demonstrated to ameliorate the progression of EAU by inhibiting uveitogenic CD4+ T cell activation and differentiation (73). In addition, impairment DC maturation with drugs prevents the generation of antigen-specific Th1 and Th17 cells and thereby attenuates EAU (74). Moreover, regulatory DCs induced in vitro suppress the development of EAU (75). These various data altogether indicate that the regulation of DC status is potentially beneficial for the treatment of uveitis.
In a previous study, conducted by the authors of the present review, we found that mouse spleen-derived DCs mediate the anti-inflammatory action of dietary ω-3 long-chain polyunsaturated fatty acids (LCPUFAs) in EAU (76). Histological analysis at 17 days after disease induction revealed retinal folds and immune cell infiltration in the eyes of EAU mice that received DCs from ω-6 LCPUFA–fed mice, and showed that such changes were markedly suppressed in EAU mice that received DCs from ω-3 LCPUFA–fed mice (Figure 1A) (77). Furthermore, DCs exposed to ω-3 LCPUFAs in vivo or in vitro suppressed T cell proliferation. This finding suggested that ω-3 LCPUFA–treated DCs attenuate inflammation mediated by T cells (Figure 1B). Cytokines released by activated DCs are essential for T cell differentiation, with IL-12 p70 promoting Th1 cell differentiation and with IL-6 and TGF–β promoting Th17 cell differentiation (78, 79). We also found that dietary ω-3 LCPUFAs acting via adoptively transferred DCs markedly inhibited IL-12 p70 and IL-6 production by T cells from EAU mice. This finding is consistent with the notion that ω-3 LCPUFAs suppress Th1 and Th17 cytokine production by CD4+ T cells, through the mediation of DCs (Figures 1C,D). Moreover, we also found that ω-3 LCPUFAs, acting via DCs, suppressed the production of proinflammatory cytokines and the anti-inflammatory cytokine IL-10. However, the DC-dependent anti-inflammatory effects of ω-3 LCPUFAs appear to outweigh their proinflammatory effects, at least in EAU.
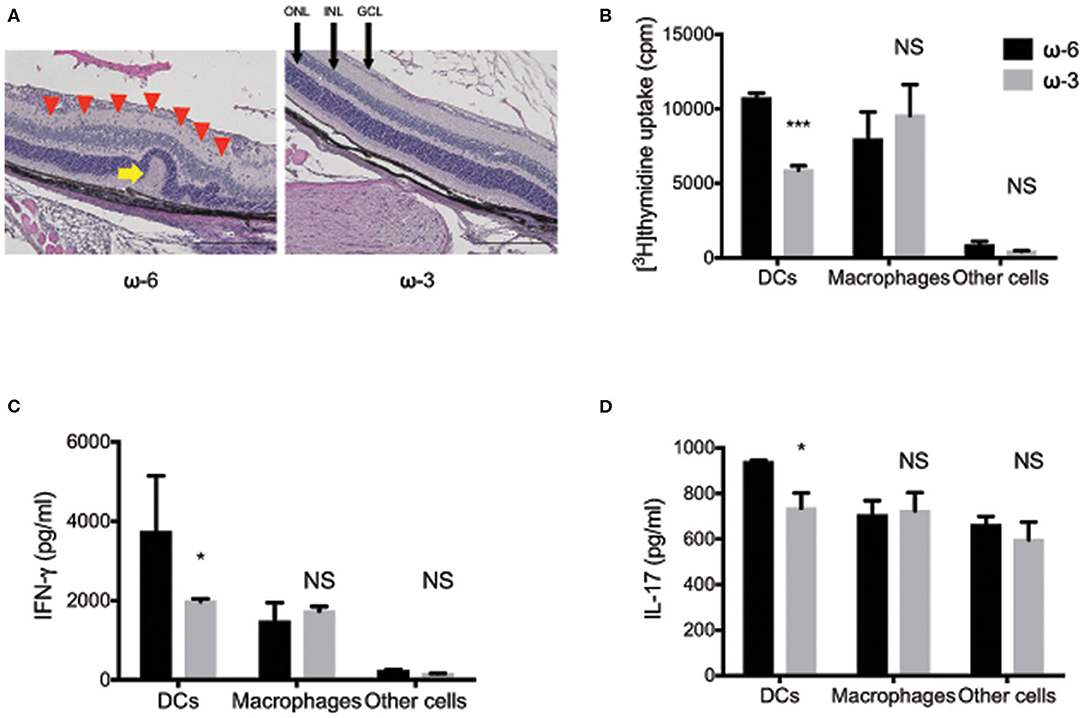
Figure 1. The effects of ω-3 long chain polyunsaturated fatty acids in experimental autoimmune uveitis model mice. (A) Hematoxylin-eosin staining of retinal sections at 17 days after disease induction in experimental autoimmune uveitis (EAU) mice maintained on a diet enriched with ω-3 or ω-6 long-chain polyunsaturated fatty acids (LCPUFAs). A red arrowhead indicate inflammatory cells in the retina. A yellow arrow indicates a retinal fold. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer. Scale bars, 200 μm. (B–D) The proliferation of T cells, as assessed by the measurement of [3H]thymidine incorporation (B), and by the production of interferon-γ (C), and interleukin-17 (D) in co-cultures of CD4+ T cells from EAU mice and the indicated antigen presenting cell fractions from mice fed with ω-3 or ω-6 LCPUFAs. Data are expressed as the means + the standard error of the mean (SEM). *P < 0.05, ***P < 0.001; NS, not significant vs. the corresponding value for the ω-6 LCPUFA diet (i.e., Sidak's multiple comparison test). The figure is reproduced from (77) with permission.
The global prevalence of diabetes mellitus (DM) tends to increase yearly, and the number of DM patients is estimated to reach 592 million within 20 years (80). Diabetic retinopathy (DR) is a representative microvascular complication of DM that causes visual impairment in working-age adults (81). Leasher et al. reported that blindness and moderate to severe visual impairment due to DR increased in 20 years (1990–2010) from 2.1 to 2.6% and from 1.3 to 1.9%, respectively (82). Vascular abnormalities such as hemorrhage, microaneurysm, capillary non-perfusion, and exudates are frequently observed in DR. Therefore, DR has been perceived as a disease that originates from vascular abnormalities. However, several lines of evidence indicate an association between inflammation and the pathophysiology of DR (12). The principal causes of visual impairment in DR are proliferative DR (PDR) and DME. In the next section, we will discuss the relationship between inflammation and PDR or DME formation.
PDR is characterized by the development of preretinal neovascularization and epiretinal fibrovascular membranes (FVMs) (81). Prolonged hyperglycemia, accumulation of advanced glycation end products, and oxidative stress under diabetic condition induce VEGF expression in the retina through protein kinase C activation. VEGF promotes leukostasis by increasing the expression of intercellular adhesion molecule-1 (ICAM-1) in retinal vascular endothelial cells (83). Low-grade inflammation initiated by leukocytes (e.g., monocytes and granulocytes) that adhere to endothelial cells via ICAM-1 induces vascular endothelial cell damage and cell death and promotes capillary loss and infiltration of leukocytes into the retina (84). Expanding inflammation and ischemia induces the further expression of VEGF and other angiogenic cytokines such as TNFα, IL-1β, IL-6, and IL-8. This negative cycle gradually advances DR (85). Van Hecke et al. showed that the prevalence of DR is positively associated with the serum levels of C-reactive protein (CRP) and soluble ICAM-1 (sICAM-1), which suggests that early vascular damage is actually caused by inflammation (86). Preventing the onset of DR is important to avoid DR-induced visual impairment. Therefore, the serum levels of CRP or sICAM-1 that indicate the level of the inflammatory activity may become useful biomarkers for predicting the onset of DR.
In a previous study with an experimental animal model, we previously reported that intravitreal injection of an anti-VEGF agent can attenuate the infiltration of leukocytes, especially macrophages, into the retina and can suppress preretinal neovascularization (87). Esser et al. demonstrated that inflammatory phase macrophages are localized in FVMs in PDR (88). This finding implied that macrophage-induced inflammation is associated with FVM formation. However, macrophages have diverse populations, and the role of each population differs (89). With regard to various populations of macrophages, Zhou et al. found that M2-like macrophages (i.e., CD163-positive macrophages) promote pre-retinal angiogenesis in DR (90). Kobayashi et al. also reported that some M2-like macrophages localized in the FVMs produce periostin (91). Periostin is a matricellular protein that is essential for FVM formation in PDR (92). Thus, we believe that M2-like macrophages may potentially become a new therapeutic target in treatment of PDR. The process from the onset of DR to the occurrence of PDR is summarized in Figure 2.
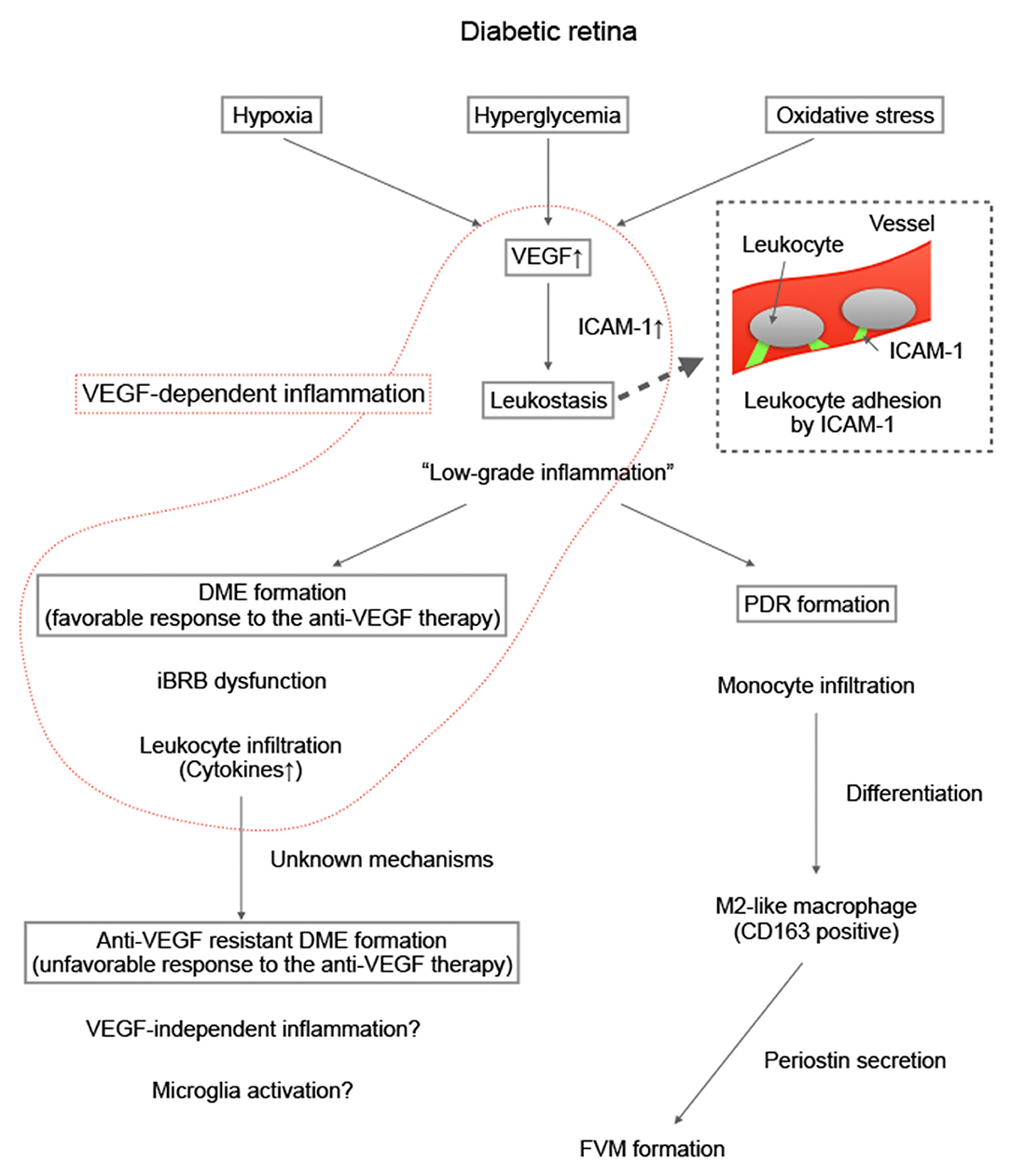
Figure 2. Summary of our hypothesis on the formation of proliferative diabetic retinopathy and antivascular endothelial growth factor resistant diabetic macular edema. Diabetic macular edema (DME) animal models such as Akimba mouse models have been developed. The details of the involvement of inflammation in the pathogenesis of DME are expected to be identified in the future.
Blood vessels in the central nervous system (CNS), including the retina have a vascular barrier function and maintain a proper neural microenvironment through strict control of vascular permeability (93). Retinal neural tissue is separated from the blood stream by the inner blood-retinal barrier (94). The collapse of the inner blood-retinal barrier under diabetic conditions results in DME, and persistent DME causes irreversible neural damage (94). Experimental investigations have proven that low-grade inflammation after leukostasis can disrupt the vascular barrier (83), and VEGF and some cytokines/chemokines that can increase vascular permeability are secreted from infiltrated leukocytes (94). Moreover, several clinical studies have demonstrated the upregulation of inflammatory cytokines/chemokines in the vitreous fluid of eyes with DME, which suggests a relationship between inflammation and the pathogenesis of DME (95).
VEGF is the most studied molecule that can increase vascular permeability (96). Regular intravitreal injections of anti-VEGF agents (i.e., anti-VEGF therapy) can improve vision and reduce the accumulation of macular fluid in DME. This therapy is the primary treatment modality for DME (3). However, clinical trials have demonstrated that ~40% of patients are anti-VEGF resistant (97). Several studies were conducted to detect additional therapeutic targets for the treatment of DME. Sfikakis et al. demonstrated the therapeutic efficacy of the intravenous injection of IFX, an anti-TNFα antibody (98). Anti-inflammatory treatment for DME produces worthwhile results; however some concerns exist regarding the adverse effects of the therapy because of the need to administer high concentrations of IFX (5 mg/kg) multiple times to patients (98). Gale et al. conducted a clinical trial to evaluate the efficacy of an oral chemokine receptor (CCR) type 2 and type 5 (CCR2/CCR5) dual antagonist for treating DME because CCR2 and CCR5 signaling pathways are associated with vascular leakage, monocyte/macrophage infiltration, and increased VEGF expression in the retina of experimental DR models (99). However, its therapeutic efficacy is inferior to that of monthly intravitreal injections of an anti-VEGF agent (99). No molecule beyond VEGF has been found, although the development of a novel treatment for anti-VEGF–resistant DME is urgently needed.
To identify a new target molecule or a novel biomarker for predicting anti-VEGF resistance, many researchers have examined the relationship between the response to anti-VEGF treatment and the concentration of intraocular inflammatory cytokines/chemokines. Hillier et al. showed that an increase in baseline aqueous ICAM-1 is associated with a favorable anatomic response, whereas an increase in baseline aqueous VEGF is associated with an unfavorable anatomic response (100). Shimura et al. concluded that a favorable response was obtained in patients with increased baseline aqueous VEGF, soluble VEGF receptor-1, monocyte chemoattractant protein-1 (MCP-1), ICAM-1, IL-6, and IP-10 (101). Felfeli et al. compared aqueous cytokine concentrations at baseline and 2 months after anti-VEGF therapy; they reported that the aqueous levels of ICAM-1, MCP-1, placenta growth factor, and TGF-β2 decreased significantly in patients with a favorable response (102).
The results of animal experiments have revealed that VEGF induces endothelial ICAM-1 expression in the early stage of DR (83), and that ICAM-1 expression decreases in endothelial cells in the chronic stage of DR (103). Therefore, DME in the “early” stage with mild vascular injury (which does not indicate a short medical history of DR) may respond well to anti-VEGF therapy. Inflammatory cytokines and VEGF decreased in patients with a favorable response to the anti-VEGF therapy, which suggests that VEGF-dependent inflammation may have primarily contributed to DME formation in these patients. The detailed mechanisms of the association between inflammation and anti-VEGF resistance are not completely understood; however, the change in the quality of inflammation may cause treatment resistance. DME animal models such as Akimba mouse models (104), will provide new insights into the involvement of inflammation in the pathogenesis of DME in future research (Figure 2).
Retinopathy of prematurity (ROP) is a retinal vasoproliferative disorder that can lead to childhood blindness (105). Blencowe et al. reported that ROP occurs in 184,700 infants annually worldwide. Among these infants, ~20,000 cases progress to a stage that requires treatment (105). The incidence of ROP onset cases has been declining because neonatal management is improving every year (106) and because of a tendency toward a declining birthrate, especially in developed countries. However, a decrease in the number of cases does not necessarily mean a decrease in the number of treatments. Improved neonatal management indicates an increase in the survival rate of preterm infants. The gestational age (GA) and birth weight (BW) of infants enrolled in major large trials are steadily decreasing (107). Low GA and low BW are common risk factors for ROP progression (108). The risk of an increase in the relative proportion of severe ROP cases that require treatment may occur in the future.
Current primary treatments for ROP are retinal photocoagulation and anti-VEGF therapy. Large clinical trials such as the Early Treatment for Retinopathy Of Prematurity study and the Bevacizumab Eliminates the Angiogenic Threat of Retinopahty of Prematurity (BEAT-ROP) study have proven that both methods are effective (109, 110). However, severe ROP is sometimes accompanied by poor mydriasis and vitreous opacity. In such cases, performing laser photocoagulation in infants with ROP is difficult. Anti-VEGF therapy can be considered the first-line therapy for these patients. The BEAT-ROP study revealed that anti-VEGF therapy (i.e., the administration of bevacizumab) is associated with significantly less recurrence than laser photocoagulation in zone I ROP cases (110). To date, bevacizumab is generally used as an off-label drug in some limited facilities. The efficacy of intravitreal injection of ranibizumab (IVR) was recently proven (111). On account that ROP has been approved as one indication for ranibizumab, more infants with ROP are expected to benefit from anti-VEGF treatment in the future.
The recurrence rate of ROP after IVR monotherapy is ~30% (111). The provision of optimal treatment at a proper time is essential to prevent the impairment of visual development in infants with ROP; therefore, the identification of a biomarker of ROP progression is very important. VEGF and many other molecules such as insulin-like growth factor-1, hypoxia-inducible factor-1, and reactive oxygen species, may be involved in the onset of ROP (108); however, Sato et al. reported that the vitreous concentrations of inflammatory cytokines/chemokines are increased in patients with ROP (13), which suggests an association between inflammation and ROP pathogenesis. Lyu et al. showed that high levels of aqueous VEGF and macrophage inflammatory protein (MIP)-1β at baseline were associated with the recurrence rate of ROP after IVR therapy (112). In a previous study, we reported that MIP-1β expression significantly increased in the retina of an ROP animal model (113). MIP-1β, also known as chemokine CC motif ligand 4, is a member of the CC chemokine family. Members of the CC chemokine family are characterized by their ability to direct the migration of leukocytes into the inflamed tissues. MIP-1β is upregulated very quickly after hypoxic stimulation in mouse retina (113). Therefore, its expression level may become a very sensitive sensor of retinal ischemia. The administration of a neutralizing antibody against MIP-1β inhibits physiological angiogenesis (113); therefore, we believe that MIP-1β has the potential to be a useful inflammatory biomarker of ROP progression or recurrence rather than a therapeutic target molecule. Moreover, Matsuda et al. reported that mast cell tryptase (MCT) released from mast cells is involved in angiogenesis in ROP (114). They demonstrated that MCT promotes angiogenesis by inducing the production of MCP-1 and other angiogenic factors from endothelial cells (114). On account that the serum MCT level is elevated in infants with ROP, MCT also has the potential to be a useful biomarker of ROP progression.
Retinitis pigmentosa refers to a subgroup of inherited retinal degenerations (IRDs) that cause progressive rod-cone degeneration (115). More than 90 causal genes have been identified for typical RP, and these genes are frequently related to the function, structure, and homeostasis of rod photoreceptor cells. Night blindness due to rod dysfunction and death is an early symptom of RP, followed by visual field constriction and loss of central vision due to secondary cone cell death. RP is a major cause of adult blindness in over one million patients globally; no effective treatment substantially delays the disease progression or restores the vision lost to RP (115).
Recent advances in gene therapy have shed light on the treatment of IRDs. Supplementation of the retinal pigment epithelium-specific 65 kDa protein (RPE65) gene in patients with Leber congenital amaurosis due to RPE65 mutations improves their light sensitivity and performance in the multi-luminance mobility test (116). This therapy has been approved in the United States and in Europe. Gene therapy for RP, which targets autosomal recessive or X-liked mutations (e.g., phosphodiesterase 6B, retinaldehyde-binding protein 1, retinitis pigmentosa GTPase regulator, MER proto-oncogene tyrosine kinase) has been assessed in clinical trials (4). However, a significant number of RP patients may not be indicated for gene therapy because of the following reasons: (1) adeno-associated viral vectors cannot accommodate large genes such as the eyes shut homolog, (2) gene correction or editing of autosomal dominant mutation in vivo is still challenging, and (3) many patients are first diagnosed in the mid- to late-stages of the disease when the rod cells are mostly lost. Therefore, the elucidation of the biological mechanisms that underlie retinal degeneration, especially in the secondary cone cell death phase, will be critical in developing novel treatments for RP, in addition to individualized gene therapy.
In RP, rods are expected to be injured because of gene mutations that are exclusively expressed or critically function in rod cells. However, why and how cones also die subsequent to rod cell death is puzzling. Accumulating evidence suggest that microenvironmental changes associated with rod cell death such as loss of trophic factors (117), metabolic alterations (118), oxidation (119), collapse of outer nuclear layer (120), and inflammation (121), which are associated with rod cell death, may contribute to the secondary cone cell death. These factors mutually influence each other. For example, cytokines or chemokines released from a collapsed retina evoke inflammatory cell activation and migration, activated microglia or macrophages produce reactive oxygen species (ROS) to enhance oxidative stress, and, alternatively, oxidative stress triggers, or augments inflammatory response. The roles of oxidative stress and metabolic dysfunction in cone cell death in RP have been summarized in a previous review (119, 120). The present review focuses on inflammation and immunobiology as potential factors that mediate and modulate cone degeneration in RP. Assessing immune involvement in RP/IRDs from a broader perspective, including RP cases in the rod degeneration phase, is beyond the scope of this review and have been described in our previous recent review (11).
Cell death and inflammation have a tight interaction with each other. Dying or dead cells stimulate phagocytes to mediate their clearance and maintain tissue homeostasis, whereas excessive activation of inflammatory cells can exert cytotoxicity and exacerbate the disease (122).
Without exception to this scenario, inflammatory cell infiltration is usually observed in the vitreous of RP patients. Using vitreous samples from post-mortem RP patients, Newsome et al. demonstrated that this cell infiltration consists of a mixed component of inflammatory cells including monocytes, NK cells, lymphocytes, and others cells (123). In a previous study, the authors of the present review graded the severity of inflammation in the anterior vitreous using vivo in slit-lamp biomicroscopy, and found that RP patients with a higher number of inflammatory cells had worse visual acuity and lower central retinal sensitivity (121). We also evaluated aqueous flare, a marker of blood-ocular barrier breakdown and inflammation. Our study data showed that aqueous flare values are increased in the eyes of RP patients, compared to these values in healthy individuals, and that aqueous flare values are negatively correlated with central visual function in RP patients (124). Consistent with our observation, independent groups have reported that aqueous flare values have a negative association with visual field area (125) and a positive correlation with inner retinal thickening, which occurs during retinal degeneration and remodeling, in RP patients (126). Daylight vision in the central and peripheral area is provided by cone cells; therefore, these findings suggest that inflammation may be implicated in retinal degeneration, especially in secondary cone cell death that occurs in RP. However, these studies were cross-sectional clinical studies and have the limitation that a cause-effect relationship could not be elucidated.
Cytokines and chemokines have critical roles in evoking the differentiation, activation, migration, and suppression of immune cells. A comprehensive measurement of inflammatory cytokines and chemokines in the aqueous humor and vitreous of the eyes of RP patients using multiplex enzyme-linked immunosorbent assay showed that IL-6, IL-8, and MCP-1 are elevated in the aqueous humor of RP patients, and that a greater variety of molecules (e.g., IL-1β, IL2, IL-4, IL-6, IL-8, IL-10, IFN-γ, MCP-1) are increased in the vitreous with more significant fold changes (121). Lu et al. also conducted multiple cytokine analyses and observed increased IL-6, IL-8, and MCP-1 levels and increased extracellular matrix-related proteins such as matrix metalloproteinases (127).
The inflammatory cytokines/chemokines elevated in RP are related to innate and acquired immunity. IL-1β, IL-8, and MCP-1 are pivotal molecules for activating and recruiting monocytes/macrophages and neutrophils to the inflammatory loci. IFN-γ, IL-2, IL-4, and IL-10 are produced primarily or partly by T lymphocytes, and they mediate the differentiation and polarization of Th cells and macrophages. These profiles of inflammatory cytokines/chemokines in RP are consistent with the infiltration of a variety of inflammatory cells into the vitreous, as described above (123). To develop an anti-inflammatory therapy for RP, further studies are needed to elucidate the key inflammatory cytokines/chemokines that critically contribute to the disease progression.
High sensitivity CRP (hs-CRP) is a serum inflammatory marker, and an increased hs-CRP is associated with age-related macular degeneration, DR, and uveitis (128, 129). The measurement of serum hs-CRP levels in RP patients without systemic disorders revealed that hs-CRP levels are ~2 times higher in RP patients than in control subjects (130). In addition, a higher hs-CRP level is associated with a faster deterioration in central retinal sensitivity in RP patients (130). Taken together, these findings suggest that peripheral immune cells and ocular resident immune cells may be implicated in the disease progression of RP.
The findings outlined previously suggest that innate and acquired immunity are activated and involved in the pathology of RP. However, the function of each inflammatory cells (e.g., microglia, macrophages, and lymphocytes) and its regulatory mechanisms remains unclear and is a topic of interest.
Microglia, a resident macrophage in the CNS that derived from the embryonic yolk sac progenitors, are the most prominent immune cells in the retina (131). Microglia are long-lived cells that persist throughout the entire lifetime of mice, and can proliferate and repopulate after experimental depletion or during retinal degeneration (132). Monocytes in the peripheral blood do not invade the CNS in healthy conditions, although they can infiltrate and differentiate into macrophages in an aging or diseased retina with a dysfunctional blood-retinal barrier (133). These monocyte-derived macrophages resemble microglia in their morphology and their long life span, but have different functional features such as lower expression of colony-stimulating factor 1 receptor and higher expression of proinflammatory molecules such as MHC-II, IL-1β, and TNF-α (134). These two myeloid cells (i.e., tissue-resident microglia and monocyte-derived macrophages) have been extensively investigated as pivotal innate immune cells that contribute to the health and disease of the retina.
Several reports have demonstrated the detrimental function of microglia/macrophages in retinal degeneration in experimental RP. In a previous study, suppression of activated microglia/macrophages with minocycline or toxin-induced depletion of CX3CR1-positive microglia/macrophages protected rod cells against cell death in retinal degeneration (rd) 10 mice (135, 136). However, attenuation of the homeostatic function of microglia by disrupting the CX3C chemokine ligand 1-CX3C chemokine receptor 1 axis or the complement component 3–complement receptor 3 axis accelerates rod degeneration, along with proinflammatory microenvironmental changes such as increased TNF-α and IL-6 levels (137, 138). Therefore, microglia/macrophages have a bidirectional function in RP, as expected from the basic understanding of the interaction between cell death and inflammation. Dissection of the protective and detrimental populations among microglia/macrophages and precise understanding of the differential function of microglia and macrophages in RP warrant further studies. In addition, clinical studies suggest a link between inflammatory markers and cone function; therefore, the effect of microglia/macrophages on cone cell death also requires further study.
Oxidative stress significantly contributes to cone cell death in RP. Campochiaro et al. postulated that rod cell loss in RP substantially reduces oxygen consumption in the retina, and the remaining cone cells are exposed to a high-level of oxygen and resultant ROS (119). They showed that oxidized proteins, lipids, and nucleic acids are accumulated in the outer retina (139), and that pharmacological or genetic suppression of oxidative stress leads to significant rescue of cone cells in animal models of RP (140).
Oxidative stress may have a direct harmful effect on cone cells, but it also affects microglial/macrophage activation in RP. In a previous study, we showed that treatment with anti-oxidant N-acetylcystein (NAC) substantially suppresses microglia/macrophage activation with reduced MCP-1, IL-1β, RANTES, and TNF-α expression (141). The anti-inflammatory effects of anti-oxidants are also observed in eyes with experimental retinal detachment that are treated with a free radical scavenger, edaravone (142). Our study further demonstrated that oxidative activation of microglia/macrophages is a key step in the augmentation of retinal inflammation and degeneration (including cone cell death) in rd10 mice. This activation is partly mediated by an oxidative DNA repair enzyme, MUTYH; an excessive activation of which leads to the formation of single strand breaks and increased expression of TNF-α in microglia/macrophages (143). The concept that oxidative stress alters homeostasis vs. neurotoxic balance of microglia and modulates cone cell survival and neuroinflammation in RP is shown in Figure 3.
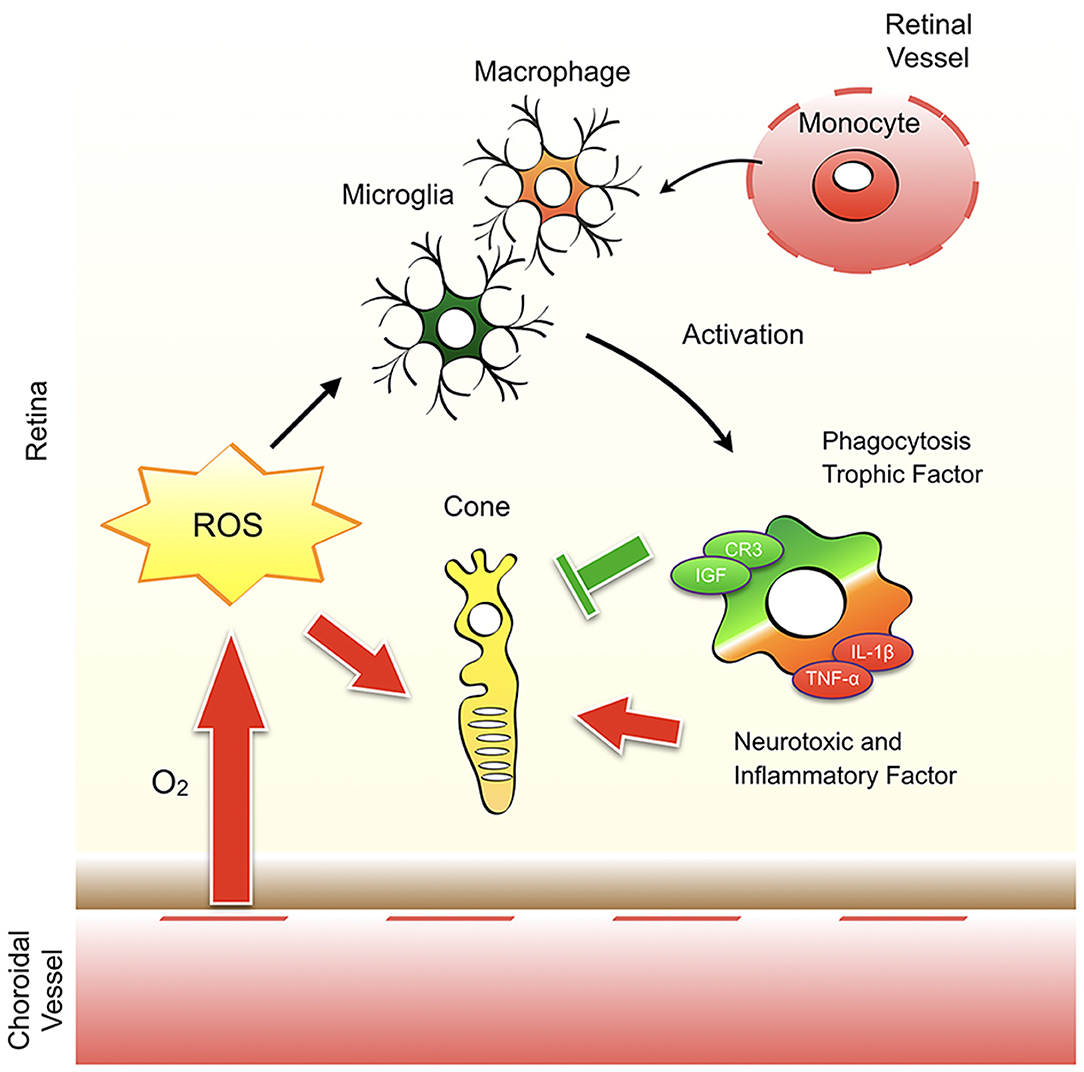
Figure 3. Oxidative stress modulates cone cell survival and neuroinflammation in retinitis pigmentosa. The remaining cones in retinitis pigmentosa (RP) are exposed to a high level of oxygen and the resultant reactive oxygen species (ROS). ROS have a direct harmful effect on cone cells and affects the activation of microglia and monocyte-derived macrophages. Activated microglia/macrophage have a bidirectional function to protect or promote cone cell death. Which environmental factors (e.g., ROS, molecules released from dead cells) and cellular factors (e.g., microglia vs. macrophage) are critical to determine the homeostatic vs. neurotoxic function of microglia/macrophage in RP is unclear.
Lymphocytes and lymphocyte-related cytokines are increased in the eyes of RP patients. In addition, several studies suggest the possible involvement of an autoimmune response and antiretinal autoantibodies in the progression of RP (144). However, the roles of acquired immunity in RP have been less investigated than the role of innate immunity.
Rohrer et al. crossed rd1 mice with Scid or Rag1−/− mice, both of which lack functional T and B lymphocytes, and showed that the deficiency of lymphocytes did not change rod cell death (145). In another study, Mishra et al. demonstrated that rod degeneration was mildly attenuated in rd1 Nod.Scid mice, which are deficient in T, B, and NK cells (146). These findings suggest that NK cells may play a minor role in rod cell death in RP, whereas lymphocytes may not have a significant function, at least solely by themselves. However, lymphocytes and their related molecules/factors are clinically observed in the mid- to late stage of RP. Thus, studying the roles of acquired immunity in cone cell death will be important in future research.
Most primary vitreoretinal lymphomas (PVRL), which were previously termed as intraocular lymphomas, are related to high-grade non-Hodgkin's lymphoma, which is a subset of primary central nervous system lymphomas (PCNSL) (147). In Japan, 21 per 100,000 patients with ocular disorders have PVRL (148). PVRL comprises 1% of non-Hodgkin's lymphoma and <1% of intraocular tumors (149). Most cases of VRL are primary or secondary to CNS disease or may present simultaneously with it; however, it can also rarely be derived from systemic metastatic lymphoma (150). PVRL usually occurs in adults from the fifth to the sixth decades of life (151). No sex or racial predilection to the disease apparently exists, although some reports proposed that PVRL occurs more frequently in females than males (152).
The clinical ocular features of VRL, termed “masquerade syndrome,” are often similar to those of chronic uveitis; therefore, a misdiagnosis of VRL sometimes leads to the administration of anti-inflammatory agents such as corticosteroids and thereby cause a delay in reaching a definitive diagnosis. The interval between the onset of the ocular or neurological findings and a definitive diagnosis is variable, and ranges from 4 to 40 months (152). The involvement of the CNS arises in 16–34% of patients with PVRL at presentation and develops in 42–92% of patients within a mean interval 8–29 months (151). Ocular involvement occurs in 15–20% of patients with PCNSL (151). Most patients with VRL have bilateral ocular involvement, but they often present with unilateral involvement at the initial visit owing to the uneven distribution of the disease. PVRL usually develops in the retina, the vitreous chamber, and/or the optic nerve, but can sometimes involve the anterior segment of the eye (153). Vitritis is the most common sign in VRL, and the findings has an “aurora borealis”-like appearance. In VRL, responses to corticosteroid therapy is initially observed but treatment resistance subsequently occurs. Multifocal whitish to yellow subretinal infiltrates are often observed. Coalescence of the lesions with “leopard skin”-like pigmentation, which is characteristic of VRL, is sometimes observed.
The overall survival of VRL patients has improved over the decades because of earlier diagnosis of the disease, which is a result of advances in molecular biological or genetic techniques. In addition, intense systemic chemotherapy and/or radiotherapy have also increased the overall survival rate. Several prospective studies have recently demonstrated that high-dose methotrexate (HD-MTX)-based chemotherapy with intravitreral MTX injections, subsequent whole brain radiotherapy (WBRT), and/or consolidation chemotherapy could improve overall survival and prevent CNS progression (5–7). Kaburaki et al. showed that a combination treatment protocol of intravitreral MTX injections, MTX-based systemic induction chemotherapy and consolidation high-dose cytarabine, and subsequent reduced-dose WBRT for the treatment of PVRL accomplished a 4-year progression-free survival of 72.7% and a 4-year overall survival of 88.9% (7), which suggests that these intensive systemic therapeutic modalities should be introduced for CNS prophylaxis. Some cases are resistant to these regimens, although no standard regimens exist for the treatment of refractory and relapsed PCNSL. Retreatment with HD-MTX-based chemotherapy has been administered in some patients. WBRT or high-dose chemotherapy and autologous stem cell transplantation in younger patients who have not undergone these treatments as part of the first-line therapy are used as a salvage therapy instead (154). However, introducing these treatments to elderly patients and patients with a poor general condition is often difficult to prevent CNS progression and to treat PCNSL because of the possible occurrence of treatment-induced adverse events such as neurotoxicity and nephrotoxicity (155, 156). In the next section, we review the association of pathogenesis, especially the genetic or immunological aspects, with B-cell VRL to exhibit new diagnostic and/or therapeutic targets for the treatment of VRL.
Most cases of PVRL can be classified as diffuse large B-cell lymphoma (DLBCL), whereas very few cases are classified as a T cell lymphoma or NK cell origin PVRL (152). Based on gene expression profiles, DLBCL is divided into three major subgroups: germinal center B-cell-like, activated B-cell-like (ABC)/non-germinal center, and primary mediastinal DLBCLs (157). The immunophenotype of most PCNSLs resembles ABC-DLBCLs, which are more aggressive and have poor prognostic outcomes, compared to the others (158).
High frequency of myeloid differentiation primary response gene 88 (MyD88) and CD79B mutations have been characterized in PCNSL (159). A single leucine-to-proline substitution at amino acid position 265 of Myd88 (MYD88 L265P), is the most common mutation and accounts for more than 60% of VRLs (160). MYD88 is the adapter protein that mediates intracellular signaling pathways downstream of the toll-like receptor and the IL-1 receptor families. The CD79B mutation occurs in 35% of patients with PVRL, and is associated with the CNS progression of PVRL (161). The B-cell receptor (BCR) complex-associated protein β chain, CD79B, forms a complex with BCR and generates after recognition of an antigen to activate chronic BCR signaling. MYD88 L265P and/or CD79B mutation contribute to the constitutive activation of NF-kB or BCR signaling, thereby promoting tumor growth.
Furthermore, in cases of PCNSL, because of the HLA locus mutation (chromosome 6q21.32), a shortage in HLA class I and II expression on tumor cells leads to escape from T or NK cell-mediated immune surveillance against tumor cells (162), which suggests that the lack of immune recognition of foreign antigens is one of the mechanisms that B-cell VRL cells preferentially retains in the eye.
PD-1, which is expressed on activated T-cells such as cytotoxic T lymphocytes (CTLs), interacts with its ligands (PD-L1 and PD-L2). These ligands are commonly expressed on tumor cells and upregulated in the tumor microenvironment (TME), thereby promoting inhibitory signaling of T cell receptors (TCRs) in CTL and subsequent tumor growth (163, 164). In PCNSL, investigations of copy number variations have revealed that frequent copy number gains at chromosome 9q24.1, which contains the PD-L1/PD-L2 locus (159). Chromosomal translocation involving the PD-L1/PD-L2 locus were also discovered in PCNSL, which indicates that immune evasion may be associated with the development of PCNSLs, including PVRL.
Cytological examination of the intraocular fluid or tissue is the gold standard for a definitive diagnosis of VRL. However, cytology alone can have a low diagnostic yield (40–60%) because of the limited amount of specimen that can be obtained, necrosis, and the fragility of VRL cells (165, 166). The vitrectomy cell block technique can improve diagnostic yield and can be utilized for immunohistochemistry of pan B-cell markers, including CD20 and CD79a, to establish a definitive diagnosis of lymphoma (167–169).
Several supplementary diagnostic methods can improve the definitive diagnosis of VRL. They include cytokine analysis to determine the ratio of IL-10 to IL-6 (i.e., the IL-10/ IL-6 ratio) (170), molecular analysis of the immunoglobulin heavy (IgH)/ TCR chain gene to confirm monoclonality, and flow cytometric identification of cell surface markers (171, 172). However, clonal expansions of lymphocytes have not been circumscribed in VRL. Therefore, molecular analysis with polymerase chain reaction and flow cytometric identification can sometimes yield false-positive results (173, 174).
Furthermore, MYD88 L265P can be screened with new genetic techniques, including allele-specific polymerase chain reaction and next generation sequencing (NGS) using an oncogene gene panels, which allows for lower cellularity or a smaller volume of samples to confirm the definitive diagnosis of VRL (175, 176).
As an exogenous factor, infection with the Epstein-Barr virus (EBV) is associated with PCNSL, specifically in immunocompromised patients such as individuals with acquired immune deficiency syndrome (AIDS) (177). EBV, which is a ubiquitous human herpes virus, affects most of the human population. EBV infects humans mostly in childhood and early adulthood, and subsequently spreads to B-lymphocytes and exists in a latent state. In patients with impaired cell-mediated immunity, such as patients with immune suppression and the elderly with immunosenescence, latent EBV may proliferate indiscriminately and drive neoplastic transformation to lymphoid malignancy (178). EBV, Toxoplasma gondii, and human herpes virus 8 are speculated as a cause of PVRL due to the detection of their gene expression in the intraocular fluids of some patients with PVRL (179).
On account that PVRL has selective tropism to CNS lesions, a theory has been proposed that chemokines and their receptors encourage the attraction and maintenance of VRL cells in intraocular tissues. In patients with B-cell chemokines, CXCL12, and CXCL13 are specifically expressed in retinal pigment epithelial cells and/or in the vitreous cavity of patients with VRL. As a consequence, B-lymphoma cells [which express CXCR4 and CXCR5 (i.e., receptors for CXCL12 and CXCL13)] are recruited into intraocular tissues such as the retina, vitreous body, and subretina (180). CXCL13 levels are also increased in the vitreous humor of patients with VRL (181). CCL19 derived from astrocytes was recently reported to promote the retention of lymphoma cells, which express CCR7, and subsequent tumor growth in chronic gliosis lesions in mice (182).
VRL cells can evade attacks by CTLs and NK cells because the eye is an immune-privileged site that possesses an immunosuppressive ocular microenvironment composed of soluble and cell surface inhibitory molecules. TGF-β is abundant in the vitreous humor to maintain an anti-inflammatory state in the eye (8). IL-10, which is widely regarded and has been analyzed as an immunosuppressive cytokine, has a pivotal role in the induction of immune tolerance in the eye (183). In cancer, regulatory cytokines in the TME such as IL-10 and TGF-β support tumor development by suppressing antitumor immunity in cancer (184, 185). Treg cells, which are a highly suppressive subset of T cells, increase regulatory cytokines secretion for the maintenance of self-tolerance and inhibition of autoimmunity, which result in tumor development (186, 187). In B-cell malignancies, including systemic DLBCL, the TME, which is formed with the reactive T-cells, macrophages, stromal cells, blood vessels, and extracellular matrix, regulates tumor cell survival or proliferation, and immune evasion for treatment resistance, associated with worse prognosis (188). Vitreous samples of B-cell VRL contain a large number of benign T cells and macrophages as well as tumor cells (174), which suggests that the infiltrating immune cells form the TME and support the suppression of anti-tumor immunity in the vitreous body.
Based on the outcome of the genetic studies described previously, several new agents are currently being investigated as a salvage therapy in clinical trials assessing in patients with refractory or relapsed PCNSL and PVRL. In a prospective French multicenter phase II trial, monotherapy with ibrutinib, which targets Bruton's tyrosine kinase downstream of BCR, was effective and had objective response rates (ORRs) of up to 70% (189). Among 14 PVRL patients, the median progression-free survival (PFS) was 22.7 months. The median overall survival was not estimated because more than one-half of the PVRL patients were alive (Table 2). Immunomodulatory agent monotherapy with lenalidomide, which inhibits the NF-kB and PI3K/AKT pathways, achieved ORRs of up to 64% (192). However, in a prospective clinical study (191), lenalidomide in combination with intravenous rituximab for refractory or relapsed PCNSL and PVRL maintained an ORR of 35.6% owing to the short response to the therapy. Among 11 PVRL patients, the median PFS was 9.2 months. Nivolumab, an anti-PD-1 agent, has been reported to have responses and maintain complete remission in patients with relapsed/refractory PCNSL (193). In the future, the combination of these agents with MTX-based chemotherapy should be assessed as the first-line therapy for PVRL.
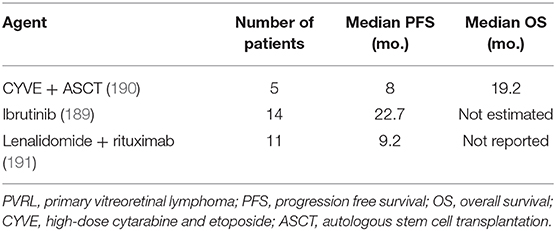
Table 2. Salvage treatment regimen for PVRL in prospective clinical trials.
Furthermore, increased expression of proinflammatory cytokines related to CTLs, such as IFN-γ, granzyme A, and IP-10 occurs in the aqueous humor and/or vitreous of VRL patients (181), which suggests that CTLs are associated with the pathogenesis of VRL. In a previous study, we revealed that subretinal infiltration of VRL cells elicits the infiltration of T-cells into the vitreous cavity (194). However, we were unable to elucidate the association of the T cells with the prognosis of the VRL patients. Little data exist that elucidate the roles of reactive T cells in DLBCL-VRL. In the future, further detailed studies on the infiltration of T cells into the eyes of VRL patients may provide new insights into the pathogenesis of the disease and deliver new therapeutic targets such as augmentation of CTL and/or NK cells function.
A profound understanding of the intricacies of immune responses will raise innovations for the management and treatment of these intractable retinal disorders. The generation of biologics, including IFX or ADA, has dramatically changed the treatment of NIU in the past few decades. However, during long-term treatment of NIU patients, a decreased response or adverse events to the biologics has emerged because of the development of antidrug antibodies or paradoxical effects. The development of selective small-molecule therapies is expected to resolve these problems. From the results of our analysis of EAU, the induction of regulatory DCs may be useful for the treatment of NIU.
In retinal vascular diseases, low-grade inflammation can destroy vascular integrity by the action of VEGF and some cytokines/chemokines from infiltrated leukocytes. Resistance to anti-VEGF therapy is sometimes observed in DR (including DME); therefore, developing a new therapy associated with low-grade inflammation as a “beyond VEGF” therapy for retinal vascular diseases, including DR and ROP, may be useful.
Immunological responses also affect the pathogenesis of RP, despite differences in genetic backgrounds. Targeting cytokines/chemokines associated with immunological responses against RP may be an attractive target for the treatment of RP, in addition to gene therapy.
The recent introduction of molecular profiling technologies, including NGS, can exhibit the molecular characterization of several cancers to provide information on tumor diagnosis and specific targeted therapy. Several agents, which were selected on the basis of the molecular characterization, have been assessed in clinical trials of cases of refractory/relapsed PCNSL; however, the utility of these molecular profiling technologies has not been established. Considering the rarity of VRL, large-scale collaborative registries, tumor molecular profiling programs, and clinical trials in institutions across the world are necessary to enhance diagnosis, prognostication, and treatment outcomes in the future.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work were supported by grants from Japan Society for the Promotion of Science (JSPS) KAKENHI Grant no. 18K09471 to AT, 18K09449 to RY, 19K09952 to YM, and 18H02956 to K-HS from the Ministry of Education, Science, Sports and Culture, Japan, and grants from Novartis Pharma Research Grants (AT; Tokyo, Japan) and Alcon Pharma Research Grants (AT; Tokyo, Japan).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Takeuchi M, Kezuka T, Sugita S, Keino H, Namba K, Kaburaki T, et al. Evaluation of the long-term efficacy and safety of infliximab treatment for uveitis in behcet's disease: a multicenter study. Ophthalmology. (2014) 121:1877–84. doi: 10.1016/j.ophtha.2014.04.042
2. Rosenfeld PJ, Brown DM, Heier JS, Boyer DS, Kaiser PK, Chung CY, et al. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. (2006) 355:1419–31. doi: 10.1056/NEJMoa054481
3. Heier JS, Korobelnik JF, Brown DM, Schmidt-Erfurth U, Do DV, Midena E, et al. Intravitreal aflibercept for diabetic macular edema: 148-week results from the VISTA and VIVID studies. Ophthalmology. (2016) 123:2376–85. doi: 10.1016/j.ophtha.2016.07.032
4. Garafalo AV, Cideciyan AV, Heon E, Sheplock R, Pearson A, WeiYang Yu C, et al. Progress in treating inherited retinal diseases: early subretinal gene therapy clinical trials and candidates for future initiatives. Prog Retin Eye Res. (2019) 30:100827. doi: 10.1016/j.preteyeres.2019.100827
5. Kreher S, Strehlow F, Martus P, Roth P, Hertenstein B, Roth A, et al. Prognostic impact of intraocular involvement in primary CNS lymphoma: experience from the G-PCNSL-SG1 trial. Ann Hematol. (2015) 94:409–14. doi: 10.1007/s00277-014-2212-z
6. Akiyama H, Takase H, Kubo F, Miki T, Yamamoto M, Tomita M, et al. High-dose methotrexate following intravitreal methotrexate administration in preventing central nervous system involvement of primary intraocular lymphoma. Cancer Sci. (2016) 107:1458–64. doi: 10.1111/cas.13012
7. Kaburaki T, Taoka K, Matsuda J, Yamashita H, Matsuda I, Tsuji H, et al. Combined intravitreal methotrexate and immunochemotherapy followed by reduced-dose whole-brain radiotherapy for newly diagnosed B-cell primary intraocular lymphoma. Br J Haematol. (2017) 179:246–55. doi: 10.1111/bjh.14848
8. Streilein JW. Ocular immune privilege: therapeutic opportunities from an experiment of nature. Nat Rev Immunol. (2003) 3:879–89. doi: 10.1038/nri1224
9. Taylor AW, Kaplan HJ. Ocular immune privilege in the year 2010: ocular immune privilege and uveitis. Ocul Immunol Inflamm. (2010) 18:488–92. doi: 10.3109/09273948.2010.525730
10. Benhar I, London A, Schwartz M. The privileged immunity of immune privileged organs: the case of the eye. Front Immunol. (2012) 3:296. doi: 10.3389/fimmu.2012.00296
11. Murakami Y, Ishikawa K, Nakao S, Sonoda KH. Innate immune response in retinal homeostasis and inflammatory disorders. Prog Retin Eye Res. (2019) 74:100778. doi: 10.1016/j.preteyeres.2019.100778
12. Antonetti DA, Barber AJ, Bronson SK, Freeman WM, Gardner TW, Jefferson LS, et al. Diabetic retinopathy: seeing beyond glucose-induced microvascular disease. Diabetes. (2006) 55:2401–11. doi: 10.2337/db05-1635
13. Sato T, Kusaka S, Shimojo H, Fujikado T. Simultaneous analyses of vitreous levels of 27 cytokines in eyes with retinopathy of prematurity. Ophthalmology. (2009) 116:2165–9. doi: 10.1016/j.ophtha.2009.04.026
14. Hori J, Yamaguchi T, Keino H, Hamrah P, Maruyama K. Immune privilege in corneal transplantation. Prog Retin Eye Res. (2019) 72:10075. doi: 10.1016/j.preteyeres.2019.04.002
15. Taylor AW, Ng TF. Negative regulators that mediate ocular immune privilege. J Leukoc Biol. (2018). 103, 1179–87. doi: 10.1002/JLB.3MIR0817-337R
16. Zhou R, Horai R, Mattapallil MJ, Caspi RR. A new look at immune privilege of the eye: dual role for the vision-related molecule retinoic acid. J Immunol. (2011) 187:4170–7. doi: 10.4049/jimmunol.1101634
17. Taylor AW. Ocular immune privilege and transplantation. Front Immunol. (2016) 7:37. doi: 10.3389/fimmu.2016.00037
18. Griffith TS, Brunner T, Fletcher SM, Green DR, Ferguson TA. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science. (1995) 270:1189–92. doi: 10.1126/science.270.5239.1189
19. Sugita S, Usui Y, Horie S, Futagami Y, Yamada Y, Ma J, et al. Human corneal endothelial cells expressing programmed death-ligand 1 (PD-L1) suppress PD-1+ T helper 1 cells by a contact-dependent mechanism. Invest Ophthalmol Vis Sci. (2009) 50:263–72. doi: 10.1167/iovs.08-2536
20. Toscano MA, Commodaro AG, Ilarregui JM, Bianco GA, Liberman A, Serra HM, et al. Galectin-1 suppresses autoimmune retinal disease by promoting concomitant Th2- and T regulatory-mediated anti-inflammatory responses. J Immunol. (2006) 176:6323–32. doi: 10.4049/jimmunol.176.10.6323
21. Dick AD, Carter D, Robertson M, Broderick C, Hughes E, Forrester JV, et al. Control of myeloid activity during retinal inflammation. J Leukoc Biol. (2003) 74:161–6. doi: 10.1189/jlb.1102535
22. Sugita S, Horie S, Nakamura O, Maruyama K, Takase H, Usui Y, et al. Acquisition of T regulatory function in cathepsin L-inhibited T cells by eye-derived CTLA-2alpha during inflammatory conditions. J Immunol. (2009) 183:5013–22. doi: 10.4049/jimmunol.0901623
23. Sugita S, Streilein JW. Iris pigment epithelium expressing CD86 (B7-2) directly suppresses T cell activation in vitro via binding to cytotoxic T lymphocyte-associated antigen 4. J Exp Med. (2003) 198:161–71. doi: 10.1084/jem.20030097
24. Sugita S, Ng TF, Lucas PJ, Gress RE, Streilein JW. B7+ iris pigment epithelium induce CD8+ T regulatory cells; both suppress CTLA-4+ T cells. J Immunol. (2006) 176:118–27. doi: 10.4049/jimmunol.176.1.118
25. Vendomele J, Khebizi Q, Fisson S. Cellular and molecular mechanisms of anterior chamber-associated immune deviation (ACAID): what we have learned from knockout mice. Front Immunol. (2017) 8:1686. doi: 10.3389/fimmu.2017.01686
26. Stein-Streilein J, Streilein JW. Anterior chamber associated immune deviation (ACAID): regulation, biological relevance, and implications for therapy. Int Rev Immunol. (2002) 21:123–52. doi: 10.1080/08830180212066
27. Goldschneider I, Cone RE. A central role for peripheral dendritic cells in the induction of acquired thymic tolerance. Trends Immunol. (2003) 24:77–81. doi: 10.1016/S1471-4906(02)00038-8
28. Simon AK, Hollander GA, McMichael A. Evolution of the immune system in humans from infancy to old age. Proc Biol Sci. (2015) 282:20143085. doi: 10.1098/rspb.2014.3085
29. Sonoda KH, Sakamoto T, Qiao H, Hisatomi T, Oshima T, Tsutsumi-Miyahara C, et al. The analysis of systemic tolerance elicited by antigen inoculation into the vitreous cavity: vitreous cavity-associated immune deviation. Immunology. (2005) 116:390–9. doi: 10.1111/j.1365-2567.2005.02239.x
30. Sakamoto T, Ishibashi T. Hyalocytes: essential cells of the vitreous cavity in vitreoretinal pathophysiology? Retina. (2011) 31:222–8. doi: 10.1097/IAE.0b013e3181facfa9
31. Qiao H, Hisatomi T, Sonoda KH, Kura S, Sassa Y, Kinoshita S, et al. The characterisation of hyalocytes: the origin, phenotype, and turnover. Br J Ophthalmol. (2005) 89:513–7. doi: 10.1136/bjo.2004.050658
32. Barisani-Asenbauer T, Maca SM, Mejdoubi L, Emminger W, Machold K, Auer H. Uveitis- a rare disease often associated with systemic diseases and infections- a systematic review of 2619 patients. Orphanet J Rare Dis. (2012) 7:57. doi: 10.1186/1750-1172-7-57
33. Takeda A, Sonoda KH, Ishibashi T. The regulation of the differentiation of Th1 and Th17 cells in uveitis. Inflam Regen. (2013) 33:261–8. doi: 10.2492/inflammregen.33.261
34. Hasegawa E, Takeda A, Yawata N, Sonoda KH. The effectiveness of adalimumab treatment for non-infectious uveitis. Immunol Med. (2019) 42:79–83. doi: 10.1080/25785826.2019.1642080
35. Foster CS, Kothari S, Anesi SD, Vitale AT, Chu D, Metzinger JL, et al. The ocular immunology and uveitis foundation preferred practice patterns of uveitis management. Surv Ophthalmol. (2016) 61:1–17. doi: 10.1016/j.survophthal.2015.07.001
36. Mesquida M, Molins B, Llorenc V, Hernandez MV, Espinosa G, Dick AD, et al. Current and future treatments for behcet's uveitis: road to remission. Int Ophthalmol. (2014) 34:365–81. doi: 10.1007/s10792-013-9788-5
37. Arida A, Sfikakis PP. Anti-cytokine biologic treatment beyond anti-TNF in behcet's disease. Clin Exp Rheumatol. (2014) 32(4 Suppl 84):S149–55.
38. Okada AA, Goto H, Ohno S, Mochizuki M. Multicenter study of infliximab for refractory uveoretinitis in behcet disease. Arch Ophthalmol. (2012) 130:592–8. doi: 10.1001/archophthalmol.2011.2698
39. Kuroyanagi K, Sakai T, Kohno H, Okano K, Akiyama G, Aoyagi R, et al. Association between the major histocompatibility complex and clinical response to infliximab therapy in patients with Behcet uveitis. Jpn J Ophthalmol. (2015) 59:401–8. doi: 10.1007/s10384-015-0404-2
40. Dick AD, Rosenbaum JT, Al-Dhibi HA, Belfort R Jr, Brezin AP, Chee SP, et al. Guidance on noncorticosteroid systemic immunomodulatory therapy in noninfectious uveitis: fundamentals of care for uveitiS (FOCUS) Initiative. Ophthalmology. (2018) 125:757–73. doi: 10.1016/j.ophtha.2017.11.017
41. Ramos-Casals M, Brito-Zeron P, Munoz S, Soria N, Galiana D, Bertolaccini L, et al. Autoimmune diseases induced by TNF-targeted therapies: analysis of 233 cases. Medicine. (2007) 86:242–51. doi: 10.1097/MD.0b013e3181441a68
42. Rutgeerts P, Feagan BG, Lichtenstein GR, Mayer LF, Schreiber S, Colombel JF, et al. Comparison of scheduled and episodic treatment strategies of infliximab in crohn's disease. Gastroenterology. (2004) 126:402–13. doi: 10.1053/j.gastro.2003.11.014
43. van der Laken CJ, Voskuyl AE, Roos JC, Stigter van Walsum M, de Groot ER, Wolbink G, et al. Imaging and serum analysis of immune complex formation of radiolabelled infliximab and anti-infliximab in responders and non-responders to therapy for rheumatoid arthritis. Ann Rheum Dis. (2007) 66:253–6. doi: 10.1136/ard.2006.057406
44. Bartelds GM, Krieckaert CL, Nurmohamed MT, van Schouwenburg PA, Lems WF, Twisk JW, et al. Development of antidrug antibodies against adalimumab and association with disease activity and treatment failure during long-term follow-up. JAMA. (2011) 305:1460–8. doi: 10.1001/jama.2011.406
45. Diedrichs-Mohring M, Kaufmann U, Wildner G. The immunopathogenesis of chronic and relapsing autoimmune uveitis - lessons from experimental rat models. Prog Retin Eye Res. (2018) 65:107–26. doi: 10.1016/j.preteyeres.2018.02.003
46. Mochizuki M, Sugita S, Kamoi K. Immunological homeostasis of the eye. Prog Retin Eye Res. (2013) 33:10–27. doi: 10.1016/j.preteyeres.2012.10.002
47. Yoshimura T, Sonoda KH, Miyazaki Y, Iwakura Y, Ishibashi T, Yoshimura A, et al. Differential roles for IFN-gamma and IL-17 in experimental autoimmune uveoretinitis. Int Immunol. (2008) 20:209–14. doi: 10.1093/intimm/dxm135
48. Luger D, Silver PB, Tang J, Cua D, Chen Z, Iwakura Y, et al. Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J Exp Med. (2008) 205:799–810. doi: 10.1084/jem.20071258
49. Amadi-Obi A, Yu CR, Liu X, Mahdi RM, Clarke GL, Nussenblatt RB, et al. TH17 cells contribute to uveitis and scleritis and are expanded by IL-2 and inhibited by IL-27/STAT1. Nat Med. (2007) 13:711–8. doi: 10.1038/nm1585
50. Okunuki Y, Mukai R, Nakao T, Tabor SJ, Butovsky O, Dana R, et al. Retinal microglia initiate neuroinflammation in ocular autoimmunity. Proc Natl Acad Sci USA. (2019) 116:9989–98. doi: 10.1073/pnas.1820387116
51. Satoh M, Namba KI, Kitaichi N, Endo N, Kitamei H, Iwata D, et al. Invariant natural killer T cells play dual roles in the development of experimental autoimmune uveoretinitis. Exp Eye Res. (2016) 153:79–89. doi: 10.1016/j.exer.2016.10.003
52. Fu Q, Man X, Wang X, Song N, Li Y, Xue J, et al. CD83(+) CCR7(+) NK cells induced by interleukin 18 by dendritic cells promote experimental autoimmune uveitis. J Cell Mol Med. (2019) 23:1827–39. doi: 10.1111/jcmm.14081
53. Lin W, Man X, Li P, Song N, Yue Y, Li B, et al. NK cells are negatively regulated by sCD83 in experimental autoimmune uveitis. Sci Rep. (2017) 7:12895. doi: 10.1038/s41598-017-13412-1
54. Chong WP, van Panhuys N, Chen J, Silver PB, Jittayasothorn Y, Mattapallil MJ, et al. NK-DC crosstalk controls the autopathogenic Th17 response through an innate IFN-gamma-IL-27 axis. J Exp Med. (2015) 212:1739–52. doi: 10.1084/jem.20141678
55. Kitaichi N, Namba K, Taylor AW. Inducible immune regulation following autoimmune disease in the immune-privileged eye. J Leukoc Biol. (2005) 77:496–502. doi: 10.1189/jlb.0204114
56. Silver PB, Horai R, Chen J, Jittayasothorn Y, Chan CC, Villasmil R, et al. Retina-specific T regulatory cells bring about resolution and maintain remission of autoimmune uveitis. J Immunol. (2015) 194:3011–9. doi: 10.4049/jimmunol.1402650
57. Muhammad F, Wang D, Montieth A, Lee S, Preble J, Foster CS, et al. PD-1(+) melanocortin receptor dependent-Treg cells prevent autoimmune disease. Sci Rep. (2019) 9:16941. doi: 10.1038/s41598-019-53297-w
58. Merida S, Palacios E, Navea A, Bosch-Morell F. Macrophages and uveitis in experimental animal models. Mediators Inflamm. (2015) 2015:671417. doi: 10.1155/2015/671417
59. Chen M, Zhao J, Ali IHA, Marry S, Augustine J, Bhuckory M, et al. Cytokine signaling protein 3 deficiency in myeloid cells promotes retinal degeneration and angiogenesis through arginase-1 up-regulation in experimental autoimmune uveoretinitis. Am J Pathol. (2018) 188:1007–20. doi: 10.1016/j.ajpath.2017.12.021
60. Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. (2005) 8:752–8. doi: 10.1038/nn1472
61. Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. (2009) 10:241–7. doi: 10.1038/ni.1703
62. Surprenant A, Rassendren F, Kawashima E, North RA, Buell G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science. (1996) 272:735–8. doi: 10.1126/science.272.5262.735
63. Di Virgilio F, Chiozzi P, Ferrari D, Falzoni S, Sanz JM, Morelli A, et al. Nucleotide receptors: an emerging family of regulatory molecules in blood cells. Blood. (2001) 97:587–600. doi: 10.1182/blood.V97.3.587
64. Takeda A, Yamada H, Hasegawa E, Arima M, Notomi S, Myojin S, et al. Crucial role of P2X7 receptor for effector T cell activation in experimental autoimmune uveitis. Jpn J Ophthalmol. (2018) 62:398–406. doi: 10.1007/s10384-018-0587-4
65. Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. (2007) 449:419–26. doi: 10.1038/nature06175
66. Steinman RM. Lasker basic medical research award. Dendritic cells: versatile controllers of the immune system. Nat Med. (2007) 13:1155–9. doi: 10.1038/nm1643
67. Lin W, Liu T, Wang B, Bi H. The role of ocular dendritic cells in uveitis. Immunol Lett. (2019) 209:4–10. doi: 10.1016/j.imlet.2019.03.016
68. Xu H, Dawson R, Forrester JV, Liversidge J. Identification of novel dendritic cell populations in normal mouse retina. Invest Ophthalmol Vis Sci. (2007) 48:1701–10. doi: 10.1167/iovs.06-0697
69. Heuss ND, Lehmann U, Norbury CC, McPherson SW, Gregerson DS. Local activation of dendritic cells alters the pathogenesis of autoimmune disease in the retina. J Immunol. (2012) 188:1191–200. doi: 10.4049/jimmunol.1101621
70. Van Brussel I, Lee WP, Rombouts M, Nuyts AH, Heylen M, De Winter BY, et al. Tolerogenic dendritic cell vaccines to treat autoimmune diseases: can the unattainable dream turn into reality? Autoimmun Rev. (2014) 13:138–50. doi: 10.1016/j.autrev.2013.09.008
71. van Rhijn-Brouwer FC, Gremmels H, Fledderus JO, Radstake TR, Verhaar MC, van Laar JM. Cellular therapies in systemic sclerosis: recent progress. Curr Rheumatol Rep. (2016) 18:12. doi: 10.1007/s11926-015-0555-7
72. Mackern-Oberti JP, Llanos C, Vega F, Salazar-Onfray F, Riedel CA, Bueno SM, et al. Role of dendritic cells in the initiation, progress and modulation of systemic autoimmune diseases. Autoimmun Rev. (2015) 14:127–39. doi: 10.1016/j.autrev.2014.10.010
73. Oh K, Kim YS, Lee DS. Maturation-resistant dendritic cells ameliorate experimental autoimmune uveoretinitis. Immune Netw. (2011) 11:399–405. doi: 10.4110/in.2011.11.6.399
74. Suzuki J, Yoshimura T, Simeonova M, Takeuchi K, Murakami Y, Morizane Y, et al. Aminoimidazole carboxamide ribonucleotide ameliorates experimental autoimmune uveitis. Invest Ophthalmol Vis Sci. (2012) 53:4158–69. doi: 10.1167/iovs.11-9323
75. Usui Y, Takeuchi M, Hattori T, Okunuki Y, Nagasawa K, Kezuka T, et al. Suppression of experimental autoimmune uveoretinitis by regulatory dendritic cells in mice. Arch Ophthalmol. (2009) 127:514–9. doi: 10.1001/archophthalmol.2009.34
76. Shoda H, Yanai R, Yoshimura T, Nagai T, Kimura K, Sobrin L, et al. Dietary omega-3 fatty acids suppress experimental autoimmune uveitis in association with inhibition of Th1 and Th17 cell function. PLoS ONE. (2015) 10:e0138241. doi: 10.1371/journal.pone.0138241
77. Uchi SH, Yanai R, Kobayashi M, Hatano M, Kobayashi Y, Yamashiro C, et al. Dendritic cells mediate the anti-inflammatory action of omega-3 long-chain polyunsaturated fatty acids in experimental autoimmune uveitis. PLoS ONE. (2019) 14:e0219405. doi: 10.1371/journal.pone.0219405
78. Heink S, Yogev N, Garbers C, Herwerth M, Aly L, Gasperi C, et al. Trans-presentation of IL-6 by dendritic cells is required for the priming of pathogenic TH17 cells. Nat Immunol. (2017) 18:74–85. doi: 10.1038/ni.3632
79. Heufler C, Koch F, Stanzl U, Topar G, Wysocka M, Trinchieri G, et al. Interleukin-12 is produced by dendritic cells and mediates T helper 1 development as well as interferon-gamma production by T helper 1 cells. Eur J Immunol. (1996) 26:659–68. doi: 10.1002/eji.1830260323
80. Guariguata L, Whiting DR, Hambleton I, Beagley J, Linnenkamp U, Shaw JE. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pract. (2014) 103:137–49. doi: 10.1016/j.diabres.2013.11.002
81. Cheung N, Mitchell P, Wong TY. Diabetic retinopathy. Lancet. (2010) 376:124–36. doi: 10.1016/S0140-6736(09)62124-3
82. Leasher JL, Bourne RR, Flaxman SR, Jonas JB, Keeffe J, Naidoo K, et al. Global estimates on the number of people blind or visually impaired by diabetic retinopathy: a meta-analysis from 1990 to 2010. Diabetes Care. (2016) 39:1643–9. doi: 10.2337/dc15-2171
83. Chibber R, Ben-Mahmud BM, Chibber S, Kohner EM. Leukocytes in diabetic retinopathy. Curr Diabetes Rev. (2007) 3:3–14. doi: 10.2174/157339907779802139
84. Capitao M, Soares R. Angiogenesis and inflammation crosstalk in diabetic retinopathy. J Cell Biochem. (2016) 117:2443–53. doi: 10.1002/jcb.25575
85. Roy S, Kern TS, Song B, Stuebe C. Mechanistic insights into pathological changes in the diabetic retina: implications for targeting diabetic retinopathy. Am J Pathol. (2017) 187:9–19. doi: 10.1016/j.ajpath.2016.08.022
86. van Hecke MV, Dekker JM, Nijpels G, Moll AC, Heine RJ, Bouter LM, et al. Inflammation and endothelial dysfunction are associated with retinopathy: the hoorn study. Diabetologia. (2005) 48:1300–6. doi: 10.1007/s00125-005-1799-y
87. Nakao S, Arima M, Ishikawa K, Kohno R, Kawahara S, Miyazaki M, et al. Intravitreal anti-VEGF therapy blocks inflammatory cell infiltration and re-entry into the circulation in retinal angiogenesis. Invest Ophthalmol Vis Sci. (2012) 53:4323–8. doi: 10.1167/iovs.11-9119
88. Esser P, Heimann K, Wiedemann P. Macrophages in proliferative vitreoretinopathy and proliferative diabetic retinopathy: differentiation of subpopulations. Br J Ophthalmol. (1993) 77:731–3. doi: 10.1136/bjo.77.11.731
89. Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. (2011) 11:750–61. doi: 10.1038/nri3088
90. Zhou Y, Yoshida S, Nakao S, Yoshimura T, Kobayashi Y, Nakama T, et al. M2 macrophages enhance pathological neovascularization in the mouse model of oxygen-induced retinopathy. Invest Ophthalmol Vis Sci. (2015) 56:4767–77. doi: 10.1167/iovs.14-16012
91. Kobayashi Y, Yoshida S, Nakama T, Zhou Y, Ishikawa K, Arita R, et al. Overexpression of CD163 in vitreous and fibrovascular membranes of patients with proliferative diabetic retinopathy: possible involvement of periostin. Br J Ophthalmol. (2015) 99:451–6. doi: 10.1136/bjophthalmol-2014-305321
92. Ishikawa K, Yoshida S, Nakao S, Nakama T, Kita T, Asato R, et al. Periostin promotes the generation of fibrous membranes in proliferative vitreoretinopathy. Faseb J. (2014) 28:131–42. doi: 10.1096/fj.13-229740
93. Dejana E, Tournier-Lasserve E, Weinstein BM. The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev Cell. (2009) 16:209–21. doi: 10.1016/j.devcel.2009.01.004
94. Klaassen I, Van Noorden CJ, Schlingemann RO. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog Retin Eye Res. (2013) 34:19–48. doi: 10.1016/j.preteyeres.2013.02.001
95. Funatsu H, Noma H, Mimura T, Eguchi S, Hori S. Association of vitreous inflammatory factors with diabetic macular edema. Ophthalmology. (2009) 116:73–9. doi: 10.1016/j.ophtha.2008.09.037
96. Siemerink MJ, Augustin AJ, Schlingemann RO. Mechanisms of ocular angiogenesis and its molecular mediators. Dev Ophthalmol. (2010) 46:4–20. doi: 10.1159/000320006
97. Bressler SB, Ayala AR, Bressler NM, Melia M, Qin H, Ferris FL, et al. Persistent macular thickening after ranibizumab treatment for diabetic macular edema with vision impairment. JAMA Ophthalmol. (2016) 134:278–85. doi: 10.1001/jamaophthalmol.2015.5346
98. Sfikakis PP, Markomichelakis N, Theodossiadis GP, Grigoropoulos V, Katsilambros N, Theodossiadis PG. Regression of sight-threatening macular edema in type 2 diabetes following treatment with the anti-tumor necrosis factor monoclonal antibody infliximab. Diabetes Care. (2005) 28:445–7. doi: 10.2337/diacare.28.2.445
99. Gale JD, Berger B, Gilbert S, Popa S, Sultan MB, Schachar RA, et al. A CCR2/5 inhibitor, PF-04634817, is inferior to monthly ranibizumab in the treatment of diabetic macular edema. Invest Ophthalmol Vis Sci. (2018) 59:2659–69. doi: 10.1167/iovs.17-22731
100. Hillier RJ, Ojaimi E, Wong DT, Mak MYK, Berger AR, Kohly RP, et al. Aqueous humor cytokine levels and anatomic response to intravitreal ranibizumab in diabetic macular edema. JAMA Ophthalmol. (2018) 136:382–8. doi: 10.1001/jamaophthalmol.2018.0179
101. Shimura M, Yasuda K, Motohashi R, Kotake O, Noma H. Aqueous cytokine and growth factor levels indicate response to ranibizumab for diabetic macular oedema. Br J Ophthalmol. (2017) 101:1518–23. doi: 10.1136/bjophthalmol-2016-309953
102. Felfeli T, Juncal VR, Hillier RJ, Mak MYK, Wong DT, Berger AR, et al. Aqueous humor cytokines and long-term response to anti-vascular endothelial growth factor therapy in diabetic macular edema. Am J Ophthalmol. (2019) 206:176–83. doi: 10.1016/j.ajo.2019.04.002
103. Noda K, Nakao S, Zandi S, Sun D, Hayes KC, Hafezi-Moghadam A. Retinopathy in a novel model of metabolic syndrome and type 2 diabetes: new insight on the inflammatory paradigm. Faseb J. (2014) 28:2038–46. doi: 10.1096/fj.12-215715
104. Rakoczy EP, Ali Rahman IS, Binz N, Li CR, Vagaja NN, de Pinho M, et al. Characterization of a mouse model of hyperglycemia and retinal neovascularization. Am J Pathol. (2010) 177:2659–70. doi: 10.2353/ajpath.2010.090883
105. Blencowe H, Lawn JE, Vazquez T, Fielder A, Gilbert C. Preterm-associated visual impairment and estimates of retinopathy of prematurity at regional and global levels for 2010. Pediatr Res. (2013) 74(Suppl 1):35–49. doi: 10.1038/pr.2013.205
106. Stoll BJ, Hansen NI, Bell EF, Walsh MC, Carlo WA, Shankaran S, et al. Trends in care practices, morbidity, and mortality of extremely preterm neonates, 1993-2012. JAMA. (2015) 314:1039–51. doi: 10.1001/jama.2015.10244
107. Quinn GE, Barr C, Bremer D, Fellows R, Gong A, Hoffman R, et al. Changes in course of retinopathy of prematurity from 1986 to 2013: comparison of three studies in the united states. Ophthalmology. (2016) 123:1595–600. doi: 10.1016/j.ophtha.2016.03.026
108. Hellstrom A, Smith LE, Dammann O. Retinopathy of prematurity. Lancet. (2013) 382:1445–57. doi: 10.1016/S0140-6736(13)60178-6
109. Good WV, Early Treatment for Retinopathy of Prematurity Cooperative G. Final results of the early treatment for retinopathy of prematurity (ETROP) randomized trial. Trans Am Ophthalmol Soc. (2004) 102:233–48; discussion 48–50.
110. Mintz-Hittner HA, Kennedy KA, Chuang AZ, Group B-RC. Efficacy of intravitreal bevacizumab for stage 3+ retinopathy of prematurity. N Engl J Med. (2011) 364:603–15. doi: 10.1056/NEJMoa1007374
111. Stahl A, Lepore D, Fielder A, Fleck B, Reynolds JD, Chiang MF, et al. Ranibizumab versus laser therapy for the treatment of very low birthweight infants with retinopathy of prematurity (RAINBOW): an open-label randomised controlled trial. Lancet. (2019) 394:1551–9. doi: 10.1016/S0140-6736(19)31344-3
112. Lyu J, Zhang Q, Jin H, Xu Y, Chen C, Ji X, et al. Aqueous cytokine levels associated with severity of type 1 retinopathy of prematurity and treatment response to ranibizumab. Graefes Arch Clin Exp Ophthalmol. (2018) 256:1469–77. doi: 10.1007/s00417-018-4034-5
113. Ishikawa K, Yoshida S, Nakao S, Sassa Y, Asato R, Kohno R, et al. Bone marrow-derived monocyte lineage cells recruited by MIP-1beta promote physiological revascularization in mouse model of oxygen-induced retinopathy. Lab Invest. (2012) 92:91–101. doi: 10.1038/labinvest.2011.141
114. Matsuda K, Okamoto N, Kondo M, Arkwright PD, Karasawa K, Ishizaka S, et al. Mast cell hyperactivity underpins the development of oxygen-induced retinopathy. J Clin Invest. (2017) 127:3987–4000. doi: 10.1172/JCI89893
115. Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. (2006) 368:1795–809. doi: 10.1016/S0140-6736(06)69740-7
116. Maguire AM, Russell S, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy, safety, and durability of voretigene neparvovec-rzyl in RPE65 mutation-associated inherited retinal dystrophy: results of phase 1 and 3 trials. Ophthalmology. (2019) 126:1273–85. doi: 10.1016/j.ophtha.2019.06.017
117. Ait-Ali N, Fridlich R, Millet-Puel G, Clerin E, Delalande F, Jaillard C, et al. Rod-derived cone viability factor promotes cone survival by stimulating aerobic glycolysis. Cell. (2015) 161:817–32. doi: 10.1016/j.cell.2015.03.023
118. Petit L, Ma S, Cipi J, Cheng SY, Zieger M, Hay N, et al. Aerobic glycolysis is essential for normal rod function and controls secondary cone death in retinitis pigmentosa. Cell Rep. (2018) 23:2629–42. doi: 10.1016/j.celrep.2018.04.111
119. Campochiaro PA, Mir TA. The mechanism of cone cell death in retinitis pigmentosa. Prog Retin Eye Res. (2018) 62:24–37. doi: 10.1016/j.preteyeres.2017.08.004
120. Punzo C, Xiong W, Cepko CL. Loss of daylight vision in retinal degeneration: are oxidative stress and metabolic dysregulation to blame? J Biol Chem. (2012) 287:1642–8. doi: 10.1074/jbc.R111.304428
121. Yoshida N, Ikeda Y, Notomi S, Ishikawa K, Murakami Y, Hisatomi T, et al. Clinical evidence of sustained chronic inflammatory reaction in retinitis pigmentosa. Ophthalmology. (2013) 120:100–5. doi: 10.1016/j.ophtha.2012.07.006
122. Gadani SP, Walsh JT, Lukens JR, Kipnis J. Dealing with danger in the CNS: the response of the immune system to injury. Neuron. (2015) 87:47–62. doi: 10.1016/j.neuron.2015.05.019
123. Newsome DA, Michels RG. Detection of lymphocytes in the vitreous gel of patients with retinitis pigmentosa. Am J Ophthalmol. (1988) 105:596–602. doi: 10.1016/0002-9394(88)90050-5
124. Murakami Y, Yoshida N, Ikeda Y, Nakatake S, Fujiwara K, Notomi S, et al. Relationship between aqueous flare and visual function in retinitis pigmentosa. Am J Ophthalmol. (2015) 159:958–63 e1. doi: 10.1016/j.ajo.2015.02.001
125. Nishiguchi KM, Yokoyama Y, Kunikata H, Abe T, Nakazawa T. Correlation between aqueous flare and residual visual field area in retinitis pigmentosa. Br J Ophthalmol. (2019) 103:475–80. doi: 10.1136/bjophthalmol-2018-312225
126. Nagasaka Y, Ito Y, Ueno S, Terasaki H. Increased aqueous flare is associated with thickening of inner retinal layers in eyes with retinitis pigmentosa. Sci Rep. (2016) 6:33921. doi: 10.1038/srep33921
127. Lu B, Yin H, Tang Q, Wang W, Luo C, Chen X, et al. Multiple cytokine analyses of aqueous humor from the patients with retinitis pigmentosa. Cytokine. (2020) 127:154943. doi: 10.1016/j.cyto.2019.154943
128. Seddon JM, Gensler G, Milton RC, Klein ML, Rifai N. Association between C-reactive protein and age-related macular degeneration. JAMA. (2004) 291:704–10. doi: 10.1001/jama.291.6.704
129. Laursen JV, Hoffmann SS, Green A, Nybo M, Sjolie AK, Grauslund J. Associations between diabetic retinopathy and plasma levels of high-sensitive C-reactive protein or von Willebrand factor in long-term type 1 diabetic patients. Curr Eye Res. (2013) 38:174–9. doi: 10.3109/02713683.2012.713153
130. Murakami Y, Ikeda Y, Nakatake S, Fujiwara K, Tachibana T, Yoshida N, et al. C-Reactive protein and progression of vision loss in retinitis pigmentosa. Acta Ophthalmol. (2018) 96:e174–e9. doi: 10.1111/aos.13502
131. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. (2010) 330:841–5. doi: 10.1126/science.1194637
132. Zhang Y, Zhao L, Wang X, Ma W, Lazere A, Qian HH, et al. Repopulating retinal microglia restore endogenous organization and function under CX3CL1-CX3CR1 regulation. Sci Adv. (2018) 4:eaap8492. doi: 10.1126/sciadv.aap8492
133. Ginhoux F, Lim S, Hoeffel G, Low D, Huber T. Origin and differentiation of microglia. Front Cell Neurosci. (2013) 7:45. doi: 10.3389/fncel.2013.00045
134. Paschalis EI, Lei F, Zhou C, Kapoulea V, Dana R, Chodosh J, et al. Permanent neuroglial remodeling of the retina following infiltration of CSF1R inhibition-resistant peripheral monocytes. Proc Natl Acad Sci USA. (2018) 115:E11359–E68. doi: 10.1073/pnas.1807123115
135. Peng Y, Zhang Y, Huang B, Luo Y, Zhang M, Li K, et al. Survival and migration of pre-induced adult human peripheral blood mononuclear cells in retinal degeneration slow (rds) mice three months after subretinal transplantation. Curr Stem Cell Res Ther. (2014) 9:124–33. doi: 10.2174/1574888X09666131219115125
136. Zhao L, Zabel MK, Wang X, Ma W, Shah P, Fariss RN, et al. Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO Mol Med. (2015) 7:1179–97. doi: 10.15252/emmm.201505298
137. Zabel MK, Zhao L, Zhang Y, Gonzalez SR, Ma W, Wang X, et al. Microglial phagocytosis and activation underlying photoreceptor degeneration is regulated by CX3CL1-CX3CR1 signaling in a mouse model of retinitis pigmentosa. Glia. (2016) 64:1479–91. doi: 10.1002/glia.23016
138. Silverman LI, Dulatova G, Tandeski T, Erickson IE, Lundell B, Toplon D, et al. In vitro and in vivo evaluation of discogenic cells, an investigational cell therapy for disc degeneration. Spine J. (2020) 20:138–49. doi: 10.1016/j.spinee.2019.08.006
139. Shen J, Yang X, Dong A, Petters RM, Peng YW, Wong F, et al. Oxidative damage is a potential cause of cone cell death in retinitis pigmentosa. J Cell Physiol. (2005) 203:457–64. doi: 10.1002/jcp.20346
140. Komeima K, Rogers BS, Lu L, Campochiaro PA. Antioxidants reduce cone cell death in a model of retinitis pigmentosa. Proc Natl Acad Sci USA. (2006) 103:11300–5. doi: 10.1073/pnas.0604056103
141. Yoshida N, Ikeda Y, Notomi S, Ishikawa K, Murakami Y, Hisatomi T, et al. Laboratory evidence of sustained chronic inflammatory reaction in retinitis pigmentosa. Ophthalmology. (2013) 120:e5–12. doi: 10.1016/j.ophtha.2012.07.008
142. Roh MI, Murakami Y, Thanos A, Vavvas DG, Miller JW. Edaravone, an ROS scavenger, ameliorates photoreceptor cell death after experimental retinal detachment. Invest Ophthalmol Vis Sci. (2011) 52:3825–31. doi: 10.1167/iovs.10-6797
143. Nakatake S, Murakami Y, Ikeda Y, Morioka N, Tachibana T, Fujiwara K, et al. MUTYH promotes oxidative microglial activation and inherited retinal degeneration. JCI Insight. (2016) 1:e87781. doi: 10.1172/jci.insight.87781
144. McMurtrey JJ, Tso MOM. A review of the immunologic findings observed in retinitis pigmentosa. Surv Ophthalmol. (2018) 63:769–81. doi: 10.1016/j.survophthal.2018.03.002
145. Rohrer B, Demos C, Frigg R, Grimm C. Classical complement activation and acquired immune response pathways are not essential for retinal degeneration in the rd1 mouse. Exp Eye Res. (2007) 84:82–91. doi: 10.1016/j.exer.2006.08.017
146. Mishra A, Das B, Nath M, Iyer S, Kesarwani A, Bhattacharjee J, et al. A novel immunodeficient NOD.SCID-rd1 mouse model of retinitis pigmentosa to investigate potential therapeutics and pathogenesis of retinal degeneration. Biol Open. (2017) 6:449–62. doi: 10.1242/bio.021618
147. Chan CC, Sen HN. Current concepts in diagnosing‘ and managing primary vitreoretinal (intraocular) lymphoma. Discov Med. (2013) 15:93–100.
148. Mochizuki M, Singh AD. Epidemiology and clinical features of intraocular lymphoma. Ocul Immunol Inflamm. (2009) 17:69–72. doi: 10.1080/09273940902957305
149. Bardenstein DS. Intraocular lymphoma. Cancer Control. (1998) 5:317–25. doi: 10.1177/107327489800500403
150. Cao X, Shen D, Callanan DG, Mochizuki M, Chan CC. Diagnosis of systemic metastatic retinal lymphoma. Acta Ophthalmol. (2011) 89:e149–54. doi: 10.1111/j.1755-3768.2009.01797.x
151. Sagoo MS, Mehta H, Swampillai AJ, Cohen VM, Amin SZ, Plowman PN, et al. Primary intraocular lymphoma. Surv Ophthalmol. (2014) 59:503–16. doi: 10.1016/j.survophthal.2013.12.001
152. Venkatesh R, Bavaharan B, Mahendradas P, Yadav NK. Primary vitreoretinal lymphoma: prevalence, impact, and management challenges. Clin Ophthalmol. (2019) 13:353–64. doi: 10.2147/OPTH.S159014
153. Fend F, Ferreri AJ, Coupland SE. How we diagnose and treat vitreoretinal lymphoma. Br J Haematol. (2016) 173:680–92. doi: 10.1111/bjh.14025
154. Grommes C, Nayak L, Tun HW, Batchelor TT. Introduction of novel agents in the treatment of primary CNS lymphoma. Neuro Oncol. (2019) 21:306–13. doi: 10.1093/neuonc/noy193
155. Correa DD, Shi W, Abrey LE, Deangelis LM, Omuro AM, Deutsch MB, et al. Cognitive functions in primary CNS lymphoma after single or combined modality regimens. Neuro Oncol. (2012) 14:101–8. doi: 10.1093/neuonc/nor186
156. Kasenda B, Loeffler J, Illerhaus G, Ferreri AJ, Rubenstein J, Batchelor TT. The role of whole brain radiation in primary CNS lymphoma. Blood. (2016) 128:32–6. doi: 10.1182/blood-2016-01-650101
157. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. (2000) 403:503–11. doi: 10.1038/35000501
158. Pasqualucci L, Dalla-Favera R. The genetic landscape of diffuse large B-cell lymphoma. Semin Hematol. (2015) 52:67–76. doi: 10.1053/j.seminhematol.2015.01.005
159. Chapuy B, Roemer MG, Stewart C, Tan Y, Abo RP, Zhang L, et al. Targetable genetic features of primary testicular and primary central nervous system lymphomas. Blood. (2016) 127:869–81. doi: 10.1182/blood-2015-10-673236
160. Bonzheim I, Giese S, Deuter C, Susskind D, Zierhut M, Waizel M, et al. High frequency of MYD88 mutations in vitreoretinal B-cell lymphoma: a valuable tool to improve diagnostic yield of vitreous aspirates. Blood. (2015) 126:76–9. doi: 10.1182/blood-2015-01-620518
161. Yonese I, Takase H, Yoshimori M, Onozawa E, Tsuzura A, Miki T, et al. CD79B mutations in primary vitreoretinal lymphoma: Diagnostic and prognostic potential. Eur J Haematol. (2019) 102:191–6. doi: 10.1111/ejh.13191
162. Riemersma SA, Jordanova ES, Schop RF, Philippo K, Looijenga LH, Schuuring E, et al. Extensive genetic alterations of the HLA region, including homozygous deletions of HLA class II genes in B-cell lymphomas arising in immune-privileged sites. Blood. (2000) 96:3569–77. doi: 10.1182/blood.V96.10.3569
163. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. (2009) 206:3015–29. doi: 10.1084/jem.20090847
164. Liu Y, Yu Y, Yang S, Zeng B, Zhang Z, Jiao G, et al. Regulation of arginase I activity and expression by both PD-1 and CTLA-4 on the myeloid-derived suppressor cells. Cancer Immunol Immunother. (2009) 58:687–97. doi: 10.1007/s00262-008-0591-5
165. Davis JL, Miller DM, Ruiz P. Diagnostic testing of vitrectomy specimens. Am J Ophthalmol. (2005) 140:822–9. doi: 10.1016/j.ajo.2005.05.032
166. Kimura K, Usui Y, Goto H. Clinical features and diagnostic significance of the intraocular fluid of 217 patients with intraocular lymphoma. Jpn J Ophthalmol. (2012) 56:383–9. doi: 10.1007/s10384-012-0150-7
167. Ito T, Takeda A, Fujiwara K, Hasegawa E, Nakao S, Ohishi Y, et al. Risk factors for failure of vitrectomy cell block technique in cytological diagnosis of vitreoretinal lymphoma. Graefes Arch Clin Exp Ophthalmol. (2019) 257:1029–36. doi: 10.1007/s00417-019-04266-6
168. Intzedy L, Teoh SC, Hogan A, Mangwana S, Mayer EJ, Dick AD, et al. Cytopathological analysis of vitreous in intraocular lymphoma. Eye. (2008) 22:289–93. doi: 10.1038/sj.eye.6702965
169. Kase S, Namba K, Iwata D, Mizuuchi K, Kitaichi N, Tagawa Y, et al. Diagnostic efficacy of cell block method for vitreoretinal lymphoma. Diagn Pathol. (2016) 11:29. doi: 10.1186/s13000-016-0479-1
170. Whitcup SM, Stark-Vancs V, Wittes RE, Solomon D, Podgor MJ, Nussenblatt RB, et al. Association of interleukin 10 in the vitreous and cerebrospinal fluid and primary central nervous system lymphoma. Arch Ophthalmol. (1997) 115:1157–60. doi: 10.1001/archopht.1997.01100160327010
171. Baehring JM, Androudi S, Longtine JJ, Betensky RA, Sklar J, Foster CS, et al. Analysis of clonal immunoglobulin heavy chain rearrangements in ocular lymphoma. Cancer. (2005) 104:591–7. doi: 10.1002/cncr.21191
172. Davis JL, Viciana AL, Ruiz P. Diagnosis of intraocular lymphoma by flow cytometry. Am J Ophthalmol. (1997) 124:362–72. doi: 10.1016/S0002-9394(14)70828-1
173. Sugita S, Takase H, Sugamoto Y, Arai A, Miura O, Mochizuki M. Diagnosis of intraocular lymphoma by polymerase chain reaction analysis and cytokine profiling of the vitreous fluid. Jpn J Ophthalmol. (2009) 53:209–14. doi: 10.1007/s10384-009-0662-y
174. Davis JL, Ruiz P Jr, Shah M, Mandelcorn ED. Evaluation of the reactive T-cell infiltrate in uveitis and intraocular lymphoma with flow cytometry of vitreous fluid (an American Ophthalmological Society thesis). Trans Am Ophthalmol Soc. (2012) 110:117–29.
175. Tan WJ, Wang MM, Ricciardi-Castagnoli P, Tang T, Chee SP, Lim TS, et al. Single-cell MYD88 sequencing of isolated B cells from vitreous biopsies aids vitreoretinal lymphoma diagnosis. Blood. (2019) 134:709–12. doi: 10.1182/blood.2019000022
176. Cani AK, Hovelson DH, Demirci H, Johnson MW, Tomlins SA, Rao RC. Next generation sequencing of vitreoretinal lymphomas from small-volume intraocular liquid biopsies: new routes to targeted therapies. Oncotarget. (2017) 8:7989–98. doi: 10.18632/oncotarget.14008
177. MacMahon EM, Glass JD, Hayward SD, Mann RB, Becker PS, Charache P, et al. Epstein-Barr virus in AIDS-related primary central nervous system lymphoma. Lancet. (1991) 338:969–73. doi: 10.1016/0140-6736(91)91837-K
178. Crombie JL, LaCasce AS. Epstein barr virus associated B-cell lymphomas and iatrogenic lymphoproliferative disorders. Front Oncol. (2019) 9:109. doi: 10.3389/fonc.2019.00109
179. Chan CC. Molecular pathology of primary intraocular lymphoma. Trans Am Ophthalmol Soc. (2003) 101:275–92.
180. Chan CC, Shen D, Hackett JJ, Buggage RR, Tuaillon N. Expression of chemokine receptors, CXCR4 and CXCR5, and chemokines, BLC and SDF-1, in the eyes of patients with primary intraocular lymphoma. Ophthalmology. (2003) 110:421–6. doi: 10.1016/S0161-6420(02)01737-2
181. Usui Y, Wakabayashi Y, Okunuki Y, Kimura K, Tajima K, Matsuda R, et al. Immune mediators in vitreous fluids from patients with vitreoretinal B-cell lymphoma. Invest Ophthalmol Vis Sci. (2012) 53:5395–402. doi: 10.1167/iovs.11-8719
182. O'Connor T, Zhou X, Kosla J, Adili A, Garcia Beccaria M, Kotsiliti E, et al. Age-related gliosis promotes central nervous system lymphoma through CCL19-mediated tumor cell retention. Cancer Cell. (2019) 36:250–67 e9. doi: 10.1016/j.ccell.2019.08.001
183. Sonoda KH, Faunce DE, Taniguchi M, Exley M, Balk S, Stein-Streilein J. NK T cell-derived IL-10 is essential for the differentiation of antigen-specific T regulatory cells in systemic tolerance. J Immunol. (2001) 166:42–50. doi: 10.4049/jimmunol.166.1.42
184. Czarneski J, Lin YC, Chong S, McCarthy B, Fernandes H, Parker G, et al. Studies in NZB IL-10 knockout mice of the requirement of IL-10 for progression of B-cell lymphoma. Leukemia. (2004) 18:597–606. doi: 10.1038/sj.leu.2403244
185. Douglas RS, Capocasale RJ, Lamb RJ, Nowell PC, Moore JS. Chronic lymphocytic leukemia B cells are resistant to the apoptotic effects of transforming growth factor-beta. Blood. (1997) 89:941–7. doi: 10.1182/blood.V89.3.941
186. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. (2008) 8:523–32. doi: 10.1038/nri2343
187. Nishikawa H, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol. (2014) 27:1–7. doi: 10.1016/j.coi.2013.12.005
188. Fowler NH, Cheah CY, Gascoyne RD, Gribben J, Neelapu SS, Ghia P, et al. Role of the tumor microenvironment in mature B-cell lymphoid malignancies. Haematologica. (2016) 101:531–40. doi: 10.3324/haematol.2015.139493
189. Soussain C, Choquet S, Blonski M, Leclercq D, Houillier C, Rezai K, et al. Ibrutinib monotherapy for relapse or refractory primary CNS lymphoma and primary vitreoretinal lymphoma: final analysis of the phase II 'proof-of-concept' iLOC study by the Lymphoma study association (LYSA) and the French oculo-cerebral lymphoma (LOC) network. Eur J Cancer. (2019) 117:121–30. doi: 10.1016/j.ejca.2019.05.024
190. Soussain C, Hoang-Xuan K, Taillandier L, Fourme E, Choquet S, Witz F, et al. Intensive chemotherapy followed by hematopoietic stem-cell rescue for refractory and recurrent primary CNS and intraocular lymphoma: societe francaise de greffe de moelle osseuse-therapie cellulaire. J Clin Oncol. (2008) 26:2512–8. doi: 10.1200/JCO.2007.13.5533
191. Ghesquieres H, Chevrier M, Laadhari M, Chinot O, Choquet S, Molucon-Chabrot C, et al. Lenalidomide in combination with intravenous rituximab (REVRI) in relapsed/refractory primary CNS lymphoma or primary intraocular lymphoma: a multicenter prospective 'proof of concept' phase II study of the French oculo-cerebral lymphoma (LOC) network and the lymphoma study association (LYSA)dagger. Ann Oncol. (2019) 30:621–8. doi: 10.1093/annonc/mdz032
192. Houillier C, Choquet S, Touitou V, Martin-Duverneuil N, Navarro S, Mokhtari K, et al. Lenalidomide monotherapy as salvage treatment for recurrent primary CNS lymphoma. Neurology. (2015) 84:325–6. doi: 10.1212/WNL.0000000000001158
193. Nayak L, Iwamoto FM, LaCasce A, Mukundan S, Roemer MGM, Chapuy B, et al. PD-1 blockade with nivolumab in relapsed/refractory primary central nervous system and testicular lymphoma. Blood. (2017) 129:3071–3. doi: 10.1182/blood-2017-01-764209
Keywords: immune privilege, non-infectious uveitis, diabetic retinopathy, retinopathy of prematurity, retinitis pigmentosa, vitreoretinal lymphoma
Citation: Takeda A, Yanai R, Murakami Y, Arima M and Sonoda K-H (2020) New Insights Into Immunological Therapy for Retinal Disorders. Front. Immunol. 11:1431. doi: 10.3389/fimmu.2020.01431
Received: 11 March 2020; Accepted: 03 June 2020;
Published: 03 July 2020.
Edited by:
Andrew W. Taylor, Boston University School of Medicine, United StatesReviewed by:
Seyed Mahmoud Hashemi, Shahid Beheshti University of Medical Sciences, IranCopyright © 2020 Takeda, Yanai, Murakami, Arima and Sonoda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Atsunobu Takeda, YXRha2VkYTQxN0B5YWhvby5jby5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.