 Jamie van Langelaar
Jamie van Langelaar Liza Rijvers
Liza Rijvers Joost Smolders
Joost Smolders Marvin M. van Luijn
Marvin M. van Luijn- 1Department of Immunology, MS Center ErasMS, Erasmus MC, University Medical Center, Rotterdam, Netherlands
- 2Department of Neurology, MS Center ErasMS, Erasmus MC, University Medical Center, Rotterdam, Netherlands
- 3Neuroimmunology Research Group, Netherlands Institute for Neuroscience, Amsterdam, Netherlands
Historically, multiple sclerosis (MS) has been viewed as being primarily driven by T cells. However, the effective use of anti-CD20 treatment now also reveals an important role for B cells in MS patients. The results from this treatment put forward T-cell activation rather than antibody production by B cells as a driving force behind MS. The main question of how their interaction provokes both B and T cells to infiltrate the CNS and cause local pathology remains to be answered. In this review, we highlight key pathogenic events involving B and T cells that most likely contribute to the pathogenesis of MS. These include (1) peripheral escape of B cells from T cell-mediated control, (2) interaction of pathogenic B and T cells in secondary lymph nodes, and (3) reactivation of B and T cells accumulating in the CNS. We will focus on the functional programs of CNS-infiltrating lymphocyte subsets in MS patients and discuss how these are defined by mechanisms such as antigen presentation, co-stimulation and cytokine production in the periphery. Furthermore, the potential impact of genetic variants and viral triggers on candidate subsets will be debated in the context of MS.
Introduction
In multiple sclerosis (MS) patients, pathogenic lymphocytes are triggered in the periphery to infiltrate the central nervous system (CNS) and cause local inflammation and demyelination. Anti-CD20 therapy has recently been approved as a novel treatment modality for MS (1–3). Although this underscores the fact that B cells play a key role in MS, the exact triggers, subsets and effector mechanisms contributing to the disease course are incompletely understood. The impact of this therapy on the antigen-presenting rather than the antibody-producing function of B cells in MS indicates that their interaction with T cells is an important driver of the pathogenesis (1, 4). Alterations in cytokine production, co-stimulation and antigen presentation most likely contribute to the development of pathogenic B and T cells that are prone to enter the CNS (4, 5). Such mechanisms might be influenced by the interplay between genetic and environmental risk factors (6). The major HLA-DRB1∗1501 locus accounts for 30% of the overall risk (6) and has been shown to promote B cell-mediated induction of brain-infiltrating T helper (Th) cells in MS patients (4). Besides for HLA-DRB1∗1501, other genetic risk variants that have been identified in the past decades also appear to potentiate B and Th cell activation, a feature that is shared amongst several autoimmune disorders (7). Furthermore, infectious triggers such as the Epstein-Barr virus (EBV) alter their function and reactivity in MS (5, 6, 8, 9). The current view is that transmigration of lymphocyte subsets into the CNS signifies relapsing disease, while compartmentalized CNS inflammation, as seen during disease progression, seems to be driven by tissue-resident populations (10, 11). Since there is a clear association of relapse occurrence and radiological disease activity early in MS with the severity of disability progression later in MS (12), it is crucial to understand what motivates these cells to invade the CNS and why these cells instigate local pathology in MS patients.
In this review, we will discuss which and how brain-infiltrating lymphocyte subsets can contribute to MS pathogenesis. These pathogenic events are characterized by: (1) peripheral escape of pathogenic B cells from T cell-mediated control, (2) mutual activation of pathogenic B and T cells within peripheral germinal centers, and (3) re-activation of infiltrating B and T cells within the CNS. We will use current knowledge to consider the extent to which genetic and viral triggers may drive these pathogenic events in MS.
Impaired T Cell-Mediated Control of Pathogenic B Cells in MS
B and T cells closely interact in secondary lymphoid organs to generate an optimal immune response against invading pathogens. Within follicles, B cells recognize antigens via the highly specific B-cell receptor (BCR), resulting in internalization, processing and presentation to T cells. This mechanism is unique and tightly coordinated involving five consecutive and interdependent steps: (1) B-cell receptor signaling, (2) actin remodeling, (3) endosomal formation and transport, (4) HLA class II synthesis and trafficking to specialized late endosomes (i.e., MIICs), and (5) antigen processing and loading onto HLA class II molecules for presentation to CD4+ Th cells (13, 14). Through their interaction with Th cells, germinal center (GC)-dependent and -independent memory B cells are formed, a process that is governed by the strength of the HLA/peptide signal (15). GC B cells respond to interleukin (IL)-21-producing follicular Th (Tfh) cells to develop into class-switched (IgG+) subsets or antibody-producing plasmablasts/plasma cells (15, 16). Memory B cells, in return, specifically trigger Th effector subsets that help CD8+ cytotoxic T cells (CTLs) to kill the infected cell (17). In MS, this crosstalk between B and T cells is likely disturbed, eventually causing pathogenic instead of protective immunity. This may already start during selection of naive autoreactive B cells in the periphery.
Normally, after removal of the majority of B-cell clones expressing polyreactive antibodies in the bone marrow (central tolerance), surviving autoreactive B cells are kept in check by peripheral tolerance checkpoints (18). In contrast to most other autoimmune diseases, only peripheral and not central B-cell tolerance checkpoints are defective in MS, which coincides with increased frequencies of naive polyreactive populations in the blood (18–21). Although the exact cause is currently unknown, the escape of pathogenic B cells from peripheral control may be related to (1) chronic T-cell stimulation and (2) T cell-intrinsic defects (see Figure 1).
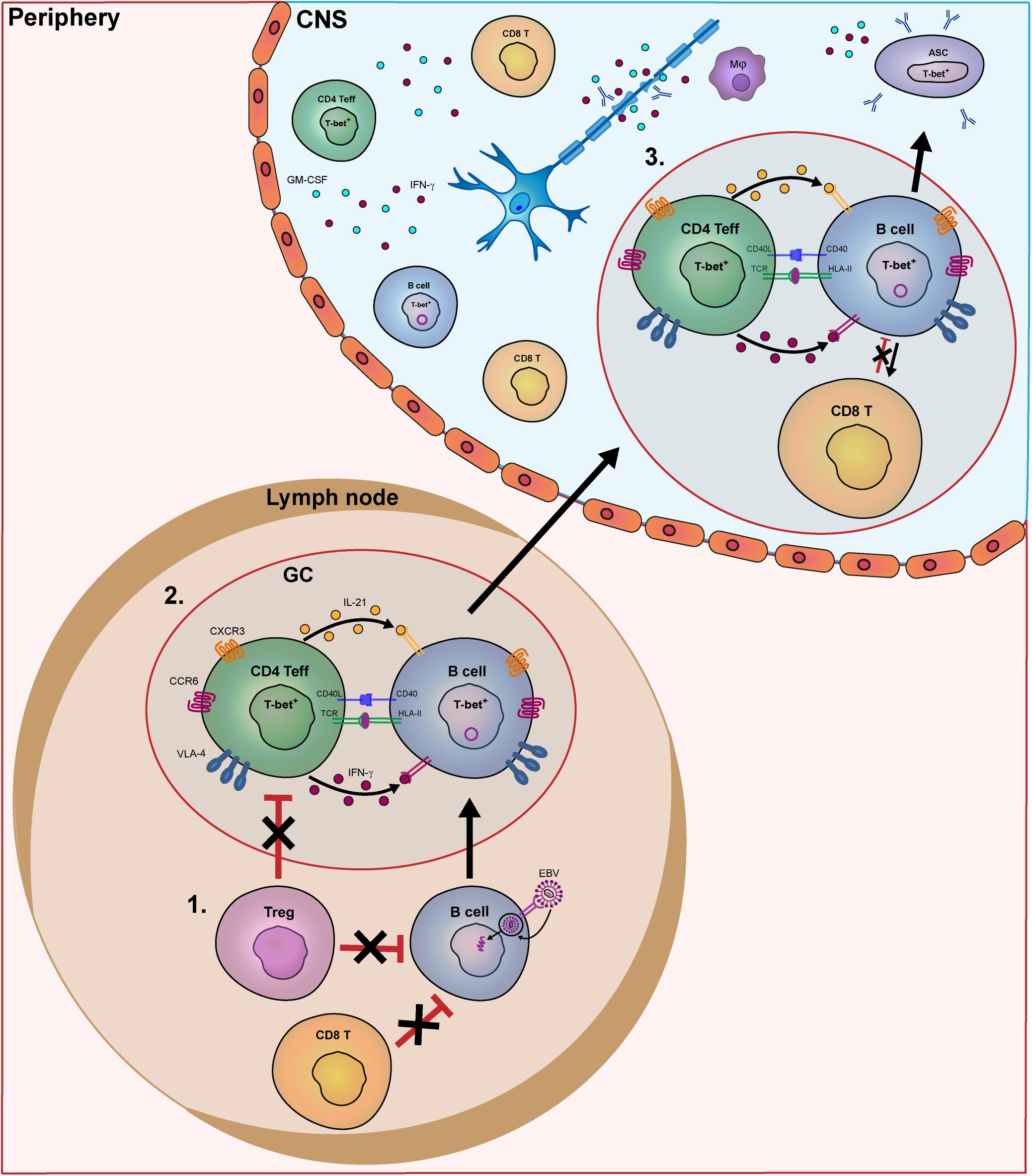
Figure 1. Model of the key pathogenic events involving human B- and T-cell subsets driving MS disease activity. In MS patients, B- and T-cells interact in the periphery and central nervous system (CNS) to contribute to disease pathogenesis. In this model, we put forward three important meeting points of pathogenic B and T cells that drive the disease course of MS. In secondary lymphoid organs, B-cell tolerance defects in MS patients allow EBV-infected B cells to escape from suppression by CD8+ and T regulatory (Treg) cells (1). Subsequently, these activated B cells enter germinal centers (GCs) and interact with follicular Th cells to further differentiate into pathogenic memory B cells. Under the influence of IFN-γ and IL-21, B cells develop into T-bet-expressing memory cells, which in turn activate Th effector cells such as Th17.1 (2). These subsets are prone for infiltrating the CNS of MS patients by distinct expression of chemokine receptors (CXCR3, CCR6), adhesion molecules (VLA-4) as well as pro-inflammatory cytokines. (3) Within the CNS, IFN-γ-, and GM-CSF-producing T cells and T-bet+ memory B cells probably come into contact in follicle-like structures, resulting in clonal expansion inflammation and demyelination. T-bet+ memory B cells further differentiate into plasmablasts/plasma cells to secrete high numbers of potentially harmful antibodies (oligoclonal bands).
Epstein-Barr virus is one of the most thoroughly investigated pathogens regarding T-cell responses in MS. Many theories have been proposed how EBV can influence MS pathogenesis (9). One hypothesis is that, due to the chronic nature of this infection, continuous antigen presentation by B cells leads to functionally impaired, so-called “exhausted” T cells (8, 22). This, together with the impact of HLA and other risk alleles (23), may result in inappropriate T cell-mediated control of EBV-infected (pathogenic) B cells. Consistent with this, peripheral CD8+ CTLs show decreased responses to EBV and not to cytomegalovirus antigens during the MS course (8). EBV antigens can also induce IL-10-producing CD4+ T regulatory cells (Tregs) capable of suppressing effector T-cell responses to recall antigens (24), as seen for other persistent viral infections such as lymphocytic choriomeningitis virus (25, 26). However, forkhead box P3 (FOXP3+) Tregs have also been described to control infections (27), suggesting that additional T cell-intrinsic defects are involved. For example, Treg populations that are enriched in MS patients produce increased levels of interferon gamma (IFN-γ), express reduced levels of FOXP3 and have defective suppressive activity in vitro (28). This is not only accompanied with less suppression of effector T cells (29, 30), but possibly also with impaired removal of pathogenic B cells, as described for other autoimmune diseases (18, 31, 32). The direct impact of Tregs on B cells in MS patients is still unknown. Treg function may be altered by variation in IL2RA and IL7RA, two known MS risk loci (33, 34). FOXP3 correlates with IL-2 receptor (IL-2R) as well as IL-7 receptor (IL-7R) expression in Tregs (35). It can thus be expected that IL2RA and IL7RA (33, 34), but also BACH2 (36) variants impair Treg development in MS. This may even influence FOXP3- and IL-2R-expressing CD8+ T cells, which can suppress pro-inflammatory CD4+ Th cells (37) and are reduced in the blood during MS relapses (38–40).
The Germinal Center as a Powerhouse of Pathogenic B- and TH-Cell Interaction in MS
Th Cells as Inducers of Pathogenic Memory B Cells
After their escape from peripheral tolerance checkpoints, naive B cells likely interact with Th cells in GCs to eventually develop into memory populations potentially capable of infiltrating the MS brain (Figure 1). Little is known about how peripheral effector Th cells mediate the development of such pathogenic B cells in MS patients. In GCs of autoimmune mice, autoreactive B cells are triggered by Tfh cells producing high levels of IFN-γ (16). IFN-γ induces the expression of the T-box transcription factor T-bet, which upregulates CXC chemokine receptor 3 (CXCR3), elicits IgG class switching and enhanced antiviral responsiveness of murine B cells (41–43). Recently, we found that B cells from MS patients preferentially develop into CXCR3+ populations that transmigrate into the CNS (44). The IFN-γ receptor (IFNGR) and downstream molecule signal transducer and activator of transcription (STAT)1 in B cells are major determinants of autoimmune GC formation in mice (45, 46). After ligation of the IFNGR, STAT1 is phosphorylated, dimerizes and translocates into the nucleus to induce genes involved in GC responses, such as T-bet and B-cell lymphoma 6 (BCL-6) (16, 47). Although IFN-γ-stimulated B cells of MS patients show enhanced pro-inflammatory capacity (44, 48), it is unclear whether alterations in the IFN-γ signaling pathway contribute to the development of T-bet+ B cells infiltrating the CNS. Interestingly, a missense SNP in IFNGR2 has been found in MS, which may alter their development (49, 50). Another target gene of the IFN-γ pathway is IFI30, which encodes for the IFN-γ-inducible lysosomal thiol reductase (GILT) and is considered one of the causal risk variants in MS (7). GILT is a critical regulator of antigen processing for presentation by HLA class II molecules (51–53). Together, these findings point to T-bet-expressing B cells as potent antigen-presenting cells that are highly susceptible to triggering by IFN-γ-producing Th effector subsets in MS (44, 54) (Figure 2).
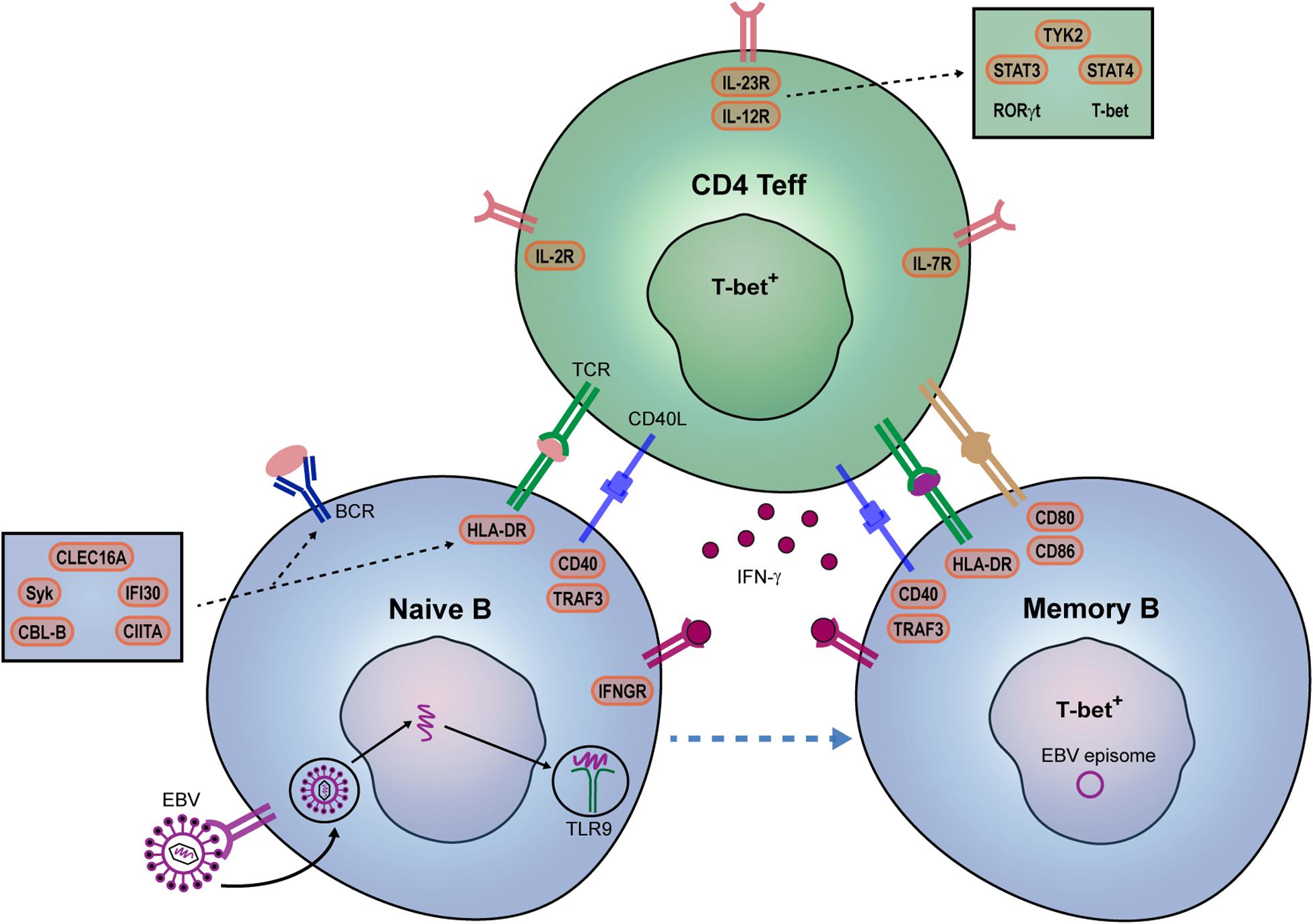
Figure 2. Potential contribution of EBV and genetic risk factors to pathogenic B- and Th-cell development in MS patients. IFN-γ is a key player in autoreactive B- and Th-cell interaction and autoimmune germinal center (GC) formation in mice. In MS, we propose that EBV infection together with specific genetic risk variants promote the IFN-γ-mediated interplay between B and T cells within GCs. EBV directly infects naive B cells and mimic GC responses. EBV DNA can also bind to TLR7/9, and together with IFN-γ, induces T-bet+ memory B cells. Their interplay may be additionally stimulated by both B cell-intrinsic (IFN-γ sensitivity: IFNGR2; B cell receptor-antigen uptake: CBLB, SYK, CLEC16A; HLA class II pathway: CLEC16A, CIITA, IFI30; co-stimulation: CD80, CD86) and Th cell-intrinsic (surface receptors: IL2RA, IL7RA, IL12RB1; downstream molecules: TYK2, STAT3, STAT4) genetic risk variants. IL12R/IL-23R complexes trigger JAK2/STAT3-dependent RORγt and TYK2/STAT4-dependent T-bet expression in Th effector cells.
Epstein-Barr virus may be an additional player in the formation of T-bet-expressing B cells. In mice, persistent viral infections sustain the development of these types of B cells, in which T-bet enhances their ability to recognize viral and self-antigens (41, 55). EBV is hypothesized to persist latently in pathogenic B cells and mimic T-cell help for further differentiation in GCs (5, 22, 56, 57). During acute infection, EBV uses a series of latency programs that drive B cells toward a GC response in an antigen-independent manner. Latent membrane protein (LMP)2A and LMP1 resemble signals coming from the BCR and CD40 receptor (56, 57). In addition to their regulation of GC responses independently of T-cell help (58), recent evidence implicates that LMP2A and LMP1 can synergize with BCR and CD40 signaling as well (59). Interestingly, downstream molecules of the BCR (e.g., Syk, CBL-B) and CD40 receptor (e.g., TRAF3) are genetic risk factors for MS (23, 60), therefore potentially cooperating with these latent proteins to enhance pathogenic B-cell development (Figure 2). This is supported by the binding of LMP2A to Syk in B cells and their escape from deletion in GCs of transgenic mice (61). Alternatively, pathogenic B cells can be induced via pathogen-associated TLR9, which binds to unmethylated CpG DNA and further integrate with BCR, CD40, and cytokine signals (62–65). Moreover, pathogenic B-cell responses in systemic autoimmune diseases such as systemic lupus erythematosus are enhanced after IFN-γ and virus-mediated induction of the T-bet (45, 55, 64, 65). In MS patients, TLR9 ligation is also a major trigger of pro-inflammatory B cells (48) and crucial for the differentiation of T-bet-expressing IgG1+ B cells during IFN-γ- and CD40-dependent GC-like cultures in vitro. Thus, under influence of specific genetic factors, EBV might join forces with IFN-γ-producing Th cells to stimulate pathogenic (T-bet+) GC B cells both in a direct (via infection and persistence in pathogenic subsets) and indirect (via TLR7/9) fashion in MS (Figure 2).
B Cells as Inducers of Pathogenic Memory TH Cells
Synchronously, within peripheral GCs, T-bet-expressing memory B cells are ideal candidates to trigger IFN-γ-producing, CNS-infiltrating Th cells in MS (Figure 1). In both mice and humans, T-bet promotes the antigen-presenting cell function of B cells. This may be related to the impact of EBV infection on B cell-intrinsic processing and presentation of antigens such as myelin oligodendrocyte glycoprotein (MOG) (5). The potent antigen-presenting cell function of B cells in MS patients is further reflected by the effective use of anti-CD20 therapy. This therapy does not affect antibody serum levels, but significantly reduces pro-inflammatory Th-cell responses in MS, both ex vivo and in vivo (1). CD20 was found to be enriched on IFN-γ-inducible T-bet-expressing IgG+ B cells in MS blood (44), pointing to this pathogenic subset as an important therapeutic target. Furthermore, genetic changes in HLA class II molecules, as well as costimulatory molecules [e.g., CD80 (66, 67) and CD86 (68)], may additionally enhance Th cell activation by such memory B cells (Figure 2). HLA class II expression on murine B cells was reported to be indispensable for EAE disease onset (69, 70). The in silico evidence that autoimmunity-associated HLA class II molecules have an altered peptide-binding groove (71, 72), together with the potential role of several minor risk variants in the HLA class II pathway [e.g., CIITA, CLEC16A, IFI30 (Figure 2)], insinuates that antigens are differently processed and presented by B cells (4, 5). This is supported by the increased ability of memory B cells to trigger CNS-infiltrating Th cells in MS patients carrying HLA-DRB1∗1501 (4). These CNS-infiltrating T cells induced by B cells showed features of both Th1 and Th17, therefore representing highly pathogenic subsets. Such subsets are characterized by master transcription factors T-bet and RORγt (73, 74), of which the latter is involved in the co-expression of IL-17 and GM-CSF in mice but not in humans (75, 76). GM-CSF is an emerging pro-inflammatory cytokine produced by Th cells in MS (33, 75, 77). Our group recently revealed that a Th subset producing high levels of IFN-γ and GM-CSF, but low levels of IL-17, termed Th17.1, plays a key role in driving early disease activity in MS patients (78). Proportions of Th17.1 cells were reduced in the blood and highly enriched in the CSF of rapid-onset MS patients. In addition, Th17.1 cells and not classical Th1 and Th17 cells accumulated in the blood of MS patients who clinically responded to natalizumab (anti-VLA-4 mAb). The increased pathogenicity of Th17.1 is further exemplified by their high levels of multidrug resistance, anti-apoptotic and cytotoxicity-associated genes ABCB1 (MDR1), FCMR (TOSO) and GZMB (granzyme B), respectively (78–81). Th17.1 cells also show pronounced expression of the IL-23 receptor (IL-23R) (78), which is essential for maintaining the pathogenicity of Th17 cells during CNS autoimmunity (82). IL-23 signals through the IL-23R and IL-12 receptor beta chain (IL-12Rβ1), resulting in JAK2-mediated STAT3 and TYK2-mediated STAT4 phosphorylation, and thereby inducing RORγt and T-bet, respectively (83). IL-12RB1, TYK2, STAT3, and STAT4 are known genetic risk variants and thus may directly induce Th effector cells in MS (Figure 2). In addition to its potential effect on Tregs (see above), MS-associated risk variant IL-2RA enhances GM-CSF production by human Th effector cells (33). To confirm the influence of these and other risk loci (84) on the induction of pathogenic Th cells such as Th17.1 in MS, functional studies need to be performed in the near future.
The increased pathogenicity of Th effector cells may additionally be skewed by IL-6-producing B cells (85, 86), which have been shown to trigger autoimmune GC formation and EAE in mice (87, 88). Blocking of IL-6 prevents the development of myelin-specific Th1 and Th17 cells in EAE (89). The IL-6-mediated resistance of pathogenic Th cells to Treg mediated suppression in MS (90, 91) further links to the abundant expression of anti-apoptotic gene FCMR in Th17.1 (78, 92). Intriguingly, B cell-derived GM-CSF can be an additional cytokine driving pathogenic Th cells in MS patients by inducing pro-inflammatory myeloid cells (93). Although the causal MS autoantigen is still unknown, previous work implies that B cell-mediated presentation of EBV antigens at least contributes to pathogenic Th-cell induction (5, 94). As mentioned above, antiviral CD8+ CTLs can become exhausted during persistent viral infections. Normally, this mechanism is compensated by the presence of cytotoxic CD4+ Th cells, which keep these types of infections under control (95). Such Th populations have been associated with MS progression (96) and are also formed after EBV infection, producing high levels of IFN-γ, IL-2, granzyme B, and perforin (97, 98). Similarly, EBV- and myelin-reactive Th cells from MS patients produce high levels of IFN-γ and IL-2 (6) and strongly respond to memory B cells presenting myelin peptides (99). These studies indicate that the involvement of EBV-infected B cells, especially those expressing T-bet (see section “Th Cells as Inducers of Pathogenic Memory B Cells”), in activating Th effector cells with cytotoxic potential (78, 100, 101) deserves further attention in MS.
Reactivation of CNS-Infiltrating B and T Cells in MS
Mechanisms of Infiltration
Under normal physiological conditions, the CNS has been considered an immune privileged environment and consists of a limited number of lymphocytes that cross the blood brain barrier (BBB) (102). However, the revelation of meningeal lymphatic structures emphasized the cross-talk between CNS and peripheral lymphocytes in secondary lymphoid organs (103). The choroid plexus has been identified as the main entry of memory cells into the CNS, which is in the case of T cells mostly mediated by CCR6 (104, 105). The normal human CSF, as is acquired from the arachnoid space by lumbar spinal taps, contains more CD4+ Th cells compared to CD8+ T cells with central memory characteristics (106–108). The arachnoid space is a continuum with the perivascular space surrounding penetrating arterial and venous structures into the parenchyma (109). Within the brain parenchyma, more CD8+ T cells than CD4+ Th cells are found, however, their numbers remain low and can be found virtually restricted to the perivascular space (11, 110). These T cells display a phenotype mostly associated with non-circulating tissue resident memory T cells. The perivenular perivascular space has been argued to be the common drainage site of antigens mobilized with the glymphatics flow (111). The exact relationship between memory T cells in the subarachnoid and perivascular space has been poorly identified in terms of replenishment and clonal association.
The BBB is dysfunctional during the early phase of MS, resulting in or is due to local recruitment of pathogenic T and B cells (112). Differential expression of pro-inflammatory cytokines, chemokine receptors and integrins by infiltrating lymphocytes have been argued to mediate disruption of the BBB in MS (104, 113). Myelin-reactive CCR6+ and not CCR6– memory Th cells from MS patients not only produce high levels of IL-17, but also IFN-γ and GM-CSF (80). Previous studies mainly focused on the migration of IL-17-producing CCR6+ Th cells through the choroid plexus in EAE and in vitro human brain endothelial cell layers in MS brain tissues (104, 114). In our recent study, we subdivided these CCR6+ memory Th cells into distinct Th17 subsets and found that especially IFN-γ producing Th17.1 (CCR6+CXCR3+CCR4–) cells were capable of infiltrating the CNS, both in ex vivo autopsied brain tissues and in in vitro transmigration assays (78). The fact that Th17.1 cells have cytotoxic potential and strongly co-express IFN-γ with GM-CSF (78) suggests that these cells are involved in disrupting the permeability of the BBB in MS (115, 116). The impact of CXCR3 on their transmigration capacity is likely the result of binding to the chemokine ligand CXCL10, which is produced by brain endothelial cells and is abundant in the CSF of MS patients (117, 118). Similar observations were made for CXCR3 (T-bet)+ B cells (44). CCR6 is also highly expressed on memory B-cell precursors within the Th cell-containing light zone of GCs (119), and on IFN-γ-producing CD8+ T cells infiltrating the MS brain (120). This implies that both populations are susceptible to enter the CNS of MS patients. In addition to chemokine receptors and pro-inflammatory cytokines, adhesion molecules such as activated leukocyte cell adhesion molecule (ALCAM) enhance transmigration of pathogenic B and T cell subsets (115, 121, 122). Furthermore, CXCR3 is co-expressed with integrin α4β1 (VLA-4), which allows both B- and T-cell populations to bind to vascular cell adhesion protein 1 (VCAM-1) on brain endothelial cells (123). This is supported by the reducing effects of VLA-4 inhibition on B- and Th17-cell infiltration into the CNS and disease susceptibility in EAE (124). Natalizumab, a monoclonal antibody against VLA-4, is used as an effective second-line treatment for MS (125). Discontinuation of this treatment often results in severe MS rebound effects (126). Hence, the peripheral entrapment of populations like Th17.1 and T-bet+ B cells in natalizumab-treated patients (44, 78) probably underlies the massive influx of blood cells causing these effects. The same is true for EBV-reactivated B cells, which are enriched in lesions from MS patients after natalizumab withdrawal (127). A previous gene network approach using several GWAS datasets further highlights the relevance of adhesion molecules on the BBB endothelium for the crossing of T and B cells (128), especially those affected by IFN-γ (115).
Local Organization and Impact
Both B and T cells accumulate in active white matter lesions of the MS brain (10, 129). In diagnostic biopsy studies, T cell-dominated inflammation is a characteristic of all lesion-types observed (130). Also in post-mortem MS lesions, white matter MS lesions with active demyelination associate with an increase in T cell numbers (10, 129). Although CD4+ Th cells are in general outnumbered by CD8+ CTLs in brain lesions as investigated in autopsy studies (10), their role as triggers of local pathology should not be overlooked in MS. This is consistent with the enrichment of CD4+ Th cells in white matter lesions with active demyelination (10). An abundant number of CD4+ Th cells were also visible in pre-active lesion sites, suggesting an involvement of these cells in the early stages of lesion formation (131). Additionally, it was demonstrated that in contrast to CD8+ CTLs, brain-associated CD4+ Th-cell clonotypes are reduced in MS blood, indicating specific recruitment (as described above) or, alternatively, clonal expansion in the CNS (132). Furthermore, dominant Th-cell clones were undetectable following reconstitution after autologous hematopoietic stem cell transplantation in MS patients, which was not seen for CD8+ T cells (133). Interestingly, T-cell clones are shared between CNS compartments within a patient, including CSF and anatomically separated brain lesions (132, 134–137). This suggests that brain-infiltrating T cells bear similar reactivity against local (auto)antigens.
In subsets of MS autopsy cases with acute and relapsing remitting MS, B cells can also be found predominantly in the perivascular space in association with active white matter lesions (10). The role of these perivascular B cells, including T-bet+ B cells (44), could be to re-activate (infiltrating) pro-inflammatory CD4+ and CD8+ T cells to cause MS pathology (Figure 1). Identical B-cell clones have been found in different CNS compartments of MS patients, including the meninges (138, 139). Within the meninges, B- and T cell-rich follicle-like structures have been found that localize next to cortical lesions, presumably mediating progressive loss of neurological function in MS (140, 141). Interestingly, MS brain-infiltrating lymphocytes express and respond to IL-21 (142), the cytokine that drives follicular T- and B-cell responses. Additionally, IFN-γ triggering of B cells promotes ectopic follicle formation in autoimmune mice (16, 45), suggesting that the structures observed in the MS CNS are induced by B cells interacting with IFN-γ-producing T cells. However, the role of IL-17 in this process should not be ruled out, as shown in EAE (143).
Besides mediating migration and organization of pathogenic lymphocytes in the MS brain, cytokines are likely relevant effector molecules. IFN-γ production by Th cells also associates with the presence of demyelinating lesions in the CNS (144–146). IFN-γ, and possibly also GM-CSF, can activate microglia or infiltrated macrophages to cause damage to oligodendrocytes (93, 147, 148). As for B cells, increased production of TNF-α, IL-6, and GM-CSF has been found (48, 87) and we have recently shown that during Tfh-like cultures, IFN-γ drives IgG-producing plasmablasts in MS (44). One could speculate that after their re-activation by IFN-γ-producing Th cells within the meningeal follicles, T-bet+ memory B cells rapidly develop into antibody-producing plasmablasts/plasma cells (Figure 1). IFN-γ-induced GC formation promotes the generation of autoantibodies in lupus mice (16, 45). The targeting of B cells and not plasmablasts/plasma cells by clinically effective anti-CD20 therapies in MS, as well as the abundance of oligoclonal bands in MS CSF, at least support the local differentiation of B cells into antibody-secreting cells (48, 149). We argue that IgG secreted by local T-bet-expressing plasmablasts/plasma cells are highly reactive in the MS brain (43, 44, 55), although the (auto)antigen specificity and pathogenicity of such antibodies remain unclear in MS, as well as their contribution as effector molecules to MS pathology.
Several antigenic targets have been proposed to contribute to MS pathology. Next to myelin, which is one of the most intensively studied antigens (150), also EBV antigens are considered as major candidates. EBNA-1 specific IgG antibodies are predictive for early disease activity (151) and are present in CSF from MS patients (152, 153). Some studies imply that reactivated B cells in ectopic meningeal follicles (154, 155) cross-present EBV peptides to activate myelin- and EBNA-1 specific Th cells (6, 156, 157). Whether EBV is detected in the brain or solely recognized in the periphery and how this contributes to local pathology is still a matter of intense debate in the field (127, 158–162). In addition to myelin (150) and EBV (6), other antigenic targets of locally produced IgG and infiltrating T cells have been suggested, such as sperm-associated antigen 16 [SPAG16 (163)], neurofilament light, RAS guanyl-releasing protein 2 [RASGRP2 (4)], αB-crystallin and GDP-l-fucose synthase (135).
Concluding Remarks
In this review, we have discussed potential triggers and mechanisms through which interacting B and T cells drive the pathogenesis of MS. In our presented model, peripheral B cells escape from tolerance checkpoints as the result of impaired control by chronically exhausted or genetically altered regulatory T cells. Subsequently, B cells interact with IFN-γ-producing effector Th cells in germinal centers of lymphoid organs to create a feedforward loop, after which highly pathogenic subsets break through blood-CNS barriers and, together with infiltrating CD8+ CTLs are locally reactivated to cause MS pathology. Although definite proof is still lacking, these pathogenic events are likely mediated by an interplay between persistent infections such as EBV and genetic risk variants. Together, these factors may alter the selection, differentiation and pathogenic features of B- and T-cell subsets. In our view, more in-depth insights into how infections and genetic burden define the CNS-infiltrating potential and antigen specificity of such subsets should be the next step to take in the near future. The development of small molecule therapeutics against subsets driving the disease course would be an effective way of generating clinically relevant benefits without harmful effects in MS patients.
Author Contributions
JL, LR, and ML designed and wrote the manuscript. ML and JS revised the manuscript.
Conflict of Interest
JS received speaker/consultancy fee from Biogen, Merck, Novartis, and Sanofi-Genzyme.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to dedicate this article to the memory of Prof. Rogier Q. Hintzen, who passed away on May 15, 2019. The research that he instigated will be further developed in our MS Center with the same drive and passion as he did.
References
1. Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-cell depletion with rituximab in relapsing–remitting multiple sclerosis. N Engl J Med. (2008) 358:676–88. doi: 10.1056/NEJMoa0706383
2. Bar-Or A, Calabresi PA, Arnold D, Markowitz C, Shafer S, Kasper LH, et al. Rituximab in relapsing-remitting multiple sclerosis: a 72-week, open-label, phase I trial. Ann Neurol. (2008) 63:395–400. doi: 10.1002/ana.21363
3. Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung, H-P Hemmer, B, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med. (2016) 376:221–34. doi: 10.1056/NEJMoa1601277
4. Jelcic I, Al Nimer F, Wang J, Lentsch V, Planas R, Jelcic I, et al. Memory B cells activate brain-homing, autoreactive CD4+ T cells in multiple sclerosis. Cell. (2018) 175:85–100.e23. doi: 10.1016/j.cell.2018.08.011
5. Morandi E, Jagessar SA, ‘t Hart BA, and Gran, B. EBV infection empowers human B cells for autoimmunity: role of autophagy and relevance to multiple sclerosis. J Immunol. (2017) 199:435–48. doi: 10.4049/jimmunol.1700178
6. Lunemann JD, Jelcic I, Roberts S, Lutterotti A, Tackenberg B, Martin R, et al. EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-gamma and IL-2. J Exp Med. (2008) 205:1763–73. doi: 10.1084/jem.20072397
7. Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. (2015) 518:337–43. doi: 10.1038/nature13835
8. Pender MP, Csurhes PA, Burrows JM, Burrows SR. Defective T-cell control of Epstein-Barr virus infection in multiple sclerosis. Clin Transl Immunol. (2017) 6:e126. doi: 10.1038/cti.2016.87
9. Bar-Or A, Pender MP, Khanna R, Steinman L, Hartung, H-P Maniar, T, et al. Epstein–Barr virus in multiple sclerosis: theory and emerging immunotherapies. Trends Mol Med. (2020) 26:296–310. doi: 10.1016/j.molmed.2019.11.003
10. Machado-Santos J, Saji E, Troscher AR, Paunovic M, Liblau R, Gabriely G, et al. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8+ T lymphocytes and B cells. Brain. (2018) 141:2066–82. doi: 10.1093/brain/awy151
11. Smolders J, Heutinck KM, Fransen NL, Remmerswaal EBM, Hombrink P, ten Berge IJM, et al. Tissue-resident memory T cells populate the human brain. Nat Commun. (2018) 9:4593. doi: 10.1038/s41467-018-07053-9
12. Rotstein D, Montalban X. Reaching an evidence-based prognosis for personalized treatment of multiple sclerosis. Nat Rev Neurol. (2019) 15:287–300. doi: 10.1038/s41582-019-0170-8
13. Yuseff, M-I, and Lennon-Duménil AM. B cells use conserved polarity cues to regulate their antigen processing and presentation functions. Front Immunol. (2015) 6:251. doi: 10.3389/fimmu.2015.00251
14. Flora C, Ronald NG. Cooperation between CD4+ and CD8+ T cells: when where, and how. Annu Rev Immunol. (2006) 24:519–40. doi: 10.1146/annurev.immunol.23.021704.115825
15. Kurosaki T, Kometani K, Ise W. Memory B cells. Nat Rev Immunol. (2015) 15:149. doi: 10.1038/nri3802
16. Rawlings DJ, Metzler G, Wray-Dutra M, Jackson SW. Altered B cell signalling in autoimmunity. Nat Rev Immunol. (2017) 17:421–36. doi: 10.1038/nri.2017.24
17. Swain SL, McKinstry KK, Strutt TM. Expanding roles for CD4? T cells in immunity to viruses. Nat Rev Immunol. (2012) 12:136–48. doi: 10.1038/nri3152
18. Kinnunen T, Chamberlain N, Morbach H, Cantaert T, Lynch M, Preston-Hurlburt P, et al. Specific peripheral B cell tolerance defects in patients with multiple sclerosis. J Clin Invest. (2013) 123:2737–41. doi: 10.1172/JCI68775
19. Samuels J, Ng YS, Coupillaud C, Paget D, Meffre E. Impaired early B cell tolerance in patients with rheumatoid arthritis. J Exp Med. (2005) 201:1659–67. doi: 10.1084/jem.20042321
20. Menard L, Saadoun D, Isnardi I, Ng YS, Meyers G, Massad C, et al. The PTPN22 allele encoding an R620W variant interferes with the removal of developing autoreactive B cells in humans. J Clin Invest. (2011) 121:3635–44. doi: 10.1172/JCI45790
21. Cotzomi E, Stathopoulos P, Lee CS, Ritchie AM, Soltys JN, Delmotte FR, et al. Early B cell tolerance defects in neuromyelitis optica favour anti-AQP4 autoantibody production. Brain. (2019) 142:1598–615. doi: 10.1093/brain/awz106
22. Pender MP. Infection of autoreactive B lymphocytes with EBV, causing chronic autoimmune diseases. Trends Immunol. (2003) 24:584–8. doi: 10.1016/j.it.2003.09.005
23. The International Multiple Sclerosis Genetics Consortium, The Wellcome Trust Case Control Consortium Sawcer, S Hellenthal, G Pirinen, M Spencer, CC, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. (2011) 476:214–9. doi: 10.1038/nature10251
24. Marshall NA, Vickers MA, Barker RN. Regulatory T cells secreting IL-10 dominate the immune response to EBV latent membrane protein 1. J Immunol. (2003) 170:6183–9. doi: 10.4049/jimmunol.170.12.6183
25. Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, Oldstone MB. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med. (2006) 12:1301–9. doi: 10.1038/nm1492
26. Blackburn SD, Wherry EJ. IL-10, T cell exhaustion and viral persistence. Trends Microbiol. (2007) 15:143–6. doi: 10.1016/j.tim.2007.02.006
27. Voo KS, Peng G, Guo Z, Fu T, Li Y, Frappier L, et al. Functional characterization of EBV-encoded nuclear antigen 1-specific CD4+ helper and regulatory T cells elicited by in vitro peptide stimulation. Cancer Res. (2005) 65:1577–86. doi: 10.1158/0008-5472.CAN-04-2552
28. Dominguez-Villar M, Baecher-Allan CM, Hafler DA. Identification of T helper type 1-like, Foxp3+ regulatory T cells in human autoimmune disease. Nat Med. (2011) 17:673–5. doi: 10.1038/nm.2389
29. Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. (2004) 199:971–9. doi: 10.1084/jem.20031579
30. Kumar M, Putzki N, Limmroth V, Remus R, Lindemann M, Knop D, et al. CD4+CD25+FoxP3+ T lymphocytes fail to suppress myelin basic protein-induced proliferation in patients with multiple sclerosis. J Neuroimmunol. (2006) 180:178–84. doi: 10.1016/j.jneuroim.2006.08.003
31. Venken K, Hellings N, Broekmans T, Hensen K, Rummens JL, Stinissen P. Natural naive CD4+CD25+CD127low regulatory T cell. (Treg) development and function are disturbed in multiple sclerosis patients: recovery of memory Treg homeostasis during disease progression. J Immunol. (2008) 180:6411–20. doi: 10.4049/jimmunol.180.9.6411
32. Kinnunen T, Chamberlain N, Morbach H, Choi J, Kim S, Craft J, et al. Accumulation of peripheral autoreactive B cells in the absence of functional human regulatory T cells. Blood. (2013) 121:1595–603. doi: 10.1182/blood-2012-09-457465
33. Hartmann FJ, Khademi M, Aram J, Ammann S, Kockum I, Constantinescu C, et al. Multiple sclerosis-associated IL2RA polymorphism controls GM-CSF production in human TH cells. Nat Commun. (2014) 5:5056. doi: 10.1038/ncomms6056
34. Kreft KL, Verbraak E, Wierenga-Wolf AF, van Meurs M, Oostra BA, Laman JD, et al. Decreased systemic IL-7 and soluble IL-7Rα in multiple sclerosis patients. Genes Immunity. (2012) 13:587. doi: 10.1038/gene.2012.34
35. Liu W, Putnam AL, Xu-yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. (2006) 203:1701–11. doi: 10.1084/jem.20060772
36. Roychoudhuri R, Hirahara K, Mousavi K, Clever D, Klebanoff CA, Bonelli M, et al. BACH2 represses effector programs to stabilize T(reg)-mediated immune homeostasis. Nature. (2013) 498:506–10. doi: 10.1038/nature12199
37. Correale J, Villa A. Role of CD8+ CD25+ Foxp3+ regulatory T cells in multiple sclerosis. Ann Neurol. (2010) 67:625–38. doi: 10.1002/ana.21944
38. Baughman EJ, Mendoza JP, Ortega SB, Ayers CL, Greenberg BM, Frohman EM, et al. Neuroantigen-specific CD8+ regulatory T-cell function is deficient during acute exacerbation of multiple sclerosis. J Autoimmun. (2011) 36:115–24. doi: 10.1016/j.jaut.2010.12.003
39. Correale J, Villa A. Isolation and characterization of CD8+ regulatory T cells in multiple sclerosis. J Neuroimmunol. (2008) 195:121–34. doi: 10.1016/j.jneuroim.2007.12.004
40. Frisullo G, Nociti V, Iorio R, Plantone D, Patanella AK, Tonali PA, et al. CD8(+)Foxp3(+) T cells in peripheral blood of relapsing-remitting multiple sclerosis patients. Hum Immunol. (2010) 71:437–41. doi: 10.1016/j.humimm.2010.01.024
41. Barnett BE, Staupe RP, Odorizzi PM, Palko O, Tomov VT, Mahan AE, et al. Cutting Edge: B cell–intrinsic T-bet expression is required to control chronic viral infection. J Immunol. (2016) 197:1017–22. doi: 10.4049/jimmunol.1500368
42. Rubtsova K, Rubtsov AV, van Dyk LF, Kappler JW, Marrack P. T-box transcription factor T-bet, a key player in a unique type of B-cell activation essential for effective viral clearance. Proc Natl Acad Sci USA. (2013) 110:E3216–24. doi: 10.1073/pnas.1312348110
43. Peng SL, Szabo SJ, Glimcher LH. T-bet regulates IgG class switching and pathogenic autoantibody production. Proc Natl Acad Sci USA. (2002) 99:5545–50. doi: 10.1073/pnas.082114899
44. van Langelaar J, Rijvers L, Janssen M, Wierenga-Wolf AF, Melief, M-J Siepman, TA, et al. Induction of brain-infiltrating T-bet–expressing B cells in multiple sclerosis. Ann Neurol. (2019) 86:264–78. doi: 10.1002/ana.25508
45. Jackson SW, Jacobs HM, Arkatkar T, Dam EM, Scharping NE, Kolhatkar NS, et al. B cell IFN-gamma receptor signaling promotes autoimmune germinal centers via cell-intrinsic induction of BCL-6. J Exp Med. (2016) 213:733–50. doi: 10.1084/jem.20151724
46. Domeier PP, Chodisetti SB, Soni C, Schell SL, Elias MJ, Wong EB, et al. IFN-gamma receptor and STAT1 signaling in B cells are central to spontaneous germinal center formation and autoimmunity. J Exp Med. (2016) 213:715–32. doi: 10.1084/jem.20151722
47. Dalpke AH, Eckerle S, Frey M, Heeg K. Triggering of Toll-like receptors modulates IFN-γ signaling: involvement of serine 727 STAT1 phosphorylation and suppressors of cytokine signaling. Eur J Immunol. (2003) 33:1776–87. doi: 10.1002/eji.200323621
48. Bar-Or A, Fawaz L, Fan B, Darlington PJ, Rieger A, Ghorayeb C, et al. Abnormal B-cell cytokine responses a trigger of T-cell–mediated disease in MS? Ann Neurol. (2010) 67:452–61. doi: 10.1002/ana.21939
49. Lill CM, Luessi F, Alcina A, Sokolova EA, Ugidos N, de la Hera B, et al. Genome-wide significant association with seven novel multiple sclerosis risk loci. J Med Genet. (2015) 52:848–55. doi: 10.1136/jmedgenet-2015-103442
50. Patsopoulos NA, Baranzini SE, Santaniello A, Shoostari P, Cotsapas C, Wong G, et al. The multiple sclerosis genomic map: role of peripheral immune cells and resident microglia in susceptibility. bioRxiv[Preprint]. (2017). doi: 10.1101/143933
51. Phipps-Yonas H, Semik V, Hastings KT. GILT expression in B cells diminishes cathepsin S steady-state protein expression and activity. Eur J Immunol. (2013) 43:65–74. doi: 10.1002/eji.201242379
52. Hastings, KT. GILT: shaping the MHC class II-restricted peptidome and CD4(+) T cell-mediated immunity. Front Immunol. (2013) 4:429. doi: 10.3389/fimmu.2013.00429
53. Hastings KT, Lackman RL, Cresswell P. Functional requirements for the lysosomal thiol reductase GILT in MHC class II-restricted antigen processing. J Immunol. (2006) 177:8569–77. doi: 10.4049/jimmunol.177.12.8569
54. Rubtsov AV, Rubtsova K, Kappler JW, Jacobelli J, Friedman RS, Marrack P. CD11c-expressing B cells are located at the T Cell/B cell border in spleen and are potent APCs. J Immunol. (2015) 195:71–9. doi: 10.4049/jimmunol.1500055
55. Piovesan D, Tempany J, Pietro, A. Di Baas, I Yiannis, C, O’Donnell K, et al. c-Myb regulates the T-Bet-dependent differentiation program in B cells to coordinate antibody responses. Cell Rep. (2017) 19:461–70. doi: 10.1016/j.celrep.2017.03.060
56. Pender, MP. The essential role of Epstein-Barr virus in the pathogenesis of multiple sclerosis. Neuroscientist. (2011) 17:351–67. doi: 10.1177/1073858410381531
57. Tracy SI, Kakalacheva K, Lünemann JD, Luzuriaga K, Middeldorp J, and Thorley-Lawson, DA. Persistence of Epstein-Barr virus in self-reactive memory B cells. J Virol. (2012) 86:12330–40. doi: 10.1128/jvi.01699-12
58. Thorley-Lawson DA, Gross A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N Engl J Med. (2004) 350:1328–37. doi: 10.1056/NEJMra032015
59. Roughan JE, and Thorley-Lawson, DA. The intersection of Epstein-Barr virus with the germinal center. J Virol. (2009) 83:3968–76. doi: 10.1128/jvi.02609-08
60. James T, Linden M, Morikawa H, Fernandes SJ, Ruhrmann S, Huss M, et al. Impact of genetic risk loci for multiple sclerosis on expression of proximal genes in patients. Hum Mol Genet. (2018) 27:912–28. doi: 10.1093/hmg/ddy001
61. Caldwell RG, Wilson JB, Anderson SJ, Longnecker R. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity. (1998) 9:405–11. doi: 10.1016/s1074-7613(00)80623-8
62. Sindhava VJ, Oropallo MA, Moody K, Naradikian M, Higdon LE, Zhou L, et al. A TLR9-dependent checkpoint governs B cell responses to DNA-containing antigens. J Clin Invest. (2017) 127:1651–63. doi: 10.1172/jci89931
63. Jegerlehner A, Maurer P, Bessa J, Hinton HJ, Kopf M, Bachmann MF. TLR9 signaling in B cells determines class switch recombination to IgG2a. J Immunol. (2007) 178:2415–20. doi: 10.4049/jimmunol.178.4.2415
64. Knox JJ, Buggert M, Kardava L, Seaton KE, Eller MA, Canaday DH, et al. T-bet+ B cells are induced by human viral infections and dominate the HIV gp140 response. JCI Insight. (2017) 2:e92943. doi: 10.1172/jci.insight.92943
65. Rubtsova K, Rubtsov AV, Thurman JM, Mennona JM, Kappler JW, Marrack P. B cells expressing the transcription factor T-bet drive lupus-like autoimmunity. J Clin Invest. (2017) 127:1392–404. doi: 10.1172/jci91250
66. Sawcer S, Ban M, Maranian M, Yeo TW, Compston A, Kirby A, et al. A high-density screen for linkage in multiple sclerosis. Am J Hum Genet. (2005) 77:454–67. doi: 10.1086/444547
67. Good-Jacobson KL, Song E, Anderson S, Sharpe AH, Shlomchik MJ. CD80 expression on B cells regulates murine T follicular helper development, germinal center B cell survival, plasma cell.generation. J Immunol. (2012) 188:4217–25. doi: 10.4049/jimmunol.1102885
68. Smets I, Fiddes B, Garcia-Perez JE, He D, Mallants K, Liao W, et al. Multiple sclerosis risk variants alter expression of co-stimulatory genes in B cells. Brain. (2018) 141:786–96. doi: 10.1093/brain/awx372
69. Molnarfi N, Schulze-Topphoff U, Weber MS, Patarroyo JC, Prod’homme T, Varrin-Doyer M, et al. MHC class II–dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J Exp Med. (2013) 210:2921–37. doi: 10.1084/jem.20130699
70. Parker Harp CR, Archambault AS, Sim J, Ferris ST, Mikesell RJ, Koni PA, et al. B cell antigen presentation is sufficient to drive neuroinflammation in an animal model of multiple sclerosis. J Immunol. (2015) 194:5077–84. doi: 10.4049/jimmunol.1402236
71. Raychaudhuri S, Sandor C, Stahl EA, Freudenberg J, Lee HS, Jia X, et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet. (2012) 44:291–6. doi: 10.1038/ng.1076
72. Patsopoulos NA, Barcellos LF, Hintzen RQ, Schaefer C, van Duijn CM, Noble JA, et al. Fine-mapping the genetic association of the major histocompatibility complex in multiple sclerosis: HLA and non-HLA effects. PLoS Genet. (2013) 9:e1003926. doi: 10.1371/journal.pgen.1003926
73. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. (2000) 100:655–69. doi: 10.1016/s0092-8674(00)80702-3
74. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. (2006) 126:1121–33. doi: 10.1016/j.cell.2006.07.035
75. Noster R, Riedel R, Mashreghi MF, Radbruch H, Harms L, Haftmann C, et al. IL-17 and GM-CSF expression are antagonistically regulated by human T helper cells. Sci Transl Med. (2014) 6:241ra80. doi: 10.1126/scitranslmed.3008706
76. El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. (2011) 12:568–75. doi: 10.1038/ni.2031
77. Galli E, Hartmann FJ, Schreiner B, Ingelfinger F, Arvaniti E, Diebold M, et al. GM-CSF and CXCR4 define a T helper cell signature in multiple sclerosis. Nat Med. (2019) 25:1290–300. doi: 10.1038/s41591-019-0521-4
78. van Langelaar J, van der RM, de Vries V, Janssen M, Wierenga-Wolf AF, Spilt IM, et al. T helper 17.1 cells associate with multiple sclerosis disease activity: perspectives for early intervention. Brain. (2018) 141:1334–49. doi: 10.1093/brain/awy069
79. Ramesh R, Kozhaya L, McKevitt K, Djuretic IM, Carlson TJ, Quintero MA, et al. Pro-inflammatory human Th17 cells selectively express P-glycoprotein and are refractory to glucocorticoids. J Exp Med. (2014) 211:89–104. doi: 10.1084/jem.20130301
80. Cao Y, Goods BA, Raddassi K, Nepom GT, Kwok WW, Love JC, et al. Functional inflammatory profiles distinguish myelin-reactive T cells from patients with multiple sclerosis. Sci Transl Med. (2015) 7:287ra74. doi: 10.1126/scitranslmed.aaa8038
81. Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. (2007) 8:639–46. doi: 10.1038/ni1467
82. Meyer Zu Horste G, Wu C, Wang C, Cong L, Pawlak M, Lee Y, et al. RBPJ controls development of pathogenic Th17 cells by regulating IL-23 receptor expression. Cell Rep. (2016) 16:392–404. doi: 10.1016/j.celrep.2016.05.088
83. Teng MW, Bowman EP, McElwee JJ, Smyth MJ, Casanova JL, Cooper AM, et al. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat Med. (2015) 21:719–29. doi: 10.1038/nm.3895
84. Hussman JP, Beecham AH, Schmidt M, Martin ER, McCauley JL, Vance JM, et al. GWAS analysis implicates NF-kappaB-mediated induction of inflammatory T cells in multiple sclerosis. Genes Immun. (2016) 17:305–12. doi: 10.1038/gene.2016.23
85. Kimura A, Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur J Immunol. (2010) 40:1830–5. doi: 10.1002/eji.201040391
86. Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. (2007) 8:967–74. doi: 10.1038/ni1488
87. Barr TA, Shen P, Brown S, Lampropoulou V, Roch T, Lawrie S, et al. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J Exp Med. (2012) 209:1001–10. doi: 10.1084/jem.20111675
88. Arkatkar T, Du SW, Jacobs HM, Dam EM, Hou B, Buckner JH, et al. B cell–derived IL-6 initiates spontaneous germinal center formation during systemic autoimmunity. J Exp Med. (2017) 214:3207–17. doi: 10.1084/jem.20170580
89. Serada S, Fujimoto M, Mihara M, Koike N, Ohsugi Y, Nomura S, et al. IL-6 blockade inhibits the induction of myelin antigen-specific Th17 cells and Th1 cells in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. (2008) 105:9041–6. doi: 10.1073/pnas.0802218105
90. Schneider A, Long SA, Cerosaletti K, Ni CT, Samuels P, Kita M, et al. In active relapsing-remitting multiple sclerosis, effector T cell resistance to adaptive T(regs) involves IL-6-mediated signaling. Sci Transl Med. (2013) 5:170ra15. doi: 10.1126/scitranslmed.3004970
91. Neurath MF, Finotto S. IL-6 signaling in autoimmunity, chronic inflammation and inflammation-associated cancer. Cytokine Growth Factor Rev. (2011) 22:83–9. doi: 10.1016/j.cytogfr.2011.02.003
92. Gaublomme JT, Yosef N, Lee Y, Gertner RS, Yang LV, Wu C, et al. Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell. (2015) 163:1400–12. doi: 10.1016/j.cell.2015.11.009
93. Li R, Rezk A, Miyazaki Y, Hilgenberg E, Touil H, Shen P, et al. Proinflammatory GM-CSF–producing B cells in multiple sclerosis and B cell depletion therapy. Sci Transl Med. (2015) 7:310ra166. doi: 10.1126/scitranslmed.aab4176
94. Lünemann JD, Kamradt T, Martin R, and Münz, C. Epstein-Barr Virus: environmental trigger of multiple sclerosis? J Virol. (2007) 81:6777–84. doi: 10.1128/jvi.00153-07
95. Takeuchi A, Saito T. CD4 CTL, a Cytotoxic subset of CD4+ T cells, their differentiation and function. Front Immunol. (2017) 8:194. doi: 10.3389/fimmu.2017.00194
96. Peeters LM, Vanheusden M, Somers V, van Wijmeersch B, Stinissen P, Broux B, et al. Cytotoxic CD4+ T cells drive multiple sclerosis progression. Front Immunol. (2017) 8:1160. doi: 10.3389/fimmu.2017.01160
97. Meckiff BJ, Ladell K, McLaren JE, Ryan GB, Leese AM, James EA, et al. Primary ebv infection induces an acute wave of activated antigen-specific cytotoxic CD4+ T Cells. J Immunol. (2019) 203:1276–87. doi: 10.4049/jimmunol.1900377
98. Lam JKP, Hui KF, Ning RJ, Xu XQ, Chan KH, Chiang AKS. Emergence of CD4+ and CD8+ polyfunctional T cell responses against immunodominant lytic and latent EBV antigens in children with primary EBV infection. Front Microbiol. (2018) 9:416. doi: 10.3389/fmicb.2018.00416
99. Harp CT, Ireland S Davis.LS, Remington G, Cassidy B, Cravens PD, et al. Memory B cells from a subset of treatment-naïve relapsing-remitting multiple sclerosis patients elicit CD4(+) T-cell proliferation and IFN-γ production in response to myelin basic protein and myelin oligodendrocyte glycoprotein. Eur J Immunol. (2010) 40:2942–56. doi: 10.1002/eji.201040516
100. Broux B, Markovic-Plese S, Stinissen P, Hellings N. Pathogenic features of CD4+CD28- T cells in immune disorders. Trends Mol Med. (2012) 18:446–53. doi: 10.1016/j.molmed.2012.06.003
101. Herich S, Schneider-Hohendorf T, Rohlmann A, Khaleghi GM, Schulte-Mecklenbeck A, Zondler L, et al. Human CCR5high effector memory cells perform CNS parenchymal immune surveillance via GZMK-mediated transendothelial diapedesis. Brain. (2019) 142:3411–27. doi: 10.1093/brain/awz301
102. Hickey, WF. Leukocyte traffic in the central nervous system: the participants and their roles. Semin Immunol. (1999) 11:125–37. doi: 10.1006/smim.1999.0168
103. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. (2015) 523:337–41. doi: 10.1038/nature14432
104. Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, et al. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol. (2009) 10:514–23. doi: 10.1038/ni.1716
105. Ransohoff RM, Kivisakk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol. (2003) 3:569–81. doi: 10.1038/nri1130
106. de Graaf MT, de Jongste AHC, Kraan J, Boonstra JG, Smitt PAES, Gratama JW. Flow cytometric characterization of cerebrospinal fluid cells. Cytometry B Clin Cytom. (2011) 80B:271–81. doi: 10.1002/cyto.b.20603
107. Giunti D, Borsellino G, Benelli R, Marchese M, Capello E, Valle MT, et al. Phenotypic and functional analysis of T cells homing into the CSF of subjects with inflammatory diseases of the CNS. J Leukoc Biol. (2003) 73:584–90. doi: 10.1189/jlb.1202598
108. Kivisakk P, Trebst C, Liu Z, Tucky BH, Sorensen TL, Rudick RA, et al. T-cells in the cerebrospinal fluid express a similar repertoire of inflammatory chemokine receptors in the absence or presence of CNS inflammation: implications for CNS trafficking. Clin Exp Immunol. (2002) 129:510–8. doi: 10.1046/j.1365-2249.2002.01947.x
109. Sorokin, L. The impact of the extracellular matrix on inflammation. Nat Rev Immunol. (2010) 10:712–23. doi: 10.1038/nri2852
110. Smolders J, Remmerswaal EB, Schuurman KG, Melief J, van Eden CG, van Lier RA, et al. Characteristics of differentiated CD8(+) and CD4. (+) T cells present in the human brain. Acta Neuropathol. (2013) 126:525–35. doi: 10.1007/s00401-013-1155-0
111. Plog BA, Nedergaard M. The glymphatic system in central nervous system health and disease: past present, and future. Annu Rev Pathol. (2018) 13:379–94. doi: 10.1146/annurev-pathol-051217-111018
112. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. (2015) 15:545. doi: 10.1038/nri3871
113. Filippi M, Bar-Or A, Piehl F, Preziosa P, Solari A, Vukusic S, et al. Multiple sclerosis. Nat Rev Dis Prim. (2018) 4:43. doi: 10.1038/s41572-018-0041-4
114. Brucklacher-Waldert V, Stuerner K, Kolster M, Wolthausen J, Tolosa E. Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain. (2009) 132:3329–41. doi: 10.1093/brain/awp289
115. Cayrol R, Wosik K, Berard JL, Dodelet-Devillers A, Ifergan I, Kebir H, et al. Activated leukocyte cell adhesion molecule promotes leukocyte trafficking into the central nervous system. Nat Immunol. (2008) 9:137–45. doi: 10.1038/ni1551
116. Rahman MT, Ghosh C, Hossain M, Linfield D, Rezaee F, Janigro D, et al. IFN-γ, IL-17A, or zonulin rapidly increase the permeability of the blood-brain and small intestinal epithelial barriers: relevance for neuro-inflammatory diseases. Biochem Biophys Res Commun. (2018) 507:274–9. doi: 10.1016/j.bbrc.2018.11.021
117. Subileau EA, Rezaie P, Davies HA, Colyer FM, Greenwood J, Male DK, et al. Expression of chemokines and their receptors by human brain endothelium: implications for multiple sclerosis. J Neuropathol Exp Neurol. (2009) 68:227–40. doi: 10.1097/NEN.0b013e318197eca7
118. Sørensen TL, Trebst C, Kivisäkk P, Klaege KL, Majmudar A, Ravid R, et al. Multiple sclerosis: a study of CXCL10 and CXCR3 co-localization in the inflamed central nervous system. J Neuroimmunol. (2002) 127:59–68. doi: 10.1016/s0165-5728(02)00097-8
119. Suan D, Krautler NJ, Maag JLV, Butt D, Bourne K, Hermes JR, et al. CCR6 defines memory B cell precursors in mouse and human germinal centers, revealing light-zone location and predominant low antigen affinity. Immunity. (2017) 47:1142–1153.e4. doi: 10.1016/j.immuni.2017.11.022
120. Annibali V, Ristori G, Angelini DF, Serafini B, Mechelli R, Cannoni S, et al. CD161(high)CD8+T cells bear pathogenetic potential in multiple sclerosis. Brain. (2011) 134(Pt 2):542–54. doi: 10.1093/brain/awq354
121. Michel L, Grasmuck C, Charabati M, Lécuyer, M-A Zandee, S Dhaeze, T, et al. Activated leukocyte cell adhesion molecule regulates B lymphocyte migration across central nervous system barriers. Sci Transl Med. (2019) 11:eaaw0475. doi: 10.1126/scitranslmed.aaw0475
122. Lecuyer MA, Saint-Laurent O, Bourbonniere L, Larouche S, Larochelle C, Michel L, et al. Dual role of ALCAM in neuroinflammation and blood-brain barrier homeostasis. Proc Natl Acad Sci USA. (2017) 114:E524–33. doi: 10.1073/pnas.1614336114
123. Elices MJ, Osborn L, Takada Y, Crouse C, Luhowskyj S, Hemler ME, et al. VCAM-1 on activated endothelium interacts with the leukocyte integrin VLA-4 at a site distinct from the VLA-4/Fibronectin binding site. Cell. (1990) 60:577–84. doi: 10.1016/0092-8674(90)90661-w
124. Lehmann-Horn K, Sagan SA, Bernard CC, Sobel RA, Zamvil SS. B-cell very late antigen-4 deficiency reduces leukocyte recruitment and susceptibility to central nervous system autoimmunity. Ann Neurol. (2015) 77:902–8. doi: 10.1002/ana.24387
125. Polman CH, O’Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. (2006) 354:899–910. doi: 10.1056/NEJMoa044397
126. Sorensen PS, Koch-Henriksen N, Petersen T, Ravnborg M, Oturai A, Sellebjerg F. Recurrence or rebound of clinical relapses after discontinuation of natalizumab therapy in highly active MS patients. J Neurol. (2014) 261:1170–7. doi: 10.1007/s00415-014-7325-8
127. Serafini B, Scorsi E, Rosicarelli B, Rigau V, Thouvenot E, Aloisi F. Massive intracerebral Epstein-Barr virus reactivation in lethal multiple sclerosis relapse after natalizumab withdrawal. J Neuroimmunol. (2017) 307:14–7. doi: 10.1016/j.jneuroim.2017.03.013
128. Damotte V, Guillot-Noel L, Patsopoulos NA, Madireddy L, Behi M. El International Multiple Sclerosis Genetics Consortium et al. A gene pathway analysis highlights the role of cellular adhesion molecules in multiple sclerosis susceptibility. Genes Immun. (2014) 15:126–32. doi: 10.1038/gene.2013.70
129. Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. (2009) 132(Pt 5):1175–89. doi: 10.1093/brain/awp070
130. Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. (2000) 47:707–17. doi: 10.1002/1531-8249(200006)47:63.0.co;2-q
131. Ramaglia V, Sheikh-Mohamed S, Legg K, Rojas OL, Zandee S, Fu F, et al. Multiplexed imaging of immune cells in staged multiple sclerosis lesions by mass cytometry. bioRxiv[Preprint]. (2019). doi: 10.1101/638015
132. Planas R, Metz I, Martin R, Sospedra M. Detailed characterization of T cell receptor repertoires in multiple sclerosis brain lesions. Front Immunol. (2018) 9:509. doi: 10.3389/fimmu.2018.00509
133. Muraro PA, Robins H, Malhotra S, Howell M, Phippard D, Desmarais C, et al. T cell repertoire following autologous stem cell transplantation for multiple sclerosis. J Clin Invest. (2014) 124:1168–72. doi: 10.1172/JCI71691
134. Junker A, Ivanidze J, Malotka J, Eiglmeier I, Lassmann H, Wekerle H, et al. Multiple sclerosis: T-cell receptor expression in distinct brain regions. Brain. (2007) 130:2789–99. doi: 10.1093/brain/awm214
135. Planas R, Santos R, Tomas-Ojer P, Cruciani C, Lutterotti A, Faigle W, et al. GDP-l-fucose synthase is a CD4+ T cell–specific autoantigen in DRB3∗02:02 patients with multiple sclerosis. Sci Transl Med. (2018) 10:eaat4301. doi: 10.1126/scitranslmed.aat4301
136. Skulina C, Schmidt S, Dornmair K, Babbe H, Roers A, Rajewsky K, et al. Multiple sclerosis: brain-infiltrating CD8+ T cells persist as clonal expansions in the cerebrospinal fluid and blood. Proc Natl Acad Sci USA. (2004) 101:2428–33. doi: 10.1073/pnas.0308689100
137. van Nierop GP, van Luijn MM, Michels SS, Melief MJ, Janssen M, Langerak AW, et al. Phenotypic and functional characterization of T cells in white matter lesions of multiple sclerosis patients. Acta Neuropathol. (2017) 134:383–401. doi: 10.1007/s00401-017-1744-4
138. Stern JNH, Yaari G, Vander Heiden JA, Church G, Donahue WF, Hintzen RQ, et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci Transl Med. (2014) 6:107. doi: 10.1126/scitranslmed.3008879
139. Lovato L, Willis SN, Rodig SJ, Caron T, Almendinger SE, Howell OW, et al. Related B cell clones populate the meninges and parenchyma of patients with multiple sclerosis. Brain. (2011) 134:534–41. doi: 10.1093/brain/awq350
140. Howell OW, Reeves CA, Nicholas R, Carassiti D, Radotra B, Gentleman SM, et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain. (2011) 134(Pt 9):2755–71. doi: 10.1093/brain/awr182
141. Magliozzi R, Howell O, Vora A, Serafini B, Nicholas R, Puopolo M, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. (2007) 130:1089–104. doi: 10.1093/brain/awm038
142. Tzartos JS, Craner MJ, Friese MA, Jakobsen KB, Newcombe J, Esiri MM, et al. IL-21 and IL-21 receptor expression in lymphocytes and neurons in multiple sclerosis brain. Am J Pathol. (2011) 178:794–802. doi: 10.1016/j.ajpath.2010.10.043
143. Peters A, Pitcher LA, Sullivan JM, Mitsdoerffer M, Acton SE, Franz B, et al. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity. (2011) 35:986–96. doi: 10.1016/j.immuni.2011.10.015
144. Olsson, T. Cytokines in neuroinflammatory disease: role of myelin autoreactive T cell production of interferon-gamma. J Neuroimmunol. (1992) 40:211–8. doi: 10.1016/0165-5728(92)90135-8
145. Renno T, Lin, J-Y Piccirillo, C Antel, J, Owens T. Cytokine production by cells in cerebrospinal fluid during experimental allergic encephalomyelitis in SJL/J mice. J Neuroimmunol. (1994) 49:1–7. doi: 10.1016/0165-5728(94)90174-0
146. Brosnan CF, Cannella B, Battistini L, Raine CS. Cytokine localization in multiple sclerosis lesions. Correlation with adhesion molecule expression and reactive nitrogen species. Neurology. (1995) 45(6 Suppl. 6):S16–21. doi: 10.1212/WNL.45.6_Suppl_6.S16
147. Bsibsi M, Peferoen LAN, Holtman IR, Nacken PJ, Gerritsen WH, Witte ME, et al. Demyelination during multiple sclerosis is associated with combined activation of microglia/macrophages by IFN-γ and alpha B-crystallin. Acta Neuropathol. (2014) 128:215–29. doi: 10.1007/s00401-014-1317-8
148. Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol. (2014) 10:217. doi: 10.1038/nrneurol.2014.38
149. Li R, Patterson KR, and Bar-Or, A. Reassessing B cell contributions in multiple sclerosis. Nat Immunol. (2018) 19:696–707. doi: 10.1038/s41590-018-0135-x
150. Genain CP, Cannella B, Hauser SL, Raine CS. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat Med. (1999) 5:170–5. doi: 10.1038/5532
151. Lunemann JD, Tintore M, Messmer B, Strowig T, Rovira A, Perkal H, et al. Elevated Epstein-Barr virus-encoded nuclear antigen-1 immune responses predict conversion to multiple sclerosis. Ann Neurol. (2010) 67:159–69. doi: 10.1002/ana.21886
152. Rand KH Houck.H, Denslow ND, Heilman KM. Epstein-Barr virus nuclear antigen-1. (EBNA-1) associated oligoclonal bands in patients with multiple sclerosis. J Neurol Sci. (2000) 173:32–9. doi: 10.1016/s0022-510x(99)00298-1
153. Castellazzi M, Contini C, Tamborino C, Fasolo F, Roversi G, Seraceni S, et al. Epstein-Barr virus-specific intrathecal oligoclonal IgG production in relapsing-remitting multiple sclerosis is limited to a subset of patients and is composed of low-affinity antibodies. J Neuroinflamm. (2014) 11:188. doi: 10.1186/s12974-014-0188-1
154. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. (2004) 14:164–74. doi: 10.1111/j.1750-3639.2004.tb00049.x
155. Serafini B, Rosicarelli B, Franciotta D, Magliozzi R, Reynolds R, Cinque P, et al. Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J Exp Med. (2007) 204:2899–912. doi: 10.1084/jem.20071030
156. Sundström P, Juto P, Wadell G, Hallmans G, Svenningsson A, Nyström L, et al. An altered immune response to Epstein-Barr virus in multiple sclerosis. A prospective study. Neurology. (2004) 62:2277–82. doi: 10.1212/01.Wnl.0000130496.51156.D7
157. Lang HLE, Jacobsen H, Ikemizu S, Andersson C, Harlos K, Madsen L, et al. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat Immunol. (2002) 3:940–3. doi: 10.1038/ni835
158. Owens GP, Bennett JL. Trigger pathogen, or bystander: the complex nexus linking Epstein- Barr virus and multiple sclerosis. Mult Scler. (2012) 18:1204–8. doi: 10.1177/1352458512448109
159. Willis SN, Stadelmann C, Rodig SJ, Caron T, Gattenloehner S, Mallozzi SS, et al. Epstein-Barr virus infection is not a characteristic feature of multiple sclerosis brain. Brain. (2009) 132(Pt 12):3318–28. doi: 10.1093/brain/awp200
160. Sargsyan SA, Shearer AJ, Ritchie AM, Burgoon MP, Anderson S, Hemmer B, et al. Absence of Epstein-Barr virus in the brain and CSF of patients with multiple sclerosis. Neurology. (2010) 74:1127–35. doi: 10.1212/WNL.0b013e3181d865a1
161. Peferoen LA, Lamers F, Lodder LN, Gerritsen WH, Huitinga I, Melief J, et al. Epstein Barr virus is not a characteristic feature in the central nervous system in established multiple sclerosis. Brain. (2010) 133:e137. doi: 10.1093/brain/awp296
162. Aloisi F, Serafini B, Magliozzi R, Howell OW, Reynolds R. Detection of Epstein-Barr virus and B-cell follicles in the multiple sclerosis brain: what you find depends on how and where you look. Brain. (2010) 133(Pt 12):e157. doi: 10.1093/brain/awq223
Keywords: Th1/Th17, T-bet+ B cells, CD8+ T cells, Epstein-Barr virus, genetic risk transmigration, germinal center, IFN-γ
Citation: van Langelaar J, Rijvers L, Smolders J and van Luijn MM (2020) B and T Cells Driving Multiple Sclerosis: Identity, Mechanisms and Potential Triggers. Front. Immunol. 11:760. doi: 10.3389/fimmu.2020.00760
Received: 10 September 2019; Accepted: 03 April 2020;
Published: 08 May 2020.
Edited by:
Sandra Amor, VU University Medical Center, NetherlandsReviewed by:
Nancy Monson, The University of Texas Southwestern Medical Center, United StatesJohann Sellner, University Hospital Salzburg, Austria
Copyright © 2020 van Langelaar Rijvers, Smolders and van Luijn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marvin M. van Luijn, m.vanluijn@erasmusmc.nl