 Zaid Abassi
Zaid Abassi Yara Knaney
Yara Knaney Tony Karram3
Tony Karram3 Samuel N. Heyman
Samuel N. Heyman
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Immunol., 05 June 2020
Sec. Viral Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01312
This article is part of the Research TopicCoronavirus Disease (COVID-19): Pathophysiology, Epidemiology, Clinical Management and Public Health ResponseView all 400 articles
Respiratory, circulatory, and renal failure are among the gravest features of COVID-19 and are associated with a very high mortality rate. A common denominator of all affected organs is the expression of angiotensin-converting enzyme 2 (ACE2), a protease responsible for the conversion of Angiotensin 1-8 (Ang II) to Angiotensin 1-7 (Ang 1-7). Ang 1-7 acts on these tissues and in other target organs via Mas receptor (MasR), where it exerts beneficial effects, including vasodilation and suppression of inflammation and fibrosis, along an attenuation of cardiac and vascular remodeling. Unfortunately, ACE2 also serves as the binding receptor of SARS viral spike glycoprotein, enabling its attachment to host cells, with subsequent viral internalization and replication. Although numerous reports have linked the devastating organ injuries to viral homing and attachment to organ-specific cells widely expressing ACE2, little attention has been given to ACE-2 expressed by the immune system. Herein we outline potential adverse effects of SARS-CoV2 on macrophages and dendritic cells, key cells of the immune system expressing ACE2. Specifically, we propose a new hypothesis that, while macrophages play an important role in antiviral defense mechanisms, in the case of SARS-CoV, they may also serve as a Trojan horse, enabling viral anchoring specifically within the pulmonary parenchyma. It is tempting to assume that diverse expression of ACE2 in macrophages among individuals might govern the severity of SARS-CoV-2 infection. Moreover, reallocation of viral-containing macrophages migrating out of the lung to other tissues is theoretically plausible in the context of viral spread with the involvement of other organs.
The reported clinical manifestations of Covid-19 keep growing steadily. Respiratory, circulatory, and renal failure are among its gravest features, and the mortality rate is very high (1–3). Other organ involvement includes the gastrointestinal tract (manifested as diarrhea and vomiting) (4, 5), gonads [impaired male fertility (6)], and nervous system (7). A common denominator of all affected organs is the expression of angiotensin-converting enzyme 2 (ACE2) (8, 9). ACE2 is a transmembranal protease responsible for the conversion of Angiotensin 1-8 (Ang II) to Angiotensin 1-7 (Ang 1-7) (10). The latter acts on these tissues and in other target organs via Mas receptor (MasR), where it exerts beneficial effects, including vasodilation and suppression of inflammation and fibrosis (8, 9). Ang 1-7 also induces diuresis/natriuresis, preserves renal function, and attenuates cardiac and vascular remodeling (11).
Unfortunately, ACE2 also serves as the binding receptor of SARS viral spike glycoprotein, enabling its attachment to host cells, with subsequent viral internalization and replication (12–14). So far, numerous reports have linked the devastating organ injuries to viral homing and attachment to organ-specific cells widely expressing ACE2; however, little attention has been given to the immune system. The following short commentary outlines potential adverse effects of SARS-CoV2 on macrophages and dendritic cells, key cells of the immune system, which also express ACE2 (15, 16).
Macrophages and dendritic cells are ubiquitous in human organs with a substantial abundance in the lungs. There are two distinct populations of pulmonary macrophages: alveolar macrophages, which reside in proximity to type I and type II epithelial alveolar cells, and interstitial macrophages, which are preferentially abundant between the microvascular endothelium and alveolar epithelium zone (17) (Figure 1). Various pathogens and noxious materials reaching the lungs provoke an innate immune response of the pulmonary parenchyma that is characterized by the differentiation of bone-marrow-derived monocytes into alveolar macrophages, which serve as a first-line defense against invading organisms. Both alveolar and interstitial macrophages can be divided into two functional phenotypes. The first is made up of classically activated macrophages (M1 macrophage), which are activated by pathogen-associated molecular patterns (PAMPs) that are also expressed by viruses. Their activity is then promoted by Th1 cells. The second population includes the alternatively activated macrophages (M2 macrophage), which are activated by Th2 cells by means of IL-4 and IL-13 (17). M1 macrophages induce recruitment of immune cells into the lung parenchyma. In contrast, activation of M2 macrophages triggers the release of anti-inflammatory cytokines, which restrict inflammation and promote tissue repair (17). Dendritic cells play a keen role in the inflammatory process as evident by their responsibility for presentation of antigens, regulation of T-cell reactions to antigen, and the intensity of the inflammatory response. Activation of dendritic cells induces their expression of co-stimulation molecules such as CD80. Viral infections provoke monocytal-enhanced proinflammatory signaling molecules and antiviral responses, as have been shown with influenza, herpes, and Zika viruses (18). It has recently been suggested that enhanced activity of pro-inflammatory macrophages in part of the COVID-19 patients leads to accelerated production of inflammatory cytokines and chemokines, and among them is CXCL10, which leads to cytokine storms. This has mostly been observed in subjects with poor prognosis (19, 20). In general, short living monocytes/macrophages are able to remarkably limit viral replication. However, that does not preclude these cells from serving as a permissive system and/or as a viral reservoir (18). Support for this notion is derived from the fact that these cells serve as the first line of defense upon encountering viral infection. However, viral infection may convert these cells into long living macrophages (Mϕ) and promote their migration into tissues where they become infected resident cells. Finally, since SARS viruses, including SARS-CoV2, utilize ACE2 as a tight binding site with high affinity (12–14), pulmonary macrophages that express ACE2 may permeate pulmonary invasion during SARS infection. Indeed, we have previously shown that monocytes/macrophages express ACE2 (15). Furthermore, monocyte-derived macrophages from patients with CHF exhibit profoundly increased ACE2 expression after treatment with spironolactone, a mineralocorticoid blocker. The beneficial impact of upregulated ACE2 in CHF patients is evident by attenuated oxidative stress, as expressed by reduced lipid peroxide content, superoxide ion release, and low-density lipoprotein oxidation. Similarly, mice treated with eplerenone, another mineralocorticoid blocker, displayed enhanced cardiac ACE2 activity in parallel to increased ACE2 activity in macrophages (15). Interestingly, macrophages also express furin and TMPRSS2, two enzymes involved in the exposure of the binding and effusion sites of the SARS virus (21, 22), as well as ADAM 17, which acts as sheddase of ACE2 (23). In the presence of all components of viral binding and activation, the virus can theoretically replicate in human macrophages and dendritic cells, triggering the aberrant production of proinflammatory cytokines/chemokines, as is the case with MERS-CoV (24). In contrast, some studies ruled out SARS-CoV viral replication in human macrophages (25). Despite abortive infection, characterized by infection without replication, SARS-CoV infection of human macrophages induced the expression of proinflammatory chemokines, whereas antiviral cytokine production was largely absent (26, 27). Studies also demonstrated that human dendritic cells are susceptible to SARS-CoV but unable to support viral replication (28).
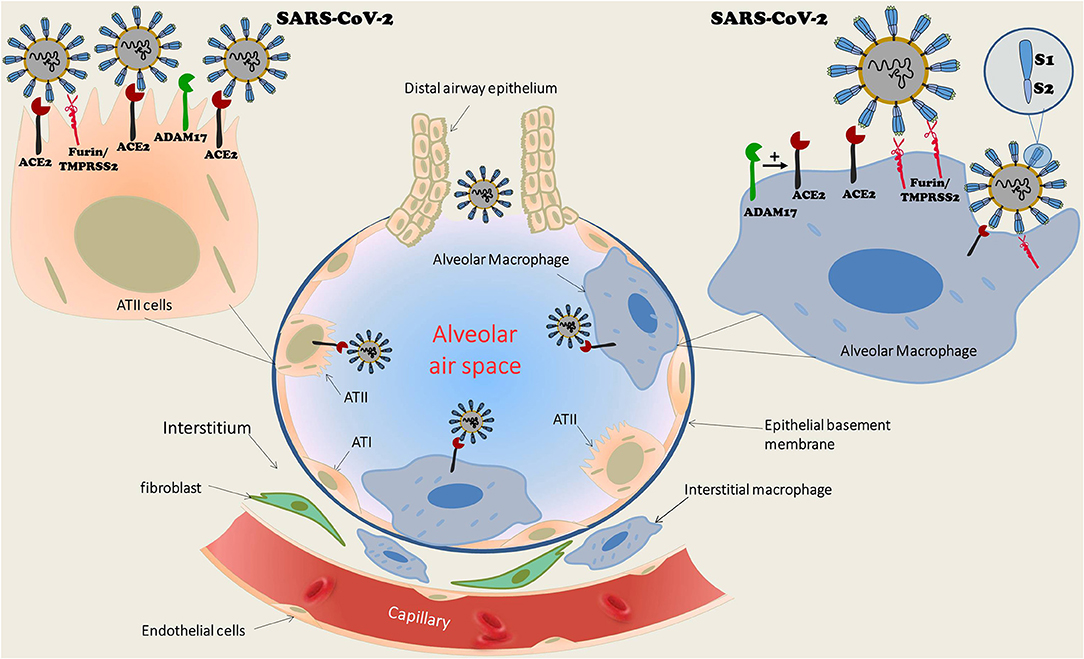
Figure 1. Schematic structure of pulmonary alveoli with diverse cell types, including cuboid ciliated epithelial cells along bronchioles, alveolar type I (ATI) and type II epithelial cells (ATII), and macrophages. The latter are ubiquitous in the lungs and consist of two distinct populations: alveolar macrophages, which reside in proximity to ATI and ATII, as well as interstitial macrophages, which are abundant between the microvascular endothelium and alveolar epithelium zone. Alveolar macrophages as well as ATII express ACE2, the binding receptor of SARS-CoV-2. In addition, both cell types express TMPRSS2/Furin, which are also required for viral attachment. It exposes the viral receptor binding protein (RBP) localized to S-glycoprotein (S1 domain of the viral spike) and reveals the effusion site on the S2 domain. Although SARS-CoV-2 replication in ATII cells is well-documented, a similar process was not confirmed in alveolar macrophages. While some studies suggested such a replication along triggering aberrant production of proinflammatory cytokines/chemokines, as is the case with MERS-CoV, others reports ruled out SARS-CoV viral replication in human macrophages. ACE2, Angiotensin converting enzyme 2; ATI, Alveolar epithelial cells type I; ATII, Alveolar epithelial cells type II; TMPRSS2, Transmembrane protease, serine 2.
COVID-19 morbidity and mortality are markedly increased in specified populations, namely aged and diabetic individuals, patients with chronic obstructive lung disease (COPD) or congestive heart failure (CHF) (3), and perhaps among patients on inhibitors of the renin angiotensin aldosterone system (RAAS) (3, 29). These observations might be linked with increased numbers of alveolar macrophages (AM) in such patients or with alterations in the AM phenotype. Indeed, increased numbers of AM in bronchoalveolar lavage (BAL) were detected in humans with COPD in proportion to their disease severity (30). Increased numbers of AM in BAL were also noted in mice following protracted exposure to diesel exhaust particles (31), and this is a consistent finding related to air pollution (32). Increased numbers of AM in BAL were noted also in aged vs. young rodents, and this difference was particularly prominent following exercise (31, 33). Furthermore, aging was associated with an altered phenotypic distribution of AM and with reduced bactericidal capacity in mice (34). AM were also more abundant in mice subjected to heart failure following augmented hypertension (35) or in models of dilated cardiomyopathy, combined with exercise (36). It was also noted in diabetic mice—associated with intensified indices of oxidative stress—yet these abnormalities were prevented by long-term treatment with angiotensin 1-7 (37). Furthermore, as with aging, experimental diabetes is associated with altered phenotype expression of AM (38) with decreased bactericidal capabilities (39). Taken together, increased susceptibility to serious COVID-19 infection occurs in clinical scenarios associated with increased AM population. It is tempting to suggest that conditions characterized by increased numbers of alveolar macrophages in the lower respiratory tract might facilitate homing of COVID-19 by their abundant expression of ACE2.
Collectively, in light of these observations, we propose a new hypothesis that while macrophages play an important role in antiviral defense mechanisms, in the case of SARS-CoV. they may also serve as a Trojan horse, enabling viral anchoring specifically within the pulmonary parenchyma. In other words, the unique expression of ACE2 in macrophages may, paradoxically, enable pulmonary invasion by SARS-CoV, facilitating engraftment, and inducing protracted local and systemic uncontrolled inflammatory responses (40). It is tempting to assume that diverse expression among individuals of ACE2 in macrophages might govern the severity of SARS-CoV-2 infection. Moreover, besides direct invasion caused by viremia, reallocation of viral-containing macrophages migrating out of the lung to other tissues is theoretically plausible in the context of viral spread with the involvement of other organs. To some extent, this setup resembles a comparable phenomenon, termed “the macrophage paradox,” were intracellular bacterial pathogens preferentially replicate within macrophages (41). Our hypothesis is further supported by a recent report of post-mortem findings in patients succumbing to SARS-CoV, showing ACE2 expression and viral nucleocaspid protein in CD169+ macrophages in lymph nodes and in the spleen (42). The attenuation of experimental lethal SARS in rodents by monocyte/macrophage depletion (43) is also to some extent in line with our hypothesis. On the other hand, a recent study demonstrated that proinflammatory monocyte-derived macrophages were abundant in bronchoalveolar lavage obtained from patients with severe SARS-CoV-2 pulmonary involvement, as compared with those with moderate disease (44). In fact, it has been suggested that monocyte-derived macrophages replace damaged infected alveolar macrophages in severe cases, and likely do not indicate the substitution of alveolar cells migrating to other tissues (44). This possibility is supported by documented death of infected macrophages in vitro. Furthermore, increasing evidence suggests that aberrant myeloid responses may underlie some of the COVID-19 hallmark manifestations, including acute respiratory distress syndrome (ARDS), cytokine release syndrome, and lymphopenia (45). In this context, recent studies in humanized hACE2 mice demonstrated that these animals exhibited characteristic alveolar interstitial pneumonia, with infiltration of lymphocytes and monocytes and accumulation of macrophages in the alveolar lumen (46), corresponding with the clinical findings (47). Moreover, primate and clinical data on SARS-CoV-1 have also shown that virus spike-specific IgG responses exacerbate ARDS due to repolarization of alveolar macrophages into pro-inflammatory phenotypes and enhanced recruitment of inflammatory monocytes via CCL2 and IL-8 (48). Collectively, it is obvious that the immune system undergoes profound and complex alterations during symptomatic COVID-19 disease, including migration of inflammatory monocytes with CD14+IL-1β+ monocytic expansion, as elegantly summarized by Vabret et al. (48) in a comprehensive review on the fast evolving field of COVID-19 immunology.
Finally, it should be emphasized that our hypothesis is not sufficiently evidence based. We still lack carefully produced data about the susceptibility of tissue macrophages to SARS-CoV-2 and their capacity to produce de novo infectious viral particles. Additional studies are also required to assess reduced ACE2 expression following macrophage invasion by SARS-CoV-2 and the plausible causative association that links modified macrophages to the evolving inflammatory storm.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
ZA, YK, TK, and SH equally participated in the design, execution of the view of point, drafted the manuscript, participated in critical discussions and revised the manuscript. YK and ZA prepared the figure. ZA supervised the project.
ZA wishes to acknowledge research support from the Israel Science Foundation (Grand No. 544/18).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Zheng YY, Ma YT, Zhang JY, Xie X. COVID-19 and the cardiovascular system. Nat Rev Cardiol. (2020) 17:259–60. doi: 10.1038/s41569-020-0360-5
2. Arentz M, Yim E, Klaff L, Lokhandwala S, Riedo FX, Chong M, et al. Characteristics and Outcomes of 21 Critically Ill Patients With COVID-19 in Washington State. JAMA. (2020) 323:1612–4. doi: 10.1001/jama.2020.4326
3. Perico L, Benigni A, Remuzzi G. Should COVID-19 concern nephrologists? Why and to What extent? The emerging impasse of angiotensin blockade. Nephron. (2020) 144:213–21. doi: 10.1159/000507305
4. Wong SH, Lui RN, Sung JJ. Covid-19 and the digestive system. J Gastroenterol Hepatol. (2020) 35:744–8. doi: 10.1111/jgh.15047
5. Jin X, Lian JS, Hu JH, Gao J, Zheng L, Zhang YM, et al. Epidemiological, clinical and virological characteristics of 74 cases of coronavirus-infected disease 2019 (COVID-19) with gastrointestinal symptoms. Gut. (2020) 69:1002–9. doi: 10.1136/gutjnl-2020-320926
6. Ma L, Xie W, Li D, Shi L, Mao Y, Xiong Y, et al. Effect of SARS-CoV-2 infection upon male gonadal function: a single center-based study. medRxiv. (2020). doi: 10.1101/2020.03.21.20037267
7. Wu Y, Xu X, Chen Z, Duan J, Hashimoto K, Yang L, et al. Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav Immun. (2020). doi: 10.1016/j.bbi.2020.03.031. [Epub ahead of print].
8. Clarke NE, Turner AJ. Angiotensin-converting enzyme 2: the first decade. Int J Hypertens. (2012) 2012:307315. doi: 10.1155/2012/307315
9. Hamming I, Cooper ME, Haagmans BL, Hooper NM, Korstanje R, Osterhaus AD, et al. The emerging role of ACE2 in physiology and disease. J Pathol. (2007) 212:1–11. doi: 10.1002/path.2162
10. Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. (2002) 277:14838–43. doi: 10.1074/jbc.M200581200
11. Santos RA, Ferreira AJ, Simões e Silva AC. Recent advances in the angiotensin-converting enzyme 2-angiotensin(1-7)-Mas axis. Exp Physiol. (2008) 93:519–27. doi: 10.1113/expphysiol.2008.042002
12. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ace2 and tmprss2 and is blocked by a clinically proven protease inhibitor. Cell. (2020) 181:271–80.e8. doi: 10.1016/j.cell.2020.02.052
13. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. (2020) 367:1444–8. doi: 10.1126/science.abb2762
14. Liu Z, Xiao X, Wei X, Li J, Yang J, Tan H, et al. Composition and divergence of coronavirus spike proteins and host ACE2 receptors predict potential intermediate hosts of SARS-CoV-2. J Med Virol. (2020) 92:595–601. doi: 10.1002/jmv.25726
15. Keidar S, Gamliel-Lazarovich A, Kaplan M, Pavlotzky E, Hamoud S, Hayek T, et al. Mineralocorticoid receptor blocker increases angiotensin-converting enzyme 2 activity in congestive heart failure patients. Circ Res. (2005) 97:946–53. doi: 10.1161/01.RES.0000187500.24964.7A
16. Nie W, Yan H, Li S, Zhang Y, Yu F, Zhu W, et al. Angiotensin-(1-7) enhances angiotensin II induced phosphorylation of ERK1/2 in mouse bone marrow-derived dendritic cells. Mol Immunol. (2009) 46:355–61. doi: 10.1016/j.molimm.2008.10.022
17. Hu G, Christman JW. Editorial: alveolar macrophages in lung inflammation and resolution. Front Immunol. (2019) 10:2275. doi: 10.3389/fimmu.2019.02275
18. Nikitina E, Larionova I, Choinzonov E, Kzhyshkowska J. Monocytes and macrophages as viral targets and reservoirs. Int J Mol Sci. (2018) 19:2821. doi: 10.3390/ijms19092821
19. Yang Y, Shen C, Li J, Yuan J, Yang M, Wang F, et al. Exuberant elevation of IP-10, MCP-3 and IL-1ra during SARS-CoV-2 infection is associated with disease severity and fatal outcome. medRxiv. (2020). doi: 10.1101/2020.03.02.20029975
20. Vaninov N. In the eye of the COVID-19 cytokine storm. Nat Rev Immunol. (2020) 20:277. doi: 10.1038/s41577-020-0305-6
21. Gagnon H, Refaie S, Gagnon S, Desjardins R, Salzet M, Day R. Proprotein convertase 1/3 (PC1/3) in the rat alveolar macrophage cell line NR8383: localization, trafficking and effects on cytokine secretion. PLoS ONE. (2013) 8:e61557. doi: 10.1371/journal.pone.0061557
22. Bertram S, Glowacka I, Blazejewska P, Soilleux E, Allen P, Danisch S, et al. TMPRSS2 and TMPRSS4 facilitate trypsin-independent spread of influenza virus in Caco-2 cells. J Virol. (2010) 84:10016–25. doi: 10.1128/JVI.00239-10
23. Nikolaidis NM, Gray JK, Gurusamy D, Fox W, Stuart WD, Huber N, et al. Ron receptor tyrosine kinase negatively regulates TNFalpha production in alveolar macrophages by inhibiting NF-kappaB activity and Adam17 production. Shock. (2010) 33:197–204. doi: 10.1097/SHK.0b013e3181ae8155
24. Zhou J, Chu H, Chan JF, Yuen KY. Middle East respiratory syndrome coronavirus infection: virus-host cell interactions and implications on pathogenesis. Virol J. (2015) 12:218. doi: 10.1186/s12985-015-0446-6
25. Yilla M, Harcourt BH, Hickman CJ, McGrew M, Tamin A, Goldsmith CS, et al. SARS-coronavirus replication in human peripheral monocytes/macrophages. Virus Res. (2005) 107:93–101. doi: 10.1016/j.virusres.2004.09.004
26. Tseng CT, Perrone LA, Zhu H, Makino S, Peters CJ. Severe acute respiratory syndrome and the innate immune responses: modulation of effector cell function without productive infection. J Immunol. (2005) 174:7977–85. doi: 10.4049/jimmunol.174.12.7977
27. Cheung CY, Poon LL, Ng IH, Luk W, Sia SF, Wu MH, et al. Cytokine responses in severe acute respiratory syndrome coronavirus-infected macrophages in vitro: possible relevance to pathogenesis. J Virol. (2005) 79:7819–26. doi: 10.1128/JVI.79.12.7819-7826.2005
28. Law HK, Cheung CY, Ng HY, Sia SF, Chan YO, Luk W, et al. Chemokine up-regulation in SARS-coronavirus-infected, monocyte-derived human dendritic cells. Blood. (2005) 106:2366–74. doi: 10.1182/blood-2004-10-4166
29. Fang L, Karakiulakis G, Roth M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir Med. (2020) 8:e21. doi: 10.1016/S2213-2600(20)30116-8
30. Agusti A, Hogg JC. Update on the pathogenesis of chronic obstructive pulmonary disease. N Engl J Med. (2019) 381:1248–56. doi: 10.1056/NEJMra1900475
31. Ribeiro Junior G, de Souza Xavier Costa N, Belotti L, Dos Santos Alemany AA, Amato-Lourenco LF, da Cunha PG, et al. Diesel exhaust exposure intensifies inflammatory and structural changes associated with lung aging in mice. Ecotoxicol Environ Saf . (2019) 170:314–23. doi: 10.1016/j.ecoenv.2018.11.139
32. Hamanaka RB, Mutlu GM. Particulate matter air pollution: effects on the cardiovascular system. Front Endocrinol (Lausanne). (2018) 9:680. doi: 10.3389/fendo.2018.00680
33. Veldhuizen RAW, McCaig LA, Pape C, Gill SE. The effects of aging and exercise on lung mechanics, surfactant and alveolar macrophages. Exp Lung Res. (2019) 45:113–22. doi: 10.1080/01902148.2019.1605633
34. Lafuse WP, Rajaram MVS, Wu Q, Moliva JI, Torrelles JB, Turner J, et al. Identification of an increased alveolar macrophage subpopulation in old mice that displays unique inflammatory characteristics and is permissive to Mycobacterium tuberculosis infection. J Immunol. (2019) 203:2252–64. doi: 10.4049/jimmunol.1900495
35. Wang H, Kwak D, Fassett J, Hou L, Xu X, Burbach BJ, et al. CD28/B7 deficiency attenuates systolic overload-induced congestive heart failure, myocardial and pulmonary inflammation, and activated T cell accumulation in the heart and lungs. Hypertension. (2016) 68:688–96. doi: 10.1161/HYPERTENSIONAHA.116.07579
36. Sugihara M, Odagiri F, Suzuki T, Murayama T, Nakazato Y, Unuma K, et al. Usefulness of running wheel for detection of congestive heart failure in dilated cardiomyopathy mouse model. PLoS ONE. (2013) 8:e55514. doi: 10.1371/journal.pone.0055514
37. Papinska AM, Soto M, Meeks CJ, Rodgers KE. Long-term administration of angiotensin (1-7) prevents heart and lung dysfunction in a mouse model of type 2 diabetes (db/db) by reducing oxidative stress, inflammation and pathological remodeling. Pharmacol Res. (2016) 107:372–80. doi: 10.1016/j.phrs.2016.02.026
38. Vance J, Santos A, Sadofsky L, Morice A, Cervantes J. Effect of high glucose on human alveolar macrophage phenotype and phagocytosis of mycobacteria. Lung. (2019) 197:89–94. doi: 10.1007/s00408-018-0181-z
39. Martinez N, Ketheesan N, West K, Vallerskog T, Kornfeld H. Impaired recognition of Mycobacterium tuberculosis by alveolar macrophages from diabetic mice. J Infect Dis. (2016) 214:1629–37. doi: 10.1093/infdis/jiw436
40. Moore JB, June C. H. Cytokine release syndrome in severe COVID-19. Science. (2020) 368:473–4. doi: 10.1126/science.abb8925
41. Price JV, Vance RE. The macrophage paradox. Immunity. (2014) 41:685–93. doi: 10.1016/j.immuni.2014.10.015
42. Feng Z, Diao B, Wang R, Wang G, Wang C, Tan Y, et al. The novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) directly decimates human spleens and lymph nodes. MedRxiv. doi: 10.1101/2020.03.27.20045427
43. Channappanavar R, Fett C, Mack M, Ten Eyck PP, Meyerholz DK, Perlman S. Sex-based differences in susceptibility to severe acute respiratory syndrome coronavirus infection. J Immunol. (2017) 198:4046–53. doi: 10.4049/jimmunol.1601896
44. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med. (2020) 12:1–3. doi: 10.1038/s41591-020-0901-9
45. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. (2020) 395:1033–4. doi: 10.1016/S0140-6736(20)30628-0
46. Bao L, Deng W, Huang B, Gao H, Liu J, Ren L, et al. The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature. (2020). doi: 10.1038/s41586-020-2312-y. [Epub ahead of print].
47. Xu H, Hou K, Xu H, Li Z, Chen H, Zhang N, et al. Acute myocardial injury of patients with coronavirus disease 2019. medRxiv. (2020). doi: 10.1101/2020.03.05.20031591
Keywords: SARS-CoV-2, macrophages, ACE2, lung, acute respiratory distress syndrome, defense, reservoir
Citation: Abassi Z, Knaney Y, Karram T and Heyman SN (2020) The Lung Macrophage in SARS-CoV-2 Infection: A Friend or a Foe? Front. Immunol. 11:1312. doi: 10.3389/fimmu.2020.01312
Received: 10 April 2020; Accepted: 26 May 2020;
Published: 05 June 2020.
Edited by:
Alexis M. Kalergis, Pontificia Universidad Católica de Chile, ChileReviewed by:
Christel Vérollet, UMR5089 Institut de Pharmacologie et de Biologie Structurale (IPBS), FranceCopyright © 2020 Abassi, Knaney, Karram and Heyman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zaid Abassi, YWJhc3NpQHR4LnRlY2huaW9uLmFjLmls
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.