 Zarina Brune
Zarina Brune Matthew R. Rice
Matthew R. Rice Betsy J. Barnes
Betsy J. Barnes
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Immunol., 03 June 2020
Sec. Cytokines and Soluble Mediators in Immunity
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01143
This article is part of the Research TopicModulating Cytokines as Treatment for Autoimmune Diseases and Cancer View all 14 articles
Interferon Regulatory Factor 5 (IRF5) is one of nine members of the IRF family of transcription factors. Although initially discovered as a key regulator of the type I interferon and pro-inflammatory cytokine arm of the innate immune response, IRF5 has now been found to also mediate pathways involved in cell growth and differentiation, apoptosis, metabolic homeostasis and tumor suppression. Hyperactivation of IRF5 has been implicated in numerous autoimmune diseases, chief among them systemic lupus erythematosus (SLE). SLE is a heterogeneous autoimmune disease in which patients often share similar characteristics in terms of autoantibody production and strong genetic risk factors, yet also possess unique disease signatures. IRF5 pathogenic alleles contribute one of the strongest risk factors for SLE disease development. Multiple models of murine lupus have shown that loss of Irf5 is protective against disease development. In an attempt to elucidate the regulatory role(s) of IRF5 in driving SLE pathogenesis, labs have begun to examine the function of IRF5 in several immune cell types, including B cells, macrophages, and dendritic cells. A somewhat untouched area of research on IRF5 is in T cells, even though Irf5 knockout mice were reported to have skewing of T cell subsets from T helper 1 (Th1) and T helper 17 (Th17) toward T helper 2 (Th2), indicating a potential role for IRF5 in T cell regulation. However, most studies attributed this T cell phenotype in Irf5 knockout mice to dysregulation of antigen presenting cell function rather than an intrinsic role for IRF5 in T cells. In this review, we offer a different interpretation of the literature. The role of IRF5 in T cells, specifically its control of T cell effector polarization and the resultant T cell-mediated cytokine production, has yet to be elucidated. A strong understanding of the regulatory role(s) of this key transcription factor in T cells is necessary for us to grasp the full picture of the complex pathogenesis of autoimmune diseases like SLE.
T cells are responsible for balancing a variety of regulatory and effector functions. Many of these roles are accomplished through the expression of a panel of cytokines controlled by a specific cohort of transcription factors. These cytokines can act to initiate, support or inhibit different T cell effector functions and, during homeostatic conditions, maintain a tight immunological balance between pro- and anti-inflammatory T cell functions. In the case of immune-mediated diseases, the balance between T cell subsets is often disrupted. For instance, patients with systemic lupus erythematosus (SLE) demonstrate an increase in T helper 1 (Th1) relative to T helper 2 (Th2) cells and a dysregulated balance between Th1 and T helper 17 (Th17) cells, while results from single cell sequencing of patients with rheumatoid arthritis (RA) have demonstrated a skewing toward Th1 effector memory CD4+ T cells, and a murine model of multiple sclerosis (MS) showed resistance to disease development due, in part, to a loss of key T cell intrinsic Th1 mediators (1–4). In SLE studies performed in humans and mice, some of the likely cytokine inflammatory mediators and immunomodulatory agents identified as participating in disease development include (but are not limited to) interferon (IFN)-α, IFN-γ, tumor necrosis factor (TNF), interleukin (IL)-1, IL-2, IL-4, IL-6, IL-8, IL-9, IL-10, IL-12, IL-13, IL-17, IL-21 and transforming growth factor (TGF)-β (5–8). Although many of these cytokines are produced by various antigen presenting cells (APCs) to help initiate effector T cell responses, all of these cytokines are also produced in varying quantities by the effector T cells themselves. Sustained T cell response, both appropriate and pathogenic following the initial priming event, depends greatly on the ability of the T cells to both produce the appropriate cytokines and reformat their transcriptional landscape at an epigenetic level to generate a positive feedback loop. Dysregulation of this positive feedback loop or inappropriate epigenetic reprogramming could result in a T cell-driven dysregulation of pro- or anti-inflammatory cytokine production, as seen in numerous autoimmune diseases (9–11). This review will delve specifically into the potential roles for IRF5 in the regulation of effector T cell decision and maintenance with a focus on Th1, Th2 and Th17 subsets, whose high interconnectivity has been demonstrated to be impacted by IRF5 deletion or hyperactivation. However, continued research into a potential role(s) for IRF5 in the other T cell subsets, particularly follicular helper T (Tfh) cells and regulatory T (Treg) cells, is an important next step in the elucidation of autoimmune disease pathogenesis via IRF5 dysregulation. For a more general review on the role of cytokines in autoimmune disease, see Raphael et al. (12).
Th1 effector cells regulate the body's defense against viruses, bacteria and intracellular pathogens and, when properly functional, are vital members of the immunological homeostasis required to maintain our health. However, dysregulation of Th1 cells has been implicated as a key player in the global immunological dysfunction that results in many autoimmune disease conditions, among them RA, SLE, MS, type 1 diabetes mellitus, idiopathic thrombocytopenic purpura, and experimental allergic encephalomyelitis (1, 2, 13–19). Th1 cells are traditionally defined by their production of IL-2 and IFN-γ and by expression of the transcription factor and epigenetic modifier, T-bet, a member of the T-box family of transcription factors (20–22). In the subsequent decades following the initial characterization of these defining factors, critical roles for the DNA-binding regulatory proteins signal transducer and activator of transcription 4 (STAT4), STAT1, and STAT5 in the development and support of Th1 subsets have also been revealed (23, 24). Briefly, naïve CD4+ T cells are stimulated to develop into Th1 effector cells by IL-12 binding to the IL-12 receptor (23, 25). Once activated, Th1 cells produce IL-2 and IFN-y. IL-2 acts as a potent inducer of both T cell proliferation and T cell effector fate decision (26). IFN-y employs both stimulatory and inhibitory roles to maintain Th1 effector dominance. IFN-y can induce the phosphorylation of STAT1, thereby increasing expression of the Th1 specific genes, IL-12 receptor beta 1 (IL12RB) and T-box transcription factor 21 (TBX21; encoding T-bet). Increased levels of T-bet results in positive feedback on T-bet expression through T-bet activation of IFNG transcription. T-bet also increases STAT1 activation and mediates the upregulation of Th1-specific genes including IL12R, which will in turn signal to increase STAT4 phosphorylation and dimerization. STAT4 itself can act as a potent transcriptional repressor of genes that would normally support Th2 differentiation (i.e., GATA3) and acts in concert with T-bet to promote the positive feedback loop resulting in increased IFN-γ production.
This feedback loop enhancing Th1 differentiation also has built in inhibitory mechanisms. T-bet can bind to and inhibit BCL-6 (B-cell lymphoma 6 protein) early in Th1 polarization, preventing transcription of genes involved in alternative effector fates (27). T-bet and BCL-6 comprise two lineage-defining factors that cooperate in the regulation of Th1 gene expression patterns (28). However, later in Th1 activation T-bet recruits BCL-6 to the IFNG promoter, resulting in inhibition of IFNG transcription and thereby shutting down one of the main drivers of the Th1 effector response (23, 28, 29). In addition, T-bet increases the transcription of the membrane protein T cell immunoglobulin mucin-3 (Tim-3) in later stages of Th1 differentiation, which acts as an inhibitor of the Th1 response upon binding to the ligand, β-galactosidase-binding lectin 9 (Gal-9) (30, 31). Gal-9 regulates Th-induced proinflammatory cytokine production (32). Further supporting the concept that dysregulation of T-bet can result in a pathologically imbalanced immune system, Tim-3 blockade has been shown to result in autoimmune disease development (33). Interestingly, most of T-bet's transcriptional regulatory capabilities have been shown to occur through epigenetic modifications of genetic loci using either H3K4 (activating) or H3K27me3 (inactivating) chromatin methylation patterns. In fact, production of the key Th1 driving cytokine IFN-γ is dependent on both chromatin remodeling by T-bet and increased IL-12R expression through direct T-bet transcriptional activity (29, 34–36). However, much less has been published with regards to the direct negative regulation of T-bet activity in activated Th1 cells and how dysregulation at the level of T-bet could result in rampant Th1 activation and the development of autoimmune disease.
As previously described, T-bet clearly plays an indispensable role in the positive feedback loop governing Th1 effector subset polarization. T-bet both positively regulates ~50% of Th1-specific genes and inhibits Th2-specific gene transduction, including GATA3, the Th2-specific transcription factor (29). Interestingly, ~70% of Th2-specific genes in Th1 cells are still bound by GATA3. In this scenario, GATA3 is bound by T-bet and inhibited from transducing Th2-specific transcripts in Th1 effector cells (37, 38). Other sources show that T-bet can also directly interact with and recruit GATA3 away from its Th2 gene loci. In either case, it is hypothesized that part of the rationale for skewing toward a Th2 phenotype upon loss of negative regulation by TBX21 is due to both increased GATA3 transcription and increased GATA3 association with Th2-specific genetic loci (29).
Th1 cells are capable of producing the cytokines granulocyte macrophage colony stimulating factor (GM-CSF), IL-2, TNF-β, and IFN-γ (39). As previously described, uncontrolled positive feedback of these cytokines on T cells can result in an imbalance between T cell subsets and their secreted cytokines, resulting in the development of autoimmune disease pathologies (40). Here we will explore the role of IRF5 in regulating an appropriate Th1 immune response and how loss of IRF5 may cause effector T cell dysregulation.
In the full-body Irf5 knockout (KO) mouse, the majority of studies have shown that there is skewing of T cells toward a Th2 effector phenotype with an accompanying decrease in Th1 effector subsets, thereby implicating a role for IRF5 in Th1 effector T cell commitment and/or maintenance (41–44). However, the T cell intrinsic IRF5-dependent molecular and genetic systems at play in these regulatory mechanisms governing Th1 feedforward and inhibitory loops have yet to be thoroughly explored. Based on previously published work, it seems likely that a main target for the dysregulation of Th1 effector T cells resulting in a substantial decrease in Th1 effector fate decision and a concomitant increase in Th2 cells would involve dysregulation of the master transcriptional regulator, T-bet. However, IRF5 does not play a role in the direct transcriptional regulation of this key transcription factor (45). This does not preclude the possibility that IRF5 interacts with T-bet on a protein level. In the following paragraphs, we propose a novel DEF6-IRF5-T-bet regulatory mechanism that controls Th1 effector T cell polarization.
The SWEF family of Rho-GTPase regulatory proteins consists of two family members, switching B cell complex subunit (SWAP70) and DEF6 (also known as IRF4 binding protein, IBP, or SWAP70-like adaptor of T cells, SLAT) (46, 47). Recent publications have shown that SWAP70 and DEF6 (also recently identified as a potential risk variant in human SLE) bind to and sequester IRF5 in the nucleus of age-associated B cells (ABCs) (46, 48). In naïve CD4+ T cells, the predominant SWEF family member expressed is DEF6 (47). As in ABCs, the importance of DEF6 as a master regulator has become increasingly evident through continuing discoveries of its roles in many aspects of T cell regulation, including IRF4 modulation, cytoskeletal kinetics and protein expression control through mammalian target of rapamycin complex 1 (mTORC1) regulation (49–53). In addition, as also observed in ABCs, upon T cell stimulation IRF5 levels are shown to dramatically increase (43, 54). If a similar pattern is followed in T cells as in ABCs, elevated levels of IRF5 may allow it to escape inhibition by DEF6 and perform its crucial regulatory role(s) in T cells (Figure 1).
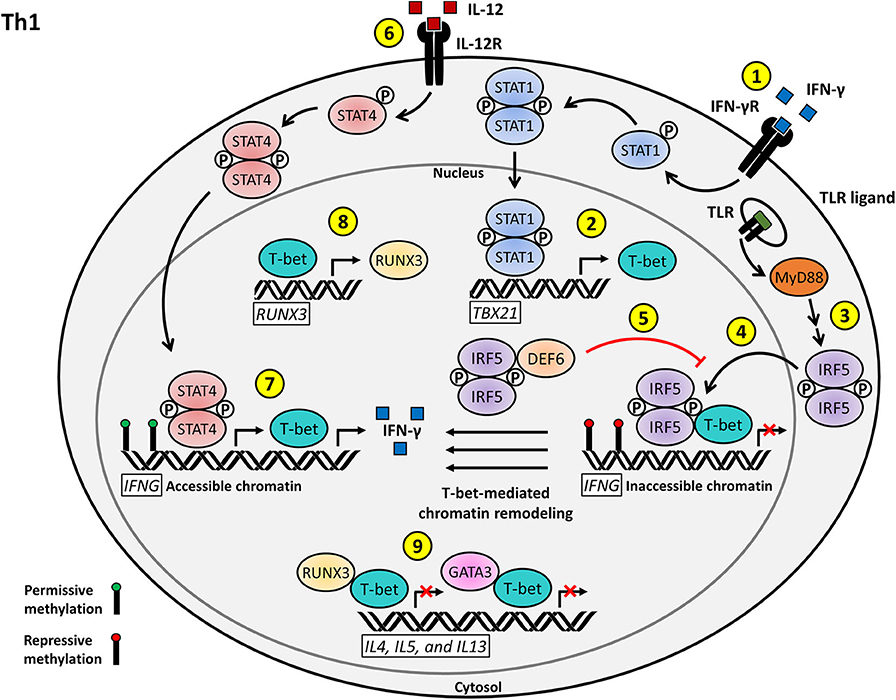
Figure 1. Proposed model for the T cell-intrinsic role of IRF5 as a positive regulator of Th1 effector function and differentiation. (1) IFN-γ stimulation of the IFN-γR on naïve CD4+ T cells induces STAT1 activation and nuclear translocation. (2) Phosphorylated STAT1 activates the transcription of TBX21, leading to the production of T-bet. (3) T cell stimulation, possibly through TLR signaling, induces IRF5 activation and nuclear translocation. (4) Nuclear IRF5 recruits T-bet to the silenced IFNG locus to facilitate permissive T-bet-mediated chromatin remodeling. (5) DEF6 binds to nuclear IRF5 in order to inhibit IRF5-mediated T-bet recruitment to the IFNG locus. (6) IL-12 signaling through the IL-12R results in STAT4 activation and nuclear translocation. (7) Phosphorylated STAT4 and T-bet induce the transcription of the accessible IFNG locus and subsequent IFN-γ signaling drives Th1 effector differentiation. (8) T-bet also acts as a positive regulator of RUNX3 transcription. (9) T-bet interacts with RUNX3 and GATA3 to inhibit the transcription of Th2 signature genes, including IL4, IL5, and IL13, to promote Th1 polarization.
So what regulatory role(s) might IRF5 play in Th1 cells? Upon stimulation of ABCs with IFN-γ and IL-21, levels of IRF5 increase, thereby allowing IRF5 to escape its negative regulation by SWEF family members, translocate to ABC transcriptional sites, and recruit T-bet to ABC-specific T-bet binding motifs (46, 55). It would be interesting to examine if this similar chromatin remodeling by T-bet followed by transcriptional activation resulting specifically from IRF5 driven T-bet recruitment in ABCs is a conserved mechanism for epigenetic and transcriptional regulation in T cells. In this scenario, IRF5 deletion would also likely result in GATA3 release from T-bet, allowing increased GATA3 translocation and binding to Th2-specific cytokine promoters, resulting in increased Th2-specific genes and cytokines (Figure 1). A mechanism similar to this one has already been alluded to in the Irf5−/− pristane-induced model of lupus, where loss of Irf5 results in an increase in the production of the Th2-specific cytokines IL-4 and IL-5 (41, 42, 56).
The proposed inclusion of IRF5 in the regulation of T-bet through a conserved interaction with DEF6 in T cells will likely have direct implications in our understanding of the control of cytokine release by T cells and the T cell-driven pathogenesis of several autoimmune diseases. An example of this is regulation of the runt-related transcription factor 3 (RUNX3) gene. RUNX3 enhances IFN-γ production and inhibits IL-4 production when recruited to the promoter regions by its interaction with T-bet or other members of the T-bet family. Upon Th1 activation, RUNX3 has been shown to be transcriptionally activated by T-bet. RUNX3 will then form a complex with T-bet and translocate to the promoters of IFN-γ and IL-4, activating and inhibiting their transcription, respectively, to maintain the Th1 positive feedback loop. Our hypothesis is that T-bet recruitment to sites of transcriptional regulation is mediated by IRF5 (Figure 1). In a 2016 paper examining effects of Runx3 polymorphisms, they identified an IRF4 binding site upstream of the Runx3 promoter (57). In 2019, this same group identified that this area in the promoter region could also mediate binding of other transcription factors, including IRF5 (57). Loss of Runx3 compromises IFN-γ production and abrogates inhibition of IL-4, thereby implying a vital role for RUNX3 in maintaining effector T cell polarization (58, 59).
Many key regulatory signaling and transcriptional proteins are expressed in both B and T cells. If IRF5 is indeed required to recruit T-bet to its transcriptional loci, loss of IRF5 would result in decreased efficiency of T-bet initiation of its Th1 transcriptional program, leading to a defect in Th1 effector subset polarization, as seen in Irf5 KO models. In addition, dysregulation of DEF6 could directly impact regulation of the key Th1 transcriptional driver, T-bet, providing a mechanism by which DEF6 polymorphisms contribute to SLE risk (Figure 1).
Th2 effector T cells are involved in the defense against parasitic infections, allergic reactions and the resolution of chronic inflammation (60). Unlike the previously discussed Th1 effector cells, the mechanisms driving Th2 differentiation are still not fully understood. Dendritic cells are thought to play a distinct role in supporting Th2 effector decision. However, they are incapable of producing the key Th2 mediating cytokine, IL-4 (61). Interestingly, IL-4 produced by CD4+ T cells themselves has been shown to be sufficient in initiating the Th2 response (62). These findings support the hypothesis that Th2 effector decision may be a default response in conditions where there is a lack of stimuli driving other Th effector fates. High levels of GATA3 expression in naïve CD4+ T cells prime the cells for Th2 differentiation, providing additional evidence for this theory. GATA3 is only downregulated upon initiation of T effector cell polarization into alternative subsets (61, 63).
Upon initiation of Th2 polarization, the principle Th2 cytokine, IL-4, acts in a stimulatory capacity through activation of STAT6 phosphorylation. Phosphorylated STAT6 increases the transcription of IL4 and GATA3. GATA3 is both a vital component of the machinery required for IL4, IL5, and IL13 transcription and is required for the global epigenetic remodeling needed to achieve Th2 polarization (64). The importance of GATA3 in Th2 effector differentiation is demonstrated by the consequences resulting from loss of GATA3. Even in the absence of the key Th1 cytokines, IFN-γ and IL-12, lack of GATA3 drives Th1 polarization (65).
Increased chromatin accessibility mediated by GATA3 both leads to the secretion of the Th2 specific cytokines, IL-4, IL-5, and IL-13, and inhibits the production of the Th1 specific cytokine, IFN-γ. Interestingly, IRF4 has recently been shown to act as an additional positive regulator of IL4 transcription during Th2 differentiation (49, 66). IRF4 forms a complex with GATA3 and the chromatin organizer special AT-rich binding protein 1 (SATB1) in order to bind to the RHS6 sequence during Th2 differentiation, located ubiquitously throughout the Th2 cytokine locus. All three of these factors are required in order for Th2-specific genes to be expressed (67). GATA3 has also been proposed to act in a positive feedback loop through the induction of IRF4 (68). The emerging roles of the complex transcriptional and regulatory networks involving the master transcription factor and epigenetic modulator, GATA3, are still being explored.
As in Th1 cells, there are regulatory mechanisms in place to inhibit the transcription of alternative T effector subset mediators upon Th2 effector commitment. One of these factors is the ubiquitous regulator, Ikaros (a hemopoietic-specific zinc finger protein also known as IKZF1). Regulatory functions for Ikaros have been implicated in almost all T helper cell subsets and loss of Ikaros has been shown to be detrimental in the maintenance of Th2 commitment. In Ikarosnull CD4+ T cells there is general hypoacetylation of the Th2 cytokine locus, increased IFN-γ production in Th2 polarizing conditions, decreased production of IL-4, IL-5, and IL-13, decreased GATA3 and c-MAF expression, and increased levels of T-bet and STAT1. All of these factors result in a skewing from Th2 to Th1 (69). Despite this growing pool of knowledge on the regulatory mechanisms ascribed to Ikaros, very little is known about the regulation of Ikaros itself in T cells (60, 70). However, a recent study of Ikaros regulation in B cells may provide insight into a conserved IRF5-dependent Ikaros regulatory mechanism in T cells (45).
As previously discussed, the regulation of Th2 cells, and thus the closely related Th1 effector subset, is still not fully understood. Pathologic skewing toward a Th2 response has been shown to result in atopic disorders, such as systemic sclerosis, and immunosuppression through the dysregulated production of their hallmark cytokines, IL-4, IL-5, IL-6, IL-9, IL-10, and IL-13 (40). IL-4, IL-10, and IL-4-induced IL-10 production in particular, has an inhibitory role on Th1 effector cells, thereby further contributing to the skewing from a Th1 to a Th2 phenotype and mediation of Th1 effector response (71, 72). Here we will explore a potential role for IRF5 in the control of Th2 subsets and how dysregulation can contribute to an enhanced pathogenic Th1 effector response.
A key role for IRF4 in Th2 subset development and, more specifically, the control of IL-4 production, has previously been identified (73). Although the precise regulatory mechanisms at play for IRF4 in T cells have yet to be fully elucidated, levels of IRF4 have been shown to be higher in resting Th2 cells compared to Th1 and Th17 (49). Inquiries into the role of IRF5 in other immune cell types have revealed alternative roles for IRF4 outside that of direct transcriptional regulation. In macrophages, IRF4 has been established as a negative regulator of Toll-like receptor (TLR) signaling by directly competing with IRF5 for binding to myeloid differentiation primary response gene 88 (MyD88) (74). MyD88 acts as a scaffold protein where IRF5 can receive its post-translational modifications from its interacting modulators. MyD88 functions downstream of all TLRs except TLR3 (75). Inhibition of the IRF5-MyD88 interaction by IRF4 results in attenuation of inflammatory cytokine production downstream of TLR signaling (48, 74, 76). However, the impact of T cell TLR signaling on intrinsic CD4+ T cell effector function and the pathological conditions that result from dysregulation are still by and large unconfirmed (77, 78).
The main body of research on the impacts that dysfunction of TLR signaling at the level of MyD88 might have on T cells examined how loss of MyD88 in upstream signaling cells (macrophages and dendritic cells) impacted Th2 differentiation. Little has been done to examine the specific roles and pathways of TLR signaling in T cells (75, 79). Mounting evidence implicates the TLR/MyD88 pathway as a potential regulatory mechanism in the Th1/Th2 effector decision. A study performed using the B. burgdorferi model of infection in a T cell-specific MyD88 deletion model demonstrated that loss of MyD88 in T cells results in an intrinsic defect in the Th1 and Th17 response. Th2 effector response was unfortunately not examined (80). However, an OVA-based murine MyD88−/- model of asthma showed significant defects in Th2 effector response upon stimulation (81). Taking these findings into account, we postulate that MyD88 plays an intrinsic role in T effector cell differentiation alongside IRF4. While IRF4 is expressed at high levels in Th2 effector cells, low levels of IRF5 are associated with a Th2 response. In Epstein-Barr Virus (EBV)-infected B cells, IRF4 was shown to be a negative transcriptional regulator of IRF5 (82). If IRF5 is no longer transcribed at high enough levels to initiate a transcriptional response tailoring an alternative T cell fate through TLR signaling, a Th2 transcriptional profile maintained by IRF4 through the previously described mechanisms can be maintained. This theory is supported by the T cell-specific MyD88 KO. Here, removal of another key player in the IRF5 TLR signaling pathway results in Th2 skewing, akin to the results seen in the Irf5 KO (80). Along with the conserved expression of IRF4 and IRF5 and the as of yet undefined mechanism by which IL-4 is initially regulated, we postulate that a conserved IRF4/IRF5/MyD88 axis in effector T cells may be playing a role in IRF5 activation and the skewing between Th1 and Th2 subsets (Figure 2).
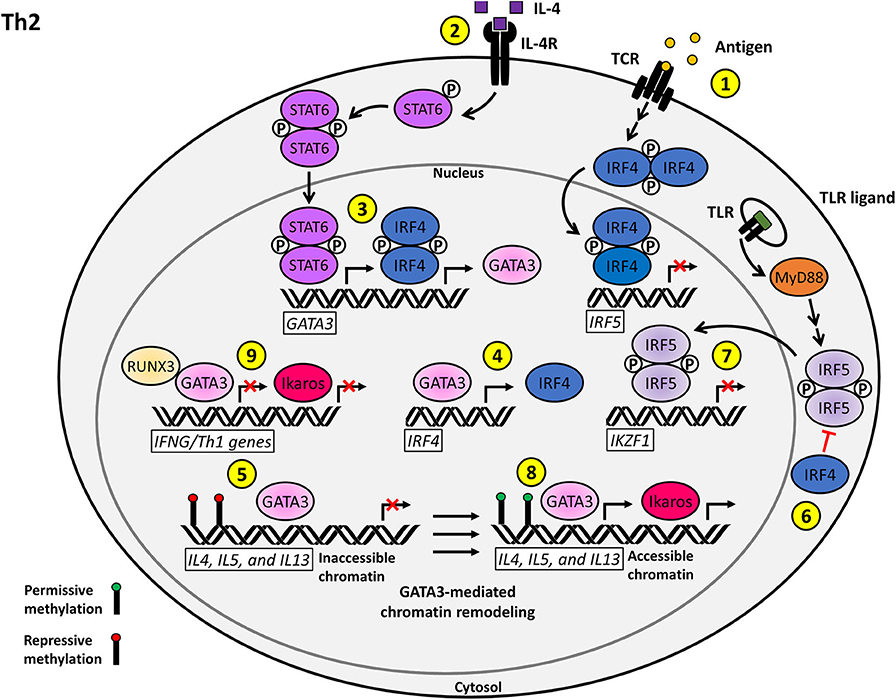
Figure 2. Proposed model for the T cell-intrinsic role of IRF5 as a negative regulator of Th2 effector function and differentiation. (1) Antigenic stimulation of the TCR induces the upregulation of IRF4, which acts as a repressor of IRF5 transcription. (2) Stimulation of the IL-4R by IL-4 on naïve CD4+ T cells induces STAT6 activation and nuclear translocation. (3) Phosphorylated STAT6 synergizes with IRF4 to activate GATA3 transcription. (4) A positive regulatory loop supported by GATA3-mediated IRF4 transcription augments Th2 polarization. (5) GATA3 induces permissive chromatin remodeling at the IL4, IL5, and lL13 Th2 cytokine locus. (6) IRF4 inhibits the TLR-induced activation of IRF5 via antagonizing the interaction between MyD88 and IRF5. (7) Nuclear IRF5 functions as a negative regulator of IKZF1 transcription, which limits the production of Ikaros. (8) Ikaros and GATA3 promote the transcription of accessible Th2 polarizing genes including IL4, IL5, and IL13. (9) Ikaros and GATA3 further reinforce the Th2 phenotype via repression of the Th1 transcriptional network.
Ikaros is another mediator that plays an important role in the maintenance of Th2 effector subset decision. Ikaros is a hematopoietic transcription factor that directly associates with Th2 regulatory gene loci and is involved in the positive regulation of Th2 gene expression (69). Ikaros has a binding site in its promoter region for IRF4, IRF5, and IRF8. In B cells, IRF8 and IRF5 both bind and regulate the Ikzf1 promoter, IRF8 acts in an activating capacity while IRF5 acts as an inhibitor. Inhibition of Ikzf1 transcription by IRF5 allows for the assumption of B cell antibody class switching to IgG2a/2c (45). Expression and function of these IRF transcription factors and Ikaros are conserved in T cells. If IRF5 were to maintain a similar negative regulatory function for Ikaros as seen in B cells, loss of IRF5 would allow unimpeded Ikaros activation, resulting in a shift toward Th2 effector polarization. On the other hand, overexpression or hyperactivation of IRF5, as seen in SLE patients, could lead to loss of Ikaros transcription and a shift from the Th2 to Th1 effector T cell subset. In support of this theory, Gene Ontology shows that IKZF1 has distal sites for T-bet and GATA3 binding (37). As previously discussed, we postulate that there is likely a role for IRF5 in the regulation of T-bet through direct interaction, as well as one for GATA3, by extension (Figure 2). Thus, there is increasing circumstantial evidence of a regulatory role for IRF5 in the control of Ikaros function, either through direct binding or through the recruitment of chromatin remodeling agents.
T cell development is highly dependent on the surrounding cytokine environment and is characterized by high degrees of plasticity which, in many cases, can serve a pathogenic role. This is especially seen in the case of dysregulated Th17 cells, which have been associated with many immunological diseases including RA, inflammatory bowel disease (IBD), SLE, MS, psoriasis and cancer (20, 83, 84). Although Th17 effector subsets have been considered for drug targets to counteract the dysfunctional immune systems that they help to support, our lack of knowledge about the pathways regulating the polarization of these cells toward pathogenic phenotypes has hindered our choice(s) of a specific target (83). Recently, the monoclonal antibody against IL-17R, marketed under the name Brodalumab (AMG827), has entered clinical trials and was shown to be effective in improving psoriasis (85). However, many other drugs on the market attempting to initiate an IL-17 blockade have been met with mixed results depending on the disease setting (86). Thus, although Th17 effector function is strongly implicated as a potential target for future drug development, we need to gain a better understanding of the mechanisms controlling Th17 pathologic phenotypes and how these can drive autoimmune disease.
In SLE patients, it has been shown that hyperactive IRF5 results in skewing toward a Th1 and Th17 phenotype. However, to say simply “Th17 phenotype” is an oversimplification of the diversity of this particular T cell subset. Th17 effector T cells exist in a gradient between classical and pathogenic which is determined in part by the cytokine milieu they are exposed to. In the pathogenic state, there are two opposing directions that Th17 cells can follow—either toward a Th1-like phenotype, which is often associated with autoimmunity or toward a more Th2-like state, which is correlated with enhanced immunosuppression (83). At steady state, Th17 cells differentiate into Tfh cells and support immunoglobulin A (IgA) production by germinal center B cells. IL-23 in particular, although not required for Th17 differentiation, is required for pathogenic Th17 maintenance and survival (87).
IL-17A, the “pathogenic” cytokine produced by Th17 cells, has been shown to be a key player in the perpetuation of inflammation associated with autoimmune tissue damage. IL-17A functions through several mechanisms including the activation of other immune cells, increasing B cell functions, recruiting neutrophils, Treg mediation and enhancing proinflammatory cytokine release (20, 88, 89). In the mouse model of human MS (murine experimental autoimmune encephalomyelitis, EAE), blocking the interaction between IL-17 and IL-17 receptors resulted in substantial attenuation of EAE development (90). Unfortunately, the picture painted by this interaction is oversimplified. To date, there have been six different IL-17 cytokines identified, IL-17A–F, and five unique versions of the IL-17 receptor, IL-17RA–RE. For a more extensive review on what is known about the functional variations of these family members, see Swaidani et al. (91) and Jin and Dong (92). Although IL-17A has been identified as the main mediator of inflammation associated with autoimmune disease, the pathways downstream of IL-17A binding to IL-17R are still not fully defined.
In non-disease states, Th17 cells serve an important function in supporting tailored immune responses to various pathogens (20). Th17 effector cells maintain a balance between the alternative Treg differentiation pathway and conversion into a Th1-like phenotype. IRF4 has been shown to be a key mediating factor in maintaining the balance between Th17 and Tregs. Irf4 KO results in an increase in the Treg FoxP3 (forkhead box P3) transcription factor and a decrease in RORγt (RAR-related orphan receptor gamma t), the major transcription factor for commitment to Th17 fate in part through transcriptional upregulation of IL-17 (93). The relatively one-sided conversion from Th17 to Th1 seems to be controlled through stimulation from circulating cytokines. Stimulation of Th17 polarized cells by IL-12 and IFN-γ results in inhibition of IL-17 secretion and conversion to a more Th1-like state, characterized by increased levels of STAT4 and T-bet expression. Increased levels of TGF-β inhibit this plasticity and result in maintenance of a stable Th17 phenotype. Early STAT transcription factors are also at play in the regulation of Th17 decision; STAT3 promotes and STAT5 inhibits Th17 differentiation (23). Because of the plasticity of the Th17 subset and its ability to interconvert between many other effector-like subsets in response to disease, the regulation of this particular subset is complex and still not well-understood. However, it has been established that maintenance of the inflammatory state that characterizes many autoimmune diseases is in part due to the IL-17-initiated positive feedback loop from defective Th17 cells (94, 95).
The role of IRF5 in Th17 effector cells is still an open field. However, based on the previous mechanisms described, especially those relating to Th1 regulation, a role for IRF5 in Th17 differentiation and plasticity seems highly likely. Several studies have supported a role for IRF5 in Th17 effector differentiation, although few, if any, studies have yet to examine an intrinsic role for IRF5 in Th17 cells. Loss of Irf5 in murine models of severe asthma resulted in decreased IFN-γ and IL-17 responses upon ovalbumin (OVA) immunization (96). In an Irf5 KO antigen-induced arthritis (AIA) model, Th1, Th17, and γδ IL-17 producing T cells were found to have significantly decreased effector responses following immunization with methylated bovine serum albumin (mBSA) in complete Freund's adjuvant (CFA). In addition, this model showed decreased levels of Ifng and Il17a mRNA and the key Th1 and Th17 cytokines IL-1β, IL-6, IL-12, and IL-23 (97).
The cytokines that are often used to characterize pro-inflammatory Th17 subsets are IL-22, GM-CSF, and IFN-γ. Interestingly, several of these inflammatory cytokine mediators are also known to induce the expression of IRF5. In macrophages, increased IRF5 expression results in an M1 (inflammatory) macrophage phenotype through the upregulation of IL-12, TNF-α, and IFN-γ, with concomitant repression of IL-10 (98). Through binding to various promoter regions, IRF5 also increases IL6, IL12, and IL23p19 transcription. Interestingly, pathogenic Th17 cells also secrete IL-12, IL-23, IL-6, and IFN-γ in addition to various other Th17-specific effector cytokines and transcription factors (66, 99). A conserved role for IRF5 in the transcriptional activation of these inflammatory cytokines in Th17 cells should be explored.
IL-10 production by Th17 cells may provide yet another avenue for a potential role for IRF5 in the regulation of Th17-mediated inflammation (100). Although the regulation of IL-10 in Th17 effector cells is not fully understood, it has been well-established that IL-10 is required for T cells to maintain control over Th17 effector function (101). One of the mechanisms by which IL-10 expression is mediated is through TGF-β and IL-6. These two factors work to activate the c-MAF transcription factor through STAT3, which in turn activates IL10 transcription by binding to the IL10 promoter (102). IL-10 acts to reduce IL-17 and IFN-γ production, thus negatively regulating pro-inflammatory Th17 effector reactions. Interestingly, in the context of Newcastle disease virus (NDV)-infected Balb-C mice, IRF5 was shown to induce Stat3 transcription in the presence of undetectable levels of the cytokines IL-6 and IL-10 (103). In addition, IRF5 has been shown to be a key mediator of IL6 transcription in human pDCs (104). IRF5 was also shown to be upregulated by the janus kinase 2 (JAK2)/STAT3 pathway in human umbilical vein endothelial cells (105). The existence of a positive feedback loop between STAT3 and IRF5 in Th17 cells, where activation of IRF5 transcription downstream of STAT3 allows for IRF5 to feedback and increase IL-6 and STAT3 expression, should be explored as a potential mechanism by which IRF5 mediates Th17 effector response (Figure 3).
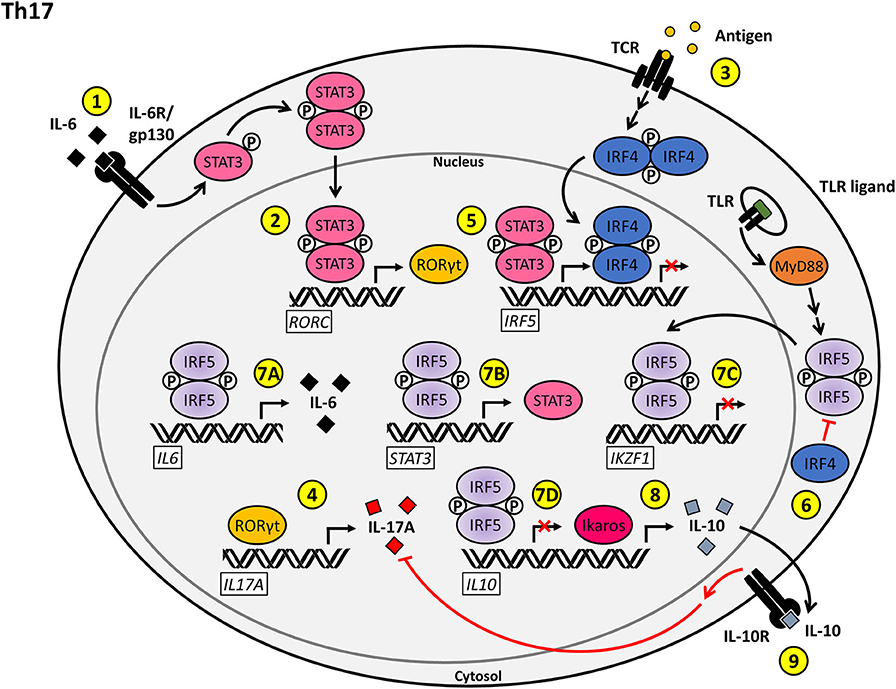
Figure 3. Proposed model for the T cell-intrinsic role of IRF5 as a positive regulator of Th17 effector function and differentiation. (1) Stimulation of the IL-6R/gp130 complex by IL-6 on naïve CD4+ T cells induces STAT3 activation and nuclear translocation. (2) Phosphorylated STAT3 induces the transcription of RORC, leading to the production of RORγt. (3) Antigenic stimulation of the TCR induces the upregulation of IRF4. (4) RORγt induces the transcription of IL17A, driving Th17-mediated inflammation. (5) The transcription of IRF5 is positively regulated by phosphorylated STAT3 and negatively regulated by IRF4. (6) IRF4 inhibits the TLR-induced activation of IRF5 via antagonizing the interaction between MyD88 and IRF5. (7A–D) Nuclear IRF5 promotes the Th17 phenotype through upregulation of IL6 (7A) and STAT3 (7B) expression and repression of IKZF1 (7C) and IL10 (7D) transcription. (8) Ikaros functions as a positive regulator of IL10 transcription. (9) IL-10 signaling through the IL-10R antagonizes the production of IL-17A, thereby inhibiting Th17 inflammatory responses.
In an alternative regulatory arm, Ikaros has been shown to be required for inhibition of heterochromatic remodeling at the gene loci for the Th17 effector program. Ikaros has also been shown to repress expression of both FOXP3 and TBX21, which both normally act to negatively regulate Th17 development (106, 107). However, T-bet was also reported to positively regulate transcription of the IL23R by binding to a specific site in the IL23R promoter sequence and inducing IFN-γ expression by Th17 cells, thus inducing a pro-inflammatory state (108, 109). Hence, depending on the location and context of T-bet expression in Th17 cells, T-bet can initiate or ameliorate inflammatory responses. The precise mechanisms through which these regulatory actions are achieved have not yet been established. In addition to binding to and regulating FOXP3 and TBX21 expression, Ikaros has a binding site specifically within the IL10 promoter and acts to positively regulate IL-10 production. It is likely that Ikaros has other, as of yet, undefined epigenetic and transcriptional regulatory roles to support Th17 effector functions (106, 110). A hint as to additional regulatory mechanisms involved in the pathways leading to Th17 effector commitment comes through literature on IRF5 regulation. In macrophages, IRF5 has been shown to have both positive and negative effects on IL10 transcription through direct binding to the IL10 promoter (98, 111). In Th17 cells, IL-10 plays a crucial role in the downregulation of the pro-inflammatory cytokines, IL-17 and IFN-γ. As loss of IRF5 results in a decrease in Th17 effector subsets, this could imply a positive regulatory role for IRF5 in a conserved pathway, either through an inhibitory role at the IL10 promoter (as seen in macrophages), a negative role in Ikaros regulation (as described in B cells) or induction of a STAT3–IRF5 positive feedback loop as previously described (Figure 3).
The role and relevance of IRF5 in immune cell dysfunction in the context of autoimmune disease and cancer progression has become a hot topic for research in recent years. However, despite our growing knowledge of functions for IRF5 in APCs, our knowledge on the role of T cell-intrinsic IRF5 function is still lacking. Most of the literature published on potential roles for IRF5 in T cells is confounded by the dysregulation of other upstream immune cell signaling pathways in the in vivo setting of an Irf5−/− mouse. The CD4+ T cell-specific Irf5 KO model attempted to address this and, in the context of CD3/CD28 TCR stimulation with IL-12, showed no defects in IFN-γ production (54). However, preliminary work from our lab utilizing RAG2−/− mice as recipients of Irf5+/+ and Irf5−/− T cells reveals a stimulus-dependent T cell-intrinsic defect that drives aberrant immune cell responses which, in the context of the hypothesized TLR driven IRF5 pathways in T cells, rather than rejecting previous work, compliments their findings (data not shown). The generation and characterization of new T cell-specific conditional Irf5 KO mice, combined with pathway-specific immune challenges, will help to delineate Irf5 intrinsic function in T cells. For example, to study an intrinsic role for Irf5 in Th17 cells, Irf5-floxed mice would be crossed to IL17(A/F)-cre mice to generate Th17-specific Irf5 conditional KO mice. A number of T cell-specific cre-reporter strains are currently available that would help prove or disprove the presented hypotheses.
In the clinical realm, SLE is characterized by a heterogeneous patient population. In each patient, the disease shares several common characteristics, but ultimately has a unique landscape and response to treatment. This is likely driven by a “multi-hit” scenario where dysfunction or dysregulation of a single (or multiple) master regulatory factor, like IRF5, will predispose individuals to developing a specific brand of immune dysregulation with many shared pathological characteristics (112). However, ultimately the path of development and resulting severity of the disease is determined by the addition of other risk allelic variations, thereby leading to the unique signature characterizing individual autoimmune conditions. This also explains the as-of-yet undefined and heterogeneous pathway-specific triggers that lead to disease development in a perfect storm of self-perpetuating dysregulated pathway activation, characterized by aberrant cytokine production. Ultimately, the goal in effective therapeutic development is to find the most specific target that ameliorates the greatest number of disease phenotypes with the fewest off-target effects. In order to accomplish this, we need a detailed understanding of the pathways that govern each immune cell implicated in disease pathogenesis. Targeting the inflammatory cytokines themselves is a difficult and non-specific therapeutic option, although early clinical trials of low dose IL-2 administration have shown some promise in patients with treatment-resistant SLE (113). However, the list of “T cell” therapeutics for autoimmune disease is brief, and many of them [i.e., secukinumab, ixekizumab, broalumab, ustekinumab, iberdomide, AMG 570 targeting ICOS-L (NCT04058028)] have either yet to be proven efficacious in the treatment of SLE, are still in early clinical trials, or broadly target the functions of other immune cells (114–116). As a result, targeted delivery of therapeutic molecules to specific immune cell subsets that drive the dysregulated release of either pro-or anti-inflammatory cytokines is the future of effective personalized treatments for autoimmune disease.
ZB, MR, and BB conceived of and wrote the manuscript.
This work was supported in part by grants from the Department of Defense LRP (W81XWH-18-1-0674) and the Lupus Research Alliance to BB.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Shah K, Lee WW, Lee SH, Kim SH, Kang SW, Craft J, et al. Dysregulated balance of Th17 and Th1 cells in systemic lupus erythematosus. Arthritis Res Ther. (2010) 12:R53. doi: 10.1186/ar2964
2. Guimaraes PM, Scavuzzi BM, Stadtlober NP, Franchi Santos L, Lozovoy MAB, Iriyoda TMV, et al. Cytokines in systemic lupus erythematosus: far beyond Th1/Th2 dualism lupus: cytokine profiles. Immunol Cell Biol. (2017) 95:824–31. doi: 10.1038/icb.2017.53
3. Fonseka CY, Rao DA, Teslovich NC, Korsunsky I, Hannes SK, Slowikowski K, et al. Mixed effects association of single cells identifies an expanded Th1-skewed cytotoxic effector CD4+ T cell subset in rheumatoid arthritis. Sci Transl Med. (2018) 10:eaaq0305. doi: 10.1126/scitranslmed.aaq0305
4. Sonar SA, Lal G. Differentiation and transmigration of CD4 T cells in neuroinflammation and autoimmunity. Front Immunol. (2017) 8:1695. doi: 10.3389/fimmu.2017.01695
5. Davis LS, Hutcheson J, Mohan C. The role of cytokines in the pathogenesis and treatment of systemic lupus erythematosus. J Interferon Cytokine Res. (2011) 31:781–9. doi: 10.1089/jir.2011.0047
6. Lourenço EV, La Cava A. Cytokines in systemic lupus erythematosus. Curr Mol Med. (2009) 9:242–54. doi: 10.2174/156652409787847263
7. Rojas M, Rodríguez Y, Leon KJ, Pacheco Y, Acosta-Ampudia Y, Monsalve DM, et al. Cytokines and inflammatory mediators in systemic lupus erythematosus. EMJ Rheumatol. (2018) 5:83–92.
8. Nambu A, Nakae S, Iwakura Y. IL-1β, but not IL-1α, is required for antigen-specific T cell activation and the induction of local inflammation in the delayed-type hypersensitivity responses. Int Immunol. (2006) 18:701–12. doi: 10.1093/intimm/dxl007
9. Moudgil KD, Choubey D. Cytokines in autoimmunity: role in induction, regulation, and treatment. J Interferon Cytokine Res. (2011) 31:695–703. doi: 10.1089/jir.2011.0065
10. Surace AEA, Hedrich CM. The role of epigenetics in autoimmune/inflammatory disease. Front Immunol. (2019) 10:1525. doi: 10.3389/fimmu.2019.01525
11. Mazzone R, Zwergel C, Artico M, Taurone S, Ralli M, Greco A, et al. The emerging role of epigenetics in human autoimmune disorders. Clin Epigenetics. (2019) 11:34. doi: 10.1186/s13148-019-0632-2
12. Wu H, Xie MM, Liu H, Dent AL. Stat3 Is important for follicular regulatory T cell differentiation. PLoS ONE. (2016) 11:e0155040. doi: 10.1371/journal.pone.0155040
13. Yamada H, Nakashima Y, Okazaki K, Mawatari T, Fukushi JI, Kaibara N, et al. Th1 but not Th17 cells predominate in the joints of patients with rheumatoid arthritis. Annu Rheumatic Dis. (2008) 67:1299–304. doi: 10.1136/ard.2007.080341
14. Oreja-Guevara C, Ramos-Cejudo J, Aroeira LS, Chamorro B, Diez-Tejedor E. TH1/TH2 cytokine profile in relapsing-remitting multiple sclerosis patients treated with Glatiramer acetate or Natalizumab. BMC Neurol. (2012) 12:95. doi: 10.1186/1471-2377-12-95
15. Azar ST, Tamim H, Beyhum HN, Habbal MZ, Almawi WY. Type I (insulin-dependent) diabetes is a Th1- and Th2-mediated autoimmune disease. Clin Diagn Lab Immunol. (1999) 6:306–10. doi: 10.1128/CDLI.6.3.306-310.1999
16. Walker LSK, von Herrath M. CD4 T cell differentiation in type 1 diabetes. Clin Exp Immunol. (2016) 183:16–29. doi: 10.1111/cei.12672
17. Domingues HS, Mues M, Lassmann H, Wekerle H, Krishnamoorthy G. Functional and pathogenic differences of Th1 and Th17 cells in experimental autoimmune encephalomyelitis. PLoS ONE. (2010) 5:e15531. doi: 10.1371/journal.pone.0015531
18. Takahashi N, Saitoh T, Gotoh N, Nitta Y, Alkebsi L, Kasamatsu, et al. The cytokine polymorphisms affecting Th1/Th2 increase the susceptibility to, and severity of, chronic ITP. BMC Immunol. (2017) 18:26. doi: 10.1186/s12865-017-0210-3
19. Yang Q, Wang B, Yu H, Zhu Y, Wang X, Jiang H, et al. TLR7 promotes Th1 polarization in immune thrombocytopenia. Thromb Res. (2011) 128:237–42. doi: 10.1016/j.thromres.2011.02.024
20. Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine. (2015) 74:5–17. doi: 10.1016/j.cyto.2014.09.011
21. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. (1986) 136:2348–57.
22. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs th1 lineage commitment. Cell. (2000) 100:655–69. doi: 10.1016/S0092-8674(00)80702-3
23. Tripathi SK, Lahesmaa R. Transcriptional and epigenetic regulation of T-helper lineage specification. Immunol Rev. (2014) 261:62–83. doi: 10.1111/imr.12204
24. Adamson AS, Collins K, Laurence A, O'Shea JJ. The current STATus of lymphocyte signaling: new roles for old players. Curr Opin Immunol. (2009) 21:161–6. doi: 10.1016/j.coi.2009.03.013
25. Kaiko GE, Horvat JC, Beagley KW, Hansbro PM. Immunological decision-making: how does the immune system decide to mount a helper T-cell response? Immunology. (2008) 123:326–38. doi: 10.1111/j.1365-2567.2007.02719.x
26. Ross SH, Cantrell DA. Signaling and function of interleukin-2 in T lymphocytes. Annu Rev Immunol. (2018) 36:411–33. doi: 10.1146/annurev-immunol-042617-053352
27. Kotov JA, Kotov DI, Linehan JL, Bardwell VJ, Gearhart MD, Jenkins MK. BCL6 corepressor contributes to Th17 cell formation by inhibiting Th17 fate suppressors. J Exp Med. (2019) 216:1450–64. doi: 10.1084/jem.20182376
28. Oestreich KJ, Huang AC, Weinmann AS. The lineage-defining factors T-bet and Bcl-6 collaborate to regulate Th1 gene expression patterns. J Exp Med. (2011) 208:1001–13. doi: 10.1084/jem.20102144
29. Zhu J, Jankovic D, Oler Andrew J, Wei G, Sharma S, et al. The transcription factor T-bet is induced by multiple pathways and prevents an endogenous Th2 cell program during th1 cell responses. Immunity. (2012) 37:660–73. doi: 10.1016/j.immuni.2012.09.007
30. Anderson AC, Lord GM, Dardalhon V, Lee DH, Sabatos-Peyton CA, Glimcher LH, et al. T-bet, a Th1 transcription factor regulates the expression of Tim-3. Eur J Immunol. (2010) 40:859–66. doi: 10.1002/eji.200939842
31. Han G, Chen G, Shen B, Li Y. Tim-3: an activation marker and activation limiter of innate immune cells. Front Immunol. (2013) 4:449. doi: 10.3389/fimmu.2013.00449
32. Su EW, Bi S, Kane LP. Galectin-9 regulates T helper cell function independently of Tim-3. Glycobiology. (2010) 21:1258–65. doi: 10.1093/glycob/cwq214
33. Sánchez-Fueyo A, Tian J, Picarella D, Domenig C, Zheng XX, Sabatos, et al. Tim-3 inhibits T helper type 1–mediated auto- and alloimmune responses and promotes immunological tolerance. Nat Immunol. (2003) 4:1093–101. doi: 10.1038/ni987
34. Mullen AC, Hutchins AS, High FA, Lee HW, Sykes KJ, Chodosh LA, et al. Hlx is induced by and genetically interacts with T-bet to promote heritable TH1 gene induction. Nat Immunol. (2002) 3:652–8. doi: 10.1038/ni807
35. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu Rev Immunol. (2010) 28:445–89. doi: 10.1146/annurev-immunol-030409-101212
36. Hatton R, Harrington L, Luther R, Wakefield T, Janowski K, Oliver J, et al. A distal conserved sequence element controls ifng gene expression by T cells and NK cells. Immunity. (2006) 25:717–29. doi: 10.1016/j.immuni.2006.09.007
37. Kanhere A, Hertweck A, Bhatia U, Gökmen MR, Perucha E, Jackson I, et al. T-bet and GATA3 orchestrate Th1 and Th2 differentiation through lineage-specific targeting of distal regulatory elements. Nat Commun. (2012) 3:1268. doi: 10.1038/ncomms2260
38. Jenner RG, Townsend MJ, Jackson I, Sun K, Bouwman RD, Young RA, et al. The transcription factors T-bet and GATA-3 control alternative pathways of T-cell differentiation through a shared set of target genes. Proc Natl Acad Sci USA. (2009) 106:17876–81. doi: 10.1073/pnas.0909357106
39. Theofilopoulos AN, Koundouris S, Kono DH, Lawson BR. The role of IFN-gamma in systemic lupus erythematosus: a challenge to the Th1/Th2 paradigm in autoimmunity. Arthritis Res. (2001) 3:136–41. doi: 10.1186/ar290
40. Romagnani S. T-cell subsets (Th1 versus Th2). Ann Allergy Asthma Immunol. (2000) 85:9–18. doi: 10.1016/S1081-1206(10)62426-X
41. Feng D, Yang L, Bi X, Stone RC, Patel P, Barnes BJ. Irf5-deficient mice are protected from pristane-induced lupus via increased Th2 cytokines and altered IgG class switching. Eur J Immunol. (2012) 42:1477–87. doi: 10.1002/eji.201141642
42. Xu Y, Lee PY, Li Y, Liu C, Zhuang H, Han S, et al. Pleiotropic IFN-dependent and -independent effects of IRF5 on the pathogenesis of experimental lupus. J Immunol. (2012) 188:4113–21. doi: 10.4049/jimmunol.1103113
43. Paun A, Bankoti R, Joshi T, Pitha PM, Stäger S. Critical role of IRF-5 in the development of T helper 1 responses to Leishmania donovani infection. PLoS Pathog. (2011) 7:e1001246. doi: 10.1371/journal.ppat.1001246
44. Watkins AA, Yasuda K, Wilson GE, Aprahamian T, Xie Y, Maganto-Garcia E, et al. IRF5 deficiency ameliorates lupus but promotes atherosclerosis and metabolic dysfunction in a mouse model of lupus-associated atherosclerosis. J Immunol. (2015) 194:1467–79. doi: 10.4049/jimmunol.1402807
45. Fang CM, Roy S, Nielsen E, Paul M, Maul R, Paun A, et al. Unique contribution of IRF-5-Ikaros axis to the B-cell IgG2a response. Genes Immun. (2012) 13:421–30. doi: 10.1038/gene.2012.10
46. Manni M, Gupta S, Ricker E, Chinenov Y, Park SH, Shi M, et al. Regulation of age-associated B cells by IRF5 in systemic autoimmunity. Nat Immunol. (2018) 19:407–19. doi: 10.1038/s41590-018-0056-8
47. Manni M, Ricker E, Pernis AB. Regulation of systemic autoimmunity and CD11c(+) Tbet(+) B cells by SWEF proteins. Cell Immunol. (2017) 321:46–51. doi: 10.1016/j.cellimm.2017.05.010
48. Tatsuma Ban TT. Regulation and role of the transcription factor IRF5 in innate immune responses and systemic lupus erythematosus. Int Immunol. (2018) 30:529–36. doi: 10.1093/intimm/dxy032
49. Biswas PS, Bhagat G, Pernis AB. IRF4 and its regulators: evolving insights into the pathogenesis of inflammatory arthritis? Immunol Rev. (2010) 233:79–96. doi: 10.1111/j.0105-2896.2009.00864.x
50. Biswas PS, Gupta S, Stirzaker RA, Kumar V, Jessberger R, Lu TT, et al. Dual regulation of IRF4 function in T and B cells is required for the coordination of T-B cell interactions and the prevention of autoimmunity. J Exp Med. (2012) 209:581–96. doi: 10.1084/jem.20111195
51. Tybulewicz VLJ, Henderson RB. Rho family GTPases and their regulators in lymphocytes. Nat Rev Immunol. (2009) 9:630–44. doi: 10.1038/nri2606
52. Kumari S, Curado S, Mayya V, Dustin ML. T cell antigen receptor activation and actin cytoskeleton remodeling. Biochim Biophys Acta. (2014) 1838:546–56. doi: 10.1016/j.bbamem.2013.05.004
53. Yi W, Gupta S, Ricker E, Manni M, Jessberger R, Chinenov Y, et al. The mTORC1-4E-BP-eIF4E axis controls de novo Bcl6 protein synthesis in T cells and systemic autoimmunity. Nat Commun. (2017) 8:254. doi: 10.1038/s41467-017-00348-3
54. Fabie A, Mai LT, Dagenais-Lussier X, Hammami A, van Grevenynghe J, Stager S. IRF-5 promotes cell death in CD4 T cells during chronic infection. Cell Rep. (2018) 24:1163–75. doi: 10.1016/j.celrep.2018.06.107
55. Zou YR, Davidson A. Age-associated B cells acquire a new wrinkle. Nat Immunol. (2018) 19:317–8. doi: 10.1038/s41590-018-0072-8
56. Byrne AJ, Weiss M, Mathie SA, Walker SA, Eames HL, Saliba D, et al. A critical role for IRF5 in regulating allergic airway inflammation. Mucosal Immunol. (2017) 10:716–26. doi: 10.1038/mi.2016.92
57. Vecellio M, Roberts AR, Cohen CJ, Cortes A, Knight JC, Bowness P, et al. The genetic association of RUNX3 with ankylosing spondylitis can be explained by allele-specific effects on IRF4 recruitment that alter gene expression. Ann Rheum Dis. (2016) 75:1534–40. doi: 10.1136/annrheumdis-2015-207490
58. Djuretic IM, Levanon D, Negreanu V, Groner Y, Rao A, Ansel KM. Transcription factors T-bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nat Immunol. (2007) 8:145–53. doi: 10.1038/ni1424
59. Yagi R, Junttila IS, Wei G, Urban JF Jr, Zhao K, Paul WE, et al. The transcription factor GATA3 actively represses RUNX3 protein-regulated production of interferon-gamma. Immunity. (2010) 32:507–17. doi: 10.1016/j.immuni.2010.04.004
60. Powell MD, Read KA, Sreekumar BK, Oestreich KJ. Ikaros zinc finger transcription factors: regulators of cytokine signaling pathways and CD4(+) T helper cell differentiation. Front Immunol. (2019) 10:1299. doi: 10.3389/fimmu.2019.01299
61. Walker JA, McKenzie ANJ. TH2 cell development and function. Nat Rev Immunol. (2018) 18:121–33. doi: 10.1038/nri.2017.118
62. Noben-Trauth N, Hu-Li J, Paul WE. Conventional, naive CD4+ T cells provide an initial source of IL-4 during Th2 differentiation. J Immunol. (2000) 165:3620–5. doi: 10.4049/jimmunol.165.7.3620
63. O'Garra A, Gabrysova L. Transcription factors directing Th2 differentiation: gata-3 plays a dominant role. J Immunol. (2016) 196:4423–5. doi: 10.4049/jimmunol.1600646
64. Yagi R, Zhu J, Paul WE. An updated view on transcription factor GATA3-mediated regulation of Th1 and Th2 cell differentiation. Int Immunol. (2011) 23:415–20. doi: 10.1093/intimm/dxr029
65. Zhu J, Min B, Hu-Li J, Watson CJ, Grinberg A, Wang Q, et al. Conditional deletion of Gata3 shows its essential function in TH1-TH2 responses. Nat Immunol. (2004) 5:1157–65. doi: 10.1038/ni1128
66. Jefferies CA. Regulating IRFs in IFN driven disease. Front Immunol. (2019) 10:325. doi: 10.3389/fimmu.2019.00325
67. Hwang SS, Jang SW, Lee KO, Kim HS, Lee GR. RHS6 coordinately regulates the Th2 cytokine genes by recruiting GATA3, SATB1, and IRF4. Allergy. (2017) 72:772–82. doi: 10.1111/all.13078
68. Zhu J. T helper 2 (Th2) cell differentiation, type 2 innate lymphoid cell (ILC2) development and regulation of interleukin-4 (IL-4) and IL-13 production. Cytokine. (2015) 75:14–24. doi: 10.1016/j.cyto.2015.05.010
69. Quirion MR, Gregory GD, Umetsu SE, Winandy S, Brown MA. Cutting edge: ikaros is a regulator of Th2 cell differentiation. J Immunol. (2009) 182:741–5. doi: 10.4049/jimmunol.182.2.741
70. Perotti EA, Georgopoulos K, Yoshida T. An ikaros promoter element with dual epigenetic and transcriptional activities. PLoS ONE. (2015) 10:e0131568. doi: 10.1371/journal.pone.0131568
71. Couper KN, Blount DG, Riley EM. IL-10: the master regulator of immunity to infection. J Immunol. (2008) 180:5771–7. doi: 10.4049/jimmunol.180.9.5771
72. Mitchell RE, Hassan M, Burton BR, Britton G, Hill EV, Verhagen J, et al. IL-4 enhances IL-10 production in Th1 cells: implications for Th1 and Th2 regulation. Sci Rep. (2017) 7:11315. doi: 10.1038/s41598-017-11803-y
73. Tominaga N, Ohkusu-Tsukada K, Udono H, Abe R, Matsuyama T, Yui K. Development of Th1 and not Th2 immune responses in mice lacking IFN-regulatory factor-4. Int Immunol. (2003) 15:1–10. doi: 10.1093/intimm/dxg001
74. Negishi H, Ohba Y, Yanai H, Takaoka A, Honma K, Yui K, et al. Negative regulation of Toll-like-receptor signaling by IRF-4. Proc Natl Acad Sci USA. (2005) 102:15989–94. doi: 10.1073/pnas.0508327102
75. Jin B, Sun T, Yu XH, Yang YX, Yeo AE. The effects of TLR activation on T-cell development and differentiation. Clin Dev Immunol. (2012) 2012:836485. doi: 10.1155/2012/836485
76. Ban T, Sato GR, Nishiyama A, Akiyama A, Takasuna M, Umehara M, et al. Lyn kinase suppresses the transcriptional activity of IRF5 in the TLR-MyD88 pathway to restrain the development of autoimmunity. Immunity. (2016) 45:319–32. doi: 10.1016/j.immuni.2016.07.015
77. Rahman AH, Taylor DK, Turka LA. The contribution of direct TLR signaling to T cell responses. Immunol Res. (2009) 45:25–36. doi: 10.1007/s12026-009-8113-x
78. Reynolds JM, Martinez GJ, Chung Y, Dong C. Toll-like receptor 4 signaling in T cells promotes autoimmune inflammation. Proc Natl Acad Sci USA. (2012) 109:13064–69. doi: 10.1073/pnas.1120585109
79. Piggott DA, Eisenbarth SC, Xu L, Constant SL, Huleatt JW, Herrick CA, et al. MyD88-dependent induction of allergic Th2 responses to intranasal antigen. J Clin Invest. (2005) 115:459–67. doi: 10.1172/JCI200522462
80. Mandraju R, Jain A, Gao Y, Ouyang Z, Norgard MV, Pasare C. MyD88 signaling in T cells is critical for effector CD4 T cell differentiation following a transitional T follicular helper cell stage. Infect Immunity. (2018) 86:e00791–17. doi: 10.1128/IAI.00791-17
81. Piggott DA. MyD88 dependent induction of Th2 responses in a mouse model of asthma. J Allergy Clin Immunol. (2004) 113:69. doi: 10.1016/j.jaci.2004.01.069
82. Xu D, Meyer F, Ehlers E, Blasnitz L, Zhang L. Interferon regulatory factor 4 (IRF-4) targets IRF-5 to regulate Epstein-Barr virus transformation. J Biol Chem. (2011) 286:18261–7. doi: 10.1074/jbc.M110.210542
83. Guery L, Hugues S. Th17 cell plasticity and functions in cancer immunity. Biomed Res Int. (2015) 2015:314620. doi: 10.1155/2015/314620
84. Kuwabara T, Ishikawa F, Kondo M, Kakiuchi T. The role of IL-17 and related cytokines in inflammatory autoimmune diseases. Mediators Inflamm. (2017) 2017:3908061. doi: 10.1155/2017/3908061
85. McMichael A, Desai SR, Qureshi A, Rastogi S, Alexis AF. Efficacy and safety of brodalumab in patients with moderate-to-severe plaque psoriasis and skin of color: results from the pooled AMAGINE-2/-3 randomized trials. Am J Clin Dermatol. (2019) 20:267–76. doi: 10.1007/s40257-018-0408-z
86. Amatya N, Garg AV, Gaffen SL. IL-17 signaling: the yin and the yang. Trends Immunol. (2017) 38:310–22. doi: 10.1016/j.it.2017.01.006
87. Stritesky GL, Yeh N, Kaplan MH. IL-23 promotes maintenance but not commitment to the Th17 lineage. J Immunol. (2008) 181:5948–55. doi: 10.4049/jimmunol.181.9.5948
88. Li Y, Wei C, Xu H, Jia J, Wei Z, Guo, et al. The immunoregulation of Th17 in host against intracellular bacterial infection. Mediators Inflamm. (2018) 2018:6587296. doi: 10.1155/2018/6587296
89. Lee GR. The balance of Th17 versus treg cells in autoimmunity. Int J Mol Sci. (2018) 19:730. doi: 10.3390/ijms19030730
90. Hofstetter HH, Ibrahim SM, Koczan D, Kruse N, Weishaupt A, Toyka KV, et al. Therapeutic efficacy of IL-17 neutralization in murine experimental autoimmune encephalomyelitis. Cell Immunol. (2005) 237:123–30. doi: 10.1016/j.cellimm.2005.11.002
91. Swaidani S, Liu C, Zhao J, Bulek K, Li X. TRAF regulation of IL-17 cytokine signaling. Front Immunol. (2019) 10:1293. doi: 10.3389/fimmu.2019.01293
92. Jin W, Dong C. IL-17 cytokines in immunity and inflammation. Emerg Microb Infect. (2013) 2:e60. doi: 10.1038/emi.2013.58
93. Brüstle A, Heink S, Huber M, Rosenplänter C, Stadelmann C, Yu P, et al. The development of inflammatory TH-17 cells requires interferon-regulatory factor 4. Nat Immunol. (2007) 8:958–66. doi: 10.1038/ni1500
94. Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev. (2008) 223:87–113. doi: 10.1111/j.1600-065X.2008.00628.x
95. Ogura H, Murakami M, Okuyama Y, Tsuruoka M, Kitabayashi C, Kanamoto M, et al. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity. (2008) 29:628–36. doi: 10.1016/j.immuni.2008.07.018
96. Oriss TB, Raundhal M, Morse C, Huff RE, Das S, Hannum R, et al. IRF5 distinguishes severe asthma in humans and drives Th1 phenotype and airway hyperreactivity in mice. JCI Insight. (2017) 2:e91019. doi: 10.1172/jci.insight.91019
97. Weiss M, Byrne AJ, Blazek K, Saliba DG, Pease JE, Perocheau D, et al. IRF5 controls both acute and chronic inflammation. Proc Natl Acad Sci USA. (2015) 112:11001–6. doi: 10.1073/pnas.1506254112
98. Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol. (2011) 12:231–8. doi: 10.1038/ni.1990
99. Kimura A, Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur J Immunol. (2010) 40:1830–5. doi: 10.1002/eji.201040391
100. Wu X, Tian J, Wang S. Insight into non-pathogenic Th17 cells in autoimmune diseases. Front Immunol. (2018) 9:1112. doi: 10.3389/fimmu.2018.01112
101. Chang KK, Liu LB, Jin LP, Zhang B, Mei J, Li H, et al. IL-27 triggers IL-10 production in Th17 cells via a c-Maf/RORgammat/Blimp-1 signal to promote the progression of endometriosis. Cell Death Dis. (2017) 8:e2666. doi: 10.1038/cddis.2017.95
102. Cao S, Liu J, Song L, Ma X. The protooncogene c-Maf is an essential transcription factor for IL-10 gene expression in macrophages. J Immunol. (2005) 174:3484–92. doi: 10.4049/jimmunol.174.6.3484
103. Barnes BJ, Richards J, Mancl M, Hanash S, Beretta L, Pitha PM. Global and distinct targets of IRF-5 and IRF-7 during innate response to viral infection. J Biol Chem. (2004) 279:45194–207. doi: 10.1074/jbc.M400726200
104. Steinhagen F, McFarland AP, Rodriguez LG, Tewary P, Jarret A, Savan R, et al. IRF-5 and NF-κB p50 co-regulate IFN-β and IL-6 expression in TLR9-stimulated human plasmacytoid dendritic cells. Eur J Immunol. (2013) 43:1896–906. doi: 10.1002/eji.201242792
105. Cai H, Yao Z, Li W. IRF-5 accelerates leukocyte adhesion to endothelial cells in ischemia-reperfusion injury through regulating the transcription of VCAM-1. Biochem Biophys Res Commun. (2017) 492:192–8. doi: 10.1016/j.bbrc.2017.08.044
106. Wong LY, Hatfield JK, Brown MA. Ikaros sets the potential for Th17 lineage gene expression through effects on chromatin state in early T cell development. J Biol Chem. (2013) 288:35170–9. doi: 10.1074/jbc.M113.481440
107. Lazarevic V, Chen X, Shim J.-H., Hwang E.-S., et al. T-bet represses TH17 differentiation by preventing Runx1-mediated activation of the gene encoding RORγt. Nat Immunol. (2011) 12:96–104. doi: 10.1038/ni.1969
108. Gocke AR, Cravens PD, Ben LH, Hussain RZ, Northrop SC, Racke MK, et al. T-bet regulates the fate of Th1 and Th17 lymphocytes in autoimmunity. J Immunol. (2007) 178:1341–8. doi: 10.4049/jimmunol.178.3.1341
109. Kallies A, Good-Jacobson KL. Transcription factor T-bet orchestrates lineage development and function in the immune system. Trends Immunol. (2017) 38:287–97. doi: 10.1016/j.it.2017.02.003
110. Umetsu SE, Winandy S. Ikaros is a regulator of Il10 expression in CD4+ T cells. J Immunol. (2009) 183:5518–25. doi: 10.4049/jimmunol.0901284
111. Hedl M, Yan J, Witt H, Abraham C. IRF5 is required for bacterial clearance in human M1-polarized macrophages, and IRF5 immune-mediated disease risk variants modulate this outcome. J Immunol. (2019) 202:920–30. doi: 10.4049/jimmunol.1800226
112. Li D, Matta B, Song S, Nelson V, Diggins K, Simpfendorfer KR, et al. IRF5 genetic risk variants drive myeloid-specific IRF5 hyperactivation and presymptomatic SLE. JCI Insight. (2020) 5:e124020. doi: 10.1172/jci.insight.124020
113. Mizui M, Tsokos GC. Targeting regulatory T cells to Treat Patients With Systemic Lupus Erythematosus. Front Immunol. (2018) 9:786. doi: 10.3389/fimmu.2018.00786
114. Comte D, Karampetsou MP, Tsokos GC. T cells as a therapeutic target in SLE. Lupus. (2015) 24:351–63. doi: 10.1177/0961203314556139
115. Schafer PH, Ye Y, Wu L, Kosek J, Ringheim G, Yang Z, et al. Cereblon modulator iberdomide induces degradation of the transcription factors Ikaros and Aiolos: immunomodulation in healthy volunteers and relevance to systemic lupus erythematosus. Ann Rheum Dis. (2018) 77:1516–23. doi: 10.1136/annrheumdis-2017-212916
Keywords: autoimmune disease, Th1, Th2, Th17, polarization
Citation: Brune Z, Rice MR and Barnes BJ (2020) Potential T Cell-Intrinsic Regulatory Roles for IRF5 via Cytokine Modulation in T Helper Subset Differentiation and Function. Front. Immunol. 11:1143. doi: 10.3389/fimmu.2020.01143
Received: 06 March 2020; Accepted: 11 May 2020;
Published: 03 June 2020.
Edited by:
Markus Neurath, University Hospital Erlangen, GermanyReviewed by:
Francesco Novelli, University of Turin, ItalyCopyright © 2020 Brune, Rice and Barnes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Betsy J. Barnes, YmJhcm5lczFAbm9ydGh3ZWxsLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.