
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 28 April 2020
Sec. Immunological Tolerance and Regulation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.00618
This article is part of the Research TopicTolerating Factor VIII: Novel Strategies to Prevent and Reverse Anti-FVIII InhibitorsView all 20 articles
 Benjamin J. Samelson-Jones1,2,3
Benjamin J. Samelson-Jones1,2,3 Valder R. Arruda1,2,3*
Valder R. Arruda1,2,3*Hemophilia A (HA) is an X-linked bleeding disorder due to deficiencies in coagulation factor VIII (FVIII). The major complication of current protein-based therapies is the development of neutralizing anti-FVIII antibodies, termed inhibitors, that block the hemostatic effect of therapeutic FVIII. Inhibitors develop in about 20–30% of people with severe HA, but the risk is dependent on the interaction between environmental and genetic factors, including the underlying F8 gene mutation. Recently, multiple clinical trials evaluating adeno-associated viral (AAV) vector liver-directed gene therapy for HA have reported promising results of therapeutically relevant to curative FVIII levels. The inclusion criteria for most trials prevented enrollment of subjects with a history of inhibitors. However, preclinical data from small and large animal models of HA with inhibitors suggests that liver-directed gene therapy can in fact eradicate pre-existing anti-FVIII antibodies, induce immune tolerance, and provide long-term therapeutic FVIII expression to prevent bleeding. Herein, we review the accumulating evidence that continuous uninterrupted expression of FVIII and other transgenes after liver-directed AAV gene therapy can bias the immune system toward immune tolerance induction, discuss the current understanding of the immunological mechanisms of this process, and outline questions that will need to be addressed to translate this strategy to clinical trials.
Hemophilia A (HA) is an X-linked bleeding disorder due to inherited deficiency in coagulation factor VIII (FVIII) activity (1, 2). Until recently, treatment in the developed world involved the intravenous administration of FVIII concentrates to treat or prevent bleeding. Recently, a FVIII-mimetic that is delivered subcutaneously, emicizumab, has been approved as prophylactic treatment for HA to prevent bleeding (3–6). Emicizumab is the first approved non-factor therapy for HA, but several others are in clinical development (7, 8). Other novel treatments for HA in clinical studies include several gene therapy approaches (9–11).
The major complication of treatment with FVIII concentrates is the development of neutralizing anti-FVIII antibodies, termed inhibitors, which substantially increase the mortality and morbidity of HA (12–19). Inhibitors are clinically quantified in Bethesda Units (BU), where 1 BU neutralizes 50% of normal FVIII activity. High-titer inhibitors greater than 5 BU generally prevent administered FVIII from having a therapeutic effect (20). On a research basis, anti-FVIII antibodies can also be quantified by immunosorbent assays that measure both non-neutralizing and neutralizing antibodies. Clinically, decreased FVIII recovery (expected peak level after FVIII protein administration) and half-life are also used as markers for non-neutralizing antibodies and evidence of an anti-FVIII immune response even in the absence of measurable Bethesda titers. Bypassing agents, which circumvent the inhibitor to provide hemostasis, are required to treat or prevent bleeding in high-titer inhibitor patients (21, 22). Due to their mechanism of action, emicizumab and other non-factor therapies are also effective in the presence of high-titer inhibitors, though they are currently only studied to prevent bleeding (5).
Inhibitors develop in about 20–30% of patients with severe hemophilia A (<1% normal FVIII activity) and in about 10% in patients with non-severe hemophilia A (1–40% normal FVIII activity) (23, 24). The risk of inhibitor development for the former is highest during their initial FVIII exposure days (25), while it is relatively constant for the latter (23). Both genetic and environmental factors influence the risk of inhibitor development (25–29). A major genetic determinant of inhibitor risk is the underlying hemophilia-causing FVIII-gene (F8) mutation (28). Patients with F8 mutations resulting in the expression of some FVIII cross-reactive material (CRM), such as missense or small in-frame deletions or insertions, are less likely to develop inhibitors while CRM-negative patients with large F8 deletions are more likely to develop inhibitors (28). Environmental factors include the manufacturing process and type of factor product, timing of first factor exposure, factor dosage, and clinical situations that result in immunological “danger signals” (24–26, 29).
Treatment of HA patients with inhibitors includes the prevention and treatment of bleeds (20, 30) and, historically, eradication of the inhibitor via the immune tolerance induction (ITI) regimens (30–33). ITI is the frequent regular infusion of FVIII concentrates over extended period-of-time (often years) with a goal of sufficiently decreasing the inhibitor to allow for the use of therapeutic FVIII, as nothing provides as effective hemostasis as FVIII in the absence of an inhibitor (20). The frequency and the dose of FVIII in ITI remain debatable, but the dosing regimens of daily or every-other-day from the International ITI Study (33) are often used (20, 30). However, ITI is only successful in about 60% of patients (32, 33). The underlying mechanism is likely to be peripheral immune tolerance induction where the activity of anti-FVIII immune cells is suppressed through tolerogenic interactions in the periphery, rather than central immune tolerance where the anti-FVIII immune cells are eliminated prior to leaving either the thymus or bone marrow.
The recent advent of emicizumab, which provides significantly improved bleeding prophylaxis compared to other bypassing agents (5, 6), has raised the question of whether inhibitor eradication remains necessary in the management of inhibitor patients (34, 35). Though the clinical consensus to this question is still forming, many experts continue to recommend ITI for new inhibitors (36) given the ongoing concerns about thrombotic complications in inhibitor patients on emicizumab receiving high cumulative doses of the bypassing agent activated Prothrombin Complex Concentrates for break-through bleeding (5, 37, 38). Long-term follow up is needed to define the “real world” safety and efficacy of indefinite emicizumab compared standard ITI.
The limited success rate of current ITI approaches has driven the pre-clinical investigations of several novel ITI strategies (39), including gene therapy approaches (40). Multiple adeno-associated viral (AAV) vector gene therapies for HA without inhibitors are in clinical development, as summarized in Table 1 (9–11). These drugs all direct the therapeutic FVIII-gene to hepatocytes expression. Though the goal of these studies is to achieve durable therapeutically relevant FVIII levels, emerging preclinical data suggest the liver-directed gene therapy can utilize the liver tolerance effect (41) to induce immune tolerance to the transgene-product (40, 42, 43). Here we review the preclinical data supporting the hypothesis that AAV liver-directed gene therapy can induce immune tolerance to FVIII and present the open questions that need to be considered when translating this approach to clinical trials.
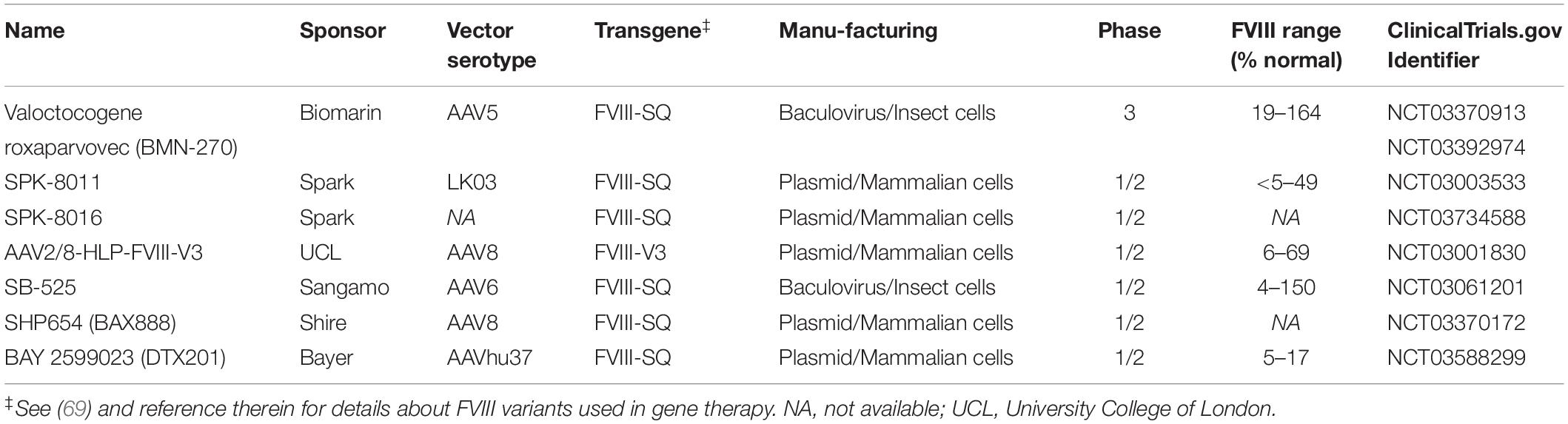
Table 1. Current FVIII AAV liver-directed gene therapy products for HA in clinical development.
The liver is a tolerogenic organ (41, 42, 44–46). Its specialized immune system limits immune reactivity against the constant flux of digested food-products as well as antigens from commensal microorganisms, while simultaneously safeguarding against gastrointestinal pathogens. From a therapeutic perspective, the liver tolerance effect was first recognized in studies of an outbred porcine liver transplant model where some allografts achieved long-term survival without immunosuppression (47); similar results were subsequently reported in other in vivo transplant models (48, 49). Moreover in animal studies, liver allotransplants also promote the immunological tolerance to other organ allografts from the same donor (47), and tolerance to renal and small bowel transplants is enhanced if the venous blood drainage of the grafts is through the portal system (50). Clinically, immunosuppression can be safely withdrawn in about 20% of liver-transplant patients (51), which is not achievable in other solid organ transplant patients.
The liver tolerance effect is also exploited by hepatotropic pathogens (44–46, 52). The Plasmodium species responsible for malaria initially target the liver after being delivered by an infected mosquito and then mature and replicate sheltered within hepatocytes before being released back into the blood stream. Malaria remains one of the most deadly human pathogens responsible for millions of annual deaths and has resisted effective vaccination strategies to date. Likewise, the protolerogenic environment of the liver likely impedes an effective adaptive anti-viral response in chronic infections by hepatitis B and hepatitis C virus (HCV) (52). Viral hepatitis remains a major source of morbidity and mortality especially in the developing world and in people with hemophilia (53, 54).
The liver tolerance effect can also be exploited therapeutically by liver-directed gene therapy to induce immune tolerance to the transgene product (40, 42, 43). This was first demonstrated in HB mice that were tolerized to human (h) factor IX (FIX) after AAV gene therapy (55), but has subsequently been demonstrated in several other disease models including HA, which is discussed in detail below. In naïve HB mice, administration of liver-directed AAV (55) or lentiviral (LV) (56) vectors encoding hFIX induces immune tolerance that is resistant to subsequent hFIX immunizations. Interestingly, induction of immune tolerance with LV vectors requires the use of genome regulatory elements that prevented transgene expression in immune cells (56), suggesting that the tolerogenic bias of liver-directed gene therapy can be overwhelmed by non-liver transgene expression. In addition to induction of immune tolerance in naïve HB mice, both liver-directed AAV and LV hFIX gene therapy can also eradicate preexisting anti-hFIX antibodies and induce immune tolerance in hFIX-immunized HB mice (57, 58).
Similar outcomes have also been observed in canine models of severe HB. Colonies of naturally occurring HB dogs with distinct F9 mutations provide highly informative models for evaluating immune responses to therapeutic canine (c) FIX. HB dogs with a F9 null mutation are inhibitor-prone and typically develop an inhibitor after a single administration of cFIX concentrate (59), while HB dogs with a F9 missense mutation are non-inhibitor-prone and very rarely develop inhibitors against cFIX, usually only in the context of a pro-inflammatory stimulus (60, 61). The use of cFIX protein, canine plasma, and cFIX encoding vectors allows the immune response to be interrogated in a species-specific manner.
No evidence of an anti-cFIX immune response has been reported after AAV (59, 62–65) or LV (66) liver-directed gene transfer with cFIX in 11 and 3 non-inhibitor-prone HB dogs, respectively, though only 1 dog has been subsequently challenged with canine plasma for bleeding (64). Moreover, AAV liver-directed gene therapy with cFIX successfully tolerized 5 out 6 naïve inhibitor-prone dogs that typically form inhibitors after cFIX protein exposure (59, 67). In this study, the immune tolerance was demonstrated to be maintained despite subsequent cFIX protein exposures in all 4 dogs evaluated, while the 5th animal was not challenged (67, 68). The singe naïve HB dog that developed an inhibitor notably had a hemolytic anemia associated with liver iron-loading and consequentially liver-fibrosis, both of which were seen on liver histology at necropsy (59). The course of this single naïve inhibitor-prone dog that was not tolerized after AAV liver-directed gene therapy with cFIX may be the exception-that-proves-the-rule as his unrelated liver pathology likely disrupted the liver tolerance effect.
Liver-directed AAV gene therapy has also eradicated preexisting anti-cFIX antibodies and induced immune tolerance in an inhibitor-prone HB dog (Wiley) (67). In this study, an inhibitor-prone HB dog was previously exposed to hFIX and developed an anti-hFIX response with cross-reactive neutralization of cFIX activity. AAV liver-directed gene transfer with the hyperactive cFIX variant, Padua (69, 70), resulted in eradication of the anti-cFIX antibodies and disappearance of the anti-hFIX neutralization within 3 months of vector administration, though a minimal residual non-neutralizing anti-hFIX response remained detectable years after vector administration (67). The persistence of non-neutralizing anti-hFIX antibodies suggests that the immune tolerance induced after liver-directed AAV gene therapy is transgene specific. Nonetheless, this dog remained tolerant to cFIX even after exposure to cFIX-WT with over 3 years of ongoing observations (VRA, unpublished data).
Several complementary immunological mechanisms underlying the liver tolerance effect have been described [reviewed in detail in (41, 42, 44, 45, 71–73)]. The liver microenvironment contains unique populations of both antigen presenting cells and suppressor and effector T cells (Figure 1). Hepatic antigen presenting cells include conventional types such as dendritic cells, but also unique non-conventional antigen presenting cells that include liver sinusoidal endothelial cells (LSECs), Kupffer cells, stellate cells, and hepatocytes. Presentation of antigens by these specialized hepatic antigen presenting cells typically results in diminished effector T cell activity and/or increased suppressor activity of regulator T cells (Tregs), both of which promote immune tolerance.
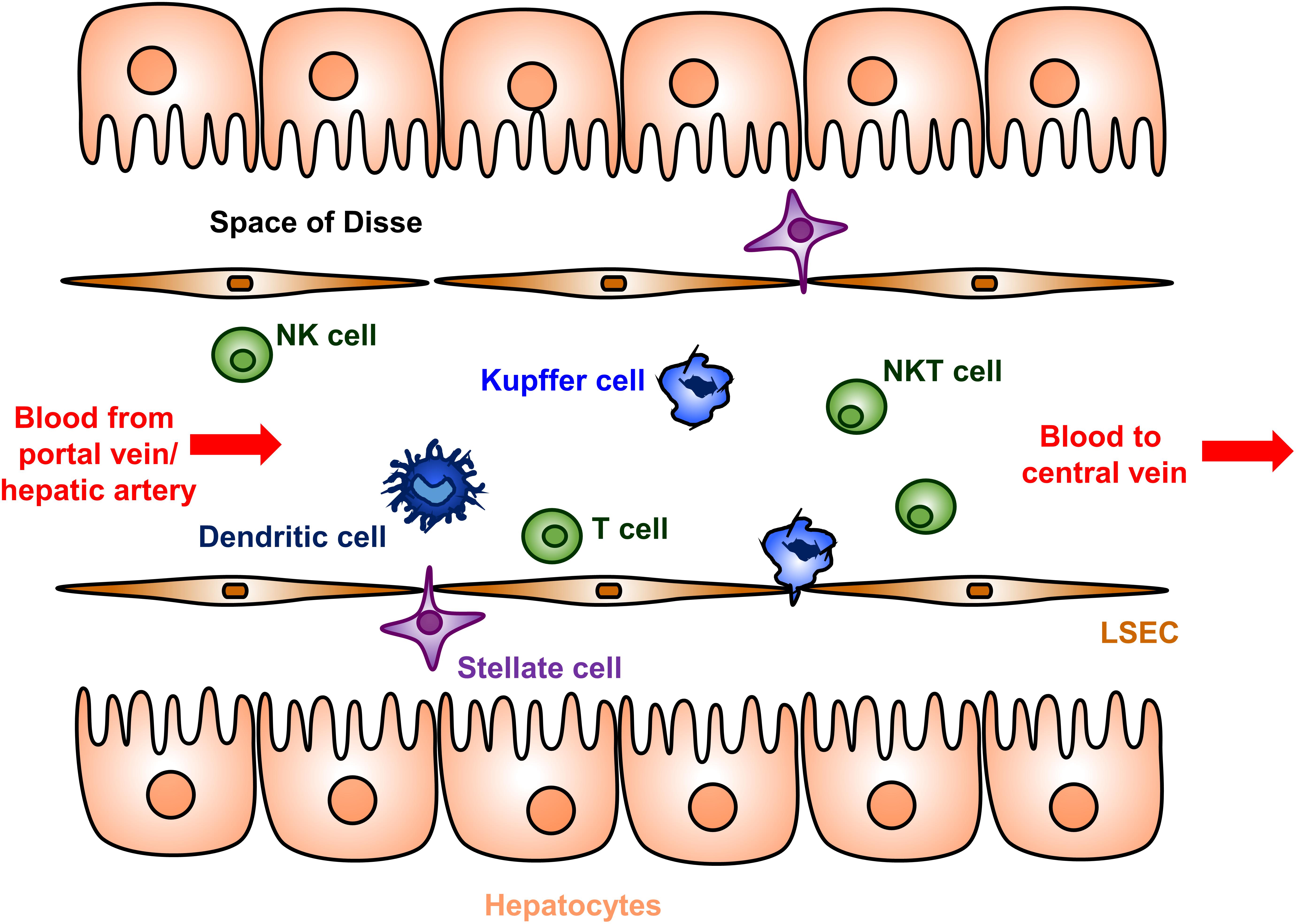
Figure 1. Cellular anatomy of the liver sinusoid. Blood enters the liver from the portal vein and hepatic artery, flows through a network of sinusoids schematically represented here, and then exits via the hepatic central vein. The sinusoids are lined by a fenestrated layer of specialized liver sinusoidal endothelial cells (LSECs), which are the endogenous site of most FVIII secretion. The LSECs shield the hepatocytes from direct sinusoidal blood flow by creating the Space of Disse, which contains the stellate cells. Dendritic cells, Kupffer cells, T cells, natural killer (NK) cells, and natural killer T (NKT) cells are abundantly present in the sinusoidal lumen. Hepatic antigen presenting cells include dendritic cells, Kupffer cells, LSECs, stellate cells, and hepatocytes. The hepatocyte microvilli can interact with luminal T cells.
Interactions between these hepatic antigen presenting cells and effector T cells often results in functional inhibition or cell death of the effector T cell. Low-expression of major histocompatibility complex (MHC) and co-stimulatory molecules on several types of hepatic antigen presenting cells contributes to abortive activation and result in T cell anergy in an antigen-specific manner (42, 73, 74). The absent production of the costimulatory cytokine IL-12 by Kupffer and dendritic cells also contributes to this process. Additionally, hepatic antigen presenting cells actively express a number of molecules that suppress effector T cell activity. Both Kupffer cells and hepatic dendritic cells produce indoleamine 2,3 dioxygenase (IDO) and prostaglandin E2, which inhibit T cell proliferation through distinct pathways (42, 74). Dendritic cells, LSECs and AAV transduced hepatocytes express Fas-L (CD95L), which allows for the direct deletion of effector T cells (42).
The liver microenvironment also promotes Treg activation and proliferation. Abundant Tregs are noted in liver allografts in mouse models and their depletion result in a loss of tolerance (75). Secretion of the suppressor cytokine IL-10 by LSECs, Kupffer cells, and hepatic dendritic cells promote Treg proliferation as well as conversion of effector T cells to Treg (42, 74). The decrease in the inhibitor titer after AAV gene therapy in the HB dog with a pre-existing inhibitor (Wiley) was associated the increasing IL-10 levels (67). Hepatocyte production of transforming growth factor-β similarly promotes Treg proliferation (76).
Immunocompetent HA mice generally develop a xenoprotein immune response against hFVIII or cFVIII after exposures via protein administration or gene therapy (77, 78), and typically not against mouse (m) FVIII (79, 80). As such, preclinical efficacy studies of non-murine FVIII gene therapy have typically utilized immunosuppression or HA mice crossed with an immunodeficient model (81, 82).
However, AAV-liver directed gene therapy with hFVIII has been demonstrated to be able to induce immune tolerance that is resistant to subsequent protein challenges in HA mice (83). The ability of this approach to induce tolerance to hFVIII was dependent on both the HA strain background and the expressed FVIII level (83, 84). Immune tolerance to hFVIII in HA mice increased with higher hFVIII levels after AAV gene therapy, achieved either through codon optimization (83) or higher vector doses (84), though the controls were likely at sub-therapeutic hFVIII levels (<1% normal).
In contrast to the murine model, outbred severe HA dogs predictably develop inhibitors against cFVIII (85–88). These dogs have a cF8 mutation analogous to the human F8 intron-22 inversion, the most common causative severe HA mutation in patients (89, 90). Intriguingly, the risk of inhibitor development in these HA dogs is in part inherited through yet undefined non-cF8 genes as the propensity of inhibitor development is increased in the progeny of certain outside breeders (86, 88). About 30% of these inhibitor-prone HA dogs, either at the colony at Queens University (QU) or University of North Carolina at Chapel Hill (UNC-CH) colony, develop anti-cFVIII inhibitors (86, 91).
None of the 15 non-inhibitor-prone HA dogs that have received cFVIII AAV-liver-directed gene therapy without immunosuppression have made an anti-cFVIII immune response (92–94). At least 6 of these animals were subsequently challenged with recombinant cFVIII protein (25 U/kg weekly for 4 weeks) 1–5 years after vector administration and did not display an anti-FVIII immune response. Recovery of the administered cFVIII protein was similar to that of naïve dogs, which confirmed complete tolerance to cFVIII (94).
However, 2 out of 9 inhibitor-prone HA dogs developed an inhibitor after cFVIII AAV-liver-directed gene therapy (91, 94, 95). A QU-housed HA dog Junior developed an inhibitor 2 weeks after administration of an AAV2 vector via the portal-vein with a peak titer of 9 BU at 4 weeks that became undetectable by week 9 (91). Similarly, an UNC-CH inhibitor-prone HA dog L51 developed an inhibitor within a week of intravenous administration of an AAV8 vector that peaked at 2.5 BU and became undetectable by week 7 (94). Notably, the eradication of L51’s inhibitor was stringently demonstrated to be immune tolerance by the lack of recurrent anti-cFVIII antibodies and expected recovery of challenges of recombinant cFVIII protein (25 U/kg weekly for 4 weeks) (94). Though limited by the small numbers, the experience of Junior and L51 suggest that AAV-liver-directed gene therapy can induce stringent immune tolerance to transgene-FVIII in large outbred models of HA.
However, the liver tolerance effect after liver-directed gene therapy in HA dogs can be disrupted in the setting of hepatotoxicity (96), similar to what was discussed above for HB dog models (59). In an early study using an adenoviral vector (rather than AAV), all 4 QU HA dogs that received liver-directed cFVIII gene therapy developed inhibitors in the setting of an early (0–4 weeks), presumably adenovirus-induced, hepatotoxicity manifested by over a 20-fold increase in liver enzymes (96). Both the increase in liver enzymes and the inhibitor titer appeared to be vector dose dependent with the 2 dogs that received the lower vector dose only developing low titer inhibitors (peak < 2 BU) that disappeared within 4 weeks; there was also no recurrence of the inhibitor after administration of canine FVIII cryoprecipitate (20 U/kg) in these 2 low-dose dogs (96). Though this study was conducted with a vector that is no longer translationally relevant for HA, the observations about the immune response in setting of acute hepatotoxicity may be germane for other gene therapy approaches.
Consistent with this hypothesis that hepatotoxicity can interfere with the pro-tolerogenic effects of liver gene therapy is the subsequent observation that none of the 3 the QU HA dogs that received liver-directed cFVIII gene therapy with a less toxic helper-dependent adenovirus vector concomitantly with steroid immunosuppression developed an anti-cFVIII immune response (97). A fourth dog in this study received cFVIII gene therapy under-the-control of a ubiquitous CMV promoter and did develop a high-titer inhibitor (peak 150 BU 1 week after vector administration) (97). This observation suggests that non-liver expressed cFVIII can potentially interfere with immune tolerance from liver-expressed transgene and is consistent with the observations from HB (56) and Pompe (98, 99) disease models.
The cumulative data from both small and large animal HA models indicates that liver-directed gene therapy, especially with AAV vectors that have minimal hepatoxicity in pre-clinical models, can induce immune tolerance in naïve animals to FVIII. Moreover, if anti-FVIII antibodies develop, the continued transgene expressed FVIII can eradicate the inhibitors and, in a limited number of examples, induce immune tolerance that is resistant to subsequent protein challenges.
The likely more challenging and clinically relevant scenario is the eradication of a pre-existing anti-FVIII immune response. We have previously reported on the successful immune tolerance induction in 4 inhibitor-prone HA dogs with pre-existing anti-cFVIII antibodies after AAV liver-directed gene therapy (Table 2) (100). In this study, 3 HA dogs from UNC-CH with peak historical inhibitor titers from 4 to 12 BU received cFVIII AAV8 liver-directed gene therapy. Notably, the anti-FVIII IgG and inhibiter titers in all 3 dogs disappeared by 5 weeks after vector administration (100).
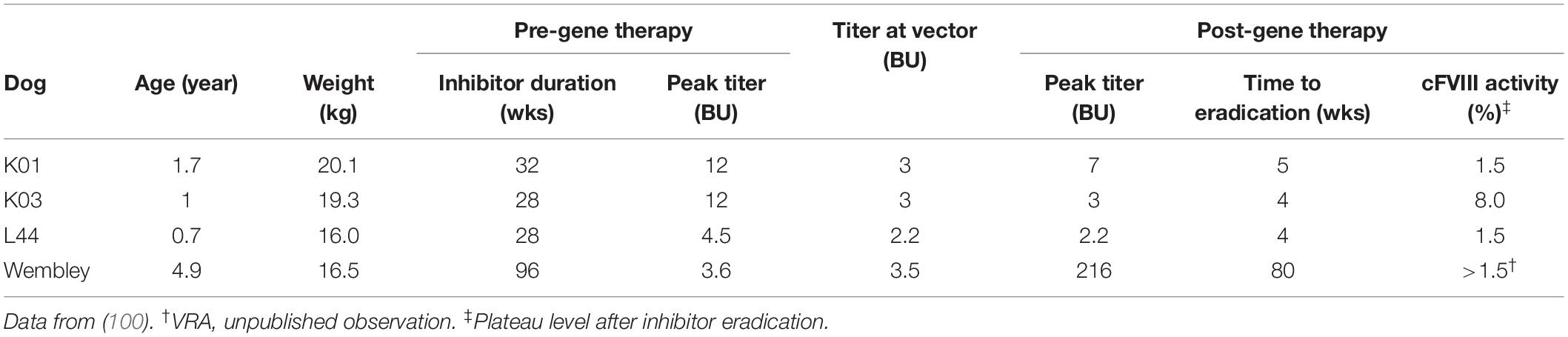
Table 2. Summary of inhibitor eradication in hemophilia A dogs following cFVIII AAV liver-directed gene therapy.
The fourth inhibitor-prone HA dog (Wembley from QU) with pre-existing anti-cFVIII antibodies treated similarly with cFVIII AAV8. At the time of vector administration, his cFVIII-inhibitor was 4 BU, which was similar to his peak titer. After vector administration, Wembley also had a large anamnestic response with dramatic increases in his anti-cFVIII IgG and inhibiter titer that peaked at 216 BU. However, both these parameters decreased until they became undetectable at about 80 weeks after vector administration (VRA, unpublished observation).
The post-gene therapy cFVIII activity after inhibitor eradications was between 1.5 and 8% of normal in these 4 dogs (100) (and VRA, unpublished observation). The immune tolerance of all 4 of these dogs was maintained despite multiple challenges with recombinant cFVIII protein that displayed typical pharmacokinetics in an ongoing study (100) (and VRA, unpublished observation). Though Wembley was tolerized to cFVIII after cFVIII AAV liver-directed gene therapy, he never fully tolerized to hFVIII, his triggering antigen (VRA, unpublished observation). Similar to the above discussion for the HB dog models (67), Wembley’s course suggests that the immune tolerance of AAV liver-directed gene therapy is transgene specific. To date, cFVIII inhibitors have been eradicated in 6 HA dogs (2 dogs naïve discussed in section “Immune Tolerance Induction in Naïve Hemophilia A Animal Models” and 4 dogs with pre-existing inhibitors discussed in section “Inhibitor Eradication and Immune Tolerance Induction in Hemophilia A Inhibitor Models With Pre-existing Anti-FVIII Immune Response”) by cFVIII AAV liver-directed gene therapy, which also provided stringent immune tolerance and long-term therapeutically relevant cFVIII levels. This limited data suggests that AAV liver-directed gene therapy has translational potential for people with HA and inhibitors.
It is an exciting time for gene therapy for HA with multiple liver-directed AAV-based products in clinical trials reporting therapeutically relevant to curative levels of FVIII (Table 1) (9–11). These current products are the result of decades of experimentation developing the necessary technologies and protocols to achieve these end points (101–104). These AAV drugs differ in their vector serotype, FVIII transgene, and manufacturing process. A variety of naturally occurring and bioengineered vector serotypes are being tested (105). Because the full-length FVIII gene exceeds the ∼4.7 kb packaging capacity of AAV vectors, all current approaches rely on B-domain deleted FVIII variants, either the standard FVIII-SQ (106) as is used in B-domain deleted protein products (e.g., Xyntha and Pfizer) or an engineered B-domain deleted variant (FVIII-V3) that is associated with increased FVIII expression (107). All transgenes have been codon-optimized, though likely by distinct algorithms.
To date, all trials stringently limit subjects at risk for inhibitor development by including only subjects with no history of inhibitors and more than 150 FVIII exposures, as inhibitors rarely develop after 50 exposure days in severe HA (25). However, a current study (NCT03734588) enrolling subjects meeting these inclusion criteria is described as a dose-finding Part 1 of a planned two part clinical development strategy focused on inhibitor patients.
No major sustained safety concerns have been reported in these studies, though several immunological obstacles to the AAV vector capsid continue to limit efficacy and widespread enrollment (108, 109). The long-term durability also remains an open question with trials reporting therapeutically relevant FVIII levels out to 3 years after vector administration, albeit with declining levels (110). Nonetheless, the success of these studies raises the question whether these drugs or similar products could be harnessed to induce immune tolerance in HA inhibitor patients by exploiting the liver tolerance effects (41, 42, 44–46).
There are several obstacles that need to be address to define the role of AAV liver gene therapy for HA complicated by the presence of inhibitors to FVIII. The experience with preclinical HA inhibitor models, especially the canine data, support the potential translation of AAV liver-directed gene therapy for people with HA and inhibitors. However, therapeutic translation of this approach will require several yet unresolved issues to be considered and specific preclinical studies designed to address unanswered questions (Table 3). Preclinical studies using the immunocompetent HA dog models that naturally develop inhibitors when exposed to cFIII in a species-specific manner will likely be the most informative.

Table 3. Translational considerations of AAV liver gene therapy for immune tolerance induction.
Foremost, is the potential detrimental role of hepatotoxicity in successful immune tolerance induction. As discussed above, preclinical studies suggest that the tolerogenic bias of liver-directed gene therapy can be disrupted in the setting of hepatotoxicity. The hepatotoxicity in these disparate canine studies was secondary to either the specific vector employed (96) or underlying liver disease in the dog model studied (59). There are several liver pathologies that occur in HA patients receiving AAV liver-directed gene therapy that may impact the liver tolerance effect.
Though rates of viral iatrogenic infections including HCV have thankfully plummeted with improved blood donor screening, highly effective virucidal procedures, and the use of recombinant FVIII products, about a third of young men with severe HA have a history of HCV infection, while the rate in older men exceeds 90% (53). Historically, about 20–30% of HCV infected patients eventually developed end-stage liver disease, though the recent approval of highly effective oral anti-viral regimens will likely radically reverse this trajectory (54). Asymptomatic liver damage from chronic HCV infection theoretically could impair the liver-tolerance effect after AAV liver-directed gene therapy. Current AAV liver-directed gene therapy clinical trials exclude patients with active HCV or clinical evidence of liver disease. Moreover, there were no appreciable differences in the clinical course of 3 out of 9 published HA subjects that received AAV5-FVIII (valoctocogene roxaparvovec, BMN 270) that had a history of resolved HCV infection (111); similarly, there has been no appreciable difference in the clinical course of HB subjects with a history of HCV receiving AAV FIX gene therapy (112–114). Other liver diseases that occur in the HA population such as non-alcoholic fatty liver disease pose the same theoretical risk of potentially disrupting the liver tolerance effect. Preclinical studies may help inform on this concern, but stringent liver disease exclusion criteria will probably be advisable in initial clinical studies with inhibitor patients.
To date, some subjects in all AAV liver-directed gene therapy trials for HA or HB have demonstrated hepatotoxicity after vector administration as evidenced by asymptomatic elevated liver enzymes [reviewed in (7, 104)]. At least 2 distinct mechanisms likely contribute to these observations. In products utilizing an AAV2, AAV8, or similar serotype vectors manufactured in mammalian cells, a well described anti-AAV capsid cellular immune response can target transduced hepatocytes leading to loss of transgene expression, which can often, though not always, be controlled with immunosuppression with steroids (108, 109, 112–116). In products utilizing AAV5 vectors manufactured in insect cells, elevated liver enzymes have also been reported, but do not appear to be associated with loss of transgene or evidence of cellular immunity (104, 111, 117); the etiology of this latter hepatotoxicity is still being investigated. Studies of both these hepatotoxicities have been hampered by lack of preclinical models that fully recapitulate the clinical observations (118–122). The potential adverse role either of these hepatotoxicities could have on inducing immune tolerance after AAV liver-directed gene therapy is unknown. However, as both hepatoxicities appear to be vector-dose dependent, avoiding them by using the lowest effective vector dose is likely sensible. The potential immunological consequence of lowering the transgene FVIII level is discussed below in section “FVIII Level and Variant Transgenes.”
The use of immunosuppression in AAV liver-directed gene therapy for HA and inhibitors will require specific preclinical studies. In HA dogs with pre-existing inhibitors, there is an early increase in CD25 + FOXP3 + CD4 + cells, assumed to be Treg cells, within the first few days after cFVIII AAV liver-directed gene therapy that is associated with the decline in anti-cFVIII antibodies that is not observed in HA dogs without inhibitors receiving similar therapy (40, 100). However, intense immunosuppression with the anti-CD25 antibody daclizumab around vector delivery increases the anti-hFIX immune response in non-human primates (NHP) receiving hFIX AAV liver-directed gene therapy and is associated with a decrease in Treg cells (123). The rationale of this study was that daclizimab could potentially decrease effector T cell activity to limit the anti-AAV capsid immune response; however, daclizumab also depleted Treg cells leading to an unanticipated increase immunity against the transgene hFIX.
These findings are not restricted to anti-CD25 antibodies. More recently we uncovered that rabbit antitymoglobulin (ATG) may also be detrimental if administered around vector delivery (BSJ and VRA, unpublished data, manuscript in preparation). Combined, this limited data does suggest that there is an early critical time-period around vector administration where Treg cell expansion may be important for immune tolerance induction. As such, immunosuppression around the time of AAV vector delivery needs to be thoughtfully considered. Reassuringly, steroids promote antigen-specific immune tolerance HA mice against concomitantly administered hFVIII protein, likely through Treg-dependent mechanisms (124). However, more intense immunosuppression therapy needs to be stringently tested in large animal models to avoid unanticipated increased immunogenicity. Ongoing mechanistic studies evaluating cFVIII AAV gene therapy in HA dogs with pre-existing inhibitors may better inform on this issue.
There is also a concern about the potential for liver toxicity due to ectopic expression of FVIII in hepatocytes after gene therapy due to specific features of FVIII secretion (125–128). However, though 2 studies in HA mice did find evidence of the unfolded protein response (UPR) after AAV liver-directed gene therapy, there was no correlation between FVIII inhibitor formation and UPR markers (126, 127). Ectopic expression of human FVIII in mouse megakaryocytes is also associated with megakaryocyte apoptosis (129). However, the theoretical concern of UPR-induced hepatotoxicity further supports the initial use of the lowest effective vector dose.
Preclinical data in HA (97) and other disease models (56, 98, 99) highlight that non-liver expression of the transgene can disrupt the pro-tolerogenic effects of liver expression. Most preclinical and clinical studies for AAV liver-directed gene therapy have utilized liver-specific promoters based on the seminal work by Dr. Kathy Ponder (130); the prototypical expression cassette contains sequences from the truncated apolipoprotein E (ApoE) hepatic control region (HCR) and the alpha-1-anti-trypsin (A1AT) promoter. Though this construct mostly limits non-liver transgene expression, we are not aware of studies that have rigorously quantified the non-liver transgene expression in a preclinical model. The non-liver expression of a particular AAV product is a function of both the transduction efficiency of the vector to other tissues and the promoter efficiency in these tissues. Recent work using a new, highly sensitive experimental system demonstrated much broader AAV transduction as well as low level and/or transient transgene expression than previously appreciated (131). This study highlights the importance of preclinical studies in immune competent animal models with preexisting inhibitors to FVIII to specifically evaluate the efficacy of immune tolerance induction of a particular vector before moving into clinical studies.
Studies in both HA (83, 84) and HB (55) mice have concluded that increasing transgene levels after AAV liver-directed gene therapy promote immune tolerance. In non-severe HA patients, increasing FVIII levels are associated with decreased bleeding frequency (132). However, as discussed above, these potential benefits should be weighed against complications that also likely increase with increasing vector dose that potentially could interfere with the liver tolerance effect, including AAV-associated hepatotoxicity and UPR in transduced hepatocytes.
By definition, antigen expression is required for immune tolerance induction. In the randomized International-ITI trial, higher and more frequent FVIII dosing was associated with a more rapid immune tolerance induction, but the lower and less frequent dose demonstrated similar overall success rate (33). The sustained cFVIII levels of the 6 HA dogs described above that had inhibitor eradication and immune tolerance induction with cFVIII AAV liver gene therapy ranged from 0.5 to 8% of normal (91, 94, 100). This canine data suggest that transgene FVIII levels in the low-mild range is sufficient for immune tolerance induction.
The use of variant FVIII transgene also needs to be carefully considered. Studies in HA and HB dogs suggest that the immune tolerance after AAV liver-directed gene therapy is transgene specific; gene therapy with cFVIII or cFIX transgenes resulted in immune tolerance toward the canine orthologs, while the anti-hFVIII or anti-FIX response persisted (67, 100). However, in this HB dog study, the transgene was the single-amino acid substituted hyperactive variant cFVIII-Padua (R338L) (69, 70) and the animal was also fully tolerized to wild-type (WT) cFIX as evidenced by the lack of neutralizing or non-neutralizing anti-cFIX-WT antibodies despite protein challenges with cFIX-WT (67) (and VRA, unpublished observation). This suggests that though the immune tolerance is transgene specific, there can be cross-tolerance between highly similar transgenes, though the degree of similarity required is undefined. The degree of similarity between variant FVIII transgenes (69) and available protein FVIII products, therefore, should be considered when designing gene therapy products for inhibitor patients.
The concern is that if the inhibitor patient tolerized only to a variant-FVIII transgene that was immunological distinct from available FVIII protein products, these FVIII protein products could be ineffective if needed for breakthrough bleeding or surgery. This would likely be salient for novel FVIII variants with multiple modifications outside the FVIII B-domain, which are likely more immunogenic than modifications restricted to the B-domain; all current FVIII transgenes in Table 1 differ only in their B-domain replacement linker (69). If a patient is tolerized to a B-domain deleted FVIII transgene after gene therapy, the clinical decision making about using alternative FVIII protein products such as extended-half-life products would be similar to switching products after ITI with FVIII protein. Preclinical studies in inhibitor dogs using the canine orthologs of FVIII variants could evaluate this scenario for specific FVIII variants, as we did for the FIX-Padua (67, 133, 134).
Emicizumab and other non-factor therapies offer the potential for better and more convenient hemostatic prophylaxis for HA patients with inhibitors (3–6), including those undergoing ITI (135). Though the experience with emicizumab and FVIII protein ITI is currently limited (135), we would anticipate that a combination of a non-factor therapy hemostatic prophylaxis and AAV FVIII gene therapy ITI would be similarly attractive. This approach would have the advantage of limiting bleeds and thus exposure to non-transgene expressed FVIII or bypassing agents. Furthermore, the use of emicizumab would allow for both inhibitor titer and transgene FVIII levels to be closely monitored.
Several AAV liver-directed gene therapy products for HA are moving through the clinical development pipeline (Table 1). To date, most clinical trials have mitigated the risk of inhibitor development by selecting only those subjects with heavy exposures to FVIII protein. Preclinical studies in a limited number of HA dogs (91, 94, 100) and other preclinical disease models (40, 42, 43) support the concept that AAV liver-directed gene therapy can harness the unique pro-tolerogenic properties of the liver, termed the liver tolerance effect (41), to induce peripheral immune tolerance to transgene FVIII. The potential advantages of our proposed AAV-mediated approach compared to standard ITI are summarized in Table 4. The ideal AAV FVIII vector for gene therapy ITI could be dosed low enough to avoid hepatotoxicities while still providing long-term hemostatic FVIII levels that, at a minimum, compare favorably to the hemostatic effect of emicizumab. Any planned immunosuppression must be evaluated in an immunocompetent animal model to determine if the specific regimen would negatively interfere with the immune tolerance induction to FVIII. This therapy would have dual benefits of eradicating the anti-FVIII antibodies while also providing continuous therapeutically relevant FVIII.
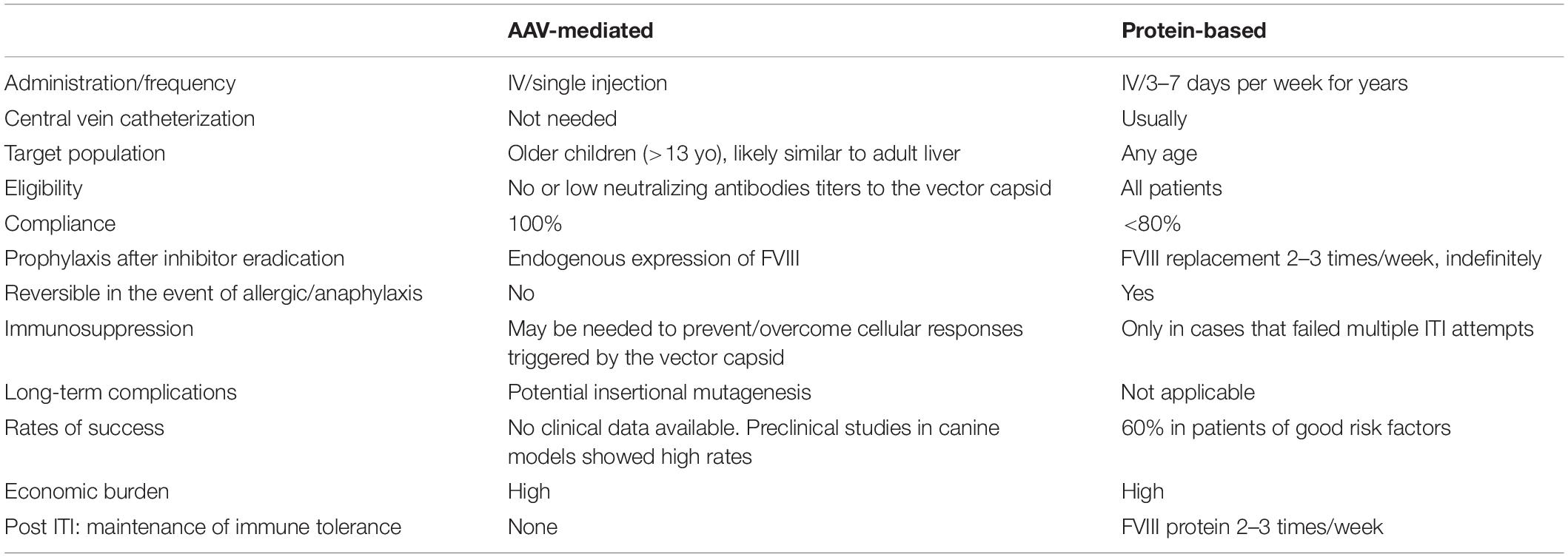
Table 4. Comparison of AAV-liver gene therapy versus protein-based FVIII for immune tolerance induction (ITI).
BS-J and VA conceived, drafted, and reviewed the manuscript.
This work was supported by the National Heart, Lung, and Blood Institute (1K08HL140078 to BS-J, R01HL137335, and 1U54HL142012 to VA).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Peyvandi, F, Garagiola, I, and Young, G. The past and future of haemophilia: diagnosis, treatments, and its complications. Lancet. (2016) 388:187–97. doi: 10.1016/S0140-6736(15)01123-X
2. Konkle, BA, Huston, H, and Nakaya Fletcher, S. Hemophilia A: GeneReviews((R)). MP Adam, HH Ardinger, RA Pagon, SE Wallace, LJH Bean, K Stephens, et al. editors Seattle, WA: University of Washington. (1993–2020).
3. Yada, K, and Nogami, K. Spotlight on emicizumab in the management of hemophilia A: patient selection and special considerations. J Blood Med. (2019) 10:171–81. doi: 10.2147/JBM.S175952
4. Mahlangu, J, Oldenburg, J, Paz-Priel, I, Negrier, C, Niggli, M, Mancuso, ME, et al. Emicizumab prophylaxis in patients who have hemophilia A without inhibitors. N Engl J Med. (2018) 379:811–22. doi: 10.1056/NEJMoa1803550
5. Oldenburg, J, Mahlangu, JN, Kim, B, Schmitt, C, Callaghan, MU, Young, G, et al. Emicizumab prophylaxis in hemophilia A with inhibitors. N Engl J Med. (2017) 377:809–18. doi: 10.1056/NEJMoa1703068
6. Young, G, Liesner, R, Chang, T, Sidonio, R, Oldenburg, J, Jiménez-Yuste, V, et al. A multicenter, open-label phase 3 study of emicizumab prophylaxis in children with hemophilia A with inhibitors. Blood. (2019) 134:2127–38. doi: 10.1182/blood.2019001869
7. Arruda, VR, Doshi, BS, and Samelson-Jones, BJ. Novel approaches to hemophilia therapy: successes and challenges. Blood. (2017) 130:2251–6. doi: 10.1182/blood-2017-08-742312
8. Callaghan, MU, Sidonio, R, and Pipe, SW. Novel therapeutics for hemophilia and other bleeding disorders. Blood. (2018) 132:23–30. doi: 10.1182/blood-2017-09-743385
9. Peyvandi, F, and Garagiola, I. Clinical advances in gene therapy updates on clinical trials of gene therapy in haemophilia. Haemophilia. (2019) 25:738–46. doi: 10.1111/hae.13816
10. Perrin, GQ, Herzog, RW, and Markusic, DM. Update on clinical gene therapy for hemophilia. Blood. (2019) 133:407–14. doi: 10.1182/blood-2018-07-82072
11. Gollomp, KL, Doshi, BS, and Arruda, VR. Gene therapy for hemophilia: progress to date and challenges moving forward. Transfus Apher Sci. (2019) 58:602–12. doi: 10.1016/j.transci.2019.08.012
12. Walsh, CE, Soucie, JM, Miller, CH and United States Hemophilia Treatment Center NetworkImpact of inhibitors on hemophilia A mortality in the United States. Am J Hematol. (2015) 90:400–5. doi: 10.1002/ajh.23957
13. Eckhardt, CL, Loomans, JI, van Velzen, AS, Peters, M, Mauser-Bunschoten, EP, Schwaab, R, et al. Inhibitor development and mortality in non-severe hemophilia A. J Thromb Haemost. (2015) 13:1217–25. doi: 10.1111/jth.12990
14. Darby, SC, Keeling, DM, Spooner, RJ, Wan, Kan S, Giangrande, PL, Collins, PW, et al. The incidence of factor VIII and factor IX inhibitors in the hemophilia population of the UK and their effect on subsequent mortality, 1977-99. J Thromb Haemost. (2004) 2:1047–54.
15. Donfield, SM, Lynn, HS, Lail, AE, Hoots, WK, Berntorp, E, Gomperts, ED, et al. Delays in maturation among adolescents with hemophilia and a history of inhibitors. Blood. (2007) 110:3656–61.
16. Witmer, C, Presley, R, Kulkarni, R, Soucie, JM, Manno, CS, and Raffini, L. Associations between intracranial haemorrhage and prescribed prophylaxis in a large cohort of haemophilia patients in the United States. Br J Haematol. (2011) 152:211–6. doi: 10.1111/j.1365-2141.2010.08469.x
17. Hoots, WK. Arthropathy in inhibitor patients: differences in the joint status. Semin Hematol. (2008) 45(2 Suppl. 1):S42–9. doi: 10.1053/j.seminhematol.2008.03.004
18. Guh, S, Grosse, SD, McAlister, S, Kessler, CM, and Soucie, JM. Healthcare expenditures for males with haemophilia and employer-sponsored insurance in the United States, 2008. Haemophilia. (2012) 18:268–75. doi: 10.1111/j.1365-2516.2011.02692.x
19. Lindvall, K, von Mackensen, S, Elmståhl, S, Khair, K, Stain, AM, Ljung, R, et al. Increased burden on caregivers of having a child with haemophilia complicated by inhibitors. Pediatr Blood Cancer. (2014) 61:706–11. doi: 10.1002/pbc.24856
20. Kempton, CL, and Meeks, SL. Toward optimal therapy for inhibitors in hemophilia. Blood. (2014) 124:3365–72. doi: 10.1182/blood-2014-05-577643
21. Leissinger, C, Gringeri, A, Antmen, B, Berntorp, E, Biasoli, C, Carpenter, S, et al. Anti-inhibitor coagulant complex prophylaxis in hemophilia with inhibitors. N Engl J Med. (2011) 365:1684–92. doi: 10.1056/NEJMoa1104435
22. Konkle, BA, Ebbesen, LS, Erhardtsen, E, Bianco, RP, Lissitchkov, T, Rusen, L, et al. Randomized, prospective clinical trial of recombinant factor VIIa for secondary prophylaxis in hemophilia patients with inhibitors. J Thromb Haemost. (2007) 5:1904–13.
23. Eckhardt, CL, van Velzen, AS, Peters, M, Astermark, J, Brons, PP, Castaman, G, et al. Factor VIII gene (F8) mutation and risk of inhibitor development in nonsevere hemophilia A. Blood. (2013) 122:1954–62. doi: 10.1182/blood-2013-02-483263
24. Peyvandi, F, Mannucci, PM, Garagiola, I, El-Beshlawy, A, Elalfy, M, Ramanan, V, et al. A randomized trial of factor VIII and neutralizing antibodies in hemophilia A. N Engl J Med. (2016) 374:2054–64. doi: 10.1056/NEJMoa1516437
25. Gouw, SC, van der Bom, JG, and Marijke van den Berg, H. Treatment-related risk factors of inhibitor development in previously untreated patients with hemophilia A: the CANAL cohort study. Blood. (2007) 109:4648–54.
26. Astermark, J. FVIII inhibitors: pathogenesis and avoidance. Blood. (2015) 125:2045–51. doi: 10.1182/blood-2014-08-535328
27. Gouw, SC, van den Berg, HM, Oldenburg, J, Astermark, J, de Groot, PG, Margaglione, M, et al. F8 gene mutation type and inhibitor development in patients with severe hemophilia A: systematic review and meta-analysis. Blood. (2012) 119:2922–34. doi: 10.1182/blood-2011-09-379453
28. Oldenburg, J, and Pavlova, A. Genetic risk factors for inhibitors to factors VIII and IX. Haemophilia. (2006) 12(Suppl. 6):15–22.
29. Garagiola, I, Palla, R, and Peyvandi, F. Risk factors for inhibitor development in severe hemophilia A. Thromb Res. (2018) 168:20–7. doi: 10.1016/j.thromres.2018.05.027
30. Young, G. How I treat children with haemophilia and inhibitors. Br J Haematol. (2019) 186:400–8. doi: 10.1111/bjh.15942
31. Brackmann, HH, and Gormsen, J. Massive factor-VIII infusion in haemophiliac with factor-VIII inhibitor, high responder. Lancet. (1977) 2:933.
32. DiMichele, DM. Immune tolerance in haemophilia: the long journey to the fork in the road. Br J Haematol. (2012) 159:123–34. doi: 10.1111/bjh.12028
33. Hay, CR, DiMichele, DM and International Immune Tolerance Study The principal results of the International immune tolerance study: a randomized dose comparison. Blood. (2012) 119:1335–44. doi: 10.1182/blood-2011-08-369132
34. Young, G. Implementing emicizumab in hemophilia inhibitor management: emicizumab should be prescribed after tolerance. Blood Adv. (2018) 2:2780–2.
35. Le Quellec, S, and Negrier, C. Emicizumab should be prescribed independent of immune tolerance induction. Blood Adv. (2018) 2:2783–6. doi: 10.1136/bmjopen-2017-020904
36. Carcao, M, Escuriola-Ettingshausen, C, Santagostino, E, Oldenburg, J, Liesner, R, Nolan, B, et al. The changing face of immune tolerance induction in haemophilia A with the advent of emicizumab. Haemophilia. (2019) 25:676–84. doi: 10.1111/hae.13762
37. Makris, M, Iorio, A, and Lenting, PJ. Emicizumab and thrombosis: the story so far. J Thromb Haemost. (2019) 17:1269–72.
38. Aledort, LM. Deaths associated with emicizumab in patients with hemophilia A. N Engl J Med. (2019) 381:1878–9.
39. Sherman, A, Biswas, M, and Herzog, RW. Innovative approaches for immune tolerance to factor VIII in the treatment of hemophilia A. Front Immunol. (2017) 8:1604. doi: 10.3389/fimmu.2017.01604
40. Arruda, VR, and Samelson-Jones, BJ. Gene therapy for immune tolerance induction in hemophilia with inhibitors. J Thromb Haemost. (2016) 14:1121–34. doi: 10.1111/jth.13331
41. Tiegs, G, and Lohse, AW. Immune tolerance: what is unique about the liver. J Autoimmun. (2010) 34:1–6. doi: 10.1016/j.jaut.2009.08.008
42. Keeler, GD, Markusic, DM, and Hoffman, BE. Liver induced transgene tolerance with AAV vectors. Cell Immunol. (2019) 342:103728. doi: 10.1016/j.cellimm.2017.12.002
43. Sack, BK, Herzog, RW, Terhorst, C, and Markusic, DM. Development of gene transfer for induction of antigen-specific tolerance. Mol Ther Methods Clin Dev. (2014) 1:14013.
45. Crispe, IN. Immune tolerance in liver disease. Hepatology. (2014) 60:2109–17. doi: 10.1002/hep.27254
46. Grant, CR, and Liberal, R. Liver immunology: how to reconcile tolerance with autoimmunity. Clin Res Hepatol Gastroenterol. (2017) 41:6–16. doi: 10.1016/j.clinre.2016.06.003
47. Calne, R, Sells, RA, Pena, JR, Davis, DR, Millard, PR, Herbertson, BM, et al. Induction of immunological tolerance by porcine liver allografts. Nature. (1969) 223:472.
48. Qian, S, Demetris, AJ, Murase, N, Rao, AS, Fung, JJ, and Starzl, TE. Murine liver allograft transplantation: tolerance and donor cell chimerism. Hepatology. (1994) 19:916–24.
49. Houssin, D, Gigou, M, Franco, D, Bismuth, H, Charpentier, B, Lang, P, et al. Specific transplantation tolerance induced by spontaneously tolerated liver allograft in inbred strains of rats. Transplantation. (1980) 29:418.
50. Gorczynski, RM, Chan, Z, Chung, S, Cohen, Z, Levy, G, Sullivan, B, et al. Prolongation of rat small bowel or renal allograft survival by pretransplant transfusion and/or by varying the route of allograft venous drainage. Transplantation. (1994) 58:816–20.
51. Lerut, J, and Sanchez−Fueyo, A. An appraisal of tolerance in liver transplantation. Am J Transplant. (2006) 6:1774–80.
52. Protzer, U, Maini, MK, and Knolle, PA. Living in the liver: hepatic infections. Nat Rev Immunol. (2012) 12:201–13. doi: 10.1038/nri3169
53. Mazepa, MA, Monahan, PE, Baker, JR, Riske, BK, Soucie, JM and Us Hemophilia Treatment Center Network. Men with severe hemophilia in the United States: birth cohort analysis of a large national database. Blood. (2016) 127:3073–81. doi: 10.1182/blood-2015-10-675140
54. Makris, M, and Konkle, BA. Hepatitis C in haemophilia: time for treatment for all. Haemophilia. (2017) 23:180–1.
55. Mingozzi, F, Liu, YL, Dobrzynski, E, Kaufhold, A, Liu, JH, Wang, Y, et al. Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J Clin Invest. (2003) 111:1347–56.
56. Brown, BD, Cantore, A, Annoni, A, Sergi, LS, Lombardo, A, Della, Valle P, et al. A microRNA-regulated lentiviral vector mediates stable correction of hemophilia B mice. Blood. (2007) 110:4144–52.
57. Markusic, DM, Hoffman, BE, Perrin, GQ, Nayak, S, Wang, X, LoDuca, PA, et al. Effective gene therapy for haemophilic mice with pathogenic factor IX antibodies. EMBO Mol Med. (2013) 5:1698–709. doi: 10.1002/emmm.201302859
58. Annoni, A, Cantore, A, Della, Valle P, Goudy, K, Akbarpour, M, Russo, F, et al. Liver gene therapy by lentiviral vectors reverses anti-factor IX pre-existing immunity in haemophilic mice. EMBO Mol Med. (2013) 5:1684–97. doi: 10.1002/emmm.201302857
59. Mount, JD, Herzog, RW, Tillson, DM, Goodman, SA, Robinson, N, McCleland, ML, et al. Sustained phenotypic correction of hemophilia B dogs with a factor IX null mutation by liver-directed gene therapy. Blood. (2002) 99:2670–6.
60. Arruda, VR, Stedman, HH, Haurigot, V, Buchlis, G, Baila, S, Favaro, P, et al. Peripheral transvenular delivery of adeno-associated viral vectors to skeletal muscle as a novel therapy for hemophilia B. Blood. (2010) 115:4678–88. doi: 10.1182/blood-2009-12-261156
61. Arruda, VR, Stedman, HH, Nichols, TC, Haskins, ME, Nicholson, M, Herzog, RW, et al. Regional intravascular delivery of AAV-2-F.IX to skeletal muscle achieves long-term correction of hemophilia B in a large animal model. Blood. (2005) 105:3458–64.
62. Wang, L, Calcedo, R, Nichols, TC, Bellinger, DA, Dillow, A, Verma, IM, et al. Sustained correction of disease in naive and AAV2-pretreated hemophilia B dogs: AAV2/8-mediated, liver-directed gene therapy. Blood. (2005) 105:3079–86.
63. Wang, L, Nichols, TC, Read, MS, Bellinger, DA, and Verma, IM. Sustained expression of therapeutic level of factor IX in hemophilia B dogs by AAV-mediated gene therapy in liver. Mol Ther. (2000) 1:154–8.
64. Snyder, RO, Miao, C, Meuse, L, Tubb, J, Donahue, BA, Lin, HF, et al. Correction of hemophilia B in canine and murine models using recombinant adeno-associated viral vectors. Nat Med. (1999) 5:64–70.
65. Harding, TC, Koprivnikar, KE, Tu, GH, Zayek, N, Lew, S, Subramanian, A, et al. Intravenous administration of an AAV-2 vector for the expression of factor IX in mice and a dog model of hemophilia B. Gene Ther. (2004) 11:204–13.
66. Cantore, A, Ranzani, M, Bartholomae, CC, Volpin, M, Valle, PD, Sanvito, F, et al. Liver-directed lentiviral gene therapy in a dog model of hemophilia B. Sci Transl Med. (2015) 7:277ra28. doi: 10.1126/scitranslmed.aaa1405
67. Crudele, JM, Finn, JD, Siner, JI, Martin, NB, Niemeyer, GP, Zhou, S, et al. AAV liver expression of FIX-Padua prevents and eradicates FIX inhibitor without increasing thrombogenicity in hemophilia B dogs and mice. Blood. (2015) 125:1553–61. doi: 10.1182/blood-2014-07-588194
68. Niemeyer, GP, Herzog, RW, Mount, J, Arruda, VR, Tillson, DM, Hathcock, J, et al. Long-term correction of inhibitor-prone hemophilia B dogs treated with liver-directed AAV2-mediated factor IX gene therapy. Blood. (2009) 113:797–806. doi: 10.1182/blood-2008-10-181479
69. Samelson-Jones, BJ, and Arruda, VR. Protein-engineered coagulation factors for hemophilia gene therapy. Mol Ther Methods Clin Dev. (2019) 12:184–201. doi: 10.1016/j.omtm.2018.12.007
70. Simioni, P, Tormene, D, Tognin, G, Gavasso, S, Bulato, C, Iacobelli, NP, et al. X-linked thrombophilia with a mutant factor IX (factor IX Padua). N Engl J Med. (2009) 361:1671–5. doi: 10.1056/NEJMoa0904377
71. Moris, D, Lu, L, and Qian, S. Mechanisms of liver-induced tolerance. Curr Opin Organ Transplant. (2017) 22:71–8. doi: 10.1097/MOT.0000000000000380
73. Horst, AK, Neumann, K, Diehl, L, and Tiegs, G. Modulation of liver tolerance by conventional and nonconventional antigen-presenting cells and regulatory immune cells. Cell Mol Immunol. (2016) 13:277–92. doi: 10.1038/cmi.2015.112
74. Thomson, AW, and Knolle, PA. Antigen-presenting cell function in the tolerogenic liver environment. Nat Rev Immunol. (2010) 10:753–66. doi: 10.1038/nri2858
75. Li, W, Carper, K, Zheng, XX, Kuhr, CS, Reyes, JD, Liang, Y, et al. The role of Foxp3+ regulatory T cells in liver transplant tolerance. Transplant Proc. (2006) 38:3205–6.
76. Lüth, S, Huber, S, Schramm, C, Buch, T, Zander, S, Stadelmann, C, et al. Ectopic expression of neural autoantigen in mouse liver suppresses experimental autoimmune neuroinflammation by inducing antigen-specific Tregs. J Clin Invest. (2008) 118:3403–10. doi: 10.1172/JCI32132
77. Qian, J, Borovok, M, Bi, L, Kazazian, HH Jr, and Hoyer, LW. Inhibitor antibody development and T cell response to human factor VIII in murine hemophilia A. Thromb Haemost. (1999) 81:240–4.
78. Scallan, CD, Liu, T, Parker, AE, Patarroyo-White, SL, Chen, H, Jiang, H, et al. Phenotypic correction of a mouse model of hemophilia A using AAV2 vectors encoding the heavy and light chains of FVIII. Blood. (2003) 102:3919–26.
79. Sarkar, R, Xiao, W, and Kazazian, HH Jr. A single adeno-associated virus (AAV)-murine factor VIII vector partially corrects the hemophilia A phenotype. J Thromb Haemost. (2003) 1:220–6.
80. Doering, C, Parker, ET, Healey, JF, Craddock, HN, Barrow, RT, and Lollar, P. Expression and characterization of recombinant murine factor VIII. Thromb Haemost. (2002) 88:450–8.
81. Ishiwata, A, Mimuro, J, Mizukami, H, Kashiwakura, Y, Takano, K, Ohmori, T, et al. Liver-restricted expression of the canine factor VIII gene facilitates prevention of inhibitor formation in factor VIII-deficient mice. J Gene Med. (2009) 11:1020–9. doi: 10.1002/jgm.1391
82. Siner, JI, Samelson-Jones, BJ, Crudele, JM, French, RA, Lee, BJ, Zhou, S, et al. Circumventing furin enhances factor VIII biological activity and ameliorates bleeding phenotypes in hemophilia models. JCI Insight. (2016) 1:e89371. doi: 10.1172/jci.insight.89371
83. Sack, BK, Merchant, S, Markusic, DM, Nathwani, AC, Davidoff, AM, Byrne, BJ, et al. Transient B cell depletion or improved transgene expression by codon optimization promote tolerance to factor VIII in gene therapy. PLoS One. (2012) 7:e37671. doi: 10.1371/journal.pone.0037671
84. Lytle, AM, Brown, HC, Paik, NY, Knight, KA, Wright, JF, Spencer, HT, et al. Effects of FVIII immunity on hepatocyte and hematopoietic stem cell-directed gene therapy of murine hemophilia A. Mol Ther Methods Clin Dev. (2016) 3:15056. doi: 10.1038/mtm.2015.56
85. Sabatino, DE, Nichols, TC, Merricks, E, Bellinger, DA, Herzog, RW, and Monahan, PE. Animal models of hemophilia. Prog Mol Biol Transl Sci. (2012) 105:151–209. doi: 10.1016/B978-0-12-394596-9.00006-8
86. Nichols, TC, Hough, C, Agersø, H, Ezban, M, and Lillicrap, D. Canine models of inherited bleeding disorders in the development of coagulation assays, novel protein replacement and gene therapies. J Thromb Haemost. (2016) 14:894–905. doi: 10.1111/jth.13301
87. Giles, AR, Tinlin, S, Hoogendoorn, H, Greenwood, P, and Greenwood, R. Development of factor VIII:C antibodies in dogs with hemophilia A (factor VIII:C deficiency). Blood. (1984) 63:451–6.
88. Tinlin, S, Webster, S, and Giles, AR. The development of homologous (canine/anti-canine) antibodies in dogs with haemophilia A (factor VIII deficiency): a ten-year longitudinal study. Thromb Haemost. (1993) 69:21–4.
89. Hough, C, Kamisue, S, Cameron, C, Notley, C, Tinlin, S, Giles, A, et al. Aberrant splicing and premature termination of transcription of the FVIII gene as a cause of severe canine hemophilia A: similarities with the intron 22 inversion mutation in human hemophilia. Thromb Haemost. (2002) 87:659–65.
90. Lozier, JN, Dutra, A, Pak, E, Zhou, N, Zheng, Z, Nichols, TC, et al. The Chapel hill hemophilia A dog colony exhibits a factor VIII gene inversion. Proc Natl Acad Sci USA. (2002) 99:12991–6.
91. Scallan, CD, Lillicrap, D, Jiang, H, Qian, X, Patarroyo-White, SL, Parker, AE, et al. Sustained phenotypic correction of canine hemophilia A using an adeno-associated viral vector. Blood. (2003) 102:2031–7.
92. Monahan, PE, Lothrop, CD, Sun, J, Hirsch, ML, Kafri, T, Kantor, B, et al. Proteasome inhibitors enhance gene delivery by AAV virus vectors expressing large genomes in hemophilia mouse and dog models: a strategy for broad clinical application. Mol Ther. (2010) 18:1907–16. doi: 10.1038/mt.2010.170
93. Sarkar, R, Mucci, M, Addya, S, Tetreault, R, Bellinger, DA, Nichols, TC, et al. Long-term efficacy of adeno-associated virus serotypes 8 and 9 in hemophilia a dogs and mice. Hum Gene Ther. (2006) 17:427–39.
94. Sabatino, DE, Lange, AM, Altynova, ES, Sarkar, R, Zhou, S, Merricks, EP, et al. Efficacy and safety of long-term prophylaxis in severe hemophilia A dogs following liver gene therapy using AAV vectors. Mol Ther. (2011) 19:442–9. doi: 10.1038/mt.2010.240
95. Jiang, H, Lillicrap, D, Patarroyo-White, S, Liu, T, Qian, X, Scallan, CD, et al. Multiyear therapeutic benefit of AAV serotypes 2, 6, and 8 delivering factor VIII to hemophilia A mice and dogs. Blood. (2006) 108: 107–15.
96. Gallo-Penn, AM, Shirley, PS, Andrews, JL, Tinlin, S, Webster, S, Cameron, C, et al. Systemic delivery of an adenoviral vector encoding canine factor VIII results in short-term phenotypic correction, inhibitor development, and biphasic liver toxicity in hemophilia A dogs. Blood. (2001) 97:107–13.
97. Brown, BD, Shi, CX, Powell, S, Hurlbut, D, Graham, FL, and Lillicrap, D. Helper-dependent adenoviral vectors mediate therapeutic factor VIII expression for several months with minimal accompanying toxicity in a canine model of severe hemophilia A. Blood. (2004) 103:804–10.
98. Franco, LM, Sun, B, Yang, X, Bird, A, Zhang, H, Schneider, A, et al. Evasion of immune responses to introduced human acid alpha-glucosidase by liver-restricted expression in glycogen storage disease type II. Mol Ther. (2005) 12:876–84.
99. Zhang, P, Sun, B, Osada, T, Rodriguiz, R, Yang, XY, Luo, X, et al. Immunodominant liver-specific expression suppresses transgene-directed immune responses in murine pompe disease. Hum Gene Ther. (2012) 23:460–72. doi: 10.1089/hum.2011.063
100. Finn, JD, Ozelo, MC, Sabatino, DE, Franck, HW, Merricks, EP, Crudele, JM, et al. Eradication of neutralizing antibodies to factor VIII in canine hemophilia A after liver gene therapy. Blood. (2010) 116:5842–8. doi: 10.1182/blood-2010-06-288001
101. Lheriteau, E, Davidoff, AM, and Nathwani, AC. Haemophilia gene therapy: progress and challenges. Blood Rev. (2015) 29:321–8. doi: 10.1016/j.blre.2015.03.002
102. Rogers, GL, and Herzog, RW. Gene therapy for hemophilia. Front Biosci (Landmark Ed). (2015) 20:556–603. doi: 10.2741/4324
103. High, KA. The gene therapy journey for hemophilia: are we there yet? Hematology Am Soc Hematol Educ Program. (2012) 2012:375–81. doi: 10.1182/asheducation-2012.1.375
104. Doshi, BS, and Arruda, VR. Gene therapy for hemophilia: what does the future hold? Ther Adv Hematol. (2018) 9:273–93. doi: 10.1177/2040620718791933
105. Wang, D, Tai, PWL, and Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov. (2019) 18:358–78. doi: 10.1038/s41573-019-0012-9
106. Lind, P, Larsson, K, Spira, J, Sydow-Bäckman, M, Almstedt, A, Gray, E, et al. Novel forms of B-domain deleted recombinant factor VIII molecules. Construction and biochemical characterization. Eur J Biochem. (1995) 232:19–27.
107. McIntosh, J, Lenting, PJ, Rosales, C, Lee, D, Rabbanian, S, Raj, D, et al. Therapeutic levels of FVIII following a single peripheral vein administration of rAAV vector encoding a novel human factor VIII variant. Blood. (2013) 121:3335–44. doi: 10.1182/blood-2012-10-462200
108. Mingozzi, F, and High, KA. Overcoming the host immune response to adeno-associated virus gene delivery vectors: the race between clearance, tolerance, neutralization, and escape. Annu Rev Virol. (2017) 4:511–34. doi: 10.1146/annurev-virology-101416-041936
109. Ertl, HCJ, and High, KA. Impact of AAV capsid-specific T-Cell responses on design and outcome of clinical gene transfer trials with recombinant adeno-associated viral vectors: an evolving controversy. Hum Gene Ther. (2017) 28:328–37. doi: 10.1089/hum.2016.172
110. Pasi, KJ, Rangarajan, S, Mitchell, N, Lester, W, Symington, E, Madan, B, et al. Multiyear Follow-up of AAV5-hFVIII-SQ Gene therapy for hemophilia A. N Engl J Med. (2020) 382:29–40. doi: 10.1056/NEJMoa1908490
111. Rangarajan, S, Walsh, L, Lester, W, Perry, D, Madan, B, Laffan, M, et al. AAV5-Factor VIII gene transfer in severe hemophilia A. N Engl J Med. (2017) 377:2519–30. doi: 10.1056/NEJMoa1708483
112. Manno, CS, Pierce, GF, Arruda, VR, Glader, B, Ragni, M, Rasko, JJ, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. (2006) 12:342–7.
113. George, LA, Sullivan, SK, Giermasz, A, Rasko, JEJ, Samelson-Jones, BJ, Ducore, J, et al. Hemophilia B Gene therapy with a high-specific-activity factor IX Variant. N Engl J Med. (2017) 377:2215–27. doi: 10.1056/NEJMoa1708538
114. Nathwani, AC, Tuddenham, EG, Rangarajan, S, Rosales, C, McIntosh, J, Linch, DC, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. (2011) 365:2357–65. doi: 10.1056/NEJMoa1108046
115. Nathwani, AC, Reiss, UM, Tuddenham, EG, Rosales, C, Chowdary, P, McIntosh, J, et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med. (2014) 371:1994–2004. doi: 10.1056/NEJMoa1407309
116. Mingozzi, F, Maus, MV, Hui, DJ, Sabatino, DE, Murphy, SL, Rasko, JE, et al. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat Med. (2007) 13:419–22.
117. Miesbach, W, Meijer, K, Coppens, M, Kampmann, P, Klamroth, R, Schutgens, R, et al. Gene therapy with adeno-associated virus vector 5-human factor IX in adults with hemophilia B. Blood. (2018) 131:1022–31. doi: 10.1182/blood-2017-09-804419
118. Martino, AT, Basner-Tschakarjan, E, Markusic, DM, Finn, JD, Hinderer, C, Zhou, S, et al. Engineered AAV vector minimizes in vivo targeting of transduced hepatocytes by capsid-specific CD8+ T cells. Blood. (2013) 121:2224–33. doi: 10.1182/blood-2012-10-460733
119. Li, H, Tuyishime, S, Wu, TL, Giles-Davis, W, Zhou, D, Xiao, W, et al. Adeno-associated virus vectors serotype 2 induce prolonged proliferation of capsid-specific CD8+ T cells in mice. Mol Ther. (2011) 19:536–46. doi: 10.1038/mt.2010.267
120. Li, C, Hirsch, M, DiPrimio, N, Asokan, A, Goudy, K, Tisch, R, et al. Cytotoxic-T-lymphocyte-mediated elimination of target cells transduced with engineered adeno-associated virus type 2 vector in vivo. J Virol. (2009) 83:6817–24. doi: 10.1128/JVI.00278-09
121. Li, C, Goudy, K, Hirsch, M, Asokan, A, Fan, Y, Alexander, J, et al. Cellular immune response to cryptic epitopes during therapeutic gene transfer. Proc Natl Acad Sci USA. (2009) 106:10770–4. doi: 10.1073/pnas.0902269106
122. Wang, L, Figueredo, J, Calcedo, R, Lin, J, and Wilson, JM. Cross-presentation of adeno-associated virus serotype 2 capsids activates cytotoxic T cells but does not render hepatocytes effective cytolytic targets. Hum Gene Ther. (2007) 18:185–94.
123. Mingozzi, F, Hasbrouck, NC, Basner-Tschakarjan, E, Edmonson, SA, Hui, DJ, Sabatino, DE, et al. Modulation of tolerance to the transgene product in a nonhuman primate model of AAV-mediated gene transfer to liver. Blood. (2007) 110:2334–41.
124. Georgescu, MT, Moorehead, PC, van Velzen, AS, Nesbitt, K, Reipert, BM, Steinitz, KN, et al. Dexamethasone promotes durable factor VIII-specific tolerance in hemophilia A mice via thymic mechanisms. Haematologica. (2018) 103:1403–13. doi: 10.3324/haematol.2018.189852
125. Malhotra, JD, Miao, H, Zhang, K, Wolfson, A, Pennathur, S, Pipe, SW, et al. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc.Natl.Acad.Sci.USA. (2008) 105:18525–30. doi: 10.1073/pnas.0809677105
126. Lange, AM, Altynova, ES, Nguyen, GN, and Sabatino, DE. Overexpression of factor VIII after AAV delivery is transiently associated with cellular stress in hemophilia A mice. Mol Ther Methods Clin Dev. (2016) 3:16064.
127. Zolotukhin, I, Markusic, DM, Palaschak, B, Hoffman, BE, Srikanthan, MA, and Herzog, RW. Potential for cellular stress response to hepatic factor VIII expression from AAV vector. Mol Ther Methods Clin Dev. (2016) 3: 16063.
128. Staber, JM, Pollpeter, MJ, Anderson, CG, Burrascano, M, Cooney, AL, Sinn, PL, et al. Long-term correction of hemophilia A mice following lentiviral mediated delivery of an optimized canine factor VIII gene. Gene Ther. (2017) 24:742–8. doi: 10.1038/gt.2017.67
129. Greene, TK, Lyde, RB, Bailey, SC, Lambert, MP, Zhai, L, Sabatino, DE, et al. Apoptotic effects of platelet factor VIII on megakaryopoiesis: implications for a modified human FVIII for platelet-based gene therapy. J Thromb Haemost. (2014) 12:2102–12. doi: 10.1111/jth.12749
130. Okuyama, T, Huber, RM, Bowling, W, Pearline, R, Kennedy, SC, Flye, MW, et al. Liver-directed gene therapy: a retroviral vector with a complete LTR and the ApoE enhancer-alpha 1-antitrypsin promoter dramatically increases expression of human alpha 1-antitrypsin in vivo. Hum Gene Ther. (1996) 7:637–45.
131. Lang, JF, Toulmin, SA, Brida, KL, Eisenlohr, LC, and Davidson, BL. Standard screening methods underreport AAV-mediated transduction and gene editing. Nat Commun. (2019) 10:3415. doi: 10.1038/s41467-019-11321-7
132. Den Uijl, IE, Mauser Bunschoten, EP, Roosendaal, G, Schutgens, RE, Biesma, DH, Grobbee, DE, et al. Clinical severity of haemophilia A: does the classification of the 1950s still stand? Haemophilia. (2011) 17:849–53. doi: 10.1111/j.1365-2516.2011.02539.x
133. Finn, JD, Nichols, TC, Svoronos, N, Merricks, EP, Bellenger, DA, Zhou, S, et al. The efficacy and the risk of immunogenicity of FIX Padua (R338L) in hemophilia B dogs treated by AAV muscle gene therapy. Blood. (2012) 120:4521–3. doi: 10.1182/blood-2012-06-440123
134. French, RA, Samelson-Jones, BJ, Niemeyer, GP, Lothrop, CD Jr., Merricks, EP, Nichols, TC, et al. Complete correction of hemophilia B phenotype by FIX-Padua skeletal muscle gene therapy in an inhibitor-prone dog model. Blood Adv. (2018) 2:505–8.
135. Batsuli, G, Zimowski, KL, Tickle, K, Meeks, SL, and Sidonio, RF Jr. Immune tolerance induction in paediatric patients with haemophilia A and inhibitors receiving emicizumab prophylaxis. Haemophilia. (2019) 25:789–96. doi: 10.1111/hae.13819
Keywords: hemophilia A, inhibitors, adeno-associated virus, gene therapy, anti-drug antibodies, immune tolerance
Citation: Samelson-Jones BJ and Arruda VR (2020) Translational Potential of Immune Tolerance Induction by AAV Liver-Directed Factor VIII Gene Therapy for Hemophilia A. Front. Immunol. 11:618. doi: 10.3389/fimmu.2020.00618
Received: 13 December 2019; Accepted: 18 March 2020;
Published: 28 April 2020.
Edited by:
Sébastien Lacroix-Desmazes, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceCopyright © 2020 Samelson-Jones and Arruda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valder R. Arruda, YXJydWRhQGVtYWlsLmNob3AuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.