 Elze Rackaityte
Elze Rackaityte Joanna Halkias
Joanna Halkias
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 09 April 2020
Sec. Immunological Tolerance and Regulation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.00588
This article is part of the Research Topic Perinatal Immunobiology View all 34 articles
The developing human fetus generates both tolerogenic and protective immune responses in response to the unique requirements of gestation. Thus, a successful human pregnancy depends on a fine balance between two opposing immunological forces: the semi-allogeneic fetus learns to tolerate both self- and maternal- antigens and, in parallel, develops protective immunity in preparation for birth. This critical window of immune development bridges prenatal immune tolerance with the need for postnatal environmental protection, resulting in a vulnerable neonatal period with heightened risk of infection. The fetal immune system is highly specialized to mediate this transition and thus serves a different function from that of the adult. Adaptive immune memory is already evident in the fetal intestine. Fetal T cells with pro-inflammatory potential are born in a tolerogenic environment and are tightly controlled by both cell-intrinsic and -extrinsic mechanisms, suggesting that compartmentalization and specialization, rather than immaturity, define the fetal immune system. Dysregulation of fetal tolerance generates an inflammatory response with deleterious effects to the pregnancy. This review aims to discuss the recent advances in our understanding of the cellular and molecular composition of fetal adaptive immunity and the mechanisms that govern T cell development and function. We also discuss the tolerance promoting environment that impacts fetal immunity and the consequences of its breakdown. A greater understanding of fetal mechanisms of immune activation and regulation has the potential to uncover novel paradigms of immune balance which may be leveraged to develop therapies for transplantation, autoimmune disease, and birth-associated inflammatory pathologies.
A healthy human pregnancy, in which the fetus shares only half of its genes with the mother, is an impressive immunological feat. Non-inherited maternal antigens and a growing repertoire of self-antigens present a unique immune challenge to survival in utero. Suppression of responses to these antigens is critical to the maintenance of pregnancy (1–3), and the semi-allogeneic fetus relies on a specialized program of immune tolerance for survival in utero. Thus, humans have evolved redundant and dominant fetal mechanisms of tolerance that override our immune system's encoded ability to mount a protective response.
Human fetal development occurs within the anatomically distinct in utero environment defined primarily by the placenta, a chimeric organ composed of both fetal and maternal cells. Maternal immune adaptation to the semi-allogeneic pregnancy includes limitations on immune cell entry, activation, and function (4) as well as the appearance of uniquely tolerogenic cellular and molecular mechanisms [reviewed in (5)]. Features of pregnancy-induced immune tolerance are driven in part by the endocrine functions of the placenta as well as the state of physiologic hypoxia derived from the vascular anatomy of this organ. Finally, the placenta creates a protected niche which filters and limits fetal exposure to external antigens and microbes. Our understanding of placental biology has evolved from a barrier organ to one of feto-maternal communication [reviewed in (6)] and there is a growing appreciation for the role of the fetal immune system in the maintenance of a healthy pregnancy.
Murine models have contributed significantly to our understanding of maternal immune responses in pregnancy, however fetal immunity is poorly modeled in the mouse. Although thymus organogenesis is remarkably similar between the species, the functional output differs drastically during development, likely influenced by the relatively short murine gestation in comparison to that of humans. The first wave of murine T cells to exit the thymus are TCRγδ thymocytes destined for the skin around embryonic day 15 (7, 8). These cells are subsequently replaced by increasing thymopoeisis of conventional TCRαβ T cells which continue to populate the periphery until the end of the first week of life (9). In humans, TCRγδ and TCRαβ T cells, including regulatory T cells, exit the fetal thymus simultaneously and comparatively earlier than in mice [around 12–14 weeks of gestation; (10–12)]. Therefore, unlike mice, most T cell development in humans occurs in utero. Mice depend on a sustained thymic output of naïve T cells throughout their lifetime (13) and neonatal thymectomy results in severe impairment of immune responses to infection and autoimmunity (14–16). In contrast, humans primarily rely on expansion of existing naïve T cells post-natally, as incidental neonatal thymectomies during cardiac surgery do not give rise to autoimmune disease or an increased susceptibility to infection (13, 17, 18). Given these differences between mice and humans, this review will primarily focus on human adaptive immune development.
In humans, all cellular components of innate and adaptive immunity are present in the developing fetus. Adaptive immunity results from antigen-specific activation of T cells and B cells followed by the generation of long-lived memory cells capable of a robust recall response. Innate immunity, triggered by molecular pattern molecules, provides rapid protection from pathogens, and clears dying or damaged self-cells. Innate immune cells are also keystone initiators of the adaptive arm of immunity. T cells are classically activated by cognate peptide-MHC interaction with the T cell receptor (TCR), a process directed by professional antigen presenting cells (APCs) in the periphery. Fetal APCs, such as dendritic cells, can be activated in response to pathogen associated molecular pattern molecules (PAMPs), migrate between tissues and lymphatics, and robustly activate naïve T cells (19). These data indicate that fetal innate immune cells possess the capacity to direct T cell activation and differentiation in utero.
Fetal immunity serves specialized evolutionary goals adapted to the unique demands of gestation and thus differs significantly from that of the adult. Immune tolerance, while critical to survival in utero, must transition to a program of protective immunity in preparation for birth. The neonatal window bridges these two programs in remarkable concert, allowing for the establishment of tolerance to commensals, while also protecting the newborn from infection. In mice, this critical window of immune development is brief: tolerance to skin commensals for example, is preferentially established during the first week of life, coinciding with an abrupt influx of regulatory T cells [Treg; (20)]. In humans, this developmental transition may be longer and likely begins in utero, as maternal environmental exposures (e.g., to farming) result in a greater abundance of highly suppressive Treg cells in neonatal umbilical cord blood (21), and childhood exposures protect from allergic disease in adult life (22–24). However, newborn protective immunity is not fully effective because neonates and infants exhibit a higher susceptibility to infection, the leading causes of mortality in these age groups (25).
This presumed failure of the infant immune system has been bolstered by studies of umbilical cord blood, in which the majority of T cells are naïve and fail to mount a pro-inflammatory response (26), and APCs exhibit an impaired ability to activate T cells (27). Umbilical cord blood, collected immediately after delivery, is composed of circulating fetal immune cells and is stereotypically distinct from the development of circulating neonatal immune cells in the first months of life (28). In contrast to immune cells in the blood, the infant intestine possesses a sizeable proportion of memory T cells (29–32) and lymphoid-derived fetal dendritic cells are capable of T cell activation (19). These recent reports of functional memory T cells capable of pro-inflammatory cytokine production in the fetal intestine indicate that the fetal immune system is not in essence immature, but is rather compartmentalized in its specialized function. Many of the unique features of the fetal immune system are lost with advancing age, for example the proclivity of naïve T cells to differentiate into Treg cells (33), the presence of innate-like CD4 and CD8 T cells (29, 34, 35), and the thymus-derived capacity for T cell IL-8 production (36, 37). The aim of this review is to summarize the latest findings on the composition and function of fetal T cell immunity with a specific focus on mechanisms of tolerance and immune regulation (Figure 1). We discuss how the intra-uterine environment influences fetal immunity through unique adaptations and also examine experimental models to investigate these paradigms. Lastly, we consider the consequences of dysregulation of fetal immunity and how current findings could be leveraged to protect the fetus from perinatal inflammatory pathologies, such as preterm birth.
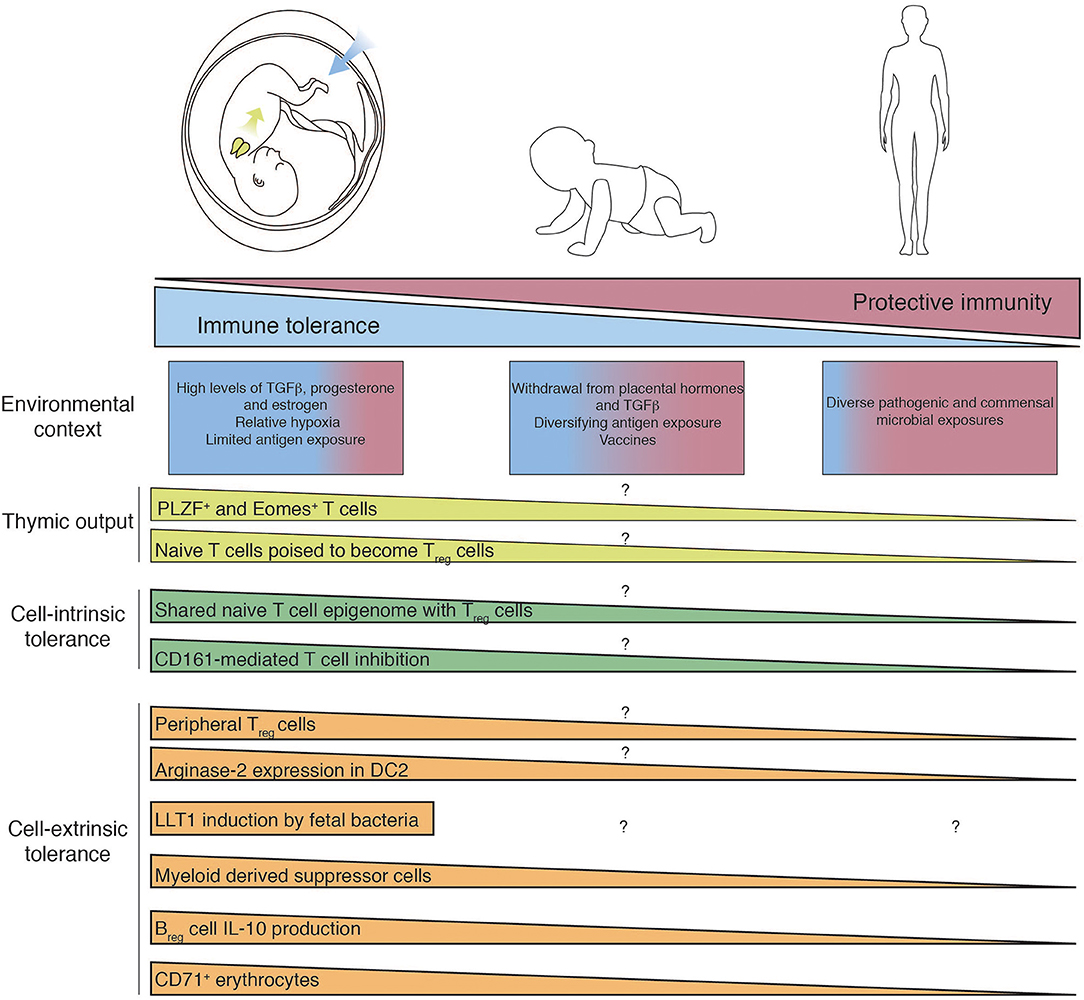
Figure 1. Mechanisms of fetal T cell tolerance and immune regulation. Recent thymic emigrants populate the periphery and encounter tissue-specific environments which differ between fetal, infant, and adult. T cell differentiation and function are governed by diverse cell-intrinsic and -extrinsic mechanisms of immune regulation, which are subsequently lost as in utero pressures for tolerance give way to the need for post-natal protective immunity. Question mark indicates features of immunity that have yet to determined.
Thymic development begins by week eight of human gestation, and the first T cells begin to populate the periphery by 12–14 weeks of gestation (10, 38, 39). Unlike mice, both γδ and αβ T cells emigrate from the thymus simultaneously (7, 8, 38) and the appearance of human Treg cells coincides with that of naïve T cells (11, 12, 16). Fetal T cell colonization in the periphery occurs in a state of relative lymphopenia in which naïve cells composed primarily of recent thymic emigrants begin to populate lymphoid and mucosal niches. Naïve T cells undergo rapid proliferation in response to homeostatic signals (40) similar to that seen in postnatal mice (41). While the vast majority of T cells in cord blood possess a naïve phenotype, healthy term cord blood contains memory T cells with adult-like inflammatory effector functions, albeit in very low proportion (42). Fetal adaptive immune memory was first reported in the fetal intestine (43–45), and memory T cells predominate in the infant and pediatric intestine (46), suggesting that early life adaptive memory is particularly abundant in mucosal tissues.
Fetal immune tolerance is essential to the maintenance of pregnancy, achieved in large part by the ability of Treg cells to suppress the activation, proliferation, and effector functions of a wide range of immune cells. Treg cells [defined in humans by expression of FoxP3, CD25, and low or absent expression of CD127 (47, 48)] are strikingly abundant in peripheral lymphoid organs during the second trimester of human gestation, in stark contrast to neonatal and adult lymph nodes and adult peripheral blood cells (2, 12, 49, 50). Although thymic output of Treg cells is similar in utero and after birth (33), fetal naïve T cells display an increased propensity to differentiate into Treg cells upon antigen encounter in the periphery [induced Treg; iTreg; (33)]. Levels of TGFβ are higher in fetal than in adult lymph nodes, which potentiates the generation of iTreg cells, and distinct fetal hematopoietic stem cells give rise to fetal T cells with the unique ability to differentiate into Treg cells (51). A recent study demonstrated that fetal naïve T cells are already poised for tolerance and share a partial transcriptome and epigenome similar to that of Treg cells, such as the heightened expression of the transcription factor Helios required for iTreg function (52). These studies indicate that both cell-intrinsic and cell-extrinsic mechanisms promote the generation of a predominantly tolerant peripheral T cell response, essential for the maintenance of pregnancy.
Mold et al. demonstrated that fetal T cells rapidly proliferate to maternal and self-antigens in the absence of Treg cells, indicating that fetal immune tolerance is an active process (33). The contribution of Treg cells to perinatal immune tolerance is highlighted by IPEX (immune dysregulation, polyendocrinopathy, entheropathy, X-linked) syndrome, resulting from the loss of the Treg lineage-defining transcription factor FOXP3. Onset of IPEX-related autoimmunity occurs in the second trimester, coinciding with the emergence of peripheral T cells and underscoring the importance of Treg- mediated tolerance to the survival of the fetus (53). That fetal T cells are actively suppressed by Treg cells points to a broader capacity of fetal responses that are kept under tight control.
The need for active tolerance to self- and maternal antigens suggests that protective Th1 responses may be detrimental to a healthy pregnancy. Indeed, human cord blood from infants born preterm exhibits an enrichment of Th1 cells (54–56), thus implicating Th1 cells in the pathophysiology of the premature termination of pregnancy (1). However, a fetal immune response with Th1 polarization is also generated in response to maternal infections (57–60), indicating that protective immunity can be elicited under specific in utero conditions and is not always associated with a negative outcome.
Contrary to the predominantly naïve phenotype of T cells in cord blood, ~50% of CD4 T cells in the human fetal intestine exhibit a memory phenotype and robustly produce the Th1 cytokines IFNγ and TNFα (29–31). We recently reported that the majority of fetal effector memory T cells in the intestine express the transcription factor PLZF, are transcriptionally distinct from either conventional memory T cells or innate-like T cells, and are absent from the adult intestine (29). A greater proportion of PLZF+ CD4+ T cells produced Th1 cytokines as compared to conventional CD4 memory T cells, and this was most evident in the small intestine. Further, IFN-γ–producing PLZF+ CD4+ T cells were enriched in the cord blood of infants born preterm as well as in infants with gastroschisis, a congenital defect defined by chronic inflammation originating from the intestine. These data suggest that dysregulation of PLZF+ CD4+ T cells may contribute to pathologic systemic fetal immune activation.
While PLZF expression is sufficient to confer a memory phenotype and effector function in murine T cells (61, 62), its contribution to human T cell lineage commitment is not as clearly defined. Unlike classic innate-like T cells, functional maturation of fetal PLZF+ CD4+ T cells is not evident in the thymus and PLZF+ CD4+ T cells display a polyclonal TCR repertoire (29). Further, the capacity for activation in response to both TCR-dependent and cytokine-dependent signaling is also present in adult intestinal memory T cells (63). Thus, fetal PLZF+ CD4+ T cells share functional and transcriptional attributes with both conventional and innate-like T cells and may serve as a link between innate and adaptive immunity in the fetus.
A number of innate-like T cells capable of Th1 cytokine production are present in the human fetus, including γδ T cells, invariant natural killer T (iNKT) cells, and mucosa-associated invariant T (MAIT) cells. These cells share certain key features: a semi-invariant TCR repertoire, non-classical MHC restriction, and rapid production of inflammatory cytokines such as IFNγ and TNFα reviewed in (64). While iNKTs are enriched in mucosal tissues such as the intestine and the liver, they represent a small proportion of the total T cell compartment (<1%) in both fetal and adult tissues (65). Low proportions of Vα7.2+ CD161+ T cells with many attributes and characteristics consistent with MAIT cells were reported in the human fetus (<1% of CD3 cells) and were enriched in the fetal liver, small intestine, and lung (66). However, subsequent studies reported that a minority of fetal Vα7.2+ CD161+ T cells reacted with the riboflavin-bound MR1 tetramer (67). These findings indicate that broader specificity may exist in semi-invariant innate-like subsets in the fetus. Fetal semi-invariant γδ T cells are functionally pre-programmed to primarily produce IFNγ and have the capacity to generate Th1 responses to CMV (68). However, recent reports suggest that the repertoire of Vγ9Vδ2 T cells is distinct to the fetus and differs from that of the adult (69). In sum, a substantial number and diversity of conventional and innate-like fetal cells with the capacity for Th1 cytokine production are present in fetal tissues, suggesting a protective role at barrier sites during the critical neonatal window of development.
Historically, the perinatal immune system has been considered skewed toward Th2 responses and away from Th1 responses. However, much of the data supporting this notion was derived from murine studies. Immunization in neonatal mice favors Th2 memory pool formation (70, 71), even when immunized with a Th1 skewing adjuvant (72), and murine thymocytes exhibit an epigenetic predisposition for Th2 differentiation (73). Mirroring findings in neonatal mice, human naïve T cells derived from fetal cord blood and infant adenoids exhibited higher expression of GATA3 than that of adult T cells, and predominantly produced an unglycosylated isoform of IL-4 that is absent from adult Th2 cells (74). Similarly, the IL-13 locus of cord blood T cells is characterized by open chromatin and permissive epigenetic marks (75), suggesting a predisposition for Th2 differentiation. Human neonatal vaccine responses elicit predominantly Th2 type immunity reviewed in (76), similar to that seen in mice. However, Th1 and Th2 cytokine responses have been generated from cord blood naïve T cells in vitro (77) and neonatal vaccination with Bacillus Calmette-Guérin (BCG) produces an adult-like Th1 response (57, 58). Similarly, helminth and mycobacterial antigens induce an adult-like CD4 Th1 skewed response in cord blood of infants in regions endemic for schistosomiasis, filariasis, and tuberculosis (59). Placental malaria results in increased IFNγ- and TNFα-producing CD4 effector memory T cells that proliferate to malaria-specific antigens upon re-stimulation (60), indicating that Th1 immunity is possible in response to the appropriate stimuli. Th2 cell function and specificity has not been extensively investigated in the fetus outside of cord blood, though IL-4 producing cells are present in the fetal intestine (29, 30). In injury contexts, Th2 cells reduce inflammation and promote tissue regeneration [reviewed in (78)], which may be co-opted in utero to promote organogenesis. Thus, while some neonatal vaccine responses induce a Th2 bias, a Th1 response can be elicited under specific conditions, broadening our prevailing understanding of human fetal adaptive immunity.
The role of Th17 cells has not been extensively studied in the context of human fetal development. Term infant cord blood is uniquely enriched in a population of CD161+ CD4 T cells which preferentially gives rise to Th17 cells under in vitro differentiation conditions (79). Further, cord blood naïve CD4 T cells exhibit an enhanced capacity for Th17 differentiation compared to adult cells (80). However, term cord blood CD4 T cells are not capable of either IL-17A or IL-17F production despite the presence of CCR6+ effector memory cells with high levels of RORC expression (42). In contrast to circulating fetal immune cells, IL-17 production is restricted to CD4+ T cells expressing the transcription factor PLZF in the mesenteric lymph node and to a lesser extent in the small intestine, supporting the spatial segregation of fetal effector function (29). Although IL-17 can be produced by human immune cell subsets other than T cells (81), IL-17 production was not appreciated in fetal γδTCR T cells (82) and the capacity of human fetal memory CD8 T cells to produce IL-17 has not yet been reported.
Cord blood naïve CD8 T cells are transcriptionally and epigenetically distinct from that of the adult with a proclivity for proliferation and innate immune responses. These cells express neutrophil-associated and antimicrobial peptide genes, but exhibit a decreased cytotoxic response in comparison to adult cells (83). Fetal CD8 T cells that express KIR/NKG2A and Eomes possess innate-like properties and produce IFNγ in response to IL-12 and IL-18 (34, 35). In parallel, the fetus is also capable of conventional CD8 T cell responses comparable to those of the adult in response to specific maternal infections. Fetal CD8 T cells mount an antiviral-specific response to hepatitis B (84), hepatitis C (85), HIV (86–88), and cytomegalovirus infection, and effectively differentiate into memory cells capable of perforin-production (89). Placental malaria also induces antigen-specific memory CD8 T cell proliferation, though this does not result in increased inflammatory cytokine production (60). Antigen-specific CD8 T cell responses in the fetus and neonate indicate that early-life immunization is possible against specific pathogens. The fetal cytokine environment may limit the activation of CD8 T cells, as TGFβ has been shown to suppress cytotoxic CD8 T cell differentiation induced by IL-15 (90). Fetal CD8 T cells may be highly atypical, as they possess the capacity to produce IL-4 when stimulated in the absence of IL-2 and IL-12, a feat reminiscent of iNKT cells that is absent from adult CD8 T cells (91). In mice, neonatal CD8 T cells originate from distinct hematopoietic stem cell sources than adult CD8 T cells and exhibit rapid and short-lived responses as compared to adults (92, 93), exhibiting a layering of immune function similar to that of human fetal CD4 T cells (51). Together, this evidence points to fetal-specific features for innate-like CD8 T cells which likely play a role in perinatal immune protection.
The remarkable potential of fetal T cells to generate inflammatory responses suggests that strong and redundant mechanisms must exist to preserve a tolerogenic environment required for a successful pregnancy. Indeed, the fetal immune system has adapted and repurposed unique cell-intrinsic and cell-extrinsic means to limit inflammation (Figure 1).
Fetal naïve T cells have a greater capacity to differentiate into Treg cells, which may in part be explained by a higher sensitivity to TGFβ indicated by high levels of SMAD2/SMAD3 phosphorylation in the unstimulated state (49). Lin28b, a repressor of let-7 microRNAs that target TGFβ signaling mediators, is highly expressed in fetal naïve T cells and is required for the increased propensity toward Treg cell differentiation (49). Unlike adult naïve T cells, fetal naïve T cells have an increased propensity to differentiate into Treg cells even in the absence of high levels of exogenous TGFβ (49), suggestive of additional cell intrinsic mechanisms such as a poised epigenome. The predisposition of fetal naïve cells to differentiate into Treg cells is driven by higher expression of and chromatin accessibility at the Helios (IKZF2) locus, and ablation of Helios in fetal naïve T cells results in poor Treg differentiation and impaired function (52). These studies point to additional cell-intrinsic differences in fetal naïve T cells that contribute to a predominant program of immune tolerance and regulation in response to antigen encounter in utero.
T cell activation is governed by productive and simultaneous TCR- and co-stimulatory molecule signaling. Molecules that inhibit T cell activation (co-inhibitory) are expressed either in conjunction with co-stimulatory molecules or following successful activation reviewed in (94). Fetal intestinal memory PLZF+ T cells express high levels of the surface C-type lectin receptor CD161 (29). In adults, engagement of CD161 inhibits human NK cells (95–97), but exerts co-stimulatory effects on iNKT cells (98), MAIT cells (99), and displays no consistent influence on adult polyclonal T cell activation (95, 100). It is thus striking that ligation of CD161 by two different monoclonal antibodies inhibited IFNγ production in response to TCR activation in fetal PLZF+ T cells (29). These reports point to a unique repurposing of the CD161 axis in the fetal context, the signaling mechanisms of which remain to be explored.
Despite strong inhibition following TCR-dependent stimulation, CD161 ligation has no effect on cytokine-mediated T cell activation, which remains a critical area of study. The differential enrichment of numerous negative T cell regulators (DUSP4, DUSP5, DUSP6, LRIG1, and DTX1) in the gene signature of fetal intestinal PLZF+ CD4+ memory T cells (29) suggests the existence of additional cell-intrinsic mechanisms of regulation to promote immune homeostasis in utero. In particular, the transcriptome of fetal PLZF+ CD4+ T cells was differentially enriched for multiple surface molecules with known inhibitory functions such as PD1 (29). Thus, determining the extent to which known and novel co-inhibitory molecules can limit fetal T cell activity is a promising therapeutic avenue for birth-related pathologies, as many inhibitors to these receptor-ligand interactions are already in use and continue to be developed for cancer immunotherapy.
T cell immunity is regulated in large part by the surrounding environment, such as the antigen presenting cells which directly engage with and efficiently activate T cells. Given their central role in T cell activation and differentiation, it is thus not surprising that they also contribute to fetal immune regulation. For example, type II fetal dendritic cells (fDC2) limit the ability of T cells to produce IFNγ, IL-2, IL-17, TNFα, and other inflammatory cytokines (19). Inhibition of TNFα production by fetal T cells is achieved through upregulation of Arginase-2 on fDC2s (19), however the mechanisms of inhibition of other inflammatory cytokines have yet to be determined. Production of IFNγ by fetal T cells may be limited by intestinal macrophages expressing high levels of LLT1, the natural ligand of CD161 (29). Exposure to fetal-specific M. luteus induces LLT1 on fetal antigen presenting cells (101), suggesting that commensal fetal bacteria may modulate this axis to promote intestinal immune tolerance. Further, suppressive functions of intestinal macrophages are well-described in adults (102–104) and it is likely that similar mechanisms are also employed by the fetal immune system.
Additional human myeloid cell populations contribute to T cell tolerance in utero. Myeloid-derived suppressor cells (MDSCs) are a heterogeneous subset of monocytic and granulocytic/neutrophilic cells that limit T cell and NK cell immunity in tumors and during the course of infection reviewed in (105). Neutrophilic MDSCs capable of suppressing Th1, Th2, and Th17 responses are enriched in cord blood and rapidly decrease to adult levels during infancy (106, 107), with levels dropping drastically after the first month of life (108). These MDSCs are capable of phagocytosis of E. coli, yet produce high levels of TGFβ and strongly suppress T cell proliferation (109), a process which requires cell-cell contact (108). Further, BCL-2 mediates lower rates of apoptosis among cord blood MDSCs, suggesting that their prolonged survival may promote a tolerizing environment even after the resolution of bacterial infection (109). It is plausible that C-type lectins may also be involved in regulating fetal MDSCs, because anti-inflammatory IL-10 production in these cells is regulated by co-triggering TLR-MyD88 and C-type lectin receptor-Syk-dependent pathways (110). Neutrophils migrate to tissues at steady-state and support organ function (111), yet their role in fetal development has not been investigated.
The lymphocyte compartment contributes to the modulation of fetal T cells through the activity of Treg cells and regulatory B (Breg) cells. Fetal tolerance is maintained in large part by active Treg cell-mediated suppression of T cell activation (33), yet whether fetal Treg cells exhibit classic or unique suppressive mechanisms has not been formally demonstrated. Further, while Treg cell specificity for non-inherited maternal antigens has been proposed, the question of antigen-specific suppression requires further study. Breg cells [reviewed in (112)] are enriched in the fetus and an immunosuppressive role for Breg cells has been proposed in the context of maternal tolerance (113, 114). In neonatal mice, IL-10-producing Breg cells limit the ability of dendritic cells to produce inflammatory cytokines, which can be induced by broad TLR stimulation of dendritic cells and is paradoxically induced by type I interferon conditioning of B cells in acute inflammatory contexts (115, 116). In humans, Breg cells are highly abundant in cord blood, produce IL-10, and are capable of suppressing CD4 T cell responses in an IL-10-dependent manner through CTLA4 and CD40 mechanisms (117). Cord blood Breg cells exhibit higher IgM expression as compared to adult blood and can inhibit both Th1 and Th2 responses (118), however IgM-expressing B cells are more prevalent in infant than in fetal intestines (32). Thus, fetal lymphocytes contribute to the maintenance of tolerance in pregnancy through the generation of Treg and Breg cells.
Human and murine neonatal peripheral blood cells exhibit high proportions of transferrin receptor expressing (CD71+) erythrocytes with the unique ability to suppress T cells (119). Similar to fetal dendritic cells (19), CD71+ erythrocytes mediate suppression through Arginase-2, suggesting that arginine depletion is a key mechanism of immune regulation in the fetal environment (119). Although the CD71 axis has not been explored in human fetal tissues, depletion of CD71+ erythrocytes results in myeloid cell activation and increased production of TNFα in the intestine of neonatal mice. Because activation of intestinal myeloid cells in response to CD71+ cell depletion does not occur in germ-free or antibiotic treated neonatal mice, this suggests that CD71+ cells may contribute to tolerance to commensal colonization, a critical immune adaptation in early life (119). Further, a subset of CD71+ erythrocytes which express high levels TGFβ may contribute to the increased conversion of naïve T cells into Treg cells. Ablation of Treg cells results in higher proportion of CD71+ erythrocytes and these erythrocytes compensate by increasing expression of TGFβ after transient CD71+ cell depletion (120). The interdependence of neonatal Treg cells and CD71+ erythrocytes points to a repurposing of conserved axes that limit T cell immunity.
The highly controlled biological environment in utero permits the development of the fetal immune system in a state of active immune suppression, while also promoting fetal-specific mechanisms of immune protection. For example, physiologic intra-uterine hypoxia could contribute to fetal immune regulation by limiting the ability of myeloid cells to activate T cells [reviewed in (121)]. Tissue hypoxia also enhances Treg numbers and function (122), pointing to multiple mechanisms by which fetal hypoxia contributes to immune tolerance in utero.
The developing human fetus is exposed to increasing levels of placental hormones with potent immunomodulatory effects throughout gestation. The placenta produces the steroid hormones estrogen [predominantly estradiol (E2) and estriol (E3)] and progesterone (P4), which are critical to maternal tolerance of the fetus (123–131). Despite high levels of placental hormones in the fetal circulation, little is known about their effect on fetal T cells. P4 promotes differentiation of naïve cord blood T cells into Treg cells and suppresses the differentiation of Th17, yet has little effect on adult T cells, suggesting that the sensitivity to P4 is lost in adulthood (125). A similar immunosuppressive role has been described for E2 in mice (123, 130, 132), yet the direct effect of E2 on human fetal T cells remains to be explored.
Recent reports suggest that the repertoire of antigens seen by the fetus is broader than previously appreciated. Fetal T cell responses to self- and maternal-antigens are more robustly skewed toward the Treg cell lineage compared to adults (33), yet observations of inflammatory recall responses in the fetus suggest a more varied antigen exposure in utero. Bacteria have been detected in amniotic fluid and the placenta through culture-independent studies of these tissues (133–136), though this remains a controversial area of research as other groups were unable to replicate these findings (137, 138). We recently reported that bacteria are variably present in the fetal intestine through high resolution scanning electron microscopy, sequencing, and viable isolation methods. The presence of Micrococcus luteus in fetal meconium was correlated with a higher proportion of intestinal PLZF+ CD161+ memory T cells and limited their ability to produce IFNγ as compared to non-fetal M. luteus species (101). This finding is supported by a study that demonstrated the requirement of a microbiome for the development of PLZF+ T cells in mice (139). Thus, maternal and fetal commensal bacteria could potentially play a role in modulating fetal immunity and limiting in utero inflammation. In addition to bacteria, the fetus is exposed to allergens in amniotic fluid (140), and cord blood T cells exhibit recall responses to ovalbumin, cockroach, mouse, house dust mite, and Fel d cat allergen (140–144). These studies underscore the ability of fetal T cells to recognize a broad range of antigens and that their responses can be regulated by highly adapted fetal bacteria. In addition to fetal-specific, cell-intrinsic mechanisms of regulation, the development of human T cell immunity is strongly shaped and influenced by a regulated set of exposures within the in utero environment.
Despite dominant and redundant mechanisms that control T cell immunity in utero, fetal T cell activation can occur in pathologic contexts and contribute to the initiation of the fetal inflammatory response. Preterm birth (PTB), defined as birth before 37 weeks of gestation, affects 1 of every 9 newborns, and is the leading cause of childhood death under the age of 5 worldwide (25, 145). Preterm labor (PTL), the main cause of PTB, is more frequent when a fetal inflammatory response is elicited (146, 147), and most of the complications associated with death and disability in preterm infants are triggered by inflammation (56, 148–151). Despite the impact of infection and inflammation on the survival and long-term outcome of preterm infants, little is known about the mechanisms that drive fetal immune responses and their contribution to perinatal immune dysregulation.
Increased proportions of activated T cells and reduced suppressive Treg cell activity are evident during fetal inflammation (54–56, 152) and maternal-reactive fetal memory Th1 cells are implicated in the pathophysiology of PTB (1). Activated CD4 and CD8 T cells derived from the cord blood of preterm infants induce uterine cell line contractility in vitro, an effect which can be rescued by anti-TNFα or anti-IFNγ antibodies (1). Thus, T cell activation and specifically Th1 cytokines may drive the fetal inflammatory response associated with PTB. Animal models of inflammatory PTB identify the fetal intestine as a site of immune activation (153–156), indicating that mucosal T cells in contact with amniotic fluid are ideally situated to contribute to the fetal inflammatory response. Indeed, fetal PLZF+ CD4 T cells, which specifically accumulate in the fetal intestine, are enriched in the cord blood of preterm infants and exhibit a heightened capacity for both TNFα and IFNγ production as compared to term infants (29). A similar pattern of PLZF T cell accumulation is evident in the CB of infants with gastroschisis, a congenital abdominal wall defect characterized by intestinal T cell activation (29). Taken together, these findings point to a role for PLZF+ CD4 T cells as mediators of the fetal inflammatory response.
Pathology associated with activation of fetal intestinal T cells is exemplified in necrotizing enterocolitis (NEC), a co-morbidity of PTB. NEC infants exhibit higher proportions of effector memory CD4 T cells in the lamina propria of the intestine and these cells have a heightened capacity to produce TNFα and reduced capacity for IL-10 production (30). While moderate TNFα levels promote intestinal epithelial maturation, dysregulation of intestinal T cells leads to tissue-damaging effects of TNFα (30). A breakdown in cell-intrinsic mechanisms of human fetal tolerance has not been investigated in this context, but lack of sufficient Treg populations may contribute to disease development in NEC (157). Murine models of NEC demonstrate that CD4 T cell activation is critical to the initiation of inflammation (158). Transfer of NEC intestinal lymphocytes was sufficient to induce Th17 inflammation in otherwise naïve mice. Decreased Treg cell levels were observed and could be rescued by retinoic acid supplementation (158), suggesting that loss of tolerance is a key tenet in this pathology. Ample evidence supports a role for dysregulation of T cell immunity in perinatal pathologies, therefore understanding the mechanisms that keep T cells in check may uncover novel therapeutics for these diseases.
The developing human immune system is not forged in an antigenic vacuum, but rather educated in the presence of self-, maternal-, and environmental antigens. It is therefore not surprising that protective adaptive immunity critical to perinatal surveillance and defense is generated concurrently with a program of immune tolerance in utero. The ability of fetal T cells to mount a protective and, in pathogenic contexts, dangerous inflammatory response reinforces the existence of fetal-specific adaptations of immune regulation. Cell intrinsic mechanisms of control are further bolstered by tolerizing pregnancy factors such as the hormonal milieu, hypoxia, and relative depravation of environmental antigens. Insight into these tolerizing mechanisms and their broader applicability to limit human immunity will likely have a major impact on human health.
Early-life immune exposures and responses strongly shape lifelong health, thus increasing the urgency to understand the development of human fetal immunity. There is a growing need to develop methods to study human samples and for innovative model systems to explore the mechanisms of disease. Discoveries in human fetal immunology for the past decades have primarily come from studies in human cord blood, and will continue to serve as an important investigative tool for biomarker and therapeutic development. The study of perinatal inflammatory pathologies relies primarily on large animal models (e.g., sheep, macaques) that possess a full adaptive immune response similar to that of the developing human to explore mechanisms of injury. While cord blood will continue to inform predictive models for disease development, a global understanding of fetal immune development and its progression into childhood and beyond is still lacking. The study of human fetal tissue has served a critical, non-redundant role in our understanding of human in utero development, leading to groundbreaking discoveries in fetal immunity and organogenesis (19, 29–33, 51, 52, 101). When obtained through ethics-board approved protocols and informed consent, donated human fetal tissue offers a wealth of information which has yet to be fully interrogated. Continuing advances in single cell phenotyping as well as cell and tissue culture will accelerate investigation of human development and the translational impact of these discoveries.
Both protective and tolerogenic human T cell immunity begins in utero and the opportunity to intervene in immune developmental trajectories before the onset of disease underscores the importance of studying fetal T cell behavior and regulation.
ER and JH researched and wrote the manuscript.
ER was supported by NSF Graduate Research Fellowship grant #1650113 and by the National Institute of Allergy and Infectious Diseases of the NIH grant number F31AI136336. JH was supported by the National Institute of Allergy and Infectious Diseases K08 Mentored Clinical Scientist Development Award (K08 AI128007) and the Burroughs Wellcome Preterm Birth Grant (# 1019828). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Science Foundation or the National Institutes of Health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Frascoli M, Coniglio L, Witt R, Jeanty C, Fleck-Derderian S, Myers DE, et al. Alloreactive fetal T cells promote uterine contractility in preterm labor via IFN-γ and TNF-α. Sci Transl Med. (2018) 10:eaan2263. doi: 10.1126/scitranslmed.aan2263
2. Michaëlsson J, Mold JE, McCune JM, Nixon DF. Regulation of T cell responses in the developing human fetus. J Immunol. (2006) 176:5741–48. doi: 10.4049/jimmunol.176.10.5741
3. Kemp MW. Preterm birth, intrauterine infection, and fetal inflammation. Front Immunol. (2014) 5:574. doi: 10.3389/fimmu.2014.00574
4. Nancy P, Tagliani E, Tay C-S, Asp P, Levy DE, Erlebacher A. Chemokine gene silencing in decidual stromal cells limits T cell access to the maternal-fetal interface. Science. (2012) 336:1317–21. doi: 10.1126/science.1220030
5. Erlebacher A. Immunology of the maternal-fetal interface. Annu Rev Immunol. (2013) 31:387–411. doi: 10.1146/annurev-immunol-032712-100003
6. PrabhuDas M, Bonney E, Caron K, Dey S, Erlebacher A, Fazleabas A, et al. Immune mechanisms at the maternal-fetal interface: Perspectives and challenges. Nat Immunol. (2015) 16:328–34. doi: 10.1038/ni.3131
7. Ikuta K, Kina T, MacNeil I, Uchida N, Peault B, Chien Y, et al. A developmental switch in thymic lymphocyte maturation potential occurs at the level of hematopoietic stem cells. Cell. (1990) 62:863–74. doi: 10.1016/0092-8674(90)90262-D
8. Havran WL, Allison JP. Developmentally ordered appearance of thymocytes expressing different T-cell antigen receptors. Nature. (1988) 335:443–5. doi: 10.1038/335443a0
9. Friedberg SH, Weissman IL. Lymphoid tissue architecture. II. Ontogeny of peripheral T and B cells in mice: evidence against Peyer's patches as the site of generation of B cells. J Immunol. (1974) 113:1477–92.
10. Lobach DF, Haynes BF. Ontogeny of the human thymus during fetal development. J Clin Immunol. (1987) 7:81–97. doi: 10.1007/BF00916002
11. Cupedo T, Nagasawa M, Weijer K, Blom B, Spits H. Development and activation of regulatory T cells in the human fetus. Eur J Immunol. (2005) 35:383–90. doi: 10.1002/eji.200425763
12. Darrasse-Jeze G. Ontogeny of CD4+CD25+ regulatory/suppressor T cells in human fetuses. Blood. (2005) 105:4715–21. doi: 10.1182/blood-2004-10-4051
13. den Braber I, Mugwagwa T, Vrisekoop N, Westera L, Mögling R, Bregje de Boer A, et al. Maintenance of peripheral naive T cells is sustained by thymus output in mice but not humans. Immunity. (2012) 36:288–97. doi: 10.1016/j.immuni.2012.02.006
14. Miller JFAP. Effect of neonatal thymectomy on the immunological responsiveness of the mouse. Proc R Soc London Ser B Biol Sci. (1962) 156:415–28. doi: 10.1098/rspb.1962.0048
15. Adkins B. T-cell function in newborn mice and humans. Immunol Today. (1999) 20:330–5. doi: 10.1016/S0167-5699(99)01473-5
16. Asano M. Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J Exp Med. (1996) 184:387–96. doi: 10.1084/jem.184.2.387
17. Mancebo E, Clemente J, Sanchez J, Ruiz-Contreras J, De Pablos P, Cortezon S, et al. Longitudinal analysis of immune function in the first 3 years of life in thymectomized neonates during cardiac surgery. Clin Exp Immunol. (2008) 154:375–83. doi: 10.1111/j.1365-2249.2008.03771.x
18. Prelog M, Keller M, Geiger R, Brandstätter A, Würzner R, Schweigmann U, et al. Thymectomy in early childhood: Significant alterations of the CD4+CD45RA+CD62L+ T cell compartment in later life. Clin Immunol. (2009) 130:123–32. doi: 10.1016/j.clim.2008.08.023
19. McGovern N, Shin A, Low G, Low D, Duan K, Yao LJ, et al. Human fetal dendritic cells promote prenatal T-cell immune suppression through arginase-2. Nature. (2017) 546:662–6. doi: 10.1038/nature22795
20. Scharschmidt TC, Vasquez KS, Truong HA, Gearty SV, Pauli ML, Nosbaum A, et al. A wave of regulatory T cells into neonatal skin mediates tolerance to commensal microbes. Immunity. (2015) 43:1011–21. doi: 10.1016/j.immuni.2015.10.016
21. Yu J, Liu X, Li Y, Meng S, Wu F, Yan B, et al. Maternal exposure to farming environment protects offspring against allergic diseases by modulating the neonatal TLR-Tregs-Th axis. Clin Transl Allergy. (2018) 8:1–13. doi: 10.1186/s13601-018-0220-0
22. Riedler J, Braun-Fahrländer C, Eder W, Schreuer M, Waser M, Maisch S, et al. Exposure to farming in early life and development of asthma and allergy: a cross-sectional survey. Lancet. (2001) 358:1129–33. doi: 10.1016/S0140-6736(01)06252-3
23. Eriksson J, Ekerljung L, Lötvall J, Pullerits T, Wennergren G, Rönmark E, et al. Growing up on a farm leads to lifelong protection against allergic rhinitis. Allergy Eur J Allergy Clin Immunol. (2010) 65:1397–403. doi: 10.1111/j.1398-9995.2010.02397.x
24. Fujimura KE, Sitarik AR, Havstad S, Lin DL, Levan S, Fadrosh D, et al. Neonatal gut microbiota associates with childhood multisensitized atopy and T cell differentiation. Nat Med. (2016) 22:1187–91. doi: 10.1038/nm.4176
25. Liu L, Oza S, Hogan D, Chu Y, Perin J, Zhu J, et al. Global, regional, and national causes of under-5 mortality in 2000–15: an updated systematic analysis with implications for the sustainable development goals. Lancet. (2016) 388:3027–35. doi: 10.1016/S0140-6736(16)31593-8
26. Chen L, Cohen AC, Lewis DB. Impaired allogeneic activation and T-helper 1 differentiation of human cord blood naive CD4 T cells. Biol Blood Marrow Transplant. (2006) 12:160–71. doi: 10.1016/j.bbmt.2005.10.027
27. Velilla PA, Rugeles MT, Chougnet CA. Defective antigen-presenting cell function in human neonates. Clin Immunol. (2006) 121:251–9. doi: 10.1016/j.clim.2006.08.010
28. Olin A, Henckel E, Chen Y, Lakshmikanth T, Pou C, Mikes J, et al. Stereotypic immune system development in newborn children. Cell. (2018) 174:1277–92.e14. doi: 10.1016/j.cell.2018.06.045
29. Halkias J, Rackaityte E, Hillman SL, Aran D, Mendoza VF, Marshall LR, et al. CD161 contributes to prenatal immune suppression of IFNγ-producing PLZF+ T cells. J Clin Invest. (2019) 129:3562–77. doi: 10.1101/305128
30. Schreurs RRCE, Baumdick ME, Sagebiel AF, Kaufmann M, Mokry M, Klarenbeek PL, et al. Human fetal TNF-α-cytokine-producing CD4+ effector memory T cells promote intestinal development and mediate inflammation early in life. Immunity. (2019) 50:278–80. doi: 10.1016/j.immuni.2018.12.010
31. Li N, Unen V, Abdelaal T, Guo N, Kasatskaya SA, Ladell K, et al. Memory CD4+ T cells are generated in the human fetal intestine. Nat Immunol. (2019) 20:301–12. doi: 10.1038/s41590-018-0294-9
32. Stras SF, Werner L, Toothaker JM, Olaloye OO, Oldham AL, McCourt CC, et al. Maturation of the human intestinal immune system occurs early in fetal development. Dev Cell. (2019) 51:357–73.e5. doi: 10.1016/j.devcel.2019.09.008
33. Mold JE, Michaelsson J, Burt TD, Muench MO, Beckerman KP, Busch MP, et al. Maternal alloantigens promote the development of tolerogenic fetal regulatory T cells in utero. Science. (2008) 322:1562–5. doi: 10.1126/science.1164511
34. Min HS, Lee YJ, Jeon YK, Kim EJ, Kang BH, Jung KC, et al. MHC class II-restricted interaction between thymocytes plays an essential role in the production of innate CD8+ T cells. J Immunol. (2011) 186:5749–57. doi: 10.4049/jimmunol.1002825
35. Jacomet F, Cayssials E, Basbous S, Levescot A, Piccirilli N, Desmier D, et al. Evidence for eomesodermin-expressing innate-like CD8+ KIR/NKG2A+ T cells in human adults and cord blood samples. Eur J Immunol. (2015) 45:1926–33. doi: 10.1002/eji.201545539
36. Gibbons D, Fleming P, Virasami A, Michel ML, Sebire NJ, Costeloe K, et al. Interleukin-8 (CXCL8) production is a signatory T cell effector function of human newborn infants. Nat Med. (2014) 20:1206–10. doi: 10.1038/nm.3670
37. Das A, Rouault-Pierre K, Kamdar S, Gomez-Tourino I, Wood K, Donaldson I, et al. Adaptive from innate: human IFN-γ+ CD4+ T cells can arise directly from CXCL8-producing recent thymic emigrants in babies and adults. J Immunol. (2017) 199:1696–705. doi: 10.4049/jimmunol.1700551
38. Haynes BF, Heinly CS. Early human T cell development: analysis of the human thymus at the time of initial entry of hematopoietic stem cells into the fetal thymic microenvironment. J Exp Med. (1995) 181:1445–58. doi: 10.1084/jem.181.4.1445
39. Farley AM, Morris LX, Vroegindeweij E, Depreter MLG, Vaidya H, Stenhouse FH, et al. Dynamics of thymus organogenesis and colonization in early human development. Dev. (2013) 140:2015–26. doi: 10.1242/dev.087320
40. Schönland SO, Zimmer JK, Lopez-Benitez CM, Widmann T, Ramin KD, Goronzy JJ, et al. Homeostatic control of T-cell generation in neonates. Blood. (2003) 102:1428–34. doi: 10.1182/blood-2002-11-3591
41. Min B, McHugh R, Sempowski GD, Mackall C, Foucras G, Paul WE. Neonates support lymphopenia-induced proliferation. Immunity. (2003) 18:131–40. doi: 10.1016/S1074-7613(02)00508-3
42. Zhang X, Mozeleski B, Lemoine S, Deriaud E, Lim A, Zhivaki D, et al. CD4 T cells with effector memory phenotype and function develop in the sterile environment of the fetus. Sci Transl Med. (2014) 6:238ra72. doi: 10.1126/scitranslmed.3008748
43. Spencer J, Dillon SB, Isaacson PG, Macdonald TT. T cell subclasses in fetal ileum. Clin Exp Immunol. (1986) 65:553–8.
44. Spencer J, MacDonald TT, Finn T, Isaacson PG. The development of gut associated lymphoid tissue in the terminal ileum of fetal human intestine. Clin Exp Immunol. (1986) 64:536–43.
45. Howie D, Spencer J, DeLord D, Pitzalis C, Wathen NC, Dogan A, et al. Extrathymic T cell differentiation in the human intestine early in life. J Immunol. (1998) 161:5862–72.
46. Thome JJC, Bickham KL, Ohmura Y, Kubota M, Matsuoka N, Gordon C, et al. Early-life compartmentalization of human T cell differentiation and regulatory function in mucosal and lymphoid tissues. Nat Med. (2016) 22:72–7. doi: 10.1038/nm.4008
47. Seddiki N, Santner-Nanan B, Martinson J, Zaunders J, Sasson S, Landay A, et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med. (2006) 203:1693–700. doi: 10.1084/jem.20060468
48. Liu W, Putnam AL, Xu-yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ Treg cells. J Exp Med. (2006) 203:1701–11. doi: 10.1084/jem.20060772
49. Bronevetsky Y, Burt TD, McCune JM. Lin28b regulates fetal regulatory T cell differentiation through modulation of TGF-β Signaling. J Immunol. (2016) 197:4344–50. doi: 10.4049/jimmunol.1601070
50. Takahata Y, Nomura A, Takada H, Ohga S, Furuno K, Hikino S, et al. CD25+CD4+ T cells in human cord blood: an immunoregulatory subset with naive phenotype and specific expression of forkhead box p3 (Foxp3) gene. Exp Hematol. (2004) 32:622–9. doi: 10.1016/j.exphem.2004.03.012
51. Mold JE, Venkatasubrahmanyam S, Burt TD, Michaelsson J, Rivera JM, Galkina SA, et al. Fetal and adult hematopoietic stem cells give rise to distinct T cell lineages in humans. Science. (2010) 330:1695–9. doi: 10.1126/science.1196509
52. Ng MSF, Roth TL, Mendoza VF, Marson A, Burt TD. Helios enhances the preferential differentiation of human fetal CD4+ naïve T cells into regulatory T cells. Sci Immunol. (2019) 4:1–20. doi: 10.1126/sciimmunol.aav5947
53. Bacchetta R, Barzaghi F, Roncarolo MG. From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation. Ann N Y Acad Sci. (2016) 1417:5–22. doi: 10.1111/nyas.13011
54. Luciano AA, Yu H, Jackson LW, Wolfe LA, Bernstein HB. Preterm labor and chorioamnionitis are associated with neonatal T cell activation. PLoS ONE. (2011) 6:e16698. doi: 10.1371/journal.pone.0016698
55. Crespo M, Martinez DG, Cerissi A, Rivera-Reyes B, Bernstein HB, Lederman MM, et al. Neonatal T-cell maturation and homing receptor responses to Toll-like receptor ligands differ from those of adult naive T cells: relationship to prematurity. Pediatr Res. (2012) 71:136–43. doi: 10.1038/pr.2011.26
56. Duggan PJ, Maalouf EF, Watts TL, Sullivan MHF, Counsell SJ, Allsop J, et al. Intrauterine T-cell activation and increased proinflammatory cytokine concentrations in preterm infants with cerebral lesions. Lancet. (2001) 358:1699–700. doi: 10.1016/S0140-6736(01)06723-X
57. Vekemans J, Amedei A, Ota MO, D'Elios MM, Goetghebuer T, Ismaili J, et al. Neonatal bacillus Calmette-Guérin vaccination induces adult-like IFN-gamma production by CD4+ T lymphocytes. Eur J Immunol. (2001) 31:1531–5. doi: 10.1002/1521-4141(200105)31:5<1531::AID-IMMU1531>3.0.CO;2-1
58. Marchant A, Goetghebuer T, Ota MO, Wolfe I, Ceesay SJ, Corrah T, et al. Newborns develop a Th1-type immune response to Mycobacterium bovis bacillus Calmette-Guerin vaccination. J Immunol. (1999) 163:2249–55.
59. Malhotra I, Ouma J, Wamachi A, Kioko J, Mungai P, Omollo A, et al. In utero exposure to helminth and mycobacterial antigens generates cytokine responses similar to that observed in adults. J Clin Invest. (1997) 99:1759–66. doi: 10.1172/JCI119340
60. Odorizzi PM, Jagannathan P, McIntyre TI, Budker R, Prahl M, Auma A, et al. In utero priming of highly functional effector T cell responses to human malaria. Sci Transl Med. (2018) 10:eaat6176. doi: 10.1126/scitranslmed.aat6176
61. Kovalovsky D, Uche OU, Eladad S, Hobbs RM, Yi W, Alonzo E, et al. The BTB–zinc finger transcriptional regulator PLZF controls the development of invariant natural killer T cell effector functions. Nat Immunol. (2008) 9:1055–64. doi: 10.1038/ni.1641
62. Savage AK, Constantinides MG, Han J, Picard D, Martin E, Li B, et al. The transcription factor PLZF directs the effector program of the NKT cell lineage. Immunity. (2008) 29:391–403. doi: 10.1016/j.immuni.2008.07.011
63. Okazawa A, Kanai T, Watanabe M, Yamazaki M, Inoue N, Ikeda M, et al. Th1-mediated intestinal inflammation in Crohn's disease may be induced by activation of lamina propria lymphocytes through synergistic stimulation of interleukin-12 and interleukin-18 without T cell receptor engagement. Am J Gastroenterol. (2002) 97:3108–17. doi: 10.1111/j.1572-0241.2002.07107.x
64. Seyda M, Elkhal A, Quante M, Falk CS, Tullius SG. T cells going innate. Trends Immunol. (2016) 37:546–56. doi: 10.1016/j.it.2016.06.004
65. Loh L, Ivarsson M, Michaëlsson J, Sandberg J, Nixon D. Invariant natural killer T cells developing in the human fetus accumulate and mature in the small intestine. Mucosal Immunol. (2014) 7:1233–43. doi: 10.1038/mi.2014.13
66. Leeansyah E, Loh L, Nixon DF, Sandberg JK. Acquisition of innate-like microbial reactivity in mucosal tissues during human fetal MAIT-cell development. Nat Commun. (2014) 5:3143. doi: 10.1038/ncomms4143
67. Youssef GB, Tourret M, Salou M, Ghazarian L, Houdouin V, Mondot S, et al. Ontogeny of human mucosal-associated invariant T cells and related T cell subsets. J Exp Med. (2018) 215:459–79. doi: 10.1084/jem.20171739
68. Vermijlen D, Brouwer M, Donner C, Liesnard C, Tackoen M, Van Rysselberge M, et al. Human cytomegalovirus elicits fetal γδ T cell responses in utero. J Exp Med. (2010) 207:807–21. doi: 10.1084/jem.20090348
69. Papadopoulou M, Tieppo P, McGovern N, Gosselin F, Chan JKY, Goetgeluk G, et al. TCR sequencing reveals the distinct development of fetal and adult human Vγ9Vδ2 T cells. J Immunol. (2019) 203:1468–79. doi: 10.4049/jimmunol.1900592
70. Barrios C, Brawand P, Berney M, Brandt C, Lambert PH, Siegrist CA. Neonatal and early life immune responses to various forms of vaccine antigens qualitatively differ from adult responses: predominance of a Th2-biased pattern which persists after adult boosting. Eur J Immunol. (1996) 26:1489–96. doi: 10.1002/eji.1830260713
71. Forsthuber T, Yip HC, Lehmann PV. Induction of TH1 and TH2 immunity in neonatal mice. Science. (1996) 271:1728–30. doi: 10.1126/science.271.5256.1728
72. Kovarik J, Bozzotti P, Pihlgren M, Lambert PH, Siegrist CA, Love-Homan L, et al. CpG oligodeoxynucleotides can circumvent the Th2 polarization of neonatal responses to vaccines but may fail to fully redirect Th2 responses established by neonatal priming. J Immunol. (1999) 162:1611–7.
73. Rose S, Lichtenheld M, Foote MR, Adkins B. Murine neonatal CD4+ cells are poised for rapid Th2 effector-like function. J Immunol. (2007) 178:2667–78. doi: 10.4049/jimmunol.178.5.2667
74. Hebel K, Weinert S, Kuropka B, Knolle J, Kosak B, Jorch G, et al. CD4+ T cells from human neonates and infants are poised spontaneously to run a nonclassical IL-4 program. J Immunol. (2014) 192:5160–70. doi: 10.4049/jimmunol.1302539
75. Webster RB, Rodriguez Y, Klimecki WT, Vercelli D. The human IL-13 locus in neonatal CD4+ T cells is refractory to the acquisition of a repressive chromatin architecture. J Biol Chem. (2007) 282:700–9. doi: 10.1074/jbc.M609501200
76. Siegrist CA. Vaccination in the neonatal period and early infancy. Int Rev Immunol. (2000) 19:195–219. doi: 10.3109/08830180009088505
77. Lewis DB, Yu CC, Meyer J, English BK, Kahn SJ, Wilson CB. Cellular and molecular mechanisms for reduced interleukin 4 and interferon-γ production by neonatal T cells. J Clin Invest. (1991) 87:194–202. doi: 10.1172/JCI114970
78. Gieseck RL, Wilson MS, Wynn TA. Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol. (2018) 18:62–76. doi: 10.1038/nri.2017.90
79. Cosmi L, De Palma R, Santarlasci V, Maggi L, Capone M, Frosali F, et al. Human interleukin 17-producing cells originate from a CD161+CD4+ T cell precursor. J Exp Med. (2008) 205:1903–16. doi: 10.1084/jem.20080397
80. Black A, Bhaumik S, Kirkman RL, Weaver CT, Randolph DA. Developmental regulation of Th17-cell capacity in human neonates. Eur J Immunol. (2012) 42:311–9. doi: 10.1002/eji.201141847
81. Cupedo T, Crellin NK, Papazian N, Rombouts EJ, Weijer K, Grogan JL, et al. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat Immunol. (2009) 10:66–74. doi: 10.1038/ni.1668
82. Dimova T, Brouwer M, Gosselin F, Tassignon J, Leo O, Donner C, et al. Effector Vγ9Vδ2 T cells dominate the human fetal γδ T-cell repertoire. Proc Natl Acad Sci USA. (2015) 112:E556–65. doi: 10.1073/pnas.1412058112
83. Galindo-Albarrán AO, López-Portales OH, Gutiérrez-Reyna DY, Rodríguez-Jorge O, Sánchez-Villanueva JA, Ramírez-Pliego O, et al. CD8+ T cells from human neonates are biased toward an innate immune response. Cell Rep. (2016) 17:2151–60. doi: 10.1016/j.celrep.2016.10.056
84. Hong M, Sandalova E, Low D, Gehring AJ, Fieni S, Amadei B, et al. Trained immunity in newborn infants of HBV-infected mothers. Nat Commun. (2015) 6:6588. doi: 10.1038/ncomms7588
85. Babik JM, Cohan D, Monto A, Hartigan-O'Connor DJ, McCune JM. The human fetal immune response to hepatitis C virus exposure in utero. J Infect Dis. (2011) 203:196–206. doi: 10.1093/infdis/jiq044
86. Luzuriaga K, Holmes D, Hereema A, Wong J, Panicali DL, Sullivan JL. HIV-1-specific cytotoxic T lymphocyte responses in the first year of life. J Immunol. (1995) 154:433–43.
87. Sanchez-Merino V, Nie S, Luzuriaga K. HIV-1-specific CD8+ T cell responses and viral evolution in women and infants. J Immunol. (2005) 175:6976–86. doi: 10.4049/jimmunol.175.10.6976
88. Legrand FA, Nixon DF, Loo CP, Ono E, Chapman JM, Miyamoto M, et al. Strong HIV-1-specific T cell responses in HIV-1-exposed uninfected infants and neonates revealed after regulatory T cell removal. PLoS ONE. (2006) 1:e102. doi: 10.1371/journal.pone.0000102
89. Marchant A, Appay V, van der Sande M, Dulphy N, Liesnard C, Kidd M, et al. Mature CD8+ T lymphocyte response to viral infection during fetal life. J Clin Invest. (2003) 111:1747–55. doi: 10.1172/JCI200317470
90. Kim YJ, Stringfield TM, Chen Y, Broxmeyer HE. Modulation of cord blood CD8+ T-cell effector differentiation by TGF-β1 and 4-1BB costimulation. Blood. (2005) 105:274–81. doi: 10.1182/blood-2003-12-4343
91. Zhang Y, Maksimovic J, Huang B, De Souza DP, Naselli G, Chen H, et al. Cord blood CD8+ T cells have a natural propensity to express IL-4 in a fatty acid metabolism and caspase activation-dependent manner. Front Immunol. (2018) 9:879. doi: 10.3389/fimmu.2018.00879
92. Wang J, Wissink EM, Watson NB, Smith NL, Grimson A, Rudd BD. Fetal and adult progenitors give rise to unique populations of CD8+ T cells. Blood. (2016) 128:3073–82. doi: 10.1182/blood-2016-06-725366
93. Smith NL, Patel RK, Reynaldi A, Grenier JK, Wang J, Watson NB, et al. Developmental origin governs CD8+ T cell fate decisions during infection. Cell. (2018) 174:117–30.e14. doi: 10.1016/j.cell.2018.05.029
94. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. (2013) 13:227–42. doi: 10.1038/nri3405
95. Aldemir H, Prod'homme V, Dumaurier MJ, Retiere C, Poupon G, Cazareth J, et al. Cutting edge: lectin-like transcript 1 is a ligand for the CD161 receptor. J Immunol. (2005) 175:7791–5. doi: 10.4049/jimmunol.175.12.7791
96. Germain C, Meier A, Jensen T, Knapnougel P, Poupon G, Lazzari A, et al. Induction of lectin-like transcript 1 (LLT1) protein cell surface expression by pathogens and interferon-γ contributes to modulate immune responses. J Biol Chem. (2011) 286:37964–75. doi: 10.1074/jbc.M111.285312
97. Rosen DB, Bettadapura J, Alsharifi M, Mathew PA, Warren HS, Lanier LL. Cutting edge: lectin-like transcript-1 is a ligand for the inhibitory human NKR-P1A receptor. J Immunol. (2005) 175:7796–9. doi: 10.4049/jimmunol.175.12.7796
98. Exley M, Porcelli S, Furman M, Garcia J, Balk S. CD161. (NKR-P1A) costimulation of CD1d-dependent activation of human T cells expressing invariant Vα24JαQ T cell receptor α chains. J Exp Med. (1998) 188:867–76. doi: 10.1084/jem.188.5.867
99. Fergusson JR, Smith KE, Fleming VM, Rajoriya N, Newell EW, Simmons R, et al. CD161 defines a transcriptional and functional phenotype across distinct human T cell lineages. Cell Rep. (2014) 9:1075–88. doi: 10.1016/j.celrep.2014.09.045
100. Rosen DB, Cao W, Avery DT, Tangye SG, Liu YJ, Houchins JP, et al. Functional consequences of interactions between human NKR-P1A and its ligand LLT1 expressed on activated dendritic cells and B cells. J Immunol. (2008) 180:6508–17. doi: 10.4049/jimmunol.180.10.6508
101. Rackaityte E, Halkias J, Fukui EM, Mendoza VF, Hayzelden C, Crawford ED, et al. Viable bacterial colonization is highly limited in the human intestine in utero. Nat Med. (2020) 86:1–44. doi: 10.1038/s41591-020-0761-3
102. Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol. (2007) 8:1086–94. doi: 10.1038/ni1511
103. Egea L, Hirata Y, Kagnoff MF. GM-CSF: a role in immune and inflammatory reactions in the intestine. Exp Rev Gastroenterol Hepatol. (2010) 4:723–31. doi: 10.1586/egh.10.73
104. Krause P, Morris V, Greenbaum JA, Park Y, Bjoerheden U, Mikulski Z, et al. IL-10-producing intestinal macrophages prevent excessive antibacterial innate immunity by limiting IL-23 synthesis. Nat Commun. (2015) 6:7055. doi: 10.1038/ncomms8055
105. Veglia F, Perego M, Gabrilovich D. Myelod derived suppressor cells coming of age. Nat Immunol. (2019) 19:108–19. doi: 10.1038/s41590-017-0022-x
106. Rieber N, Gille C, Köstlin N, Schäfer I, Spring B, Ost M, et al. Neutrophilic myeloid-derived suppressor cells in cord blood modulate innate and adaptive immune responses. Clin Exp Immunol. (2013) 174:45–52. doi: 10.1111/cei.12143
107. He YM, Li X, Perego M, Nefedova Y, Kossenkov AV, Jensen EA, et al. Transitory presence of myeloid-derived suppressor cells in neonates is critical for control of inflammation. Nat Med. (2018) 24:224–31. doi: 10.1038/nm.4467
108. Gervassi A, Lejarcegui N, Dross S, Jacobson A, Itaya G, Kidzeru E, et al. Myeloid derived suppressor cells are present at high frequency in neonates and suppress in vitro t cell responses. PLoS ONE. (2014) 9:e107816. doi: 10.1371/journal.pone.0107816
109. Leiber A, Schwarz J, Köstlin N, Spring B, Fehrenbach B, Katava N, et al. Neonatal myeloid derived suppressor cells show reduced apoptosis and immunosuppressive activity upon infection with Escherichia coli. Eur J Immunol. (2017) 47:1009–021. doi: 10.1002/eji.201646621
110. Zhang X, Majlessi L, Deriaud E, Leclerc C, Lo-Man R. Coactivation of Syk kinase and MyD88 adaptor protein pathways by bacteria promotes regulatory properties of neutrophils. Immunity. (2009) 31:761–71. doi: 10.1016/j.immuni.2009.09.016
111. Casanova-Acebes M, Nicolás-Ávila JA, Yao Li JL, García-Silva S, Balachander A, et al. Neutrophils instruct homeostatic and pathological states in naive tissues. J Exp Med. (2018) 215:2778–95. doi: 10.1084/jem.20181468
112. Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol. (2012) 30:221–41. doi: 10.1146/annurev-immunol-020711-074934
113. Rolle L, Memarzadeh Tehran M, Morell-García A, Raeva Y, Schumacher A, et al. Cutting Edge: IL-10-producing regulatory B cells in early human pregnancy. Am J Reprod Immunol. (2013) 70:448–53. doi: 10.1111/aji.12157
114. Fettke F, Schumacher A, Costa SD, Zenclussen AC. B cells: the old new players in reproductive immunology. Front Immunol. (2014) 5:285. doi: 10.3389/fimmu.2014.00285
115. Sun CM, Deriaud E, Leclerc C, Lo-Man R. Upon TLR9 signaling, CD5+ B cells control the IL-12-dependent Th1-priming capacity of neonatal DCs. Immunity. (2005) 22:467–77. doi: 10.1016/j.immuni.2005.02.008
116. Zhang X, Deriaud E, Jiao X, Braun D, Leclerc C, Lo-Man R. Type I interferons protect neonates from acute inflammation through interleukin 10-producing B cells. J Exp Med. (2007) 204:1107–18. doi: 10.1084/jem.20062013
117. Sarvaria A, Basar R, Mehta RS, Shaim H, Muftuoglu M, Khoder A, et al. IL-10+ regulatory B cells are enriched in cord blood and may protect against cGVHD after cord blood transplantation. Blood. (2016) 128:1346–61. doi: 10.1182/blood-2016-01-695122
118. Esteve-Solé A, Teixidó I, Deyà-Martínez A, Yagüe J, Plaza-Martín AM, Juan M, et al. Characterization of the highly prevalent regulatory CD24hiCD38hi B-Cell population in human cord blood. Front Immunol. (2017) 8:201. doi: 10.3389/fimmu.2017.00201
119. Elahi S, Ertelt JM, Kinder JM, Jiang TT, Zhang X, Xin L, et al. Immunosuppressive CD71+ erythroid cells compromise neonatal host defence against infection. Nature. (2013) 504:158–62. doi: 10.1038/nature12675
120. Shahbaz S, Bozorgmehr N, Koleva P, Namdar A, Jovel J, Fava RA, et al. CD71+ VISTA+ erythroid cells promote the development and function of regulatory T cells through TGF-β. PLoS Biol. (2018) 16:1–26. doi: 10.1371/journal.pbio.2006649
121. Sitkovsky M, Lukashev D. Regulation of immune cells by local-tissue oxygen tension: HIF1α and adenosine receptors. Nat Rev Immunol. (2005) 5:712–21. doi: 10.1038/nri1685
122. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and Treg cells. Nature. (2011) 475:226–30. doi: 10.1038/nature10169
123. Polanczyk MJ, Carson BD, Subramanian S, Afentoulis M, Vandenbark AA, Ziegler SF, Offner H. Cutting edge: estrogen drives expansion of the CD4+ CD25+ regulatory T cell compartment. J Immunol. (2004) 173:2227–30. doi: 10.4049/jimmunol.173.4.2227
124. Salem ML. Estrogen, a double-edged sword: modulation of TH1- and TH2-mediated inflammations by differential regulation of TH1/TH2 cytokine production. Curr Drug Targets Inflamm Allergy. (2004) 3:97–104. doi: 10.2174/1568010043483944
125. Lee JH, Ulrich B, Cho J, Park J, Kim CH. Progesterone promotes differentiation of human cord blood fetal T cells into T regulatory cells but suppresses their differentiation into Th17 cells. J Immunol. (2011) 187:1778–87. doi: 10.4049/jimmunol.1003919
126. Miyaura H, Iwata M. Direct and indirect inhibition of Th1 development by progesterone and glucocorticoids. J Immunol. (2002) 168:1087–94. doi: 10.4049/jimmunol.168.3.1087
127. Hughes GC, Clark EA, Wong AH. The intracellular progesterone receptor regulates CD4+ T cells and T cell-dependent antibody responses. J Leukoc Biol. (2013) 93:369–75. doi: 10.1189/jlb.1012491
128. Gilmore W, Weiner LP, Correale J. Effect of estradiol on cytokine secretion by proteolipid protein-specific T cell clones isolated from multiple sclerosis patients and normal control subjects. J Immunol. (1997) 158:446–51.
129. Maret A, Coudert JD, Garidou L, Foucras G, Gourdy P, Krust A, et al. Estradiol enhances primary antigen-specific CD4 T cell responses and Th1 development in vivo. Essential role of estrogen receptor α expression in hematopoietic cells. Eur J Immunol. (2003) 33:512–21. doi: 10.1002/immu.200310027
130. Tai P, Wang J, Jin H, Song X, Yan J, Kang Y, et al. Induction of regulatory T cells by physiological level estrogen. J Cell Physiol. (2008) 214:456–64. doi: 10.1002/jcp.21221
131. Fox HS, Bond BL, Parslow TG. Estrogen regulates the IFN-gamma promoter. J Immunol. (1991) 146:4362–7.
132. Chen R-Y, Fan Y-M, Zhang Q, Liu S, Li Q, Ke G-L, et al. Estradiol inhibits Th17 cell differentiation through inhibition of RORγT transcription by recruiting the ERα/REA complex to estrogen response elements of the RORγT promoter. J Immunol. (2015) 194:4019–28. doi: 10.4049/jimmunol.1400806
133. Aagaard K, Ma J, Antony KM, Ganu R, Petrosino J, Versalovic J. The placenta harbors a unique microbiome. Sci Transl Med. (2014) 6:237ra65. doi: 10.1126/scitranslmed.3008599
134. Parnell LA, Briggs CM, Cao B, Delannoy-bruno O, Andrew E, Mysorekar IU. Microbial communities in placentas from term normal pregnancy exhibit spatially variable profiles. Sci Rep. (2017) 7:11200. doi: 10.1038/s41598-017-11514-4
135. Collado MC, Rautava S, Aakko J, Isolauri E, Salminen S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci Rep. (2016) 6:23129. doi: 10.1038/srep23129
136. Steel JH, Malatos S, Kennea N, Edwards AD, Miles L, Duggan P, et al. Bacteria and inflammatory cells in fetal membranes do not always cause preterm labor. Pediatr Res. (2005) 57:404–11. doi: 10.1203/01.PDR.0000153869.96337.90
137. de Goffau MC, Lager S, Sovio U, Gaccioli F, Cook E, Peacock SJ, et al. Human placenta has no microbiome but can contain potential pathogens. Nature. (2019) 574:E15. doi: 10.1038/s41586-019-1628-y
138. Lauder AP, Roche AM, Sherrill-mix S, Bailey A, Laughlin AL, Bittinger K, et al. Comparison of placenta samples with contamination controls does not provide evidence for a distinct placenta microbiota. Microbiome. (2016) 4:29. doi: 10.1186/s40168-016-0172-3
139. Ennamorati M, Vasudevan C, Clerkin K, Halvorsen S, Verma S, Ibrahim S, et al. Intestinal microbes influence development of thymic lymphocytes in early life. Proc Natl Acad Sci U S A. (2020) 117:2570–2578. doi: 10.1073/pnas.1915047117
140. Cullinan P, Harris JM, Taylor AJN, Hole AM, Jones M, Barnes F, et al. Detection of house-dust-mite allergen in amniotic fluid and umbilical-cord blood. Lancet. (2000) 356:1899–900. doi: 10.1016/S0140-6736(00)03264-5
141. Hagendorens MM, Ebo DG, Bridts CH, Van De Water L, De Clerck LS, Stevens WJ. Prenatal exposure to house dust mite allergen. (Der p 1), cord blood T cell phenotype and cytokine production and atopic dermatitis during the first year of life. Pediatr Allergy Immunol. (2004) 15:308–15. doi: 10.1111/j.1399-3038.2004.00169.x
142. Kondo N, Nishida T, Shinoda S, Agata H, Fukutomi O, Takenaka R, et al. Cord blood lymphocyte responses to antigens for the prediction of allergy. Pediatr Asthma Allergy Immunol. (1998) 12:61–6. doi: 10.1089/pai.1998.12.61
143. Miller RL, Chew GL, Bell CA, Biedermann SA, Aggarwal M, Kinney PL, et al. Prenatal exposure, maternal sensitization, and sensitization in utero to indoor allergens in an inner-city cohort. Am J Respir Crit Care Med. (2001) 164:995–1001. doi: 10.1164/ajrccm.164.6.2011107
144. Prescott SL, Macaubas C, Holt BJ, Smallacombe TB, Loh R, Sly PD, et al. Transplacental priming of the human immune system to environmental allergens: universal skewing of initial T cell responses toward the Th2 cytokine profile. J Immunol. (1998) 160:4730–7.
145. Beck S, Wojdyla D, Say L, Betran AP, Merialdi M, Requejo JH, et al. The worldwide incidence of preterm birth: a systematic review of maternal mortality and morbidity. Bull World Health Organ. (2010) 88:31–38. doi: 10.2471/BLT.08.062554
146. Romero R, Gomez R, Ghezzi F, Bo Hyun Yoon, Mazor M, Edwin SS, et al. A fetal systemic inflammatory response is followed by the spontaneous onset of preterm parturition. Am J Obstet Gynecol. (1998) 179:186–93. doi: 10.1016/S0002-9378(98)70271-6
147. Romero R, Miranda J, Chaiworapongsa T, Korzeniewski SJ, Chaemsaithong P, Gotsch F, et al. Prevalence and clinical significance of sterile intra-amniotic inflammation in patients with preterm labor and intact membranes. Am J Reprod Immunol. (2014) 72:458–74. doi: 10.1111/aji.12296
148. Dammann O, Leviton A. Brain damage in preterm newborns: might enhancement of developmentally regulated endogenous protection open a door for prevention? Pediatrics. (1999) 104:541–50. doi: 10.1542/peds.104.3.541
149. Dammann O, Leviton A. Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr Res. (1997) 42:1–8. doi: 10.1203/00006450-199707000-00001
150. Viscardi RM. Perinatal inflammation and lung injury. Semin Fetal Neonatal Med. (2012) 17:30–35. doi: 10.1016/j.siny.2011.08.002
151. Takahashi N, Uehara R, Kobayashi M, Yada Y, Koike Y, Kawamata R, et al. Cytokine profiles of seventeen cytokines, growth factors and chemokines in cord blood and its relation to perinatal clinical findings. Cytokine. (2010) 49:331–7. doi: 10.1016/j.cyto.2009.11.024
152. Rueda CM, Wells CB, Gisslen T, Jobe AH, Kallapur SG, Chougnet CA. Effect of chorioamnionitis on regulatory T cells in moderate/late preterm neonates. Hum Immunol. (2015) 76:65–73. doi: 10.1016/j.humimm.2014.10.016
153. Wolfs TGAM, Kramer BW, Thuijls G, Kemp MW, Saito M, Willems MGM, et al. Chorioamnionitis-induced fetal gut injury is mediated by direct gut exposure of inflammatory mediators or by lung inflammation. Am J Physiol Gastrointest Liver Physiol. (2014) 306:G382–93. doi: 10.1152/ajpgi.00260.2013
154. Wolfs TGAM, Kallapur SG, Polglase GR, Pillow JJ, Nitsos I, Newnham JP, et al. IL-1α mediated chorioamnionitis induces depletion of FoxP3+ cells and ileal inflammation in the ovine fetal gut. PLoS ONE. (2011) 6:e18355. doi: 10.1371/journal.pone.0018355
155. Maneenil G, Kemp MW, Kannan PS, Kramer BW, Saito M, Newnham JP, et al. Oral, nasal and pharyngeal exposure to lipopolysaccharide causes a fetal inflammatory response in sheep. PLoS ONE. (2015) 10:e0119281. doi: 10.1371/journal.pone.0119281
156. Nikiforou M, Jacobs EMR, Kemp MW, Hornef MW, Payne MS, Saito M, et al. Intra-amniotic Candida albicans infection induces mucosal injury and inflammation in the ovine fetal intestine. Sci Rep. (2016) 6:29806. doi: 10.1038/srep29806
157. Weitkamp JH, Koyama T, Rock MT, Correa H, Goettel JA, Matta P, et al. Necrotising enterocolitis is characterised by disrupted immune regulation and diminished mucosal regulatory (FOXP3)/effector (CD4, CD8) T cell ratios. Gut. (2013) 62:73–82. doi: 10.1136/gutjnl-2011-301551
Keywords: fetal immunity, T cells, PLZF, Treg cell, inflammation, preterm birth, fetal inflammatory response
Citation: Rackaityte E and Halkias J (2020) Mechanisms of Fetal T Cell Tolerance and Immune Regulation. Front. Immunol. 11:588. doi: 10.3389/fimmu.2020.00588
Received: 11 December 2019; Accepted: 13 March 2020;
Published: 09 April 2020.
Edited by:
Sandra Maria Blois, Charité–Universitätsmedizin Berlin, GermanyReviewed by:
Tatjana Nikolic, Leiden University, NetherlandsCopyright © 2020 Rackaityte and Halkias. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joanna Halkias, am9hbm5hLmhhbGtpYXNAdWNzZi5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.