 Priyanshu Srivastava
Priyanshu Srivastava Ankit Kumar
Ankit Kumar Abdul Hasan
Abdul Hasan Divya Mehta
Divya Mehta Ramesh Kumar
Ramesh Kumar Chetan Sharma
Chetan Sharma Sujatha Sunil
Sujatha Sunil- Vector-Borne Diseases Group, International Centre for Genetic Engineering and Biotechnology (ICGEB), New Delhi, India
Chikungunya disease (CHIKD) is a viral infection caused by an alphavirus, chikungunya virus (CHIKV), and triggers large outbreaks leading to epidemics. Despite the low mortality rate, it is a major public health concern owing to high morbidity in affected individuals. The complete spectrum of this disease can be divided into four phases based on its clinical presentation and immunopathology. When a susceptible individual is bitten by an infected mosquito, the bite triggers inflammatory responses attracting neutrophils and initiating a cascade of events, resulting in the entry of the virus into permissive cells. This phase is termed the pre-acute or the intrinsic incubation phase. The virus utilizes the cellular components of the innate immune system to enter into circulation and reach primary sites of infection such as the lymph nodes, spleen, and liver. Also, at this point, antigen-presenting cells (APCs) present the viral antigens to the T cells thereby activating and initiating adaptive immune responses. This phase is marked by the exhibition of clinical symptoms such as fever, rashes, arthralgia, and myalgia and is termed the acute phase of the disease. Viremia reaches its peak during this phase, thereby enhancing the antigen-specific host immune response. Simultaneously, T cell-mediated activation of B cells leads to the formation of CHIKV specific antibodies. Increase in titres of neutralizing IgG/IgM antibodies results in the clearance of virus from the bloodstream and marks the initiation of the post-acute phase. Immune responses mounted during this phase of the infection determine the degree of disease progression or its resolution. Some patients may progress to a chronic arthritic phase of the disease that may last from a few months to several years, owing to a compromised disease resolution. The present review discusses the immunopathology of CHIKD and the factors that dictate disease progression and its resolution.
Introduction
Chikungunya disease (CHIKD), caused by an arthritogenic alphavirus, chikungunya virus (CHIKV), is becoming a major public health hazard (1, 2). The past few decades have seen the re-emergence of this viral infection as evidenced by epidemics in Africa, Asia, Europe and, in recent years, the Americas (3, 4). The first report of this infection dates back to the 1900s; it was then confused with dengue and was largely underreported (5). The latest outbreaks started during 2005 in the La Reunion islands where Aedes albopictus was the primary vector (6–8). It was identified that specific mutations in the viral E1 glycoprotein provided fitness to the virus by reducing its extrinsic incubation period within the mosquito and thereby was transmitted by the vector over a longer period of time (9, 10). Post-2005, the virus spread to different parts of the globe either by travelers or autochthonous outbreaks in tropical and temperate climates involving both mosquito species (9, 11).
CHIKD is primarily a viral infection manifested as a fever with severe arthritic joint involvement. Rightly so, the disease was named “Chikungunya,” which in Makonde means “that which bends up,” emphasizing the excruciating joint pain experienced by the affected individuals which disables movement (12). Even though the infection is classically characterized by fever and joint pains that last for up to a week and sequelae involving joint pains for a few weeks (13), the disease can last up to 2–3 years in a small proportion of patients, affecting the joints and resulting in arthritis-like conditions (14, 15). Recent outbreaks have reported neurological complications in patients stemming from the involvement of the central nervous system (16, 17) as well as an impaired immune system (18, 19). Furthermore, mortality owing to co-morbid conditions was also observed (20, 21). The virus has also been reported to infect through vertical transmission between mother and unborn child and results in complications such as encephalopathy (22, 23).
CHIKD establishes itself and progresses to a prolonged malady involving the joints over a period of time. As the virus infects an individual, there is a period of intrinsic incubation before the clinical symptoms appear and the disease progresses. The disease can then be clinically categorized into acute, post-acute, and chronic phases that can last for a few days up to several months, mainly depending on the individual's immune response to the virus (24, 25). Several studies have established that host immunity can play an important role in disease progression and its resolution after an acute phase of the disease (26–28). The innate immune system has shown to be protective, which may result in early resolution of the disease as evaluated in some reports (29, 30). At the same time, components of adaptive immunity have been reported to be instrumental in mounting the severity of the disease and resulting in the chronic arthritic condition that could last for years (26, 31, 32). Recent research has attributed several viral factors that could also contribute to alleviated chronic conditions, such as persistence of defective viral particles in the host (33) and prolonged infection of synovial macrophages (24).
The current review is a chronological compilation of the immunopathological events that take place during the acute and post-acute phases of CHIKD that may either lead to resolution of the disease or contribute to an exaggerated immune response, resulting in a full-blown arthritic chronic phase lasting up-to several years (Figure 1).
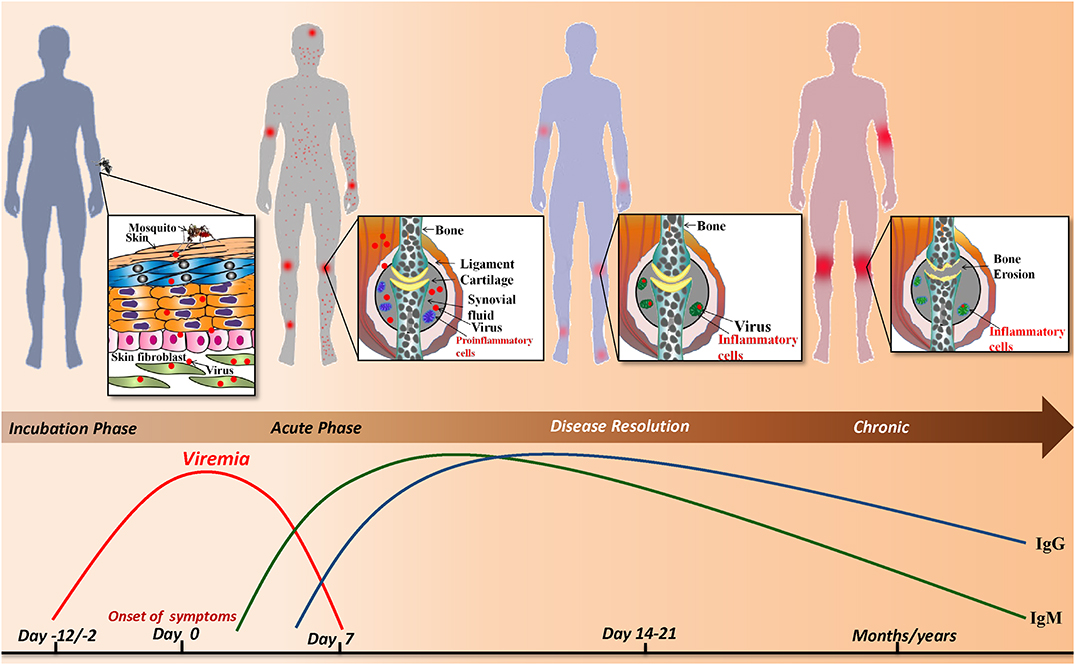
Figure 1. Sequence of events during CHIKV infection: infected Aedes mosquito deposits the virus into the dermis and epidermis of the skin. The viral infection is characterized by an incubation period of which is followed by the acute phase during which a rapid rise in viremia occurs and clinical symptoms such as joint pain, fever, maculopapular and petechial rash appear. As the viral load increases the host innate and adaptive immune responses are evoked simultaneously. IgM and IgG levels rise and the virus is resolved from the host. Despite the robust host immune response, viral particles persist in the synovial fluid of joints and alter joint pathology resulting in a chronic phase that lasts for months/years.
Acute Phase
Early Events Leading to the Establishment of CHIKV Infection
The pre-acute phase or the incubation period of CHIKD ranges from 2 to 12 days (13, 34–38). As an infected mosquito takes a blood meal, it deposits the virus into the epidermis and dermis layers of the skin (39). At the same time, the mosquito also punctures nearby blood vessels, allowing the virus to enter the bloodstream as evidenced by the presence of virus in the sera as early as within 5 min of a mosquito bite (40). At the site of a mosquito bite, the resident cells of the cutaneous immune system defend the host from the pathogen. The cutaneous immune system acts as the first line of defense against the pathogen and is comprised of a heterogeneous mix of cells, such as the lymphocytes, dendritic cells (DCs), and monocytes (39). Gamma delta T lymphocytes (γδ T cells) constitute the most abundant skin-resident T lymphocytes (41) and have demonstrated to play a critical role in cytotoxicity, cytokine secretion, and DC maturation and enhance their function (42, 43). These cells are present in the skin, the local site of infection for CHIKV, and lack the major histocompatibility complex (MHC) restriction; therefore they do not require conventional antigen processing and thus can react to the unprocessed antigens (43). Infection of CHIKV in mice (footpad) led to a significant increase in the prevalence of this T cell subset in foot and popliteal lymph nodes (44). Further, mice deficient in γδ T cells had an increased disease severity accompanied by histopathologic changes (45, 46). Skin resident dendritic cells such as langerhans cells encounter antigens at the site of infection and may disseminate the virus to draining lymph nodes where they interact with the effector T cells, resulting in the initiation of the adaptive immune response (38). Various studies (although contradictory in the explanation of mechanism) have documented the role of bite-induced enhancement of arboviral infection (47–50). Studies have indicated that mosquito saliva has immune-modulatory properties that enhance pathogen establishment within the host by limiting the host immune response (39, 47, 51, 52). One such study of CHIKV infection in mice models observed changes in cutaneous cytokine profiles when inoculating the virus with a needle or mimicking natural infection using mosquitoes. Needle inoculation of the virus into mice resulted in an increased expression of TLR3 and Th1 cytokines (IFN-γ and IL2) whereas infection by mosquito bite elicited a Th2 cytokine response with an increase in IL4 and a reduction observed in TLR3, IFN-γ and IL2 (53). In a different study carried out using dengue virus, similar IFN-γ reduction was observed when the virus was inoculated with mosquito saliva as compared to direct needle inoculation of the virus (54). Similarly, research on the immune modulatory properties of mosquito saliva showed that mice previously sensitized with a salivary gland extract of mosquitoes have more robust cellular infiltration than non-sensitized mice. This study suggests that mosquito saliva has the ability to differentially regulate cellular microenvironment around the bite site (55). Another recent study correlated the initial events of mosquito bite and viral inoculation with subsequent development of systemic disease, highlighting an important role for cutaneous innate immune response. The study observed that stromal cells and macrophages were the main cellular targets for semliki forest virus replication at the initial bite sites, but these cells did not express markers of interferon induction upon infection. Dermal DC's, that are less abundant in the dermis, were major activators of type I IFN response at the initial inoculation site and therefore were major players in limiting viral replication and clinical progression (56).
However, other studies have argued that bite-induced enhancement is too early a process to activate and engage cells of the adaptive immune response (47, 51). Infections with Semliki forest virus aided or unaided by mosquito saliva showed no reduction in IFN-γ levels, suggesting no perturbations of cutaneous antiviral immunity (47). The authors showed that in the presence of saliva, two major events were observed. First, the development of edema that retained the virus at the bite site thus delaying the dissemination of the virus to draining lymph nodes, slowing the initial innate immune response activation and enhancing cutaneous cell replication. Second, an influx of neutrophils attracted by cutaneous cells was observed beginning from 90 min of infection in the presence of mosquito saliva (47). Although neutrophils were themselves refractory to arboviral infection, they attracted cells of the myeloid lineage as new targets for viral replication. Myeloid cells are also responsible for spreading the virus from the initial site of infection, and neutrophil depletion at the bite site reduced viral dissemination (47). The importance of type I IFNs during early infection is evidenced by IFNAR-deficient mice models of CHIKV infection in which mice died within 3 days post CHIKV infection. Skin fibroblast cells upon infection by CHIKV virus were found to be major producers of type I IFNs (Specifically IFN-β). IFN production in these cells was triggered by the activation of extracellular as well as intracellular pathogen sensing receptors, such as TLR3 and MyD88, as a result of infection, thus leading to a strong anti-viral defense early during CHIKV infection (57).
Establishment of the Acute Phase of Infection
The beginning of the acute phase is marked by viral dissemination from the local site of replication to the primary sites of CHIKV infection in the host body (24). Hallmarks of the acute phase are peak viremia, manifestation of clinical symptoms, and production of neutralizing antibodies (58–60). The acute phase of CHIKD typically lasts for 7–14 days (37, 61, 62). In both humans and murine models, the general observation is that CHIKV-induced immunopathology is considered the primary mediator of damage and persistent pain.
Viral replication in fibroblast cells (primary site of infection) is initially limited by a rapid and robust induction of interferons (IFN-β) and downstream signaling molecules. Interferons help to recruit immune cells that sense the viruses within the infected cells. This sensing may involve the specific pathogen-associated motifs like viral nucleic acids, that are recognized by pattern recognition molecules/receptors (PPRs); these receptors can be cytoplasmic [e.g., Class I—NOD-like Receptors (NLRs), retinoic inducible gene-I like receptors (RLRs), melanoma differentiation factor (MDA)-5, laboratory of genetics and physiology (LGP)-2, cyclic GMP-AMP synthase (cGAS), Gamma-interferon-inducible (IFI) protein-16 and Class II—Protein kinase R (PKR), DNA sensor AIM2 and 2′-5′-oligoadenylate synthase (OAS)-3] (63), or endosomal (e.g., Toll-like receptors; TLRs) (64). It was observed that inflammasome-specific molecules such as NLRP3 of the NLR family were associated with peak inflammatory symptoms during CHIKV infection and these effects could be reversed upon the usage of caspase-1 or NLRP3 inhibitors (65). Previously, the role of inflammasomes in CHIKV infection was explored through in-vitro studies using siRNA targeting caspase-1 that showed enhancement in CHIKV replication with silencing (66).
Members of RIG-1-like helicase families such as RIG-1 and MDA5 play a significant role in the activation of IFN-I response (67). These PPRs from Class I detect dsRNA or 5′-triphosphate RNA in the cytosol and induce IFN production. It was shown that stimulation with 5′-triphosphate RNA generates a robust antiviral response against CHIKV through RIG-1 pathway with the help of transcription factor IRF3 (67). Simultaneously, TLR3 gene expression was found to be significantly elevated and associated with up-regulation of several downstream molecules including IRF-1, IRF-3, IRF-7 and OAS-3, IFN-β, and TFN-α production (68). An association of TLR3 with CHIKV replication was shown by an agonist of TRIF-dependent signaling of interferon induction (69) and TRIF knockout mice showed high viral titer with increased foot swelling in comparison to WT infected mice (70). The role of TLR7 was also investigated by MyD88 knockout mice that showed high viremia but no difference in the severity of disease in comparison to WT infected mice (70), implying that MyD88 dependent pathway may not be involved in disease progression. However, another study showed a significant role of MyD88 signaling in controlling viral dissemination (57). Transcriptional analyses of peripheral blood from CHIKV infected patients showed a high expression of viperin in monocyte (71). The role of viperin was further evaluated using Rsad2 knockout mice that showed a significant direct correlation of the expression of this molecule with respect to higher viremia and joint inflammation (71).
Despite the robust and rapid immune response against the virus in permissive cells, CHIKV effectively evades the cellular control mechanisms. In fibroblast cells, for example, CHIKV dodges the interferon response by inducing a translational shut off (27). Additionally, the cytopathic nature of CHIKV induces apoptosis (72). Fibroblast and stromal cells, for example, undergo apoptosis within 24 h post-infection (72, 73). The virus utilizes this cellular response to increase its rate of infection and since the viral particles are sequestered within the apoptotic blebs, it escapes recognition by the immune system. Engulfment of the apoptotic blebs by neighboring phagocytic cells such as macrophages promotes infection in a non-inflammatory or dormant manner (74). As the virus replicates, it simultaneously invades nearby blood vessels. Evidence has demonstrated the infection of blood monocytes by CHIKV virus (75). The virus continues replication, achieving a peak in viremia during the acute phase.
Immunopathology of Acute Phase
As CHIKV infection spreads, host immune responses are activated by engaging various subsets of myeloid cells as well as lymphocytes to control this spread. A major event in the acute phase is infiltration of macrophages, neutrophils, natural killer (NK) cells, and other lymphocytes to the primary sites of infection, mainly joints and muscles, leading to hypertrophy in the cells of synovial lining as well as surrounding synovial vessels (76) that manifests as arthralgia in the patients (24, 77). This cellular infiltration is mediated by pro-inflammatory cytokines and chemokines rich milieu at the sites of infection (30) (Figure 2). CCL2 (MCP-1) is a major chemokine that is produced by various cell types like endothelial, fibroblasts, epithelial, smooth muscle, mesangial, astrocytic, monocytic, and microglial cells (78–81). Macrophages, being the major source of CCL2, regulate the infiltration of monocytes and NK cells to the site of infection. Cessation in macrophage recruitment by using Bindarit, a modulator of CCL2 production, has shown reduced inflammation in synovial and skeletal muscle tissues (82). Another study showed contrasting results in which CCR2−/− mice exhibited increased foot swelling and cartilage erosion upon CHIKV infection (83). Macrophages are also known to be the reservoirs of CHIKV in later stages of infection (24), which possibly plays an important role in prolonged inflammation. CCR2 plays an important role in controlling inflammation and musculoskeletal pathology by maintaining the balance of monocytes and neutrophil infiltration to the infected tissues (83). CCR2−/− mice upon CHIKV infection showed enhanced arthritic symptoms without any significant change in viremia, as compared to wild type mice. Also, the monocyte/macrophage infiltrates were replaced by an enhanced neutrophil infiltration, followed by eosinophils infiltrates as well (83). These results suggest that neutrophils are one of the key mediators of arthritic symptoms and tissue damage in CHIKV infection by releasing granules carrying antimicrobial molecules and producing reactive oxygen species (ROS) due to an oxidative burst, which is also a well-established feature of neutrophils in other models of inflammation (84, 85). Neutrophils are also known to produce an anti-viral effect in an alternate way, by forming neutrophils extracellular traps (NETs) through a process called NETosis. In vitro CHIKV infection in neutrophils isolated from mice resulted in the formation of NETs through TLR7 and ROS dependent mechanism. Also, the correlation of NETosis with susceptibility and viral load has been shown in IFNAR−/− mice model and CHIKV infected patients, respectively, suggesting the anti-CHIKV role of NETosis (86). NETs are also known to activate plasmacytoid dendritic cells (pDCs), a subtype of dendritic cells, which secrete high levels of IFN-I (87, 88). In CHIKV infection, the virus is sensed by pDCs in an indirect manner that results in selective IRF7 activation and type-I IFN production without the involvement of other inflammatory cytokine responses (89).
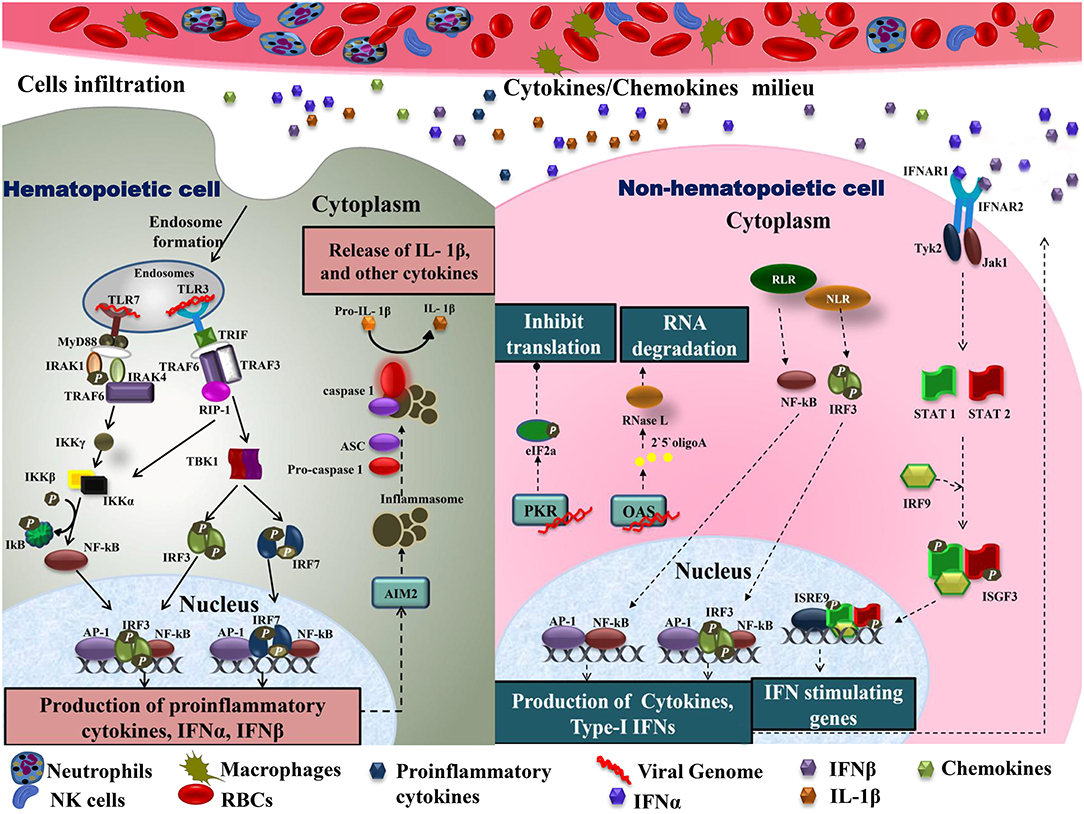
Figure 2. Immune cell infiltration in CHIKV infected tissues: the primary target organs during CHIKV infection include liver, spleen, joints, and kidney. As the virus is disseminated to these sites during acute phase, it infects non-hematopoietic cells. Active viral replication within these cells generate viral dsRNA and ssRNA which is recognized by RLR and NLR leading to downstream activation of NF-κB and phosphorylation of IRF3 results in type-I IFNs and cytokine production within cells. Increase amount of IFNs in extracellular matrix activates IFNARs leading phosphorylation and dimerization of STAT1 and STAT2 in the presence of IRF9 results in activation of IFN stimulating genes. The presence of viral dsRNA and pro-inflammatory cytokines within these cells also activates OAS and PKR. Downstream of PKR activation is the generation of initiation factor eIF2a leading to a translational arrest. On the other hand, OAS once triggered generates RNase L which degrades viral RNA. In case of hematopoietic cells, endosomal TLRs recognize viral dsRNA. Downstream of TLR7, MyD88-IRAK-1-IRAF4-TRAF6 complex is recruited leading to phosphorylation of IkB. Likewise, signaling downstream of TLR3 involves the recruitment of TRIF. TRIF further interacts with TRAF6 and TRAF3. This complex recruits kinase RIP-1 triggering IRF3 and IRF7 phosphorylation downstream of TBK1 activation as well as IkB phosphorylation. These events lead to NF-κB dependent induction of pro-inflammatory cytokines, IFN-α and IFN-β. Cytokines within these cells also produce inflammasomes downstream of AIM2, which promotes apoptosis by cleaving pro-caspase1 to caspase1. Active IL-1β production is also enhanced during this event. The pro-inflammatory cytokines are also released extracellularly, generating a cytokine rich milieu and attracting other immune cells causing cellular infiltration. TLR, Toll-Like receptor; MyD88, myeloid differentiation primary response 88; TRAF, tumor necrosis factor receptor (TNFR)-associated factors; IKB, nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor; TRIF, TIR-domain-containing adapter-inducing interferon; IRF, interferon regulatory factor; TBK1, TANK-binding kinase 1; NF-κB, nuclear factor k-light-chain enhancer of activated B cells; IFN, interferons; RLR, RIG-1 like receptor; NLR, NOD-like receptor; OAS, 2′5′ oligoadenylate synthetase; PKR, dsRNA-dependent protein kinase R; eIF2a, eukaryotic initiation factor 2 alpha subunit; RNase L, 2-5A dependent ribonuclease L; AIM2, absent in melanoma; IL-1β, Interleukine 1β; STAT, signal transducer and activator of transcription; IRF9, interferon regulatory factor 9; ISGF, IFN-stimulated gene factor; IFNAR, interferon-alpha/beta receptor.
NK cells are also a major line of defense against the virus, displaying a marked change in surface receptor repertoire with increased expression of NKp44, CD57, ILT2, CD8α, and NKG2C and decreased expression of NKp30, NKp46, NKG2A, and CD16. Specifically, NKG2C cells undergo rapid expansion during the early acute phase of CHIKD and exert strong cytolytic response upon infected cells (90). As the disease progresses in CHIKD, terminally differentiated NK cells mature into CD56dimCD57+ phenotype and show a strong cytolytic response and enhanced resistance to cytokine stimulation (90, 91). It has been observed that the number of mature NK cells peak in the early acute phase (i.e., day 3 post-onset of symptoms) and their persistence is correlated with the viral load. Additionally, the persistence of these cells is also associated with chronic CHIKD (90).
In addition to the above mentioned pro-inflammatory mechanisms, the acute phase is also mediated by CD8+ T cells responses at the early stages of infection while in the later stages it's the CD4+ T cells that predominate the repertoire of the immune cells in humans (92). Murine studies have presented with the evidence that both CD4+ and CD8+ T cells infiltrate CHIKV-infected tissues during the course of infection (93, 94). These infiltrating T cells are understandably involved in mounting site-specific antiviral immune responses, contrary to which CD4+ T cells have recently been shown to contribute toward the pathogenesis during CHIKV infection in mice without altering much of the viral titers along with IFN-γ production (95).
During the acute phase, anti-CHIKV IgM could be detected in sera samples of patients and mice from day 3 onwards after the onset of clinical symptoms (1, 96, 97) whereas the anti-CHIKV IgG with neutralizing activity is produced from day 4 (1, 97). Detection of Anti-CHIKV IgG is mostly rare during the initial days (<4 days) of infection (97). Several studies have also established a correlation between antibodies response with cytokine levels, viral titres, and disease progression during the acute and chronic phases (97, 98). Patients with high virus titres in the acute phase or viremic phase triggered the production of Anti-CHIKV IgG3 antibodies which are strongly involved in protection against chronicity or arthralgia conditions whereas patients with low virus titres associated with chronic arthralgia (99). This suggested that high virus titres in the acute phase seem to induce high titer of Anti-CHIKV antibodies which protect against chronic stages of CHIKD (99). Additionally, CD4+ null mice showed lower levels of anti-CHIKV antibodies along with significantly reduced neutralizing activity (100).
CHIKV infected cells are killed by the induction of cytolytic mechanisms along with the several antiviral factors by the immune cells, mainly CD8+ T cells (92). Naïve CD8+ T cells induce a development environment upon activation leading to effector and memory T-cell proliferation and differentiation (1, 59). Once activated, effector CD8+ T cells exhibit functions, such as cytotoxicity and cytokine production, against the virus, resulting in its elimination (101). Furthermore, it is known that CD8+ T lymphocytes mediate cytolytic activity against target cells in two major pathways either by the release of cytolytic granules through exocytosis or granule independent pathway, in which they bind to the death receptors of the target cells (102). Studies have reported that the peripheral blood of acute and chronic CHIKD patients had no differences in the percentage of CD8+ T cells in relation to healthy individuals. Analysis of early activation marker CD69 for CD8+ T cells revealed increased CD8+ T cell activation in both acute and chronic diseases (103). The study further observed that CD8+ T lymphocytes in patients with acute CHIKD have increased secretion of CD107A along with granzyme and perforin expression in comparison to the control group, suggesting that CD8+ T cells may have a role in mediating cytolytic killing of CHIKV infected cells during the acute phase of CHIKD (103). Interestingly, the CD8+ T cells failed to present any such increase in the expression of granzyme and perforins in patients with chronic disease in comparison to healthy controls (103). Such observations point to the possibility that these mechanisms slow down or even shut off, owing to the process of T-cell exhaustion during the later stages of the disease.
Post-Acute Phase
CHIKV Clearance
Clearance of CHIKV from blood paves the path to recovery in CHIKD patients and is comprised of various immune pathways constituting the post-acute phase of CHIKD. This phase is marked by complete clearance of active CHIKV from the body by neutralizing antibodies in the blood and destruction of infected cells by infiltrating NK cells, CD8+ T cells, and neutrophils. Detection of neutralizing antibodies could begin from day 4 to 7 post-onset of symptoms and could last up to 3 weeks post clinical presentation. IgM antibodies could be detected during the acute phase of infection, whereas IgG antibodies are detected in the post-acute phase and may last for months (104–106). Titres of neutralizing antibodies during the acute phase could predict the severity of CHIKD (97). Some patients are completely asymptomatic during the post-acute phase and recover completely. However, a majority of patients show temporary improvement in their clinical state and deterioration occurs after a short “healing” period (61). Also, a recent study using a C57BL/6J mice model has correlated viral clearance and development of IgG antibodies with age. It suggests that CHIKV neutralization and IgG titres were higher in 8-week-old mice during the acute phase, whereas CHIKV clearance was not observed until post-acute phase in 20 weeks old mice (107).
In the context of antibodies, detection of anti-CHIKV IgG is mostly rare during the acute phase of infection while detection occurs in the post-acute phase and persists for months or even years (108). Persistence of CHIKV specific IgM antibodies in the blood and synovial fluid is associated with severe chronic CHIKD (109–111). IgG2c isotype is the major class of antibody produced in the case of CHIKV infected mice model due to IFN-γ produced from CD4+ T cells (95). Numerous reports have sought to characterize the neutralizing ability of CHIKV specific antibodies, and it has been reported in mice models as a therapeutic treatment against chikungunya persistence (104, 112–118). In accordance with that, mice mutant Rag1−/− which lack B and T cells, leading to high viral titres in various tissues, concluded that chikungunya infection persists in tissues that are controlled by adaptive immune cells (119). Administration of polyclonal anti-CHIKV virus antisera in CHIKV infected RAG1−/− and B cell-deficient μMT mice resulted in clearance of the virus and could be not detected in blood (25). Similarly, administration of human monoclonal CHIKV-specific antibodies in chikungunya infected rhesus monkeys led to inhibition of viral spread and inflammatory responses in various tissues like joints and muscles (116).
Clearance of circulating pathogens from the system, though, generally involves opsonins like pathogen-specific antibodies and complement component 3 (C3) (120–122). However, a recent study reports that clearance of CHIKV and RRV remains unaffected in the C3−/− and B cell-deficient μMT mice, suggesting the role of an uncharacterized innate immune pathway (123–125). The other mechanism of clearance involves scavenger receptors (SRs), which is a non-opsonic mechanism known for the clearance of endogenous and microbial ligands (126, 127). It was found that Kupffer cells found in the liver carry several SRs, out of which SR-A6 (MARCO) was identified as the key factor responsible for CHIKV clearance (123).
Disease Resolution
Chikungunya disease resolution is a combination of viral clearance from the host and resolution of inflammation, as all the major symptoms witnessed by the CHIKD patients are due to the robust pro-inflammatory host response. As discussed in the earlier parts of the review, viral clearance in the circulatory system is led by neutralizing antibodies, and virus replicating in the tissues is tackled by infiltrating pro-inflammatory cells like macrophages, CD8+ T cells, neutrophils, and NK cells. The other arm of disease resolution, resolution of inflammation, is a complex network of events that strives to attain homeostasis post-inflammation and is regulated by a wide range of mediators (128–133), and inadequate release of these mediators-of-resolution may lead to persistence of inflammation (134). In the case of CHIKV, proteomic profiling of CHIKV infected patients revealed a number of factors that mediate chemotaxis of neutrophils and phagocytes to be downregulated. Azurocidin (AZU1), annexin A1 (ANXA1), CTSG, S-100 calcium-binding proteins S100A7, S100A8, S100A9, and transforming growth factor beta 2 (TGFB2) are significantly downregulated mediators of inflammation found in CHIKV patients with 1–10 days post-onset of symptoms (133). However, a phase-specific analysis of these mediators in CHIKV patients would provide better insight into the mechanism underneath the resolution of inflammation upon CHIKV infection. Also, RNA-seq analysis of feet and lymph nodes of CHIKV infected mice highlighted granzyme A as a major promoter of arthritic inflammation (135).
An important feature that decides resolution of the disease, mainly through regulation of inflammation of joints and attainment of homeostasis, is macrophage polarization and phenotype switching from M1 (pro-inflammatory) to M2 (anti-inflammatory). M1 macrophages restrict the proliferation of surrounding cells and damage infected tissues by releasing pro-inflammatory cytokines, whereas M2 macrophages promote multiplication of contiguous cells and tissue repair (136). This polarization and phenotype switching is regulated majorly by IRF/STAT signaling, tissue microenvironment (hypoxia), oligomerization domain (NOD)-like receptors, cytokine GM-CSF, and NF-κB signaling (136, 137) and has been discussed in detail by Dr. Nan Wang (136). Macrophages exhibiting M2-like activation patterns are found in the CHIKV induced musculoskeletal inflammatory lesions (138) and are known to limit the pro-inflammatory immune response. Arginase 1 (Arg1) is an enzyme associated with M2 macrophages; upon genetic deletion of this enzyme tissue pathology was reduced and enhanced clearance of Ross River virus (RRV) was observed in mice model (138). These results suggest that M2 macrophages play an essential role in the resolution of inflammation, but at the same time M2 macrophages might support the persistence of CHIKV in the system leading to chronic disease. This hypothesis is supported by a study performed on a non-human primate model that suggests long term CHIKV persistence in the macrophages (24).
RNA interference (RNAi) controls the spatial and temporal regulation of the complex gene network involved in the immune response (139). Besides a significant function of silencing of genes related to diseases (140), the viral infection is an example of the complex interplay between viral RNA, endogenous miRNA, and host immune factors (141–144). A recent report suggested that CHIKV establishes infections by regulating miRNA expression profile (145). miR15, miR-16, miR-17, let-7e, miR-125, miR-99, and miR-23a are altered during CHIKV infection which indicates that these act as biomarkers in chikungunya infection (146). Target analysis suggests that targets of these miRNAs are involved in the RIG-1 pathway, TGF-beta-signaling pathway, JAK–STAT-signaling pathway, MAPK-signaling pathway, cytokine–cytokine receptor interactions, and Fc gamma R-mediated phagocytosis (145). In addition, CHIKV infection in human synovial fibroblasts triggers the expression of miR-146a which downregulates the expression of TRAF6, IRAK1, and IRAK2 that in turn leads to decreased activation of NF-κB pathway by a negative feedback loop and promotes CHIKV replication (147, 148). Likewise, miRNAs are also known to participate in macrophage polarization. For example, miR-146a directly binds to the IRF transcription factors and (149, 150) leads to suppression of inflammatory responses by promoting M2 switching. Similarly, miR-210 targets the NF-κB factors and elicits the suppression of M1 switching, resulting in suppression of inflammation (151–153).
Other key players of CHIKD resolution are subsets of T cells, the regulatory T cells (Tregs), and Th17 cells. Differentiation of naïve Th cells into Th17 and Tregs is a common signaling pathway arbitrated by TGF-β; however, the activation of these cells depends on the pro-inflammatory signals which also regulate the fate of these cells in a reciprocal manner (154). In the presence of IL-6 or IL-21, along with TGF-β, naïve Th cells differentiate into Th17 cells, but in the absence of proinflammatory signals, TGF-β drives Treg differentiation (155–157). Th17 cell produces IL-17, IL-22, and IL-23 that attracts neutrophils and hence promotes inflammation at the site of infection (158). In contrast, Treg produces cytokines IL-10 and TGF-β, which suppresses the activity of various immune cells and thereby induces anti-inflammatory responses. Expansion of Tregs, governed by enhanced IL-2 levels and antibody-mediated signaling, also inhibits CD4+ effector T cell repertoire, resulting in alleviation of joint pathology (159). In this way, these two cells function in a yin and yang manner and maintain immune homeostasis during infection. It has been shown that the imbalance in cytokines, such as the increase in the concentration of IL-6 along with TGF-β which results in naïve Th cells differentiating into Th17 cells (160), is a key player in arthritis and rheumatism (161). Several studies have shown a significant elevation in the levels of IL-1β, IL-6, and IL-17 associated with Th17 cells in both CHIKV infected humans and CHIKD mice models (92, 93, 162, 163). Conversely, a study conducted on the patient sera samples from Thailand 2009–2010 outbreak showed no significant variations in IL-17 levels as compared to the healthy controls (164). Recent research on flavivirus using mouse model displaying the genetic diversity of the human population has, however, shown that the susceptibility of the host is not only affected by the viral sequence itself but is also controlled by multiple genes of the host (165). These studies suggest the need for detailed research unveiling the fundamental mechanisms involving immune cells and host genetic diversity as a whole in CHIKD resolution and inflammation.
Another class of regulatory cells that modulates the immune response by producing anti-inflammatory cytokines such as interleukin-10 (IL-10), IL-35, and transforming growth factor β (TGF-β) is a subset of B cells called regulatory B cells (Bregs) (166, 167). Bregs cells inhibit the expansion of pro-inflammatory lymphocytes and other pathogenic T cells (166) by exerting the inhibitory effect on production of pro-inflammatory molecules, such as TNF-α, IFN-γ, and IL-17 from CD4+ T cells (167). However, Bregs also regulate the immune response by modulating the induction of cell death and IgM production, as reported both in humans and mice models (168–171). CD9+ Bregs stimulate differentiation of CD4+ T cells into Tregs by producing high levels of TGF-β (171) which further alleviates the production of TNF-α and induces apoptosis in pro-inflammatory CD4+ T cells. Production of IgM persuades elimination of apoptotic bodies, leading to reduction of pro-inflammatory mediators (172, 173). Bregs have been studied for their role in rheumatoid arthritis (RA) and its regulation seems to be one of the key players that enhance RA disease (174, 175). Considering that chikungunya-induced arthritis is an inflammatory response triggered by the virus that persists in joint tissues resulting in chronic rheumatoid disease (1), it is possible that Bregs might be involved in the regulation of chikungunya-induced RA via modulating inflammatory responses.
Chronic Phase
In several CHIKD cases, inflammation causing joints pain might persist for several months or years, resulting in symptoms similar to rheumatoid arthritis, and is termed as the chronic phase of CHIKD (176–178). Typically, the chronic phase is characterized by joint swelling, joint stiffness, arthralgia, and tendonitis/tenosynovitis (119). Serologically, it marks a significant reduction of the virus as well as of viral RNA, reduced osteoprotegerin, and increased IL-6 and RANKL (receptor activator of NF-κB) ferritin, CRP (C-reactive protein), CXCL9, CXCL1, CHIKV-specific IgG titer IL-12 GM-CSF, IL-17, IL-27, IL-29, IL-8, MCP-1, MIP, CD95/CD95L, CXCL-9, and CXCL-10 (26, 92, 103, 162, 179–182). At this stage, CHIKV virus particles/viral RNA is cleared from the blood; however, studies have reported the persistence of CHIKV RNA in the macrophages (24) and fibroblast during chronic phase (183). Further, the viral dsRNA (intermediate stage during replication) found in the monocytes/macrophages of joints has been reported to trigger an arthritogenic response by activating NF-κB, both in in vitro studies (76), as well as in ankles and wrists of mice Rag−/− (119). Apart from RNA intermediates, studies have reported the persistence of CHIKV specific proteins in host cells over an extended period of time. CHIKV-nsP3 was found to be present as granules along the cell membrane in cells in persistent infection (33), while in another study, CHIKV-capsid proteins were evident in CHIKV infected mice as late as 60 days post-infection, suggesting the active translation of viral proteins during the chronic phase of CHIKD (25).
CD4+ and CD8+ T cells infiltrate the joints in CHIKD, causing inflammation. Specifically, CD4+ T cells are majorly responsible for joint swelling and inflammation and possibly mediate inflammatory pathways through CD4+ T-cell lineages, TH1 and TH2 cells that secrete anti-osteoclastogenic cytokines, IFN-γ, and IL-4. The infiltration of TH 17 cells into the inflammatory joint links the abnormal T-cell response to bone damage in arthritis (184, 185). One study found increased expression of CD95/CD95L in CD4+ cells (92). In CD8+ cells, although cells are nearly constant from healthy to acute to chronic phase, the persistent exposure of antigens to CD8+ T cells leads to altered expression of biomarkers like CD69A, CD107A, and IL-17A (103). As CD8+ T cells only express CD95L after activation (186), it can be safely assumed that these cells stay activated through the chronic phase, hence having a role in disease resolution and chronicity. Furthermore, effective immune cell activation drives the initial antiviral response leading to T-cell exhaustion, and this may be linked to viral persistence observed in the chronic phase (26). Additionally, the absence of CD8+ T cells in synovial tissue may also be a contributing factor toward the persistence of CHIKV (26).
Another hallmark of the chronic phase is bone erosion and degradation of extracellular matrix in joints, causing chronic arthralgia in CHIKD patients. Key players of the mechanism involved in bone erosion are resident macrophages, CCL2, IL-6, RANKL (187), osteoprotegerin, TRAF6, NFATc1, calcitonin receptors, cathepsin K, and β3 integrin (184, 188); degradation of the extracellular matrix, meanwhile, is mediated by several factors such as fibroblast-like synoviocytes IL-1, TNF-α, Janus kinase, ERK, c-jun, and c-fos. These molecules, in turn, activate matrix metalloproteinases (MMP) in chondrocytes that promote extracellular matrix degradation during the chronic phase (189) (Figure 3).
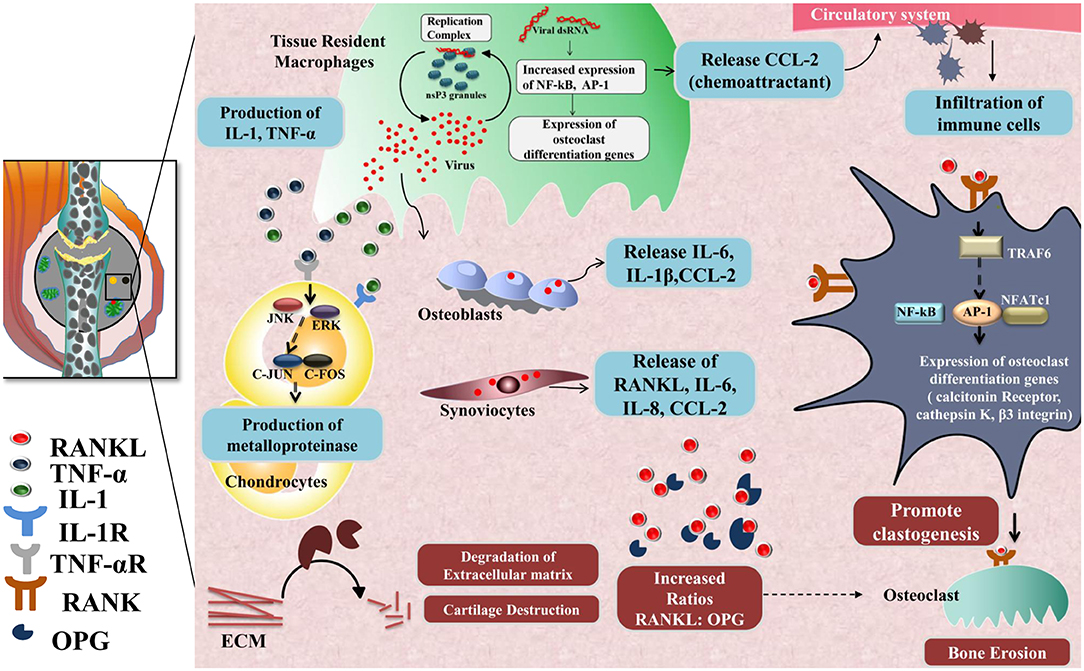
Figure 3. Chronic phase-CHIKV mediated joint pathology: in the joints, osteoblast cells (Bone-forming cells) are susceptible to CHIKV infection causing over-expression of IL-6 and RANKL and inhibition of osteoprotegerin (OPG). Also, fibroblast-like synoviocytes (HFLS) that are present in the synovial membrane lining the synovial joints, secrete enhanced levels of RANKL, IL-6, IL-8, and (MCP-1), upon CHIKV infection resulting in higher RANKL: OPG ratio, which favors osteoclast formation from precursor cells. RANKL and OPG maintain bone homeostasis as RANKL promotes osteoclastogenesis. RANKL recruits TRAF6 which induces auto amplification of NFATc1 (a transcription factor) via NF-κB and c-fos pathway and dependent on calcium signaling of ITAMs clastogenesis. NFATc1 in complex with AP-1 regulates the expression of genes involved in osteoclast differentiation e.g., calcitonin receptor, cathepsin K, and β3 integrin. Secretion of IL-1 and TNF-α by CHIKV infected resident macrophages phosphorylate JNK and ERK which in turn activates AP-1 family member, c-jun, which dimerize with c-fos and induce transcription of matrix metalloproteinases (MMP) that degrades components of extracellular matrix by collagenase activity.
Conclusion
Complexities in CHIKD immunopathology in the host begins from the point of mosquito saliva that may contain both the virus as well as the viral antibodies that may affect the virus pathogenicity when delivered into the host during a bite. Furthermore, the immunomodulatory role of mosquito saliva is strikingly an understudied phenomenon in CHIKV infection. Does the mosquito saliva possess factors that can actively modulate viral pathogenesis? Can these molecules be utilized for developing novel antiviral strategies? How exactly does the presence of viral antibodies from previous bites affect CHIKV pathogenicity during the next bite to a naïve individual? Even though some of these aspects have been looked into, in-depth studies are necessary to address these concerns more effectively. Additionally, answering some of these questions may help in arriving on novel transmission blocking strategies.
Once the infection is established, the innate and adaptive host immunity plays definitive roles in the progression of the disease that we know to be both protective as well as pathogenic in its response. These responses decide the clinical presentation of the disease at the febrile acute phase in terms of peak viremia, small joint and large joint involvement, as well as disease resolution during the post-acute phase. The current understanding of the subject suggests that arthritogenic symptoms in the acute phase of CHIKD are primarily due to pro-inflammatory host responses that could lead to the possible association of severe chronic cases with autoimmunity. Can we derive more knowledge regarding this association by looking into other similar autoimmune disorders such as rheumatoid arthritis? Will employing animal models specific to autoimmune disorders help in answering some immunological questions pertaining to immune response of CHIKV-induced arthritis? Can these systems recapitulate the actual pathogenesis seen in humans? How well are we using the clinical evidence to understand disease progression in the case of CHIKD? How do we use the information pertaining to inflammation more usefully in understanding CHIKD immunopathology? Can we devise intervention approaches based on this information pertaining to inflammation specifically to alleviate the arthritic phase?
The resolution of CHIKD is not yet fully understood and would be the most interesting area to be studied in future, as there are several well-established mediators of inflammation that have not been studied in the context of CHIKV chronic infections. This review has strived to emphasize the various immunological aspects that may lead to disease resolution/progression in CHIKD and highlight the lacuna present currently in the field.
Author Contributions
SS: conceptualization, supervision, and funding acquisition. PS, AK, AH, DM, RK, CS, and SS: methodology, data curation, writing-original draft preparation, and writing-review and editing.
Funding
This research was funded by ICGEB Core funds.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Suhrbier A, Jaffar-Bandjee C, Gasque P. Arthritogenic alphaviruses—an overview. Nat Rev Rheumatol. (2012) 8:420. doi: 10.1038/nrrheum.2012.64
2. Wahid B, Ali A, Rafique S, Idrees M. Global expansion of chikungunya virus: mapping the 64-year history. Int J Infect Dis. (2017) 58:69–76. doi: 10.1016/j.ijid.2017.03.006
3. Powers AM, Logue CH. Changing patterns of chikungunya virus: re-emergence of a zoonotic arbovirus. J Genrl Virol. (2007) 88:2363–77. doi: 10.1099/vir.0.82858-0
4. Leparc-Goffart I, Nougairede A, Cassadou S, Prat C, de Lamballerie X. Chikungunya in the Americas. Lancet. (2014) 383:514. doi: 10.1016/S0140-6736(14)60185-9
5. Rao TR, Carey DE, Pavri KM. Preliminary isolation and identification of chikungunya virus from cases of dengue-like illness in Madras city. Indian J Med Res. (1965) 53:689–93.
6. Schuffenecker I, Iteman I, Michault A, Murri S, Frangeul L, Vaney C, et al. Genome microevolution of chikungunya viruses causing the Indian Ocean outbreak. PLoS Med. (2006) 3:e263. doi: 10.1371/journal.pmed.0030263
7. Vazeille M, Moutailler S, Coudrier D, Rousseaux C, Khun H, Huerre M, et al. Two chikungunya isolates from the outbreak of La Reunion (Indian Ocean) exhibit different patterns of infection in the mosquito, Aedes albopictus. PLoS ONE. (2007) 2:e1168. doi: 10.1371/journal.pone.0001168
8. Vazeille M, Jeannin C, Martin E, Schaffner FA, Failloux B. Chikungunya: a risk for Mediterranean countries? Acta Tropica. (2008) 105:200–2. doi: 10.1016/j.actatropica.2007.09.009
9. Tsetsarkin KA, Vanlandingham DL, McGee CE, Higgs S. A single mutation in chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog. (2007) 3:e201. doi: 10.1371/journal.ppat.0030201
10. Weaver SC, Forrester NL. Chikungunya: evolutionary history and recent epidemic spread. Antiviral Res. (2015) 120:32–9. doi: 10.1016/j.antiviral.2015.04.016
11. Pialoux GB, Gaüzère A, Jauréguiberry S, Strobel M. Chikungunya, an epidemic arbovirosis. Lancet Infect Dis. (2007) 7:319–27. doi: 10.1016/S1473-3099(07)70107-X
12. Robinson MC. An epidemic of virus disease in Southern Province, Tanganyika territory, in 1952–1953. Trans R Soc Trop Med Hyg. (1955) 49:28–32. doi: 10.1016/0035-9203(55)90080-8
13. Brighton S, Prozesky O, De La Harpe A. Chikungunya virus infection-A retrospective study of 107 cases. South African Med J. (1983) 68:313–5.
14. Borgherini G, Poubeau P, Jossaume A, Gouix A, Cotte L, Michault A, et al. Persistent arthralgia associated with chikungunya virus: a study of 88 adult patients on reunion island. Clin Infect Dis. (2008) 47:469–75. doi: 10.1086/590003
15. Javelle E, Ribera A, Degasne I, Gaüzère A, Marimoutou C, Simon F. Specific management of post-chikungunya rheumatic disorders: a retrospective study of 159 cases in Reunion Island from 2006-2012. PLoS Neglect Trop D. (2015) 9:e0003603. doi: 10.1371/journal.pntd.0003603
16. Tournebize P, Charlin C, Lagrange M. Neurological manifestations in chikungunya: about 23 cases collected in Reunion Island. Rev Neurol. (2009) 165:48–51. doi: 10.1016/j.neurol.2008.06.009
17. Rampal MS, Meena H. Neurological complications in chikungunya fever. J Assoc Physicians India. (2007) 55:765–9. Available online at: https://europepmc.org/article/MED/31312203#free-full-text
18. Balavoine S, Pircher M, Hoen B, Herrmann-Storck C, Najioullah F, Madeux B, et al. Guillain–barré syndrome and chikungunya: description of all cases diagnosed during the 2014. outbreak in the French West Indies. Am J Trop Med Hyg. (2017) 97:356–60. doi: 10.4269/ajtmh.15-0753
19. Mehta R, Gerardin P, de Brito CAA, Soares CN, Ferreira LB, Solomon T. The neurological complications of chikungunya virus: a systematic review. Rev Med Virol. (2018) 28:e1978. doi: 10.1002/rmv.1978
20. Renault P, Solet JL, Sissoko D, Balleydier E, Larrieu S, Filleul L, et al. A major epidemic of chikungunya virus infection on Reunion Island, France, 2005-2006. Am J Trop Med Hyg. (2007) 77:727–31. doi: 10.4269/ajtmh.2007.77.727
21. Hsu CH, Cruz-Lopez F, Torres DV, Perez-Padilla J, Lorenzi OD, Rivera A, et al. Risk factors for hospitalization of patients with chikungunya virus infection at sentinel hospitals in Puerto Rico. PLoS Neglect Trop D. (2019) 13:e0007084. doi: 10.1371/journal.pntd.0007084
22. Ramful D, Carbonnier M, Pasquet M, Bouhmani B, Ghazouani J, Noormahomed T, et al. Mother-to-child transmission of chikungunya virus infection. Pediatr Infect Dis J. (2007) 26:811–5. doi: 10.1097/INF.0b013e3180616d4f
23. Gérardin P, Barau G, Michault A, Bintner M, Randrianaivo H, Choker G, et al. Multidisciplinary prospective study of mother-to-child chikungunya virus infections on the island of La Reunion. PLoS Med. (2008) 5:e60. doi: 10.1371/journal.pmed.0050060
24. Labadie K, Larcher T, Joubert C, Mannioui A, Delache B, Brochard P, et al. Chikungunya disease in nonhuman primates involves long-term viral persistence in macrophages. J Clin Invest. (2010) 120:894–906. doi: 10.1172/JCI40104
25. Poo YS, Rudd PA, Gardner J, Wilson JA, Larcher T, Colle A, et al. Multiple immune factors are involved in controlling acute and chronic chikungunya virus infection. PLoS Neglect Trop Dis. (2014) 8:e3354. doi: 10.1371/journal.pntd.0003354
26. Hoarau JJ, Bandjee MCJ, Trotot PK, Das T, Li-Pat-Yuen G, Dassa B, et al. Persistent chronic inflammation and infection by chikungunya arthritogenic alphavirus in spite of a robust host immune response. J Immunol. (2010) 184:5914–27. doi: 10.4049/jimmunol.0900255
27. White LK, Sali T, Alvarado D, Gatti E, Pierre P, Streblow D, et al. Chikungunya virus induces IPS-1-dependent innate immune activation and protein kinase R-independent translational shutoff. J Virol. (2011) 85:606–20. doi: 10.1128/JVI.00767-10
28. Akahata W, Yang Y, Andersen H, Sun S, Holdaway HA, Kong P, et al. A virus-like particle vaccine for epidemic chikungunya virus protects nonhuman primates against infection. Nat Med. (2010) 16:334. doi: 10.1038/nm.2105
29. Carpentier KS, Morrison TE. Innate immune control of alphavirus infection. Curr Opin Virol. (2018) 28:53–60. doi: 10.1016/j.coviro.2017.11.006
30. Long KM, Heise MT. Protective and pathogenic responses to chikungunya virus infection. Curr Trop Med Rep. (2015) 2:13–21. doi: 10.1007/s40475-015-0037-z
31. Gérardin P, Fianu A, Michault A, Mussard C, Boussaïd K, Rollot O, et al. Predictors of chikungunya rheumatism: a prognostic survey ancillary to the TELECHIK cohort study. Arthritis Res Ther. (2013) 15:R9. doi: 10.1186/ar4137
32. Hoarau J-J, Gay F, Pellé O, Samri A, Jaffar-Bandjee M-C, Gasque P, et al. Identical strength of the T cell responses against E2, nsP1 and capsid CHIKV proteins in recovered and chronic patients after the epidemics of 2005-2006 in La Reunion Island. PLoS ONE. (2013) 8:e84695. doi: 10.1371/journal.pone.0084695
33. Remenyi R, Gao Y, Hughes RE, Curd A, Zothner C, Peckham M, et al. Persistent replication of a chikungunya virus replicon in human cells is associated with presence of stable cytoplasmic granules containing nonstructural protein 3. J Virol. (2018) 92:e00477–18. doi: 10.1128/JVI.00477-18
34. Staples JE, Breiman RF, Powers AM. Chikungunya fever: an epidemiological review of a re-emerging infectious disease. Clin Infect Dis. (2009) 49:942–8. doi: 10.1086/605496
35. Mohan A, Kiran D, Manohar IC, Kumar DP. Epidemiology, clinical manifestations, and diagnosis of chikungunya fever: lessons learned from the re-emerging epidemic. Indian J Dermatol. (2010) 55:54. doi: 10.4103/0019-5154.60355
36. Tanabe IS, Tanabe EL, Santos EC, Martins WV, Araújo IM, Cavalcante MC, et al. Cellular and molecular immune response to chikungunya virus infection. Front Cell Infect Microbiol. (2018) 8:345. doi: 10.3389/fcimb.2018.00345
37. Álvarez-Argüelles ME, Alba SR, Pérez MR, Riveiro JAB, García SM. Diagnosis and molecular characterization of chikungunya virus infections. In: Rodriguez-Morales AJ, editor. Current Topics in Neglected Tropical Diseases. Colombia: Intechopen (2019). p. 34–57.
38. Paul BJ, Sadanand S. Chikungunya infection: a re-emerging epidemic. Rheumatol Ther. (2018) 5:317–26. doi: 10.1007/s40744-018-0121-7
39. Briant L, Desprès P, Choumet V, Missé D. Role of skin immune cells on the host susceptibility to mosquito-borne viruses. Virology. (2014) 464:26–32. doi: 10.1016/j.virol.2014.06.023
40. Styer LM, Kent KA, Albright RG, Bennett CJ, Kramer LD, Bernard KA. Mosquitoes inoculate high doses of West Nile virus as they probe and feed on live hosts. PLoS Pathog. (2007) 3:1262–70. doi: 10.1371/journal.ppat.0030132
41. Hayday AC. [gamma][delta] cells: a right time and a right place for a conserved third way of protection. Annu Rev Immunol. (2000) 18:975–1026. doi: 10.1146/annurev.immunol.18.1.975
42. Born W, Cady C, Jones-Carson J, Mukasa A, Lahn M, O'brien R. Immunoregulatory functions of γδ T cells. Adv Immunol. (1998). 71:77–144. doi: 10.1016/S0065-2776(08)60400-9
43. Carding SR, Egan PJ. γδ T cells: functional plasticity and heterogeneity. Nat Rev Immunol. (2002) 2:336. doi: 10.1038/nri797
44. Long KM, Ferris MT, Whitmore AC, Montgomery SA, Thurlow LR, McGee CE, et al. γδ T cells play a protective role in chikungunya virus-induced disease. J Virol. (2016) 90:433–43. doi: 10.1128/JVI.02159-15
45. Fu YX, Roark CE, Kelly K, Drevets D, Campbell P, O'Brien R, et al. Immune protection and control of inflammatory tissue necrosis by gamma delta T cells. J Immunol. (1994) 153:3101–15.
46. Souza DCD, Cooper AM, Frank AA, Mazzaccaro RJ, Bloom BR, Orme IM. An anti-inflammatory role for gamma delta T lymphocytes in acquired immunity to Mycobacterium tuberculosis. J Immunol. (1997) 158:1217–21.
47. Pingen M, Bryden SR, Pondeville E, Schnettler E, Kohl A, Merits A, et al. Host inflammatory response to mosquito bites enhances the severity of arbovirus infection. Immunity. (2016) 44:1455–69. doi: 10.1016/j.immuni.2016.06.002
48. Reagan KL, Machain-Williams C, Wang T, Blair CD. Immunization of mice with recombinant mosquito salivary protein D7 enhances mortality from subsequent West Nile virus infection via mosquito bite. PLoS Neglect Trop Dis. (2012) 6:e1935. doi: 10.1371/journal.pntd.0001935
49. Pingen M, Schmid MA, Harris E, McKimmie CS. Mosquito biting modulates skin response to virus infection. Trends Parasitol. (2017) 33:645–57. doi: 10.1016/j.pt.2017.04.003
50. Schneider BS, Higgs S. The enhancement of arbovirus transmission and disease by mosquito saliva is associated with modulation of the host immune response. Trans R Soc Trop Med Hyg. (2008) 102:400–8. doi: 10.1016/j.trstmh.2008.01.024
51. Styer LM, Lim PY, Louie KL, Albright RG, Kramer LD, Bernard KA. Mosquito saliva causes enhancement of West Nile virus infection in mice. J Virol. (2011) 85:1517–27. doi: 10.1128/JVI.01112-10
52. Schneider BS, Soong L, Zeidner NS, Higgs S. Aedes aegypti salivary gland extracts modulate anti-viral and TH1/TH2 cytokine responses to sindbis virus infection. Viral Immunol. (2004) 17:565–73. doi: 10.1089/vim.2004.17.565
53. Thangamani S, Higgs S, Ziegler S, Vanlandingham D, Tesh R, Wikel S. Host immune response to mosquito-transmitted chikungunya virus differs from that elicited by needle inoculated virus. PLoS ONE. (2010) 5:e12137. doi: 10.1371/journal.pone.0012137
54. McCracken M, Christofferson R, Grasperge B, Calvo E, Chisenhall D, Mores C. Aedes aegypti salivary protein “aegyptin” co-inoculation modulates dengue virus infection in the vertebrate host. Virology. (2014) 468:133–39. doi: 10.1016/j.virol.2014.07.019
55. Henrique MO, Neto LS, Assis JB, Barros MS, Capurro ML, Lepique AP, et al. Evaluation of inflammatory skin infiltrate following Aedes aegypti bites in sensitized and non-sensitized mice reveals saliva-dependent and immune-dependent phenotypes. Immunology. (2019) 158:47–59. doi: 10.1111/imm.13096
56. Bryden SR, Pingen M, Lefteri DA, Miltenburg J, Delang L, Jacobs S, et al. Pan-viral protection against arboviruses by activating skin macrophages at the inoculation site. Sci Transl Med. (2020) 12:eaax2421. doi: 10.1126/scitranslmed.aax2421
57. Schilte C, Couderc T, Chretien F, Sourisseau M, Gangneux N, Guivel-Benhassine F, et al. Type I IFN controls chikungunya virus via its action on nonhematopoietic cells. J Exp Med. (2010) 207:429–42. doi: 10.1084/jem.20090851
58. Harrison V, Binn L, Randall R. Comparative immunogenicities of chikungunya vaccines prepared in avian and mammalian tissues. Am J Trop Med Hyg. (1967) 16:786–91. doi: 10.4269/ajtmh.1967.16.786
59. Schwartz O, Albert ML. Biology and pathogenesis of chikungunya virus. Nat Rev Microbiol. (2010) 8:491. doi: 10.1038/nrmicro2368
60. Jin J, Galaz-Montoya JG, Sherman MB, Sun SY, Goldsmith CS, O'Toole ET, et al. Neutralizing antibodies inhibit chikungunya virus budding at the plasma membrane. Cell Host Microbe. (2018) 24:417–28. doi: 10.1016/j.chom.2018.07.018
61. da Cunha RV, Trinta KS. Chikungunya virus: clinical aspects and treatment-a review. Mem Inst Oswaldo Cruz. (2017) 112:523–31. doi: 10.1590/0074-02760170044
62. Marques DL, Duarte ALBP, Ranzolin A, Dantas AT, Cavalcanti NG, Gonçalves SG, et al. Recommendations of the Brazilian Society of Rheumatology for diagnosis and treatment of chikungunya fever. Part 1-Diagnosis and special situations. Rev Bras Reumatol Engl Ed. (2017) 57:s421–37. doi: 10.1016/j.rbre.2017.05.006
63. Habjan M, Pichlmair A. Cytoplasmic sensing of viral nucleic acids. Curr Opin Virol. (2015) 11:31–7. doi: 10.1016/j.coviro.2015.01.012
64. Jensen S, Thomsen AR. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J Virol. (2012) 86:2900–10. doi: 10.1128/JVI.05738-11
65. Chen W, Foo SS, Zaid A, Teng TS, Herrero LJ, Wolf S, et al. Specific inhibition of NLRP3 in chikungunya disease reveals a role for inflammasomes in alphavirus-induced inflammation. Nat Microbiol. (2017) 2:1435–45. doi: 10.1038/s41564-017-0015-4
66. Ekchariyawat P, Hamel R, Bernard E, Wichit S, Surasombatpattana P, Talignani L, et al. Inflammasome signaling pathways exert antiviral effect against chikungunya virus in human dermal fibroblasts. Infect Genet Evol. (2015) 32:401–8. doi: 10.1016/j.meegid.2015.03.025
67. Olagnier D, Scholte FE, Chiang C, Albulescu IC, Nichols C, He Z, et al. Inhibition of dengue and chikungunya virus infections by RIG-I-mediated type I interferon-independent stimulation of the innate antiviral response. J Virol. (2014) 88:4180–94. doi: 10.1128/JVI.03114-13
68. Priya R, Patro I, Parida M. TLR3 mediated innate immune response in mice brain following infection with chikungunya virus. Virus Res. (2014) 189:194–205. doi: 10.1016/j.virusres.2014.05.010
69. Pryke KM, Abraham J, Sali TM, Gall BJ, Archer I, Liu A, et al. A novel agonist of the TRIF pathway induces a cellular state refractory to replication of Zika, chikungunya, and dengue viruses. MBio. (2017) 8:e00452–17. doi: 10.1128/mBio.00452-17
70. Rudd PA, Wilson J, Gardner J, Larcher T, Babarit C, Le TT, et al. Interferon response factors 3 and 7 protect against chikungunya virus hemorrhagic fever and shock. J Virol. (2012) 86:9888–98. doi: 10.1128/JVI.00956-12
71. Teng TS, Foo S, Simamarta D, Lum FM, Teo TH, Lulla A, et al. Viperin restricts chikungunya virus replication and pathology. J Clin Invest. (2012) 122:4447–60. doi: 10.1172/JCI63120
72. Sourisseau M, Schilte C, Casartelli N, Trouillet C, Guivel-Benhassine F, Rudnicka D, et al. Characterization of reemerging chikungunya virus. PLoS Pathog. (2007) 3:e89. doi: 10.1371/journal.ppat.0030089
73. Wikan N, Sakoonwatanyoo P, Ubol S, Yoksan S, Smith DR. Chikungunya virus infection of cell lines: analysis of the East, Central and South African lineage. PLoS ONE. (2012) 7:e31102. doi: 10.1371/journal.pone.0031102
74. Krejbich-Trotot P, Gay B, Li-Pat-Yuen G, Hoarau JJ, Jaffar-Bandjee MC, Briant L, et al. Chikungunya triggers an autophagic process which promotes viral replication. Virol J. (2011) 8:432. doi: 10.1186/1743-422X-8-432
75. Her Z, Malleret B, Chan M, Ong EK, Wong S-C, Kwek DJ, et al. Active infection of human blood monocytes by chikungunya virus triggers an innate immune response. J Immunol. (2010) 184:5903–13. doi: 10.4049/jimmunol.0904181
76. Zare F, Bokarewa M, Nenonen N, Bergstrom T, Alexopoulou L, Flavell RA, et al. Arthritogenic properties of double-stranded (viral) RNA. J Immunol. (2004) 172:5656–63. doi: 10.4049/jimmunol.172.9.5656
77. Dupuis-Maguiraga L, Noret M, Brun S, Le Grand R, Gras G, Roques P. Chikungunya disease: infection-associated markers from the acute to the chronic phase of arbovirus-induced arthralgia. PLoS Negl Trop Dis. (2012) 6:e1446. doi: 10.1371/journal.pntd.0001446
78. Cushing SD, Berliner JA, Valente AJ, Territo MC, Navab M, Parhami F, et al. Minimally modified low density lipoprotein induces monocyte chemotactic protein 1 in human endothelial cells and smooth muscle cells. Proc Natl Acad Sci USA. (1990) 87:5134–8. doi: 10.1073/pnas.87.13.5134
79. Standiford TJ, Kunkel SL, Phan SH, Rollins BJ, Strieter RM. Alveolar macrophage-derived cytokines induce monocyte chemoattractant protein-1 expression from human pulmonary type II-like epithelial cells. J Biol Chem. (1991) 266:9912–8.
80. Brown Z, Strieter RM, Neild GH, Thompson RC, Kunkel SL, Westwick J. IL-1 receptor antagonist inhibits monocyte chemotactic peptide 1 generation by human mesangial cells. Kidney Int. (1992) 42:95–101. doi: 10.1038/ki.1992.266
81. Barna BP, Pettay J, Barnett GH, Zhou P, Iwasaki K, Estes ML. Regulation of monocyte chemoattractant protein-1 expression in adult human non-neoplastic astrocytes is sensitive to tumor necrosis factor (TNF) or antibody to the 55-kDa TNF receptor. J Neuroimmunol. (1994) 50:101–7. doi: 10.1016/0165-5728(94)90220-8
82. Rulli NE, Rolph MS, Srikiatkhachorn A, Anantapreecha S, Guglielmotti A, Mahalingam S. Protection from arthritis and myositis in a mouse model of acute chikungunya virus disease by bindarit, an inhibitor of monocyte chemotactic protein-1 synthesis. J Infect Dis. (2011) 204:1026–30. doi: 10.1093/infdis/jir470
83. Poo YS, Nakaya H, Gardner J, Larcher T, Schroder WA, Le TT, et al. CCR2 deficiency promotes exacerbated chronic erosive neutrophil-dominated chikungunya virus arthritis. J Virol. (2014) 88:6862–72. doi: 10.1128/JVI.03364-13
84. Schönrich G, Raftery MJ. Neutrophil extracellular traps go viral. Front Immunol. (2016) 7:366. doi: 10.3389/fimmu.2016.00366
85. Bian Z, Guo Y, Ha B, Zen K, Liu Y. Regulation of the inflammatory response: enhancing neutrophil infiltration under chronic inflammatory conditions. J Immunol. (2012) 188:844–53. doi: 10.4049/jimmunol.1101736
86. Hiroki CH, Toller-Kawahisa JE, Fumagalli MJ, Colón DF, Figueiredo LT, Fonseca BA, et al. Neutrophil extracellular traps effectively control acute chikungunya virus infection. Front Immunol. (2019) 10:3108. doi: 10.3389/fimmu.2019.03108
87. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA–peptide complexes in systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra19. doi: 10.1126/scitranslmed.3001180
88. Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol. (2015) 15:471. doi: 10.1038/nri3865
89. Webster B, Werneke SW, Zafirova B, This S, Coléon S, Decembre E, et al. Plasmacytoid dendritic cells control dengue and chikungunya virus infections via IRF7-regulated interferon responses. Elife. (2018) 7:e34273. doi: 10.7554/eLife.34273
90. Petitdemange C, Becquart P, Wauquier N, Beziat V, Debre P, Leroy EM, et al. Unconventional repertoire profile is imprinted during acute chikungunya infection for natural killer cells polarization toward cytotoxicity. PLoS Pathog. (2011) 7:e1002268. doi: 10.1371/journal.ppat.1002268
91. Nielsen CM, White MJ, Goodier MR, Riley EM. Functional significance of CD57 expression on human NK cells and relevance to disease. Front Immunol. (2013) 4:422. doi: 10.3389/fimmu.2013.00422
92. Wauquier N, Becquart P, Nkoghe D, Padilla C, Ndjoyi-Mbiguino A, Leroy EM. The acute phase of chikungunya virus infection in humans is associated with strong innate immunity and T CD8 cell activation. J Infect Dis. (2011) 204:115–23. doi: 10.1093/infdis/jiq006
93. Gardner J, Anraku I, Le TT, Larcher T, Major L, Roques P, et al. Chikungunya virus arthritis in adult wild-type mice. J Virol. (2010) 84:8021–32. doi: 10.1128/JVI.02603-09
94. Morrison TE, Oko L, Montgomery SA, Whitmore AC, Lotstein AR, Gunn BM, et al. A mouse model of chikungunya virus–induced musculoskeletal inflammatory disease: evidence of arthritis, tenosynovitis, myositis, and persistence. Am J Pathol. (2011) 178:32–40. doi: 10.1016/j.ajpath.2010.11.018
95. Teo TH, Lum M, Claser C, Lulla V, Lulla A, Merits A, et al. A pathogenic role for CD4+ T cells during chikungunya virus infection in mice. J Immunol. (2013) 190:259–69. doi: 10.4049/jimmunol.1202177
96. Panning M, Grywna K, Van Esbroeck M, Emmerich P, Drosten C. Chikungunya fever in travelers returning to Europe from the Indian Ocean region, (2006). Emerg Infect Dis. (2008) 14:416. doi: 10.3201/eid1403.070906
97. Jain J, Nayak K, Tanwar N, Gaind R, Gupta B, Shastri J, et al. Clinical, serological, and virological analysis of 572 chikungunya patients from 2010 to 2013 in India. Clin Infect Dis. (2017) 65:133–40. doi: 10.1093/cid/cix283
98. Kam YW, Lum FM, Teo TH, Lee WW, Simarmata D, Harjanto S, et al. Early neutralizing IgG response to chikungunya virus in infected patients targets a dominant linear epitope on the E2 glycoprotein. EMBO Mol Med. (2012) 4:330–43. doi: 10.1002/emmm.201200213
99. Kam W, Simarmata D, Chow A, Her Z, Teng S, Ong EK, et al. Early appearance of neutralizing immunoglobulin G3 antibodies is associated with chikungunya virus clearance and long-term clinical protection. J Infect Dis. (2012) 205:1147–54. doi: 10.1093/infdis/jis033
100. Lum M, Teo H, Lee WW, Kam W, Rénia L, Ng LF. An essential role of antibodies in the control of chikungunya virus infection. J Immunol. (2013) 190:6295–302. doi: 10.4049/jimmunol.1300304
101. Fox JM, Diamond MS. Immune-mediated protection and pathogenesis of chikungunya virus. J Immunol. (2016) 197:4210–8. doi: 10.4049/jimmunol.1601426
102. Wolint P, Betts MR, Koup RA, Oxenius A. Immediate cytotoxicity but not degranulation distinguishes effector and memory subsets of CD8+ T cells. J Exp Med. (2004) 199:925–36. doi: 10.1084/jem.20031799
103. Dias NS, Gois BM, Lima VS, Guerra-Gomes IC, Araujo MG, Gomes AS, et al. Human CD8 T-cell activation in acute and chronic chikungunya infection. Immunology. (2018) 155:499–504. doi: 10.1111/imm.12992
104. Couderc T, Khandoudi N, Grandadam M, Visse C, Gangneux N, Bagot S, et al. Prophylaxis and therapy for chikungunya virus infection. J Infect Dis. (2009) 200:516–23. doi: 10.1086/600381
105. Tandale BV, Sathe PS, Arankalle VA, Wadia RS, Kulkarni R, Shah SV, et al. Systemic involvements and fatalities during chikungunya epidemic in India, (2006). J Clin Virol. (2009) 46:145–9. doi: 10.1016/j.jcv.2009.06.027
106. Johnson BW, Russell BJ, Goodman CH. Laboratory diagnosis of chikungunya virus infections and commercial sources for diagnostic assays. J Infect Dis. (2016) 214:S471–4. doi: 10.1093/infdis/jiw274
107. Jain J, Narayanan V, Kumar A, Shrinet J, Srivastava P, Chaturvedi S, et al. Establishment and comparison of pathogenicity and related neurotropism in two age groups of immune competent mice, C57BL/6J using an Indian isolate of chikungunya virus (CHIKV). Viruses. (2019) 11:578. doi: 10.3390/v11060578
108. Pierro A, Rossini G, Gaibani P, Finarelli AC, Moro ML, Landini MP, et al. Persistence of anti–chikungunya virus–specific antibodies in a cohort of patients followed from the acute phase of infection after the 2007 outbreak in Italy. N Microbes N Infect. (2015) 7:23–5. doi: 10.1016/j.nmni.2015.04.002
109. Malvy D, Ezzedine K, Mamani-Matsuda M, Autran B, Tolou H, Receveur MC, et al. Destructive arthritis in a patient with chikungunya virus infection with persistent specific IgM antibodies. BMC Infect Dis. (2009) 9:200. doi: 10.1186/1471-2334-9-200
110. Mizuno Y, Kato Y, Takeshita N, Ujiie M, Kobayashi T, Kanagawa S, et al. Clinical and radiological features of imported chikungunya fever in Japan: a study of six cases at the National Center for Global Health and Medicine. J Infect Chemother. (2011) 17:419–3. doi: 10.1007/s10156-010-0124-y
111. Schilte C, Staikowsky F, Couderc T, Madec Y, Carpentier F, Kassab S, et al. Chikungunya virus-associated long-term arthralgia: a 36-month prospective longitudinal study. PLoS Negl Trop Dis. (2013) 7:e2137. doi: 10.1371/journal.pntd.0002137
112. Long F, Fong RH, Austin SK, Chen Z, Klose T, Fokine A, et al. Cryo-EM structures elucidate neutralizing mechanisms of anti-chikungunya human monoclonal antibodies with therapeutic activity. Proc Natl Acad Sci USA. (2015) 112:13898–903. doi: 10.1073/pnas.1515558112
113. Fox JM, Long F, Edeling MA, Lin H, van Duijl-Richter MK, Fong RH, et al. Broadly neutralizing alphavirus antibodies bind an epitope on E2 and inhibit entry and egress. Cell. (2015) 163:1095–107. doi: 10.1016/j.cell.2015.10.050
114. Smith SA, Silva LA, Fox JM, Flyak AI, Kose N, Sapparapu G, et al. Isolation and characterization of broad and ultrapotent human monoclonal antibodies with therapeutic activity against chikungunya virus. Cell Host Microbe. (2015) 18:86–95. doi: 10.1016/j.chom.2015.06.009
115. Hawman DW, Fox JM, Ashbrook AW, May NA, Schroeder KM, Torres RM, et al. Pathogenic chikungunya virus evades B cell responses to establish persistence. Cell Rep. (2016) 16:1326–38. doi: 10.1016/j.celrep.2016.06.076
116. Broeckel R, Fox JM, Haese N, Kreklywich CN, Sukulpovi-Petty S, Legasse A, et al. Therapeutic administration of a recombinant human monoclonal antibody reduces the severity of chikungunya virus disease in rhesus macaques. PLoS Neglect Trop Dis. (2017) 11:e0005637. doi: 10.1371/journal.pntd.0005637
117. Pal P, Dowd KA, Brien JD, Edeling MA, Gorlatov S, Johnson S, et al. Development of a highly protective combination monoclonal antibody therapy against chikungunya virus. PLoS Pathog. (2013) 9:e1003312. doi: 10.1371/journal.ppat.1003312
118. Fric J, Bertin-Maghit S, Wang CI, Nardin A, Warter L. Use of human monoclonal antibodies to treat chikungunya virus infection. J Infect Dis. (2013) 207:319–22. doi: 10.1093/infdis/jis674
119. Hawman DW, Stoermer KA, Montgomery SA, Pal P, Oko L, Diamond MS, et al. Chronic joint disease caused by persistent chikungunya virus infection is controlled by the adaptive immune response. J Virol. (2013) 87:13878–88. doi: 10.1128/JVI.02666-13
120. Guilliams M, Bruhns P, Saeys Y, Hammad H, Lambrecht BN. The function of Fcgamma receptors in dendritic cells and macrophages. Nat Rev Immunol. (2014) 14:94–108. doi: 10.1038/nri3582
121. Kubes P, Jenne C. Immune responses in the liver. Annu Rev Immunol. (2018) 36:247–77. doi: 10.1146/annurev-immunol-051116-052415
122. van Lookeren Campagne M, Wiesmann C, Brown EJ. Macrophage complement receptors and pathogen clearance. Cell Microbiol. (2007) 9:2095–102. doi: 10.1111/j.1462-5822.2007.00981.x
123. Carpentier KS, Davenport BJ, Haist KC, McCarthy MK, May NA, Robison A, et al. Discrete viral E2 lysine residues and scavenger receptor MARCO are required for clearance of circulating alphaviruses. Elife. (2019) 8:e49163. doi: 10.7554/eLife.49163
124. Kitamura D, Roes J, Kuhn R, Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature. (1991) 350:423–6. doi: 10.1038/350423a0
125. Wessels MR, Butko P, Ma M, Warren HB, Lage AL, Carroll MC. Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc Natl Acad Sci USA. (1995) 92:11490–4. doi: 10.1073/pnas.92.25.11490
126. Canton J, Neculai D, Grinstein S. Scavenger receptors in homeostasis and immunity. Nat Rev Immunol. (2013) 13:621–34. doi: 10.1038/nri3515
127. Mukhopadhyay S, Gordon S. The role of scavenger receptors in pathogen recognition and innate immunity. Immunobiology. (2004) 209:39–49. doi: 10.1016/j.imbio.2004.02.004
128. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. (2014) 510:92. doi: 10.1038/nature13479
129. Perretti M, D'acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol. (2009) 9:62. doi: 10.1038/nri2470
130. Wallace JL, Ianaro A, Flannigan KL, Cirino G. Gaseous mediators in resolution of inflammation. Sem Immunol. (2015). 227–33. doi: 10.1016/j.smim.2015.05.004
131. Mirakaj V, Dalli J, Granja T, Rosenberger P, Serhan CN. Vagus nerve controls resolution and pro-resolving mediators of inflammation. J Exp Med. (2014) 211:1037–48. doi: 10.1084/jem.20132103
132. Pavlov VA, Tracey KJ. The vagus nerve and the inflammatory reflex—linking immunity and metabolism. Nat Rev Endocrinol. (2012) 8:743. doi: 10.1038/nrendo.2012.189
133. Puttamallesh VN, Sreenivasamurthy SK, Singh PK, Harsha HC, Ganjiwale A, Broor S, et al. Proteomic profiling of serum samples from chikungunya-infected patients provides insights into host response. Clin Proteomics. (2013) 10:14. doi: 10.1186/1559-0275-10-14
134. Sugimoto MA, Sousa LP, Pinho V, Perretti M, Teixeira MM. Resolution of inflammation: what controls its onset? Front Immunol. (2016) 7:160. doi: 10.3389/fimmu.2016.00160
135. Wilson JA, Prow NA, Schroder WA, Ellis JJ, Cumming HE, Gearing LJ, et al. RNA-Seq analysis of chikungunya virus infection and identification of granzyme A as a major promoter of arthritic inflammation. PLoS Pathog. (2017) 13:e1006155. doi: 10.1371/journal.ppat.1006155
136. Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage M1–M2 polarization balance. Front Immunol. (2014) 5:614. doi: 10.3389/fimmu.2014.00614
137. Imtiyaz HZ, Williams EP, Hickey MM, Patel SA, Durham AC, Yuan J, et al. Hypoxia-inducible factor 2α regulates macrophage function in mouse models of acute and tumor inflammation. J Clin Invest. (2010) 120:2699–714. doi: 10.1172/JCI39506
138. Stoermer KA, Burrack A, Oko L, Montgomery SA, Borst LB, Gill RG, et al. Genetic ablation of arginase 1 in macrophages and neutrophils enhances clearance of an arthritogenic alphavirus. J Immunol. (2012) 189:4047–59. doi: 10.4049/jimmunol.1201240
139. Lindsay MA. microRNAs and the immune response. Trends Immunol. (2008) 29:343–51. doi: 10.1016/j.it.2008.04.004
140. Hobert O. Common logic of transcription factor and microRNA action. Trends Biochem Sci. (2004) 29:462–8. doi: 10.1016/j.tibs.2004.07.001
141. Ghosh Z, Mallick B, Chakrabarti J. Cellular versus viral microRNAs in host–virus interaction. Nucleic Acids Res. (2008) 37:1035–48. doi: 10.1093/nar/gkn1004
142. Gottwein E, Cullen BR. Viral and cellular microRNAs as determinants of viral pathogenesis and immunity. Cell Host Microbe. (2008) 3:375–87. doi: 10.1016/j.chom.2008.05.002
143. Chatzikyriakidou A, Voulgari PV, Georgiou I, Drosos AA. miRNAs and related polymorphisms in rheumatoid arthritis susceptibility. Autoimmun Rev. (2012) 11:636–41. doi: 10.1016/j.autrev.2011.11.004
144. Kurowska-Stolarska M, Alivernini S, Ballantine LE, Asquith DL, Millar NL, Gilchrist DS, et al. MicroRNA-155 as a proinflammatory regulator in clinical and experimental arthritis. Proc Natl Acad Sci USA. (2011) 108:11193–8. doi: 10.1073/pnas.1019536108
145. Parashar D, Paingankar MS, More A, Patil P, Amdekar S. Altered microRNA expression signature in chikungunya-infected mammalian fibroblast cells. Virus Genes. (2018) 54:502–13. doi: 10.1007/s11262-018-1578-8
146. Saxena T, Tandon B, Sharma S, Chameettachal S, Ray P, Ray AR, et al. Combined miRNA and mRNA signature identifies key molecular players and pathways involved in chikungunya virus infection in human cells. PLoS ONE. (2013) 8:e79886. doi: 10.1371/journal.pone.0079886
147. Selvamani SP, Mishra R, Singh SK. Chikungunya virus exploits miR-146a to regulate NF-κB pathway in human synovial fibroblasts. PLoS ONE. (2014) 9:e103624. doi: 10.1371/journal.pone.0103624
148. Hill JM, Clement C, Zhao Y, Lukiw WJ. Induction of the pro-inflammatory NF-kB-sensitive miRNA-146a by human neurotrophic viruses. Front Microbiol. (2015) 6:43. doi: 10.3389/fmicb.2015.00043
149. Peng L, Zhang H, Hao Y, Xu F, Yang J, Zhang R, et al. Reprogramming macrophage orientation by microRNA 146b targeting transcription factor IRF5. EBio Med. (2016) 14:83–96. doi: 10.1016/j.ebiom.2016.10.041
150. Curtale G, Rubino M, Locati M. MicroRNAs as molecular switches in macrophage activation. Front Immunol. (2019) 10:799. doi: 10.3389/fimmu.2019.00799
151. Li H, Jiang T, Li Q, Zheng L, Zhao J. Transcriptional regulation of macrophages polarization by MicroRNAs. Front Immunol. (2018) 9:1175. doi: 10.3389/fimmu.2018.01175
152. Zhang D, Cao X, Li J, Zhao G. MiR-210 inhibits NF-κB signaling pathway by targeting DR6 in osteoarthritis. Sci Rep. (2015) 5:12775. doi: 10.1038/srep12775
153. Qi J, Qiao Y, Wang P, Li S, Zhao W, Gao C. microRNA-210 negatively regulates LPS-induced production of proinflammatory cytokines by targeting NF-kappaB1 in murine macrophages. FEBS Lett. (2012) 586:1201–7. doi: 10.1016/j.febslet.2012.03.011
154. Zhu J, Paul WE. Heterogeneity and plasticity of T helper cells. Cell Res. (2010) 20:4. doi: 10.1038/cr.2009.138
155. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. (2006) 24:179–89. doi: 10.1016/j.immuni.2006.01.001
156. Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. (2006) 441:231–4. doi: 10.1038/nature04754
157. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. (2006) 441:235–8. doi: 10.1038/nature04753
158. Lee GR. The balance of Th17 versus Treg cells in autoimmunity. Int J Mol Sci. (2018) 19:730. doi: 10.3390/ijms19030730
159. Lee WW, Teo TH, Her Z, Lum FM, Kam YW, Haase D, et al. Expanding regulatory T cells alleviates chikungunya virus-induced pathology in mice. J Virol. (2015) 89:7893–904. doi: 10.1128/JVI.00998-15
160. Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. (2009) 9:556–67. doi: 10.1038/nri2586
161. Koenders MI, Marijnissen RJ, Devesa I, Lubberts E, Joosten LA, Roth J, et al. Tumor necrosis factor-interleukin-17 interplay induces S100A8, interleukin-1beta, and matrix metalloproteinases, and drives irreversible cartilage destruction in murine arthritis: rationale for combination treatment during arthritis. Arthritis Rheum. (2011) 63:2329–39. doi: 10.1002/art.30418
162. Chaaitanya IK, Muruganandam N, Sundaram SG, Kawalekar O, Sugunan AP, Manimunda SP, et al. Role of proinflammatory cytokines and chemokines in chronic arthropathy in CHIKV infection. Viral Immunol. (2011) 24:265–71. doi: 10.1089/vim.2010.0123
163. Ng LF, Chow A, Sun YJ, Kwek DJ, Lim PL, Dimatatac F, et al. IL-1beta, IL-6, and RANTES as biomarkers of chikungunya severity. PLoS ONE. (2009) 4:e4261. doi: 10.1371/journal.pone.0004261
164. Lohachanakul J, Phuklia W, Thannagith M, Thonsakulprasert T, Ubol S. High concentrations of circulating interleukin-6 and monocyte chemotactic protein-1 with low concentrations of interleukin-8 were associated with severe chikungunya fever during the 2009-2010 outbreak in Thailand. Microbiol Immunol. (2012) 56:134–8. doi: 10.1111/j.1348-0421.2011.00417.x
165. Manet C, Simon-Lorière E, Jouvion G, Hardy D, Prot M, Conquet L, et al. Genetic diversity of collaborative cross mice controls viral replication, clinical severity and brain pathology induced by zika virus infection, independently of Oas1b. J Virol. (2019) 94:e01034–19. doi: 10.1101/677484
166. Rosser EC, Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity. (2015) 42:607–12. doi: 10.1016/j.immuni.2015.04.005
167. Rincon-Arevalo H, Sanchez-Parra CC, Castano D, Yassin L, Vasquez G. Regulatory B cells and mechanisms. Int Rev Immunol. (2016) 35:156–76. doi: 10.3109/08830185.2015.1015719
168. Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol. (2002) 3:944–50. doi: 10.1038/ni833
169. Khoder A, Sarvaria A, Alsuliman A, Chew C, Sekine T, Cooper N, et al. Regulatory B cells are enriched within the IgM memory and transitional subsets in healthy donors but are deficient in chronic GVHD. Blood. (2014) 124:2034–45. doi: 10.1182/blood-2014-04-571125
170. Mauri C, Menon M. The expanding family of regulatory B cells. Int Immunol. (2015) 27:479–86. doi: 10.1093/intimm/dxv038
171. Chien CH, Chiang BL. Regulatory T cells induced by B cells: a novel subpopulation of regulatory T cells. J Biomed Sci. (2017) 24:86. doi: 10.1186/s12929-017-0391-3
172. Miles K, Simpson J, Brown S, Cowan G, Gray D, Gray M. Immune tolerance to apoptotic self is mediated primarily by regulatory B1a cells. Front Immunol. (2017) 8:1952. doi: 10.3389/fimmu.2017.01952
173. Hardy RS, Filer A, Cooper MS, Parsonage G, Raza K, Hardie DL, et al. Differential expression, function and response to inflammatory stimuli of 11beta-hydroxysteroid dehydrogenase type 1 in human fibroblasts: a mechanism for tissue-specific regulation of inflammation. Arthritis Res Ther. (2006) 8:R108. doi: 10.1186/ar1993
174. Cui D, Zhang L, Chen J, Zhu M, Hou L, Chen B, et al. Changes in regulatory B cells and their relationship with rheumatoid arthritis disease activity. Clin Exp Med. (2015) 15:285–92. doi: 10.1007/s10238-014-0310-9
175. Lorenzetti R, Janowska I, Smulski CR, Frede N, Henneberger N, Walter L, et al. Abatacept modulates CD80 and CD86 expression and memory formation in human B-cells. J Autoimmun. (2019) 101:145–52. doi: 10.1016/j.jaut.2019.04.016
176. Sissoko D, Malvy D, Ezzedine K, Renault P, Moscetti F, Ledrans M, et al. Post-epidemic chikungunya disease on Reunion Island: course of rheumatic manifestations and associated factors over a 15-month period. PLoS Negl Trop Dis. (2009) 3:e389. doi: 10.1371/journal.pntd.0000389
177. Chopra A, Anuradha V, Ghorpade R, Saluja M. Acute chikungunya and persistent musculoskeletal pain following the 2006 Indian epidemic: a 2-year prospective rural community study. Epidemiol Infect. (2012) 140:842–50. doi: 10.1017/S0950268811001300
178. Waymouth HE, Zoutman DE, Towheed TE. Chikungunya-related arthritis: case report and review of the literature. Semin Arthritis Rheum. (2013) 43:273–8. doi: 10.1016/j.semarthrit.2013.03.003
179. Anfasa F, Provacia L, GeurtsvanKessel C, Wever R, Gerstenbluth I, Osterhaus AD, et al. Hyperferritinemia is a potential marker of chronic chikungunya: a retrospective study on the Island of Curacao during the 2014-2015 outbreak. J Clin Virol. (2017) 86:31–8. doi: 10.1016/j.jcv.2016.11.003
180. Kelvin AA, Banner D, Silvi G, Moro ML, Spataro N, Gaibani P, et al. Inflammatory cytokine expression is associated with chikungunya virus resolution and symptom severity. PLoS Negl Trop Dis. (2011) 5:e1279. doi: 10.1371/journal.pntd.0001279
181. Chow A, Her Z, Ong EK, Chen JM, Dimatatac F, Kwek DJ, et al. Persistent arthralgia induced by chikungunya virus infection is associated with interleukin-6 and granulocyte macrophage colony-stimulating factor. J Infect Dis. (2011) 203:149–57. doi: 10.1093/infdis/jiq042
182. Gualberto Cavalcanti N, MeloVilar K, Branco Pinto Duarte AL, de Jesus Barreto M, Cristiny Pereira M, da Rocha Pitta I, et al. IL-27 in patients with chikungunya fever: a possible chronicity biomarker? Acta Trop. (2019) 196:48–51. doi: 10.1016/j.actatropica.2019.05.005
183. Young AR, Locke MC, Cook LE, Hiller BE, Zhang R, Hedberg ML, et al. Dermal and muscle fibroblasts and skeletal myofibers survive chikungunya virus infection and harbor persistent RNA. PLoS Pathog. (2019). 15:e1007993. doi: 10.1371/journal.ppat.1007993
184. Danks L, Takayanagi H. Immunology and bone. J Biochem. (2013) 154:29–39. doi: 10.1093/jb/mvt049
185. Amdekar S, Parashar D, Alagarasu K. Chikungunya virus-induced arthritis: role of host and viral factors in the pathogenesis. Viral Immunol. (2017) 30:691–702. doi: 10.1089/vim.2017.0052
186. Guegan JP, Legembre P. Nonapoptotic functions of Fas/CD95 in the immune response. FEBS J. (2018) 285:809–27. doi: 10.1111/febs.14292
187. Noret M, Herrero L, Rulli N, Rolph M, Smith PN, Li RW, et al. Interleukin 6, RANKL, and osteoprotegerin expression by chikungunya virus-infected human osteoblasts. J Infect Dis. (2012) 206:455–7:7–9. doi: 10.1093/infdis/jis368
188. Takayanagi H. The role of NFAT in osteoclast formation. Ann NY Acad Sci. (2007) 1116:227–37. doi: 10.1196/annals.1402.071
Keywords: chikungunya virus, incubation phase, acute phase, immunopathology, disease resolution, chronic phase, chikungunya induced arthritis, viral clearance
Citation: Srivastava P, Kumar A, Hasan A, Mehta D, Kumar R, Sharma C and Sunil S (2020) Disease Resolution in Chikungunya—What Decides the Outcome? Front. Immunol. 11:695. doi: 10.3389/fimmu.2020.00695
Received: 31 July 2019; Accepted: 27 March 2020;
Published: 28 April 2020.
Edited by:
Lisa F. P. Ng, Singapore Immunology Network (A*STAR), SingaporeCopyright © 2020 Srivastava, Kumar, Hasan, Mehta, Kumar, Sharma and Sunil. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sujatha Sunil, c3VqYXRoYUBpY2dlYi5yZXMuaW4=
†These authors have contributed equally to this work