 Shubhranshu S. Gupta
Shubhranshu S. Gupta Jin Wang3,4
Jin Wang3,4 Min Chen
Min Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 22 May 2020
Sec. Immunological Memory
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01013
CD8+ T cells represent one of the most versatile immune cells critical for clearing away viral infections. Due to their important role, CD8+ T cell activation and memory formation during viral infection have been the focus of several studies recently. Although CD8+ T cell activation and memory formation have been associated with metabolic alterations, the molecular understanding behind T cells choosing one type of metabolism over others based on their differentiation stage is still unclear. This review focuses on how the signaling molecules and cellular processes that are characteristic of CD8+ T cell activation and memory formation also play a critical role in selecting specific type of metabolism during viral infections. In addition, we will summarize the epigenetic factors regulating these metabolic alterations.
CD8+ T cells constitute one of the critical mediators of adaptive immune response. T cells are further classified into naïve, effector and memory T cells based on their state of differentiation. While naïve CD8+ T cells represent the population that has not been activated by cognate antigen and are yet to receive the activation signal, effector and memory CD8+ T cells are antigen experienced. In the context of viral infection, naïve CD8+ T cells are activated when antigen-presenting cells (APC) present cognate antigens (derived from viral peptides) in the context of MHC I along with CD4+ T cell help (1). After activation, these virus-specific CD8+ T cells eventually clear out the infection and enter a ‘contraction phase’ wherein majority of them undergo apoptosis (2). However, a small percentage of these activated CD8+ T cells survive and form memory CD8+ T cells. Activation of naïve CD8+ T cells results in a myriad of activation signals, which not only expand the pool of antigen-specific cells, but also reprogram them metabolically. Metabolic reprogramming is critical to ensure that the activated CD8+ T cells not only fulfill the increasing energy demand during clonal proliferation, but also generate the metabolic intermediates they require to synthesize cellular components for the daughter cells.
Among different metabolic pathways, aerobic glycolysis, glutaminolysis and oxidative phosphorylation are the most studied types in CD8+ T cells; as determined using several cellular metabolism assays including extracellular flux analysis, stable isotope labeling, liquid chromatography-mass spectrometry (LC-MS) and gene expression studies (3). Aerobic glycolysis, a cytosolic pathway, is characterized by conversion of glucose into pyruvate with net production of 2 ATP molecules per glucose molecule (4). Oxidative phosphorylation, a mitochondrial pathway, is characterized by transfer of electrons from electron donors (such as NADH and FADH2) to electron transport chain (ETC) that ultimately results in the generation of 2.5 ATP per NADH and 1.5 ATP per FADH2 molecule (5). Bulk of NADH and FADH2 entering ETC reaction in T cells are generated either during tricarboxylic acid (TCA) cycle or from fatty acid oxidation (4). Finally, glutaminolysis involves breakdown of glutamine into glutamate, followed by conversion of glutamate into anaplerotic substrate α-ketoglutarate (α-KG) that fuels cellular proliferation (6–8). While naïve CD8+ T cells depend predominantly on basal glycolysis and oxidative phosphorylation fueled by fatty acid oxidation to generate ATP for their survival (9–12), activated T cells upregulate aerobic glycolysis (10, 12–14) and glutaminolysis (15). However, when the activated effector CD8+ T cells differentiate into memory CD8+ T cells, metabolism is switched back to fatty acid oxidation fueled by long-chain and short/branched-chain fatty acids, as recently shown by us and others (16–18).
Progress in T cell immunology research in the past decade has led to significant advancement in our understanding about various molecular factors driving T cell activation and memory formation. However, it is still unclear as to how those molecular factors also determine metabolic fate in virus-specific CD8+ T cells. This review focuses on bridging that knowledge gap in CD8+ T cells during the course of anti-viral immune response.
Long term survival of naïve CD8+ T cells depends on continuous contact with self-peptide/MHC complex and IL-7 signaling (19). The expression of IL-7 receptor (IL-7R) is modulated by availability of IL-7 in T cell milieu. Depletion of IL-7 or loss-of-function mutation in IL-7R has been shown to severely restrict the survival of naïve T cells (20, 21), suggesting that signaling via IL-7R is critical for their long-term survival. Even though it is not clear if there is any cross-talk between signaling through self-peptide/MHC complex and IL-7 receptor (IL-7R), it has been proposed that signaling through self-peptide/MHC complex leads to the formation of lipid rafts which are then integrated into the cell membrane of naïve T cells; followed by recruitment of IL-7R to the cell surface by lipid rafts (19). In addition, expression of IL-7R in naïve CD8+ T cells can also be induced by FOXO1, a transcription factor recently identified to be responsible for quiescence in naïve T cells (22). Naïve CD8+ T cells, possessing a metabolically resting phenotype, rely primarily on fatty acid oxidation and basal glycolysis (9–13, 20) to meet their bioenergetic needs. Deletion of IL-7R leads to a reduction in glycolytic flux in naïve T cells (19, 20). This reduction in glycolysis is independent of any alteration in glucose uptake, since the expression of GLUT1 transporting glucose into T cells remained unchanged. Furthermore, IL-7R-mediated signaling also induces fatty acid oxidation-dependent oxidative phosphorylation in naive CD8+ T cells (23). Although the exact mechanism that underlies IL-7R-mediated modulation of basal glycolysis and oxidative phosphorylation still remains unclear, it's possible that upon activation of IL-7R, the resulting phosphorylation of STAT5 (pSTAT5) (20) and subsequent translocation of pSTAT5 to the nucleus causes it to activate transcription of glycolytic and fatty acid metabolism genes (24, 25). The dependence on both glycolysis and fatty acid oxidation could also be interconnected. The product of glycolysis, pyruvate, is converted into acetyl-CoA by pyruvate dehydrogenase (PDH), which can be then used to synthesize fatty acids. Fatty acids can further serve as electron donors in mitochondria during fatty acid oxidation (ß-oxidation) to generate ATP. Because naïve CD8+ T cells are resting in nature, their ATP needs are meant to largely sustain basal cellular processes.
IL-7 signaling-mediated activation of STAT1, STAT3, and STAT5 can further activate DNA and histone methyltransferases that methylate DNA and histone residues, respectively, in order to regulate gene expression (26–28). DNA methyltransferases (DNMTs) can methylate promoter regions associated with target genes and silence them; while histone methyltransferases (HMTs) can trimethylate lysine 4 on histone 3 (H3K4me3) or lysine 27 on histone 3 (H3K27me3) around target genes, resulting in gene activation or gene repression, respectively (29). So far, the metabolic targets of DNMTs and HMTs are largely unknown in naïve CD8+ T cells. Although DNMT such as DNMT1 has been reported to perform DNA methylation at Ifng locus in naive CD8+ T cells (30), whether it also methylates genes associated with naive CD8+ T cell metabolism is hitherto unknown. As far as HMTs are concerned, whether they differentially methylate H3K4 and H3K27 to maintain low levels of GLUT1 expression typical of naïve CD8+ T cells should also be examined. In addition, role of protein arginine N-methyltransferase 1 (PRMT1), an HMT which activates FOXO1 (31), could be tested in naive CD8+ T cells.
Taken together, although naive CD8+ T cells use IL-7 signaling to mediate basal glycolysis and oxidative phosphorylation, the genetic and epigenetic causes for their metabolic quiescence need to be further elucidated.
Viral infection is typically followed by activation of innate immune system, where viral antigens are systemically taken up by antigen-presenting cells (APCs). Presentation of viral peptides in the context of MHC-I results in activation of naïve CD8+ T cells; although viral peptides could also be loaded onto MHC-II molecules in an autophagy- or endosome-dependent manner to activate naïve CD4+ T cells (32–35). Activation of naïve CD8+ T cells involves engagement of TCR by APCs across the immunological synapse. TCR engagement results in phosphorylation of immunoreceptor tyrosine-based activation motif (ITAM) by LCK, that further propagates TCR signaling and results in T cell stimulation. In addition to T cell stimulation, co-stimulation via CD28 eventually results in activation of AKT. Activated AKT induces nuclear translocation of NF-κB and expression of anti-apoptotic BCL-XL, resulting in T cell proliferation and production of IL-2 (36). AKT in conjunction with TCR signal activates mTOR (23, 24); which promotes T cell activation, proliferation and production of effector cytokines such as IFN-γ and TNF-α (37). mTOR activation further leads to expression of MYC and HIF1α (15, 38), which control expression of cell cycle progression genes (cyclin A, CDK2/4, cdc25a) (11) as well as activation markers and cytokines (including IFN- γ, TNF-α, TIM3, OX40, CD137, and Granzyme B) (39, 40). MYC and HIF1α can alternatively be induced by NFAT as well (41). NFAT is activated by calcium-dependent activation of calcineurin resulting from TCR signaling. Upon activation, NFAT translocates to the nucleus and induces the expression of T cell activation markers and cytokines such as CD40L, IL-2, and TNF-α; and promotes T cell proliferation via expression of cell cycle genes such as CDK4/6 and cyclin D1/D3 (42).
Metabolism in activated CD8+ T cells is characterized by elevated level of glycolytic flux (Figure 1). The induction of glycolysis after TCR stimulation begins as soon as 15 min after TCR engagement (10). However, additional co-stimulatory signal via CD28 is required to keep glycolysis upregulated for longer duration (13, 14). This upregulation in glycolysis results from an increased expression of GLUT1 on the cell membrane due to AKT activation triggered by TCR and CD28 co-signaling (12, 14, 43). Increased expression of GLUT1 promotes glucose uptake into activated CD8+ T cells, which triggers glycolytic flux to produce pyruvate and synthesize ATP needed to meet increasing energy demands. Increased production of pyruvate during glycolysis is followed by its conversion into acetyl CoA, which enters TCA cycle to produce anaplerotic substrate α-KG required for the production of various cell components for new daughter cells (11, 44, 45).
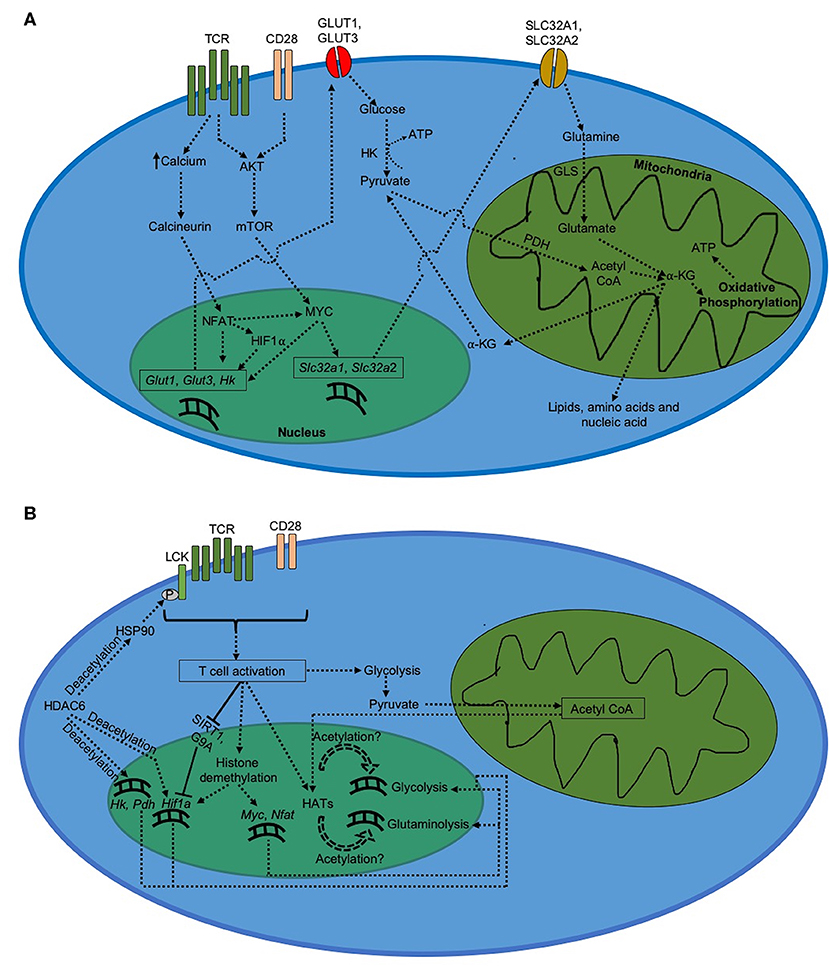
Figure 1. Metabolism in CD8+ T cells after activation. (A) Regulation of metabolism during CD8+ T cell activation. Activated CD8+ T cells demonstrate a drastic increase in glycolysis and glutaminolysis, which serve to generate ATP as well as biogenic precursors for daughter cells. (B) Epigenetic regulation of metabolism during CD8+ T cell activation.
MYC induced by AKT-dependent mTOR signaling can induce the upregulation of glycolysis in CD8+ T cells (15, 46, 47). MYC-dependent glycolytic regulation has been shown to be mediated through expression of miR-17~92 (48), a polycistronic microRNA cluster that increases the sensitivity of virus-specific CD8+ T cells to TCR stimulation (49). MYC also enhances glutaminolysis upon CD8+ T cell activation (11, 15). In addition, MYC increases the transcription of glutamine transporters, such as SLC32A1 and SLC32A2, required for the uptake of glutamine into activated T cells (50). This is followed by increased glutaminolysis, where glutamine is catabolized into glutamate by glutaminase (GLS). Glutamate is then converted into α-KG via the TCA cycle, which eventually results in biosynthesis of lipids, nucleotides and proteins necessary for T cell proliferation (45, 51). Via TCA cycle, α-KG further generates NADH and FADH2 that undergo oxidative phosphorylation to produce ATP in mitochondria. In addition, pyruvate formed during glycolysis can enter mitochondria and generate acetyl CoA with help of pyruvate dehydrogenase (PDH) (45).
During CD8+ T cell activation, glycolysis is also activated by NFAT via increased expression of GLUT1 and GLUT3 (41). In addition, NFAT upregulates expression of hexokinase (HK), which is a rate-determining factor in glycolysis in eukaryotes (41, 52). Increase in NFAT activation further increases expression of MYC and HIF1α in CD8+ T cells (53). Increased HIF1α-mediated glycolysis in activated CD8+ T cells (10) further suggests that NFAT plays a critical role in the expression of glycolytic activators during CD8+ T cell activation (Figure 1A).
Although the critical roles of glucose and glutamine uptake have been elucidated in regulating glycolysis and glutaminolysis, respectively, it is still unknown whether these metabolic pathways are interdependent in activated CD8+ T cells during acute viral infections. In fact, in activated alloreactive T cells, glutamine has been shown to undergo gluconeogenesis (54), a pathway characterized by the synthesis of glucose from non-sugar precursors. Hence, it is possible that glucose, which virus-specific activated CD8+ T cells acquire via GLUT1- and GLUT3-mediated uptake, could also be generated from glutamine via gluconeogenesis. This gap in knowledge, however, remains to be addressed.
The switch in metabolism from a quiescent state in naïve CD8+ T cells to an activated state after activation is accompanied by significant increase in cellular respiration, which results from a rapid upregulation of glycolysis and glutaminolysis. Changes in epigenetic regulation of metabolic genes could be responsible for this outcome. Increased expression of genes depends upon how accessible they are to cellular transcription factors. Eukaryotic cells typically make genes more accessible by loosening the chromatin-histone interaction around the target genes, which occurs by modifications at the histone sites. Gcn5, a histone acetyltransferase (HAT) induced by TCR signaling, has been shown to be recruited by NFAT to Il-2 promoter and facilitate T cell activation via H3K9 acetylation (55). However, in order to do so, HATs require acetyl groups whose availability depends on acetyl CoA levels (56). Acetyl CoA is formed from pyruvate which is an end product of glycolysis. An initial upregulation in glycolysis due to TCR signaling feeds more acetyl CoA to HATs, resulting in glycolytic and glutaminolytic genes becoming more accessible to transcription factors like MYC, NFAT, and HIF1α. This could further increase glycolysis and glutaminolysis after CD8+ T cell activation. In addition, several studies have shown critical roles played by different subtypes of histone deacetylases (HDAC) in regulating CD8+ T cell activation and metabolism. HDAC6, a cytoplasmic isoform of HDAC (57), has been shown to deacetylate heat shock protein 90 (HSP90) resulting in LCK phosphorylation upon TCR engagement and activation of CD8+ T cells (58). Inhibition of HDAC6 reduces expression of critical glycolytic genes such as HK, PDH, and HIF1α, thereby impairing glycolytic flux during activation of lymphocytes (59). Furthermore, HDAC5 has also been shown to mediate optimal IFN-γ production during CD8+ T cell activation (60). On the other hand, HDAC2 represses optimal IL-2 production during CD8+ T cell activation via histone H3 deacetylation; and hence CD8+ T cells downregulate HDAC2 activity at Il2ra locus early during the activation in order to sustain optimal IL-2 production during anti-viral primary response (61). Likewise, SIRT1, a class III HDAC whose expression is inhibited by IL-2 signaling, also negatively regulates CD8+ T cell activation via deacetylation of Hif1a; leading to downregulation of SIRT1 expression by activated T cells to sustain optimal glycolysis and IFN-γ production (62–66).
Differential methylation of histone proteins associated with target genes has also been reported to play critical role in regulating gene expression during CD8+ T cell activation (Figure 1B). For instance, histone demethylation remodels chromatin into an open conformation. After viral infection, Myc, Nfat and Hif1a genes get increasingly demethylated upon activation of naïve CD8+ T cells (67). DNA methyltransferases (DNMTs) can also silence gene expression via methylation of cytosine residues of CpG dinucleotides in the promoter region. During viral infection, CD8+ T cells upregulate DNMT3a expression which ultimately leads to silencing of Tcf7 gene in virus-specific activated CD8+ T cells (68). Absence of TCF7 in activated CD8+ T cells has been reported to downregulate Hif1a expression (69), suggesting that preventing DNMT3a-dependent Tcf7 silencing during T cell activation could potentially improve HIF1α-mediated glycolysis in virus-specific CD8+ T cells. This hypothesis, however, needs to be tested under experimental setting. In addition, histone methyltransferases (HMTs) can methylate histones associated with genes involved in T cell activation. For example, differential methylation at lysine 9 site on histone H3 (H3K9 methylation) by G9a leads to repressive chromatin conformation around Hif1a gene (70, 71). Because of its inhibitory role in regulating T cell activation, virus-specific CD8+ T cells restrict G9a activity at Il2ra locus early during activation (61, 70, 71). Furthermore, Enhancer of Zeste Homolog 2 (EZH2), which represses gene expression via H3K27 trimethylation, is induced upon T cell activation and is critical for repression of FOXO1, an inhibitor of glycolysis (28, 72), in virus-specific activated CD8+ T cells (73).
Despite the availability of plethora of information about epigenetic regulators of metabolism in virus-specific activated CD8+ T cells, several questions remain unanswered. For instance, although it is known that Myc, Nfat, and Hif1a genes are increasingly demethylated during CD8+ T cell activation (67), the epigenetic mechanism underlying that demethylation process is still unclear. Either DNA demethylases or histone demethylases (or both) may play a role here, but the specifics around it are still lacking. Moreover, whether preventing DNMT3a-dependent Tcf7 silencing during CD8+ T cell activation could potentially improve HIF1α-mediated glycolysis also needs to be experimentally demonstrated. In addition, the epigenetic regulation underlying MYC-driven glutaminolysis in virus-specific activated CD8+ T cells also remains to be addressed.
During CD8+ T cell primary response, viral infection is eventually cleared off. This is followed by most of the virus-specific CD8+ T cells undergoing programmed cell death, a phase termed as contraction phase (2). IL-15 signaling plays a critical role in controlling the survival of virus-specific activated effector CD8+ T cells through the contraction phase (74, 75) via induction of Nix and Runx2 expression (18, 76); thereby regulating the formation of T cell memory against viral infections (77). The indispensable role of IL-15 signaling is further demonstrated by its ability to induce p70 S6 kinase-mediated homeostatic proliferation in virus-specific memory CD8+ T cells; and prime them to rapidly enter cell proliferation upon future viral re-infection (78). In addition to IL-15 signaling, anti-viral T cell memory formation is also regulated by autophagy, a cellular recycling process wherein cells break down their own components via autophagolysosomal fusion (79, 80). The requirement of autophagy during T cell memory formation is linked to its role in clearing off dysfunctional mitochondria in virus-specific CD8+ T cells (18, 80). In this section we will discuss the mechanisms through which IL-15 signaling and autophagy regulate metabolism during anti-viral CD8+ T cell memory formation.
During the contraction phase, the surviving cells that would eventually form memory CD8+ T cells are characterized by gradual transitioning to a more quiescent metabolic phenotype from an activated metabolism. Despite their metabolic quiescence, virus-specific memory CD8+ T cells are metabolically more fit compared to activated effector CD8+ T cells, as demonstrated by higher expression of metabolic fitness-associated genes (81). Memory CD8+ T cells use long-chain fatty acid oxidation instead of glycolysis (16, 43). This reduced dependence on glycolysis during T cell memory formation is due to increased expression of FOXO1 (31, 72, 82). On the other hand, dependence on fatty acid oxidation during formation of CD8+ T cell memory is mediated by both TRAF6, a negative regulator of T cell activation, as well as IL-15 signaling (16, 83, 84). During memory formation, IL-15 is either trans-presented by APCs (85, 86) or produced by CD8+ T cells themselves (87). IL-15 signaling activates mitochondrial biogenesis mediated by transcription factor A, mitochondrial (TFAM); thereby increasing mitochondrial density in CD8+ T cells (83). We recently showed that the quality of these mitochondria during CD8+ T cell memory formation is regulated by NIX, a mitochondrial outer membrane protein, which is induced by IL-15 signaling during the contraction phase (18). In addition, IL-15 signaling during CD8+ T cell memory formation has been shown to increase expression of CPT1A, a metabolic enzyme that transports long-chain fatty acids into mitochondria thereby mediating fatty acid oxidation (83). We showed that this IL-15-dependent induction of long-chain fatty acid oxidation in virus-specific memory CD8+ T cells is regulated by NIX via prevention of HIF1α accumulation during the contraction phase (18). Long-chain fatty acid oxidation plays a critical role during anti-viral T cell memory formation by maintaining optimal ATP levels necessary for memory precursor effector CD8+ T cells (MPECs) to evade apoptosis during the contraction phase, leading to successful differentiation into memory CD8+ T cells (18).
Long-chain fatty acid metabolism by T cell memory-associated molecular factors is critical for memory T cell formation (Figure 2A). The increased demand for long-chain fatty acids during CD8+ T cell memory formation is met through lysosomal lipolysis (17). Although it is not clear as to what triggers lysosomal lipolysis during memory formation, one possible pathway could be autophagy. Consistent with this possibility, Cai et al. (88) showed that mitochondria receive long-chain fatty acids for fatty acid oxidation from the phospholipids degraded via autophagy. Hepatocytes have also been shown to use autophagy to deliver cellular lipids to lysosomes and break them down into free long-chain fatty acids in order to carry out mitochondrial fatty acid oxidation (89). As to where does the cellular lipids come from within CD8+ T cells during memory formation, O'Sullivan et al. (17) showed that CD8+ T cells can synthesize their own lipids using fatty acid synthase (FASN). Besides cellular lipid biosynthesis, another source of fatty acids is active degradation of cell organelles themselves via autophagy. Mammalian cells degrade dysfunctional mitochondria via mitochondrial autophagy (mitophagy) that is induced by NIX in virus-specific CD8+ T cells to fuel long-chain fatty acid oxidation inside healthy mitochondria (18, 90). Moreover, we and others have also shown that absence of mitochondrial autophagy during the contraction phase impairs the ability of CD8+ T cells to actively degrade dysfunctional mitochondria, thereby leading to a defective memory formation and long-chain fatty acid oxidation (18, 79, 80). In addition, we recently found that temporal upregulation of Nix, but not other mitophagy molecules such as Bnip3, Pink1, or Parkin, occurs during effector memory formation in virus-specific CD8+ T cells (18). This upregulation of Nix expression leads to clearance of dysfunctional mitochondria selectively during effector memory, but not central memory formation (18). These results suggest that CD8+ T cells use autophagy to prevent accumulation of dysfunctional mitochondria during the contraction phase to sustain optimal long-chain fatty acid oxidation during memory formation. This also explains why increasing autophagy via rapamycin and spermidine treatment enhances memory formation in virus-specific CD8+ T cells (16, 80).
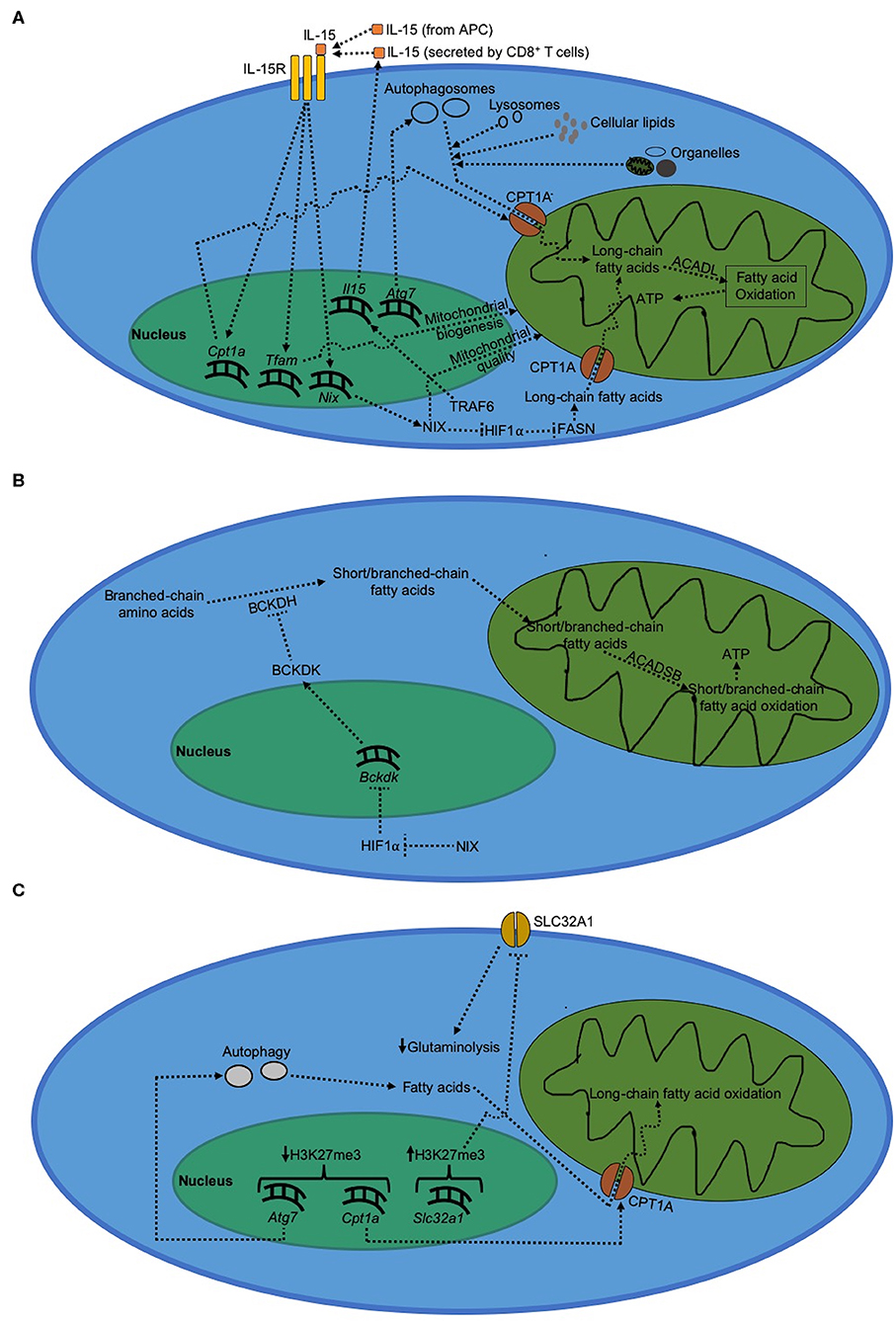
Figure 2. Metabolism in CD8+ T cells during memory formation. (A) Regulation of long-chain fatty acid metabolism during CD8+ T cell during memory formation. Immunological memory formation is characterized by metabolic shift to fatty acid oxidation driven by long-chain fatty acids, which serves to primarily generate ATP. (B) Regulation of short/branched-chain fatty acid metabolism during CD8+ T cell memory formation. (C) Epigenetic regulation of metabolism during CD8+ T cell memory formation.
In spite of these advances in the field, there are some emerging questions that still stand out. Although lysosomal lipolysis and autophagy are known to independently play a role in regulating CD8+ T cell memory formation (17, 18, 79, 80), whether the two pathways overlap is still unknown. Even if they are interdependent, it's still unclear as to how cellular lipids are selectively targeted by autophagosomes to be delivered to lysosomes; given the fact that research in the past decade increasingly suggest autophagy being a selective process in contrast to what was thought earlier (91). Furthermore, roles played by other mitophagy-mediating molecules such as FUNDC1 and BCL2L13, which are also outer mitochondrial membrane proteins that mediate mitophagy in non-immune cells (92, 93), are still unknown in the context of T cell memory formation during acute viral infections. Even though NIX is critical for the formation of T cell memory against acute viral infections, it will be interesting to determine whether NIX overexpression during the contraction phase would promote T cell memory formation against latent viral infections.
In contrast to long-chain fatty acids, short/branched-chain fatty acids are synthesized de novo from branched-chain amino acids by T cells (18, 94, 95); and their supply is upregulated under conditions where the availability of long-chain fatty acids becomes limited such as absence of NIX during the contraction phase (18). Short/branched-chain fatty acid metabolism involves ß-oxidation of 2-methylbutyrate, isobutyrate and isovalerate by short/branched-chain specific acyl-CoA dehydrogenase (ACADSB) inside mitochondria to generate ATP molecules (Figure 2B). We recently reported that short/branched-chain fatty acid oxidation plays a critical role in immunological memory formation in virus-specific CD8+ T cells in mice (18). Synthesis of short/branched-chain fatty acids is regulated by branched-chain-α-keto acid dehydrogenase kinase (BCKDK) (94), an enzyme that inhibits the flux of short/branched-chain fatty acid oxidation during T cell memory formation (18). We further showed that BCKDK expression is negatively regulated by HIF1α; and accumulation of HIF1α during the contraction phase results in reduced BCKDK expression, thereby increasing the flux of short/branched-chain fatty acid oxidation during anti-viral T cell memory formation (18).
Short/branched-chain fatty acid oxidation generates less ATP than long-chain fatty acid oxidation during memory formation in virus-specific CD8+ T cells; and is hence bioenergetically less efficient (18). Upon deletion of NIX in virus-specific CD8+ T cells, reduction in long-chain fatty acid oxidation during viral infection is accompanied by an upregulation of short/branched-chain fatty acid oxidation to partially compensate for the loss in ATP synthesis (18). It is still unclear why short/branched-chain fatty acid oxidation is selectively upregulated despite the availability of bioenergetically more efficient options such as medium and very long chain fatty acids. One possible explanation could be that medium-chain fatty acids have sterically bulkier carbon chain which may restrict their diffusion into the mitochondria unlike short/branched-chain fatty acids. Very long chain fatty acids, on the other hand, require ATP-dependent active transporters- a situation which may further deplete ATP during the contraction phase wherein availability of optimal ATP is critical for virus-specific MPECs to successfully differentiate into memory CD8+ T cells (18, 96). These hypotheses, however, will need to be experimentally tested. In addition, the molecular mechanism underlying negative regulation of BCKDK expression by HIF1α is still unknown; and it will be interesting to study whether HIF1α can directly repress the transcription of the Bckdk gene.
Although the current understanding of the molecular mechanisms driving the expression of various genes during T cell memory formation is limited, analysis of epigenetic landscape shows that during T cell memory formation, trimethylation on H3K27 sites (transcriptionally restrictive) reduces on Atg7 (97), an essential gene for autophagy extensively studied in lymphocytes (79, 80, 98). The corresponding increase in autophagy also corelates with a decrease in the repressive H3K27 trimethylation around Cpt1a gene during memory formation (97). This likely results in a more open chromatin structure around autophagy and long-chain fatty acid oxidation genes that could explain increased dependence on fatty acid metabolism during CD8+ T cell memory formation (Figure 2C). In the same study, H3K27 trimethylation around Slc32a1 gene, which mediates glutaminolysis in activated CD8+ T cells, was shown to increase in memory CD8+ T cells. This suggests that in conjunction with an increase in fatty acid oxidation, effector CD8+ T cells make glutaminolytic genes less accessible during differentiation into memory CD8+ T cells; thereby funneling available metabolic intermediates toward fatty acid oxidation rather than cellular biogenesis that is otherwise typical of T cell activation. In addition, SIRT1-mediated activation of autophagy is critical for maintaining optimal mitochondrial quality in mammalian tissues (99), suggesting that deacetylation of autophagy genes may also play an important role in positively regulating mitochondrial metabolism. However, whether this role of SIRT1 also applies to differentiation of memory CD8+ T cells needs to be experimentally verified through future studies. In addition, RUNX3, a RUNX2 paralog (100), has been shown to increase chromatin accessibility during T cell memory formation; thereby promoting the differentiation of virus-specific memory CD8+ T cells (101). Although the exact epigenetic mechanism through which RUNX3 increases the chromatin accessibility during T cell memory formation is still not clear, it has been reported that Runx3 itself is increasingly demethylated in virus-specific memory precursor CD8+ T cells presumably via downregulation of DNA methyltransferase DNMT3a (68, 102–104).
Depending on the stage of T cell differentiation, a transcription factor could induce different metabolic fates. For example, HIF1α upregulates glycolytic genes (as described above) during CD8+ T cell activation (10) but activates short/branched-chain fatty acid metabolism during CD8+ T cell memory formation (18). The molecular mechanism regulating transcription factor's ability to induce different metabolic pathways at different stages of T cell differentiation is still unknown. However, it's possible that the changing epigenetic landscape during T cell memory formation increases the accessibility of new genes to these transcription factors. Consistent with this possibility, trimethylation of H3K27, deacetylation and demethylation around genes regulating short/branched-chain fatty acid oxidation could play a role here, but this needs to be experimentally demonstrated. Furthermore, the epigenetic mechanism behind contrasting regulation of HIF1α protein level during T cell activation vs memory formation needs to be further elucidated. Although it has been shown that methylation of HIF1α protein by methyltransferases G9a and GLP prevents HIF1α protein accumulation in neuroblastoma cells (71), it is still unknown whether HIF1α downregulation during the contraction phase is also mediated via increased G9a/GLP-dependent HIF1α protein methylation. Further studies are necessary to understand the complete epigenetic story underlying the proclivity of virus-specific memory CD8+ T cells to choose long- and short/branched-chain fatty acid oxidation.
The choice of metabolic pathway in naïve, effector and memory CD8+ T cells depends very much on their state of differentiation and functions. Since naïve CD8+ T cells are less differentiated compared to their successor T cell repertoires, their ultimate objective is long-term survival so as to be available for differentiation into activated effector T cells when necessary. Mere survival requires basal levels of metabolism, which results in a low demand for energy. This is likely responsible for choosing basal levels of glycolysis and fatty acid oxidation in naïve CD8+ T cells. On the other hand, effector CD8+ T cells must not only proliferate but also produce exponentially higher level of ATP to feed the process of clonal proliferation and effector functions by funneling energy into a myriad of enzymatic processes occurring during T cell activation. This results in upregulated glycolysis and glutaminolysis to produce biosynthetic intermediates needed for cellular proliferation and meet the increasing energy demands. Since the main function of memory CD8+ T cells is long-term survival in order to protect against future re-infection, their quiescent metabolism is driven by autophagy-mediated fatty acid oxidation. Autophagy may serve two purposes: to extract fatty acids from cellular lipid stores for fatty acid oxidation; and degrade dysfunctional organelles so that the memory T cells can replace them with quality ones in order to survive long-term and re-activate optimally upon re-encountering pathogens.
SG wrote the manuscript. MC and JW revised the manuscript.
This work was supported by grants from the American Heart Association (15GRNT25700357) and Department of Defense (PR140593) to MC, Cancer Prevention Research Institute of Texas (CPRIT) to MC and JW (RP160384), NIH to JW (R01 AI116644 and R01 AI123221), and Lupus Research Institute to MC and JW.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Smith CM, Wilson NS, Waithman J, Villadangos JA, Carbone FR, Heath WR, et al. Cognate CD4+ T cell licensing of dendritic cells in CD8+ T cell immunity. Nat Immunol. (2004) 5:1143–8. doi: 10.1038/ni1129
2. Weant AE, Michalek RD, Khan IU, Holbrook BC, Willingham MC, Grayson JM. Apoptosis regulators bim and fas function concurrently to control autoimmunity and CD8+ T cell contraction. Immunity. (2008) 28:218–30. doi: 10.1016/j.immuni.2007.12.014
3. Zhang L, Romero P. Metabolic control of CD8+ T cell fate decisions and antitumor immunity. Trends Mol Med. (2018) 24:30–48. doi: 10.1016/j.molmed.2017.11.005
4. Raud B, McGuire PJ, Jones RG, Sparwasser T, Berod L. Fatty acid metabolism in CD8 + T cell memory: challenging current concepts. Immunol Rev. (2018) 283:213–31. doi: 10.1111/imr.12655
5. Deshpande OA, Mohiuddin SS. Biochemistry, Oxidative Phophorylation. (2020). Available online at: http://www.ncbi.nlm.nih.gov/pubmed/31985985 (accessed April 8, 2020).
6. Gaber T, Chen Y, Krauß PL, Buttgereit F. “Metabolism of T Lymphocytes in Health and Disease. Int Rev Cell Mol Biol. (2019) 342:95–148. doi: 10.1016/bs.ircmb.2018.06.002
7. Yang L, Venneti S, Nagrath D. Glutaminolysis: a hallmark of cancer metabolism. Annu Rev Biomed Eng. (2017) 19:163–94. doi: 10.1146/annurev-bioeng-071516-044546
8. Singh D, Vishnoi T, Kumar A. Effect of alpha-ketoglutarate on growth and metabolism of cells cultured on three-dimensional cryogel matrix. Int J Biol Sci. (2013) 9:521–30. doi: 10.7150/ijbs.4962
9. Krauss S, Brand MD, Buttgereit F. Signaling takes a breath - new quantitative perspectives on bioenergetics and signal transduction. Immunity. (2001) 15:497–502. doi: 10.1016/S1074-7613(01)00205-9
10. Menk AV, Scharping NE, Moreci RS, Zeng X, Guy C, Salvatore S, et al. Early TCR signaling induces rapid aerobic glycolysis enabling distinct acute T cell effector functions. Cell Rep. (2018) 22:1509–21. doi: 10.1016/j.celrep.2018.01.040
11. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The Transcription factor myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. (2011) 35:871–82. doi: 10.1016/j.immuni.2011.09.021
12. Jones RG, Thompson CB. Revving the engine: signal transduction fuels T cell activation. Immunity. (2007) 27:173–8. doi: 10.1016/j.immuni.2007.07.008
13. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. (2002) 16:769–77. doi: 10.1016/S1074-7613(02)00323-0
14. Jacobs SR, Herman CE, MacIver NJ, Wofford JA, Wieman HL, Hammen JJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated akt-dependent and independent pathways. J Immunol. (2008) 180:4476–86. doi: 10.4049/jimmunol.180.7.4476
15. Waickman AT, Powell JD. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol Rev. (2012) 249:43–58. doi: 10.1111/j.1600-065X.2012.01152.x
16. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. (2009) 460:103–7. doi: 10.1038/nature08097
17. Sullivan DO, Van der Windt GJW, Huang SC, Jonathan D, Chang C, Buck MD, et al. Memory CD8+ T Cells use cell intrinsic lipolysis. Immunity. (2014) 41:75–88. doi: 10.1016/j.immuni.2014.06.005
18. Gupta SS, Sharp R, Hofferek C, Kuai L, Dorn GW, Wang J, et al. NIX-mediated mitophagy promotes effector memory formation in antigen-specific CD8+ T cells. Cell Rep. (2019) 29:1862-77.e7. doi: 10.1016/j.celrep.2019.10.032
19. Carrette F, Surh CD. IL-7 signaling and CD127 receptor regulation in the control of T cell homeostasis. Semin Immunol. (2012) 24:209–17. doi: 10.1016/j.smim.2012.04.010
20. Jacobs SR, Michalek RD, Rathmell JC. IL-7 is essential for homeostatic control of T cell metabolism in vivo. J Immunol. (2010) 184:3461–9. doi: 10.4049/jimmunol.0902593
21. Rathmell JC, Farkash EA, Gao W, Thompson CB. IL-7 Enhances the survival and maintains the size of naive T cells. J Immunol. (2001) 167:6869–76. doi: 10.4049/jimmunol.167.12.6869
22. Kerdiles YM, Beisner DR, Tinoco R, Dejean AS, Castrillon DH, DePinho RA, et al. Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat Immunol. (2009) 10:176–84. doi: 10.1038/ni.1689
23. Patsoukis N, Bardhan K, Weaver J, Herbel C, Seth P, Li L, et al. The role of metabolic reprogramming in T cell fate and function. Curr Trends Immunol. (2016) 17:1–12.
24. Jones N, Vincent EE, Cronin JG, Panetti S, Chambers M, Holm SR, et al. Akt and STAT5 mediate naïve human CD4+ T-cell early metabolic response to TCR stimulation. Nat Commun. (2019) 10:2042. doi: 10.1038/s41467-019-10023-4
25. Al-Khami AA, Zheng L, Del Valle L, Hossain F, Wyczechowska D, Zabaleta J, et al. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology. (2017) 6:e1344804. doi: 10.1080/2162402X.2017.1344804
26. Abdelsamed HA, Zebley CC, Youngblood B. Epigenetic maintenance of acquired gene expression programs during memory CD8 T cell homeostasis. Front Immunol. (2018) 9:6. doi: 10.3389/fimmu.2018.00006
27. Wingelhofer B, Neubauer HA, Valent P, Han X, Constantinescu SN, Gunning PT, et al. Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia. (2018) 32:1713–26. doi: 10.1038/s41375-018-0117-x
28. Mandal M, Powers SE, Maienschein-Cline M, Bartom ET, Hamel KM, Kee BL, et al. Epigenetic repression of the Igk locus by STAT5-mediated recruitment of the histone methyltransferase Ezh2. Nat Immunol. (2011) 12:1212–20. doi: 10.1038/ni.2136
29. Araki Y, Wang Z, Zang C, Iii WHW, Schones D, Cui K, et al. Genome-wide analysis of histone methylation reveals chromatin state-based complex regulation of differential gene transcription and function of CD8 memory T cells Yasuto. Immunity. (2010) 30:912–25. doi: 10.1016/j.immuni.2009.05.006
30. Chen Y, Zander R, Khatun A, Schauder DM, Cui W. Transcriptional and epigenetic regulation of effector and memory CD8 T cell differentiation. Front Immunol. (2018) 9:2826. doi: 10.3389/fimmu.2018.02826
31. Hedrick SM, Michelini RH, Doedens AL, Goldrath AW, Stone EL. FOXO transcription factors throughout T cell biology. Nat Rev Immunol. (2012) 12:649–61. doi: 10.1038/nri3278
32. Crotzer VL, Blum JS. Autophagy and its role in MHC-mediated antigen presentation. J Immunol. (2009) 182:3335–41. doi: 10.4049/jimmunol.0803458
33. Zhou D, Li P, Lin Y, Lott JM, Hislop AD, Canaday DH, Brutkiewicz RR, et al. Lamp-2a facilitates MHC class II presentation of cytoplasmic antigens. Immunity. (2005) 22:571–81. doi: 10.1016/j.immuni.2005.03.009
34. Schmid D, Pypaert M, Münz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. (2007) 26:79–92. doi: 10.1016/j.immuni.2006.10.018
35. Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol. (2013) 31:443–73. doi: 10.1146/annurev-immunol-032712-095910
36. Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. (2009) 27:591–619. doi: 10.1146/annurev.immunol.021908.132706
37. Pollizzi KN, Patel CH, Sun IH, Oh MH, Waickman AT, Wen J, et al. mTORC1 and mTORC2 selectively regulate CD8+ T cell differentiation. J Clin Invest. (2015) 125:2090–108. doi: 10.1172/JCI77746
38. Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. (2010) 39:171–83. doi: 10.1016/j.molcel.2010.06.022
39. Palazon A, Tyrakis PA, Macias D, Veliça P, Rundqvist H, Fitzpatrick S, et al. An HIF-1α/VEGF-A axis in cytotoxic T cells regulates tumor progression. Cancer Cell. (2017) 32:669–83.e5. doi: 10.1016/j.ccell.2017.10.003
40. Phan AT, Doedens AL, Palazon A, Tyrakis PA, Cheung KP, Johnson RS, et al. Constitutive glycolytic metabolism supports CD8 + T cell effector memory differentiation during viral infection. Immunity. (2016) 45:1024–37. doi: 10.1016/j.immuni.2016.10.017
41. Klein-Hessling S, Muhammad K, Klein M, Pusch T, Rudolf R, Flöter J, et al. NFATc1 controls the cytotoxicity of CD8+ T cells. Nat Commun. (2017) 8:1–15. doi: 10.1038/s41467-017-00612-6
42. Mognol GP, Carneiro FRG, Robbs BK, Faget DV, Viola JPB. Cell cycle and apoptosis regulation by NFAT transcription factors: new roles for an old player. Cell Death Dis. (2016) 7:e2199. doi: 10.1038/cddis.2016.97
43. Windt GJW, Pearce EL. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol Rev. (2012) 249:27–42. doi: 10.1111/j.1600-065X.2012.01150.x
44. Kumari A. Citric acid cycle. Sweet Biochem. (2018) 7–11. doi: 10.1016/B978-0-12-814453-4.00002-9
45. Lane AN, Fan TWM. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. (2015) 43:2466–85. doi: 10.1093/nar/gkv047
46. Dan HC, Ebbs A, Pasparakis M, Van Dyke T, Basseres DS, Baldwin AS. Akt-dependent activation of mTORC1 complex involves phosphorylation of mTOR (mammalian target of rapamycin) by IκB kinase α (IKKα). J Biol Chem. (2014) 289:25227–40. doi: 10.1074/jbc.M114.554881
47. Gerriets VA, Rathmell JC. Metabolic pathways in T cell fate and function. Trends Immunol. (2012) 33:168–73. doi: 10.1016/j.it.2012.01.010
48. Izreig S, Samborska B, Johnson RM, Sergushichev A, Ma EH, Lussier C, et al. The miR-17 ~ 92 microRNA cluster is a global regulator of tumor metabolism. Cell Rep. (2016) 16:1915–28. doi: 10.1016/j.celrep.2016.07.036
49. Baumann F, Yuzefpolskiy Y, Sarkar S, Kalia V. Mir-17~92 promotes terminal differentiation of effector CD8 T cells by lowering TCR stimulation threshold (LYM4P.750). J Immunol. (2014) 192:65.7.
50. Gnanaprakasam JNR, Wang R. MYC in regulating immunity: metabolism and beyond. Genes. (2017) 8:14–6. doi: 10.3390/genes8030088
51. Vatrinet R, Leone G, De Luise M, Girolimetti G, Vidone M, Gasparre G, et al. The α-ketoglutarate dehydrogenase complex in cancer metabolic plasticity. Cancer Metab. (2017) 5:1–14. doi: 10.1186/s40170-017-0165-0
52. Li X, Gu J, Zhou Q. Review of aerobic glycolysis and its key enzymes - new targets for lung cancer therapy. Thorac Cancer. (2015) 6:17–24. doi: 10.1111/1759-7714.12148
53. Vaeth M, Maus M, Klein-Hessling S, Freinkman E, Yang J, Eckstein M, et al. Store-operated Ca2+ entry controls clonal expansion of T cells through metabolic reprogramming. Immunity. (2017) 47:664–79.e6. doi: 10.1016/j.immuni.2017.09.003
54. Glick GD, Rossignol R, Lyssiotis CA, Wahl D, Lesch C, Sanchez B, et al. Anaplerotic metabolism of alloreactive T cells provides a metabolic approach to treat graft-versus-host disease. J Pharmacol Exp Ther. (2014) 351:298–307. doi: 10.1124/jpet.114.218099
55. Gao B, Kong Q, Zhang Y, Yun C, Dent SYR, Song J, et al. The histone acetyltransferase Gcn5 positively regulates T cell activation. J Immunol. (2017) 198:3927–38. doi: 10.4049/jimmunol.1600312
56. Galdieri L, Vancura A. Acetyl-CoA carboxylase regulates global histone acetylation. J Biol Chem. (2012) 287:23865–76. doi: 10.1074/jbc.M112.380519
57. Akimova T, Beier UH, Liu Y, Wang L, Hancock WW. Histone/protein deacetylases and T-cell immune responses. Blood. (2012) 119:2443–51. doi: 10.1182/blood-2011-10-292003
58. Tsuji G, Okiyama N, Villarroel VA, Katz SI. Histone deacetylase 6 inhibition impairs effector CD8 T-cell functions during skin inflammation. J Allergy Clin Immunol. (2015) 135:1228–39. doi: 10.1016/j.jaci.2014.10.002
59. Ren J, Catalina MD, Eden K, Liao X, Read KA, Luo X, et al. Selective histone deacetylase 6 inhibition normalizes B cell activation and germinal center formation in a model of systemic lupus erythematosus. Front Immunol. (2019) 10:2512. doi: 10.3389/fimmu.2019.02512
60. Xiao H, Jiao J, Wang L, O'Brien S, Newick K, Wang LCS, et al. HDAC5 controls the functions of Foxp3 + T-regulatory and CD8 + T cells. Int J Cancer. (2016) 138:2477–86. doi: 10.1002/ijc.29979
61. Shin HM, Kapoor VN, Guan T, Kaech SM, Welsh RM, Berg LJ. Epigenetic modifications induced by Blimp-1 regulate CD8+ T cell memory progression during acute virus infection. Immunity. (2013) 39:661–75. doi: 10.1016/j.immuni.2013.08.032
62. Kuroda S, Yamazaki M, Abe M, Sakimura K, Takayanagi H, Iwai Y. Basic leucine zipper transcription factor, ATF-like (BATF) regulates epigenetically and energetically effector CD8 T-cell differentiation via Sirt1 expression. Proc Natl Acad Sci USA. (2011) 108:14885–9. doi: 10.1073/pnas.1105133108
63. Jeng MY, Hull PA, Fei M, Kwon HS, Tsou CL, Kasler H, et al. Metabolic reprogramming of human CD8 + memory T cells through loss of SIRT1. J Exp Med. (2018) 215:51–62. doi: 10.1084/jem.20161066
64. Wang Y, Bi Y, Chen X, Li C, Li Y, Zhang Z, et al. Histone Deacetylase SIRT1 negatively regulates the differentiation of Interleukin-9-producing CD4 + T cells. Immunity. (2016) 44:1337–49. doi: 10.1016/j.immuni.2016.05.009
65. Gao B, Kong Q, Kemp K, Zhao YS, Fang D. Analysis of sirtuin 1 expression reveals a molecular explanation of IL-2-mediated reversal of T-cell tolerance. Proc Natl Acad Sci USA. (2012) 109:899–904. doi: 10.1073/pnas.1118462109
66. Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1α. Mol Cell. (2010) 38:864–78. doi: 10.1016/j.molcel.2010.05.023
67. Scharer CD, Barwick BG, Youngblood BA, Ahmed R, Boss JM. Global DNA methylation remodeling accompanies CD8 T cell effector function. J Immunol. (2013) 191:3419–29. doi: 10.4049/jimmunol.1301395
68. Ladle BH, Li KP, Phillips MJ, Pucsek AB, Haile A, Powell JD, et al. De novo DNA methylation by DNA methyltransferase 3a controls early effector CD8 + T-cell fate decisions following activation. Proc Natl Acad Sci USA. (2016) 113:10631–6. doi: 10.1073/pnas.1524490113
69. Mielke LA, Liao Y, Clemens EB, Firth MA, Duckworth B, Huang Q, et al. TCF-1 limits the formation of Tc17 cells via repression of the MAF–RORγt axis. J Exp Med. (2019) 216:1682–99. doi: 10.1084/jem.20181778
70. Casciello F, Windloch K, Gannon F, Lee JS. Functional Role of G9a Histone methyltransferase in cancer. Front Immunol. (2015) 6:487. doi: 10.3389/fimmu.2015.00487
71. Bao L, Chen Y, Lai HT, Wu SY, Wang JE, Hatanpaa KJ, et al. Methylation of hypoxia-inducible factor (HIF)-1α by G9a/GLP inhibits HIF-1 transcriptional activity and cell migration. Nucleic Acids Res. (2018) 46:6576–91. doi: 10.1093/nar/gky449
72. Zhang W, Patil S, Chauhan B, Guo S, Powell DR, Le J, et al. FoxO1 regulates multiple metabolic pathways in the liver. J Biol Chem. (2006) 281:10105–17. doi: 10.1074/jbc.M600272200
73. Gray SM, Amezquita RA, Guan T, Kleinstein SH, Kaech SM. Polycomb repressive complex 2-mediated chromatin repression guides effector CD8 + T cell terminal differentiation and loss of multipotency. Immunity. (2017) 46:596–608. doi: 10.1016/j.immuni.2017.03.012
74. Yajima T, Yoshihara K, Nakazato K, Kumabe S, Koyasu S, Sad S, et al. IL-15 Regulates CD8 + T cell contraction during primary infection. J Immunol. (2006) 176:507–15. doi: 10.4049/jimmunol.176.1.507
75. Kalia V, Sarkar S, Ahmed R. CD8 T-cell memory differentiation during acute and chronic viral infections. In: Zanetti M, Schoenberger SP, editors. Memory T Cells. (2010) New York, NY: Springer New York, 79–95. doi: 10.1007/978-1-4419-6451-9_7
76. Olesin E, Nayar R, Saikumar-Lakshmi P, Berg LJ. The transcription factor Runx2 is required for long-term persistence of antiviral CD8 + memory T cells. ImmunoHorizons. (2018) 2:251–61. doi: 10.4049/immunohorizons.1800046
77. Schluns KS, Williams K, Ma A, Zheng XX, Lefrançois L. Cutting edge: requirement for IL-15 in the generation of primary and memory antigen-specific CD8 T cells. J Immunol. (2002) 168:4827–31. doi: 10.4049/jimmunol.168.10.4827
78. Richer MJ, Pewe LL, Hancox LS, Hartwig SM, Varga SM, Harty JT. Inflammatory IL-15 is required for optimal memory T cell responses. J Clin Invest. (2015) 125:3477–90. doi: 10.1172/JCI81261
79. Xu X, Araki K, Li S, Han J, Ye L, Tan WG, et al. Autophagy is essential for effector CD8+ T cell survival and memory formation. Nat Immunol. (2014) 15:1152–61. doi: 10.1038/ni.3025
80. Puleston DJ, Zhang H, Powell TJ, Lipina E, Sims S, Panse I, et al. Autophagy is a critical regulator of memory CD8(+) T cell formation. Elife. (2014) 3:e03706. doi: 10.7554/eLife.03706
81. Sarkar S, Kalia V, Haining WN, Konieczny BT, Subramaniam S, Ahmed R. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J Exp Med. (2008) 205:625–40. doi: 10.1084/jem.20071641
82. Michelini RH, Doedens AL, Goldrath AW, Hedrick SM. Differentiation of CD8 memory T cells depends on Foxo1. J Exp Med. (2013) 210:1189–200. doi: 10.1084/jem.20130392
83. van der Windt GJW, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. (2012) 36:68–78. doi: 10.1016/j.immuni.2011.12.007
84. King CG, Kobayashi T, Cejas PJ, Kim T, Yoon K, Kim GK, et al. TRAF6 is a T cell-intrinsic negative regulator required for the maintenance of immune homeostasis. Nat Med. (2006) 12:1088–92. doi: 10.1038/nm1449
85. Mortier E, Advincula R, Kim L, Chmura S, Barrera J, Reizis B, et al. Macrophage- and dendritic-cell-derived Interleukin-15 receptor alpha supports homeostasis of distinct CD8+ T cell subsets. Immunity. (2009) 31:811–22. doi: 10.1016/j.immuni.2009.09.017
86. Stonier SW, Ma LJ, Castillo EF, Schluns KS. Dendritic cells drive memory CD8 T-cell homeostasis via IL-15 transpresentation. Blood. (2008) 112:4546–54. doi: 10.1182/blood-2008-05-156307
87. Miranda-Carús ME, Benito-Miguel M, Llamas MA, Balsa A, Martín-Mola E. Human T cells constitutively express IL-15 that promotes ex vivo T cell homeostatic proliferation through autocrine/juxtacrine loops. J Immunol. (2005) 175:3656–62. doi: 10.4049/jimmunol.175.6.3656
88. Cai M, He J, Xiong J, Tay LWR, Wang Z, Rog C, et al. Phospholipase D1-regulated autophagy supplies free fatty acids to counter nutrient stress in cancer cells. Cell Death Dis. (2016) 7:1–10. doi: 10.1038/cddis.2016.355
89. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature. (2009) 458:1131–5. doi: 10.1038/nature07976
90. Kristensen AR, Schandorff S, Høyer-Hansen M, Nielsen MO, Jäättelä M, Dengjil J, et al. Ordered organelle degradation during starvation-induced autophagy. Mol Cell Proteomics. (2008) 7:2419–28. doi: 10.1074/mcp.M800184-MCP200
91. Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. (2011) 7:279–96. doi: 10.4161/auto.7.3.14487
92. Murakawa T, Yamaguchi O, Hashimoto A, Hikoso S, Takeda T, Oka T, et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun. (2015) 6:7527. doi: 10.1038/ncomms8527
93. Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. (2012) 14:177–85. doi: 10.1038/ncb2422
94. Shimomura Y, Honda T, Shiraki M, Murakami T, Sato J, Kobayashi H, et al. Branched-chain amino acid catabolism in exercise and liver disease. J Nutr. (2006) 136:250S−3S. doi: 10.1093/jn/136.1.250S
95. Cole JT. Metabolism of BCAAs. In: Rajendram R, Preedy VR, Patel V, editors. Branched Chain Amino Acids in Clinical Nutrition. (2015). New York, NY: Springer New York, 13–24. doi: 10.1007/978-1-4939-1923-9_2
96. Jia Z, Moulson CL, Pei Z, Miner JH, Watkins PA. Fatty acid transport protein 4 is the principal very long chain fatty Acyl-CoA synthetase in skin fibroblasts. J Biol Chem. (2007) 282:20573–83. doi: 10.1074/jbc.M700568200
97. Russ BE, Olshanksy M, Smallwood HS, Li J, Denton AE, Prier JE, et al. Distinct epigenetic signatures delineate transcriptional programs during virus-specific CD8+ T cell differentiation. Immunity. (2014) 41:853–65. doi: 10.1016/j.immuni.2014.11.001
98. Chen M, Hong MJ, Sun H, Wang L, Shi X, Gilbert BE, et al. Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nat Med. (2014) 20:503–10. doi: 10.1038/nm.3521
99. Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci USA. (2008) 105:3374–9. doi: 10.1073/pnas.0712145105
100. Bruno L, Ramlall V, Studer RA, Sauer S, Bradley D, Dharmalingam G, et al. Selective deployment of transcription factor paralogs with submaximal strength facilitates gene regulation in the immune system. Nat Immunol. (2019) 20:1372–80. doi: 10.1038/s41590-019-0471-5
101. Wang D, Diao H, Getzler AJ, Rogal W, Frederick MA, Milner J, et al. The transcription factor Runx3 establishes chromatin accessibility of cis-regulatory landscapes that drive memory cytotoxic T lymphocyte formation. Immunity. (2018) 48:659–74.e6. doi: 10.1016/j.immuni.2018.03.028
102. Youngblood B, Hale JS, Kissick HT, Ahn E, Xu X, Wieland A, et al. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature. (2017) 552:404–9. doi: 10.1038/nature25144
103. Serroukh Y, Gu-Trantien C, Hooshiar Kashani B, Defrance M, Vu Manh TP, Azouz A, et al. The transcription factors Runx3 and ThPOK cross-regulate acquisition of cytotoxic function by human Th1 lymphocytes. Elife. (2018) 7:e30496. doi: 10.7554/eLife.30496
Keywords: CD8+ T cells, metabolism, glycolysis, fatty acid oxidation, glutaminolysis, epigenetics, viral infections
Citation: Gupta SS, Wang J and Chen M (2020) Metabolic Reprogramming in CD8+ T Cells During Acute Viral Infections. Front. Immunol. 11:1013. doi: 10.3389/fimmu.2020.01013
Received: 14 February 2020; Accepted: 28 April 2020;
Published: 22 May 2020.
Edited by:
Vandana Kalia, University of Washington School of Medicine, United StatesReviewed by:
Jason Kyle Whitmire, University of North Carolina at Chapel Hill, United StatesCopyright © 2020 Gupta, Wang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Chen, bWluY0BiY20uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.