 Pratibha Bhalla1
Pratibha Bhalla1 Christian A. Wysocki
Christian A. Wysocki Nicolai S. C. van Oers
Nicolai S. C. van Oers
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 05 May 2020
Sec. T Cell Biology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.00830
This article is part of the Research TopicNew Insights into Thymic Functions during Stress, Aging, and in Disease SettingsView all 17 articles
22q11.2 deletion syndrome (DiGeorge), CHARGE syndrome, Nude/SCID and otofaciocervical syndrome type 2 (OTFCS2) are distinct clinical conditions in humans that can result in hypoplasia and occasionally, aplasia of the thymus. Thymic hypoplasia/aplasia is first suggested by absence or significantly reduced numbers of recent thymic emigrants, revealed in standard-of-care newborn screens for T cell receptor excision circles (TRECs). Subsequent clinical assessments will often indicate whether genetic mutations are causal to the low T cell output from the thymus. However, the molecular mechanisms leading to the thymic hypoplasia/aplasia in diverse human syndromes are not fully understood, partly because the problems of the thymus originate during embryogenesis. Rodent and Zebrafish models of these clinical syndromes have been used to better define the underlying basis of the clinical presentations. Results from these animal models are uncovering contributions of different cell types in the specification, differentiation, and expansion of the thymus. Cell populations such as epithelial cells, mesenchymal cells, endothelial cells, and thymocytes are variably affected depending on the human syndrome responsible for the thymic hypoplasia. In the current review, findings from the diverse animal models will be described in relation to the clinical phenotypes. Importantly, these results are suggesting new strategies for regenerating thymic tissue in patients with distinct congenital disorders.
Thymic hypoplasia is a common transient condition seen in newborns, particularly for premature babies (1, 2). A short-lived hypoplasia of the thymus can occur at any age due to infections, diverse forms of stress, pregnancy, alcoholism, malnutrition, and radiation exposure (3–5). In the elderly, a severe and everlasting involution of the thymic tissue is a well-recognized consequence of the aging process (6, 7). There are several genetic disorders in humans that result in permanent hypoplasia or occasional aplasia of the thymus evident at birth. These genetic disorders often lead to severe combined immunodeficiency (SCID) (8). The mutations can be monogenic or multigenic, impacting either the patterning of the thymic anlage, the thymic stromal cell populations, and/or the developing thymocytes. The stromal cell populations include mesenchymal cells, TECs and endothelial cells. Clinical conditions known to impact these stromal cell populations are chromosome 22q11.2 deletion syndrome (22q11.2del), also referred to as DiGeorge syndrome, Coloboma-heart defects-atresia choanae-retardation of growth-genital abnormalities-ear (CHARGE) syndrome arising from mutations in Chromodomain Helicase DNA Binding Protein 7 (CHD7), Nude/SCID due to autosomal recessive mutations in Forkhead Box N1 (FOXN1), otofaciocervical syndrome type 2 (OTFCS2) due to mutations in PAX1, as well as mutations in TBX1 (located within the chromosome 22q11.2 locus) and TBX2 (Table 1) (9–17). Hypoplasia/aplasia of the thymus can also arise in a developing fetus via teratogen exposures; diabetic- or retinoic acid- induced embryopathies (18–21). In the current review, the genetic mutations that affect the stromal cell populations needed for the formation and/or function of the thymus are described. Since these mutations often influence the specification of the thymus during embryogenesis, detailed mechanistic insights have come from mouse, rat and even zebrafish models.
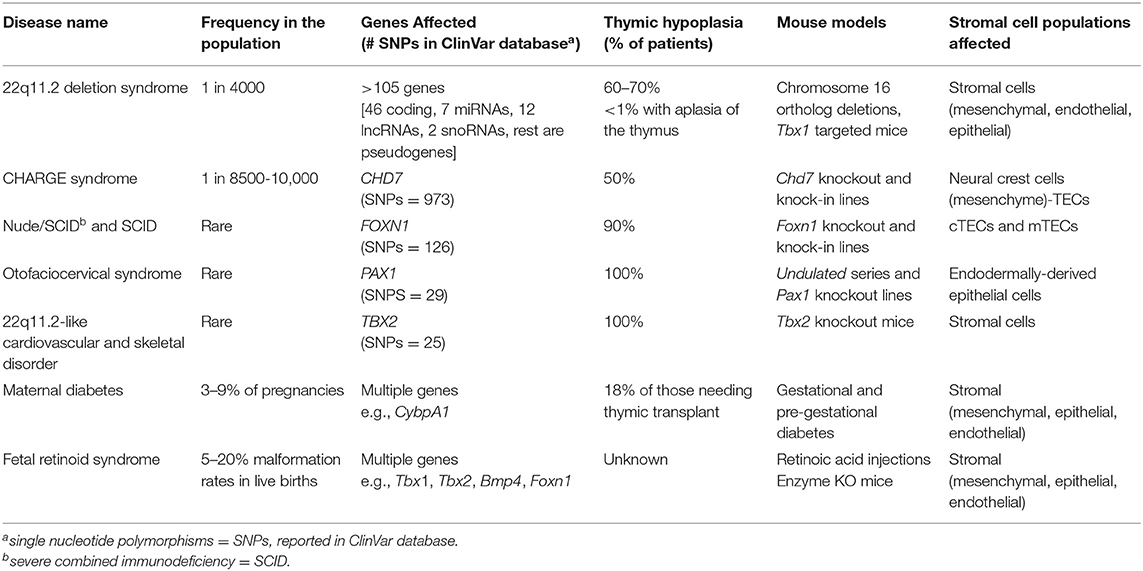
Table 1. Stromal cell intrinsic causes of thymic hypoplasia (T−/loB+NK+) from specific human clinical disorders.
The thymus and parathyroid glands develop from the 3rd pharyngeal pouch (PP), a temporary embryonic structure that begins as an evagination of endothelial cells from the gut tube between e9.5–10.5 (Figures 1A,B) (23). The formation of the 3rd PP requires several transcription factors including Paired box gene family members, Sin oculus homolog 1 (Six1), and Eyes absent 1 (Eya1) (23–25). As the 3rd PP forms, an endothelial layer within this region is surrounded by an area of neural crest-derived mesenchymal cells. Ectodermal in origin, these mesenchymal cells secrete bone morphogenic protein 4 (bmp4) and bone morphogenic protein 2 (bmp2) to support the patterning of the 3rd PP (26). The targeted deletion of bmp4 in neural crest cells leads to a reduced contraction of the mesenchymal cells in the 3rd PP (26). This results in morphogenesis defects of both the thymus and parathyroid domains, which are delineated by the expression of Forkhead box n1 (Foxn1) and Glia cells missing 2 (Gcm2), respectively. The demarcation of the thymus domain by bmp4 is balanced by Sonic hedgehog (Shh), which establishes the dorsal parathyroid region (27). Interestingly, the initial specification of the thymus and parathyroid regions can occur in the absence of neural crest cells, which are lacking in splotch mutant mice, which have mutations in the Paired box gene 3 (Pax3) transcription factor (28). Paired box gene 1 (Pax1) is a related family member also involved in the development of the thymic anlage (15, 29). A Pax1 deficiency in mice leads to mild hypoplasia of the thymus (29, 30). Interestingly, PAX1 autosomal recessive mutations in humans leads to a more severe hypoplasia of the thymus (13, 15, 17).
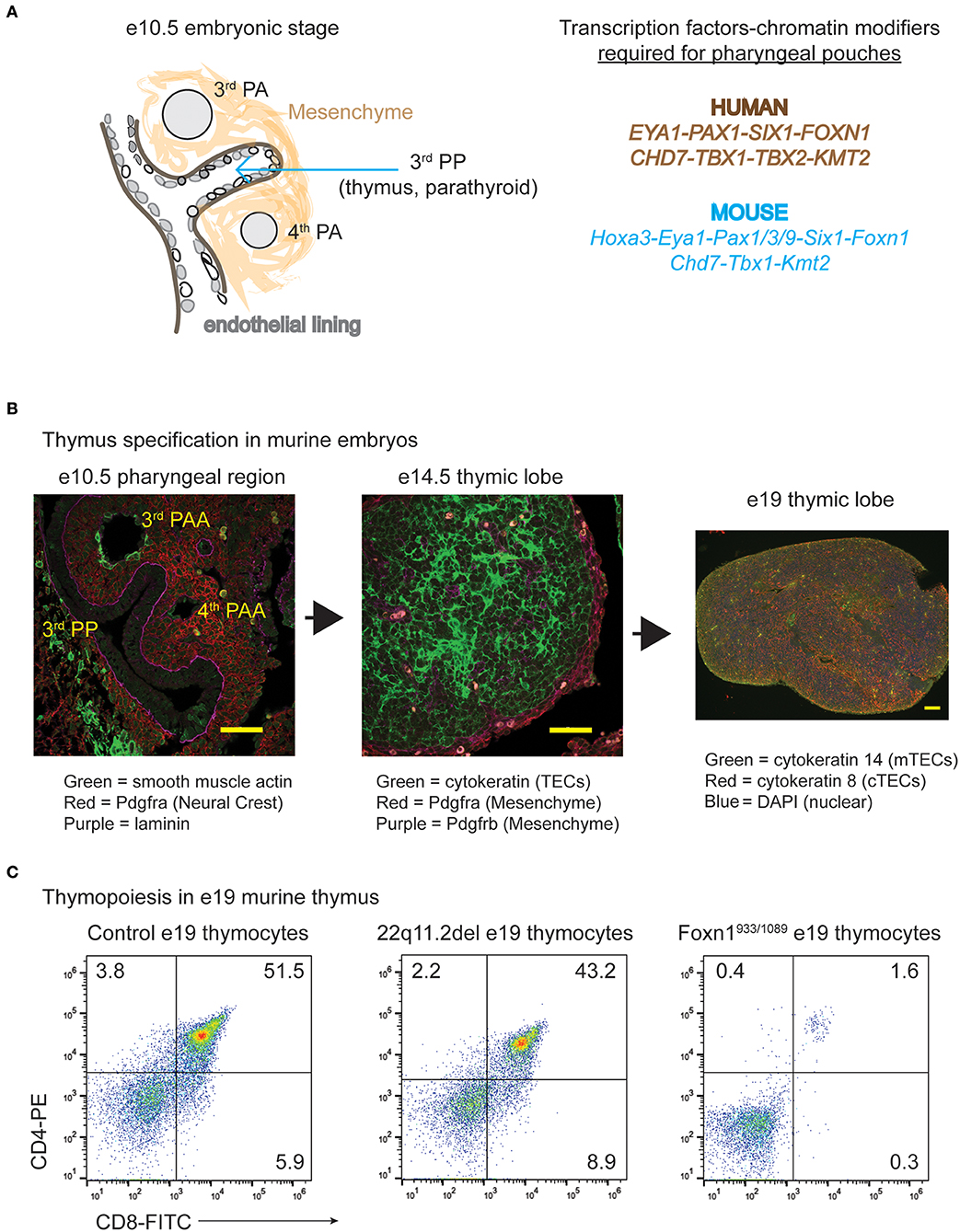
Figure 1. The specification and expansion of the thymus during embryogenesis in normal and disease states. (A) Cartoon diagram illustrating the development process of the thymus along with the various transcription factors and gene products required. The genes that have roles in the specification of the human pharyngeal apparatus that affects the 3rd pharyngeal pouch (thymus and parathyroid) are shown in brown, while those confirmed importance for these processes in mice are in blue. (B) Transverse tissue sections or intact thymic lobes were isolated from normal embryos at the indicated ages of gestation. The transverse sections or whole mounts of the tissue were prepared for immunohistochemistry and H&E staining. Antibodies against vascular smooth muscle, pdgfr-a (alpha) marking the mesenchymal cells and thymic capsule, pdgfr-b (beta) delineating mesenchymal cells and the vasculature, cytokeratin (TECs) and laminin were used, with the colors indicated below the image. (C) Thymocyte subset distributions present in e19–19.5 embryonic thymuses from control C57BL/6 mice, those modeling 22q11.2 deletion syndrome (Tbx1neo2/neo2) and those with compound heterozygous mutations in Foxn1 (Foxn1933/1089) are shown. The Foxn1 mutations genocopy that identified in a human patient (22). Both control and 22q11.2del thymuses have similar distributions of CD4 and CD8 thymocyte subset percentages, suggesting normal TEC functions. The Foxn1 mutant mice are blocked at the CD4−CD8− subset, indicating a severe TEC dysfunction.
With regards to the stromal cell populations, the neural crest-derived mesenchymal cells have at least three distinct roles in the development of the thymic tissue. First, these cells form the thymic capsule and vasculature, establishing the overall structure of the thymus. Noteworthy, the mechanical removal of the mesenchymal capsule using e12.5 fetal thymic lobes renders the tissue hypoplastic (31–33). Yet, the development and proportions of thymocytes subsets are normal in these mesenchymal-depleted hypoplastic tissues, revealing intact TEC functions in the setting of their reduced numbers. Second, the mesenchymal cells enable the expanding thymic lobes to detach from the pharynx between e11.5–12.5, with each lobe from the right and left 3rd PP pairing and descending into the mediastinum. This process requires both Pax3 and Homeobox a3 (Hoxa3) transcription factors, with the targeted deletion of Hoxa3 in neural crest cells resulting in smaller sized thymic lobes remaining attached to the pharynx (28, 34). Third, the mesenchymal cells support thymic epithelial cell (TECs) expansion and differentiation. This involves a combination of ligands and growth factors produced by mesenchymal cells; bmp4, bmp2, fibroblast and insulin growth factors, wnt proteins, and retinoic acid (32, 33, 35–38). Cross-talk between the mesenchymal cells and TECs facilitates thymic tissue expansion, differentiation of TECs into cortical and medullary subsets and recruitment of hematopoietic thymic seeding progenitors (39, 40). The hematopoietic progenitors arrive in timed waves, with the first cells appearing prior to the vascularization of the thymic tissue (41, 42). Following tissue vascularization and remodeling of the epithelia into a 3-dimensional meshwork, the thymic seeding progenitors enter through the cortical-medullary junction (41). These progenitor cells, through a process of cell-cell interactions with TECs, develop into thymocytes. Consistent with the theme of cross-communication among the various cell types in the thymus, ligands expressed by the thymocytes further support the differentiation and expansion of TECs. For example, immature thymocytes are needed for the proper expansion of cTECs during late stages of embryogenesis (43). The cortical TECs positively select T cells expressing the correct T cell receptor (Tcr) specificity for self-peptides embedded by major histocompatibility molecules (44–46). In addition, the emergence of mature SP thymocytes enhances mTEC differentiation and proliferation by releasing epidermal growth factor (Egf) and lymphotoxin and expressing CD40L and RANKL (47–49). The mTECs ensure deletion of potentially autoreactive T cells and enable T regulatory cell selection (44–46). Of note, there are some distinctions between mouse and human thymic tissue specification during embryogenesis (25). Differing contributions of Pax1 and Pax9 is one such example, as detailed in the section on otofaciocervical syndrome type 2 (17). In addition, unlike mice, both humans and rats express MHC class II on developing thymocytes and these cells can support the selection and maturation of CD4 single positive cells (50–52). Several articles in the current series “new insights into thymic functions during stress, aging, and in disease settings” as well as other reviews have provided detailed information about the development and contribution of TECs in thymopoiesis (53, 54). The current review will focus on the TECs and other stromal cell types affected by selected clinical disorders.
Chromosome 22q11.2 deletion syndrome (22q11.2del; OMIM #188400) is a common human disorder (frequency of 1/4000), resulting in variable and complex congenital malformations (8, 55–58). The congenital defects can include thymic hypoplasia, outflow track problems of the heart, hypoparathyroidism, dysmorphic facial features, and/or other midline organ involvement (Table 1). Additional complications for children with 22q11.2del include developmental delay, and over time, neurological problems such as schizophrenia and autism, malignancy, and/or autoimmunity (57–61). Most individuals with 22q11.2del have a 3 Mb microdeletion on chromosome 22, resulting in a hemizygosity of nearly 106 genes (8, 58, 60, 62, 63). A smaller, nested deletion of 1.5 Mb creates a haploinsufficiency of 30 genes, which occurs in 5–8% of 22q11.2del patients (8, 58, 60).
Thymic hypoplasia is reported for 60–70% of individuals with 22q11.2del (56, 58, 64). Due to their thymic hypoplasia, 22q11.2del patients have an average 5-fold reduction in the number of T cell receptor excision circles (TRECs) compared to matched controls, with TRECs measuring the circulating naïve T cells emerging from the thymus (56, 65, 66). In rare cases a patient with 22q11.2del may have complete thymic aplasia, resulting in near-complete absence of autologous T cells, defined by <50 naïve CD3+ T cells per microliter of peripheral blood (14). An effective clinical treatment option for such a patient is an allogeneic thymic tissue transplant, first depleted of thymocytes prior to the placement of small fragments of this tissue within the quadricep muscles (67–70). The thymic stromal tissue consists of TECs, mesenchymal cells, and endothelial cells (71). Upon transplant, the stromal tissue recruits host-derived hematopoietic cells that mature into thymocytes (70, 72). A remarkable feature of this thymus transplantation procedure is the successful selection of TCR-expressing T cells recognizing peptides presented by host (recipient) antigen-presenting cells (12, 70, 72, 73). However, the processes of both positive and negative selection and that of MHC restriction of the developing T cells are not completely understood in these thymic tissue transplants. The positive selection of host T cells in a donor thymus MHC (HLA) background could be caused by recipient-derived epithelial progenitor cells (74). Alternatively, the developing human thymocytes could promote positive selection as these cells express MHC class II molecules (50). When MHC class II is forcibly expressed on murine thymocytes, such cells can now positively select CD4 T cells (51, 52). The thymocyte-selected CD4 single positive cells formed in these mouse models are different than conventional CD4 T cells (75). They express the promyelocytic leukemia zinc finger protein (plzf) and produce both gamma-interferon and IL-4, reflecting more innate-like responses (75). The thymic transplants for 22q11.2del patients can additionally enable T regulatory cell development (69, 76). In normal thymopoiesis, these Tregs develop through interactions with medullary TECs. Negative selection is similarly not well-understood following thymus tissue grafting, with the developing T cells tolerant to both the donor and host MHC (76). It is likely that host dendritic cells along with donor mTECs tolerizing/eliminating any developing T cells targeting either host and donor peptide-MHC complexes (53, 68, 76).
Not all 22q11.2del patients who have a severe hypoplasia of the thymus are grafted with an allogeneic thymus (77, 78). Thus, matched sibling and sometimes unrelated bone marrow transplants have been successfully used to treat 22q11.2del patients who have a severe thymic hypoplasia (limited TRECs) (77–81). The recipient 22q11.2del patients have normal T cell functions and humoral immunity, suggesting T cell reconstitution. However, the majority of the donor T cells have a memory phenotype and a limited TCR repertoire (77, 78). In the short term, there is no difference reported in the mortality for the patients receiving a thymic tissue vs. those with a bone marrow transplant (80, 81). This conclusion will require a long-term longitudinal study comparing infection and survival rates with a larger cohort. However, the lack of naïve T cell development in the bone marrow recipients is of clinical concern for 22q11.2del patients and as described in subsequent sections, individuals with FOXN1 and PAX1 mutations (82).
An important take-home message from the clinical approaches to treat 22q11.2del patients is that the deletion primarily impacts the stromal cells of the thymus. Yet, which stromal cell type(s) is affected by 22q11.2del remains unknown. One group has analyzed thymuses isolated from 22q11.2del patients, available since this tissue is often removed to allow surgical access to the heart (83). The most distinguishing feature of the thymuses from 22q11.2del patients is its smaller size compared to age-matched control tissues (83). Thymopoiesis appears normal, as the percentage of CD4−CD8− (DN), CD4+CD8+ (DP), and CD4+CD8−, and CD4−CD8+ (SP) thymocyte subsets in the hypoplastic tissues is similar to that seen with control samples. The medullary region does appear smaller in the 22q11.2del samples, although the levels of a key gene expressed in medullary TECs, Autoimmune regulator (AIRE), is not statistically different from controls (83). Yet, the number of thymic CD4+Foxp3+ T regulatory cells (Tregs) is diminished in the hypoplastic lobes and these cells have less suppressive capabilities compared to controls (83). It remains unknown why this difference exists but may explain the higher prevalence of autoimmune cytopenias in the 22q11.2del cohort (56, 84, 85). The number of these CD4+ Tregs is also decreased in peripheral tissues, but this arises from the generalized T cell lymphopenia affecting most T cell subsets in the setting of 22q11.2del (11, 85–89).
The congenital hypoplasia/aplasia of the thymus caused by 22q11.2del occurs during the patterning of the pharyngeal apparatus in embryos (58, 90–92). This is best revealed in mice, as comparative analyses between normal embryos and those obtained from 22q11.2del mouse models suggest patterning defects of the pharyngeal pouches and arches (61, 90, 91, 93–95). The 22q11.2del mouse lines were initially developed with orthologous deletions on murine chromosome 16 to identify genes causal to the congenital malformations (Table 2). This led to the realization that the principal cause of the congenital defects was linked to a haploinsufficiency of the T-box Transcription Factor 1 (TBX1) (90, 91, 93, 94, 96). TBX1 interacts with members of the Histone-lysine N-methyltransferase (KMT2)-family, activating the low level transcription of over 2,000 genes (97). Interestingly, the penetrance and severity of the congenital malformations due to a haploinsufficiency of TBX1 varies considerably in the mouse models, which recapitulates the wide range of differences among individual 22q11.2del patients (Table 2). Emerging evidence suggests this variability is due to a combination of genetic and epigenetic regulators, both within and outside chromosome 22q11.2, which influence all the clinical phenotypes of 22q11.2del (8, 98, 99).
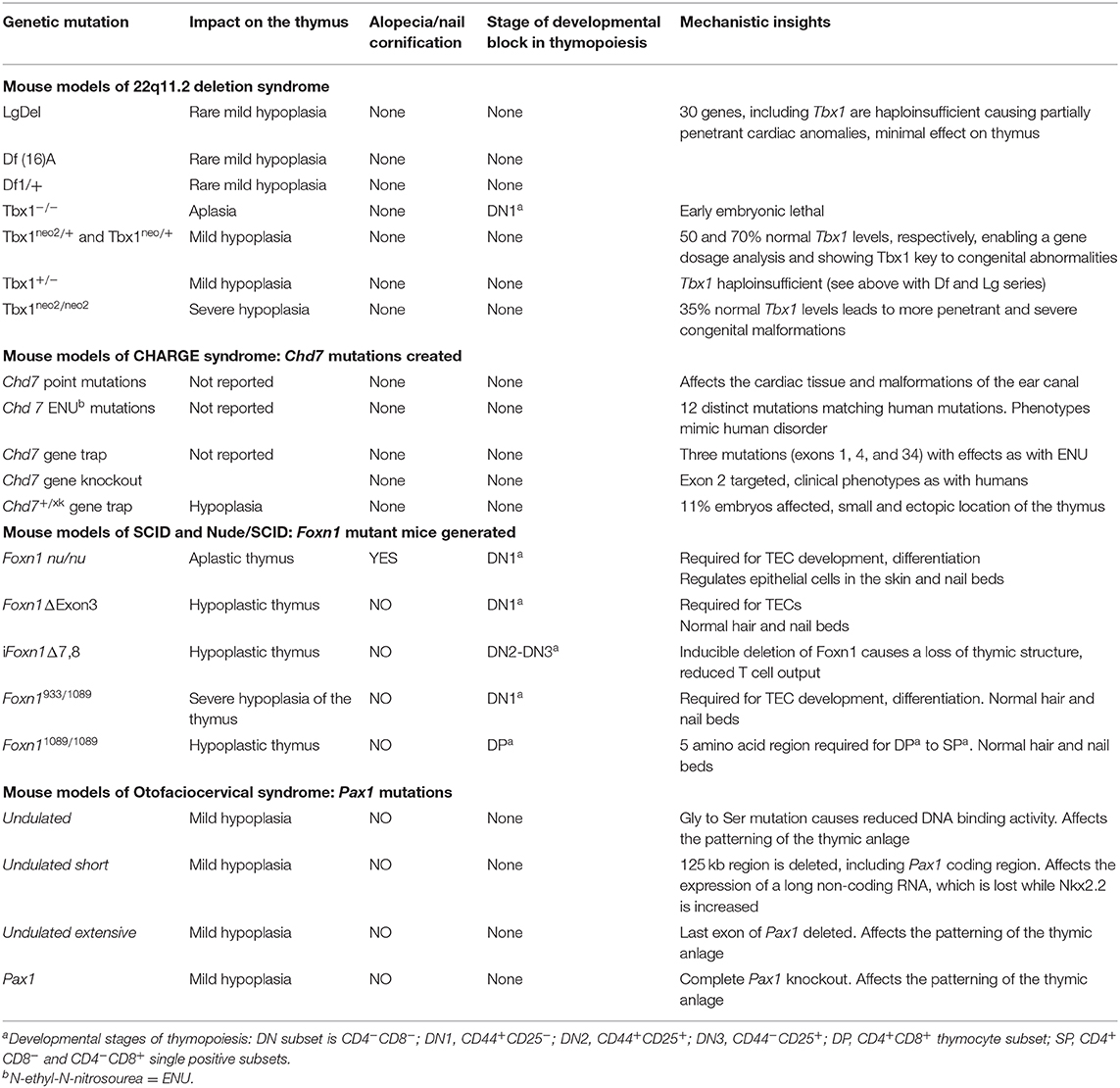
Table 2. Mouse models of human clinical disorders leading to hypoplasia or aplasia of the thymus.
In the mouse models, haploinsufficiency of Tbx1 is generally not very penetrant in eliciting hypoplasia/aplasia of the thymus (90, 91, 94, 96, 100). By comparing embryos expressing varying levels of Tbx1, expression of this transcription factor at or below 35% normal values results in a more frequent and damaging thymic hypoplasia (101). Thymic hypoplasia resulting from the reduced levels of Tbx1 are likely caused by developmental abnormalities in the pharyngeal region. However, the studies published to date have not concentrated on the 3rd PP. What is noticeably different are the 4th pharyngeal arches (PA), adjacent to the 3rd PPs, which are absent or developmentally delayed between day e9.5-11.5 of embryogenesis (Figure 1B) (101, 102). This impacts the patterning of the structures originating from the right and left 4th PA, causing a displaced right subclavian artery and interrupted aortic arch type B, respectively. Both cardiac presentations are common clinical phenotypes of human 22q11.2del (102). Tbx1 is specifically expressed in the regions comprising the pharyngeal arches as well as in the endothelial layer that juxtaposes the developing parathyroid (103). It is not expressed in the thymic anlage, suggesting that Tbx1 haploinsufficiency does not directly impact TECs, consistent with the observations that enforced expression of Tbx1 within the 3rd PP actually represses TEC development (103). A plausible explanation for the thymic hypoplasia in 22q11.2del is that reduced levels of Tbx1 in the pharyngeal region impact the neural crest-derived mesenchymal cells that surround the 3rd PP. The importance of these mesenchymal cells and other cell types has been more clearly revealed in Tbx1-null embryos. An immunohistochemical analysis of these embryos reveals an abnormal distribution of proteins involved in the formation of the extracellular matrix, cell adhesions, and cell-cell contact (vinculin, paxillin and collagen) (104). Changes in the expression patterns of these proteins affects the NCC-derived mesenchyme along with the epithelial cells in the second heart field (104). Such results strongly suggest that the NCC-derived mesenchymal cells surrounding the 3rd PP may also have abnormal mesenchymal and endothelial cell distributions required for the proper patterning of the 3rd PP. This is likely what causes a size restriction on the developing thymus. In one mouse model of 22q11.2del (Tbx1neo2/neo2), the embryonic thymus is size restricted yet still supports normal T cell development (Table 2, Figure 1C). This indicates that the TECs are functional, matching the phenotype noted in the hypoplastic thymic lobes from 22q11.2del patients (83). In summary, mouse models of 22q11.2del strongly suggest that the initial developmental problems leading to thymic hypoplasia are coupled to mesenchymal cell defects. As the mesenchymal cells provide critical support functions for TECs, the consequence is reduced TEC expansion. Comparing 22q11.2del with other human clinical syndromes further supports this notion, as described next.
Coloboma-heart defects-atresia choanae-retardation of growth-genital abnormalities-ear abnormalities (CHARGE) is a multi-syndromic congenital disease (11, 105, 106). Approximately 90% of CHARGE patients have mutations in Chromodomain Helicase DNA Binding Protein 7 (CHD7) (OMIM# 0214800) (107). CHD7 is an ATP-dependent nucleosome remodeling factor, regulating chromatin accessibility and consequently, gene expression (108). CHD7 also positively regulates ribosomal RNA biogenesis in the nucleolus (109). Affecting an estimated 1 in every 10,000 humans, 953 mutations have so far been discovered in CHD7 (ClinVar database). These include missense, non-sense, deletion, splicing, and frame-shift mutations, resulting in a loss-of-function of varying severity depending on the location and/or effect of the mutation on the protein (105, 110). Patients with the CHARGE syndrome have immune system problems that contribute to their recurrent infections; otitis media, sinusitis, upper airway infections, pneumonia, and/or sepsis (106, 111). These infections are most often attributed to malformations of the craniofacial region, the upper respiratory tract, and the 7th cranial nerve (facial innervation). Of note, the first descriptions of CHARGE suggested that the infectious issues were of low incidence (105). More recent reports reveal that immune system complications are far more prevalent, with developmental problems of the thymus additionally reported as causal to the increased susceptibility to infections (106, 112). An immunological assessment of 59 CHARGE patients revealed that about 50% had a T−/loB+NK+ phenotype (106). Immunoglobulin levels and subclasses were normal in most of these CHARGE patients. The absolute numbers of B cells, including memory cells, were very similar to that in controls. The low T cell numbers were a consequence of a thymic dysfunction as TREC levels for these patients were reduced relative to normal controls. Chart reviews for 36 CHARGE patients who had cardiac surgeries revealed 16 of 36 had a hypoplasia or aplasia of the thymus (106). The prevalence of the thymic hypoplasia may be higher in embryos, as a small/absent thymus was noted in seven of 10 CHARGE fetuses described in one study (113).
Chd7 is required for the formation of the multipotent migratory neural crest cells that migrate throughout the body, establishing the bone, cartilage, peripheral nervous system, and cardiac structures (114). To understand the role of Chd7 in CHARGE, especially given the varied congenital problems that can arise, various mouse models have been developed (115–118). The mouse models include those generated with gene-trapped ES cell lines, N-ethyl-N-nitrosourea (ENU) mutagenesis, targeted mutations in Chd7, and various floxed alleles of the gene (Table 2). Embryological analyses indicate that Chd7 is expressed in the pharyngeal region, including the 3rd PP, the 4th PA, and the 1st PP, the latter forming the auditory tube and middle ear canal (119). As early as e10.5, the 4th PA is malformed or absent in 50% of the Chd7 mutant embryos, resulting in an interrupted aortic arch type B and displaced right subclavian artery, just as with 22q11.2del (90). While most of the studies did not focus on thymus abnormalities, one group did report on this tissue. In this study, about 11% of e14.5 Chd7+/xk embryos were found to have irregularly shaped thymic lobes, smaller and more oblong in appearance along with some ectopic positioning (119). Chd7 is expressed in the surrounding mesenchyme and at higher levels in TECs, suggesting that the Chd7 mutations impact both neural crest-derived mesenchymal cells and TECs (115). Interestingly, modulation of retinoic acid (RA) levels in utero can limit the severity of the phenotypes resulting from the Chd7 mutations (120, 121). The clinical phenotypes due to retinoic acid embryopathies, including hypoplasia of the thymus, are discussed in a later section.
Complementing the murine models, Zebrafish studies have provided additional insights into how Chd7 impacts thymic tissue specification. One technology commonly used in Zebrafish is a gene knockdown approach with morpholino oligonucleotides (MOs), creating morphants that have a block in transcript expression. Chd7 morphants have a disrupted morphogenesis of the 3rd PP, with the migration and function of neural crest cells (NCCs) in this area impaired (122). Both bmp4 and bmp2 levels are significantly diminished in the chd7 morphants, again revealing the importance of these soluble proteins in establishing the thymus and parathyroid domains. At later developmental time points, the Chd7 knockdown impairs the formation of the thymic capsule and vasculature. This is coupled with a reduced formation/expansion of the TECs that may involve impaired differentiation of the endothelial layer. Finally, the TECs have a substantial loss of Foxn1 expression, providing a mechanistic basis for the hypoplasia due to TEC abnormalities (122). In summary, the Chd7 knockdown impacts the NCC-derived mesenchymal cells along with the TECs, which suggests that CHARGE affects more stromal cell populations than 22q11.2del.
Autosomal recessive mutations in the Forkhead Box N1 (FOXN1) transcription factor cause a T−B+NK+ SCID phenotype due to a thymic aplasia as well as alopecia universalis and nail plate dystrophy (OMIM #601705) (123–127). Three distinct autosomal recessive mutations in FOXN1 have been reported for 10 patients to date, and these mutations result in a complete loss of protein function, impacting TECs and skin epithelial cells. Patients with compound heterozygous mutations in FOXN1 have also been reported with an atypical phenotype, a thymic hypoplasia without the co-presenting alopecia and nail dystrophy (22). With the increasing number of infants noted to have low TRECs, the subsequent use of exome sequencing for them has uncovered many individuals with single allelic mutations in FOXN1 (22, 128). While such affected individuals will likely recover normal T cell numbers as one allele remains functional, it is unclear what impact such single allelic mutations will have on T cell output later in life (129). To date, about 131 distinct mutations in human FOXN1 have been reported, and while many are benign, there are >20 that have either complete or partial loss of function consequences (ClinVar database). The best clinical treatment option for patients with autosomal recessive or specific compound heterozygous mutations that contribute to a loss-of-function for FOXN1 is a thymic tissue transplant (12). Yet, while bone marrow transplants have also been undertaken for such patients, the underlying defect lies with the TECs of the thymus (22). Paralleling the clinical findings with 22q11.2del, a thymic tissue transplant is the best option as this directly resolves the TEC anomalies.
In the thymus, Foxn1 is the master transcriptional regulator of TEC development, supporting the differentiation of both cortical and medullary TEC subsets (45, 130–132). These TEC subsets are critical for establishing the repertoire of TCR-expressing T cells that are selected to recognize but not respond to self-peptide/MHC complexes (44, 45, 131). Foxn1 is a 648 amino acid long transcription factor that contains DNA binding and transactivation domains, both required for protein function (133, 134). The DNA binding domain of Foxn1 comprises three alpha helices, three beta sheets, and two loops (wings) (130, 135). The 3rd helix and the 2nd winged segment interact with the major and minor grooves of DNA, respectively (130, 135). The DNA binding sequence bound by Foxn1 is GAa/cGC, present in about 500 target genes (132). The genes regulated by Foxn1 include keratins, keratin-associated proteins, cytokeratins, thymo-proteasome components, and cell surface proteins (132, 136). These proteins are important for both cortical and medullary TEC functions along with the extrusion of the hair shaft through the dermal layers of the skin and for nail bed formation (132, 137, 138). In many of the promoter/enhancer elements bound by Foxn1, there are CREB and Tp63 binding sites, suggesting cooperative gene regulation by multiple transcription factors (132).
Mouse and rat models have greatly aided in delineating the functions of Foxn1. First and foremost was a spontaneously arising mutant mouse line, discovered in 1966, with a pronounced nude phenotype (nu/nu). Almost three decades passed before the mapping of the nu/nu allele to autosomal recessive mutations in Foxn1 (130, 134). The nu mutation results in a single base pair deletion in exon 3, causing a frameshift and almost no protein expression (130). The mice lack fur, whiskers, and nails (130, 139, 140). The thymus in the nu/nu mouse is a small cystic tissue that is unable to support TEC and consequently, thymocyte development (23, 141). Such nude mice are commonly referred to as Nude/SCID given their combined lack of fur and T−B+NK+ immune profile. An analysis of embryos from these mice show that Foxn1 is not required for the initial specification of the thymic region within the 3rd PP, but rather for the vascularization of this tissue along with TEC differentiation/expansion (142, 143). Nude rats (rnuN, rnu) and cats (nu/nu) with autosomal recessive mutations in Foxn1 have similarly been described, with the first nude rat actually found in 1953, prior to the mouse reports (130, 144–146). While the nu and rnuN mutations prevent translation of the DNA binding and transactivation domains, much like the autosomal recessive FOXN1 mutations in humans, rnu rats carry a mutation within exon 8, which creates a stop codon. This leads to the expression of a truncated protein (amino acids 1-473) lacking the transactivation domain. Characterizing this region revealed several aspartic acid residues essential for protein function (133). In an unrelated study, the introduction of a truncated Foxn1 construct, wherein only exon 3 is deleted, blocks TEC development/expansion while allowing for hair extrusion and nail formation (147). It remains unclear how this occurs as both DNA binding and transactivation domains remain intact. In a separate cohort of mice developed to genocopy the compound heterozygous FOXN1 mutations identified in an infant, the mice (Foxn1933/1089) had T−B+NK+ immune profile with normal hair growth and nail extensions (22). Unlike 22q11.2del and CHARGE, these FOXN1 mutations directly impact TEC development, causing a loss of both DP and SP thymocytes (Figure 1C). One of the mutations in FOXN1 (FOXN11089) causes a loss of 5 amino acids at the very end of the DNA binding domain (p.W363C with a 5 amino acid loss). Knock-in mice harboring this mutation on both alleles (Foxn11089/1089) have a selective block in thymopoiesis at the DP stage, with hair follicles and nails appearing normal (22). This 5-amino acid sequence is highly conserved with Foxn4, an ancestral ortholog of Foxn1 (148). Interestingly, the cephalochordate species (lancelets) lack a thymus, and have a divergent sequence within this 5-amino acid stretch (22, 148). This suggests this small sequence is important for the expansion of DP thymocytes and their positive selection into CD4+ and CD8+ subsets (22). There is a 2nd patient described with distinct compound heterozygous mutations in FOXN1 (FOXN11288/1465). In functional assays, one of the mutations (Foxn11465) leads to a p.R489fsX61 truncation of the protein, resulting in 18% normal transcriptional activity (22). This mutation prevents the translation of the transactivation domain, revealing a requirement for this region to maintain normal TEC functions. Of note, an increasing number of single allelic FOXN1 mutations are being reported for patients initially presenting with low TRECs (22, 128). The subsequent characterization of these novel mutations will likely reveal the basis for the differential functions of FOXN1 in TEC subsets vs. skin epithelial cells. Of note, one research group has identified a cis-regulatory element (RE) in the 1st intron of Foxn1, the targeting of which reduces TEC numbers and functions without any impact on skin epithelial cells (149). This RE is a target of the Foxn1 DNA binding domain, revealing a positive autoregulatory loop (150). The possibility exists that human patients may contain such intronic FOXN1 mutations, but these have not been reported to date as whole genome sequencing, which is not commonly done, would be required to uncover them.
Post-natally, Foxn1 needs to be continuously expressed in TECs to maintain normal T cell output from the thymus (132). Thus, the inducible deletion of Foxn1 in adult mice reduces thymic cellularity, and impacts the expansion of the DN1-DN4 subsets of thymocytes (132). In “old” mice, Foxn1 levels in the thymus are reduced significantly, which partly explains the tissue involution (151–153). Restoring Foxn1 in the aged thymus significantly improves thymic cellularity and T cell output (152–155). Taken together, the numerous human reports regarding single allelic mutations in FOXN1 and the diverse mouse models are beginning to reveal key regulatory features of this critical transcription factor needed for T cell output throughout life.
Loss-of-function mutations in PAX1 lead to skeletal defects along with thymic hypoplasia in patients, the latter contributing to the T−/loB+NK+ phenotype (13, 15, 17). Four such patients received bone marrow transplants (prior to identification of the PAX1 homozygous mutations) in an attempt to correct their SCID presentations. Notably, the bone marrow transplants were unable to restore T cell development [reviewed in (17)]. The T cells, characterized in the patients after their bone marrow transplants, were of donor origin and exhibited a memory phenotype. Such findings are consistent with the current knowledge that PAX1 regulates the patterning of the pharyngeal region, thereby impacting the stromal cell populations that would not be corrected by a bone marrow transplant.
Using embryos isolated from pregnant mice, Pax1 transcripts are evident in the four pharyngeal pouches as early as e10.5, and become confined to mesenchymal condensations as embryogenesis progresses (29). This transcription factor is present in the 3rd PP endoderm and is subsequently detected in a subset of TECs during embryogenesis (29). Its expression is retained in the adult thymus. The deletion of Pax1 results in a marginal hypoplasia of the thymus (29, 30). This was reported in the undulated series of mouse lines that had varying mutations within Pax1 or with surrounding regulatory elements. These mice were initially described in the 1940's due to their kinked tails and vertebral deformities (Table 2) (29, 156). All undulated mutants have a smaller thymus about 2/3rd normal size (29, 30). Interestingly, only in the context of a Hoxa3 haploinsufficiency does the thymus in the Pax1 mutant lines exhibit a more severe hypoplasia, with the two lobes ectopically positioned (30). The mild thymus phenotypes in the mouse models comprising various Pax1 mutations sharply contrast the severe hypoplasia in humans with PAX1 autosomal recessive mutations. One possible explanation is a compensatory contribution by murine Pax9 when Pax1 is lacking. Pax9 overlaps in expression with Pax1 in the endodermal-derived epithelium of the pharyngeal pouches (157). In mice, a complete deficiency of Pax9 causes a thymic aplasia and a lack of teeth, while in humans, autosomal recessive mutations in PAX9 cause selective tooth agenesis (158, 159).
Maternal diabetes and systemic use of retinoic acid (RA) derivatives during pregnancy can cause long-term thymic hypoplasia in newborns (18–21). What's more, gestational diabetes leads to congenital malformations in the developing fetus which overlap with those noted in individuals with 22q11.2del; hypoplasia/aplasia of the thymus, cardiac outflow tract defects, and hypoparathyroidism (160–163). Estimates suggest that 18% of infants who required a thymic tissue transplant due to an aplasia of this tissue, and did not have 22q11.2del, were born to mothers who had maternal diabetes (72). In spite of the obvious overlap in clinical presentations between 22q11.2del and diabetic embryopathy, it remains unknown how blood sugar dysregulation affects the pharyngeal apparatus.
In rodent studies, the induction of diabetes in pregnant mice and rats causes thymic hypoplasia along with intrauterine growth impairment (164). While intrauterine growth delay will certainly contribute to thymic hypoplasia, there is some evidence that the hypoplasia can result from patterning defects within the pharyngeal apparatus. As gestational diabetes in rodent models is difficult to regulate, the use of a pregestational diabetes mouse model has revealed that retinoic acid production is dysregulated in the developing embryos. Thus, pregestational diabetes reduces the expression of Cytochrome P450 family 26 subfamily A member 1 (Cyp26a1), an enzyme that catabolizes retinoic acid (RA) in the caudal region (tailbud) of developing embryos (165). RA is a derivative of Vitamin A, which functions as a natural morphogen regulating the patterning of the 3rd PP along with the 4th PA (166–170). Both reductions and elevations in RA can lead to hypoplasia of the thymus along with the other congenital malformations that overlap remarkably with 22q11.2del and CHARGE phenotypes (8). While the levels of Cyb26a1 or related family members within the pharyngeal region were not assessed in the pregestational diabetes model, their loses would increase RA, which could cause the problems of the thymus. Consistent with this, injecting high levels of RA in pregnant mice at e9.5 results in the formation of a hypoplastic/aplastic thymus, examined at e11.5–e12.5 (21). It is known that high levels of RA can impair the migration of the NCCs in the region surrounding the 3rd PP (21). Moreover, high levels of RA can reduce the expression of Pax1 within the 3rd PP and Tbx1 throughout the pharyngeal apparatus (171–173). RA likewise represses Bmp4 activity, impacting thymic tissue specification and development by ultimately reducing the levels of Foxn1 (174). These changes have some similarity to that described for embryos developing in the setting of Chd7 mutations.
The second medical condition that can lead to permanent hypo- or aplasia of the thymus in newborns is exposure to elevated levels of RA during pregnancy. Drugs such as tretinoin or isotretinoin are retinoids prescribed to patients to both reduce the severity of their acne and smoothen the skin. However, if taken during pregnancy, the higher levels of RA can trigger 22q11.2-like congenital malformations in the developing embryos (18, 72, 160–162, 175, 176). The mechanism for this hypoplasia is a combination of Tbx1, Pax1, and Foxn1 suppression, as described in the preceding sections of this review (Figure 1A). The profound damage caused by RA has led to a generalized warning from the FDA for women to avoid treatments with RA derivatives during pregnancy.
A number of clinical conditions impact the specification of the thymus during embryogenesis. Interestingly, those that affect the stromal cell populations have overlapping phenotypes, revealing that many of the affected genes function in related developmental pathways. 22q11.2del appears to impact one of the earlier stromal cell types involved in this process, the NCC-derived mesenchymal cells. These cells regulate the patterning and formation of the thymic anlage. CHARGE affects mesenchymal cells, endothelial cells and the TECs, while FOXN1 mutations selectively affect the TECs. It is becoming obvious that the three stromal cell types have considerable cross-talk to coordinate the formation and expansion of the thymus. A balanced interplay among all three is essential for the normal specification and expansion of the thymic tissue. Variations in the functions of any one of these stromal cell populations will impact the other, which likely explains the overlapping clinical phenotypes noted among affected individuals.
Animal work described in this article has been approved and conducted under the oversight of the UT Southwestern Institutional Animal Care and Use Committee (APN numbers: 2015-101163 and 2015-101247).
PB and NO generated the figures. PB, CW, and NO wrote and modified the manuscript.
Our work was supported, in part, by grants from the National Institutes of Health R01 (R01 AI114523, R21 AI144140 NO), Beecherl funds from the Department of Immunology at UT Southwestern Medical Center (NO), and the Jeffery Modell Foundation (CW).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Ms. Fatma Coskun and Mr. Austin Thompson for critically reviewing the manuscript, the figures, and for helpful discussions (UT Southwestern Medical Center). We also thanks Drs. Maria Teresa de la Morena (Seattle Children's Hospital, Seattle, WA) and M. Louise Markert (Duke University) for ongoing information and feedback concerning clinical presentations for patients with 22q11.2del and FOXN1 mutations.
1. Kwan A, Abraham RS, Currier R, Brower A, Andruszewski K, Abbott JK, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA. (2014) 312:729–38. doi: 10.1001/jama.2014.9132
2. Amatuni GS, Currier RJ, Church JA, Bishop T, Grimbacher E, Nguyen A, et al. Newborn screening for severe combined immunodeficiency and T-cell Lymphopenia in California, 2010–2017. Pediatrics. (2019) 143:e20182300. doi: 10.1542/peds.2018-2300
3. Gruver AL, Sempowski GD. Cytokines, leptin, and stress-induced thymic atrophy. J Leukocyte Biol. (2008) 84:915–23. doi: 10.1189/jlb.0108025
4. Belkaya S, Silge RL, Hoover AR, Medeiros JJ, Eitson JL, Becker AM, et al. Dynamic modulation of thymic microRNAs in response to stress. PLoS ONE. (2011) 6:e27580. doi: 10.1371/journal.pone.0027580
5. Chaudhry MS, Velardi E, Dudakov JA, Van Den Brink MRM. Thymus: the next (re)generation. Immunol Rev. (2016) 271:56–71. doi: 10.1111/imr.12418
6. Gui J, Mustachio L, Su D-M, Craig RW. Thymus size and age-related thymic involution: early programming, sexual dimorphism, progenitors and stroma. Aging Dis. (2012) 3:280−90.
7. Palmer D. The effect of age on thymic function. Front Immunol. (2013) 4:316. doi: 10.3389/fimmu.2013.00316
8. Du Q, De La Morena MT, Van Oers NS. The genetics and epigenetics of 22q11.2 deletion syndromes. Front Genet. (2020) 10:1365. doi: 10.3389/fgene.2019.01365
9. Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet. (2003) 362:1366–73. doi: 10.1016/s0140-6736(03)14632-6
10. Zweier C, Sticht H, Aydin-Yaylagul I, Campbell CE, Rauch A. Human TBX1 missense mutations cause gain of function resulting in the same phenotype as 22q11.2 deletions. Am J Hum Genet. (2007) 80:510–7. doi: 10.1086/511993
11. Gennery AR, Slatter MA, Rice J, Hoefsloot LH, Barge D, Mclean-Tooke A, et al. Mutations in CHD7 in patients with CHARGE syndrome cause T–B + natural killer cell + severe combined immune deficiency and may cause Omenn-like syndrome. Clin Exp Immunol. (2008) 153:75–80. doi: 10.1111/j.1365-2249.2008.03681.x
12. Markert ML, Marques JG, Neven B, Devlin BH, Mccarthy EA, Chinn IK, et al. First use of thymus transplantation therapy for FOXN1 deficiency (nude/SCID): a report of 2 cases. Blood. (2011) 117:688–96. doi: 10.1182/blood-2010-06-292490
13. Pohl E, Aykut A, Beleggia F, Karaca E, Durmaz B, Keupp K, et al. A hypofunctional PAX1 mutation causes autosomal recessively inherited otofaciocervical syndrome. Hum Genet. (2013) 132:1311–20. doi: 10.1007/s00439-013-1337-9
14. Morsheimer M, Brown Whitehorn TF, Heimall J, Sullivan KE. The immune deficiency of chromosome 22q11.2 deletion syndrome. Am J Med Genet A. (2017) 173:2366–72. doi: 10.1002/ajmg.a.38319
15. Paganini I, Sestini R, Capone GL, Putignano AL, Contini E, Giotti I, et al. A novel PAX1 null homozygous mutation in autosomal recessive otofaciocervical syndrome associated with severe combined immunodeficiency. Clin Genet. (2017) 92:664–8. doi: 10.1111/cge.13085
16. Liu N, Schoch K, Luo X, Pena LDM, Bhavana VH, Kukolich MK, et al. Functional variants in TBX2 are associated with a syndromic cardiovascular and skeletal developmental disorder. Hum Mol Genet. (2018) 27:2454–65. doi: 10.1093/hmg/ddy146
17. Yamazaki Y, Urrutia R, Franco LM, Giliani S, Zhang K, Alazami AM, et al. PAX1 is essential for development and function of the human thymus. Sci Immunol. (2020) 5:eaax1036. doi: 10.1126/sciimmunol.aax1036
18. Lammer EJ, Chen DT, Hoar RM, Agnish ND, Benke PJ, Braun JT, et al. Retinoic acid embryopathy. N Engl J Med. (1985) 313:837–41. doi: 10.1056/NEJM198510033131401
19. Novak RW, Robinson HB. Coincident DiGeorge anomaly and renal agenesis and its relation to maternal diabetes. Am J Med Genet. (1994) 50:311–2. doi: 10.1002/ajmg.1320500402
20. Digilio MC, Marino B, Formigari R, Giannotti A. Maternal diabetes causing DiGeorge anomaly and renal agenesis. Am J Med Genet. (1995) 55:513–4. doi: 10.1002/ajmg.1320550427
21. Mulder GB, Manley N, Maggio-Price L. Retinoic acid-induced thymic abnormalities in the mouse are associated with altered pharyngeal morphology, thymocyte maturation defects, and altered expression of Hoxa3 and Pax1. Teratology. (1998) 58:263–75. doi: 10.1002/(sici)1096-9926(199812)58:6<263::Aid-tera8>3.0.Co;2-a
22. Du Q, Huynh LK, Coskun F, Molina E, King MA, Raj P, et al. FOXN1 compound heterozygous mutations cause selective thymic hypoplasia in humans. J Clin Invest. (2019) 129:4724–38. doi: 10.1172/JCI127565
23. Gordon J, Manley NR. Mechanisms of thymus organogenesis and morphogenesis. Development. (2011) 138:3865–78. doi: 10.1242/dev.059998
24. Xu PX, Zheng W, Laclef C, Maire P, Maas RL, Peters H, et al. Eya1 is required for the morphogenesis of mammalian thymus, parathyroid and thyroid. Development. (2002) 129:3033–44.
25. Farley AM, Morris LX, Vroegindeweij E, Depreter ML, Vaidya H, Stenhouse FH, et al. Dynamics of thymus organogenesis and colonization in early human development. Development. (2013) 140:2015–26. doi: 10.1242/dev.087320
26. Gordon J, Patel SR, Mishina Y, Manley NR. Evidence for an early role for BMP4 signaling in thymus and parathyroid morphogenesis. Dev Biol. (2010) 339:141–54. doi: 10.1016/j.ydbio.2009.12.026
27. Moore-Scott BA, Manley NR. Differential expression of Sonic hedgehog along the anterior–posterior axis regulates patterning of pharyngeal pouch endoderm and pharyngeal endoderm-derived organs. Dev Biol. (2005) 278:323–35. doi: 10.1016/j.ydbio.2004.10.027
28. Griffith AV, Cardenas K, Carter C, Gordon J, Iberg A, Engleka K, et al. Increased thymus- and decreased parathyroid-fated organ domains in Splotch mutant embryos. Dev Biol. (2009) 327:216–27. doi: 10.1016/j.ydbio.2008.12.019
29. Wallin J, Eibel H, Neubuser A, Wilting J, Koseki H, Balling R. Pax1 is expressed during development of the thymus epithelium and is required for normal T-cell maturation. Development. (1996) 122:23–30.
30. Su D-M, Manley NR. Hoxa3 and Pax1 transcription factors regulate the ability of fetal thymic epithelial cells to promote thymocyte development. J Immunol. (2000) 164:5753–60. doi: 10.4049/jimmunol.164.11.5753
31. Auerbach R. Morphogenetic interactions in the development of the mouse thymus gland. Dev Biol. (1960) 2:271–84. doi: 10.1016/0012-1606(60)90009-9
32. Jenkinson WE, Jenkinson EJ, Anderson G. Differential requirement for mesenchyme in the proliferation and maturation of thymic epithelial progenitors. J Exp Med. (2003) 198:325–32. doi: 10.1084/jem.20022135
33. Jenkinson WE, Rossi SW, Parnell SM, Jenkinson EJ, Anderson G. PDGFRalpha-expressing mesenchyme regulates thymus growth and the availability of intrathymic niches. Blood. (2007) 109:954–60. doi: 10.1182/blood-2006-05-023143
34. Chen L, Zhao P, Wells L, Amemiya CT, Condie BG, Manley NR. Mouse and zebrafish Hoxa3 orthologues have non-equivalent in vivo protein function. Proc Natl Acad Sci. (2010) 107:10555–60. doi: 10.1073/pnas.1005129107
35. Frank DU, Fotheringham LK, Brewer JA, Muglia LJ, Tristani-Firouzi M, Capecchi MR, et al. An Fgf8 mouse mutant phenocopies human 22q11 deletion syndrome. Development. (2002) 129:4591–603.
36. Itoi M, Tsukamoto N, Yoshida H, Amagai T. Mesenchymal cells are required for functional development of thymic epithelial cells. Int Immunol. (2007) 19:953–64. doi: 10.1093/intimm/dxm060
37. Sitnik KM, Kotarsky K, White AJ, Jenkinson WE, Anderson G, Agace WW. Mesenchymal cells regulate retinoic acid receptor-dependent cortical thymic epithelial cell homeostasis. J Immunol. (2012) 188:4801–9. doi: 10.4049/jimmunol.1200358
38. Bleul CC, Boehm T. BMP signaling is required for normal thymus development. J Immunol. (2005) 175:5213–21. doi: 10.4049/jimmunol.175.8.5213
39. Itoi M, Kawamoto H, Katsura Y, Amagai T. Two distinct steps of immigration of hematopoietic progenitors into the early thymus anlage. Int Immunol. (2001) 13:1203–11. doi: 10.1093/intimm/13.9.1203
40. Jenkinson WE, Bacon A, White AJ, Anderson G, Jenkinson EJ. An epithelial progenitor pool regulates thymus growth. J Immunol. (2008) 181:6101–8. doi: 10.4049/jimmunol.181.9.6101
41. Luis TC, Luc S, Mizukami T, Boukarabila H, Thongjuea S, Woll PS, et al. Initial seeding of the embryonic thymus by immune-restricted lympho-myeloid progenitors. Nat Immunol. (2016) 17:1424–35. doi: 10.1038/ni.3576
42. Le Douarin NM, Jotereau FV. Tracing of cells of the avian thymus through embryonic life in interspecific chimeras. J Exp Med. (1975) 142:17–40. doi: 10.1084/jem.142.1.17
43. Klug DB, Carter C, Gimenez-Conti IB, Richie ER. Cutting Edge: thymocyte-independent and thymocyte-dependent phases of epithelial patterning in the fetal thymus. J Immunol. (2002) 169:2842–5. doi: 10.4049/jimmunol.169.6.2842
44. Anderson G, Takahama Y. Thymic epithelial cells: working class heroes for T cell development and repertoire selection. Trends Immunol. (2012) 33:256–63. doi: 10.1016/j.it.2012.03.005
45. Vaidya HJ, Briones Leon A, Blackburn CC. FOXN1 in thymus organogenesis and development. Eur J Immunol. (2016) 46:1826–37. doi: 10.1002/eji.201545814
46. Abramson J, Anderson G. Thymic epithelial cells. Annu Rev Immunol. (2017) 35:85–118. doi: 10.1146/annurev-immunol-051116-052320
47. Hikosaka Y, Nitta T, Ohigashi I, Yano K, Ishimaru N, Hayashi Y, et al. The cytokine RANKL produced by positively selected thymocytes fosters medullary thymic epithelial cells that express autoimmune regulator. Immunity. (2008) 29:438–50. doi: 10.1016/j.immuni.2008.06.018
48. Satoh R, Kakugawa K, Yasuda T, Yoshida H, Sibilia M, Katsura Y, et al. Requirement of Stat3 signaling in the postnatal development of thymic medullary epithelial cells. PLoS Genet. (2016) 12:e5776. doi: 10.1371/journal.pgen.1005776
49. Wu W, Shi Y, Xia H, Chai Q, Jin C, Ren B, et al. Epithelial LTbetaR signaling controls the population size of the progenitors of medullary thymic epithelial cells in neonatal mice. Sci Rep. (2017) 7:44481. doi: 10.1038/srep44481
50. Park SH, Bae YM, Kim TJ, Ha IS, Kim S, Chi JG, et al. HLA-DR expression in human fetal thymocytes. Hum Immunol. (1992) 33:294–8. doi: 10.1016/0198-8859(92)90338-n
51. Choi EY, Jung KC, Park HJ, Chung DH, Song JS, Yang SD, et al. Thymocyte-thymocyte interaction for efficient positive selection and maturation of CD4 T cells. Immunity. (2005) 23:387–96. doi: 10.1016/j.immuni.2005.09.005
52. Li W, Kim MG, Gourley TS, Mccarthy BP, Sant'angelo DB, Chang CH. An alternate pathway for CD4 T cell development: thymocyte-expressed MHC class II selects a distinct T cell population. Immunity. (2005) 23:375–86. doi: 10.1016/j.immuni.2005.09.002
53. Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don't see). Nat Rev Immunol. (2014) 14:377–91. doi: 10.1038/nri3667
54. Takada K, Kondo K, Takahama Y. Generation of peptides that promote positive selection in the thymus. J Immunol. (2017) 198:2215–22. doi: 10.4049/jimmunol.1601862
55. Morrow BE, Mcdonald-Mcginn DM, Emanuel BS, Vermeesch JR, Scambler PJ. Molecular genetics of 22q11.2 deletion syndrome. Am J Med Genet A. (2018) 176:2070–81. doi: 10.1002/ajmg.a.40504
56. Sullivan KE. Chromosome 22q11.2 deletion syndrome and DiGeorge syndrome. Immunol Rev. (2019) 287:186–201. doi: 10.1111/imr.12701
57. Zinkstok JR, Boot E, Bassett AS, Hiroi N, Butcher NJ, Vingerhoets C, et al. Neurobiological perspective of 22q11.2 deletion syndrome. Lancet Psychiatry. (2019) 6:951–60. doi: 10.1016/S2215-0366(19)30076-8
58. Mcdonald-Mcginn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JA, et al. 22q11.2 deletion syndrome. Nat Rev Dis Primers. (2015) 1:15071. doi: 10.1038/nrdp.2015.71
59. Merico D, Costain G, Butcher NJ, Warnica W, Ogura L, Alfred SE, et al. MicroRNA dysregulation, gene networks, and risk for schizophrenia in 22q11.2 deletion syndrome. Front Neurol. (2014) 5:238. doi: 10.3389/fneur.2014.00238
60. Guna A, Butcher NJ, Bassett AS. Comparative mapping of the 22q11.2 deletion region and the potential of simple model organisms. J Neurodev Disord. (2015) 7:18. doi: 10.1186/s11689-015-9113-x
61. Meechan DW, Maynard TM, Tucker ES, Fernandez A, Karpinski BA, Rothblat LA, et al. Modeling a model: mouse genetics, 22q11.2 deletion syndrome, and disorders of cortical circuit development. Prog Neurobiol. (2015) 130:1–28. doi: 10.1016/j.pneurobio.2015.03.004
62. Botto LD, May K, Fernhoff PM, Correa A, Coleman K, Rasmussen SA, et al. A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics. (2003) 112:101–7. doi: 10.1542/peds.112.1.101
63. Oskarsdottir S, Vujic M, Fasth A. Incidence and prevalence of the 22q11 deletion syndrome: a population-based study in Western Sweden. Arch Dis Child. (2004) 89:148–51. doi: 10.1136/adc.2003.026880
64. Sullivan KE. The clinical, immunological, and molecular spectrum of chromosome 22q11.2 deletion syndrome and DiGeorge syndrome. Curr Opin Allergy Clin Immunol. 4:505–12. doi: 10.1097/00130832-200412000-00006
65. Douek DC, Mcfarland RD, Keiser PH, Gage EA, Massey JM, Haynes BF, et al. Changes in thymic function with age and during the treatment of HIV infection. Nature. (1998) 396:690–5. doi: 10.1038/25374
66. Ferrando-Martínez S, Lorente R, Gurbindo D, José M, Leal M, Muñoz-Fernández M, et al. Low thymic output, peripheral homeostasis deregulation, and hastened regulatory T cells differentiation in children with 22q11.2 deletion syndrome. J Pediatrics. (2014) 164:882–9. doi: 10.1016/j.jpeds.2013.12.013
67. Markert ML, Boeck A, Hale LP, Kloster AL, Mclaughlin TM, Batchvarova MN, et al. Transplantation of thymus tissue in complete DiGeorge syndrome. N Engl J Med. (1999) 341:1180–9. doi: 10.1056/NEJM199910143411603
68. Markert ML, Sarzotti M, Ozaki DA, Sempowski GD, Rhein ME, Hale LP, et al. Thymus transplantation in complete DiGeorge syndrome: immunologic and safety evaluations in 12 patients. Blood. (2003) 102:1121–30. doi: 10.1182/blood-2002-08-2545
69. Chinn IK, Milner JD, Scheinberg P, Douek DC, Markert ML. Thymus transplantation restores the repertoires of forkhead box protein 3 (FoxP3)+ and FoxP3- T cells in complete DiGeorge anomaly. Clin Exp Immunol. (2013) 173:140–9. doi: 10.1111/cei.12088
70. Davies EG. Immunodeficiency in DiGeorge syndrome and options for treating cases with complete athymia. Front Immunol. (2013) 4:322. doi: 10.3389/fimmu.2013.00322
71. Kernfeld EM, Genga RMJ, Neherin K, Magaletta ME, Xu P, Maehr R. A single-cell transcriptomic atlas of thymus organogenesis resolves cell types and developmental maturation. Immunity. (2018) 48:1258–70.e1256. doi: 10.1016/j.immuni.2018.04.015
72. Markert ML, Devlin BH, Alexieff MJ, Li J, Mccarthy EA, Gupton SE, et al. Review of 54 patients with complete DiGeorge anomaly enrolled in protocols for thymus transplantation: outcome of 44 consecutive transplants. Blood. (2007) 109:4539–47. doi: 10.1182/blood-2006-10-048652
73. Markert ML, Devlin BH, Mccarthy EA. Thymus transplantation. Clin Immunol. (2010) 135:236–46. doi: 10.1016/j.clim.2010.02.007
74. Takaki T, Hosaka N, Miyake T, Cui W, Nishida T, Inaba M, et al. Presence of donor-derived thymic epithelial cells in [B6 → MRL/lpr] mice after allogeneic intra-bone marrow–bone marrow transplantation (IBM–BMT). J Autoimmunity. (2008) 31:408–15. doi: 10.1016/j.jaut.2008.09.003
75. Lee YJ, Jeon YK, Kang BH, Chung DH, Park CG, Shin HY, et al. Generation of PLZF+ CD4+ T cells via MHC class II-dependent thymocyte-thymocyte interaction is a physiological process in humans. J Exp Med. (2010) 207:237–46. doi: 10.1084/jem.20091519
76. Chinn IK, Markert ML. Induction of tolerance to parental parathyroid grafts using allogeneic thymus tissue in patients with DiGeorge anomaly. J Allergy Clin Immunol. (2011) 127:1351–5. doi: 10.1016/j.jaci.2011.03.033
77. Janda A, Sedlacek P, Hönig M, Friedrich W, Champagne M, Matsumoto T, et al. Multicenter survey on the outcome of transplantation of hematopoietic cells in patients with the complete form of DiGeorge anomaly. Blood. (2010) 116:2229–36. doi: 10.1182/blood-2010-03-275966
78. Kojima D, Muramatsu H, Okuno Y, Kataoka S, Murakami N, Tanahashi Y, et al. Successful T-cell reconstitution after unrelated cord blood transplantation in a patient with complete DiGeorge syndrome. J Allergy Clin Immunol. (2016) 138:1471–3.e1474. doi: 10.1016/j.jaci.2016.04.048
79. Goldsobel AB, Haas A, Stiehm ER. Bone marrow transplantation in DiGeorge syndrome. J Pediatr. (1987) 111:40–4. doi: 10.1016/s0022-3476(87)80339-6
80. Matsumoto T, Amamoto N, Kondoh T, Nakayama M, Takayanagi T, Tsuji Y. Complete-type DiGeorge syndrome treated by bone marrow transplantation. Bone Marrow Transpl. (1998) 22:927–30. doi: 10.1038/sj.bmt.1701475
81. Land MH, Garcia-Lloret MI, Borzy MS, Rao PN, Aziz N, Mcghee SA, et al. Long-term results of bone marrow transplantation in complete DiGeorge syndrome. J Allergy Clin Immunol. (2007) 120:908–15. doi: 10.1016/j.jaci.2007.08.048
82. Markert ML. Treatment of infants with complete DiGeorge anomaly. J Allergy Clin Immunol. (2008) 121:1063. doi: 10.1016/j.jaci.2007.12.1181
83. Marcovecchio GE, Bortolomai I, Ferrua F, Fontana E, Imberti L, Conforti E, et al. Thymic epithelium abnormalities in DiGeorge and down syndrome patients contribute to dysregulation in T cell development. Front Immunol. (2019) 10:447. doi: 10.3389/fimmu.2019.00447
84. Montin D, Marolda A, Licciardi F, Robasto F, Di Cesare S, Ricotti E, et al. Immunophenotype anomalies predict the development of autoimmune cytopenia in 22q11.2 deletion syndrome. J Allergy Clin Immunol. (2019) 7:2369–76. doi: 10.1016/j.jaip.2019.03.014
85. Mcdonald-Mcginn DM, Reilly A, Wallgren-Pettersson C, Hoyme HE, Yang SP, Adam MP, et al. Malignancy in chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Am J Med Genet A. (2006) 140:906–9. doi: 10.1002/ajmg.a.31199
86. Piliero LM, Sanford AN, Mcdonald-Mcginn DM, Zackai EH, Sullivan KE. T-cell homeostasis in humans with thymic hypoplasia due to chromosome 22q11.2 deletion syndrome. Blood. (2004) 103:1020–5. doi: 10.1182/blood-2003-08-2824
87. Staple L, Andrews T, Mcdonald-Mcginn D, Zackai E, Sullivan KE. Allergies in patients with chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome) and patients with chronic granulomatous disease. Pediatr Allergy Immunol. (2005) 16:226–30. doi: 10.1111/j.1399-3038.2005.00259.x
88. Kanaya Y, Ohga S, Ikeda K, Furuno K, Ohno T, Takada H, et al. Maturational alterations of peripheral T cell subsets and cytokine gene expression in 22q11.2 deletion syndrome. Clin Exp Immunol. (2006) 144:85–93. doi: 10.1111/j.1365-2249.2006.03038.x
89. Zemble R, Luning Prak E, Mcdonald K, Mcdonald-Mcginn D, Zackai E, Sullivan K. Secondary immunologic consequences in chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Clin Immunol. (2010) 136:409–18. doi: 10.1016/j.clim.2010.04.011
90. Lindsay EA, Botta A, Jurecic V, Carattini-Rivera S, Cheah YC, Rosenblatt HM, et al. Congenital heart disease in mice deficient for the DiGeorge syndrome region. Nature. (1999) 401:379–83. doi: 10.1038/43900
91. Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, et al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. (2001) 104:619–29. doi: 10.1016/s0092-8674(01)00247-1
92. Digeorge A. Congenital Absence of the thymus and its immunological consequences: concurrence with congenital hypoparathyroidism. Birth Defects Orig ArtSerIV. (1968) 1:116–21.
93. Puech A, Saint-Jore B, Merscher S, Russell RG, Cherif D, Sirotkin H, et al. Normal cardiovascular development in mice deficient for 16 genes in 550 kb of the velocardiofacial/DiGeorge syndrome region. Proc Natl Acad Sci USA. (2000) 97:10090–5. doi: 10.1073/pnas.97.18.10090
94. Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. (2001) 410:97–101. doi: 10.1038/35065105
95. Liao J, Kochilas L, Nowotschin S, Arnold JS, Aggarwal VS, Epstein JA, et al. Full spectrum of malformations in velo-cardio-facial syndrome/DiGeorge syndrome mouse models by altering Tbx1 dosage. Hum Mol Genet. (2004) 13:1577–85. doi: 10.1093/hmg/ddh176
96. Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. (2001) 27:286–91. doi: 10.1038/85845
97. Fulcoli FG, Franzese M, Liu X, Zhang Z, Angelini C, Baldini A. Rebalancing gene haploinsufficiency in vivo by targeting chromatin. Nat Commun. (2016) 7:11688. doi: 10.1038/ncomms11688
98. Zhao Y, Diacou A, Johnston HR, Musfee FI, Mcdonald-Mcginn DM, Mcginn D, et al. Complete sequence of the 22q11.2 allele in 1,053 subjects with 22q11.2 deletion syndrome reveals modifiers of conotruncal heart defects. Am J Hum Genet. (2020) 106:26–40. doi: 10.1016/j.ajhg.2019.11.010
99. De La Morena MT, Eitson JL, Dozmorov IM, Belkaya S, Hoover AR, Anguiano E, et al. Signature MicroRNA expression patterns identified in humans with 22q11.2 deletion/DiGeorge syndrome. Clin Immunol. (2013) 147:11–22. doi: 10.1016/j.clim.2013.01.011
100. Kimber WL, Hsieh P, Hirotsune S, Yuva-Paylor L, Sutherland HF, Chen A, et al. Deletion of 150 kb in the minimal DiGeorge/velocardiofacial syndrome critical region in mouse. Hum Mol Genet. (1999) 8:2229–37. doi: 10.1093/hmg/8.12.2229
101. Zhang Z, Baldini A. In vivo response to high-resolution variation of Tbx1 mRNA dosage. Hum Mol Genet. (2008) 17:150–7. doi: 10.1093/hmg/ddm291
102. Lindsay EA, Baldini A. Recovery from arterial growth delay reduces penetrance of cardiovascular defects in mice deleted for the DiGeorge syndrome region. Hum Mol Genet. (2001) 10:997–1002. doi: 10.1093/hmg/10.9.997
103. Reeh KA, Cardenas KT, Bain VE, Liu Z, Laurent M, Manley NR, et al. Ectopic TBX1 suppresses thymic epithelial cell differentiation and proliferation during thymus organogenesis. Development. (2014) 141:2950–8. doi: 10.1242/dev.111641
104. Alfano D, Altomonte A, Cortes C, Bilio M, Kelly RG, Baldini A. Tbx1 regulates extracellular matrix-cell interactions in the second heart field. Hum Mol Genet. (2019) 28:2295–308. doi: 10.1093/hmg/ddz058
105. Bergman JE, Janssen N, Van Der Sloot AM, De Walle HE, Schoots J, Rendtorff ND, et al. A novel classification system to predict the pathogenic effects of CHD7 missense variants in CHARGE syndrome. Hum Mutat. (2012) 33:1251–60. doi: 10.1002/humu.22106
106. Wong MTY, Schölvinck EH, Lambeck AJA, Van Ravenswaaij-Arts CMA. CHARGE syndrome: a review of the immunological aspects. Eur J Hum Genet. (2015) 23:1451–9. doi: 10.1038/ejhg.2015.7
107. Van Ravenswaaij-Arts C, Martin DM. New insights and advances in CHARGE syndrome: diagnosis, etiologies, treatments, and research discoveries. Am J Med Genet C. (2017) 175:397–406. doi: 10.1002/ajmg.c.31592
108. Bouazoune K, Kingston RE. Chromatin remodeling by the CHD7 protein is impaired by mutations that cause human developmental disorders. Proc Natl Acad Sci USA. (2012) 109:19238–43. doi: 10.1073/pnas.1213825109
109. Zentner GE, Hurd EA, Schnetz MP, Handoko L, Wang C, Wang Z, et al. CHD7 functions in the nucleolus as a positive regulator of ribosomal RNA biogenesis. Hum Mol Genet. (2010) 19:3491–501. doi: 10.1093/hmg/ddq265
110. Zentner GE, Layman WS, Martin DM, Scacheri PC. Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am J Med Genet A. (2010) 152A:674–86. doi: 10.1002/ajmg.a.33323
111. Jyonouchi S, Mcdonald-Mcginn DM, Bale S, Zackai EH, Sullivan KE. CHARGE (Coloboma, Heart Defect, Atresia Choanae, Retarded Growth and Development, Genital Hypoplasia, Ear Anomalies/Deafness) syndrome and chromosome 22q11.2 deletion syndrome: a comparison of immunologic and non-immunologic phenotypic features. Pediatrics. (2009) 123:e871–7. doi: 10.1542/peds.2008-3400
112. Wong MTY, Lambeck AJA, Van Der Burg M, La Bastide-Van Gemert S, Hogendorf LA, Van Ravenswaaij-Arts CMA, et al. Immune dysfunction in children with CHARGE syndrome: a cross-sectional study. PLoS ONE. (2015) 10:e0142350. doi: 10.1371/journal.pone.0142350
113. Sanlaville D, Etchevers HC, Gonzales M, Martinovic J, Clément-Ziza M, Delezoide A-L, et al. Phenotypic spectrum of CHARGE syndrome in fetuses with CHD7 truncating mutations correlates with expression during human development. J Med Genet. (2006) 43:211–317. doi: 10.1136/jmg.2005.036160
114. Bajpai R, Chen DA, Rada-Iglesias A, Zhang J, Xiong Y, Helms J, et al. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature. (2010) 463:958–62. doi: 10.1038/nature08733
115. Bosman EA, Penn AC, Ambrose JC, Kettleborough R, Stemple DL, Steel KP. Multiple mutations in mouse Chd7 provide models for CHARGE syndrome. Hum Mol Genet. (2005) 14:3463–76. doi: 10.1093/hmg/ddi375
116. Hurd EA, Poucher HK, Cheng K, Raphael Y, Martin DM. The ATP-dependent chromatin remodeling enzyme CHD7 regulates pro-neural gene expression and neurogenesis in the inner ear. Development. (2010) 137:3139–50. doi: 10.1242/dev.047894
117. Ogier JM, Carpinelli MR, Arhatari BD, Symons RCA, Kile BT, Burt RA. CHD7 deficiency in “looper,” a new mouse model of CHARGE syndrome, results in ossicle malformation, otosclerosis and hearing impairment. PLoS ONE. (2014) 9:e97559. doi: 10.1371/journal.pone.0097559
118. Gage PJ, Hurd EA, Martin DM. Mouse models for the dissection of CHD7 functions in eye development and the molecular basis for ocular defects in CHARGE syndrome. Invest Ophthalmol Vis Sci. (2015) 56:7923–30. doi: 10.1167/iovs.15-18069
119. Randall V, Mccue K, Roberts C, Kyriakopoulou V, Beddow S, Barrett AN, et al. Great vessel development requires biallelic expression of Chd7 and Tbx1 in pharyngeal ectoderm in mice. J Clin Invest. (2009) 119:3301–10. doi: 10.1172/JCI37561
120. Micucci JA, Layman WS, Hurd EA, Sperry ED, Frank SF, Durham MA, et al. CHD7 and retinoic acid signaling cooperate to regulate neural stem cell and inner ear development in mouse models of CHARGE syndrome. Hum Mol Genet. (2013) 23:434–48. doi: 10.1093/hmg/ddt435
121. Yao H, Hill SF, Skidmore JM, Sperry ED, Swiderski DL, Sanchez GJ, et al. CHD7 represses the retinoic acid synthesis enzyme ALDH1A3 during inner ear development. JCI Insight. (2018) 3:e97440. doi: 10.1172/jci.insight.97440
122. Liu Z-Z, Wang Z-L, Choi T-I, Huang W-T, Wang H-T, Han Y-Y, et al. Chd7 is critical for early T-cell development and thymus organogenesis in zebrafish. Am J Pathol. (2018) 188:1043–58. doi: 10.1016/j.ajpath.2017.12.005
123. Pignata C, Fiore M, Guzzetta V, Castaldo A, Sebastio G, Porta F, et al. Congenital Alopecia and nail dystrophy associated with severe functional T-cell immunodeficiency in two sibs. Am J Med Genet. (1996) 65:167–70. doi: 10.1002/(SICI)1096-8628(19961016)65:2<167::AID-AJMG17>3.0.CO;2-O
124. Li B, Li J, Devlin BH, Markert ML. Thymic microenvironment reconstitution after postnatal human thymus transplantation. Clin Immunol. (2011) 140:244–59. doi: 10.1016/j.clim.2011.04.004
125. Vigliano I, Gorrese M, Fusco A, Vitiello L, Amorosi S, Panico L, et al. FOXN1 mutation abrogates prenatal T-cell development in humans. J Med Genet. (2011) 48:413–6. doi: 10.1136/jmg.2011.089532
126. Chou J, Massaad MJ, Wakim RH, Bainter W, Dbaibo G, Geha RS. A novel mutation in FOXN1 resulting in SCID: a case report and literature review. Clin Immunol. (2014) 155:30–2. doi: 10.1016/j.clim.2014.08.005
127. Radha Rama Devi A, Panday NN, Naushad SM. FOXN1 Italian founder mutation in Indian family: implications in prenatal diagnosis. Gene. (2017) 627:222–5. doi: 10.1016/j.gene.2017.06.033
128. Bosticardo M, Yamazaki Y, Cowan J, Giardino G, Corsino C, Scalia G, et al. Heterozygous FOXN1 variants cause low TRECs and severe T cell lymphopenia, revealing a crucial role of FOXN1 in supporting early thymopoiesis. Am J Hum Genet. (2019) 105:549–61. doi: 10.1016/j.ajhg.2019.07.014
129. Dixit VD. Thymic fatness and approaches to enhance thymopoietic fitness in aging. Curr Opin Immunol. (2010) 22:521–8. doi: 10.1016/j.coi.2010.06.010
130. Nehls M, Pfeifer D, Schorpp M, Hedrich H, Boehm T. New member of the winged-helix protein family disrupted in mouse and rat nude mutations. Nature. (1994) 372:103–7. doi: 10.1038/372103a0
131. Romano R, Palamaro L, Fusco A, Giardino G, Gallo V, Del Vecchio L, et al. FOXN1: a master regulator gene of thymic epithelial development program. Front Immunol. (2013) 4:e187. doi: 10.3389/fimmu.2013.00187
132. Žuklys S, Handel A, Zhanybekova S, Govani F, Keller M, Maio S, et al. Foxn1 regulates key target genes essential for T cell development in postnatal thymic epithelial cells. Nat Immunol. (2016) 17:1206–15. doi: 10.1038/ni.3537
133. Schüddekopf K, Schorpp M, Boehm T. The whn transcription factor encoded by the nude locus contains an evolutionarily conserved and functionally indispensable activation domain. Proc Natl Acad Sci USA. (1996) 93:9661–4. doi: 10.1073/pnas.93.18.9661
134. Schlake T, Schorpp M, Nehls M, Boehm T. The nude gene encodes a sequence-specific DNA binding protein with homologs in organisms that lack an anticipatory immune system. Proc Natl Acad Sci USA. (1997) 94:3842–7. doi: 10.1073/pnas.94.8.3842
135. Newman JA, Aitkenhead H, Gravard A, Rota IA, Handel AE, Hollander GA, et al. The structural basis for forkhead box family specificity revealed by the crystal structure of human FOXN1 in complex with DNA. bioRxiv. (2018) 428011. doi: 10.1101/428011
136. Uddin MM, Ohigashi I, Motosugi R, Nakayama T, Sakata M, Hamazaki J, et al. Foxn1-beta5t transcriptional axis controls CD8(+) T-cell production in the thymus. Nat Commun. (2017) 8:14419. doi: 10.1038/ncomms14419
137. Schorpp M, Schlake T, Kreamalmeyer D, Allen PM, Boehm T. Genetically separable determinants of hair keratin gene expression. Dev Dyn. (2000) 218:537–43. doi: 10.1002/1097-0177(200007)218:3<537::AID-DVDY1007>3.0.CO;2-P
138. Potter CS, Pruett ND, Kern MJ, Baybo MA, Godwin AR, Potter KA, et al. The nude mutant gene Foxn1 is a HOXC13 regulatory target during hair follicle and nail differentiation. J Invest Dermatol. (2011) 131:828–37. doi: 10.1038/jid.2010.391
139. Flanagan SP. “Nude,” a new hairless gene with pleiotropic effects in the mouse. Genet Res. (1966) 8:295–309.
140. Wortis HH, Nehlsen S, Owen JJ. Abnormal development of the thymus in “nude” mice. J Exp Med. (1971) 134:681–92.
141. Nehls M, Kyewski B, Messerle M, Waldschütz R, Schüddekopf K, Smith AJH, et al. Two genetically separable steps in the differentiation of thymic epithelium. Science. (1996) 272:886–9. doi: 10.1126/science.272.5263.886
142. Blackburn CC, Augustine CL, Li R, Harvey RP, Malin MA, Boyd RL, et al. The nu gene acts cell-autonomously and is required for differentiation of thymic epithelial progenitors. Proc Natl Acad Sci USA. (1996) 93:5742–6.
143. Mori K, Itoi M, Tsukamoto N, Amagai T. Foxn1 is essential for vascularization of the murine thymus anlage. Cell Immunol. (2010) 260:66–9. doi: 10.1016/j.cellimm.2009.09.007
144. Festing MF, May D, Connors TA, Lovell D, Sparrow S. An athymic nude mutation in the rat. Nature. (1978) 274:365–6. doi: 10.1038/274365a0
145. Schuurman H-J, Hougen HP, Van Loveren H. The rnu (Rowett Nude) and rnuN (nznu, New Zealand Nude) Rat: an update. ILAR J. (1992) 34:3–12. doi: 10.1093/ilar.34.1-2.3
146. Abitbol M, Bosse P, Thomas A, Tiret L. A deletion in FOXN1 is associated with a syndrome characterized by congenital hypotrichosis and short life expectancy in Birman cats. PLoS ONE. (2015) 10:e0120668. doi: 10.1371/journal.pone.0120668
147. Su D-M, Navarre S, Oh W-J, Condie BG, Manley NR. A domain of Foxn1 required for crosstalk-dependent thymic epithelial cell differentiation. Nat Immunol. (2003) 4:983. doi: 10.1038/ni983
148. Swann JB, Weyn A, Nagakubo D, Bleul CC, Toyoda A, Happe C, et al. Conversion of the thymus into a bipotent lymphoid organ by replacement of FOXN1 with its paralog, FOXN4. Cell Rep. (2014) 8:1184–97. doi: 10.1016/j.celrep.2014.07.017
149. Larsen BM, Cowan JE, Wang Y, Tanaka Y, Zhao Y, Voisin B, et al. Identification of an intronic regulatory element necessary for tissue-specific expression of Foxn1 in thymic epithelial cells. J Immunol. (2019) 203:686–95. doi: 10.4049/jimmunol.1801540
150. Handel AE, Holländer GA. Comment on “Identification of an intronic regulatory element necessary for tissue-specific expression of Foxn1 in thymic epithelial cells.” J Immunol. (2019) 203:2355. doi: 10.4049/jimmunol.1900948
151. Chen L, Xiao S, Manley NR. Foxn1 is required to maintain the postnatal thymic microenvironment in a dosage-sensitive manner. Blood. (2009) 113:567–74. doi: 10.1182/blood-2008-05-156265
152. Zook EC, Krishack PA, Zhang S, Zeleznik-Le NJ, Firulli AB, Witte PL, et al. Overexpression of Foxn1 attenuates age-associated thymic involution and prevents the expansion of peripheral CD4 memory T cells. Blood. (2011) 118:5723–31. doi: 10.1182/blood-2011-03-342097
153. Reis MDDS, Csomos K, Dias LPB, Prodan Z, Szerafin T, Savino W, et al. Decline of FOXN1 gene expression in human thymus correlates with age: possible epigenetic regulation. Immunity Ageing. (2015) 12:18–18. doi: 10.1186/s12979-015-0045-9
154. Bredenkamp N, Nowell CS, Blackburn CC. Regeneration of the aged thymus by a single transcription factor. Development. (2014) 141:1627–37. doi: 10.1242/dev.103614
155. Song Y, Su M, Zhu J, Di W, Liu Y, Hu R, et al. FOXN1 recombinant protein enhances T-cell regeneration after hematopoietic stem cell transplantation in mice. Eur J Immunol. (2016) 46:1518–28. doi: 10.1002/eji.201546196
156. Wright ME. Undulated: a new genetic factor in mus musculus affecting the spine and tail. Heredity. (1947) 1:137–41. doi: 10.1038/hdy.1947.10
157. Peters H, Wilm B, Sakai N, Imai K, Maas R, Balling R. Pax1 and Pax9 synergistically regulate vertebral column development. Development. (1999) 126:5399–408.
158. Peters H, Neubüser A, Kratochwil K, Balling R. Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev. (1998) 12:2735–47. doi: 10.1101/gad.12.17.2735
159. Lee W-C, Yamaguchi T, Watanabe C, Kawaguchi A, Takeda M, Kim Y-I, et al. Association of common PAX9 variants with permanent tooth size variation in non-syndromic East Asian populations. J Hum Genet. (2012) 57:654–9. doi: 10.1038/jhg.2012.90
160. Kucera J. Rate and type of congenital anomalies among offspring of diabetic women. J Reprod Med. (1971) 7:73–82.
161. Mills JL. Malformations in infants of diabetic mothers. Teratology. (2010) 25:385–94. doi: 10.1002/bdra.20757
162. Sacks DA, Hadden DR, Maresh M, Deerochanawong C, Dyer AR, Metzger BE, et al. Frequency of gestational diabetes mellitus at collaborating centers based on IADPSG consensus panel-recommended criteria: the Hyperglycemia and Adverse Pregnancy Outcome (HAPO) Study. Diabetes Care. (2012) 35:526–8. doi: 10.2337/dc11-1641
163. Dornemann R, Koch R, Mollmann U, Falkenberg MK, Mollers M, Klockenbusch W, et al. Fetal thymus size in pregnant women with diabetic diseases. J Perinat Med. (2017) 45:595–601. doi: 10.1515/jpm-2016-0400
164. Padmanabhan R, Shafiullah M. Effect of maternal diabetes and ethanol interactions on embryo development in the mouse. Mol Cell Biochem. (2004) 261:43–56. doi: 10.1023/b:mcbi.0000028736.00532.1e
165. Lee LM, Leung MB, Kwok RC, Leung YC, Wang CC, Mccaffery PJ, et al. Perturbation of retinoid homeostasis increases malformation risk in embryos exposed to pregestational diabetes. Diabetes. (2017) 66:1041–51. doi: 10.2337/db15-1570
166. Niederreither K, Vermot J, Le Roux I, Schuhbaur B, Chambon P, Dolle P. The regional pattern of retinoic acid synthesis by RALDH2 is essential for the development of posterior pharyngeal arches and the enteric nervous system. Development. (2003) 130:2525–34. doi: 10.1242/dev.00463
167. Vermot J, Niederreither K, Garnier JM, Chambon P, Dolle P. Decreased embryonic retinoic acid synthesis results in a DiGeorge syndrome phenotype in newborn mice. Proc Natl Acad Sci USA. (2003) 100:1763–8. doi: 10.1073/pnas.0437920100
168. Cipollone D, Amati F, Carsetti R, Placidi S, Biancolella M, D'amati G, et al. A multiple retinoic acid antagonist induces conotruncal anomalies, including transposition of the great arteries, in mice. Cardiovasc Pathol. (2006) 15:194–202. doi: 10.1016/j.carpath.2006.04.004
169. Voss AK, Vanyai HK, Collin C, Dixon MP, Mclennan TJ, Sheikh BN, et al. MOZ regulates the Tbx1 locus, and Moz mutation partially phenocopies DiGeorge syndrome. Dev Cell. (2012) 23:652–63. doi: 10.1016/j.devcel.2012.07.010
170. Rhinn M, Dollé P. Retinoic acid signalling during development. Development. (2012) 139:843–58. doi: 10.1242/dev.065938
171. Roberts C, Ivins SM, James CT, Scambler PJ. Retinoic acid down-regulates Tbx1 expression in vivo and in vitro. Dev Dyn. (2005) 232:928–38. doi: 10.1002/dvdy.20268
172. Ryckebusch L, Bertrand N, Mesbah K, Bajolle F, Niederreither K, Kelly RG, et al. Decreased levels of embryonic retinoic acid synthesis accelerate recovery from arterial growth delay in a mouse model of DiGeorge syndrome. Circ Res. (2010) 106:686–94. doi: 10.1161/CIRCRESAHA.109.205732
173. Smith CL, Tallquist MD. PDGF function in diverse neural crest cell populations. Cell Adh Migr. (2010) 4:561–6. doi: 10.4161/cam.4.4.12829
174. Thompson DL, Gerlach-Bank LM, Barald KF, Koenig RJ. Retinoic acid repression of bone morphogenetic protein 4 in inner ear development. Mol Cell Biol. (2003) 23:2277–86. doi: 10.1128/MCB.23.7.2277-2286.2003
175. Lipson AH, Collins F, Webster WS. Multiple congenital defects associated with maternal use of topical tretinoin. Lancet. (1993) 341:1352–3. doi: 10.1016/0140-6736(93)90868-h
Keywords: thymus development, thymic hypoplasia, TECs, mesenchymal cells, 22q11.2 deletion syndrome, PAX1, FOXN1, CHD7
Citation: Bhalla P, Wysocki CA and van Oers NSC (2020) Molecular Insights Into the Causes of Human Thymic Hypoplasia With Animal Models. Front. Immunol. 11:830. doi: 10.3389/fimmu.2020.00830
Received: 28 February 2020; Accepted: 14 April 2020;
Published: 05 May 2020.
Edited by:
Ichiro Taniuchi, RIKEN Center for Integrative Medical Sciences (IMS), JapanReviewed by:
Takeshi Nitta, The University of Tokyo, JapanCopyright © 2020 Bhalla, Wysocki and van Oers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicolai S. C. van Oers, bmljb2xhaS52YW5vZXJzQHV0c291dGh3ZXN0ZXJuLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.