 Margaret A. McBride1
Margaret A. McBride1 Allison M. Owen2
Allison M. Owen2 Cody L. Stothers1Antonio Hernandez2Liming Luan2
Cody L. Stothers1Antonio Hernandez2Liming Luan2 Katherine R. Burelbach2Tazeen K. Patil2
Katherine R. Burelbach2Tazeen K. Patil2 Julia K. Bohannon1,2Edward R. Sherwood1,2
Julia K. Bohannon1,2Edward R. Sherwood1,2 Naeem K. Patil2*
Naeem K. Patil2*- 1Department of Pathology, Microbiology and Immunology, Vanderbilt University Medical Center, Nashville, TN, United States
- 2Department of Anesthesiology, Vanderbilt University Medical Center, Nashville, TN, United States
Critically ill, severely injured and high-risk surgical patients are vulnerable to secondary infections during hospitalization and after hospital discharge. Studies show that the mitochondrial function and oxidative metabolism of monocytes and macrophages are impaired during sepsis. Alternatively, treatment with microbe-derived ligands, such as monophosphoryl lipid A (MPLA), peptidoglycan, or β-glucan, that interact with toll-like receptors and other pattern recognition receptors on leukocytes induces a state of innate immune memory that confers broad-spectrum resistance to infection with common hospital-acquired pathogens. Priming of macrophages with MPLA, CPG oligodeoxynucleotides (CpG ODN), or β-glucan induces a macrophage metabolic phenotype characterized by mitochondrial biogenesis and increased oxidative metabolism in parallel with increased glycolysis, cell size and granularity, augmented phagocytosis, heightened respiratory burst functions, and more effective killing of microbes. The mitochondrion is a bioenergetic organelle that not only contributes to energy supply, biosynthesis, and cellular redox functions but serves as a platform for regulating innate immunological functions such as production of reactive oxygen species (ROS) and regulatory intermediates. This review will define current knowledge of leukocyte metabolic dysfunction during and after sepsis and trauma. We will further discuss therapeutic strategies that target leukocyte mitochondrial function and might have value in preventing or reversing sepsis- and trauma-induced immune dysfunction.
Introduction
Serious infection is a major threat to critically ill patients and frequently precipitates sepsis, a complex disease spectrum that includes systemic inflammation and organ dysfunction. As such, sepsis is the leading cause of death in non-cardiac intensive care units (ICU) and accounts for 40% of ICU expenditures (1). Early investigators postulated that systemic inflammation was the underlying factor driving the pathogenesis of sepsis and septic shock (2–4). High concentrations of pro-inflammatory mediators such as tumor necrosis factor, IL-1, and platelet activating factor were present in plasma and fluids of septic animals and humans (3, 5). Blockade of pro-inflammatory mediators in experimental animals attenuated or prevented the development of septic shock (6, 7). Those observations prompted clinical trials aimed at blocking cytokine and non-cytokine mediators of inflammation, which were not successful at improving survival in patients with severe sepsis or septic shock (8). Specifically, a trial of anakinra, a recombinant IL-1 receptor antagonist, was not found to be effective in improving mortality in sepsis (9). However, a subgroup analysis found that the use of anakinra improved survival in patients with concurrent hepatobiliary dysfunction and disseminated intravascular coagulation, which are specific features of macrophage activation syndrome (10). Therefore, subgroup analysis of diverse sepsis patients for underlying conditions needs to be considered in studies evaluating different sepsis treatments to better understand the therapeutic benefit in different sub-populations of sepsis patients. Later investigations showed that septic patients had impaired innate and adaptive antimicrobial immunity, which resulted in their inability to control primary and secondary infections. Likewise, patients that survive sepsis and severe trauma have long-term physical and cognitive disabilities and frequently require readmission to the hospital due to recurrent infections (11). Research indicates that the septic or severely injured host responds to severe inflammation by activating anti-inflammatory pathways to mitigate further inflammatory injury. Among those pathways are increased production of anti-inflammatory cytokines such as IL-10 and transforming growth factor-β (TGFβ) and upregulation of checkpoint inhibitors such as PD-1, CTLA-4, BTLA, and PDL1 by leukocytes (12, 13). Other investigators have shown large-scale apoptosis and dysfunction of lymphocytes and the proliferation of myeloid-derived suppressor cells, which act to suppress innate and adaptive antimicrobial responses (14, 15). Most recently, the concept of metabolic dysfunction has emerged as a factor underlying impaired function of the innate and adaptive immune systems of septic and severely injured patients. This paper will review current knowledge of leukocyte metabolic dysfunction in the setting of sepsis and severe injury and discuss interventions to improve leukocyte metabolism and function.
Overview of Sepsis-Induced Mitochondrial Dysfunction
Glycolysis and mitochondrial oxidative phosphorylation form the backbone of cellular metabolism. Glucose is primarily metabolized to pyruvate through glycolysis, along with a net generation of two ATP molecules. Cells transport pyruvate into mitochondria where it is metabolized to acetyl-CoA via the enzymatic action of the pyruvate dehydrogenase complex (PDH). Acetyl- CoA is metabolized through a series of enzymatic reactions in the mitochondrial tricarboxylic acid (TCA) cycle to produce reducing intermediates including NADH and FADH2, which feed electrons into the TCA cycle-linked electron transport chain (ETC). Optimal flow of electrons through ETC complexes (I-IV) is required for maintenance of mitochondrial membrane potential and proton gradient, which ultimately facilitate ATP generation (16). Recent studies show that mitochondria not only generate adenosine triphosphate (ATP), but also are intricately involved in cellular signaling pathways that regulate calcium homeostasis, reactive oxygen species (ROS) generation, redox signaling, and maintenance of immune cell competence, all of which are critical for our survival (17–19).
The 3rd International Consensus Conference defined sepsis as organ dysfunction caused by a dysregulated host response to infection (20). Evidence indicates that mitochondrial dysfunction is a key player in induction and propagation of sepsis-induced organ injury, which is demonstrated in both animal and human studies (21, 22). Brealey et al., were among the first to demonstrate that sepsis leads to significant impairment of skeletal muscle mitochondrial ETC activity (specifically complex I), which correlates with the severity of septic shock in humans (23). Furthermore, decreased skeletal muscle ATP concentrations were predictive of increased mortality among sepsis patients. A clinical study by Matkovich et al., showed a striking 43% decline in levels of mRNA that encode proteins involved in mitochondrial TCA cycle and ETC complexes in the hearts of septic patients (24). Numerous animal studies also demonstrate a role for mitochondrial dysfunction in sepsis pathology. Using animal models, sepsis has been shown to cause a significant impairment of mitochondrial function in multiple organs including heart, kidney, liver, and skeletal muscle (25–28). Although these studies demonstrate a role for mitochondrial dysfunction in sepsis pathology, discrepancies in various studies also show a highly variable mitochondrial function in multiple organs depending on the sepsis model used, severity of sepsis induced, time course studied, and methodology used for measurement of mitochondrial function (29). Therefore, there remains some controversy in the field as to whether mitochondria are the actual initiators or concurrent amplifiers of organ dysfunction during sepsis (29).
Sepsis-Induced Mitochondrial Dysfunction in Leukocytes
Recent studies demonstrate that sepsis-induced impairment of leukocyte mitochondrial function contributes to impaired antimicrobial immune responses and increased susceptibility to secondary infections (30, 31). The majority of the studies implicating a role for sepsis-induced leukocyte mitochondrial dysfunction used Peripheral Blood Mononuclear Cells (PBMCs) isolated from septic patients (summarized in Table 1). Adrie et al., demonstrated significant sepsis-induced depolarization of mitochondrial membrane potential and increased expression of cell death markers in peripheral blood monocytes. Eventual non-survivors demonstrated higher depolarization of the mitochondrial membrane as compared to survivors (32). Other studies showed a reduction in mitochondrial respiration in the presence of high ADP and Pi (also known as state 3 respiration), ATP synthase complex activity and mitochondrial spare respiratory capacity in PBMCs from sepsis patients (33, 34, 39). Reduced mitochondrial respiration in leukocytes was associated with increased incidence of organ failure (34). Garrabou et al., demonstrated a significant impairment of mitochondrial ETC complexes I, III, and IV in PBMCs of patients with confirmed systemic infection but without septic shock (35).
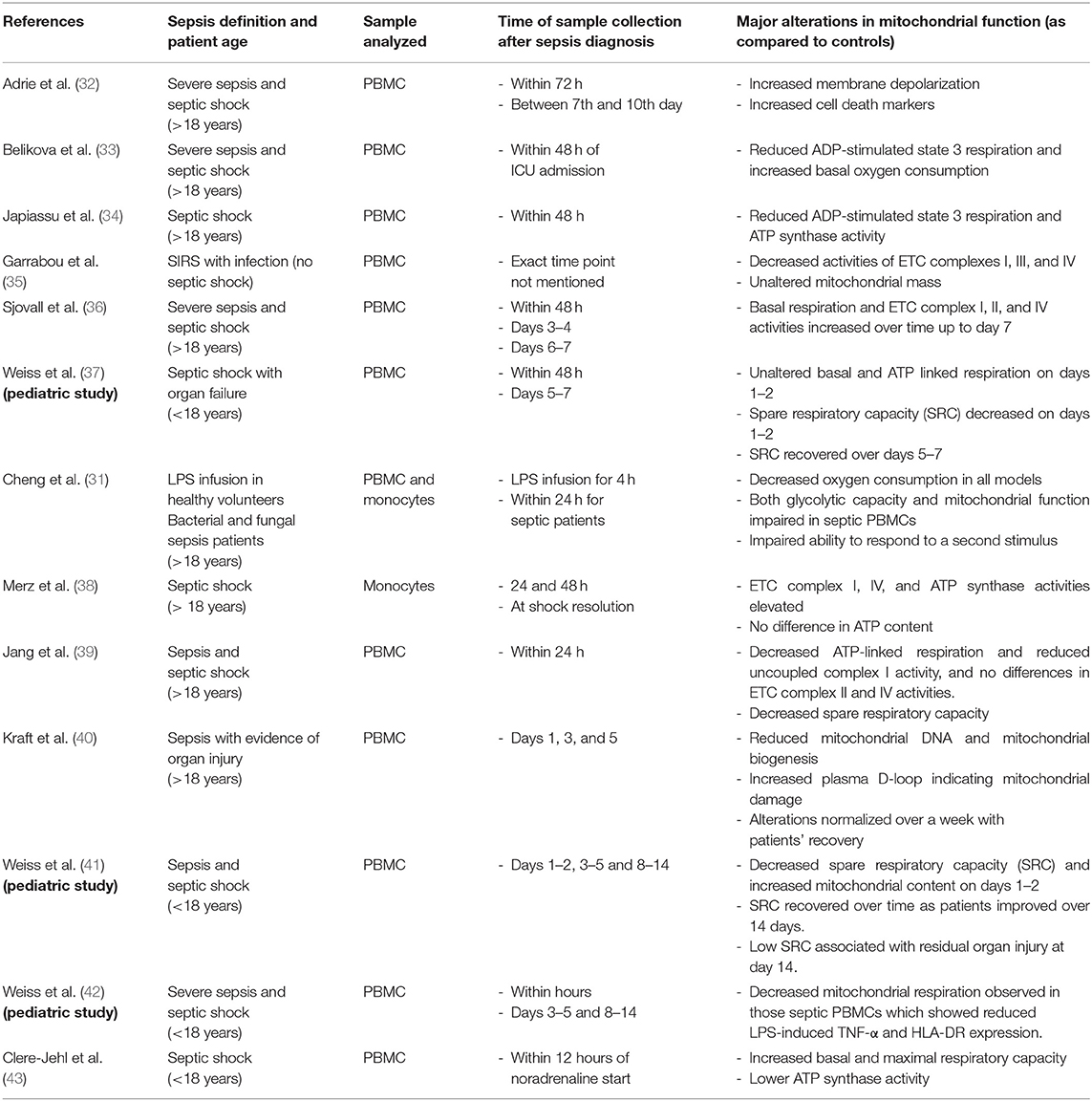
Table 1. Summary of clinical studies showing sepsis-induced alterations in leukocyte mitochondrial function.
In a major study, Cheng et al., showed that both bacterial and fungal sepsis leads to a shift in cellular metabolism toward glycolysis (Warburg effect), and leukocytes isolated from septic patients, as well as those treated with lipopolysaccharide (LPS), demonstrated a reduced oxygen consumption capacity signifying mitochondrial defects (31, 44). Furthermore, these metabolic defects were associated with impaired ability of leukocytes to produce pro-inflammatory cytokines in response to a secondary stimulus, which the authors refer to as a state of immunoparalysis (31). A study by Kraft et al., brings to light an important observation that effective reversal of the initial sepsis-induced leukocyte mitochondrial damage via early activation of mitochondrial biogenesis improved clinical outcomes among septic patients (40). They showed that mRNA levels of genes related to mitochondrial biogenesis, including PGC-1α, NRF1, and TFAM, were significantly reduced 1 day after the initiation of sepsis along with a decrease in mitochondrial DNA copy number. Recovery of these parameters was paralleled by improved clinical outcome and discharge from the ICU over a 1 week period (40). In multiple pediatric studies using PBMCs, Weiss et al., demonstrated that sepsis leads to a significant decrease in mitochondrial respiration and spare respiratory capacity implying a decreased bioenergetic reserve and mitochondrial dysfunction (37, 41, 42).
In contrast to these studies demonstrating sepsis-induced impairment of mitochondrial respiration, some studies show unaffected or increased mitochondrial respiration. Using PBMCs and monocytes from patients with severe sepsis and septic shock, Sjovall et al., and Merz et al., showed a significant increase in activities of mitochondrial ETC complexes I, II, and IV and did not observe a difference in these parameters among survivors vs. non-survivors (36, 38). In line with these studies, Clere-Jehl et al., showed that sepsis leads to a significant increase in mitochondrial respiratory capacity of PBMCs (43). However, mitochondrial respiration was impaired upon suspending the PBMCs in septic plasma, implying a role for a soluble plasma factor, which the authors attributed to a high level of HMGB1 (43). The contrasting findings might be attributed to the vast heterogeneity in sepsis patient populations, differing time points selected for measurements and underlying co-morbidities. Leukocyte-specific mitochondrial function in freshly isolated systemic immune cells has not been assessed in animal models.
In summary, the majority of studies implicate mitochondrial dysfunction as an important contributor toward sepsis-induced leukocyte and organ dysfunction. Importantly, early recovery of mitochondrial function correlates positively with improved clinical outcomes in septic patients (40, 45). Therefore, therapies targeting recovery of mitochondrial function hold potential for reversing leukocyte dysfunction during sepsis. Agents that target the AMP kinase pathway, such as AICAR (5-aminoimidazole-4-carboxamide ribonucleotide), or the mTOR signaling pathway, such as metformin, could provide benefit. Recent studies demonstrate that activation of pattern recognition receptors of innate leukocytes, especially monocytes and macrophages, augments mitochondrial function and rewires mitochondrial metabolism leading to accumulation of specific TCA cycle intermediates such as citrate, itaconate, succinate, fumarate, and others. Prophylactic treatment with TLR4 agonists can protect against severe infections for up to 14 days (46–48). That benefit is due, in part, to heightened mitochondrial and antimicrobial functions in macrophages Therefore, TLR agonist-induced mitochondrial metabolic reprogramming in innate leukocytes is associated with the generation of distinct innate immune memory. Mitochondrial reprogramming and innate immune memory are now being widely investigated as novel strategies for developing mitochondria-targeted therapies for protection against infections and sepsis in critically ill patients.
The Impact of Trauma on Leukocyte Metabolism
Although similar to sepsis, trauma provides a different set of signals to the immune system. While infection and sepsis can be a complication of trauma, the direct impact of trauma on immune system function is generated through tissue injury, inflammation, and tissue ischemia and reperfusion (49, 50). The effect of trauma on immune function is variable and largely dependent on the severity of injury (51, 52). The release of endogenous cell products, such as mitochondrial DNA, oxidized phospholipids, and ATP can activate toll-like receptors and inflammasomes to precipitate immune system activation (53, 54). Excessive or inappropriate immune system activation following major trauma could lead to immune dysfunction. Impairment of neutrophil and monocyte chemotaxis and antimicrobial functions have been described (55–57) as have alterations in lymphocyte function (58). However, little is known about the impact of major trauma on the metabolic state of leukocytes, which raises an area for research.
Potential Therapeutic Strategies Targeting Leukocyte Mitochondrial Function During Sepsis and Trauma
Effective mitochondrial biogenesis requires a coordinated action of complex intracellular pathways including both nuclear and mitochondrial genome encoded proteins (59, 60). PGC-1α is recognized as one of the most important and inducible transcription factor that drives mitochondrial biogenesis in response to external stimuli for maintaining mitochondrial homeostasis (61). The activity of PGC-1α is regulated by post-translational modifications. Sirtuin 1 (SIRT1)-induced deacetylation and adenosine monophosphate-activated protein kinase (AMPK)-induced phosphorylation are known to activate PGC-1α (62). Along with PGC-1α, other cellular transcription factors and mediators, including NRF1 and NRF2, PGC-1β, TFAM, ERRα, CREB, also play an important role in regulating mitochondrial biogenesis (63). The following section will discuss some of the promising therapeutic strategies targeting augmentation of mitochondrial biogenesis, which could be applicable for protecting or restoring leukocyte mitochondrial function during sepsis and trauma.
Pharmacological Agents Targeting Mitochondrial Biogenesis and Function
Studies included in this section are summarized in Table 2.
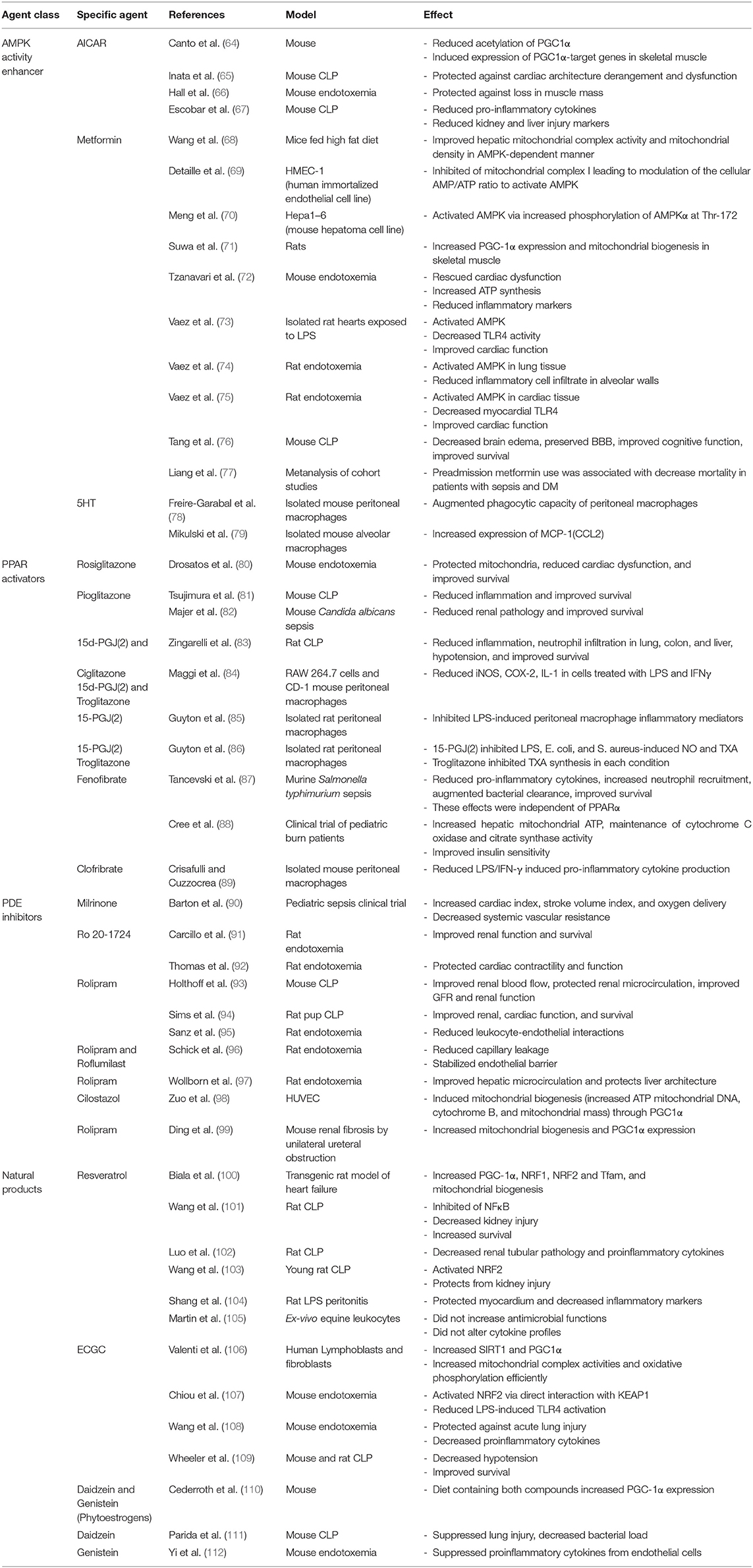
Table 2. Pharmacologic agents targeting mitochondrial biogenesis and function.
Modulators of AMPK Activity
AMPK is one of the key cellular mediators required for maintaining cellular energy homeostasis. AMPK exists in multiple isoforms and it is a heterotrimeric complex composed of one alpha subunit (either α1 or α2), beta subunit (either β1 or β2), and gamma subunit (either γ1, γ2, or γ3) (113). Previous studies show that AMPK induced transcriptional upregulation of genes involved in mitochondrial metabolism require PGC-1α (114) and overexpression of AMPK increases PGC-1α expression (115). AMPK regulates PGC-1α activity via direct phosphorylation at threonine-177 and serine-538, and the effect of AMPK on increased expression on mitochondrial proteins and function is regulated via PGC-1α (62, 114). AMPK has also been shown to activate SIRT1, an enzyme which catalyzes deacetylation and activation of PGC-1α leading to mitochondrial biogenesis (116). Therefore, activation of the AMPK pathway is a promising approach to stimulate mitochondrial biogenesis in various disease conditions, such as sepsis, that negatively affect mitochondrial function.
Treatment with AICAR will induce mitochondrial biogenesis and function in skeletal muscle cells, an effect mediated through activation of SIRT1, which leads to deacetylation and activation of PGC-1α (64). In a murine cecal ligation and puncture (CLP) model, AICAR protected against the sepsis-induced derangements in cardiac architecture and dysfunction (65). AICAR treatment also protected against LPS-induced loss in muscle mass (66) and reduced pro-inflammatory cytokine production and sepsis-induced increases in markers of kidney and liver injury during CLP-induced sepsis. Inhibition of AMPK by compound C exacerbated sepsis-associated tissue injury (67).
Metformin, a clinically used biguanide anti-diabetic drug, improves mitochondrial function via activation of AMPK (68). The mechanisms leading to metformin-induced activation of AMPK include increased phosphorylation of AMPKα at Thr-172 and via inhibition of mitochondrial complex I leading to modulation of the cellular AMP/ATP ratio (69, 70). Studies by Suwa et al. recognized that metformin, a first line oral drug for the treatment of type 2 diabetes, increases PGC1-α and mitochondrial protein content in muscle through AMPK activation (71). Metformin has been shown to be protective in studies employing animal models of sepsis (117). During LPS- and CLP-induced sepsis, metformin protected against sepsis-induced injury in brain, heart, liver, and lung. These benefits were mediated through inhibition of oxidative stress and inflammation, reduced infiltration of neutrophils, maintenance of mitochondrial membrane potential, and preservation of mitochondrial function (72–76, 118). In humans, a metanalysis including five observational cohort studies found that pre-admission use of metformin was associated with decreased mortality among patients with sepsis and diabetes mellitus (77). This association warrants further study of causality and the mechanism behind this association to assess the therapeutic benefit of metformin during sepsis.
Despite the described benefits of AICAR and metformin in reducing inflammation and providing organ protection in experimental models of sepsis, little is known about the impact of these drugs on immune function in the septic or severely injured host, which provides fertile ground for future research.
5-Hydroxytryptamine Receptor (5HT) Agonists
Specific agonists of the 5HT receptor family have been shown to induce mitochondrial biogenesis (119). 5HT is the chemical name for endogenous neurotransmitter serotonin. 5HT receptors are G-protein coupled receptors with serotonin functioning as its endogenous ligand. It remains to be determined if 5HT receptor agonists could provide therapeutic benefit to protect against sepsis-induced organ injury. Immune cells including macrophages, monocytes and T cells express 5HT receptors (120). Serotonin has been shown to augment the phagocytic capacity of murine peritoneal macrophages via 5HT1A receptor subtype (78). Serotonin has also been shown to activate alveolar macrophages via 5HT2c receptor leading to increased expression of the monocyte chemoattractant MCP-1 (79). Various studies have shown the stimulatory effect of serotonin on other immune cells including Natural Killer cells, dendritic cells, and T cells (120, 121). Studies evaluating the effect of serotonin and synthetic 5HT receptor agonists on mitochondrial biogenesis in leukocytes is currently lacking.
Peroxisome Proliferator-Activated Receptor (PPAR) Activators
PPARs are a class of nuclear receptors/transcription factors that are comprised of three isotypes including PPARα, PPARβ/δ, and PPARγ (122). PPARs are known to regulate various metabolic functions including triglyceride and lipoprotein metabolism, fatty acid synthesis, and oxidation and energy homeostasis to name a few (123). PGC1-α, the aforementioned transcription factor known for its role in mitochondrial biogenesis, also functions as a coactivator PPARγ (124). Thiazolidinediones are clinically used anti-diabetic drugs, which increase insulin sensitivity through activation of PPARγ (125). Rosiglitazone, a thiazolidinedione class drug, was shown to attenuate LPS-induced cardiac dysfunction and protect mitochondria leading to improved survival (80). Pioglitazone, another PPARγ agonist, has been shown to reduce inflammation and improve survival in a murine CLP and Candida albicans-induced sepsis (81, 82). Zingarelli et al. showed that treatment with PPARγ ligands, 15-deoxy-Delta(12,14)-PGJ(2) (15d-PGJ(2)), and ciglitazone attenuated inflammation, reduced excess neutrophil influx into various organs, decreased hypotension and improved survival through regulation of NF-κB and AP-1 signaling pathways using murine CLP model of sepsis (83). Other studies have also shown similar anti-inflammatory effects of synthetic PPARγ ligands including 15d-PGJ(2) and troglitazone on macrophages (84–86, 126). Fenofibrate, a known PPARα agonist used clinically for the management of dyslipidemia, reduced pro-inflammatory cytokines levels, promoted neutrophil recruitment to the site of infection and augmented bacterial clearance leading to improved survival in a murine model of Salmonella typhimurium-induced sepsis (87). The beneficial effect of fenofibrate was shown to be independent of PPARα but dependent on the preservation of neutrophil CXCR2 expression (87). Using another PPARα agonist, Crisafulli et al. demonstrated that clofibrate reduces LPS/IFNγ induced pro-inflammatory cytokine production in murine peritoneal macrophages (89). Treatment of pediatric burn patients with fenofibrate within the first week after burn injury has been shown to increase hepatic mitochondrial ATP production, maintain cytochrome c oxidase levels and citrate synthase activity along with improving insulin sensitivity, thereby indicating the therapeutic utility of fenofibrate-induced augmentation of mitochondrial function after burn injury (88). A study by Standage et al. showed that PPARα expression is decreased in the whole blood of pediatric sepsis patients and this correlated with the severity of sepsis outcomes and PPARα is required for maintaining optimal immune function during sepsis (127). In summary, PPAR agonists might have therapeutic potential in attenuation of sepsis induced inflammation and organ injury. However, the specific effect of various PPAR agonists on mitochondrial biogenesis and function in various organs and leukocytes in context of sepsis and trauma has not been investigated in detail and needs to be evaluated in future studies.
Phosphodiesterase (PDE) Inhibitors
Phosphodiesterases serve to hydrolyze cAMP and cGMP, increase levels of which reduces vascular tone, tightens endothelial junctions, and increases cardiac contractility. The cAMP-response-element-binding protein (CREB) is involved in transcriptional activation of PGC1α (128). In pediatric sepsis patients, treatment with PDE3 inhibitors increase both cAMP and cGMP levels and not only improve cardiac function (90, 129, 130) but also increase survival (131, 132). PDE4 inhibitors such as rolipram and Ro 20-1724 are selective for cAMP (133). Inhibition of PDE4 using Ro 20-1724 reduced systemic vascular resistance and improved cardiac and renal function in LPS model of sepsis in rats (91, 92). Treatment with rolipram improves renal blood flow, protects renal microcirculation and improves glomerular filtrate rate and renal function in a murine model of CLP-induced sepsis, even when administered 6 h after CLP (93). Rolipram treatment also improved renal and cardiac function leading to improved survival in septic rat pups (94). PDE4 inhibitors, rolipram and roflumilast, have been shown to reduce leukocyte-endothelial interactions which inhibits inflammatory cell influx, and reduce capillary leakage during LPS-induced inflammation (95, 96). Wollborn et al. showed that treatment with rolipram improves hepatic microcirculation and protects liver architecture in a rat model of LPS induced inflammation (97). Pharmacological agents such as rolipram and cilastozol which are specifically inhibit PDE4 and PDE3, respectively, and have been shown to increase CREB phosphorylation, upregulate PGC-1α expression and contribute to the induction of mitochondrial biogenesis (98, 99, 134). Future studies addressing the impact of PDE inhibitors on mitochondrial function in organs and leukocytes in context of sepsis and trauma are warranted.
Natural Products That Induce Mitochondrial Biogenesis
Resveratrol, a polyphenol compound found in grapes and red wine, has been shown to activate PGC1α and mitochondrial biogenesis through SIRT1 or AMPK signaling (135). Resveratrol upregulates PGC-1α, NRF1, NRF2 and Tfam leading to potentiation of mitochondrial biogenesis (100). In multiple studies using a CLP model of polymicrobial sepsis in rats, resveratrol treatment results in increased survival as well as decreased kidney injury associated with inhibition of NFκB (101, 102). In a similar model of pediatric sepsis-induced kidney injury in young rats, resveratrol was shown to activate NRF2 and protect from injury (103). Shang et al. report that resveratrol is protective in LPS-induced cardiomyopathy in rats also through inhibition of NFκB (104). In horses, however, Martin et al. showed that a 3 week course of resveratrol did not increase antimicrobial function or alter cytokine release profiles of ex vivo stimulated leukocytes (105).
Epigallocatechin gallate (ECGC), a natural compound found in tea, promotes cAMP dependent signaling and increases SIRT1 and consequently PGC1α (106). In murine LPS-induced endotoxemia, ECGC protected against acute lung injury and decreased proinflammatory cytokine production (108). ECGC has been shown to induce the NRF2 antioxidant response element through direct interaction with its inhibitor KEAP1 thereby leading NRF2 activation (107). NRF2, like PGC1α, is known to be involved in mitochondrial biogenesis. In the CLP model, ECGC attenuated hypotension and improved survival (109).
Estrogen receptors are known to regulate mitochondrial biogenesis, so it follows that phytoestrogens may also induce mitochondrial biogenesis and have protective affects in sepsis. A diet high in two phytoestrogens daidzein and genistein has been shown to increase PGC-1α expression, and these two compounds were separately shown to decreases proinflammatory cytokines in LPS-induced endotoxemia, and increase survival and bacterial clearance in CLP-induced sepsis respectively (110–112).
Metabolic Reprogramming of Innate Leukocytes by Microbial Ligands
Stimulation of innate immune cells with microbial ligands such as LPS, peptidoglycan, or β-glucan reprograms their metabolism, which supports the increased physiological demands needed to augment antimicrobial capacity to combat invading infections (47, 136, 137). The reprogrammed phenotype of innate leukocytes manifests as distinct augmentation of glycolysis and mitochondrial tricarboxylic acid cycle flux and oxidative phosphorylation, as detailed below (Figure 1).
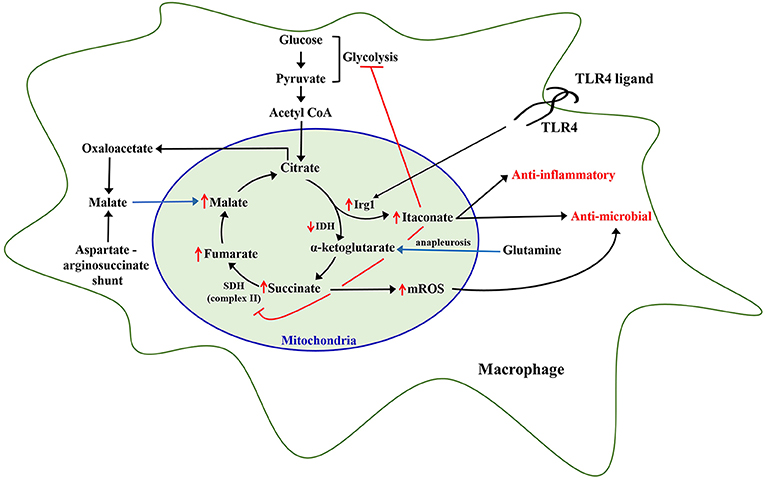
Figure 1. Metabolic reprogramming of leukocytes. Inflammatory stimulation of leukocytes, specifically monocytes and macrophages, with Toll-like receptor 4 (TLR4) ligands like lipopolysaccharide, has been shown to rewire mitochondrial metabolic pathways including upregulation of immunoresponsive gene 1 (Irg1) leading to increased itaconate generation, and increased accumulation of other TCA cycle metabolites including succinate, fumarate, malate, and citrate which continue to be replenished via additional pathways including glutamine anapleurosis and aspartate-arginosuccinate shunt. Itaconate produced by Irg1 inhibits succinate dehydrogenase, which causes an increase in mitochondrial reactive oxygen species (mROS). Itaconate and mROS augment antimicrobial capacity of leukocytes.
Reprogramming of Glycolysis
Hard et al. discovered that immune macrophages, defined as those from peritoneal cavities of mice injected with bacteria, produced more lactate and consumed less oxygen than controls (138). Further investigations showed that macrophages stimulated with LPS manifest increased glucose uptake, an elevated glycolytic rate and augmentation of the pentose phosphate pathway (139, 140). These findings were reminiscent of the aerobic glycolysis noted by Warburg et al. in cancer cells, which preferentially utilize glycolysis, even in aerobic conditions that should favor oxidative phosphorylation as more energetically efficient (141). Aerobic glycolysis in macrophages in facilitated, in part, by stabilization of hypoxia-inducible factor (HIF)-1α. Early macrophage activation induces accumulation of succinate and itaconate, which are transported out of mitochondria in the cytosol where it acts to stabilize HIF-1α by impairing the activity of prolyl hydroxylases (142, 143). HIF-1α facilitates increased expression of numerous gene products that regulate inflammation including enzymes that promote glycolysis (140). Though this effect is notable in multiple types of murine macrophages, Vijayan et al. reported that LPS does not increase glycolysis in human PBMCs (144). Multiple purposes for this increase in glycolysis, over oxidative phosphorylation at the expense of energy efficiency, have been hypothesized. West et al. described that classically activated macrophages require mitochondrial reactive oxygen species for effective bacterial clearance (145). The contributions of mitochondrial complex I to ATP synthesis during oxidative phosphorylation may detract from mROS generation (145). As suggested in Viola et al., glycolysis may also be advantageous because it supplies biosynthetic intermediates important for rapid cellular adaptations, as well as NADPH through the pentose phosphate shunt, which is important for generation of ROS. The Warburg effect in macrophage activation is specific to the classical M1 phenotype, but not in alternatively activated M2 macrophages, which rely on oxidative phosphorylation (146). Interestingly, increases in oxidative phosphorylation and glycolysis occur in macrophages activated by the TLR4 agonist MPLA 72 h after exposure, resulting in a hybrid phenotype with metabolic characteristics common to both M1 and M2 macrophages (47).
LPS also induces the TCA cycle metabolite itaconate, in both murine and human macrophages (147) (Figure 1). It has been recently shown that itaconate inhibits glycolysis via inhibiting glycolytic enzymes aldolase A and glyceraldehyde-3-phosphate hydrogenase in RAW 264.7 macrophage cell lines (148, 149). Itaconate has also been shown to inhibit succinate dehydrogenase, which might reprogram citric acid cycle function and facilitate mROS generation due to reverse electron transport secondary to inhibition of SDH-dependent complex II (150).
Reprogramming of Mitochondrial Metabolism
The majority of recent studies demonstrate significant alterations in the generation of TCA cycle intermediates upon TLR agonist-induced inflammatory stimulation of monocytes and macrophages. Studies from our laboratory, and others, show that citrate, itaconate, and succinate accumulate during metabolic rewiring of macrophages and monocytes (47, 140, 151, 152). Recent studies have elucidated a unique role for each of these metabolites in the context of cellular metabolic and antimicrobial functions.
Citrate is converted to α-ketoglutarate by isocitrate dehydrogenase (IDH) through the intermediate cis-aconitate. Michelucci et al., demonstrated that stimulation of macrophages with LPS leads to significant upregulation of immunoresponsive gene 1 (Irg1) enzyme, which catalyzes the production of itaconate from cis-aconitate in the mitochondria, thus diverting pyruvate-derived citrate production away from energy generation and toward production of itaconate (153). Jha et al., also showed that LPS induces downregulation of IDH and succinate dehydrogenase (SDH) function in macrophages leading to a significant accumulation of citrate and succinate (151). In line with this, studies from our laboratory show that MPLA treatment reduces TCA cycle flux between citrate and α-ketoglutarate at 24 h after stimulation in association with induction of Irg1 expression and large scale itaconate production (47). Therefore, it is evident that inflammatory stimulation of macrophages drives citrate toward production of itaconate. Itaconate has now been shown to be a critical regulator of macrophage and monocytic function after LPS stimulation. Intracellular itaconate concentrations of up to 8 mM have been shown in macrophages at 6 h after LPS stimulation (153), which subsequently steadily decline over time (152). There are multiple known downstream cellular effects of this dramatic increase in itaconate. First, itaconate inhibits mitochondrial complex II or SDH function in a dose-dependent manner leading to succinate accumulation (154), which is supported by the observation that Irg1 knockout macrophages do not accumulate succinate following LPS stimulation (151). The implications of succinate accumulation are discussed later. Itaconate also plays a major role in potentiating cellular anti-inflammatory and anti-oxidant effects through activation and nuclear translocation of NRF2via alkylation of KEAP1, a known physiological inhibitor of NRF2 (147). Through activation of NRF2, 4-octyl-itacoante (a cell permeable analog of itaconate) increases expression of key anti-inflammatory genes including heme oxygenase 1 and potently inhibits proinflammatory cytokine release (147). Macrophages lacking the Irg1 enzyme produce increased proinflammatory cytokines, including IL-6, IL-18, and IL-1β, in response to LPS relative to wild type macrophages and treatment with a cell permeable itaconate derivative decreases proinflammatory cytokines in response to LPS (147, 155).
Itaconate is also known to be secreted by macrophages into the extracellular milieu and have direct antibacterial effects (156). Itaconate competitively inhibits the microbial enzyme isocitrate lyase, a required step in the glyoxylate shunt, thereby limiting bacterial growth under nutrient poor conditions as occur at the site of infection (157). The microbial glyoxylate shunt bypasses two decarboxylation steps in the tricarboxylic acid cycle, facilitating the assimilation of carbon when only two-carbon sources such as ethanol or acetate are available (151, 158–160). Pathogens that have shown sensitivity to itaconate-induced microbial growth inhibition include Mycobacterium tuberculosis, Staphylococcus aureus, Legionella pneumonia, Acinetobacter baumanii, and Salmonella enterica (153, 161, 162). Therefore, itaconate affects cellular metabolism and affords anti-inflammatory and anti-microbial protection upon inflammatory activation of immune cells. As such, our knowledge of the role of itaconate is currently limited to macrophages and monocytes, and future studies addressing its effects on other leukocytes such as neutrophils and dendritic cells will shed more light on the novel aspects of this critical metabolite. Nonetheless, based on studies, therapeutic utility of itaconate to protect against life-threatening infections and sepsis merits further investigation.
Succinate is another TCA cycle metabolite that significantly accumulates in LPS-stimulated macrophages and monocytes (150, 152, 163). Succinate is the principal substrate for succinate dehydrogenase, which not only participates in the TCA cycle but also in ETC complex II. Oxidation of succinate to fumarate results in reduction of FAD+ and ultimately Coenzyme Q, which continues in the ETC via complex III and IV, leading to ATP generation via ATP synthase (16). Itaconate-induced inhibition of SDH and facilitation of glutamine anapleurosis are the major sources of intracellular succinate accumulation upon LPS stimulation of macrophages (150, 151). High levels of succinate and succinate dehydrogenase activity are associated with inducing a pro-inflammatory phenotype in innate leukocytes as result of succinate-mediated hypoxia inducible factor α (HIF-1α) stabilization, increased mitochondrial ROS generation, and protein succinylation (137, 163). LPS-induced succinate accumulation is associated with stabilization of HIF-1α, leading to increased IL-1β production and inflammation (140, 164). Rapid oxidation of increased succinate to fumarate by SDH requires CoQ, which is consumed under LPS stimulation, thereby driving reverse electron transport leading to a substantial generation of mitochondrial ROS (165). Although uncontrolled generation of mitochondrial ROS can have deleterious effects on cellular functions, it has also been shown to play an important role in microbial clearance (145). However, further studies are needed to establish the antimicrobial role of SDH-generated ROS in in vivo models of infection.
Inflammation-induced increases in intracellular accumulation of citrate also affects cellular metabolism and functions. Activated macrophages accumulate citrate due to decreased isocitrate dehydrogenase activity (47, 151). De Souza and colleagues recently demonstrated that LPS-mediated increase in IFN-γ limits isocitrate dehydrogenase activity in an autocrine manner in macrophages, implying a role for IFN-γ in LPS-mediated increase in citrate levels (166). Accumulated citrate is not only converted to itaconate (153) in the mitochondria but also transported from the mitochondria into the cytosol via mitochondrial citrate carrier (CIC) (167). Increased CIC and cytosolic citrate has been shown to fuel the LPS-induced generation of pro-inflammatory mediators such as nitric oxide, ROS, and prostaglandins in macrophages (168). Our studies also show that MPLA-stimulated citrate transported into the cytosol is ultimately converted to malate and pyruvate, and the cytosolic malate replenishes mitochondrial oxaloacetate pools to further fuel a sustained increase in mitochondrial TCA cycle flux (47). Importantly, these alterations in citrate metabolism are associated with a sustained augmentation of mitochondrial density and oxygen consumption, along with increased macrophage phagocytic capacity (47). Therefore, citrate accumulation not only plays an important role in fueling acute inflammation but also potentiates a sustained increase in TCA cycle flux and antimicrobial functions, which need further evaluation.
Evidence for Metabolic Reprogramming in Murine and Human Sepsis Studies
The majority of studies demonstrating the effect of inflammatory activation on metabolic reprogramming of innate leukocytes such as macrophages and monocytes have been performed in vitro. Corroborating the changes described in the in vitro studies described above, metabolic reprogramming of innate immune cells in response to TLR activation has also been observed in some in vivo murine and human studies. Sterile endotoxemia (LPS administration) in mice causes peritoneal macrophages to more than double glucose uptake, suggesting an increase in glycolysis in this model (169). Functionally, monocytes from septic patients were found to have increased basal glycolysis compared to healthy controls (170). Shalova et al. performed a gene ontology analysis to compare monocytes from septic patients relative to healthy controls, and reported that the top 10 most significantly downregulated gene clusters were all related to cellular metabolism (171). Consistent with this, Cheng et al. found diminished glycolysis and oxidative phosphorylation in peripheral blood mononuclear cells (PBMCs) in septic patients with immunoparalysis as compared to control subjects (31). Genome-wide microarray analysis of PBMCs from patients with both bacterial and fungal sepsis in this study identified that genes for oxidative phosphorylation and glycolysis were both increased along with evidence of mitochondrial dysfunction pathways, suggesting that immune cell metabolism is significantly affected during sepsis. Further studies to separate the adaptive from the pathogenic changes in leukocyte metabolism could guide the development of therapies to augment or suppress these metabolic changes. For example, a study by Pan et al. demonstrated that a known anti-inflammatory compound, deoxyelephantophin, both blocks LPS-induced glycolytic increase and protects mice against endotoxemia (172).
There are limited in vivo studies analyzing the effect of sepsis on alterations of specific mitochondrial TCA cycle intermediates during sepsis. A murine study by Chao et al. employing scrub typhus infection demonstrated a 60-fold increase in plasma itaconate levels at 10 days after infection (173). A clinical study by Meiser et al. reported absence of any detectable itaconate in the plasma and urine of septic patients, in which the authors concluded that itaconate may not be a suitable systemic biomarker for predicting sepsis outcomes (174). That study evaluated the levels of itaconate at a single time point among sepsis patients and failed to elaborate on the clinical condition of patients during sample collection and the exact time point for collection. A recent study by Beloborodova et al. detected low concentrations of itaconic acid (0.5–2.3 μM) in the plasma of septic shock patients collected within 24 h and none was detected in patients at later stages of sepsis (175). The levels of succinate were higher in the late stage sepsis patients as compared to early stage, but lower than the control healthy group. It must, however, be noted that the early and late stage sepsis patients included in this study were entirely different patient cohorts and the authors do not report the changes in plasma itaconate levels as sepsis progressed in each septic patient subset. It is critical to follow septic patients and study the alterations in itaconate levels at various time points after sepsis induction to derive a definitive conclusion for the use of itaconate as a biomarker for sepsis outcomes or for supporting itaconate's use for therapeutic purpose to combat sepsis. Future studies evaluating sepsis-induced alterations in the levels of mitochondrial metabolites would be critical to further the field of metabolic reprogramming toward discovery of novel therapeutics to protect against infections and sepsis.
Innate Immune Memory and Trained Immunity
Classically, the role of the innate immune system is to recognize pathogens and mount a non-specific yet rapid response, whereas immunological memory has been traditionally considered a unique hallmark of the adaptive immune system. However, recent studies indicate that innate immune cells adapt upon exposure to a pathogen or pathogen-derived ligand, triggering augmentation of cell physiology and antimicrobial functions which allows for robust responses to a subsequent challenge either by the same or different pathogen (176). This phenomenon by which innate antimicrobial efficiency is increased due to the priming effect of prior exposure is termed “innate immune memory” or “trained immunity” (Figure 2). This immunoregulatory process confers host resistance to infection in plants and invertebrates that do not have adaptive immunity but also in mammals (177). The cell type (myeloid, natural killer, and innate lymphoid cells), stimuli (pattern recognition receptors and cytokines), genetic mechanism (epigenetic rewiring), and time scale (persisting weeks to months) are unique to innate immune memory, independent of those involved in classical immunological memory (178). An important player in health and disease, trained immunity may also serve as an innovative therapeutic strategy for protecting vulnerable patients from life-threatening infections in the future.
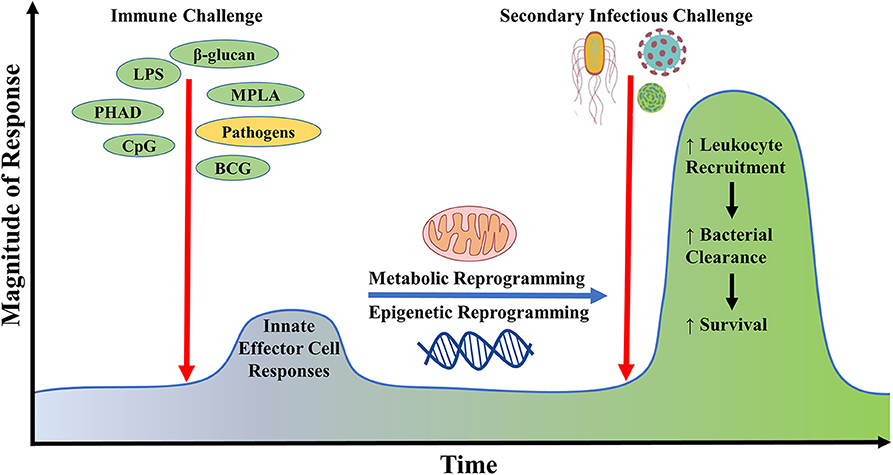
Figure 2. Generation of innate immune memory using microbial ligands. Initial challenge with microbial ligands such as lipopolysaccharide, monophosphoryl lipid A, CpG, β-glucan potently stimulates host innate effector immune responses in cells such as neutrophils, monocytes, and macrophages, leading to the reprogramming of their metabolic and epigenetic status. Upon re-exposure of the initially primed host with a secondary inflammatory stimulus or infectious challenge, there occurs a heightened innate immune response against invading microbes via increased immune cell recruitment leading to improved microbial clearance and survival. This phenomenon is termed as innate immune memory.
Metabolic Reprogramming and Innate Immune Memory
Recent findings strongly indicate that metabolic reprogramming is a key process underlying development of innate immune memory. Several studies have revealed that expression of key pro-inflammatory proteins and an effective immune response relies on intact mitochondrial respiration (179, 180), and the study of the metabolic demands of mounting an immune response has been a topic of increasing interest (181). It has become widely appreciated that metabolism dynamics regulate innate immunity via production of metabolite intermediates which influence cellular phenotype and function (182). β-glucan immunomodulation has been associated with upregulated glycolysis in trained macrophages (183) and monocytes (184), likely to support pro-inflammatory macrophage antimicrobial functions (182, 185). This has been shown to be dependent on a shift from oxidative phosphorylation toward glycolysis through an Akt/mTOR/HIF-1α dependent pathway (183, 186). We recently reviewed regulation and function of HIF-1α in myeloid cells (187). On the other hand, TLR ligands (such as LPS, MPLA, and CPG) increase aerobic glycolysis in concert with increased antimicrobial functions (such as respiratory burst and phagocytosis) as well as induce mitochondrial biogenesis and increased oxidative metabolism (47). These metabolic alterations allow immediate leukocyte activation, cytokine secretion, and a more effective innate immune response to infection (46, 47, 188, 189). Our study using HIF-1α deficient macrophages demonstrated that HIF-1α is required for these metabolic alterations (46). Another study from our group showed that the inhibition of mTOR, which stabilizes HIF-1α, diminishes the protective response of TLR4 ligands (47).
Despite the apparent benefits of inducing innate immune memory, reprogramming of leukocyte oxidative metabolism could be a double-edged sword. As noted above, current research indicates that priming the immune system with microbial ligands at doses that do not cause damaging systemic inflammation induces protective immunity in association with an increase in leukocyte oxidative metabolism (47, 48). It appears that the heightened metabolic state induced under those conditions is utilized to facilitate augmented leukocyte antimicrobial functions such as phagocytosis, oxidative burst, and microbial killing. However, in cases of tissue injury, reprogrammed leukocytes could funnel energy to drive hyperinflammation. A recent paper by Di Gioia and colleagues showed that oxidized phospholipids derived from 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphorylcholine (oxPAPC) can induce increased leukocyte oxidative metabolism and hyperinflammation, especially in the presence of microbial ligands such as LPS (190). Oxidized phospholipids are damage associated molecular patterns (DAMPS) that are released following tissue injury. Di Gioia and colleagues reported that oxPAPC and LPS strongly drive production of pro-IL-1β in macrophages, which is cleaved and secreted as the mature protein upon activation of the inflammasome by DAMPS such as ATP (190). However, the ramifications of these alterations in models of acute inflammation remain to be fully elucidated since a study by Chu and colleagues showed that oxPAPC inhibits non-canonical inflammasome activation and is protective in an experimental model of septic shock (191).
Innate Immune Memory—a Novel Therapeutic Target to Protect Against Infections and Sepsis
The non-specific protection conferred by trained immunity lends itself to an exciting novel therapeutic approach by which patients could be primed and protected from a wide array of infections thus preventing sepsis and subsequent mortality. Several microbial ligands have immunomodulatory potential, most notably, TLR and dectin-1 agonists. Rowley first reported in 1956 that priming mice with the TLR4 agonist lipopolysaccharide (LPS), a structural component of the cell wall of Gram-negative bacteria, conferred host protection to subsequent exposure to Gram-negative pathogens (192). Following this discovery, it has been found that LPS challenge protects against a wide array of pathogens, including fungal (193), Gram-positive Staphylococcus aureus (194), and several Gram-negative pathogens, including Escherichia coli (192), Salmonella enterica serovar typhimurium (195), and Pseudomonas aeruginosa (196, 197), as well as polymicrobial sepsis (198). Priming with LPS induces enhanced bacterial clearance (196, 199) and leukocyte recruitment (194, 200).
Leukocytes primed with LPS can also be described as not only trained, but also “endotoxin tolerant,” which is defined by an attenuated pro-inflammatory response upon secondary challenge with the stimulus. A body of literature suggests that the phenomenon of endotoxin tolerance is a state of immunoparalysis during which the host is more susceptible to infection (201, 202), and results in poorer patient outcomes (203–206). However, the clear relationship between endotoxin tolerance and susceptibility to later infections has not been established. In fact, our group recently demonstrated that the cytokine response to LPS is not indicative of antimicrobial immunity (46), and a body of literature illustrates that altering proinflammatory cytokines during infection has had no protective benefit (207–210) thereby bringing into question whether proinflammatory cytokine levels are an essential element in determining immune competence.
TLR4 Agonist-Induced Innate Immune Memory and Protection Against Infection
As LPS is toxic to humans, experimental studies have progressed to investigate other agonists that confer this attractive phenotype of host resistance to infection after priming. Intriguingly, prophylactic administration of the vaccine adjuvant MPLA, which is derived by cleaving the C1 phosphate group from lipid A and is 100-fold less toxic than LPS (211–213) improves bacterial clearance, attenuates physiologic dysfunction, induces leukocyte expansion and recruitment to sites of infection, enhances antimicrobial functions, and profoundly improves survival during infection with a wide array of clinically relevant pathogens (47, 188, 214–217). TLR4 is unique among TLRs as it can generally signal through both the myeloid differentiation primary response gene 88 (MyD88)-dependent and the TIR-domain-containing adapter inducing interferon-β (TRIF)-dependent pathways. A study of human neutrophils, however, revealed that TLR4 activation by LPS does not activate the TRIF-dependent pathway in neutrophils, postulated to be due to neutrophil's more prominent role in bacterial responses compared to viral (218). Our group is investigating the relative contribution of these pathways in TLR-mediated trained innate immunity, and has shown that MyD88 deficient mice fail to augment leukocyte recruitment or G-CSF production in response to infection following priming with MPLA, both of which are known to play a critical role in MPLA-mediated protection (188, 189). Further, the MyD88-selective TLR9 agonist CpG oligodeoxynucleotide (CpG) preserves physiologic function and improves bacterial clearance following infectious challenge with Pseudomonas aeruginosa (46). CpG similarly provided protection in a model of intracerebral Escherichia coli (219), which implies that TLR-mediated resistance to infection is dependent on MyD88 signaling.
TLR4 agonist-induced antimicrobial properties are independent of antibiotic therapy. This is of particular importance due to the current rise in global antibiotic resistance (220–222). The rate of antibiotic resistance has been far exceeding the rate of new antibiotic class development, and current market trends suggests pharmaceutical companies will not be able to support new antibiotic development programs (220, 223). Thus, there is an increasing need for novel antimicrobial therapeutic strategies, lending to the possibility of adopting agents that induce trained immunity as independent or adjunct antimicrobial therapeutic agents. Several synthetic ligands that target TLRs and dectin-1 are under development. Novel synthetic phosphorylated hexaacyl disaccharides (PHADs), which target TLR4, are equipotent with MPLA as agents to augment antimicrobial immunity and have strong potential to be developed into drug candidates (48). PHADs are synthesized de novo and are currently under investigation as immunopotentiating agents (48, 213). The antimicrobial functions of PHADs are linked to the increased recruitment of innate leukocytes to the sites of infection and augmentation of their antimicrobial activity.
Therapeutic Utility of Other Microbial Ligands
The class of TLR agonists that have strong potential for clinical translation extend beyond TLR4 ligands. The TLR9-selective agonist CpG oligodeoxynucleotide (CpG-ODN) is a short single-stranded synthetic bacterial DNA molecule that has been shown to confer host resistance to an array of pathogens including the parasite Leishmania major (224), the Gram-negative pathogens Francisella tularensis (225), Pseudomonas aeruginosa (226), and Burkholderia pseudomallei (227–229), Gram-positive Listeria monocytogenes (230), and viral HSV infections (231). Further, CpG-ODN also has promise as a vaccine adjuvant (232) and antitumor therapeutic (233, 234). There are several classes of CpG-ODN based on their variety of sequence and structure which elicit specific immunomodulatory profiles (232). Unlike TLR4, which signals through both MyD88- and TRIF-dependent pathways, activation of TLR9 triggers MyD88-dependent signaling alone. CpG-mediated host protection to infection seems to be dependent on downstream induction of Th1-type immune response, specifically the production of Interferon-β (224, 235). Further work is necessary to define the cellular and molecular underlying mechanisms by which CpG boosts antimicrobial responses and protects against infection.
Other microbial ligands and infections themselves can induce innate immune memory and enhance antimicrobial functions through different signaling mechanisms. β-glucans are structurally diverse polysaccharide components found mainly in fungal cell walls that are key pathogen-associated molecular patterns that trigger an immune response and are the quintessential inducers of trained immunity (236). Glucans are potent immunomodulators that augment host resistance against Gram-negative [Escherichia coli; (237, 238)], Gram-positive (Staphylococcus aureus) (239, 240), fungal [Candida albicans; (241)], and parasitic (Leishmania braziliensis) (242) infections. Glucan binds Dectin-1, which triggers downstream Raf-1/Akt-dependent signaling to augment phagocytosis, ROS production, microbial killing, and cytokine production (243–245). Further, glucan has been shown to decrease infectious complications in high risk surgical patients (246). The biological mechanisms underlying the immunomodulatory effects of glucan remain to be fully understood but glucan strongly induces metabolic reprogramming and epigenetic changes that alter gene expression and augment leukocyte function (236). Interestingly, trained immunity can also be induced by Bacillus Calmette-Guerin (BCG), which has conferred resistance to Schistosoma mansoni (247) and Candida albicans (248) infections in mice. These studies found that BCG-primed macrophages show increased phagocytosis and ROS production and improved clearance of pathogens. Epidemiological studies show that BCG, among other vaccines such as measles and oral polio vaccine, confer beneficial protective effects to unrelated pathogens in humans (249–251). Furthermore, evidence suggests that certain viral infections, such as malaria (252) and murine cytomegalovirus (253, 254), and parasitic infections [Nippostrongylus brasiliensis; (255)] induce a state of cross-protection to different pathogens through increased innate antimicrobial efficiency.
Conclusions
Here, we have reviewed the impact of sepsis on the mitochondrial function of innate leukocytes, and potential therapeutic strategies for reprogramming leukocyte metabolism to induce innate immune memory and restore host immune competency. Studies in both animal sepsis models and human septic patients reveal significant mitochondrial dysfunction in various organ systems, which correlates with sepsis severity and outcomes. In particular, sepsis-induced mitochondrial dysfunction in leukocytes is a key driver of impaired immune responses leading to increased susceptibility to secondary infections in septic patients. Studies show that early recovery of mitochondrial function in leukocytes correlates with improved septic patient outcomes.
TLR agonists are a class of microbial ligands with attractive immunomodulatory properties. Recent studies demonstrate that TLR agonists can mediate non-specific protection against infection with protective effects lasting up to 2 weeks, independent of the adaptive immune system. This induction of apparent innate immune memory is mediated by TLR agonist-induced metabolic reprogramming of leukocytes. The altered metabolic phenotype is characterized by increased glycolysis, oxidative phosphorylation, and intra-cellular concentrations of key metabolic intermediates such as itaconate and succinate, which influence cellular antimicrobial and anti-inflammatory functions. Current studies show that administration of drugs such as TLR ligands which boost leukocyte oxidative metabolism days prior to infectious challenge improve survival. Therefore, pre-treatment of critically, who are at risk for acquiring life-threatening infections, with immunomodulators that induce metabolic reprogramming and innate immunity might augment host resistance to infection and improve survival. In vitro data demonstrates that oxidative metabolism is boosted ~3 days after treatment. Though it is impossible to predict exactly which patients will face an infectious challenge when, patients at risk for hospital acquired infections could be dosed at admission or prior to an event that may lead to infection, such as abdominal surgery. A recent study by Casilag et al. shows that combination therapy with MPLA significantly augmented the efficacy of antibiotics leading to reduced bacterial burden and improved survival in a murine model of bacterial pneumonia, even when administered after induction of pneumonia (256). Therefore, treatment with immunomodulators such as TLR agonists and others may also be beneficial later in the course of sepsis to augment host innate immunity and improve outcomes.
With the increasing development of antimicrobial resistance, host-directed immunotherapies offer a promising approach to combat the risk of deadly infections in critically ill and injured patients. Immunomodulatory strategies aimed at augmenting host immunity provide a means of mediating sustained broad protection against a variety of common nosocomial pathogens. This review highlights the prospect of developing microbial ligands as novel therapeutics with the aim of augmenting leukocyte mitochondrial function and inducing innate immune memory for protection against life-threatening infections in critically ill patients.
Author Contributions
All authors contributed toward writing of the manuscript sections and conceptualization of figures. NP and ES critically revised the manuscript for important intellectual concepts. All authors have read and approved the submitted version.
Funding
This work was supported by National Institutes of Health (NIH) grants R01 GM104306, R01 GM119197 and R01 AI151210 to ES, R01 GM121711 to JB, T32 GM108554-05 and Shock Society Faculty Research Award to NP, K08 GM123345 to AH, AHA grant 19PRE34430054 to CS, and T32 GM007347-41 to MM and CS. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kahn JM, Le T, Angus DC, Cox CE, Hough CL, White DB, et al. The epidemiology of chronic critical illness in the United States*. Crit Care Med. (2015) 43:282–7. doi: 10.1097/CCM.0000000000000710
2. Filkins JP. Monokines and the metabolic pathophysiology of septic shock. Fed Proc. (1985) 44:300–4.
3. Beutler B, Cerami A. Cachectin/tumor necrosis factor: an endogenous mediator of shock and inflammation. Immunol Res. (1986) 5:281–93. doi: 10.1007/bf02935501
4. Tracey KJ, Lowry SF, Fahey TJ III, Albert JD, Fong Y, Hesse D, et al. Cachectin/tumor necrosis factor induces lethal shock and stress hormone responses in the dog. Surg Gynecol Obstet. (1987) 164:415–22.
5. Hesse DG, Tracey KJ, Fong Y, Manogue KR, Palladino MAJr, Cerami A, et al. Cytokine appearance in human endotoxemia and primate bacteremia. Surg Gynecol Obstet. (1988) 166:147–53.
6. Tracey KJ, Fong Y, Hesse DG, Manogue KR, Lee AT, Kuo GC, et al. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature. (1987) 330:662–4. doi: 10.1038/330662a0
8. Baumgartner JD, Calandra T. Treatment of sepsis: past and future avenues. Drugs. (1999) 57:127–32.
9. Opal SM, Fisher CJ Jr, Dhainaut JF, Vincent JL, Brase R, Lowry SF, et al. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: a phase III, randomized, double-blind, placebo-controlled, multicenter trial. The Interleukin-1 Receptor Antagonist Sepsis Investigator Group. Crit Care Med. (1997) 25:1115–24. doi: 10.1097/00003246-199707000-00010
10. Shakoory B, Carcillo JA, Chatham WW, Amdur RL, Zhao H, Dinarello CA, et al. Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome: reanalysis of a prior phase III trial. Crit Care Med. (2016) 44:275–81. doi: 10.1097/CCM.0000000000001402
11. Mira JC, Gentile LF, Mathias BJ, Efron PA, Brakenridge SC, Mohr AM, et al. Sepsis pathophysiology, chronic critical illness, and persistent inflammation-immunosuppression and catabolism syndrome. Crit Care Med. (2017) 45:253–62. doi: 10.1097/CCM.0000000000002074
12. Patera AC, Drewry AM, Chang K, Beiter ER, Osborne D, Hotchkiss RS. Frontline Science: defects in immune function in patients with sepsis are associated with PD-1 or PD-L1 expression and can be restored by antibodies targeting PD-1 or PD-L1. J Leukoc Biol. (2016) 100:1239–54. doi: 10.1189/jlb.4HI0616-255R
13. Patil NK, Guo Y, Luan L, Sherwood ER. Targeting immune cell checkpoints during sepsis. Int J Mol Sci. (2017) 18:2413. doi: 10.3390/ijms18112413
14. Hotchkiss RS, Sherwood ER. Immunology. Getting sepsis therapy right. Science. (2015) 347:1201–2. doi: 10.1126/science.aaa8334
15. Alkhateeb T, Kumbhare A, Bah I, Youssef D, Yao ZQ, McCall CE, et al. S100A9 maintains myeloid-derived suppressor cells in chronic sepsis by inducing miR-21 and miR-181b. Mol Immunol. (2019) 112:72–81. doi: 10.1016/j.molimm.2019.04.019
16. Zhao RZ, Jiang S, Zhang L, Yu ZB. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int J Mol Med. (2019) 44:3–15. doi: 10.3892/ijmm.2019.4188
17. McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol. (2006) 16:R551–60. doi: 10.1016/j.cub.2006.06.054
18. Galluzzi L, Kepp O, Kroemer G. Mitochondria: master regulators of danger signalling. Nat Rev Mol Cell Biol. (2012) 13:780–8. doi: 10.1038/nrm3479
19. Mills EL, Kelly B, O'Neill LAJ. Mitochondria are the powerhouses of immunity. Nat Immunol. (2017) 18:488–98. doi: 10.1038/ni.3704
20. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA. (2016) 315:801–10. doi: 10.1001/jama.2016.0287
21. Lee I, Huttemann M. Energy crisis: the role of oxidative phosphorylation in acute inflammation and sepsis. Biochim Biophys Acta. (2014) 1842:1579–86. doi: 10.1016/j.bbadis.2014.05.031
22. Singer M. The role of mitochondrial dysfunction in sepsis-induced multi-organ failure. Virulence. (2014) 5:66–72. doi: 10.4161/viru.26907
23. Brealey D, Brand M, Hargreaves I, Heales S, Land J, Smolenski R, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. (2002) 360:219–23. doi: 10.1016/S0140-6736(02)09459-X
24. Matkovich SJ, Al Khiami B, Efimov IR, Evans S, Vader J, Jain A, et al. Widespread down-regulation of cardiac mitochondrial and sarcomeric genes in patients with sepsis. Crit Care Med. (2017) 45:407–14. doi: 10.1097/CCM.0000000000002207
25. Brealey D, Karyampudi S, Jacques TS, Novelli M, Stidwill R, Taylor V, et al. Mitochondrial dysfunction in a long-term rodent model of sepsis and organ failure. Am J Physiol Regul Integr Comp Physiol. (2004) 286:R491–7. doi: 10.1152/ajpregu.00432.2003
26. Vanasco V, Magnani ND, Cimolai MC, Valdez LB, Evelson P, Boveris A, et al. Endotoxemia impairs heart mitochondrial function by decreasing electron transfer, ATP synthesis and ATP content without affecting membrane potential. J Bioenerg Biomembr. (2012) 44:243–52. doi: 10.1007/s10863-012-9426-3
27. Patil NK, Parajuli N, MacMillan-Crow LA, Mayeux PR. Inactivation of renal mitochondrial respiratory complexes and manganese superoxide dismutase during sepsis: mitochondria-targeted antioxidant mitigates injury. Am J Physiol Renal Physiol. (2014) 306:F734–43. doi: 10.1152/ajprenal.00643.2013
28. Eyenga P, Roussel D, Morel J, Rey B, Romestaing C, Gueguen-Chaignon V, et al. Time course of liver mitochondrial function and intrinsic changes in oxidative phosphorylation in a rat model of sepsis. Intensive Care Med Exp. (2018) 6:31. doi: 10.1186/s40635-018-0197-y
29. Arulkumaran N, Deutschman CS, Pinsky MR, Zuckerbraun B, Schumacker PT, Gomez H, et al. Mitochondrial function in sepsis. Shock. (2016) 45:271–81. doi: 10.1097/SHK.0000000000000463
30. Vachharajani V, Liu T, McCall CE. Epigenetic coordination of acute systemic inflammation: potential therapeutic targets. Expert Rev Clin Immunol. (2014) 10:1141–50. doi: 10.1586/1744666X.2014.943192
31. Cheng SC, Scicluna BP, Arts RJ, Gresnigt MS, Lachmandas E, Giamarellos-Bourboulis EJ, et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat Immunol. (2016) 17:406–13. doi: 10.1038/ni.3398
32. Adrie C, Bachelet M, Vayssier-Taussat M, Russo-Marie F, Bouchaert I, Adib-Conquy M, et al. Mitochondrial membrane potential and apoptosis peripheral blood monocytes in severe human sepsis. Am J Respir Crit Care Med. (2001) 164:389–95. doi: 10.1164/ajrccm.164.3.2009088
33. Belikova I, Lukaszewicz AC, Faivre V, Damoisel C, Singer M, Payen D. Oxygen consumption of human peripheral blood mononuclear cells in severe human sepsis. Crit Care Med. (2007) 35:2702–8. doi: 10.1097/01.ccm.0000295593.25106.c4
34. Japiassu AM, Santiago AP, d'Avila JC, Garcia-Souza LF, Galina A, Castro Faria-Neto HC, et al. Bioenergetic failure of human peripheral blood monocytes in patients with septic shock is mediated by reduced F1Fo adenosine-5'-triphosphate synthase activity. Crit Care Med. (2011) 39:1056–63. doi: 10.1097/CCM.0b013e31820eda5c
35. Garrabou G, Moren C, Lopez S, Tobias E, Cardellach F, Miro O, et al. The effects of sepsis on mitochondria. J Infect Dis. (2012) 205:392–400. doi: 10.1093/infdis/jir764
36. Sjovall F, Morota S, Persson J, Hansson MJ, Elmer E. Patients with sepsis exhibit increased mitochondrial respiratory capacity in peripheral blood immune cells. Crit Care. (2013) 17:R152. doi: 10.1186/cc12831
37. Weiss SL, Selak MA, Tuluc F, Perales Villarroel J, Nadkarni VM, Deutschman CS, et al. Mitochondrial dysfunction in peripheral blood mononuclear cells in pediatric septic shock. Pediatr Crit Care Med. (2015) 16:e4–12. doi: 10.1097/PCC.0000000000000277
38. Merz TM, Pereira AJ, Schurch R, Schefold JC, Jakob SM, Takala J, et al. Mitochondrial function of immune cells in septic shock: a prospective observational cohort study. PLoS ONE. (2017) 12:e0178946. doi: 10.1371/journal.pone.0178946
39. Jang DH, Orloski CJ, Owiredu S, Shofer FS, Greenwood JC, Eckmann DM. Alterations in mitochondrial function in blood cells obtained from patients with sepsis presenting to an emergency department. Shock. (2019) 51:580–4. doi: 10.1097/SHK.0000000000001208
40. Kraft BD, Chen L, Suliman HB, Piantadosi CA, Welty-Wolf KE. Peripheral blood mononuclear cells demonstrate mitochondrial damage clearance during sepsis. Crit Care Med. (2019) 47:651–8. doi: 10.1097/CCM.0000000000003681
41. Weiss SL, Zhang D, Bush J, Graham K, Starr J, Murray J, et al. Mitochondrial dysfunction is associated with an immune paralysis phenotype in pediatric sepsis. Shock. (2019). doi: 10.1097/SHK.0000000000001486. [Epub ahead of print].
42. Weiss SL, Zhang D, Bush J, Graham K, Starr J, Tuluc F, et al. Persistent mitochondrial dysfunction linked to prolonged organ dysfunction in pediatric sepsis. Crit Care Med. (2019) 47:1433–41. doi: 10.1097/CCM.0000000000003931
43. Clere-Jehl R, Helms J, Kassem M, Le Borgne P, Delabranche X, Charles AL, et al. Septic shock alters mitochondrial respiration of lymphoid cell-lines and human peripheral blood mononuclear cells: the role of plasma. Shock. (2019) 51:97–104. doi: 10.1097/SHK.0000000000001125
44. Palsson-McDermott EM, O'Neill LA. The Warburg effect then and now: from cancer to inflammatory diseases. Bioessays. (2013) 35:965–73. doi: 10.1002/bies.201300084
45. Carre JE, Orban JC, Re L, Felsmann K, Iffert W, Bauer M, et al. Survival in critical illness is associated with early activation of mitochondrial biogenesis. Am J Respir Crit Care Med. (2010) 182:745–51. doi: 10.1164/rccm.201003-0326OC
46. Fensterheim BA, Guo Y, Sherwood ER, Bohannon JK. The cytokine response to lipopolysaccharide does not predict the host response to infection. J Immunol. (2017) 198:3264–73. doi: 10.4049/jimmunol.1602106
47. Fensterheim BA, Young JD, Luan L, Kleinbard RR, Stothers CL, Patil NK, et al. The TLR4 agonist monophosphoryl lipid a drives broad resistance to infection via dynamic reprogramming of macrophage metabolism. J Immunol. (2018) 200:3777–89. doi: 10.4049/jimmunol.1800085
48. Hernandez A, Luan L, Stothers CL, Patil NK, Fults JB, Fensterheim BA, et al. Phosphorylated hexa-acyl disaccharides augment host resistance against common nosocomial pathogens. Crit Care Med. (2019) 47:e930–8. doi: 10.1097/CCM.0000000000003967
49. Ioannou A, Dalle Lucca J, Tsokos GC. Immunopathogenesis of ischemia/reperfusion-associated tissue damage. Clin Immunol. (2011) 141:3–14. doi: 10.1016/j.clim.2011.07.001
50. Leung CH, Caldarone CA, Wang F, Venkateswaran S, Ailenberg M, Vadasz B, et al. Remote ischemic conditioning prevents lung and liver injury after hemorrhagic shock/resuscitation: potential role of a humoral plasma factor. Ann Surg. (2014) 261:1215–25. doi: 10.1097/SLA.0000000000000877
51. Chow CC, Clermont G, Kumar R, Lagoa C, Tawadrous Z, Gallo D, et al. The acute inflammatory response in diverse shock states. Shock. (2005) 24:74–84. doi: 10.1097/01.shk.0000168526.97716.f3
52. McGhan LJ, Jaroszewski DE. The role of toll-like receptor-4 in the development of multi-organ failure following traumatic haemorrhagic shock and resuscitation. Injury. (2012) 43:129–36. doi: 10.1016/j.injury.2011.05.032
53. Yao X, Carlson D, Sun Y, Ma L, Wolf SE, Minei JP, et al. Mitochondrial ROS induces cardiac inflammation via a pathway through mtDNA damage in a pneumonia-related sepsis model. PLoS ONE. (2015) 10:e0139416. doi: 10.1371/journal.pone.0139416
54. Chen R, Zhu S, Zeng L, Wang Q, Sheng Y, Zhou B, et al. AGER-mediated lipid peroxidation drives caspase-11 inflammasome activation in sepsis. Front Immunol. (2019) 10:1904. doi: 10.3389/fimmu.2019.01904
55. Hazeldine J, Dinsdale RJ, Harrison P, Lord JM. Traumatic injury and exposure to mitochondrial-derived damage associated molecular patterns suppresses neutrophil extracellular trap formation. Front Immunol. (2019) 10:685. doi: 10.3389/fimmu.2019.00685
56. Kartchner LB, Gode CJ, Dunn JLM, Glenn LI, Duncan DN, Wolfgang MC, et al. One-hit wonder: Late after burn injury, granulocytes can clear one bacterial infection but cannot control a subsequent infection. Burns. (2019) 45:627–40. doi: 10.1016/j.burns.2018.08.019
57. Sakuma M, Khan MAS, Yasuhara S, Martyn JA, Palaniyar N. Mechanism of pulmonary immunosuppression: extrapulmonary burn injury suppresses bacterial endotoxin-induced pulmonary neutrophil recruitment and neutrophil extracellular trap (NET) formation. FASEB J. (2019) 33:13602–16. doi: 10.1096/fj.201901098R
58. Patil NK, Luan L, Bohannon JK, Hernandez A, Guo Y, Sherwood ER. Frontline Science: anti-PD-L1 protects against infection with common bacterial pathogens after burn injury. J Leukoc Biol. (2018) 103:23–33. doi: 10.1002/JLB.5HI0917-360R
59. Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. (2008) 88:611–38. doi: 10.1152/physrev.00025.2007
60. Dominy JE, Puigserver P. Mitochondrial biogenesis through activation of nuclear signaling proteins. Cold Spring Harb Perspect Biol. (2013) 5:a015008. doi: 10.1101/cshperspect.a015008
61. Scarpulla RC. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta. (2011) 1813:1269–78. doi: 10.1016/j.bbamcr.2010.09.019
62. Gureev AP, Shaforostova EA, Popov VN. Regulation of mitochondrial biogenesis as a way for active longevity: interaction between the Nrf2 and PGC-1alpha signaling pathways. Front Genet. (2019) 10:435. doi: 10.3389/fgene.2019.00435
63. Hock MB, Kralli A. Transcriptional control of mitochondrial biogenesis and function. Annu Rev Physiol. (2009) 71:177–203. doi: 10.1146/annurev.physiol.010908.163119
64. Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. (2009) 458:1056–60. doi: 10.1038/nature07813
65. Inata Y, Piraino G, Hake PW, O'Connor M, Lahni P, Wolfe V, et al. Age-dependent cardiac function during experimental sepsis: effect of pharmacological activation of AMP-activated protein kinase by AICAR. Am J Physiol Heart Circ Physiol. (2018) 315:H826–37. doi: 10.1152/ajpheart.00052.2018
66. Hall DT, Griss T, Ma JF, Sanchez BJ, Sadek J, Tremblay AMK, et al. The AMPK agonist 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), but not metformin, prevents inflammation-associated cachectic muscle wasting. EMBO Mol Med. (2018) 10:e8307. doi: 10.15252/emmm.201708307
67. Escobar DA, Botero-Quintero AM, Kautza BC, Luciano J, Loughran P, Darwiche S, et al. Adenosine monophosphate-activated protein kinase activation protects against sepsis-induced organ injury and inflammation. J Surg Res. (2015) 194:262–72. doi: 10.1016/j.jss.2014.10.009
68. Wang Y, An H, Liu T, Qin C, Sesaki H, Guo S, et al. Metformin improves mitochondrial respiratory activity through activation of AMPK. Cell Rep. (2019) 29:1511–23 e1515. doi: 10.1016/j.celrep.2019.09.070
69. Detaille D, Guigas B, Chauvin C, Batandier C, Fontaine E, Wiernsperger N, et al. Metformin prevents high-glucose-induced endothelial cell death through a mitochondrial permeability transition-dependent process. Diabetes. (2005) 54:2179–87. doi: 10.2337/diabetes.54.7.2179
70. Meng S, Cao J, He Q, Xiong L, Chang E, Radovick S, et al. Metformin activates AMP-activated protein kinase by promoting formation of the alphabetagamma heterotrimeric complex. J Biol Chem. (2015) 290:3793–802. doi: 10.1074/jbc.M114.604421
71. Suwa M, Egashira T, Nakano H, Sasaki H, Kumagai S. Metformin increases the PGC-1alpha protein and oxidative enzyme activities possibly via AMPK phosphorylation in skeletal muscle in vivo. J Appl Physiol. (2006) 101:1685–92. doi: 10.1152/japplphysiol.00255.2006
72. Tzanavari T, Varela A, Theocharis S, Ninou E, Kapelouzou A, Cokkinos DV, et al. Metformin protects against infection-induced myocardial dysfunction. Metabolism. (2016) 65:1447–58. doi: 10.1016/j.metabol.2016.06.012
73. Vaez H, Najafi M, Rameshrad M, Toutounchi NS, Garjani M, Barar J, et al. AMPK activation by metformin inhibits local innate immune responses in the isolated rat heart by suppression of TLR 4-related pathway. Int Immunopharmacol. (2016) 40:501–7. doi: 10.1016/j.intimp.2016.10.002
74. Vaez H, Najafi M, Toutounchi NS, Barar J, Barzegari A, Garjani A. Metformin Alleviates lipopolysaccharide-induced acute lung injury through suppressing toll-like receptor 4 signaling. Iran J Allergy Asthma Immunol. (2016) 15:498–507.
75. Vaez H, Rameshrad M, Najafi M, Barar J, Barzegari A, Garjani A. Cardioprotective effect of metformin in lipopolysaccharide-induced sepsis via suppression of toll-like receptor 4 (TLR4) in heart. Eur J Pharmacol. (2016) 772:115–23. doi: 10.1016/j.ejphar.2015.12.030
76. Tang G, Yang H, Chen J, Shi M, Ge L, Ge X, et al. Metformin ameliorates sepsis-induced brain injury by inhibiting apoptosis, oxidative stress and neuroinflammation via the PI3K/Akt signaling pathway. Oncotarget. (2017) 8:97977–89. doi: 10.18632/oncotarget.20105
77. Liang H, Ding X, Li L, Wang T, Kan Q, Wang L, et al. Association of preadmission metformin use and mortality in patients with sepsis and diabetes mellitus: a systematic review and meta-analysis of cohort studies. Crit Care. (2019) 23:50. doi: 10.1186/s13054-019-2346-4
78. Freire-Garabal M, Nunez MJ, Balboa J, Lopez-Delgado P, Gallego R, Garcia-Caballero T, et al. Serotonin upregulates the activity of phagocytosis through 5-HT1A receptors. Br J Pharmacol. (2003) 139:457–63. doi: 10.1038/sj.bjp.0705188
79. Mikulski Z, Zaslona Z, Cakarova L, Hartmann P, Wilhelm J, Tecott LH, et al. Serotonin activates murine alveolar macrophages through 5-HT2C receptors. Am J Physiol Lung Cell Mol Physiol. (2010) 299:L272–80. doi: 10.1152/ajplung.00032.2010
80. Drosatos K, Khan RS, Trent CM, Jiang H, Son NH, Blaner WS, et al. Peroxisome proliferator-activated receptor-γ activation prevents sepsis-related cardiac dysfunction and mortality in mice. Circ Heart Fail. (2013) 6:550–62. doi: 10.1161/CIRCHEARTFAILURE.112.000177
81. Tsujimura Y, Matsutani T, Matsuda A, Kutsukake M, Uchida E, Sasajima K, et al. Effects of pioglitazone on survival and omental adipocyte function in mice with sepsis induced by cecal ligation and puncture. J Surg Res. (2011) 171:e215–221. doi: 10.1016/j.jss.2011.08.012
82. Majer O, Bourgeois C, Zwolanek F, Lassnig C, Kerjaschki D, Mack M, et al. Type I interferons promote fatal immunopathology by regulating inflammatory monocytes and neutrophils during Candida infections. PLoS Pathog. (2012) 8:e1002811. doi: 10.1371/journal.ppat.1002811
83. Zingarelli B, Sheehan M, Hake PW, O'Connor M, Denenberg A, Cook JA. Peroxisome proliferator activator receptor-gamma ligands, 15-deoxy-Delta(12,14)-prostaglandin J2 and ciglitazone, reduce systemic inflammation in polymicrobial sepsis by modulation of signal transduction pathways. J Immunol. (2003) 171:6827–37. doi: 10.4049/jimmunol.171.12.6827
84. Maggi LB Jr, Sadeghi H, Weigand C, Scarim AL, Heitmeier MR, Corbett JA. Anti-inflammatory actions of 15-deoxy-delta 12,14-prostaglandin J2 and troglitazone: evidence for heat shock-dependent and -independent inhibition of cytokine-induced inducible nitric oxide synthase expression. Diabetes. (2000) 49:346–55. doi: 10.2337/diabetes.49.3.346
85. Guyton K, Bond R, Reilly C, Gilkeson G, Halushka P, Cook J. Differential effects of 15-deoxy-delta(12,14)-prostaglandin J2 and a peroxisome proliferator-activated receptor gamma agonist on macrophage activation. J Leukoc Biol. (2001) 69:631–8. doi: 10.1189/jlb.69.4.631
86. Guyton K, Zingarelli B, Ashton S, Teti G, Tempel G, Reilly C, et al. Peroxisome proliferator-activated receptor-gamma agonists modulate macrophage activation by gram-negative and gram-positive bacterial stimuli. Shock. (2003) 20:56–62. doi: 10.1097/01.shk.0000070903.21762.f8
87. Tancevski I, Nairz M, Duwensee K, Auer K, Schroll A, Heim C, et al. Fibrates ameliorate the course of bacterial sepsis by promoting neutrophil recruitment via CXCR2. EMBO Mol Med. (2014) 6:810–20. doi: 10.1002/emmm.201303415
88. Cree MG, Zwetsloot JJ, Herndon DN, Qian T, Morio B, Fram R, et al. Insulin sensitivity and mitochondrial function are improved in children with burn injury during a randomized controlled trial of fenofibrate. Ann Surg. (2007) 245:214–21. doi: 10.1097/01.sla.0000250409.51289.ca
89. Crisafulli C, Cuzzocrea S. The role of endogenous and exogenous ligands for the peroxisome proliferator-activated receptor alpha (PPAR-alpha) in the regulation of inflammation in macrophages. Shock. (2009) 32:62–73. doi: 10.1097/shk.0b013e31818bbad6
90. Barton P, Garcia J, Kouatli A, Kitchen L, Zorka A, Lindsay C, et al. Hemodynamic effects of i.v. milrinone lactate in pediatric patients with septic shock. A prospective, double-blinded, randomized, placebo-controlled, interventional study. Chest. (1996) 109:1302–12. doi: 10.1378/chest.109.5.1302
91. Carcillo JA, Herzer WA, Mi Z, Thomas NJ, Jackson EK. Treatment with the type IV phosphodiesterase inhibitor Ro 20-1724 protects renal and mesenteric blood flow in endotoxemic rats treated with norepinephrine. J Pharmacol Exp Ther. (1996) 279:1197–204.
92. Thomas NJ, Carcillo JA, Herzer WA, Mi Z, Tofovic SP, Jackson EK. Type IV phosphodiesterase inhibition improves cardiac contractility in endotoxemic rats. Eur J Pharmacol. (2003) 465:133–9. doi: 10.1016/s0014-2999(03)01456-0
93. Holthoff JH, Wang Z, Patil NK, Gokden N, Mayeux PR. Rolipram improves renal perfusion and function during sepsis in the mouse. J Pharmacol Exp Ther. (2013) 347:357–64. doi: 10.1124/jpet.113.208520
94. Sims CR, Singh SP, Mu S, Gokden N, Zakaria D, Nguyen TC, et al. Rolipram improves outcome in a rat model of infant sepsis-induced cardiorenal syndrome. Front Pharmacol. (2017) 8:237. doi: 10.3389/fphar.2017.00237
95. Sanz MJ, Cortijo J, Taha MA, Cerda-Nicolas M, Schatton E, Burgbacher B, et al. Roflumilast inhibits leukocyte-endothelial cell interactions, expression of adhesion molecules and microvascular permeability. Br J Pharmacol. (2007) 152:481–92. doi: 10.1038/sj.bjp.0707428
96. Schick MA, Wunder C, Wollborn J, Roewer N, Waschke J, Germer CT, et al. Phosphodiesterase-4 inhibition as a therapeutic approach to treat capillary leakage in systemic inflammation. J Physiol. (2012) 590:2693–708. doi: 10.1113/jphysiol.2012.232116
97. Wollborn J, Wunder C, Stix J, Neuhaus W, Bruno RR, Baar W, et al. Phosphodiesterase-4 inhibition with rolipram attenuates hepatocellular injury in hyperinflammation in vivo and in vitro without influencing inflammation and HO-1 expression. J Pharmacol Pharmacother. (2015) 6:13–23. doi: 10.4103/0976-500X.149138
98. Zuo L, Li Q, Sun B, Xu Z, Ge Z. Cilostazol promotes mitochondrial biogenesis in human umbilical vein endothelial cells through activating the expression of PGC-1alpha. Biochem Biophys Res Commun. (2013) 433:52–7. doi: 10.1016/j.bbrc.2013.02.068
99. Ding H, Bai F, Cao H, Xu J, Fang L, Wu J, et al. PDE/cAMP/Epac/C/EBP-beta signaling cascade regulates mitochondria biogenesis of tubular epithelial cells in renal fibrosis. Antioxid Redox Signal. (2018) 29:637–52. doi: 10.1089/ars.2017.7041
100. Biala A, Tauriainen E, Siltanen A, Shi J, Merasto S, Louhelainen M, et al. Resveratrol induces mitochondrial biogenesis and ameliorates Ang II-induced cardiac remodeling in transgenic rats harboring human renin and angiotensinogen genes. Blood Press. (2010) 19:196–205. doi: 10.3109/08037051.2010.481808
101. Wang N, Mao L, Yang L, Zou J, Liu K, Liu M, et al. Resveratrol protects against early polymicrobial sepsis-induced acute kidney injury through inhibiting endoplasmic reticulum stress-activated NF-κB pathway. Oncotarget. (2017) 8:36449–61. doi: 10.18632/oncotarget.16860
102. Luo CJ, Luo F, Bu QD, Jiang W, Zhang W, Liu XM, et al. Protective effects of resveratrol on acute kidney injury in rats with sepsis. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. (2019) 164:49–56. doi: 10.5507/bp.2019.006
103. Wang Y, Feng F, Liu M, Xue J, Huang H. Resveratrol ameliorates sepsis-induced acute kidney injury in a pediatric rat model via Nrf2 signaling pathway. Exp Ther Med. (2018) 16:3233–40. doi: 10.3892/etm.2018.6533
104. Shang X, Lin K, Yu R, Zhu P, Zhang Y, Wang L, et al. Resveratrol protects the myocardium in sepsis by activating the phosphatidylinositol 3-kinases (PI3K)/AKT/Mammalian target of rapamycin (mTOR) pathway and inhibiting the nuclear factor-κB (NF-κB) signaling pathway. Med Sci Monit. (2019) 25:9290–8. doi: 10.12659/MSM.918369
105. Martin LM, Johnson PJ, Amorim JR, DeClue AE. Effects of orally administered resveratrol on TNF, IL-1β, leukocyte phagocytic activity and oxidative burst function in horses: a prospective, randomized, double-blinded, placebo-controlled study. Int J Mol Sci. (2020) 21:1453. doi: 10.3390/ijms21041453
106. Valenti D, De Rasmo D, Signorile A, Rossi L, de Bari L, Scala I, et al. Epigallocatechin-3-gallate prevents oxidative phosphorylation deficit and promotes mitochondrial biogenesis in human cells from subjects with Down's syndrome. Biochim Biophys Acta. (2013) 1832:542–52. doi: 10.1016/j.bbadis.2012.12.011
107. Chiou YS, Huang Q, Ho CT, Wang YJ, Pan MH. Directly interact with Keap1 and LPS is involved in the anti-inflammatory mechanisms of (-)-epicatechin-3-gallate in LPS-induced macrophages and endotoxemia. Free Radic Biol Med. (2016) 94:1–16. doi: 10.1016/j.freeradbiomed.2016.02.010
108. Wang J, Fan SM, Zhang J. Epigallocatechin-3-gallate ameliorates lipopolysaccharide-induced acute lung injury by suppression of TLR4/NF-κB signaling activation. Braz J Med Biol Res. (2019) 52:e8092. doi: 10.1590/1414-431X20198092
109. Wheeler DS, Lahni PM, Hake PW, Denenberg AG, Wong HR, Snead C, et al. The green tea polyphenol epigallocatechin-3-gallate improves systemic hemodynamics and survival in rodent models of polymicrobial sepsis. Shock. (2007) 28:353–9. doi: 10.1097/shk.0b013e3180485823
110. Cederroth CR, Vinciguerra M, Gjinovci A, Kühne F, Klein M, Cederroth M, et al. Dietary phytoestrogens activate AMP-activated protein kinase with improvement in lipid and glucose metabolism. Diabetes. (2008) 57:1176–85. doi: 10.2337/db07-0630
111. Parida S, Singh TU, Thangamalai R, Narasimha Reddy CE, Panigrahi M, Kandasamy K, et al. Daidzein pretreatment improves survival in mouse model of sepsis. J Surg Res. (2015) 197:363–73. doi: 10.1016/j.jss.2015.03.059
112. Yi L, Zhou Z, Zheng Y, Chang M, Huang X, Guo F, et al. Suppressive effects of GSS on lipopolysaccharide-induced endothelial cell injury and ALI via TNF-α and IL-6. Mediators Inflamm. (2019) 2019:4251394. doi: 10.1155/2019/4251394
113. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. (2018) 19:121–35. doi: 10.1038/nrm.2017.95
114. Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci USA. (2007) 104:12017–22. doi: 10.1073/pnas.0705070104
115. Garcia-Roves PM, Osler ME, Holmstrom MH, Zierath JR. Gain-of-function R225Q mutation in AMP-activated protein kinase gamma3 subunit increases mitochondrial biogenesis in glycolytic skeletal muscle. J Biol Chem. (2008) 283:35724–34. doi: 10.1074/jbc.M805078200
116. Ruderman NB, Xu XJ, Nelson L, Cacicedo JM, Saha AK, Lan F, et al. AMPK and SIRT1: a long-standing partnership? Am J Physiol Endocrinol Metab. (2010) 298:E751–60. doi: 10.1152/ajpendo.00745.2009
117. Ismail Hassan F, Didari T, Khan F, Niaz K, Mojtahedzadeh M, Abdollahi M. A review on the protective effects of metformin in sepsis-induced organ failure. Cell J. (2020) 21:363–70. doi: 10.22074/cellj.2020.6286
118. Liu G, Wu K, Zhang L, Dai J, Huang W, Lin L, et al. Metformin attenuated endotoxin-induced acute myocarditis via activating AMPK. Int Immunopharmacol. (2017) 47:166–72. doi: 10.1016/j.intimp.2017.04.002
119. Cameron RB, Beeson CC, Schnellmann RG. Development of therapeutics that induce mitochondrial biogenesis for the treatment of acute and chronic degenerative diseases. J Med Chem. (2016) 59:10411–34. doi: 10.1021/acs.jmedchem.6b00669
120. Wu H, Denna TH, Storkersen JN, Gerriets VA. Beyond a neurotransmitter: the role of serotonin in inflammation and immunity. Pharmacol Res. (2019) 140:100–14. doi: 10.1016/j.phrs.2018.06.015
121. Ahern GP. 5-HT and the immune system. Curr Opin Pharmacol. (2011) 11:29–33. doi: 10.1016/j.coph.2011.02.004
122. Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. (2002) 53:409–35. doi: 10.1146/annurev.med.53.082901.104018
123. Han L, Shen WJ, Bittner S, Kraemer FB, Azhar S. PPARs: regulators of metabolism and as therapeutic targets in cardiovascular disease. Part I: PPAR-alpha. Future Cardiol. (2017) 13:259–78. doi: 10.2217/fca-2016-0059
124. Liang H, Ward WF. PGC-1alpha: a key regulator of energy metabolism. Adv Physiol Educ. (2006) 30:145–51. doi: 10.1152/advan.00052.2006
125. Lebovitz HE. Thiazolidinediones: the forgotten diabetes medications. Curr Diab Rep. (2019) 19:151. doi: 10.1007/s11892-019-1270-y
126. Ruiz PA, Kim SC, Sartor RB, Haller D. 15-deoxy-delta12,14-prostaglandin J2-mediated ERK signaling inhibits gram-negative bacteria-induced RelA phosphorylation and interleukin-6 gene expression in intestinal epithelial cells through modulation of protein phosphatase 2A activity. J Biol Chem. (2004) 279:36103–11. doi: 10.1074/jbc.M405032200
127. Standage SW, Caldwell CC, Zingarelli B, Wong HR. Reduced peroxisome proliferator-activated receptor alpha expression is associated with decreased survival and increased tissue bacterial load in sepsis. Shock. (2012) 37:164–9. doi: 10.1097/SHK.0b013e31823f1a00
128. Fernandez-Marcos PJ, Auwerx J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr. (2011) 93:884S–90. doi: 10.3945/ajcn.110.001917
129. Irazuzta J, Sullivan KJ, Garcia PC, Piva JP. Pharmacologic support of infants and children in septic shock. J Pediatr. (2007) 83:S36–45. doi: 10.2223/JPED.1623
130. Meyer S, Gortner L, Brown K, Abdul-Khaliq H. The role of milrinone in children with cardiovascular compromise: review of the literature. Wien Med Wochenschr. (2011) 161:184–91. doi: 10.1007/s10354-011-0869-7
131. de Oliveira CF, de Oliveira DS, Gottschald AF, Moura JD, Costa GA, Ventura AC, et al. ACCM/PALS haemodynamic support guidelines for paediatric septic shock: an outcomes comparison with and without monitoring central venous oxygen saturation. Intensive Care Med. (2008) 34:1065–75. doi: 10.1007/s00134-008-1085-9
132. Brierley J, Carcillo JA, Choong K, Cornell T, Decaen A, Deymann A, et al. Clinical practice parameters for hemodynamic support of pediatric and neonatal septic shock: 2007 update from the American College of Critical Care Medicine. Crit Care Med. (2009) 37:666–88. doi: 10.1097/CCM.0b013e31819323c6
133. Maurice DH, Ke H, Ahmad F, Wang Y, Chung J, Manganiello VC. Advances in targeting cyclic nucleotide phosphodiesterases. Nat Rev Drug Discov. (2014) 13:290–314. doi: 10.1038/nrd4228
134. MacKenzie SJ, Houslay MD. Action of rolipram on specific PDE4 cAMP phosphodiesterase isoforms and on the phosphorylation of cAMP-response-element-binding protein (CREB) and p38 mitogen-activated protein (MAP) kinase in U937 monocytic cells. Biochem J. (2000) 347(Pt 2):571–8. doi: 10.1042/0264-6021:3470571
135. Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. (2006) 127:1109–22. doi: 10.1016/j.cell.2006.11.013
136. Van Wyngene L, Vandewalle J, Libert C. Reprogramming of basic metabolic pathways in microbial sepsis: therapeutic targets at last? EMBO Mol Med. (2018) 10:e8712. doi: 10.15252/emmm.201708712
137. Patil NK, Bohannon JK, Hernandez A, Patil TK, Sherwood ER. Regulation of leukocyte function by citric acid cycle intermediates. J Leukoc Biol. (2019) 106:105–17. doi: 10.1002/JLB.3MIR1118-415R
139. Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. (2012) 15:813–26. doi: 10.1016/j.cmet.2012.04.023
140. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. (2013) 496:238–42. doi: 10.1038/nature11986
142. Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. (2005) 7:77–85. doi: 10.1016/j.ccr.2004.11.022
143. Seim GL, Britt EC, John SV, Yeo FJ, Johnson AR, Eisenstein RS, et al. Two-stage metabolic remodelling in macrophages in response to lipopolysaccharide and interferon-gamma stimulation. Nat Metab. (2019) 1:731–42. doi: 10.1038/s42255-019-0083-2
144. Vijayan V, Pradhan P, Braud L, Fuchs HR, Gueler F, Motterlini R, et al. Human and murine macrophages exhibit differential metabolic responses to lipopolysaccharide - A divergent role for glycolysis. Redox Biol. (2019) 22:101147. doi: 10.1016/j.redox.2019.101147
145. West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. (2011) 472:476–80. doi: 10.1038/nature09973
146. Wang F, Zhang S, Vuckovic I, Jeon R, Lerman A, Folmes CD, et al. Glycolytic stimulation is not a requirement for M2 macrophage differentiation. Cell Metab. (2018) 28:463–75.e464. doi: 10.1016/j.cmet.2018.08.012
147. Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. (2018) 556:113–7. doi: 10.1038/nature25986
148. Liao ST, Han C, Xu DQ, Fu XW, Wang JS, Kong LY. 4-Octyl itaconate inhibits aerobic glycolysis by targeting GAPDH to exert anti-inflammatory effects. Nat Commun. (2019) 10:5091. doi: 10.1038/s41467-019-13078-5
149. Qin W, Qin K, Zhang Y, Jia W, Chen Y, Cheng B, et al. S-glycosylation-based cysteine profiling reveals regulation of glycolysis by itaconate. Nat Chem Biol. (2019) 15:983–91. doi: 10.1038/s41589-019-0323-5
150. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. (2016) 167:457–70 e413. doi: 10.1016/j.cell.2016.08.064
151. Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. (2015) 42:419–30. doi: 10.1016/j.immuni.2015.02.005
152. Zhu X, Meyers A, Long D, Ingram B, Liu T, Yoza BK, et al. Frontline Science: monocytes sequentially rewire metabolism and bioenergetics during an acute inflammatory response. J Leukoc Biol. (2019) 105:215–28. doi: 10.1002/JLB.3HI0918-373R
153. Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci USA. (2013) 110:7820–5. doi: 10.1073/pnas.1218599110
154. Cordes T, Wallace M, Michelucci A, Divakaruni AS, Sapcariu SC, Sousa C, et al. Immunoresponsive gene 1 and itaconate inhibit succinate dehydrogenase to modulate intracellular succinate levels. J Biol Chem. (2016) 291:14274–84. doi: 10.1074/jbc.M115.685792
155. Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. (2016) 24:158–66. doi: 10.1016/j.cmet.2016.06.004
156. Sugimoto M, Sakagami H, Yokote Y, Onuma H, Kaneko M, Mori M, et al. Non-targeted metabolite profiling in activated macrophage secretion. Metabolomics. (2012) 8:624–33. doi: 10.1007/s11306-011-0353-9
157. Cordes T, Michelucci A, Hiller K. Itaconic acid: the surprising role of an industrial compound as a mammalian antimicrobial metabolite. Annu Rev Nutr. (2015) 35:451–73. doi: 10.1146/annurev-nutr-071714-034243
158. McFadden BA, Purohit S. Itaconate, an isocitrate lyase-directed inhibitor in Pseudomonas indigofera. J Bacteriol. (1977) 131:136–44.
159. Dolan SK, Welch M. The glyoxylate shunt, 60 years on. Annu Rev Microbiol. (2018) 72:309–30. doi: 10.1146/annurev-micro-090817-062257
160. Williams NC, O'Neill LAJ. A role for the krebs cycle intermediate citrate in metabolic reprogramming in innate immunity and inflammation. Front Immunol. (2018) 9:141. doi: 10.3389/fimmu.2018.00141
161. Sasikaran J, Ziemski M, Zadora PK, Fleig A, Berg IA. Bacterial itaconate degradation promotes pathogenicity. Nat Chem Biol. (2014) 10:371–7. doi: 10.1038/nchembio.1482
162. Naujoks J, Tabeling C, Dill BD, Hoffmann C, Brown AS, Kunze M, et al. IFNs modify the proteome of legionella-containing vacuoles and restrict infection via IRG1-derived itaconic acid. PLoS Pathog. (2016) 12:e1005408. doi: 10.1371/journal.ppat.1005408
163. Mills E, O'Neill LA. Succinate: a metabolic signal in inflammation. Trends Cell Biol. (2014) 24:313–20. doi: 10.1016/j.tcb.2013.11.008
164. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MAR, Sheedy FJ, Gleeson LE, et al. Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. (2015) 21:347. doi: 10.1016/j.cmet.2015.01.017
165. Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. (2014) 515:431–5. doi: 10.1038/nature13909
166. De Souza DP, Achuthan A, Lee MK, Binger KJ, Lee MC, Davidson S, et al. Autocrine IFN-I inhibits isocitrate dehydrogenase in the TCA cycle of LPS-stimulated macrophages. J Clin Invest. (2019) 129:4239–44. doi: 10.1172/jci127597
167. Palmieri F. The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Arch. (2004) 447:689–709. doi: 10.1007/s00424-003-1099-7
168. Infantino V, Convertini P, Cucci L, Panaro MA, Di Noia MA, Calvello R, et al. The mitochondrial citrate carrier: a new player in inflammation. Biochem J. (2011) 438:433–6. doi: 10.1042/BJ20111275
169. Fukuzumi M, Shinomiya H, Shimizu Y, Ohishi K, Utsumi S. Endotoxin-induced enhancement of glucose influx into murine peritoneal macrophages via GLUT1. Infect Immun. (1996) 64:108–12.
170. Schenz J, Tamulyte S, Nusshag C, Brenner T, Poschet G, Weigand MA, et al. Population-specific metabolic alterations in professional antigen-presenting cells contribute to sepsis-associated immunosuppression. Shock. (2020) 53:5–15. doi: 10.1097/shk.0000000000001337
171. Shalova IN, Lim JY, Chittezhath M, Zinkernagel AS, Beasley F, Hernandez-Jimenez E, et al. Human monocytes undergo functional re-programming during sepsis mediated by hypoxia-inducible factor-1alpha. Immunity. (2015) 42:484–98. doi: 10.1016/j.immuni.2015.02.001
172. Pan L, Hu L, Zhang L, Xu H, Chen Y, Bian Q, et al. Deoxyelephantopin decreases the release of inflammatory cytokines in macrophage associated with attenuation of aerobic glycolysis via modulation of PKM2. Int Immunopharmacol. (2020) 79:106048. doi: 10.1016/j.intimp.2019.106048
173. Chao CC, Ingram BO, Lurchachaiwong W, Ching WM. Metabolic characterization of serum from mice challenged with Orientia tsutsugamushi-infected mites. New Microbes New Infect. (2018) 23:70–6. doi: 10.1016/j.nmni.2018.01.005
174. Meiser J, Kraemer L, Jaeger C, Madry H, Link A, Lepper PM, et al. Itaconic acid indicates cellular but not systemic immune system activation. Oncotarget. (2018) 9:32098–107. doi: 10.18632/oncotarget.25956
175. Beloborodova N, Pautova A, Sergeev A, Fedotcheva N. Serum levels of mitochondrial and microbial metabolites reflect mitochondrial dysfunction in different stages of sepsis. Metabolites. (2019) 9:196. doi: 10.3390/metabo9100196
176. Netea MG. Training innate immunity: the changing concept of immunological memory in innate host defence. Eur J Clin Invest. (2013) 43:881–4. doi: 10.1111/eci.12132
177. Kurtz J. Specific memory within innate immune systems. Trends Immunol. (2005) 26:186–92. doi: 10.1016/j.it.2005.02.001
178. Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, et al. Trained immunity: a program of innate immune memory in health and disease. Science. (2016) 352:aaf1098. doi: 10.1126/science.aaf1098
179. Karlsson H, Nassberger L. In vitro metabolic inhibition of the human lymphocyte: influence on the expression of interleukin-2 receptors. Immunol Cell Biol. (1992) 70 (Pt 5):309–13. doi: 10.1038/icb.1992.39
180. Sanchez-Alcazar JA, Hernandez I, De la Torre MP, Garcia I, Santiago E, Munoz-Yague MT, et al. Down-regulation of tumor necrosis factor receptors by blockade of mitochondrial respiration. J Biol Chem. (1995) 270:23944–50. doi: 10.1074/jbc.270.41.23944
181. Buttgereit F, Burmester GR, Brand MD. Bioenergetics of immune functions: fundamental and therapeutic aspects. Immunol Today. (2000) 21:192–9. doi: 10.1016/s0167-5699(00)01593-0
182. O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. (2016) 16:553–65. doi: 10.1038/nri.2016.70
183. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. (2014) 345:1250684. doi: 10.1126/science.1250684
184. Arts RJ, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. (2016) 24:807–19. doi: 10.1016/j.cmet.2016.10.008
185. Dominguez-Andres J, Netea MG. Long-term reprogramming of the innate immune system. J Leukoc Biol. (2019) 105:329–38. doi: 10.1002/JLB.MR0318-104R
186. Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. (2014) 345:1251086. doi: 10.1126/science.1251086
187. Stothers CL, Luan L, Fensterheim BA, Bohannon JK. Hypoxia-inducible factor-1alpha regulation of myeloid cells. J Mol Med. (2018) 96:1293–306. doi: 10.1007/s00109-018-1710-1
188. Bohannon JK, Luan L, Hernandez A, Afzal A, Guo Y, Patil NK, et al. Role of G-CSF in monophosphoryl lipid A-mediated augmentation of neutrophil functions after burn injury. J Leukoc Biol. (2016) 99:629–40. doi: 10.1189/jlb.4A0815-362R
189. Hernandez A, Bohannon JK, Luan L, Fensterheim BA, Guo Y, Patil NK, et al. The role of MyD88- and TRIF-dependent signaling in monophosphoryl lipid A-induced expansion and recruitment of innate immunocytes. J Leukoc Biol. (2016) 100:1311–22. doi: 10.1189/jlb.1A0216-072R
190. Di Gioia M, Spreafico R, Springstead JR, Mendelson MM, Joehanes R, Levy D, et al. Endogenous oxidized phospholipids reprogram cellular metabolism and boost hyperinflammation. Nat Immunol. (2020) 21:42–53. doi: 10.1038/s41590-019-0539-2
191. Chu LH, Indramohan M, Ratsimandresy RA, Gangopadhyay A, Morris EP, Monack DM, et al. The oxidized phospholipid oxPAPC protects from septic shock by targeting the non-canonical inflammasome in macrophages. Nat Commun. (2018) 9:996. doi: 10.1038/s41467-018-03409-3
192. Landy M, Pillemer L. Increased resistance to infection and accompanying alteration in properidin levels following administration of bacterial lipopolysaccharides. J Exp Med. (1956) 104:383–409. doi: 10.1084/jem.104.3.383
193. Rayhane N, Fitting C, Lortholary O, Dromer F, Cavaillon JM. Administration of endotoxin associated with lipopolysaccharide tolerance protects mice against fungal infection. Infect Immun. (2000) 68:3748–53. doi: 10.1128/iai.68.6.3748-3753.2000
194. Murphey ED, Fang G, Sherwood ER. Endotoxin pretreatment improves bacterial clearance and decreases mortality in mice challenged with Staphylococcus aureus. Shock. (2008) 29:512–8. doi: 10.1097/shk.0b013e318150776f
195. Lehner MD, Ittner J, Bundschuh DS, van Rooijen N, Wendel A, Hartung T. Improved innate immunity of endotoxin-tolerant mice increases resistance to Salmonella enterica serovar typhimurium infection despite attenuated cytokine response. Infect Immun. (2001) 69:463–71. doi: 10.1128/IAI.69.1.463-471.2001
196. Varma TK, Durham M, Murphey ED, Cui W, Huang Z, Lin CY, et al. Endotoxin priming improves clearance of Pseudomonas aeruginosa in wild-type and interleukin-10 knockout mice. Infect Immun. (2005) 73:7340–7. doi: 10.1128/IAI.73.11.7340-7347.2005
197. Murphey ED, Fang G, Varma TK, Sherwood ER. Improved bacterial clearance and decreased mortality can be induced by LPS tolerance and is not dependent upon IFN-gamma. Shock. (2007) 27:289–95. doi: 10.1097/01.shk.0000245024.93740.28
198. Wheeler DS, Lahni PM, Denenberg AG, Poynter SE, Wong HR, Cook JA, et al. Induction of endotoxin tolerance enhances bacterial clearance and survival in murine polymicrobial sepsis. Shock. (2008) 30:267–73. doi: 10.1097/shk.0b013e318162c190
199. Deng M, Scott MJ, Loughran P, Gibson G, Sodhi C, Watkins S, et al. Lipopolysaccharide clearance, bacterial clearance, and systemic inflammatory responses are regulated by cell type-specific functions of TLR4 during sepsis. J Immunol. (2013) 190:5152–60. doi: 10.4049/jimmunol.1300496
200. Shahin RD, Engberg I, Hagberg L, Svanborg Eden C. Neutrophil recruitment and bacterial clearance correlated with LPS responsiveness in local gram-negative infection. J Immunol. (1987) 138:3475–80
201. Wolk K, Docke WD, von Baehr V, Volk HD, Sabat R. Impaired antigen presentation by human monocytes during endotoxin tolerance. Blood. (2000) 96:218–23. doi: 10.1182/blood.V96.1.218
202. Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. (2009) 30:475–87. doi: 10.1016/j.it.2009.07.009
203. Ward NS, Casserly B, Ayala A. The compensatory anti-inflammatory response syndrome (CARS) in critically ill patients. Clin Chest Med. (2008) 29:617–25, viii. doi: 10.1016/j.ccm.2008.06.010
204. Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. (2013) 13:862–74. doi: 10.1038/nri3552
205. Pena OM, Hancock DG, Lyle NH, Linder A, Russell JA, Xia J, et al. An endotoxin tolerance signature predicts sepsis and organ dysfunction at initial clinical presentation. EBiomedicine. (2014) 1:64–71. doi: 10.1016/j.ebiom.2014.10.003
206. Davenport EE, Burnham KL, Radhakrishnan J, Humburg P, Hutton P, Mills TC, et al. Genomic landscape of the individual host response and outcomes in sepsis: a prospective cohort study. Lancet Respir Med. (2016) 4:259–71. doi: 10.1016/S2213-2600(16)00046-1
207. Fisher CJ Jr, Dhainaut JF, Opal SM, Pribble JP, Balk RA, Slotman GJ, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. JAMA. (1994) 271:1836–43.
208. Fisher CJJr, Agosti JM, Opal SM, Lowry SF, Balk RA, Sadoff JC, et al. Treatment of septic shock with the tumor necrosis factor receptor:Fc fusion protein. The Soluble TNF Receptor Sepsis Study Group. N Engl J Med. (1996) 334:1697–702. doi: 10.1056/NEJM199606273342603
209. Reinhart K, Karzai W. Anti-tumor necrosis factor therapy in sepsis: update on clinical trials and lessons learned. Crit Care Med. (2001) 29:S121–5. doi: 10.1097/00003246-200107001-00037
210. Deans KJ, Haley M, Natanson C, Eichacker PQ, Minneci PC. Novel therapies for sepsis: a review. J Trauma. (2005) 58:867–74. doi: 10.1097/01.ta.0000158244.69179.94
211. Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. (2009) 458:1191–5. doi: 10.1038/nature07830
212. Bohannon JK, Hernandez A, Enkhbaatar P, Adams WL, Sherwood ER. The immunobiology of toll-like receptor 4 agonists: from endotoxin tolerance to immunoadjuvants. Shock. (2013) 40:451–62. doi: 10.1097/SHK.0000000000000042
213. Hernandez A, Patil NK, Stothers CL, Luan L, McBride MA, Owen AM, et al. Immunobiology and application of toll-like receptor 4 agonists to augment host resistance to infection. Pharmacol Res. (2019) 150:104502. doi: 10.1016/j.phrs.2019.104502
214. Chase JJ, Kubey W, Dulek MH, Holmes CJ, Salit MG, Pearson FC III, et al. Effect of monophosphoryl lipid A on host resistance to bacterial infection. Infect Immun. (1986) 53:711–2.
215. Hirano T, Kodama S, Kawano T, Maeda K, Suzuki M. Monophosphoryl lipid A induced innate immune responses via TLR4 to enhance clearance of nontypeable Haemophilus influenzae and Moraxella catarrhalis from the nasopharynx in mice. FEMS Immunol Med Microbiol. (2011) 63:407–17. doi: 10.1111/j.1574-695X.2011.00866.x
216. Romero CD, Varma TK, Hobbs JB, Reyes A, Driver B, Sherwood ER. The Toll-like receptor 4 agonist monophosphoryl lipid a augments innate host resistance to systemic bacterial infection. Infect Immun. (2011) 79:3576–87. doi: 10.1128/IAI.00022-11
217. Roquilly A, Broquet A, Jacqueline C, Gautreau L, Segain JP, de Coppet P, et al. Toll-like receptor-4 agonist in post-haemorrhage pneumonia: role of dendritic and natural killer cells. Eur Respir J. (2013) 42:1365–78. doi: 10.1183/09031936.00152612
218. Tamassia N, Le Moigne V, Calzetti F, Donini M, Gasperini S, Ear T, et al. The MyD88-independent pathway is not mobilized in human neutrophils stimulated via TLR4. J Immunol. (2007) 178:7344–56. doi: 10.4049/jimmunol.178.11.7344
219. Ribes S, Meister T, Ott M, Redlich S, Janova H, Hanisch UK, et al. Intraperitoneal prophylaxis with CpG oligodeoxynucleotides protects neutropenic mice against intracerebral Escherichia coli K1 infection. J Neuroinflammation. (2014) 11:14. doi: 10.1186/1742-2094-11-14
220. Hampton T. Report reveals scope of US antibiotic resistance threat. JAMA. (2013) 310:1661–3. doi: 10.1001/jama.2013.280695
221. Hampton T. Novel programs and discoveries aim to combat antibiotic resistance. JAMA. (2015) 313:2411–3. doi: 10.1001/jama.2015.4738
222. Marston HD, Dixon DM, Knisely JM, Palmore TN, Fauci AS. Antimicrobial resistance. JAMA. (2016) 316:1193–204. doi: 10.1001/jama.2016.11764
223. Jacobs A. Crisis Looms in Antibiotics as Drug Makers Go Bankrupt. (2019) Available online at: https://www.nytimes.com/2019/12/25/health/antibiotics-new-resistance.html (accessed May 5, 2020).
224. Zimmermann S, Egeter O, Hausmann S, Lipford GB, Rocken M, Wagner H, et al. CpG oligodeoxynucleotides trigger protective and curative Th1 responses in lethal murine leishmaniasis. J Immunol. (1998) 160:3627–30.
225. Elkins KL, Rhinehart-Jones TR, Stibitz S, Conover JS, Klinman DM. Bacterial DNA containing CpG motifs stimulates lymphocyte-dependent protection of mice against lethal infection with intracellular bacteria. J Immunol. (1999) 162:2291–8.
226. Jiang M, Yao J, Feng G. Protective effect of DNA vaccine encoding pseudomonas exotoxin A and PcrV against acute pulmonary P. aeruginosa infection. PLoS ONE. (2014) 9:e96609. doi: 10.1371/journal.pone.0096609
227. Wongratanacheewin S, Kespichayawattana W, Intachote P, Pichyangkul S, Sermswan RW, Krieg AM, et al. Immunostimulatory CpG oligodeoxynucleotide confers protection in a murine model of infection with Burkholderia pseudomallei. Infect Immun. (2004) 72:4494–502. doi: 10.1128/IAI.72.8.4494-4502.2004
228. Rozak DA, Gelhaus HC, Smith M, Zadeh M, Huzella L, Waag D, et al. CpG oligodeoxyribonucleotides protect mice from Burkholderia pseudomallei but not Francisella tularensis Schu S4 aerosols. J Immune Based Ther Vaccines. (2010) 8:2. doi: 10.1186/1476-8518-8-2
229. Judy BM, Taylor K, Deeraksa A, Johnston RK, Endsley JJ, Vijayakumar S, et al. Prophylactic application of CpG oligonucleotides augments the early host response and confers protection in acute melioidosis. PLoS ONE. (2012) 7:e34176. doi: 10.1371/journal.pone.0034176
230. Krieg AM, Love-Homan L, Yi AK, Harty JT. CpG DNA induces sustained IL-12 expression in vivo and resistance to Listeria monocytogenes challenge. J Immunol. (1998) 161:2428–34.
231. Harandi AM, Eriksson K, Holmgren J. A protective role of locally administered immunostimulatory CpG oligodeoxynucleotide in a mouse model of genital herpes infection. J Virol. (2003) 77:953–62. doi: 10.1128/jvi.77.2.953-962.2003
232. Vollmer J, Krieg AM. Immunotherapeutic applications of CpG oligodeoxynucleotide TLR9 agonists. Adv Drug Deliv Rev. (2009) 61:195–204. doi: 10.1016/j.addr.2008.12.008
233. Krieg AM. Antitumor applications of stimulating toll-like receptor 9 with CpG oligodeoxynucleotides. Curr Oncol Rep. (2004) 6:88–95. doi: 10.1007/s11912-004-0019-0
234. Wang XS, Sheng Z, Ruan YB, Guang Y, Yang ML. CpG oligodeoxynucleotides inhibit tumor growth and reverse the immunosuppression caused by the therapy with 5-fluorouracil in murine hepatoma. World J Gastroenterol. (2005) 11:1220–4. doi: 10.3748/wjg.v11.i8.1220
235. Herbst MM, Pyles RB. Immunostimulatory CpG treatment for genital HSV-2 infections. J Antimicrob Chemother. (2003) 52:887–9. doi: 10.1093/jac/dkg481
236. Camilli G, Tabouret G, Quintin J. The complexity of fungal beta-glucan in health and disease: effects on the mononuclear phagocyte system. Front Immunol. (2018) 9:673. doi: 10.3389/fimmu.2018.00673
237. Williams DL, Browder W, McNamee R, Di Luzio NR. Glucan immunomodulation in experimental E. coli sepsis. Adv Exp Med Biol. (1982) 155:701–6. doi: 10.1007/978-1-4684-4394-3_77
238. Williams DL, Sherwood ER, Browder IW, McNamee RB, Jones EL, Di Luzio NR. Pre-clinical safety evaluation of soluble glucan. Int J Immunopharmacol. (1988) 10:405–14. doi: 10.1016/0192-0561(88)90127-0
239. Di Luzio NR, Williams DL. Protective effect of glucan against systemic Staphylococcus aureus septicemia in normal and leukemic mice. Infect Immun. (1978) 20:804–10.
240. Marakalala MJ, Williams DL, Hoving JC, Engstad R, Netea MG, Brown GD. Dectin-1 plays a redundant role in the immunomodulatory activities of beta-glucan-rich ligands in vivo. Microbes Infect. (2013) 15:511–5. doi: 10.1016/j.micinf.2013.03.002
241. Lagrange PH, Fourgeaud M, Neway T, Pilet C. Mycobacterial polar glycopeptidolipids enhance resistance to experimental murine candidiasis. C R Acad Sci III. (1995) 318:359–65.
242. Dos Santos JC, Barroso de Figueiredo AM, Teodoro Silva MV, Cirovic B, de Bree LCJ, Damen M, et al. Beta-glucan-induced trained immunity protects against Leishmania braziliensis infection: a crucial role for IL-32. Cell Rep. (2019) 28:2659–72 e2656. doi: 10.1016/j.celrep.2019.08.004
243. Di Luzio NR, Williams DL, Sherwood ER, Browder IW. Modification of diverse experimental immunosuppressive states by glucan. Surv Immunol Res. (1985) 4:160–7. doi: 10.1007/bf02918811
244. Williams DL, Sherwood ER, Browder IW, McNamee RB, Jones EL, Rakinic J, et al. Effect of glucan on neutrophil dynamics and immune function in Escherichia coli peritonitis. J Surg Res. (1988) 44:54–61. doi: 10.1016/0022-4804(88)90122-9
245. Sherwood ER, Varma TK, Fram RY, Lin CY, Koutrouvelis AP, Toliver-Kinsky TE. Glucan phosphate potentiates endotoxin-induced interferon-gamma expression in immunocompetent mice, but attenuates induction of endotoxin tolerance. Clin Sci. (2001) 101:541–50.
246. Babineau TJ, Marcello P, Swails W, Kenler A, Bistrian B, Forse RA. Randomized phase I/II trial of a macrophage-specific immunomodulator (PGG-glucan) in high-risk surgical patients. Ann Surg. (1994) 220:601–9. doi: 10.1097/00000658-199411000-00002
247. Tribouley J, Tribouley-Duret J, Appriou M. [Effect of Bacillus Callmette Guerin (BCG) on the receptivity of nude mice to Schistosoma mansoni]. C R Seances Soc Biol Fil. (1978) 172:902–4.
248. van 't Wout JW, Poell R, van Furth R. The role of BCG/PPD-activated macrophages in resistance against systemic candidiasis in mice. Scand J Immunol. (1992) 36:713–9. doi: 10.1111/j.1365-3083.1992.tb03132.x
249. Kleinnijenhuis J, Quintin J, Preijers F, Joosten LA, Ifrim DC, Saeed S, et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci USA. (2012) 109:17537–42. doi: 10.1073/pnas.1202870109
250. Benn CS, Netea MG, Selin LK, Aaby P. A small jab - a big effect: nonspecific immunomodulation by vaccines. Trends Immunol. (2013) 34:431–9. doi: 10.1016/j.it.2013.04.004
251. Jensen KJ, Larsen N, Biering-Sorensen S, Andersen A, Eriksen HB, Monteiro I, et al. Heterologous immunological effects of early BCG vaccination in low-birth-weight infants in Guinea-Bissau: a randomized-controlled trial. J Infect Dis. (2015) 211:956–67. doi: 10.1093/infdis/jiu508
252. Ataide MA, Andrade WA, Zamboni DS, Wang D, Souza Mdo C, Franklin BS, et al. Malaria-induced NLRP12/NLRP3-dependent caspase-1 activation mediates inflammation and hypersensitivity to bacterial superinfection. PLoS Pathog. (2014) 10:e1003885. doi: 10.1371/journal.ppat.1003885
253. Nabekura T, Girard JP, Lanier LL. IL-33 receptor ST2 amplifies the expansion of NK cells and enhances host defense during mouse cytomegalovirus infection. J Immunol. (2015) 194:5948–52. doi: 10.4049/jimmunol.1500424
254. Schlums H, Cichocki F, Tesi B, Theorell J, Beziat V, Holmes TD, et al. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity. (2015) 42:443–56. doi: 10.1016/j.immuni.2015.02.008
255. Chen F, Wu W, Millman A, Craft JF, Chen E, Patel N, et al. Neutrophils prime a long-lived effector macrophage phenotype that mediates accelerated helminth expulsion. Nat Immunol. (2014) 15:938–46. doi: 10.1038/ni.2984
Keywords: sepsis, infection, trauma, trained immunity, mitochondria, metabolic reprogramming
Citation: McBride MA, Owen AM, Stothers CL, Hernandez A, Luan L, Burelbach KR, Patil TK, Bohannon JK, Sherwood ER and Patil NK (2020) The Metabolic Basis of Immune Dysfunction Following Sepsis and Trauma. Front. Immunol. 11:1043. doi: 10.3389/fimmu.2020.01043
Received: 17 March 2020; Accepted: 30 April 2020;
Published: 29 May 2020.
Edited by:
Thomas Griffith, University of Minnesota Twin Cities, United StatesReviewed by:
Charles C. Caldwell, University of Cincinnati, United StatesBruce Walcheck, University of Minnesota Twin Cities, United States
Copyright © 2020 McBride, Owen, Stothers, Hernandez, Luan, Burelbach, Patil, Bohannon, Sherwood and Patil. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Naeem K. Patil, bmFlZW0ucGF0aWxAdnVtYy5vcmc=