
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 02 July 2020
Sec. Inflammation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01409
 Vincent Gies1,2,3*
Vincent Gies1,2,3* Nassima Bekaddour4,5
Nassima Bekaddour4,5 Yannick Dieudonné1,2,6
Yannick Dieudonné1,2,6 Aurélien Guffroy1,2,6
Aurélien Guffroy1,2,6 Quentin Frenger1,7
Quentin Frenger1,7 Frédéric Gros1,7
Frédéric Gros1,7 Mathieu Paul Rodero4,5
Mathieu Paul Rodero4,5 Jean-Philippe Herbeuval4,5*†
Jean-Philippe Herbeuval4,5*† Anne-Sophie Korganow1,2,6*†
Anne-Sophie Korganow1,2,6*†As the world is severely affected by COVID-19 pandemic, the use of chloroquine and hydroxychloroquine in prevention or for the treatment of patients is allowed in multiple countries but remained at the center of much controversy in recent days. This review describes the properties of chloroquine and hydroxychloroquine, and highlights not only their anti-viral effects but also their important immune-modulatory properties and their well-known use in autoimmune diseases, including systemic lupus and arthritis. Chloroquine appears to inhibit in vitro SARS virus' replication and to interfere with SARS-CoV2 receptor (ACE2). Chloroquine and hydroxychloroquine impede lysosomal activity and autophagy, leading to a decrease of antigen processing and presentation. They are also known to interfere with endosomal Toll-like receptors signaling and cytosolic sensors of nucleic acids, which result in a decreased cellular activation and thereby a lower type I interferons and inflammatory cytokine secretion. Given the antiviral and anti-inflammatory properties of chloroquine and hydroxychloroquine, there is a rational to use them against SARS-CoV2 infection. However, the anti-interferon properties of these molecules might be detrimental, and impaired host immune responses against the virus. This duality could explain the discrepancy with the recently published studies on CQ/HCQ treatment efficacy in COVID-19 patients. Moreover, although these treatments could be an interesting potential strategy to limit progression toward uncontrolled inflammation, they do not appear per se sufficiently potent to control the whole inflammatory process in COVID-19, and more targeted and/or potent therapies should be required at least in add-on.
Chloroquine (CQ) and hydroxychloroquine (HCQ) are “old” drugs but are still widely used in very diverse situations, including infectious diseases (1–3), rheumatic/inflammatory diseases (4), or in clinical research protocols as add-on cancer therapy (5). Indeed, they are cheap and safe considering rare effective ocular toxicity (<2%) and acute cardiac toxicity. However, clinicians should be warned that CQ/HCQ treatments, especially with high doses, can be complicated with heart failure, or non-reversible conduction disorders (6).
As the world is severely affected by COVID-19 pandemic, the use of CQ/HCQ in prevention or for the treatment of patients is at the center of much controversy in recent days. We thus believe appropriate to make a short review and discuss about their pharmacological properties and the ways CQ/HCQ are able to interfere with innate or adaptative immune system.
HCQ and CQ are weak bases with a common flat aromatic core structure. These decades-old drugs are in fact synthetic antimalarial drugs, although HCQ is above all a major treatment for systemic lupus (7). The story of antimalarials starts with the cinchona bark that was already used by the Incas for its antipyretic property rather than malaria treatment itself. It was only in 1820, that Pelletier and Caventou, french pharmacists, isolated the fundamental antimalarial alkaloid: quinine. During world war II, American soldiers who fought in the pacific region received antimalarials (quinacrine) in prophylaxis, showing a beneficial effect of this compound on lupus and rheumatoid arthritis. CQ was subsequently introduced in 1943, showing its beneficial effect in systemic lupus erythematosus in 1953 (8). CQ cardiac and retinal side effects led to the development, in 1955, of a hydroxylated derivative: HCQ, a little less active but above all less toxic molecule (9, 10). HCQ holds actually a major place in the treatment of autoimmune/ inflammatory rheumatic or dermatological diseases.
Autophagy is a catabolic homeostatic or induced process that involves the sequestration of cytoplasmic components in double-membraned autophagosomes. Autophagosomes ultimately fuse with lysosomes leading to the degradation of their content. Autophagy is a major player in immunity (11). It contributes to reduce inflammation by modulating type I interferon production and inflammasome activity. Autophagy is a countermeasure to infectious diseases clearing intracellular pathogens but autophagic membrane can conversely constitute hubs for viral replication. Autophagy also contributes to generate antigens processed on MHC class II (12).
An important mode of action of HCQ is the inhibition of lysosomal activity (Figure 1 and Table 1). As weak bases (Figure S1A), CQ and HCQ accumulate in lysosomes (lysosomotropism) and inhibit their function. In vitro, CQ can disrupt the endolysosomal system and therefore destabilize the lysosomal membranes leading to intracellular release of lysosomal enzymes and impairment of autophagosome–lysosome fusion (13, 14). As a consequence LC3-II (microtubule-associated protein light chain 3-II), a lipidated protein normally associated with autophagosomes until degradation/recycling after fusion with lysosomes, accumulates under HCQ incubation (Figure S1B) (13, 25). Lysosomes are involved not only in the recycling of cellular substrates but also in the processing of antigens and their presentation on MHC class II proteins (4, 26–29). Thus, CQ/HCQ decrease, within 1 h in vitro, antigen processing and presentation by antigen presenting cells (4, 15, 16). Other mechanisms are invoked for the impact of HCQ or CQ on autophagy. Recently Rebecca et al. identified that inhibition of palmitoyl-protein thioesterase 1 (PPT1) is, at least in part, responsible for the observed anti-autophagy effect (30).
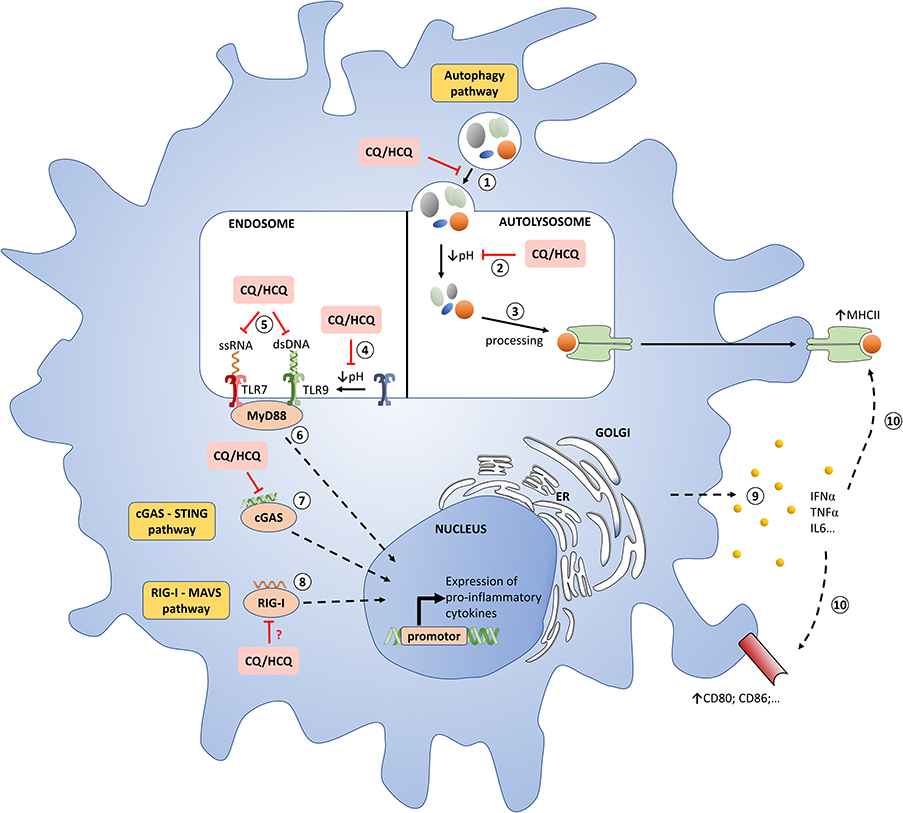
Figure 1. Molecular mechanisms of chloroquine/hydroxychloroquine in pDCs. (1) Autophagosome-lysosome fusion. CQ/HCQ impair this step. (2) Degradation of cargo from autophagosome. CQ/HCQ accumulate in lysosomes (lysosomotropism) and inhibit their function by increasing the pH. (3) Antigens are processed on MHC class II. (4) Proteolysis of TLRs by acid-dependent proteases is an essential step for the recognition of ligands. CQ/HCQ inhibit acid-dependent proteases by increasing the pH. (5) TLRs interact with nucleic acids presented to endosomal compartments. HCQ and CQ can bind directly to nucleic acids, preventing their recognition and inhibiting TLR-ligand interactions. (6) TLRs activation lead to MyD88 recrutement with a subsequent synthesis of pro-inflammatory cytokines, especially IFN-I. (7) Cytosolic DNA binds to cGAS, which then synthesizes the second messenger cGAMP to mediate STING-dependent transcription of IFN-I. HCQ and CQ block the binding of dsDNA / cGAS, thus attenuating the underlying activation of the STING pathway mediated by cGAMP. (8) Cytosolic RNA is recognized by RIG-I, the signal is transferred to MAVS, then to subsequent interactors leading to expression of IFN-I. CQ/HCQ may impair this process. (9 and 10) The release of IFNα, among other cytokines, stimulates a feedback activation with notably MHCII and co-stimulatory molecules upregulation. CQ: chloroquine; dsDNA, double-stranded DNA; ER, endoplasmic reticulum; HCQ: hydroxychlroquine; IFN-I: type I interferons; MHC: major histocompatibily complex; pDC: plasmacytoid dendritic cell; TLR : toll like receptor.
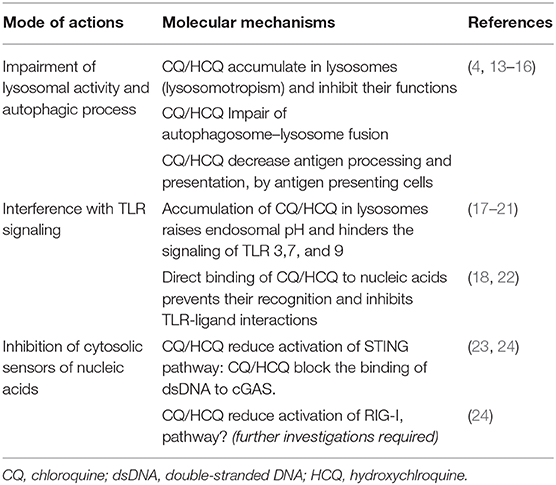
Table 1. Main mechanisms of actions of CQ/HCQ.
Perhaps the most important advance in our understanding of CQ/HCQ has been the discovery of their inhibiting effects on toll like receptors (TLRs), one of our first line of defense against bacterial and viral agents. HCQ and CQ inhibit some endosomal TLRs, mostly TLR7 and TLR9, which are able to recognize viral, bacterial and endogenous nucleic acids (Figure 1 and Table 1) (4, 31). These TLRs are located in the intracellular compartments to minimize accidental exposure to self-nucleic material (31). TLR7 is activated by ssRNA compounds, while TLR9 is activated by unmethylated CpG DNA (32). Activation of these endosomal TLRs can significantly contribute to promoting inflammation and/or the development of autoimmune diseases, such as systemic lupus erythematosus (SLE) (33, 34). Therefore, inhibition of endosomal TLRs, and consequently type I interferons (IFN-I) production, holds great therapeutic potential for the treatment of autoimmune diseases (35). CQ/HCQ have shown their ability to inhibit, in mice and in vitro, the TLR9 stimulation induced by CpG and the underlying production of IL-6 and TNFα (36, 37). These drugs can also inhibits RNA-mediated activation of TLR7 signaling and the production of IFNα (38, 39). In addition to cytokines secretion, TLR7 and 9 inhibition will also impair costimulatory molecules expression, such as CD86 on B cells, contrary to stimulation with pokeweed mitogen (Figure S1C) (40). Two modes of action are currently suggested:
• Raising endosomal pH has been proposed as the explanation for defective maturation and antigen presentation as well as impaired responses to TLR activation. Indeed, acidic conditions are necessary for strong interactions of nucleic acids with TLR9 (17). In this way, blocking endosomal acidification hinders the signaling of TLR 3, 7, and 9 (18–20). Ewald et al. showed that cleavage of TLR9 does not take place in the absence of acidification of the endolysosomal compartments. However, the proteolysis of TLR9 is an essential step for the recognition of ligands (CpG for TLR9) and the recruitment of MyD88, and consequently the signaling of TLR9 (21).
• CQ/HCQ can also directly bind to nucleic acids and such interferences lead to structural changes in the TLR ligands, preventing their recognition and inhibiting TLR-ligand interactions (18, 22).
The capacity of plasmacytoid dendritic cells (pDCs) to produce massive quantities of type I IFN has driven our understanding about the biology of these cells and their major role in both immunity against virus and in inflammation/autoimmunity (41).
pDCs can be acutely activated through different cell surface receptors and cytosolic sensors but TLR7/9 are likely their dominant mode of activation for endogenous or exogenous nucleic acids, with respect to IFN-I production (41–45). When triggered, pDCs expressed costimulatory molecules, and are able to prime subsequently T cells. Within 10 h following the sensing of nucleic acids, more than 80% of genes expressed in pDCs are IFNs or driven by IFNs (41, 46, 47). In addition, pDCs secrete cytokines as IL-6, TNFα and IL-12 and control the expression of many inflammatory cytokines. Because of these properties, they are considered as one of the main cellular actor of anti-viral defense (45).
In parallel, pDCs and IFN-I, have been largely studied in chronic inflammatory/autoimmune diseases and systemic lupus (SLE) (48). IFN-I are involved in SLE physiopathology through multiple mechanisms, focusing recent therapeutic research and trials (49, 50). HCQ is essential in the treatment of the disease and in prevention of flares, and CQ/HCQ are able to decrease IFNs production by pDCs (Figure S1D) (22, 51). In addition, HCQ/CQ effects are not limited to pDCs and result in several other cytokines inhibition (Figures S1E,F) (22, 52–54).
Apart from TLRs, HCQ/CQ interfere with other pattern recognition receptors (PRRs) essential to the anti-viral response namely the cGAS–STING and RIG-I–MAVS pathways (Figure 1 and Table 1). These cytosolic PRRs recognize and respond to DNA and RNA, respectively (23, 55). Cytosolic DNA is recognized by the cGAS-STING signaling axis which stimulates antiviral immunity by inducing IFN-I (56, 57). More precisely, cytosolic DNA binds to cGAS, which then synthesizes the second messenger cGAMP to mediate STING-dependent transcription of IFN-I (57). CQ/HCQ block the binding of dsDNA to cGAS, thus attenuating the underlying activation of the STING pathway mediated by cGAMP (23, 24).
Cytosolic RNA is recognized by RIG-I, the signal is transferred to MAVS, then to subsequent interactors leading to expression of IFN-I (55). An et al. showed that CQ/HCQ may inhibit RIG-I–stimulated induction of IFNs (24). However, the potential impacts of CQ/HCQ on this pathway need further investigations.
Considering CQ/HCQ and pathogens direct interactions, only the interference of CQ and Plasmodium, via hemozoin crystallization inhibition leading to parasite death, has been clearly documented (58). Immunity against infectious agents, including bacteria, virus or fungus, also involve vessels, and inflammation at the endothelium level (59). In this view, CQ/HCQ has been suggested to prevent prothrombotic state and endothelial dysfunction (60, 61). Finally, neutrophil extracellular traps (NETs) and the ability to mount NETosis is essential in innate immunity against pathogens. However, CQ is also effective as an early upstream inhibitor of NET formation in murine models of inflammation (62).
Considering SARS-CoV2, which is at the center of current concerns, some studies argue for lower nasopharyngeal carriage after 6 days (63) and an overall improvement of patients with CQ/HCQ (63–66), while more recent studies do not show rapid viral clearance or lower mortality (67–73).
Little is known about the mechanisms of action of CQ/HCQ during SARS-CoV2 infection. CQ has repeatedly demonstrated its ability, in vitro or in mouse models, to inhibit the replication of various strains of coronavirus, including those responsible for severe acute respiratory syndrome (SARS) in 2002 and 2003 (74–76). Additionally, in vitro studies carried out on SARS-CoV2 responsible for COVID-19 infection also confirm the potential interest of CQ/HCQ (77–79). Indeed, the increased endosomal pH due to CQ/HCQ may affect early stage of viral replication, via inhibition of virus-endosome fusion (80), and seem to stall the release of viral genome (77, 79). Although a direct interaction between HCQ/CQ and the virus has yet not been described, in vitro CQ interferes with terminal glycosylation of the SARS coronavirus receptor ACE2 (75, 81). Strikingly ACE2 is an interferon stimulated gene (82, 83), as CQ/HCQ decrease IFN-I secretion, they may also hinder the expression of the viral receptor in the neighboring airway epithelial cells (83).
Thanks to its known capacity to inhibit both TLRs and cGAS-STING pathway, HCQ is a first line treatment in SLE and other autoimmune/inflammatory conditions. More than 70% of SLE patients around the world are under HCQ therapy which represents today indeed thousands of people. From its wide use, we know that HCQ efficiency takes a few weeks to impact clinical symptoms, as arthritis, or cutaneous manifestations (7). However, despite the first use of CQ and HCQ in immune related disease almost 70 years ago, their exact mechanisms of action are only beginning to be understood, whether considering pharmacokinetic or molecular mechanisms. In any case, in vitro CQ/HCQ inhibit the production of IFN-I and inflammatory cytokines by innate immune cells. However, considering cytokines profile in SLE patients under diverse immunosuppressive therapies compared to untreated patients (84), HCQ per se is far from being the most efficient.
Recently, a new coronavirus SARS-CoV2 infection (or COVID-19) has started to spread around world with the development of pneumonia and severe acute respiratory syndrome (SARS). CQ/HCQ remain at the center of a therapy controversy as several publications describe an improvement of patients with CQ/HCQ (63–66), while others give results showing rather their ineffectiveness (67–73). Undoubtedly, several mechanistical arguments are in favor of HCQ/CQ interfering with coronavirus infection and virus spreading. However, we know that COVID-19 is not only an acute viral infection. Some patients experience a biphasic evolution/two-step disease progression. They are first infected, viremic, with flu-symptoms. At this stage, they may develop mild respiratory manifestations. Some days later, for yet unclear reasons, disease can dramatically worsen with an inflammatory procession leading to a subsequent “cytokine storm,” a pro-thrombotic state and even a macrophage activation syndrome with heavy lung damage and/or multiorgan failure (85–87).
Looking back to SARS-CoV models, it has been suggested and documented in mice that the first phase of the disease could be accompanied by an initial virus mediated and adapted IFN-I response. The second phase of the disease would occur secondary to an inappropriate delayed IFN-I production in infected lungs. This leads to a subsequent and excessive innate immune response with pathogenic inflammatory monocyte-macrophages and sub-optimal T cell response, partly due to T cell apoptosis, which is not enough to dampen immune innate system overactivation (88). Accordingly, a recent study from Hadjadj et al. identified an impaired IFN-I activity, increased T cell apoptosis and exacerbated inflammatory responses in severe COVID-19 patients (89). Thus, like what has been suggested for SARS-CoV-1 infection (88), the driving clinical features of severe COVID-19 patients stem from a dysregulated immune response in patients with notably a delayed and abnormal production of IFN-I (82, 89).
Hence, what are our aims when we treat SARS-CoV2 infection with CQ/HCQ ? Do we wish to decrease a beginning viremia by endolysosomal pathway inhibition with CQ/HCQ, which may further delay the anti-viral IFN-I response? or do we wish to control the inflammatory storm by interfering with PRRs and endosomal TLRs signaling, leading to a decreased cellular activation and thereby cytokine secretion? Then, if we initiate the treatment in the second phase of the disease, when the disease becomes somehow an inflammatory disease, are we really in frame with CQ/HCQ already demonstrated properties?
It appears from our clinical experience and from national or international series of COVID19 infected patients, that men are more affected than women (69, 90) and patients with autoimmune diseases, including SLE who are under HCQ or other immunosuppressive drugs, are not drastically affected. This could have other explanations than HCQ efficiency: (i) most of patients with autoimmunity are women, (ii) some of them present an intrinsic IFN-I response which is not completely abolished by HCQ and remains more intense than healthy donors, (iii) immunosuppressive therapies could impact or prevent high auto-inflammatory conditions.
At this time, we do not have any definitive clue concerning CQ/HCQ impacts on the first anti-viral phase of COVID-19, and the use of CQ/HCQ must remain cautious as results from recent studies do not support these treatments (67, 69, 72). Considering published data on COVID-19 disease and older data about SARS physiopathology, and although CQ/HCQ remain an interesting potential strategy to limit progression toward uncontrolled inflammation, they do not appear per se to be sufficient to control the whole inflammatory process in COVID-19, and more targeted and/or potent therapies should be required at least in add-on.
VG, AG, MR, J-PH, and A-SK wrote the manuscript. VG, AG, YD, FG, J-PH, and A-SK performed the literature search. VG, YD, and NB designed the figures. NB, VG, AG, YD, and QF performed the experiments. NB, VG, YD, AG, QF, FG, MR, J-PH, and A-SK interpreted the data. All authors reviewed and approved the manuscript.
Publication fees were supported by Hôpitaux Universitaires de Strasbourg.
AG received funding from Shire Takeda, outside the submitted work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Benoit Beitz (BIOASTER, LYON, France) for expert technical assistance for the mass cytometry (CYTOF) analysis.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.01409/full#supplementary-material
CQ, chloroquine; HCQ, hydroxychloroquine; IFN, interferon; IFN-I, type I interferons; NET, neutrophil extracellular trap; pDC, plasmacytoid dendritic cell; SARS, severe acute respiratory syndrome; SLE, systemic lupus erythematosus; TLR, toll like receptor.
1. Raoult D, Houpikian P, Tissot Dupont H, Riss JM, Arditi-Djiane J, Brouqui P. Treatment of Q fever endocarditis: comparison of 2 regimens containing doxycycline and ofloxacin or hydroxychloroquine. Arch Intern Med. (1999) 159:167–73. doi: 10.1001/archinte.159.2.167
2. Lagier J-C, Raoult D. Whipple's disease and tropheryma whipplei infections: when to suspect them and how to diagnose and treat them. Curr Opin Infect Dis. (2018) 31:463–70. doi: 10.1097/QCO.0000000000000489
3. Tang Y-Q, Ye Q, Huang H, Zheng W-Y. An overview of available antimalarials: discovery, mode of action and drug resistance. Curr Mol Med. (2020). doi: 10.2174/1566524020666200207123253. [Epub ahead of print].
4. Schrezenmeier E, Dörner T. Mechanisms of action of hydroxychloroquine and chloroquine: implications for rheumatology. Nat Rev Rheumatol. (2020) 16:155–66. doi: 10.1038/s41584-020-0372-x
5. Xu R, Ji Z, Xu C, Zhu J. The clinical value of using chloroquine or hydroxychloroquine as autophagy inhibitors in the treatment of cancers: a systematic review and meta-analysis. Medicine. (2018) 97:e12912. doi: 10.1097/MD.0000000000012912
6. Chatre C, Roubille F, Vernhet H, Jorgensen C, Pers Y-M. cardiac complications attributed to chloroquine and hydroxychloroquine: a systematic review of the literature. Drug Saf. (2018) 41:919–31. doi: 10.1007/s40264-018-0689-4
7. Fanouriakis A, Kostopoulou M, Alunno A, Aringer M, Bajema I, Boletis JN, et al. 2019 update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann Rheum Dis. (2019) 78:736–45. doi: 10.1136/annrheumdis-2019-215089
8. Shee JC. Lupus erythematosus treated with chloroquine. Lancet. (1953) 265:201–2. doi: 10.1016/S0140-6736(53)90138-X
9. Wallace DJ. The history of antimalarials. Lupus. (1996) 5(Suppl. 1):S2–3. doi: 10.1177/0961203396005001021
10. Lee S-J, Silverman E, Bargman JM. The role of antimalarial agents in the treatment of SLE and lupus nephritis. Nat Rev Nephrol. (2011) 7:718–29. doi: 10.1038/nrneph.2011.150
11. Arbogast F, Gros F. Lymphocyte autophagy in homeostasis, activation, and inflammatory diseases. Front Immunol. (2018) 9:1801. doi: 10.3389/fimmu.2018.02627
12. Choi AMK, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. (2013) 368:651–62. doi: 10.1056/NEJMra1205406
13. Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema K-J, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. (2018) 14:1435–55. doi: 10.1080/15548627.2018.1474314
14. Circu M, Cardelli J, Barr MP, O'Byrne K, Mills G, El-Osta H. Modulating lysosomal function through lysosome membrane permeabilization or autophagy suppression restores sensitivity to cisplatin in refractory non-small-cell lung cancer cells. PLoS ONE. (2017) 12:e0184922. doi: 10.1371/journal.pone.0184922
15. Ziegler HK, Unanue ER. Decrease in macrophage antigen catabolism caused by ammonia and chloroquine is associated with inhibition of antigen presentation to T cells. Proc Natl Acad Sci USA. (1982) 79:175–8. doi: 10.1073/pnas.79.1.175
16. Fox RI. Mechanism of action of hydroxychloroquine as an antirheumatic drug. Semin Arthritis Rheu. (1993) 23:82–91. doi: 10.1016/S0049-0172(10)80012-5
17. Mellman I, Fuchs R, Helenius A. Acidification of the endocytic and exocytic pathways. Annu Rev Biochem. (1986) 55:663–700. doi: 10.1146/annurev.bi.55.070186.003311
18. Häcker H, Mischak H, Miethke T, Liptay S, Schmid R, Sparwasser T, et al. CpG-DNA-specific activation of antigen-presenting cells requires stress kinase activity and is preceded by non-specific endocytosis and endosomal maturation. EMBO J. (1998) 17:6230–40. doi: 10.1093/emboj/17.21.6230
19. Garcia-Cattaneo A, Gobert F-X, Müller M, Toscano F, Flores M, Lescure A, et al. Cleavage of Toll-like receptor 3 by cathepsins B and H is essential for signaling. Proc Natl Acad Sci USA. (2012) 109:9053–8. doi: 10.1073/pnas.1115091109
20. Ewald SE, Engel A, Lee J, Wang M, Bogyo M, Barton GM. Nucleic acid recognition by Toll-like receptors is coupled to stepwise processing by cathepsins and asparagine endopeptidase. J Exp Med. (2011) 208:643–51. doi: 10.1084/jem.20100682
21. Ewald SE, Lee BL, Lau L, Wickliffe KE, Shi G-P, Chapman HA, et al. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature. (2008) 456:658–62. doi: 10.1038/nature07405
22. Kužnik A, Benčina M, Švajger U, Jeras M, Rozman B, Jerala R. Mechanism of endosomal tlr inhibition by antimalarial drugs and imidazoquinolines. J Immunol. (2011) 186:4794–804. doi: 10.4049/jimmunol.1000702
23. Gaidt MM, Ebert TS, Chauhan D, Ramshorn K, Pinci F, Zuber S, et al. The DNA inflammasome in human myeloid cells is initiated by a STING-cell death program upstream of NLRP3. Cell. (2017) 171:1110–24.e18. doi: 10.1016/j.cell.2017.09.039
24. An J, Woodward JJ, Sasaki T, Minie M, Elkon KB. Cutting edge: antimalarial drugs inhibit IFN-β production through blockade of cyclic GMP-AMP synthase–DNA interaction. J Immunol. (2015) 194:4089–93. doi: 10.4049/jimmunol.1402793
25. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. (2016) 12:1–222. doi: 10.1080/15548627.2015.1100356
26. Lotteau V, Teyton L, Peleraux A, Nilsson T, Karlsson L, Schmid SL, et al. Intracellular transport of class II MHC molecules directed by invariant chain. Nature. (1990) 348:600–5. doi: 10.1038/348600a0
27. Ghislat G, Lawrence T. Autophagy in dendritic cells. Cell Mol Immunol. (2018) 15:944–52. doi: 10.1038/cmi.2018.2
28. Münz C. Autophagy beyond intracellular MHC class II antigen presentation. Trends Immunol. (2016) 37:755–63. doi: 10.1016/j.it.2016.08.017
29. Ballabio A, Bonifacino JS. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol. (2020) 21:101–18. doi: 10.1038/s41580-019-0185-4
30. Rebecca VW, Nicastri MC, Fennelly C, Chude CI, Barber-Rotenberg JS, Ronghe A, et al. PPT1 promotes tumor growth and is the molecular target of chloroquine derivatives in cancer. Cancer Discov. (2019) 9:220–9. doi: 10.1158/2159-8290.CD-18-0706
31. Wallace DJ, Gudsoorkar VS, Weisman MH, Venuturupalli SR. New insights into mechanisms of therapeutic effects of antimalarial agents in SLE. Nat Rev Rheumatol. (2012) 8:522–33. doi: 10.1038/nrrheum.2012.106
32. Barrat FJ, Elkon KB, Fitzgerald KA. Importance of nucleic acid recognition in inflammation and autoimmunity. Annu Rev Med. (2016) 67:323–36. doi: 10.1146/annurev-med-052814-023338
33. Barrat FJ, Meeker T, Gregorio J, Chan JH, Uematsu S, Akira S, et al. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J Exp Med. (2005) 202:1131–9. doi: 10.1084/jem.20050914
34. Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. (2006) 6:823–35. doi: 10.1038/nri1957
35. Hennessy EJ, Parker AE, O'Neill LAJ. Targeting Toll-like receptors: emerging therapeutics? Nat Rev Drug Discov. (2010) 9:293–307. doi: 10.1038/nrd3203
36. Hong Z, Jiang Z, Liangxi W, Guofu D, Ping L, Yongling L, et al. Chloroquine protects mice from challenge with CpG ODN and LPS by decreasing proinflammatory cytokine release. Int Immunopharmacol. (2004) 4:223–34. doi: 10.1016/j.intimp.2003.12.006
37. Macfarlane DE, Manzel L. Antagonism of immunostimulatory CpG-oligodeoxynucleotides by quinacrine, chloroquine, and structurally related compounds. J Immunol. (1998) 160:1122–31.
38. Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, et al. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med. (2005) 202:1171–7. doi: 10.1084/jem.20050630
39. Vollmer J, Tluk S, Schmitz C, Hamm S, Jurk M, Forsbach A, et al. Immune stimulation mediated by autoantigen binding sites within small nuclear RNAs involves Toll-like receptors 7 and 8. J Exp Med. (2005) 202:1575–85. doi: 10.1084/jem.20051696
40. Gies V, Schickel J-N, Jung S, Joublin A, Glauzy S, Knapp A-M, et al. Impaired TLR9 responses in B cells from patients with systemic lupus erythematosus. JCI Insight. (2018) 3:e96795. doi: 10.1172/jci.insight.96795
41. Barrat FJ, Su L. A pathogenic role of plasmacytoid dendritic cells in autoimmunity and chronic viral infection. J Exp Med. (2019) 216:1974–85. doi: 10.1084/jem.20181359
42. Liu Y-J. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol. (2005) 23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633
43. Saitoh S-I, Abe F, Kanno A, Tanimura N, Saitoh YM, Fukui R, et al. TLR7 mediated viral recognition results in focal type I interferon secretion by dendritic cells. Nat Commun. (2017) 8:1–12. doi: 10.1038/s41467-017-01687-x
44. Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol. (2015) 15:471–85. doi: 10.1038/nri3865
45. Reizis B. Plasmacytoid dendritic cells: development, regulation, and function. Immunity. (2019) 50:37–50. doi: 10.1016/j.immuni.2018.12.027
46. Duramad O, Fearon KL, Chan JH, Kanzler H, Marshall JD, Coffman RL, et al. IL-10 regulates plasmacytoid dendritic cell response to CpG-containing immunostimulatory sequences. Blood. (2003) 102:4487–92. doi: 10.1182/blood-2003-07-2465
47. Ito T, Kanzler H, Duramad O, Cao W, Liu Y-J. Specialization, kinetics, and repertoire of type 1 interferon responses by human plasmacytoid predendritic cells. Blood. (2006) 107:2423–31. doi: 10.1182/blood-2005-07-2709
48. Kaul A, Gordon C, Crow MK, Touma Z, Urowitz MB, van Vollenhoven R, et al. Systemic lupus erythematosus. Nat Rev Dis Primers. (2016) 2:16039. doi: 10.1038/nrdp.2016.39
49. Rönnblom L, Leonard D. Interferon pathway in SLE: one key to unlocking the mystery of the disease. Lupus Sci Med. (2019) 6:e000270. doi: 10.1136/lupus-2018-000270
50. Smith N, Rodero MP, Bekaddour N, Bondet V, Ruiz-Blanco YB, Harms M, et al. Control of TLR7-mediated type I IFN signaling in pDCs through CXCR4 engagement—a new target for lupus treatment. Sci Adv. (2019) 5:eaav9019. doi: 10.1126/sciadv.aav9019
51. Sacre K, Criswell LA, McCune JM. Hydroxychloroquine is associated with impaired interferon-alpha and tumor necrosis factor-alpha production by plasmacytoid dendritic cells in systemic lupus erythematosus. Arthritis Res Ther. (2012) 14:R155. doi: 10.1186/ar3895
52. Picot S, Peyron F, Vuillez JP, Polack B, Ambroise-Thomas P. Chloroquine inhibits tumor necrosis factor production by human macrophages in vitro. J Infect Dis. (1991) 164:830. doi: 10.1093/infdis/164.4.830
53. Jang C-H, Choi J-H, Byun M-S, Jue D-M. Chloroquine inhibits production of TNF-alpha, IL-1beta and IL-6 from lipopolysaccharide-stimulated human monocytes/macrophages by different modes. Rheumatology. (2006) 45:703–10. doi: 10.1093/rheumatology/kei282
54. van den Borne BE, Dijkmans BA, de Rooij HH, le Cessie S, Verweij CL. Chloroquine and hydroxychloroquine equally affect tumor necrosis factor-alpha, interleukin 6, and interferon-gamma production by peripheral blood mononuclear cells. J Rheumatol. (1997) 24:55–60.
55. Pichlmair A, Schulz O, Tan CP, Näslund TI, Liljeström P, Weber F, et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5'-phosphates. Science. (2006) 314:997–1001. doi: 10.1126/science.1132998
56. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. (2008) 455:674–8. doi: 10.1038/nature07317
57. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. (2013) 339:786–91. doi: 10.1126/science.1232458
58. Kapishnikov S, Staalsø T, Yang Y, Lee J, Pérez-Berná AJ, Pereiro E, et al. Mode of action of quinoline antimalarial drugs in red blood cells infected by Plasmodium falciparum revealed in vivo. Proc Natl Acad Sci USA. (2019) 116:22946–52. doi: 10.1073/pnas.1910123116
59. Konradt C, Hunter CA. Pathogen interactions with endothelial cells and the induction of innate and adaptive immunity. Eur J Immunol. (2018) 48:1607–20. doi: 10.1002/eji.201646789
60. Espinola RG, Pierangeli SS, Gharavi AE, Harris EN, Ghara AE. Hydroxychloroquine reverses platelet activation induced by human IgG antiphospholipid antibodies. Thromb Haemost. (2002) 87:518–22. doi: 10.1055/s-0037-1613033
61. Rand JH, Wu X-X, Quinn AS, Ashton AW, Chen PP, Hathcock JJ, et al. Hydroxychloroquine protects the annexin A5 anticoagulant shield from disruption by antiphospholipid antibodies: evidence for a novel effect for an old antimalarial drug. Blood. (2010) 115:2292–9. doi: 10.1182/blood-2009-04-213520
62. Denning N-L, Aziz M, Gurien SD, Wang P. DAMPs and NETs in Sepsis. Front Immunol. (2019) 10:2536. doi: 10.3389/fimmu.2019.02536
63. Gautret P, Lagier J-C, Parola P, Hoang VT, Meddeb L, Mailhe M, et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int J Antimicrob Agents. (2020) 105949. doi: 10.1016/j.ijantimicag.2020.105949. [Epub ahead of print].
64. Chen Z, Hu J, Zhang Z, Jiang S, Han S, Yan D, et al. Efficacy of hydroxychloroquine in patients with COVID-19: results of a randomized clinical trial. medRxiv. [Preprint]. (2020). doi: 10.1101/2020.03.22.20040758. [Epub ahead of print].
65. Sarma P, Kaur H, Kumar H, Mahendru D, Avti P, Bhattacharyya A, et al. Virological and clinical cure in covid-19 patients treated with hydroxychloroquine: a systematic review and meta-analysis. J Med Virol. (2020). doi: 10.1002/jmv.25898
66. Yu B, Li C, Chen P, Zhou N, Wang L, Li J, et al. Low dose of hydroxychloroquine reduces fatality of critically ill patients with COVID-19. Sci China Life Sci. (2020). doi: 10.1007/s11427-020-1732-2. [Epub ahead of print].
67. Mahévas M, Tran V-T, Roumier M, Chabrol A, Paule R, Guillaud C, et al. Clinical efficacy of hydroxychloroquine in patients with covid-19 pneumonia who require oxygen: observational comparative study using routine care data. BMJ. (2020) 369:m1844. doi: 10.1136/bmj.m1844
68. Molina JM, Delaugerre C, Le Goff J, Mela-Lima B, Ponscarme D, Goldwirt L, et al. No evidence of rapid antiviral clearance or clinical benefit with the combination of hydroxychloroquine and azithromycin in patients with severe COVID-19 infection. Med Mal Infect. (2020) 50:384. doi: 10.1016/j.medmal.2020.03.006
69. Rosenberg ES, Dufort EM, Udo T, Wilberschied LA, Kumar J, Tesoriero J, et al. Association of treatment with hydroxychloroquine or azithromycin with in-hospital mortality in patients with COVID-19 in New York State. JAMA. (2020). doi: 10.1001/jama.2020.8630. [Epub ahead of print].
70. Tang W, Cao Z, Han M, Wang Z, Chen J, Sun W, et al. Hydroxychloroquine in patients with mainly mild to moderate coronavirus disease 2019: open label, randomised controlled trial. BMJ. (2020) 369:m1849. doi: 10.1136/bmj.m1849
71. Geleris J, Sun Y, Platt J, Zucker J, Baldwin M, Hripcsak G, et al. Observational study of hydroxychloroquine in hospitalized patients with covid-19. N Engl J Med. (2020) 382:2411–8. doi: 10.1056/NEJMoa2012410
72. Boulware DR, Pullen MF, Bangdiwala AS, Pastick KA, Lofgren SM, Okafor EC, et al. A randomized trial of hydroxychloroquine as postexposure prophylaxis for Covid-19. N Engl J Med. (2020). doi: 10.1056/NEJMoa2016638
73. Magagnoli J, Narendran S, Pereira F, Cummings T, Hardin JW, Sutton SS, et al. Outcomes of hydroxychloroquine usage in United States veterans hospitalized with Covid-19. medRxiv. (2020). doi: 10.1016/j.medj.2020.06.001
74. Keyaerts E, Vijgen L, Maes P, Neyts J, Van Ranst M. In vitro inhibition of severe acute respiratory syndrome coronavirus by chloroquine. Biochem Biophys Res Commun. (2004) 323:264–8. doi: 10.1016/j.bbrc.2004.08.085
75. Vincent MJ, Bergeron E, Benjannet S, Erickson BR, Rollin PE, Ksiazek TG, et al. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol J. (2005) 2:69. doi: 10.1186/1743-422X-2-69
76. Barnard DL, Day CW, Bailey K, Heiner M, Montgomery R, Lauridsen L, et al. Evaluation of immunomodulators, interferons and known in vitro SARS-coV inhibitors for inhibition of SARS-coV replication in BALB/c mice. Antivir Chem Chemother. (2006) 17:275–84. doi: 10.1177/095632020601700505
77. Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. (2020) 30:269–71. doi: 10.1038/s41422-020-0282-0
78. Yao X, Ye F, Zhang M, Cui C, Huang B, Niu P, et al. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Clin Infect Dis. (2020) ciaa237. doi: 10.1093/cid/ciaa237
79. Liu J, Cao R, Xu M, Wang X, Zhang H, Hu H, et al. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. (2020) 6:1–4. doi: 10.1038/s41421-020-0156-0
80. Yang Z-Y, Huang Y, Ganesh L, Leung K, Kong W-P, Schwartz O, et al. pH-dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC-SIGN. J Virol. (2004) 78:5642–50. doi: 10.1128/JVI.78.11.5642-5650.2004
81. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. (2020) 367:1444–8. doi: 10.1126/science.abb2762
82. Blanco-Melo D, Nilsson-Payant BE, Liu W-C, Uhl S, Hoagland D, Møller R, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. (2020) 181:1036–45.e9. doi: 10.1016/j.cell.2020.04.026
83. Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN, Tzouanas CN, et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell. (2020) 181:1016–35.e19. doi: 10.1016/j.cell.2020.04.035
84. Banchereau R, Hong S, Cantarel B, Baldwin N, Baisch J, Edens M, et al. Personalized immunomonitoring uncovers molecular networks that stratify lupus patients. Cell. (2016) 165:551–65. doi: 10.1016/j.cell.2016.03.008
85. Lescure F-X, Bouadma L, Nguyen D, Parisey M, Wicky P-H, Behillil S, et al. Clinical and virological data of the first cases of COVID-19 in Europe: a case series. Lancet Infect Dis. (2020) 20:697–706. doi: 10.1016/S1473-3099(20)30200-0
86. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. (2020) 395:1033–4. doi: 10.1016/S0140-6736(20)30628-0
87. Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and immunologic featsures in severe and moderate coronavirus disease (2019). J Clin Invest. (2020) 130:2620–9. doi: 10.1101/2020.02.16.20023903
88. Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe. (2016) 19:181–93. doi: 10.1016/j.chom.2016.01.007
89. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Pere H, et al. Impaired type I interferon activity and exacerbated inflammatory responses in severe Covid-19 patients. medRxiv. [Preprint]. (2020) 1. doi: 10.1101/2020.04.19.20068015
Keywords: chloroquine, hydroxychloroquine, COVID-19, SARS-CoV2, interferon, TLR, STING, RIG-I
Citation: Gies V, Bekaddour N, Dieudonné Y, Guffroy A, Frenger Q, Gros F, Rodero MP, Herbeuval J-P and Korganow A-S (2020) Beyond Anti-viral Effects of Chloroquine/Hydroxychloroquine. Front. Immunol. 11:1409. doi: 10.3389/fimmu.2020.01409
Received: 04 May 2020; Accepted: 02 June 2020;
Published: 02 July 2020.
Edited by:
Massimo Gadina, National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), United StatesReviewed by:
Fabrizio Conti, Sapienza University of Rome, ItalyCopyright © 2020 Gies, Bekaddour, Dieudonné, Guffroy, Frenger, Gros, Rodero, Herbeuval and Korganow. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vincent Gies, di5naWVzQHVuaXN0cmEuZnI=; Jean-Philippe Herbeuval, amVhbi1waGlsaXBwZS5IZXJiZXV2YWxAcGFyaXNkZXNjYXJ0ZXMuZnI=; Anne-Sophie Korganow, a29yZ2Fub3dAdW5pc3RyYS5mcg==
†These authors have contributed equally to the work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.