
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 24 June 2020
Sec. Autoimmune and Autoinflammatory Disorders
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01300
 Shangling Zhu1†
Shangling Zhu1† Yuanmei Ye1†
Yuanmei Ye1† Yiming Shi2†
Yiming Shi2† Junlong Dang3
Junlong Dang3 Xiaoxue Feng1Yingdi Chen1
Xiaoxue Feng1Yingdi Chen1 Fang Liu1Nancy Olsen4
Fang Liu1Nancy Olsen4 Jianlin Huang1*
Jianlin Huang1* Song Guo Zheng5*
Song Guo Zheng5*Activated fibroblast-like synoviocytes (FLSs) play a central role in the formation of synovial pannus and joint destruction in rheumatoid arthritis (RA). Targeting FLSs could be a potential therapeutic strategy. The objective of this study is to explore the role of c-Jun N-terminal kinase (JNK) in proliferation, migration and invasion of FLSs promoted by the sonic hedeghog (SHH) signaling pathway in patients with RA. Activation of SHH signaling was evaluated by real-time PCR and Western Blot. Levels of phosphorylation of JNK and c-Jun were detected by Western Blot. FLSs proliferation was quantified by Cell Counting Kit-8 (CCK-8) assay and flow cytometry. Cell migration and invasion were assessed by wound healing assay and Transwell chamber assay. Invasiveness of FLSs in vivo was evaluated using a humanized synovitis animal model. We observed that treatment of SHH agonist (SAG) significantly increased the levels of phosphorylation of JNK and c-Jun, while SHH antagonist (cyclopamine) significantly decreased the expression of phospho-JNK and phospho-c-Jun in FLSs. The elevated level of phospho-c-Jun stimulated by SAG was decreased in the presence of JNK inhibitor (SP600125) (P < 0.001). FLSs proliferation, migration and invasion were promoted by SHH agonist (P < 0.05). However, the enhanced aggressiveness of FLSs was abolished in the presence of JNK inhibitor (P < 0.05). In vivo study showed that the invasion of FLSs into cartilage was increased by SHH overexpression and the excessive invasiveness was inhibited by blockade of JNK signaling (P < 0.01). These results suggest that JNK is one of the downstream molecules mediating the effect of SHH signaling in FLSs. These findings indicate that SHH-JNK signaling could be a potential therapeutic target to suppress the aggressiveness of FLSs and prevent articular damage of RA.
Rheumatoid arthritis (RA) is an immune-mediated inflammatory disease characterized by inflammation and damage in the joints (1). Activated fibroblast-like synoviocytes (FLSs), the major cell population in the hyperplastic synovial intimal lining, have a central role in the formation of synovial pannus and joint destruction (2). In RA, FLSs interact with immune cells and produce abundant inflammatory cytokines and chemokines, contributing to the perpetuation of inflammation (3–8). In addition, FLSs derived from RA patients display an aggressive phenotype of excessive proliferation, resistance to apoptosis and enhanced capacities to migrate and invade, the latter considered as the main cause of tissue damage (9). Therefore, understanding underlying mechanism(s) of aggressive features of FLSs has the potential to find novel targets for the treatment of RA, which may reduce inflammation as well as bone erosion (10–12).
The sonic hedgehog (SHH) signaling pathway is a conserved pathway and plays a critical role in embryonic development, stem cell maintenance and tissue homeostasis. SHH signaling is tightly regulated by various molecules. Briefly, the SHH ligand binds to the membrane receptor Patched1 and activates the G-protein coupled receptor-like protein named Smoothened (SMO). Subsequently, downstream molecules of SMO facilitate the signaling transduction from cytoplasm to nucleus, leading to the activation of the transcriptional factors GLI family zinc finger 1–3 (GLI1-3). Among the three GLI factors, GLI1 functions as a transcriptional activator and a target gene, while GLI2 and GLI3 are the mediators of signaling transduction and can act as activators or repressors according to the cellular context (13). Suppressor of Fused (SUFU) can directly bind to GLI transcription factors and act as a major negative regulator by promoting the generation of the repressor form of GLI or by preventing translocation to the nucleus (14, 15).
Inappropriate activation of SHH signaling was implicated in the tumorigenesis and metastasis of various human tumors. The underlying molecular mechanism is controversial; however, activated SHH signaling is proven to contribute to increased cell proliferation, differentiation and epithelial-to-mesenchymal transition which is correlated with cell invasion and metastatic abilities of tumors (16, 17). We have previously reported that SHH signaling was activated in the synovial tissue of RA patients (18). In addition, SHH signaling promoted FLSs proliferation and migration (19, 20), indicating that SHH signaling contributes to the aggressive behavior of FLSs. However, the mechanism by which SHH signaling promotes the tumor-like behavior of FLSs is still not yet fully elucidated.
Increasing evidence has suggested that mitogen-activated protein kinase (MAPK) signaling, including extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38, is implicated in the inflammation and joint destruction of RA (21, 22). We have recently reported that SHH signaling promoted FLSs proliferation and migration through activating ERK/MAPK (23). Therefore, we sought to determine the role of other MAPK signaling in SHH-induced FLSs proliferation, migration and invasion in the current study.
The study was approved by the Medical Ethics Committee of the Sixth Affiliated Hospital of Sun Yat-sen University. All patients provided written informed consent to participate in the study.
Nine patients with active RA, including 2 males and 7 females (aged from 46–59 years) were recruited, and synovial tissue samples were obtained during knee arthroscopy or synovectomy. RA patients were classified according to the 1987 American College of Rheumatology (ACR) revised classification criteria (24) and exhibited moderate to severe disease activity (Disease Activity Score of 28 joint counts >3.2). Normal cartilages were obtained from patients with traumatic injuries who were undergoing hip surgery. Animal study was approved by Guangdong Laboratory Animals Monitoring Institute.
The synovial tissue biopsies isolated from RA patients were finely minced into small pieces and transferred to a tissue culture flask in Dulbecco's modified Eagle's medium (DMEM) (Hyclone Laboratories, LOGAN, UT, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco, Grand Island, NY, USA), 100 U/mL penicillin (Gibco) and 100 μg/mL streptomycin (Gibco) in a humidified incubator at 37°C under 5% CO2. FLSs usually migrated out from the tissue and grew in monolayer in 10–14 days. After growth to ~95% confluency, cells were trypsinized and planted for proliferation. FLSs from passages 3–5 were used for planned experiments after being confirmed as being FLSs by morphology and purity analysis (<1% CD11b-positive, <1% CD 68-positive, <1% FcgRII and FcgRIII receptor-positive and >96% CD90-positive) (Supplementary Figure 1).
FLSs were treated with SHH agonist SAG (1 μM, Calbiochem, Darmstadt, Germany) or antagonist cyclopamine (10 μM, Sigma-Aldrich, Saint Louis, MO, USA) for 24 h. For the examination of production of matrix metalloproteinases (MMPs) and expression of IL-6 and IL-8, FLSs were treated with SAG (1 μM) alone or SAG with JNK inhibitor SP600125 (20 μM, Sigma-Aldrich, Saint Louis, MO, USA) for 24 h.
Total RNA from FLSs was prepared using E.Z.N.A. ®Total RNA Kit (OMEGA, Omega Bio-tek, Norcross, GA, USA) according to the manufacturer's protocol. cDNAs were synthesized using the PrimeScript™ RT reagent Kit with gDNA Eraser (Takara Biotechnology, Dalian, China) according to the manufacturer's instructions. To quantify the relative expressions of target genes, real time PCR was performed using TB Green® Premix Ex Taq™ II Kit (Takara Biotechnology) on a QuantStudio 5 Real-Time PCR System (Thermo Fisher Scientific Inc.,Waltham, MA, USA). All experiments were performed in triplicate and repeated at least three times with similar results. Relative levels were quantified by the comparative delta Ct method.
Primers for amplification were as follows (forward, reverse): SMO: (CCTGCTCACCTGGTCACTC, CACGGTATCGGTAGTTCTTGTAG), SUFU: (GCACATGCTGCTGACAGAGGAC, AGACACCAACGATCTGGAGGAAGG), GLI1: (AGGGAGTGCAGCCAATACAG, CCGGAGTTGATGTAGCTGGT), MMP1: (TTGCAGCTCATGAACTCGGC, CCGATGGGCTGGACAGGATT), MMP13: (TCCTGGCTGCCTTCCTCTTCTTG, AGTCATGGAGCTTGCTGCATTCTC), IL-6: (GGTGTTGCCTGCTGCCTTCC, GTTCTGAAGAGGTGAGTGGCTGTC), IL-8: (TCTCTTGGCAGCCTTCCTGA, TTTCTGTGTTGGCGCAGTGT), β-actin: (GGACTTCGAGCAAGAGATGG, AGCACTGTGTTGGCGTACAG).
To further verify the effects of SMO agonist and antagonist on SHH signaling, the protein expression of SMO, SUFU, and GLI1 was determined by Western Blot analysis. The activation of JNK signaling was measured by quantifying phosphorylation of JNK and c-Jun after SAG or cyclopamine treatment. For the detection of MMP1 and MMP 13, FLSs were treated with SAG (1 μM) alone or SAG with JNK inhibitor SP600125 (20 μM) for 48 h. Briefly, total protein was extracted using RIPA buffer (Sigma, Shanghai, China) supplemented with a cocktail of protease inhibitor (Roche, Mannheim, Germany), phosphatase inhibitor (Roche, Mannheim, Germany) and Phenylmethanesulfonyl fluoride (Sigma, Shanghai, China). For the detection of GLI1, nuclear protein was isolated using NE-PER Nuclear and Cytoplasmic Extraction Reagents (ThermoFisher Scientific, Rockford, IL, USA) according to the manufacturer's instructions. Polyacrylamide gel electrophoresis (PAGE) was performed to separate the proteins using 4–12% SurePAGE™ Gels (GeneScript, Piscataway, NJ, USA). The proteins were blotted onto a polyvinylidene fluoride (PVDF) membrane. After blocking at room temperature for 1 h, the membranes were incubated overnight at 4°C with primary antibodies. Primary antibodies were diluted 1:1000 for anti-SMO (Abcam, Cambridge, UK), anti-SUFU (Cell Signaling Technology, Shanghai, China) and anti-GLI1 antibody (Abcam). For the detection of JNK signaling, primary antibodies including anti-phospho-JNK, anti-JNK, anti-phospho-c-Jun and anti-c-Jun antibodies (1:1000, Cell Signaling Technology) were used. To detect the expression of MMP1 and MMP13, anti-MMP1 and anti-MMP13 antibody (1:1000, Abcam) were used. Subsequently membranes were incubated for 1 h at room temperature with secondary antibodies (1:5000, Cell Signaling Technology) conjugated with horseradish peroxidase. Immobilized proteins were measured by the enhanced chemiluminescent (ECL) detection system. The quantification of band density was performed using ImageJ software and the results were representative of at least four independent experiments.
To examine the effect of SAG and cyclopamine on the expression and distribution of GLI1, immunofluorescence was performed. FLSs were planted at the density of 2 × 104 cells/mL in glass bottom culture dishes (NEST, San Diego, CA, USA) and treated with SAG or cyclopamine for 48 h. Cells were fixed with 4% paraformaldehyde for 20 min and permeabilized with 0.1% Triton X-100 in phosphate buffered saline (PBS) for 15 min. After blocking at room temperature for 30 min, the cells were incubated with anti-GLI1 antibody (1:200, Abcam) overnight at 4°C. The cells were then incubated with Cy3-conjugated secondary antibody (1:500, Abcam) for 1 h and subsequently stained with DAPI for 15 min. The expression of GLI1 was determined using a confocal fluorescence microscope (Leica Microsystems-TCS SP8, Mannheim, Germany).
FLSs were seeded at a density of 3 × 104/mL in 96-well plates. Cells were treated with SAG or SP600125 for 48 h. Cell viability was subsequently assessed using the cell counting kit-8 (CCK-8, Dojindo, Tokyo, Japan) according to the manufacturer's instructions. Cells in the control group were treated with vehicle (DMSO in DMEM supplemented with 10% FBS). The experiments were performed in triplicate and repeated four times.
For cell cycle phase analysis, FLSs were plated at a density of 5 × 104/mL in 6-well plates and serum-starved for 24 h before incubated with SAG or SP600125. After treated for 18 h, FLSs were harvested and fixed in 70% cold ethanol overnight at −20°C. Fixed cells were subsequently stained using Cell Cycle Analysis Kit (Beyotime, Shanghai, China) according to the manufacturer's instructions. Stained cells were analyzed using a BD LSRFortessa™ Cell Analyzer (Becton Dickinson, San Jose, CA, USA). For each analysis, 10000 events were evaluated with the Flowjo software.
For Ki-67 staining, FLSs were first treated with SAG or SP600125 for 18 h. After being fixed and permeabilized with Foxp3/Transcription Factor Buffer Set (Invitrogen, Carslbad, CA, USA), cells were stained with anti-human Ki-67 PE antibody (1:100, Invitrogen, Carslbad, CA, USA) overnight at 4°C. Stained cells were washed with PBS and analyzed by a BD LSRFortessa™ Cell Analyzer (Becton Dickinson). Ki-67 expression was evaluated using Flowjo software.
For the detection of cell apoptosis, FLSs (5 × 104 cells/mL) were seeded into 6-well plates and treated with SAG or SP600125 for 24 h. Cell apoptosis was determined by AnnexinV/Dead Cell Apoptosis Kit (ESscience, Shanghai, China) according to the manufacturer's protocol. Stained cells were analyzed by a BD LSRFortessa™ Cell Analyzer (Becton Dickinson). AnnexinV-positive/PI-negative staining was regarded as apoptosis and the percentages of apoptotic cells were calculated using Flowjo software. The data were obtained from at least four independent experiments.
FLSs were cultured in 24-well plates and wounded with a 200 μL micropipette tip at 90% confluence. After washed three times with serum-free DMEM to remove the debris, the remaining cells were incubated with DMEM containing 2.5% FBS. SAG with or without SP600125 was added to the culture medium. After 12 h of incubation, FLSs were fixed in 4% paraformaldehyde for 15 min and stained with 0.1% crystal violet for 15 min. The number of cells that migrated beyond the scratching line was calculated. The experiments were repeated independently at least four times.
The ability of FLS migration was detected using Boyden chambers with 8.0 μm pore size Cell Culture Inserts (Corning Inc., Corning, NY, USA). Briefly, FLSs were trypsinized, collected and re-suspended in serum-free DMEM and planted at a density of 1 × 105 cells/mL in the upper chambers. DMEM containing 10% FBS (600 μL) was used as a chemoattractant. SAG with or without SP600125 was added to the lower compartment after the cells were adherent to the surface. After 16 h of incubation, cells remaining on the upper surface of the filter were removed with a cotton swab. The cells adhering beneath the filter were fixed in 4% paraformaldehyde for 15 min and stained with 0.1% crystal violet for 15 min. Migration ability of FLSs was quantified by calculating the mean number of migrated cells in five random fields at 50 magnifications for each assay.
To examine cell invasion, matrigel-coated membrane was used in the invasion assay. The matrigel basement membrane matrix (Corning Inc., Corning, NY, USA) was diluted with serum-free DMEM and coated on the inserts according to the manufacturer's instructions. FLSs were planted at a density of 2.5 × 105 cells/mL in the upper chambers. The following experiments were similar to the chemotaxis assay. The experiments were performed in duplicate and repeated at least three times.
To examine the level of active MMP1 in cell culture supernatants, FLSs were treated with SAG alone or SAG in the presence of SP600125 for 48 h before the cell culture supernatants were collected. The expression of active MMP1 was determined by Fluorokine® E Human Active MMP1 Fluorescent Assay Kit (R&D, MN, USA). All the procedures were performed according to the manufacturer's instructions. The samples were obtained from three independent experiments and assayed in duplicate.
FLSs were treated with SAG alone or in combination with SP600125 for 48 h and human IL-6 (R&D) and IL-8 (Invitrogen, Vienna, Austria) in cell culture supernatants were examined by ELISA Kits. All the procedures were performed according to the manufacturer's instructions.
AAV overexpressing SHH (CMV-SHH, serotype 5) and blank control virus (CMV-GFP, serotype 5) were purchased from Applied Biological Materials Inc. (Jiangsu, China). FLSs were infected with AAV at a MOI of 10000 and cultured for 7 days before harvested for further experiments. The overexpression of SHH after AAV infection in FLSs was verified by the real time PCR and Western Blot analysis.
We evaluated the invasiveness of FLSs in vivo using a humanized synovitis animal model as previously reported (25, 26). The severe combined immunodeficiency (SCID) mice were employed in the experiment. Briefly, a cartilage-gelatin sponge sandwich was made before implanted subcutaneously into the left side of SCID mouse in the first operation. Two weeks later, cartilage-sponge complex containing FLSs (5 × 105) was inserted under the right flank skin of SCID mouse. FLSs were infected with AAV overexpressing SHH or blank control AAV before implantation. For inhibition of JNK, mice were intraperitoneally injected with SP600125 (15 mg/kg) every 3 days for 45 days. The implants were harvested at day 60 and hematoxylin and eosin (H&E) staining was subsequently performed to evaluate the invasion of FLSs into cartilage. The clinical score including invaded distance and cartilage degradation was assessed by two trained researchers.
SPSS statistical software, version 20.0, was used for all statistical analyses. Values are presented as Means ± standard deviation (S.D.). Data were obtained from at least 3 independent experiments for the in vitro study. For the analysis of protein and mRNA expression, data were normalized and presented as fold change over the controls. The normality of data was examined by Shapiro-Wilk test and homogeneity of variances was examined by Levene test. Comparisons in two groups were performed using independent sample Student's t-test. Statistical differences among groups were tested by one-way analysis of variance (ANOVA). Post hoc comparisons were made by Dunnett's test. Statistical significance was set at P < 0.05.
SHH signaling is regulated by several key components of the pathway named smoothened (SMO), Suppressor of Fused (SUFU), and GLI zinc-finger proteins. To measure the effects of agonist and antagonist of SHH signaling, SMO, SUFU, and GLI1 expression was determined in the study since GLI1, which is regulated by SMO and SUFU, serves as the transcription activator of SHH signaling and controls the downstream target genes (13). As shown in Figure 1A, treatment of SHH agonist SAG significantly increased the expression of SMO and GLI1 mRNA, while treatment of SHH antagonist cyclopamine reduced the expression of SMO and GLI1 in FLSs. As SUFU is a major negative regulator of SHH signaling, this is expected that SUFU mRNA expression was inhibited by SAG, while the expression of SUFU was promoted by blocking SHH signaling with cyclopamine. The effect of SAG and cyclopamine on the expression of SMO and SUFU protein was similar to the results of mRNA expression (Figure 1B).
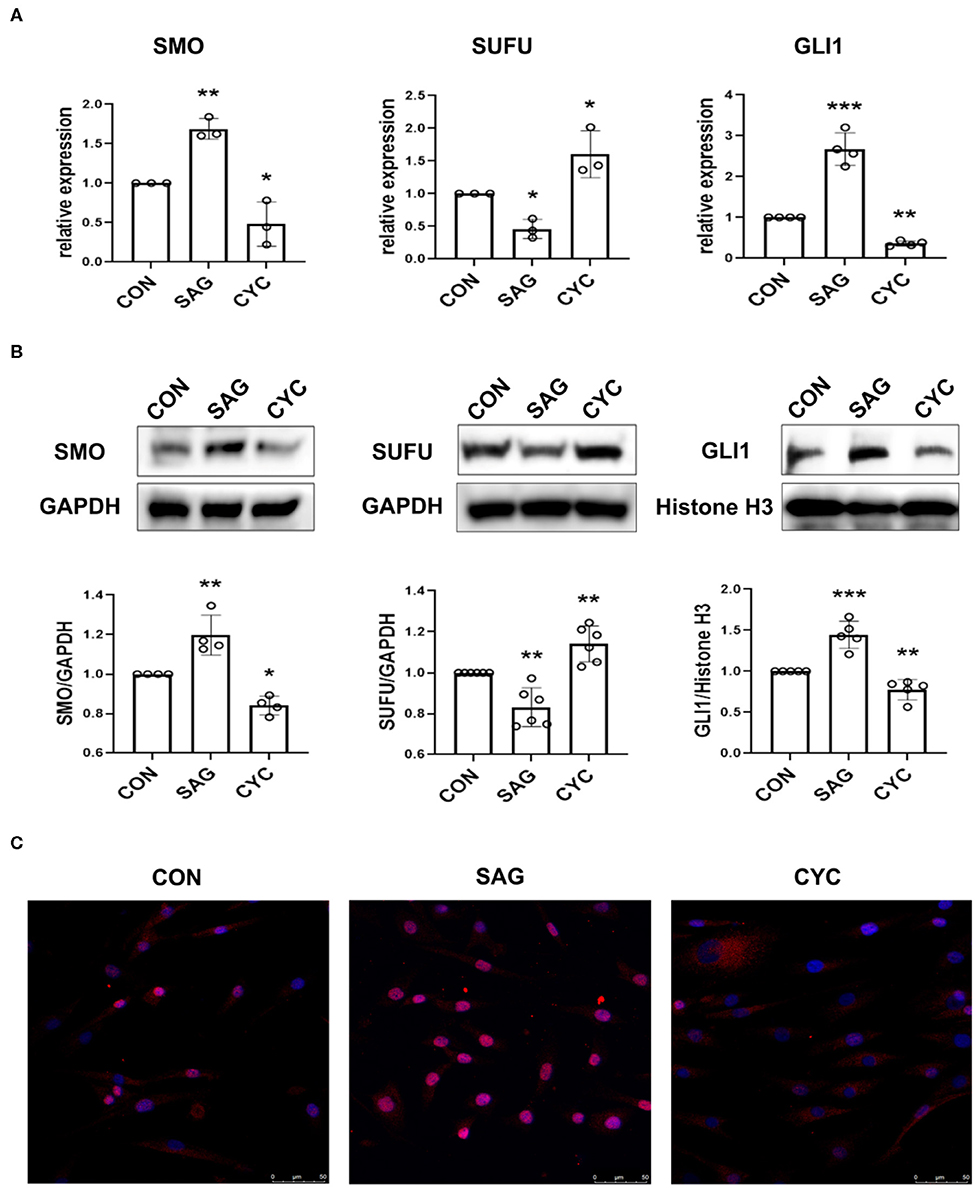
Figure 1. SAG and cyclopamine modulate the activation of SHH signaling. (A) SMO, SUFU and GLI1 mRNA expression was evaluated by real-time PCR after FLSs were treated with SAG (1 μM) or cyclopamine (10 μM) for 24 h. Relative quantification of gene expression was performed by the 2−ΔΔCt method. (B) The expression of SMO, SUFU and GLI1 was measured by Western Blot after FLSs were treated with SAG (1 μM) or cyclopamine (10 μM) for 48 h. Total protein was isolated for detection of SMO and SUFU and nuclear extracts were prepared for detection of GLI1. Results were normalized to loading control (GAPDH for total protein and Histone H3 for nuclear extracts). The results are shown as the mean ± S.D. (C) The distribution of GLI1 protein was measured by immunofluorescence (n = 3). FLSs were treated with SAG or cyclopamine for 48 h and stained with anti-GLI1 antibody (red signal) and DAPI (blue signal). Representative images are shown at 200 × magnification. FLSs in control group were treated with vehicle (DMSO supplemented in DMEM with 10% FBS). Data were analyzed using one-way analysis of variance (ANOVA). post hoc comparisons were made by Dunnett's test (A,B). *P < 0.05 vs. control group. **P < 0.01 vs. control group. ***P < 0.001 vs. control group. CON, control group; CYC, cyclopamine; SP, SP600125.
To further confirm the effects of SAG and cyclopamine on the activation of SHH signaling, nuclear protein was extracted and the expression of transcription factor GLI1 was determined. In addition, the distribution of GLI1 in FLSs after treated with SAG and cyclopamine was also evaluated in the study. Indeed, we observed that SAG significantly increased the expression of GLI1 protein in the nucleus and inhibition of SHH signaling with cyclopamine decreased the level of GLI1 expression (Figure 1B). The immunofluorescence staining showed that FLSs treated with SAG displayed translocation of GLI1 protein from cytoplasm to nucleus, while cells treated with cyclopamine demonstrated lower proportion of GLI1 accumulation in the nucleus (Figure 1C). These results suggest that SAG and cyclopamine effectively but differently modulate the activation of SHH signaling pathway.
To determine the effects of SAG and cyclopamine on the activation of JNK-MAPK signaling, the phosphorylation of JNK and c-Jun was measured. The results showed that SAG significantly increased the level of phosphorylation of JNK and c-Jun in FLSs, compared with the controls (Figures 2A,C). We also observed that the phosphorylation of JNK and c-Jun was significantly inhibited in the presence of SHH antagonist (Figures 2B,D). Moreover, we also noted that phosphorylation of c-Jun stimulated by SAG was significantly inhibited in the presence of JNK specific inhibitor SP600125 (Figure 2E).
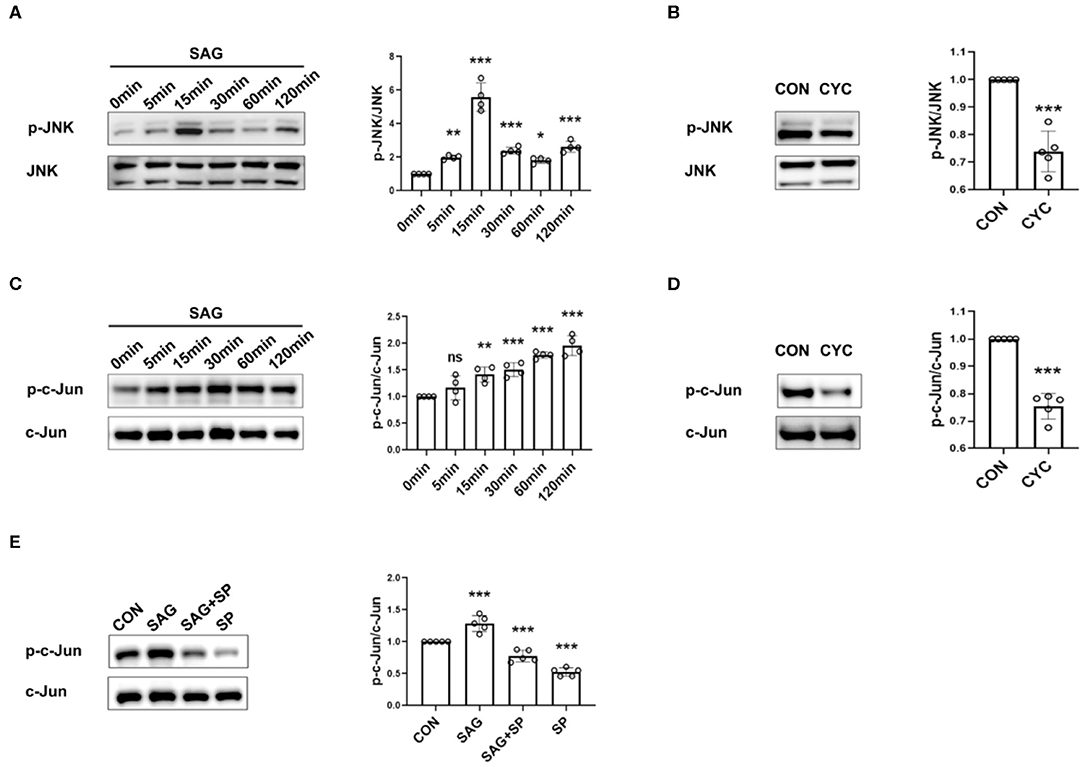
Figure 2. SHH agonist and antagonist regulate JNK signaling. (A–E) Effects of SHH agonist (SAG) and antagonist (cyclopamine) on the phosphorylation of JNK and c-Jun were evaluated by Western Blot. FLSs were stimulated with SAG (1 μM) at different time points (5, 15, 30, 60, 120 min) and the levels of phospho-JNK and phospho-c-Jun were determined by Western Blot (n = 4) (A,C). To detect the effect of SHH antagonist on JNK signaling, FLSs were treated with cyclopamine (10 μM) for 24 and 48 h before the levels of phospho-JNK (B) and phospho-c-Jun (D) were detected, respectively (n = 5). The expression of phospho-c-Jun was evaluated after FLSs were treated with SAG in the presence of JNK inhibitor SP600125 for 15 min (n = 5) (E). Results were normalized to the levels of total JNK or c-Jun. FLSs in control group were treated with vehicle (DMSO supplemented in DMEM with 10%FBS). The results are shown as the mean ± S.D. Data were analyzed using ANOVA with Dunnett's post hoc test (A,C,E) or independent sample Student's t-test (B,D). *P < 0.05 vs. control group. **P < 0.01 vs. control group. ***P < 0.001 vs. control group. ns, non-significant; CON, control group; CYC, cyclopamine; SP, SP600125.
To elucidate the effects of SHH-JNK signaling on FLSs proliferation, the cell counting kit-8 (CCK-8) assay was performed to evaluate the cell viability. We found that SAG significantly increased cell viability compared with controls (P < 0.01). However, the effect of SAG on cell viability was abolished by JNK inhibition (P < 0.01, Figure 3A). To exclude the non-specific cytotoxicity of JNK inhibition on cell viability, we further determined the effect of SHH-JNK signaling on cell apoptosis. As shown in Figures 3F,G, SHH-JNK signaling did not affect the cell apoptosis status of FLSs. Therefore, we propose that the effect of SHH-JNK signaling on cell viability is attributed to its impact on cell proliferation.
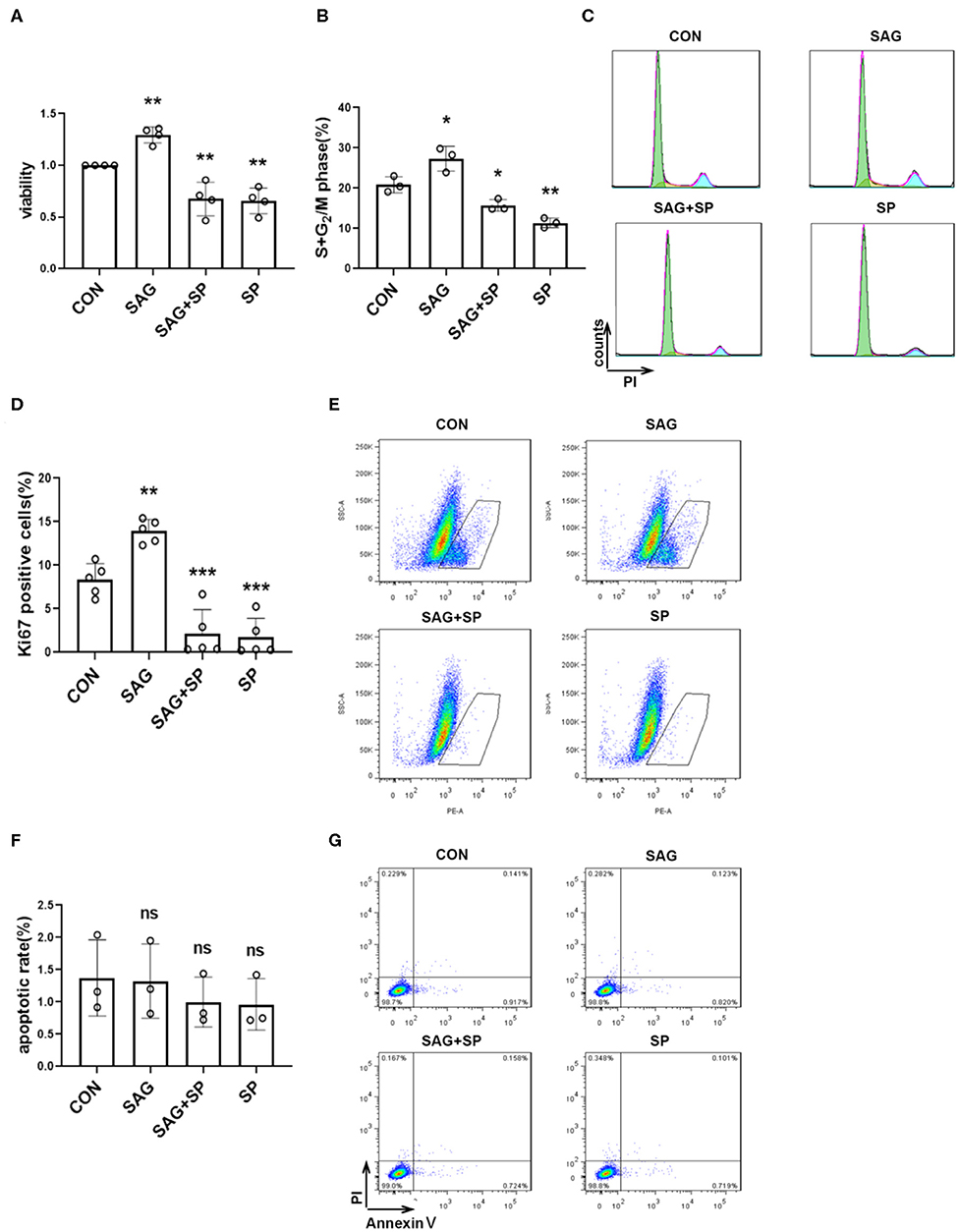
Figure 3. SHH agonist promotes FLSs proliferation via JNK. (A) FLSs viability was assessed by Cell Counting Kit-8 assay after FLSs were treated with SHH agonist (SAG, 1 μM) alone or SAG with JNK inhibitor (SP600125, 20 μM) for 48 h (n = 4). (B–G) Cell cycle analysis, Ki-67 expression and cell apoptosis were assessed by flow cytometry. For the detection of cell cycle distribution (B,C) and expression of Ki-67 (D,E), FLSs were treated with SAG (1 μM) with or without SP600125 (20 μM) for 18 h. For the detection of cell apoptosis (F,G), FLSs were treated with SAG (1 μM) with or without SP600125 (20 μM) for 24 h. Percentage of apoptosis was defined by the number of AnnexinV+PI− cells. FLSs in control group were treated with vehicle (DMSO supplemented in DMEM with 10%FBS). The results are shown as the mean ± S.D. Data were analyzed using ANOVA and post hoc comparisons were made by Dunnett's test (A,B,D,F). *P < 0.05 vs. control group. **P < 0.01 vs. control group. ***P < 0.001 vs. control group. ns, non-significant; CON, control group; SP, SP600125.
Additionally, the effect of SHH-JNK signaling on cell cycle distribution was assessed by flow cytometry. The results showed that stimulation of SAG for 18 h significantly increased the percentage of cells in the S and G2 phases compared to controls (27.35 ± 3.06% vs. 20.86 ± 1.98%, P < 0.05). We also found that percentages of cells in the S and G2 phases promoted by SAG were significantly decreased in the presence of SP600125 (15.73 ± 1.42%, Figures 3B,C).
Furthermore, we detected the effect of SHH-JNK on the expression of Ki-67 by flow cytometry. As shown in Figures 3D,E, incubation with SAG for 18 h resulted in an increase of Ki-67 expression in FLSs compared to that of control group (13.92 ± 1.33% vs. 8.34 ± 1.83%, P < 0.01). The percentage of Ki-67 positive cells was significantly decreased after incubation with SAG and SP600125 (2.17 ± 2.74%, P < 0.001). These results indicate that SHH promotes FLSs proliferation in a JNK-dependent manner.
To examine whether SHH signaling regulates migration of FLSs via JNK, we treated FLSs with SAG in the presence of JNK specific inhibitor SP600125 in wound healing and cell chemotaxis assays. We observed that the migration ability of FLSs was significantly enhanced after treatment with SAG, compared with controls (P < 0.01). However, we also noticed that the SAG-induced migration ability of FLSs was inhibited by blocking JNK with SP600125 (Figures 4A–D).
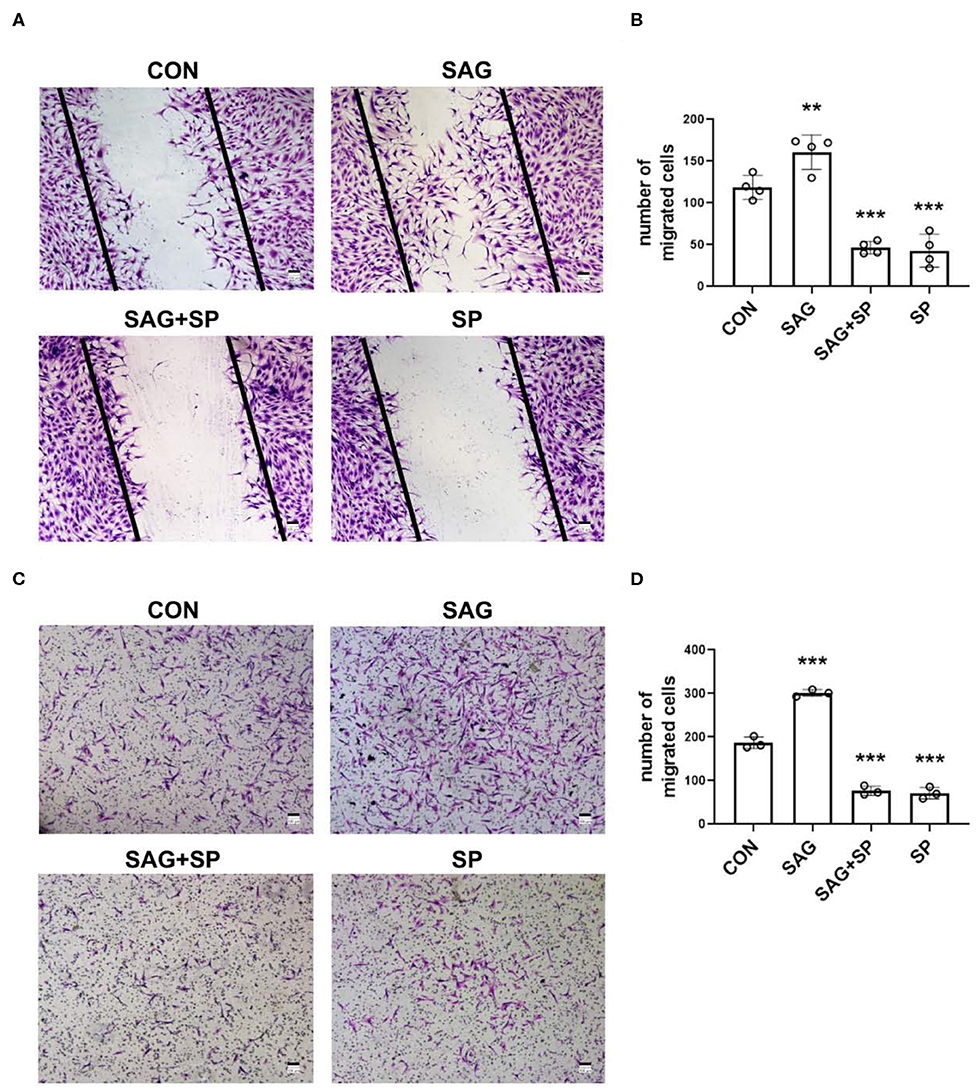
Figure 4. SHH agonist promotes FLSs migration via JNK. (A–D) FLSs migration was evaluated by wound healing assay and chemotaxis assay. In the wound healing assay, FLSs were treated with SAG (1 μM) or SAG in the presence of SP600125 (20 μM) for 12 h after scratching (n = 4) (A,B). In the chemotaxis assay, FLSs were planted in the upper chambers. DMEM containing 10% FBS, SAG (1 μM) with or without SP600125 (20 μM) was added to the lower compartment. Cells were treated for 16 h (n = 3) (C,D). The numbers of migrated cells in wound healing assay and chemotaxis assay were calculated. FLSs in control group were treated with vehicle (DMSO supplemented in DMEM with 2.5% FBS in wound healing assay and 10% FBS in chemotaxis assay). The results are shown as the mean ± S.D. Data were analyzed using ANOVA and post hoc comparisons were made by Dunnett's test (B,D). **P < 0.01 vs. control group. ***P < 0.001 vs. control group. CON, control group; SP, SP600125.
As shown in Figure 5A, FLSs stimulated with SHH agonist showed increased invasiveness. To determine whether the SAG-induced increase in cell invasiveness was dependent upon JNK signaling, invasion through matrigel was studied in the presence SP600125 in the lower chambers. The results demonstrated that blockage of JNK with SP600125 significantly decreased the SAG-induced cell invasion (Figures 5A,B).
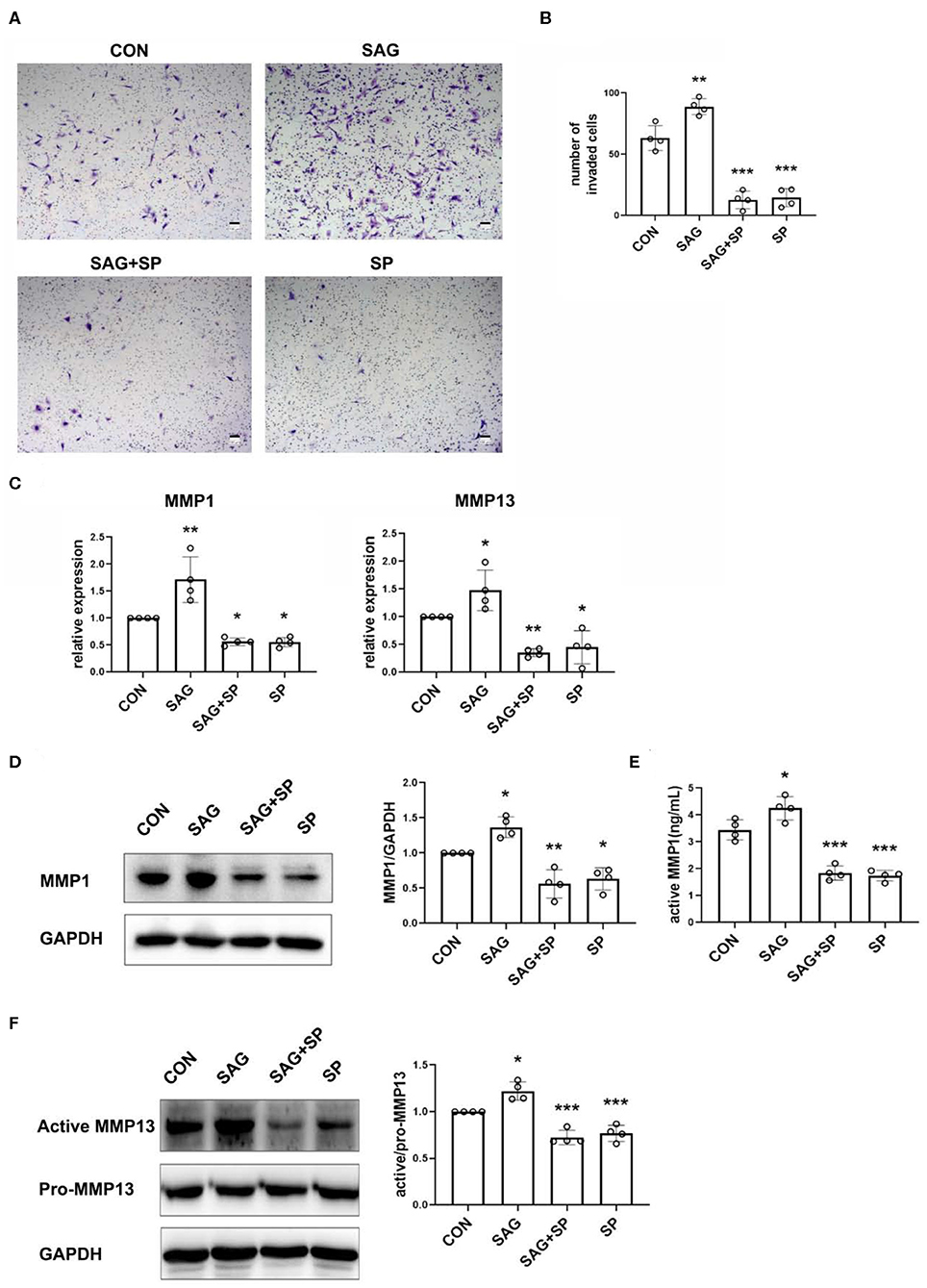
Figure 5. SHH agonist increases FLSs invasion via JNK. (A,B) FLSs invasion was evaluated by Boyden chambers with matrigel-coated membrane used in the system (n = 4). FLSs were planted in the upper chambers. DMEM containing 10% FBS, SAG (1 μM) with or without SP600125 (20 μM) was added to the lower compartment. Cells were treated for 16 h. The numbers of invaded cells were calculated. (C) MMP1 and MMP13 mRNA expression was evaluated by real-time PCR after FLSs were treated with SAG (1 μM) or SAG in the presence of SP600125 (20 μM) for 24 h (n = 4). (D,E) Level of MMP1 protein expression was determined by Western Blot and active MMP1 level in cell culture supernatants was quantified by Fluorokine E activity assay after FLSs were treated with SAG (1 μM) or SAG in the presence of SP600125 (20 μM) for 48 h (n = 4). (F) The proenzyme and active form of MMP13 were examined by Western Blot (n = 4). The results are shown as the mean ± S.D. Data were analyzed using ANOVA and post hoc comparisons were made by Dunnett's test (B–F). *P < 0.05 vs. control group. **P < 0.01 vs. control group. ***P < 0.001 vs. control group. CON, control group; SP, SP600125.
To further detect the mechanism of SHH-JNK on cell invasion, we examined the levels of MMP1 and MMP13 mRNA and protein expression in FLSs after being treated with SAG alone or SAG in the presence of SP600125. The results showed that mRNA expression of MMP1 and MMP13 was significantly increased in FLSs treated with SAG compared to the controls, and the effect of SAG on MMP1, MMP13 mRNA expression was inhibited in the presence of SP600125 (Figure 5C). In addition, the expression of pro-MMP1 in cell lysates and level of active MMP1 in cell supernatants were also significantly increased by SAG. However, the levels of pro- and active MMP1 were dramatically reduced in the presence of SP600125 (Figures 5D,E). Similarly, SAG also promoted the expression of active MMP13 in FLSs, with an increase in the ratio of active and pro-MMP13. The co-treatment of SAG and SP600125 led to a remarkable decrease in active MMP13 production (Figure 5F).
To determine the role of SHH-JNK signaling in expression of pro-inflammatory cytokines by FLSs, the mRNA expression of IL-6 and IL-8 was detected after the FLSs were treated with SAG alone or in combination with SP600125. Upregulated expression of IL-6 and IL-8 was observed after stimulation of SAG; however, the elevated expression of IL-6 and IL-8 was not suppressed in the presence of SP600125 (Supplementary Figure 2A). In addition, we detected the production of IL-6 and IL-8 in cell culture supernatants and found that SAG stimulation significantly promoted the expression of IL-6 and IL-8, which was similar to the results of mRNA expression. Of note, we aslo observed that JNK inhibitor did not significantly suppress the production of IL-6 and IL-8 under normal conditions (Supplementary Figure 2B).
To further assess the effect of SHH-JNK signaling on FLSs invasiveness in vivo, we employed a humanized synovitis animal model and FLSs were infected with AAV overexpressing SHH before implantation (Figure 6A). As shown in Figures 6B,C, FLSs overexpressing SHH significantly increased the invaded distance and cartilage degradation in the primary and contralateral cartilage, indicating SHH significantly promoted the invasiveness of FLSs. Of note, the destruction of contralateral cartilages which are not directly exposed to FLSs has been attributed to FLSs migration and invasion abilities through the marine body. Therefore, the invasion and destruction of contralateral cartilages not only displayed the invasiveness of FLSs, but also revealed the migration of FLSs in vivo. Moreover, we observed that administration of JNK inhibitor to the mice significantly decreased the cartilage invasion by FLSs carrying overexpressed SHH.
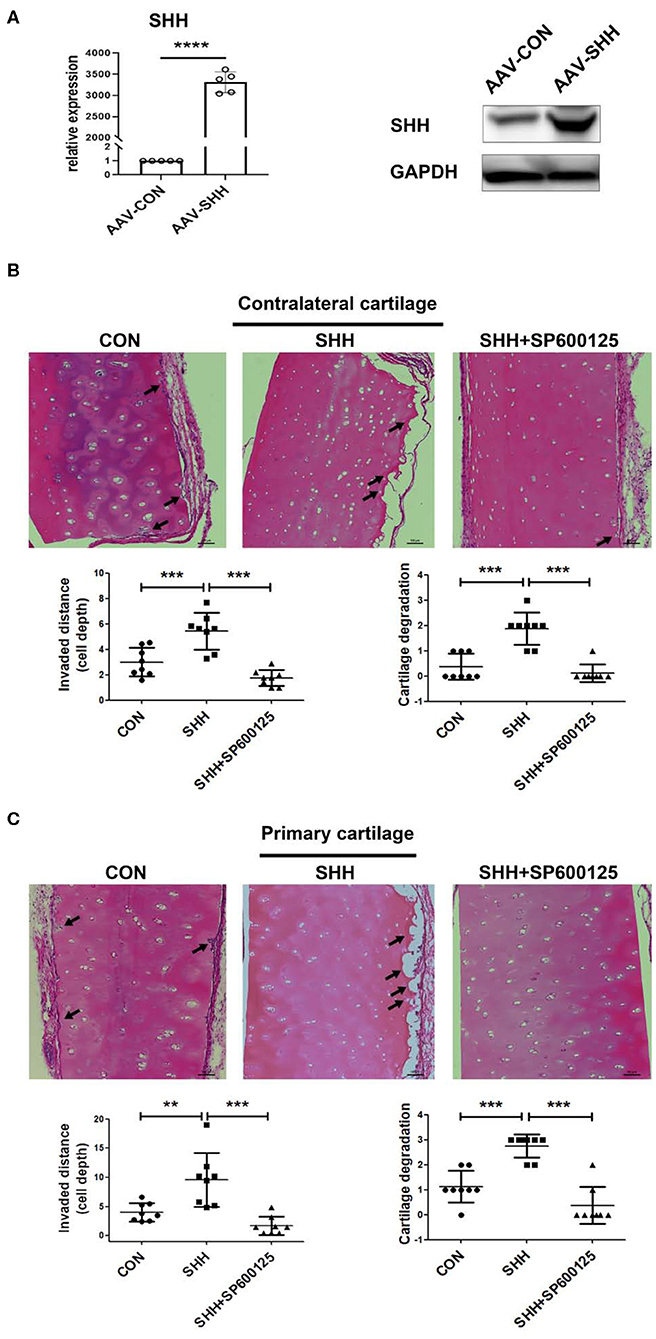
Figure 6. Inhibition of JNK alleviates FLSs invasiveness in vivo promoted by SHH. The invasiveness of FLSs in vivo was evaluated using a humanized synovitis animal model. SCID mice were used in the experiments (n = 5 in each group). A cartilage-sponge complex without FLSs was implanted under the left flank skin of a SCID mouse (contralateral cartilage), and cartilage-sponge complex containing FLSs (5 × 105) was inserted under the right flank skin (primarily cartilage) 2 weeks later. FLSs were infected with AAV virus overexpressing SHH (CMV-SHH, serotype 5) or blank control virus (CMV-GFP, serotype 5) before implantation. The overexpression of SHH in FLSs after AAV virus infection was verified by the real time PCR and Western Blot analysis (A). Mice were injected with SP600125 (15 mg/kg) every 3 days for 45 days. At day 60, the cartilages from mice were harvested and subjected to H&E staining. Representative images were shown at 100 × magnification. The arrow indicated the lesion of cartilage invasion by FLSs (B,C). Data were analyzed using ANOVA and post hoc comparisons were made by Dunnett's test. Comparisons in two groups were performed using independent sample Student's t-test. **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. SHH group. CON, control group.
Aggressive behavior of FLSs as dysregulated proliferation, increased ability for migration and invasiveness is responsible for pannus formation of RA patients and mediates inflammation and destruction of the joint (27). We have previously reported that SHH signaling promoted FLSs proliferation and migration (19, 20). In line with our findings, a recent study also revealed that inhibition of GLI1 significantly decreased FLSs proliferation (28). However, the mechanism by which SHH signaling promotes the aggressive phenotype of FLSs is not completely clarified.
MAPK cascade is one of the most widely studied signaling pathways in inflammatory diseases. Of the three pathways of classical MAPK family, JNK is a significant contributor to the pathological changes of RA for its ability to phosphorylate c-Jun, which then initiates the expression of MMPs. In vivo study showed that inhibition of JNK with antagonist SP600125 alleviated the severity of arthritis and prevented bone destruction in adjuvant-induced arthritis (29). As we previously found that ERK/MAPK was activated by SHH signaling and involved in SHH-driven proliferation and migration of FLSs (23), we are aware that ERK/MAPK is not the only mediator of abnormal behavior of FLSs, and in this study we sought to determine whether JNK contributes to FLSs aggressiveness promoted by SHH signaling.
In the present study, we showed that activation of SHH signaling increased phosphorylation of JNK and c-Jun, conversely, SHH blockade significantly inhibited the activation of JNK signaling. The phosphorylation of c-Jun stimulated by SHH agonist was inhibited in the presence of JNK inhibitor. Additionally, we reported that treating FLSs with SHH agonist significantly increased proliferation, migration, and invasion, and FLSs overexpressing SHH showed increased invasiveness into cartilage in vivo, indicating SHH signaling is involved in the activated and aggressive phenotype of FLSs in RA. We further provided evidence that inhibiting JNK with antagonist SP600125 reversed the aggressive behavior of FLSs in vitro and in vivo. Our observations suggest that SHH signaling promotes FLSs proliferation, migration and invasion in a JNK dependent manner. Although activation of SHH signaling promotes the expression of pro-inflammatory cytokines such as IL-6 and IL-8 in FLSs, the role of JNK signaling in SHH-mediated cytokine expression appears to be not essential.
Previous studies have focused on the crosstalk between SHH-GLI signaling and several oncogenic pathways including MAPKs, PI3K/AKT/mTOR, and TGFβ/SMAD signaling pathways, and implicated the strategy of targeting SHH-GLI and MEK1/2 for the therapy of cancers (30). However, evidence about the interaction between SHH-GLI1 signaling and JNK-MAPK signaling is limited. Previous studies reported that overexpression of SHH elevated phosphorylation of c-Jun in keratinocytes (31). Other studies have shown that JNK activation was involved in bFGF-mediated inhibition of SHH-induced proliferation in granule cell precursors (32). Studies on chemoresistant cancer cells suggested that JNK was activated by SMO through Gβγ and stimulated GLI activity, consequently contributing to chemoresistance (33). Therefore, the mechanism of cross-talk between SHH-GLI and JNK is still unclear and may differ depending on cellular contexts. Our results now for the first time demonstrate that activation of SHH signaling promotes activation of JNK-MAPK and it is likely that JNK is one of the downstream molecules mediating the effect of SHH signaling in FLSs. However, whether JNK activation occurs in upstream of SHH signaling target GLI1 in FLSs remains to be further investigated.
Specifically, MMPs are considered to be major players in destruction of extracellular matrix (ECM) components in RA and other inflammatory diseases (34–37). Additionally, the collagenases MMP1 and MMP13 are associated with the invasiveness of FLSs and articular damage (38, 39). In this study, we have observed SHH signaling significantly increased the expression of MMP1 and MMP13, and blockage of JNK showed a decrease in MMP1 and MMP13 expression. Our study suggests that SHH-JNK signaling may promote invasiveness of FLSs through secretion of MMP1 and MMP13, and could be a potential target to reduce cartilage damage in RA.
We have identified new evidence implicating the interaction between SHH signaling and JNK-MAPK signaling pathway and its role in the regulation of aggressive phenotype of FLSs from RA patients. These findings suggest that SHH signaling could be a potential therapeutic target to suppress the aggressiveness of RA-FLSs, and prevent or even treat articular damage in RA.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
The studies involving human participants were reviewed and approved by Medical Ethics Committee of the Sixth Affiliated Hospital of Sun Yat-sen University. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Guangdong Laboratory Animals Monitoring Institute.
JH and SGZ: conceptualization. XF, YC, and FL: formal analysis. SGZ, JH, and SZ: funding acquisition. SZ, YY, YS, and JD: investigation. SZ, YY, YS, and JD: methodology. JH and SGZ: supervision. SZ, YY, and YS: writing—original draft. NO: writing—review & editing. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Natural Science Foundation of China (81571584, 81671611, 81701609, 81871224, and 81871270). SGZ was in part supported from NIH R01AR059103, NIH STAR Award, and NIH R61/3 AR073409.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.01300/full#supplementary-material
Supplementary Figure 1. Cell morphology and detection of surface molecules of FLSs. (A) FLSs at passage 3 were characterized as spindle-cell morphology under a light microscope (100 × magnification). (B) The surface molecules CD90, CD68, CD11b, FcgRII, and FcgRIII receptor were detected by flow cytometry.
Supplementary Figure 2. Effect of SHH-JNK signaling on IL-6 and IL-8 expression in FLSs. (A) The mRNA expression of IL-6 and IL-8 was determined by real-time PCR after FLSs were treated with SAG (1 μM) or SAG in the presence of SP600125 (20 μM) for 24 h. (B) The expression of IL-6 and IL-8 in cell culture supernatants was measured by an ELISA. The results are shown as the mean ± S.D. Data were analyzed using ANOVA and post hoc comparisons were made by Dunnett's test. *P < 0.05 vs. control group. **P < 0.01 vs. control group. ns, non-significant; CON, control group; SP, SP600125.
AAV, adeno-associated virus; ACR, American College of Rheumatology; CCK-8, cell counting kit-8; DMEM, Dulbecco's modified Eagle's medium; DMSO, dimethyl sulfoxide; ECL, enhanced chemiluminescent; ECM, extracellular matrix; ERK, extracellular signal-regulated kinase; FBS, fetal bovine serum; FLSs, fibroblast-like synoviocytes; GLI1, GLI family zinc finger 1; JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; MMP, matrix metalloproteinase; PAGE, polyacrylamide gel electrophoresis; PBS, phosphate buffered saline; PVDF, polyvinylidene fluoride; RA, rheumatoid arthritis; SCID, severe combined immunodeficiency; SHH, sonic hedgehog; SMO, smoothened; SUFU, suppressor of Fused.
1. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. (2016) 388:2023–38. doi: 10.1016/S0140-6736(16)30173-8
2. Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev. (2010) 233:233–55. doi: 10.1111/j.0105-2896.2009.00859.x
3. Zou Y, Xu S, Xiao Y, Qiu Q, Shi M, Wang J, et al. Long noncoding RNA LERFS negatively regulates rheumatoid synovial aggression and proliferation. J Clin Invest. (2018) 128:4510–24. doi: 10.1172/JCI97965
4. Mo BY, Guo XH, Yang MR, Liu F, Bi X, Liu Y, et al. Long non-coding RNA GAPLINC promotes tumor-like biologic behaviors of fibroblast-like synoviocytes as microRNA sponging in rheumatoid arthritis patients. Front Immunol. (2018) 9:702. doi: 10.3389/fimmu.2018.00702
5. Liu Y, Pan YF, Xue YQ, Fang LK, Guo XH, Guo X, et al. uPAR promotes tumor-like biologic behaviors of fibroblast-like synoviocytes through PI3K/Akt signaling pathway in patients with rheumatoid arthritis. Cell Mol Immunol. (2018) 15:171–81. doi: 10.1038/cmi.2016.60
6. Li N, Wang JC, Liang TH, Zhu MH, Wang JY, Fu XL, et al. Pathologic finding of increased expression of interleukin-17 in the synovial tissue of rheumatoid arthritis patients. Int J Clin Exp Pathol. (2013) 6:1375–9.
7. Luo Y, Wu W, Gu J, Zhang X, Dang J, Wang J, et al. Human gingival tissue-derived MSC suppress osteoclastogenesis and bone erosion via CD39-adenosine signal pathway in autoimmune arthritis. EBioMedicine. (2019) 43:620–31. doi: 10.1016/j.ebiom.2019.04.058
8. Luo Y, Zheng SG. Hall of fame among pro-inflammatory cytokines: interleukin-6 gene and its transcriptional regulation mechanisms. Front Immunol. (2016) 7:604. doi: 10.3389/fimmu.2016.00604
9. Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol. (2013) 9:24–33. doi: 10.1038/nrrheum.2012.190
10. Kong N, Lan Q, Su W, Chen M, Wang J, Yang Z, et al. Induced T regulatory cells suppress osteoclastogenesis and bone erosion in collagen-induced arthritis better than natural T regulatory cells. Ann Rheum Dis. (2012) 71:1567–72. doi: 10.1136/annrheumdis-2011-201052
11. Chen W, Wang J, Xu Z, Huang F, Qian W, Ma J, et al. Apremilast ameliorates experimental arthritis via suppression of Th1 and Th17 cells and enhancement of CD4+Foxp3+ regulatory T cells differentiation. Front Immunol. (2018) 9:1662. doi: 10.3389/fimmu.2018.01662
12. Zhou L, Wang J, Li J, Li T, Chen Y, June RR, et al. 1,25-Dihydroxyvitamin D3 ameliorates collagen-induced arthritis via suppression of Th17 cells through miR-124 mediated inhibition of IL-6 signaling. Front Immunol. (2019) 10:178. doi: 10.3389/fimmu.2019.00178
13. Hui CC, Angers S. Gli proteins in development and disease. Annu Rev Cell Dev Biol. (2011) 27:513–37. doi: 10.1146/annurev-cellbio-092910-154048
14. Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. (2009) 9:631–43. doi: 10.1038/nrc2713
15. Infante P, Faedda R, Bernardi F, Bufalieri F, Lospinoso Severini L, Alfonsi R, et al. Itch/β-arrestin2-dependent non-proteolytic ubiquitylation of SuFu controls Hedgehog signalling and medulloblastoma tumorigenesis. Nat Commun. (2018) 9:976. doi: 10.1038/s41467-018-03339-0
16. Louro ID, Bailey EC, Li X, South LS, McKie-Bell PR, Yoder BK, et al. Comparative gene expression profile analysis of GLI and c-MYC in an epithelial model of malignant transformation. Cancer Res. (2002) 62:5867–73.
17. Yue D, Li H, Che J, Zhang Y, Tseng HH, Jin JQ, et al. Hedgehog/Gli promotes epithelial-mesenchymal transition in lung squamous cell carcinomas. J Exp Clin Cancer Res. (2014) 33:34. doi: 10.1186/1756-9966-33-34
18. Wang M, Zhu S, Peng W, Li Q, Li Z, Luo M, et al. Sonic hedgehog signaling drives proliferation of synoviocytes in rheumatoid arthritis: a possible novel therapeutic target. J Immunol Res. (2014) 2014:401903. doi: 10.1155/2014/401903
19. Zhu SL, Huang JL, Peng WX, Wu DC, Luo MQ, Li QX, et al. Inhibition of smoothened decreases proliferation of synoviocytes in rheumatoid arthritis. Cell Mol Immunol. (2017) 14:214–22. doi: 10.1038/cmi.2015.67
20. Peng WX, Zhu SL, Zhang BY, Shi YM, Feng XX, Liu F, et al. Smoothened regulates migration of fibroblast-like synoviocytes in rheumatoid arthritis via activation of Rho GTPase signaling. Front Immunol. (2017) 8:159. doi: 10.3389/fimmu.2017.00159
21. Schett G, Tohidast-Akrad M, Smolen JS, Schmid BJ, Steiner CW, Bitzan P, et al. Activation, differential localization, and regulation of the stress-activated protein kinases, extracellular signal-regulated kinase, c-JUN N-terminal kinase, and p38 mitogen-activated protein kinase, in synovial tissue and cells in rheumatoid arthritis. Arthritis Rheum. (2000) 43:2501–12. doi: 10.1002/1529-0131(200011)43:11<2501::AID-ANR18>3.0.CO;2-K
22. Schett G, Zwerina J, Firestein G. The p38 mitogen-activated protein kinase (MAPK) pathway in rheumatoid arthritis. Ann Rheum Dis. (2008) 67:909–16. doi: 10.1136/ard.2007.074278
23. Liu F, Feng XX, Zhu SL, Huang HY, Chen YD, Pan YF, et al. Sonic hedgehog signaling pathway mediates proliferation and migration of fibroblast-like synoviocytes in rheumatoid arthritis via MAPK/ERK Signaling pathway. Front Immunol. (2018) 9:2847. doi: 10.3389/fimmu.2018.02847
24. Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. (1988) 31:315–24. doi: 10.1002/art.1780310302
25. Dang J, Zhu S, Wang J. A protocol for humanized synovitis mice model. Am J Clin Exp Immunol. (2019) 8:47–52.
26. Lefèvre S, Knedla A, Tennie C, Kampmann A, Wunrau C, Dinser R, et al. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat Med. (2009) 15:1414–20. doi: 10.1038/nm.2050
27. Bi X, Guo XH, Mo BY, Wang ML, Luo XQ, Chen YX, et al. LncRNA PICSAR promotes cell proliferation, migration and invasion of fibroblast-like synoviocytes by sponging miRNA-4701–5p in rheumatoid arthritis. EBioMedicine. (2019) 50:408–20. doi: 10.2139/ssrn.3381127
28. Qin S, Sun D, Li X, Kong F, Yu Q, Hua H, et al. GANT61 alleviates arthritic symptoms by targeting fibroblast-like synoviocytes in CIA rats. J Orthop Sci. (2019) 24:353–60. doi: 10.1016/j.jos.2018.09.003
29. Han Z, Boyle DL, Chang L, Bennett B, Karin M, Yang L, et al. c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J Clin Invest. (2001) 108:73–81. doi: 10.1172/JCI12466
30. Rovida E, Stecca B. Mitogen-activated protein kinases and Hedgehog-GLI signaling in cancer: a crosstalk providing therapeutic opportunities. Semin Cancer Biol. (2015) 35:154–67. doi: 10.1016/j.semcancer.2015.08.003
31. Bigelow RL, Jen EY, Delehedde M, Chari NS, McDonnell TJ. Sonic hedgehog induces epidermal growth factor dependent matrix infiltration in HaCaT keratinocytes. J Invest Dermatol. (2005) 124:457–65. doi: 10.1111/j.0022-202X.2004.23590.x
32. Fogarty MP, Emmenegger BA, Grasfeder LL, Oliver TG, Wechsler-Reya RJ. Fibroblast growth factor blocks Sonic hedgehog signaling in neuronal precursors and tumor cells. Proc Natl Acad Sci USA. (2007) 104:2973–8. doi: 10.1073/pnas.0605770104
33. Zhan X, Wang J, Liu Y, Peng Y, Tan W. GPCR-like signaling mediated by smoothened contributes to acquired chemoresistance through activating Gli. Mol Cancer. (2014) 13:4. doi: 10.1186/1476-4598-13-4
34. Abeles AM, Pillinger MH. The role of the synovial fibroblast in rheumatoid arthritis: cartilage destruction and the regulation of matrix metalloproteinases. Bull NYU Hosp Jt Dis. (2006) 64:20–4.
35. Burrage PS, Mix KS, Brinckerhoff CE. Matrix metalloproteinases: role in arthritis. Front Biosci. (2006) 11:529–43. doi: 10.2741/1817
36. Lu L, Feng M, Gu J, Xia Z, Zhang H, Zheng S, et al. Restoration of intrahepatic regulatory T cells through MMP-9/13-dependent activation of TGF-β is critical for immune homeostasis following acute liver injury. J Mol Cell Biol. (2013) 5:369–79. doi: 10.1093/jmcb/mjt042
37. Chen X, Chang L, Li X, Huang J, Yang L, Lai X, et al. Tc17/IL-17A Up-regulated the expression of MMP-9 via NF-κB pathway in nasal epithelial cells of patients with chronic rhinosinusitis. Front Immunol. (2018) 9:2121. doi: 10.3389/fimmu.2018.02121
38. Huang TL, Mu N, Gu JT, Shu Z, Zhang K, Zhao JK, et al. DDR2-CYR61-MMP1 signaling pathway promotes bone erosion in rheumatoid arthritis through regulating migration and invasion of fibroblast-like synoviocytes. J Bone Miner Res. (2019) 34:779–80. doi: 10.1002/jbmr.3688
Keywords: rheumatoid arthritis, fibroblast-like synoviocyte, proliferation, migration, invasion, sonic hedgehog, c-Jun N-terminal kinase
Citation: Zhu S, Ye Y, Shi Y, Dang J, Feng X, Chen Y, Liu F, Olsen N, Huang J and Zheng SG (2020) Sonic Hedgehog Regulates Proliferation, Migration and Invasion of Synoviocytes in Rheumatoid Arthritis via JNK Signaling. Front. Immunol. 11:1300. doi: 10.3389/fimmu.2020.01300
Received: 09 December 2019; Accepted: 22 May 2020;
Published: 24 June 2020.
Edited by:
Myung-Shik Lee, Yonsei University Health System, South KoreaReviewed by:
Jeehee Youn, Hanyang University, South KoreaCopyright © 2020 Zhu, Ye, Shi, Dang, Feng, Chen, Liu, Olsen, Huang and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianlin Huang, amlhbmxpbl9oQDE2My5jb20=; Song Guo Zheng, c29uZ2d1b3poZW5nMjAxM0B5YWhvby5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.