 Elisa Martini
Elisa Martini Silvia Giugliano
Silvia Giugliano Maria Rescigno2,3
Maria Rescigno2,3 Marinos Kallikourdis
Marinos Kallikourdis
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Immunol. , 31 March 2020
Sec. T Cell Biology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.00509
This article is part of the Research Topic Control of Regulatory T cell Stability, Plasticity and Function in Health and Disease View all 21 articles
The evolution of the full range of functions of regulatory T cells (Treg) coincides with the evolution of mammalian pregnancy. Accordingly, Treg function has been shown to be crucial for maternal-fetal tolerance and implantation. As reproduction is a key point of selective pressure, mammalian pregnancy may represent an evolutionary driver for the development of Treg. Yet beyond the chronological boundaries of mammalian pregnancy, several key physiological and pathological events are being gradually uncovered as involving the immunomodulating functions of Treg cells. These include autoimmunity, age-related inflammation in males and in post-menopausal females, but also oncological and cardiovascular diseases. The latter two sets of diseases collectively compose the main causes of mortality world-wide. Emerging data point to Treg-modulable effects in these diseases, in a departure from the relatively narrower perceived role of Treg as master regulators of autoimmunity. Yet recent evidence also suggests that changes in intestinal microbiota can affect the above pathological conditions. This is likely due to the finding that, whilst the presence and maintenance of intestinal microbiota requires active immune tolerance, mediated by Treg, the existence of microbiota per se profoundly affects the polarization, stability, and balance of pro- and anti-inflammatory T cell populations, including Treg and induced Treg cells. The study of these “novel,” but possibly highly relevant from an ontogenesis perspective, facets of Treg function may hold great potential for our understanding of the mechanisms underlying human disease.
Biological systems develop as serendipitous solutions to selective pressure, according to evolutionary theory. The evolution of regulatory T (Treg) cells and their master regulator transcription factor, foxp3 (1), must have occurred in response to selective pressure that conferred an advantage to vertebrates that possessed them. Whilst an early form of foxp3 does exist in zebrafish (2), the full set of domains of foxp3 only appear in the non-placental mammal platypus (3). Additionally, the enhancer element that is necessary for the induction of induced Treg (iTreg) in the periphery, also first appears in the platypus (4).
The platypus is an egg-laying mammal, and the egg creates a barrier separating the (non-self) paternal antigens from the maternal adaptive immune system. Absence of a barrier would necessitate a mechanism of suppression of maternal anti-fetal responses, a requirement termed “immunological paradox of pregnancy” by transplantation pioneer Medawar (5). On the other hand, all subsequent (in terms of speciation) mammals are placental, having dispensed with the egg, benefiting from the advantage of a continuous flow of nutrients to the fetus. Thus, one can speculate that the serendipitous acquisition of an immunosuppressive T cell subpopulation could have enabled the elimination of the egg barrier.
In support of such a speculation we and others have shown that placental pregnancy with a genetically different father is not possible in the absence of regulatory T cells (6–8). Defects in Treg cells are associated with increased early-stage miscarriage and preeclampsia in humans (9, 10). In summary, whilst a robust adaptive immune system, as developed in vertebrates, is essential in maintaining defense of the self against pathogens (11), the evolution of Treg cells in placental mammals may have enabled the more complex management of the distinction between the self vs. the “non-self of the same species”. The recognition of non-self of the same species, which is central in placental pregnancy, is ironically a much older problem, as sea-dwelling protochordate Botryllus had to fend off -and not tolerate- competition from neighboring individuals of the same species, using molecular processes not too dissimilar from those of Natural Killer (NK) cells (12). In mammalian pregnancy, maternal uterine NK cells interacting with non-classical Class I Major Histocompatibility molecules, such as HLA-G, independent of presence or absence of alloantigen, are essential for vascularization of the placenta, especially at the start of pregnancy (13, 14).
Evidence from mice and humans demonstrates that the abundance of Treg cells is modified during events linked to placental pregnancy. Periodic fluctuations in uterine (15) or peripheral (16) Treg levels render the cells more abundant during the fertile window of the estrus/menstrual cycle, so that suppression can take place should a pregnancy occur. These fluctuations are possibly estrogen-driven, as estrogen has been shown to boost Treg function (17, 18), whilst estrogen-depleting ovariectomy reduces Treg cell abundance (19). Once fertilization occurs, a much more substantial expansion of Treg cells can be observed (6). In this expansion, a role for paternal and male antigen-driven expansion of Treg has been demonstrated; initially in response to seminal fluid antigens (20), as well as paternal antigens (8, 21), which may explain the clonal expansion of Treg cells in the decidua but not the periphery in pregnant women (10).
Intriguingly, the pregnancy-associated expansion can be interrupted, should a uterine infection appear that could jeopardize fetus and mother (22). From a speculative evolutionary perspective, pathogen-induced reductions in Treg functionality would be selected for, as they would spare the mother from pathogens that could expand uncontrollably in an immunosuppressed environment. A putative mechanism may involve recognition of the pathogen by IL-6-producing innate immune cells, blocking the suppressive potential of Treg (23). Indeed, IL-6 is associated with fertility and pregnancy-related pathologies (24), and the cytokine is also known to mediate a conversion of Treg cells into Th17 pro-inflammatory cells in autoimmune arthritis (25). It should be noted that danger signal-induced fetal rejection can be mediated by invariant/semi-invariant lymphocytes, such as iNKT cells (26), Mucosal-Associated Invariant T cells (27) or γδ T cells (28).
It is reasonable to ask how the function most often ascribed to Treg cells, the control of autoimmunity, fits with their role in placental reproduction. Pregnancy is known to temporarily alleviate the symptoms of rheumatoid arthritis in a majority of patients (29). In a murine model of autoimmune arthritis we have shown that the pregnancy-driven expansion of Treg is indeed responsible for this amelioration (30).
A few studies on peripheral blood Treg levels in human patients have shown a maintenance of their elevated numbers in the short period post-partum (31). Nonetheless, in antigen-specific murine models, which can be monitored with more precision, long-term increases in Treg levels from previous pregnancies were much lower than the peak reached during pregnancy (8). Consequently, Treg levels must be dropping after every pregnancy. The same could occur at menopause, as the ovariectomy-induced Treg contraction suggests (19). The time period post-partum and menopause are the main windows of incidence of most -though not all (32)—autoimmune diseases, which often affect far more women than men. In a logical corollary of an evolutionary selection-driven role for Treg in pregnancy, this higher incidence of autoimmunity in women may be an unwanted consequence of an estrogen-responsive Treg population that is necessary for an improved, placental reproduction. Such unwanted deleterious effects of selected-for earlier benefits fall within the term antagonistic pleiotropy (Figure 1), often applied to describe the benefit of infection-fighting immunity in young age leading to deleterious inflammation in old age (33). As pregnancy is the timepoint of genetic heredity, conditions after pregnancy may potentially feature antagonistic pleiotropy effects.
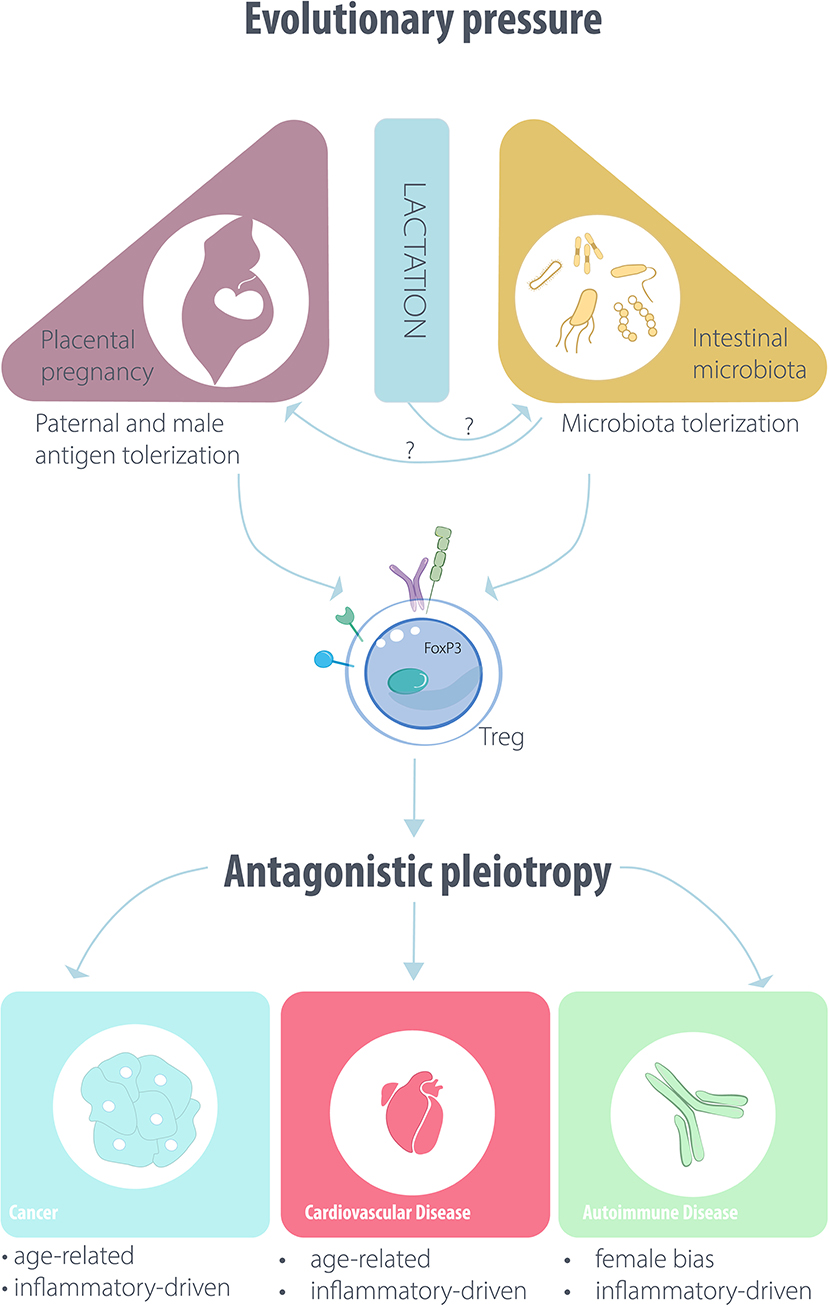
Figure 1. Outline of the putative evolutionary drivers affecting Treg/iTreg function and disease pathogenesis.
The absence of estrogen fluctuation in ovariectomized female mice induces not only a reduction in Treg levels but also a shortening of their lifespan, which approaches that of male mice (19). This is compatible with an overall beneficial effect of Treg in females undergoing estrus/menstrual cycles. If this conjecture were to be true, it would lead to a propensity for inflammation-associated disease in post-menopause women and in all men. Murine models may not be the optimal subjects of study for this question, as mice do not undergo menopause; only humans and cetaceans do (34), possibly as a result of socially-driven selection. Further, modern humans have another unique feature: compared to other primates or even human hunter-gatherer populations, modern humans live substantially longer, with longevity data forming a branching point corresponding to the first industrialized societies (circa 1850), when sanitation was applied in a large scale (35). We can therefore hypothesize that if Treg function has been selected, in all mammals from the platypus onwards, thanks to its beneficial effects in enabling pregnancy, it would still nonetheless be involved, beneficially or detrimentally, in the regulation of any inflammation-associated ailment that occurs in our recently-acquired long lifespans. The reported decay of thymic Treg and iTreg with advanced age (36) could indeed contribute to a loss of self-tolerance to antigens expressed by aging tissue, promoting inflammation-associated disease pathogenesis. The ailments that predominantly affect older humans and lead to the majority of human mortality in the developed world are cancer and cardiovascular disease (37). Fitting our conjecture above on a Treg-related benefit prior to menopause, cardiovascular disease has higher incidence in men than in women, with a difference that decreases with older age (38). For all these reasons, thus, we would expect to find regulatory roles for Treg cells within these groups of diseases.
The link between inflammation and cancer is two-pronged. On the one hand, extensive findings have demonstrated that pro-inflammatory cytokines may enhance the chances of carcinogenesis and genetic instability (39). Treg-mediated suppression of such oncogenic inflammation would be beneficial. Such events clearly happen away from the clinically observable conditions of cancer patients, whose diagnosis occurs long after the carcinogenic event; this may be limiting the incentive to study the role of Treg cells in carcinogenesis. Yet even in growing tumors, evidence has shown that formation of tumor-promoting fibrotic capsules around prostate tumors occurs only in the presence of pro-inflammatory T cells (40), selective suppression of which would be beneficial.
On the other hand, the most clinically important interaction between immunity and cancer is the anti-tumoral, pro-inflammatory function of immunosurveillance (41), which has enabled the development of tumor immunotherapy. The latter, in its most applicable form of immune checkpoint blockade immunotherapy, is based on antibody-mediated reactivation of pro-inflammatory T cells. Yet Treg cells express and utilize the immunotherapy target molecules CTLA-4 (42) and PD-1 (43), and the suppressive action of the Treg may be inhibiting beneficial anti-tumor immunity (44). Why would Treg cells inhibit an anti-tumor response? Interpreted according to the signals a Treg cell may have evolved to deal with, a tumor expressing self-antigens and neo-antigens may be not that different from a fetus, the putative driver of the Treg cells' selection. Genes and processes that help fight non-pediatric, growing, solid tumors cannot have been inherited and selected for in mammals, as until very recently it was not possible to survive and reproduce following cancer incidence.
And yet an obvious solution does arise from the, admittedly speculative, study of the evolutionary drivers of Treg function. As hypothesized above, Treg suppression could collapse in order to reject an infected fetus, in order to protect the mother from the infecting pathogen. In this context, as pioneered in principle by Coley's toxin (45), vaccination strategies that fool the immune system into identifying the tumor as an infected fetus may represent tools that are aligned with the evolutionary drivers of the biological components that we are trying to modulate (46).
Cardiovascular diseases, ranging from atherosclerosis to myocardial infarction (MI) and heart failure, are not traditionally thought of as linked to immunity. Over the recent years, experiments showing that stressed cardiomyocytes release pro-inflammatory cytokines (47) led to clinical trials aiming to therapeutically inhibit the cytokine activity via monoclonal antibodies. As these failed (48), renewed efforts centered on identifying the adaptive immune cells involved in the progression of pathogenesis in atherosclerosis and heart failure. This was based on the premise that a chronic immune response may well be under the control of adaptive immunity. Accordingly, a role for Treg was identified in atherosclerosis (49), whilst therapeutic effects by the experimental administration of Treg cells in a model of pressure overload induced heart failure were also reported (50). In a more translational approach, we have used a molecule derived from Treg, CTLA-4, in soluble fusion protein form (CTLA-4-Ig/Abatacept) to treat advanced-stage heart failure in the pressure overload model. Surprisingly, the treatment with the drug, which is FDA-approved for use in Rheumatoid Arthritis patients, was almost 3-fold more effective than the current standard therapy for heart failure, demonstrating the potential of Treg-inspired therapeutic strategies (51).
More recently we identified, via single cell RNA sequencing, that Treg cells found to be infiltrating the ailing myocardium, express PD-1. Inhibition of PD-1 in healthy hearts blocked the Treg-mediated suppression, releasing cardiac inflammation, which in turn led to a significant reduction in heart function (52). This is intriguing, as anti-PD-1 treatment in human cancer patients has been shown to occasionally lead to T cell-mediated fulminant myocarditis (53). Luckily a solution exists, as CTLA-4-Ig treatment of tumor immunotherapy-induced myocarditis patients has a rescuing effect (54).
In MI that progresses to chronic ischemic heart failure, very recent evidence suggests that Treg cells may lose their immunosuppressive properties, becoming pro-inflammatory and worsening disease outcome (55). Their role is somewhat less clear in the early phase of post-MI repair, where the pro-inflammatory conventional T cells may be useful in the short term in order to deal with the extensive tissue damage (56, 57).
Treg can dampen the response against pathogens during an infection, limiting collateral damage. As a consequence, this also leads to pathogen persistence, which in turn boosts the persistence of protective immunity against the pathogen itself (58). Yet, simultaneously, the inflammation associated with the response limits the functionality of Treg cells (59), a finding that matches the inhibition of Treg function induced by danger signals, mentioned above (23), or indeed by inflammation per se, including in contexts of cardiovascular disease (60).
The fetus expressing paternal antigens is not the only “non-self” that our adaptive immune system has to tolerate via Treg cells. Intestinal microbiota are essential for our survival and are not rejected (61), despite reaching very high cell numbers in the gut (62). The tolerization of “useful” bacteria may be mediated via Treg-mediated suppression (63), whereas “harmful” bacteria may be attacked by pro-inflammatory T cell subpopulations (64). Conversely, both anti-inflammatory iTreg and pro-inflammatory Th17 cells are induced in the gut, displaying a plasticity that depends on the microbiota (65, 66). For example, the immunomodulatory capsule polyaccharide A (PSA) of Bacteroidetes fragilis has been shown to induce IL-10-secreting Treg cells in the gut, restraining gut inflammation (67). Further, bacterial metabolites such as short-chain fatty acids (SCFAs), are involved in Treg differentiation (68–70).
Consequences of the microbiota-induced plasticity may affect disease pathogenesis. The anti-tumoral, pro-inflammatory effect of anti-CTLA-4 or anti-PD-1, described above, was abolished in experimental systems where the intestinal microbiota were eliminated (71, 72), demonstrating the potency of the microbiota-mediated effects. In agreement with these striking results, multiple translational studies have now highlighted how the microbiome of patients that respond to anti-PD-1 treatment is significantly different from that of non-responders (73), and how antibiotic treatment in combination with anti-PD-1/anti-PD-L1 immunotherapy can have a direct effect on patient survival rate (74).
In an analogous manner, emerging evidence demonstrates that the microbiome can significantly affect the pathogenesis and outcome of cardiovascular disease. Alteration of the gut microbiota has been associated with atherosclerotic lesion formation, as revealed by gut metagenome analysis in patients (75). Gut microbiota-produced SCFAs have even been shown to affect blood pressure regulation (76). The above findings exemplify how gut microbiota, possibly also via their effects on iTreg/Th17 populations, have substantial, though still largely unexplored, regulatory roles on the major disease groups that drive mortality world-wide.
The hypothesis that pregnancy may have been a driver for the selection and survival of estrogen-responsive, immunosuppressive Treg cells that enable us to reproduce via a placenta and not using eggs is attractive. The enhanced ability to provide a continuous flow of nutrients to the fetus could even be hypothesized to have helped select bigger brains, capable of abstract thought. However, the interaction between Treg cells and the intestinal microbiota, which is central to our physiology and pathology, raises an intriguing possibility. The evolution of fully-functional foxp3 may have indeed been a prerequisite for the evolution of placental mammalian pregnancy. At least until further species are sequenced, the full set of foxp3 domains appears to have first appeared in the platypus. Yet Treg evolution in the platypus cannot have had pregnancy as a driver of selective pressure, as the platypus is lactating but does not have a placenta. We could, however, hypothesize that, prior to pregnancy, the intestinal microbiota required to digest milk in mammals, which first appeared in the platypus, offering clear advantages as a source of readily-available calories for the litter, could be distinctly different from those found in non-mammalian vertebrates. Could such a bacterial diversification drive the selection of a dedicated tolerizing immune cell population? Could this immunosuppressive population only then enable the evolution of pregnancy in the next speciation step? Compatible with this hypothetical conjecture, the intestinal microbiota of lactating animals has been recently found to be significantly different (77). Further experimentation will be required to assess the validity of these hypotheses.
The interaction between intestinal microbiota and Treg, especially if the conjecture that the former may have been a driver for the evolution of the latter is valid, offers novel means of investigating the functional aspects of Treg cells. In the long term, one would hope that this will lead to innovative therapeutic strategies, in the context of autoimmunity, cancer and cardiovascular disease.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors are supported by AIRC 5x1000 grant 22757.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. (2003) 299:1057–61. doi: 10.1126/science.1079490
2. Quintana FJ, Iglesias AH, Farez MF, Caccamo M, Burns EJ, Kassam N, et al. Adaptive autoimmunity and Foxp3-based immunoregulation in zebrafish. PLoS ONE. (2010) 5:e9478. doi: 10.1371/journal.pone.0009478
3. Andersen KG, Nissen JK, Betz AG. Comparative genomics reveals key gain-of-function events in Foxp3 during regulatory T cell evolution. Front Immunol. (2012) 3:113. doi: 10.3389/fimmu.2012.00113
4. Samstein RM, Josefowicz SZ, Arvey A, Treuting PM, Rudensky AY. Extrathymic generation of regulatory T cells in placental mammals mitigates maternal-fetal conflict. Cell. (2012) 150:29–38. doi: 10.1016/j.cell.2012.05.031
5. Medawar PB. Some immunological and endocrinological problems raised by the evolution of viviparity in vertebrates. Symp Soc Exp Biol Med. (1953) 7:320–38.
6. Aluvihare VR, Kallikourdis M, Betz AG. Regulatory T cells mediate maternal tolerance to the fetus. Nat Immunol. (2004) 5:266–71. doi: 10.1038/ni1037
7. Kahn DA, Baltimore D. Pregnancy induces a fetal antigen-specific maternal T regulatory cell response that contributes to tolerance. Proc Natl Acad Sci USA. (2010) 107:9299–304. doi: 10.1073/pnas.1003909107
8. Rowe JH, Ertelt JM, Xin L, Way SS. Pregnancy imprints regulatory memory that sustains anergy to fetal antigen. Nature. (2012) 490:102–6. doi: 10.1038/nature11462
9. Sasaki Y, Sakai M, Miyazaki S, Higuma S, Shiozaki A, Saito S. Decidual and peripheral blood CD4+CD25+ regulatory T cells in early pregnancy subjects and spontaneous abortion cases. Mol Hum Reprod. (2004) 10:347–53. doi: 10.1093/molehr/gah044
10. Tsuda S, Zhang X, Hamana H, Shima T, Ushijima A, Tsuda K, et al. Clonally expanded decidual effector regulatory t cells increase in late gestation of normal pregnancy, but not in preeclampsia, in humans. Front Immunol. (2018) 9:1934. doi: 10.3389/fimmu.2018.01934
11. Kallikourdis M, Viola A, Benvenuti F. Human immunodeficiencies related to defective APC/T cell interaction. Front Immunol. (2015) 6:433. doi: 10.3389/fimmu.2015.00433
12. Taketa DA, De Tomaso AW. Botryllus schlosseri allorecognition: tackling the enigma. Dev Comp Immunol. (2015) 48:254–65. doi: 10.1016/j.dci.2014.03.014
13. Ashkar AA, Di Santo JP, Croy BA. Interferon gamma contributes to initiation of uterine vascular modification, decidual integrity, and uterine natural killer cell maturation during normal murine pregnancy. J Exp Med. (2000) 192:259–70. doi: 10.1084/jem.192.2.259
14. Clark DA, Chaouat G, Wong K, Gorczynski RM, Kinsky R. Tolerance mechanisms in pregnancy: a reappraisal of the role of class I paternal MHC antigens. Am J Reprod Immunol. (2010) 63:93–103. doi: 10.1111/j.1600-0897.2009.00774.x
15. Kallikourdis M, Betz AG. Periodic accumulation of regulatory T cells in the uterus: preparation for the implantation of a semi-allogeneic fetus? PLoS ONE. (2007) 2:e382. doi: 10.1371/journal.pone.0000382
16. Arruvito L, Sanz M, Banham AH, Fainboim L. Expansion of CD4+CD25+and FOXP3+ regulatory T cells during the follicular phase of the menstrual cycle: implications for human reproduction. Immunol J. (2007) 178:2572–8. doi: 10.4049/jimmunol.178.4.2572
17. Luo CY, Wang L, Sun C, Li DJ. Estrogen enhances the functions of CD4+CD25+Foxp3+ regulatory T cells that suppress osteoclast differentiation and bone resorption in vitro. Cell Mol Immunol. (2011) 8:50–8. doi: 10.1038/cmi.2010.54
18. Adurthi S, Kumar MM, Vinodkumar HS, Mukherjee G, Krishnamurthy H, Acharya KK, et al. Oestrogen receptor-α binds the FOXP3 promoter and modulates regulatory T-cell function in human cervical cancer. Sci Rep. (2017) 7:17289. doi: 10.1038/s41598-017-17102-w
19. Benedusi V, Martini E, Kallikourdis M, Villa A, Meda C, Maggi A. Ovariectomy shortens the life span of female mice. Oncotarget. (2015) 6:10801–11. doi: 10.18632/oncotarget.2984
20. Guerin LR, Moldenhauer LM, Prins JR, Bromfield JJ, Hayball JD, Robertson SA. Seminal fluid regulates accumulation of FOXP3+ regulatory T cells in the preimplantation mouse uterus through expanding the FOXP3+ cell pool and CCL19-mediated recruitment. Biol Reprod. (2011) 85:397–408. doi: 10.1095/biolreprod.110.088591
21. Zhao JX, Zeng YY, Liu Y. Fetal alloantigen is responsible for the expansion of the CD4(+)CD25(+) regulatory T cell pool during pregnancy. J Reprod Immunol. (2007) 75:71–81. doi: 10.1016/j.jri.2007.06.052
22. Rowe JH, Ertelt JM, Xin L, Way SS. Listeria monocytogenes cytoplasmic entry induces fetal wastage by disrupting maternal Foxp3+ regulatory T cell-sustained fetal tolerance. PLoS Pathog. (2012) 8:e1002873. doi: 10.1371/journal.ppat.1002873
23. Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. (2003) 299:1033–6. doi: 10.1126/science.1078231
24. Prins JR, Gomez-Lopez N, Robertson SA. Interleukin-6 in pregnancy and gestational disorders. J Reprod Immunol. (2012) 95:1–14. doi: 10.1016/j.jri.2012.05.004
25. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med. (2014) 20:62–8. doi: 10.1038/nm.3432
26. Li L, Shi L, Yang X, Ren L, Yang J, Lin Y. Role of invariant natural killer T cells in lipopolysaccharide-induced pregnancy loss. Cell Immunol. (2013) 286:1–10. doi: 10.1016/j.cellimm.2013.10.007
27. Solders M, Gorchs L, Erkers T, Lundell AC, Nava S, Gidlöf S, et al. MAIT cells accumulate in placental intervillous space and display a highly cytotoxic phenotype upon bacterial stimulation. Sci Rep. (2017) 7:6123. doi: 10.1038/s41598-017-06430-6
28. Arck PC, Ferrick DA, Steele-Norwood D, Croitoru K, Clark DA. Regulation of abortion by gamma delta T cells. Am J Reprod Immunol. (1997) 37:87–93. doi: 10.1111/j.1600-0897.1997.tb00196.x
29. Hench PS. The ameliorating effect of pregnancy on chronic atrophic (infectious rheumatoid) arthritis, fibrositis, and intermittent hydrarthrosis. Mayo Clin Proc. (1938) 13 161–167.
30. Munoz-Suano A, Kallikourdis M, Sarris M, Betz AG. Regulatory T cells protect from autoimmune arthritis during pregnancy. Autoimmun J. (2012) 38:J103–8. doi: 10.1016/j.jaut.2011.09.007
31. Lima J, Martins C, Nunes G, Sousa MJ, Branco JC, Borrego LM. Regulatory T cells show dynamic behavior during late pregnancy, delivery, and the postpartum period. Reprod Sci. (2017) 24:1025–32. doi: 10.1177/1933719116676395
32. Zen M, Ghirardello A, Iaccarino L, Tonon M, Campana C, Arienti S, et al. Hormones, immune response, pregnancy in healthy women patients, SLE. Swiss Med Wkly. (2010) 140:187–201. doi: 10.4414/smw.2010.12597
33. Ottaviani E, Malagoli D, Capri M, Franceschi C. Ecoimmunology: is there any room for the neuroendocrine system. Bioessays. (2008) 30:868–74. doi: 10.1002/bies.20801
34. Johnstone RA, Cant MA. The evolution of menopause in cetaceans and humans: the role of demography. Proc Biol Sci. (2010) 277:3765–71. doi: 10.1098/rspb.2010.0988
35. Colchero F, Rau R, Jones OR, Barthold JA, Conde DA, Lenart A, et al. The emergence of longevous populations. Proc Natl Acad Sci USA. (2016) 113:E7681–90. doi: 10.1073/pnas.1612191113
36. Darrigues J, van Meerwijk JPM, Romagnoli P. Age-dependent changes in regulatory T lymphocyte development and function: a mini-review. Gerontology. (2018) 64:28–35. doi: 10.1159/000478044
37. Braunwald E. The war against heart failure: the lecture. Lancet. (2015) 385:812–24. doi: 10.1016/S0140-6736(14)61889-4
38. Bots SH, Peters SAE, Woodward M. Sex differences in coronary heart disease and stroke mortality: a global assessment of the effect of ageing between 1980 and 2010. BMJ Glob Health. (2017) 2:e000298. doi: 10.1136/bmjgh-2017-000298
39. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
40. Garetto S, Sardi C, Martini E, Roselli G, Morone D, Angioni R, et al. Tailored chemokine receptor modification improves homing of adoptive therapy T cells in a spontaneous tumor model. Oncotarget. (2016) 7:43010–26. doi: 10.18632/oncotarget.9280
41. Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. (2001) 410:1107–11. doi: 10.1038/35074122
42. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. (2008) 322:271–5. doi: 10.1126/science.1160062
43. Wang Q, He J, Flies DB, Luo L, Chen L. Programmed death one homolog maintains the pool size of regulatory T cells by promoting their differentiation and stability. Sci Rep. (2017) 7:6086. doi: 10.1038/s41598-017-06410-w
44. Barbee MS, Ogunniyi A, Horvat TZ, Dang TO. Current status and future directions of the immune checkpoint inhibitors ipilimumab, pembrolizumab, and nivolumab in oncology. Ann Pharmacother. (2015) 49:907–37. doi: 10.1177/1060028015586218
45. Park JM, Fisher DE. Testimony from the bedside: from Coley's toxins to targeted immunotherapy. Cancer Cell. (2010) 18:9–10. doi: 10.1016/j.ccr.2010.06.010
46. Avogadri F, Martinoli C, Petrovska L, Chiodoni C, Transidico P, Bronte V, et al. Cancer immunotherapy based on killing of Salmonella-infected tumor cells. Cancer Res. (2005) 65:3920–7. doi: 10.1158/0008-5472.CAN-04-3002
47. Ancey C, Corbi P, Froger J, Delwail A, Wijdenes J, Gascan H, et al. Secretion of IL-6, IL-11 and LIF by human cardiomyocytes in primary culture. Cytokine. (2002) 18:199–205. doi: 10.1006/cyto.2002.1033
48. Bozkurt B, Torre-Amione G, Warren MS, Whitmore J, Soran OZ, Feldman AM, et al. Results of targeted anti-tumor necrosis factor therapy with etanercept (ENBREL) in patients with advanced heart failure. Circulation. (2001) 103:1044–7. doi: 10.1161/01.CIR.103.8.1044
49. Ait-Oufella H, Salomon BL, Potteaux S, Robertson AK, Gourdy P, Zoll J, et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med. (2006) 12:178–80. doi: 10.1038/nm1343
50. Kanellakis P, Dinh TN, Agrotis A, Bobik A. CD4+CD25+Foxp3+ regulatory T cells suppress cardiac fibrosis in the hypertensive heart. Hypertens J. (2011) 29:1820–8. doi: 10.1097/HJH.0b013e328349c62d
51. Kallikourdis M, Martini E, Carullo P, Sardi C, Roselli G, Greco CM, et al. T cell costimulation blockade blunts pressure overload-induced heart failure. Nat Commun. (2017) 8:14680. doi: 10.1038/ncomms14680
52. Martini E, Kunderfranco P, Peano C, Carullo P, Cremonesi M, Schorn T, et al. Single cell sequencing of mouse heart immune infiltrate in pressure overload-driven heart failure reveals extent of immune activation. Circulation. (2019) 140:2089–107. doi: 10.1161/CIRCULATIONAHA.119.041694
53. Johnson DB, Balko JM, Compton ML, Chalkias S, Gorham J, Xu Y, et al. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med. (2016) 375:1749–55. doi: 10.1056/NEJMoa1609214
54. Salem JE, Allenbach Y, Vozy A, Brechot N, Johnson DB, Moslehi JJ, et al. Abatacept for severe immune checkpoint inhibitor-associated myocarditis. N Engl J Med. (2019) 380:2377–9. doi: 10.1056/NEJMc1901677
55. Bansal SS, Ismahil MA, Goel M, Patel B, Hamid T, Rokosh G, et al. Activated T lymphocytes are essential drivers of pathological remodeling in ischemic heart failure. Circ Heart Fail. (2017) 10:e003688. doi: 10.1161/CIRCHEARTFAILURE.116.003688
56. Hofmann U, Beyersdorf N, Weirather J, Podolskaya A, Bauersachs J, Ertl G, et al. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation. (2012) 125:1652–63. doi: 10.1161/CIRCULATIONAHA.111.044164
57. Rieckmann M, Delgobo M, Gaal C, Büchner L, Steinau P, Reshef D, et al. Myocardial infarction triggers cardioprotective antigen-specific T helper cell responses. J Clin Invest. (2019) 130:4922–4936. doi: 10.1172/JCI123859
58. Belkaid Y. Role of Foxp3-positive regulatory T cells during infection. Eur Immunol J. (2008) 38:918–21. doi: 10.1002/eji.200738120
59. Oldenhove G, Bouladoux N, Wohlfert EA, Hall JA, Chou DL, Dos Santos L, et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity. (2009) 31:772–86. doi: 10.1016/j.immuni.2009.10.001
60. Garetto S, Trovato AE, Lleo A, Sala F, Martini E, Betz AG, et al. Peak inflammation in atherosclerosis, primary biliary cirrhosis and autoimmune arthritis is counter-intuitively associated with regulatory T cell enrichment. Immunobiology. (2015) 220:1025–9. doi: 10.1016/j.imbio.2015.02.006
61. Quercia S, Candela M, Giuliani C, Turroni S, Luiselli D, Rampelli S, et al. Rom lifetime to evolution: timescales of human gut microbiota adaptation. Front Microbiol. (2014) 5:587. doi: 10.3389/fmicb.2014.00587
62. Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. (2016) 14:e1002533. doi: 10.1371/journal.pbio.1002533
63. Cebula A, Seweryn M, Rempala GA, Pabla SS, McIndoe RA, Denning TL, et al. Thymus-derived regulatory T cells contribute to tolerance to commensal microbiota. Nature. (2013) 497:258–62. doi: 10.1038/nature12079
64. Kato LM, Kawamoto S, Maruya M, Fagarasan S. The role of the adaptive immune system in regulation of gut microbiota. Immunol Rev. (2014) 260:67–75. doi: 10.1111/imr.12185
65. Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell. (2010) 140:845–58. doi: 10.1016/j.cell.2010.02.021
66. Tanoue T, Atarashi K, Honda K. Development and maintenance of intestinal regulatory T cells. Nat Rev Immunol. (2016) 16:295–309. doi: 10.1038/nri.2016.36
67. Shen Y, Giardino Torchia ML, Lawson GW, Karp CL, Ashwell JD, Mazmanian SK. Outer membrane vesicles of a human commensal mediate immune regulation and disease protection. Cell Host Microbe. (2012) 12:509–20. doi: 10.1016/j.chom.2012.08.004
68. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, Deroos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. (2013) 504:451–5. doi: 10.1038/nature12726
69. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. (2013) 504:446–50. doi: 10.1038/nature12721
70. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. (2013) 341:569–73. doi: 10.1126/science.1241165
71. Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. (2015) 350:1084–9. doi: 10.1126/science.aac4255
72. Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. (2015) 350:1079–84. doi: 10.1126/science.aad1329
73. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. (2018) 359:97–103. doi: 10.1126/science.aan4236
74. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. (2018) 359:91–7. doi: 10.1126/science.aan3706
75. Karlsson FH, Fåk F, Nookaew I, Tremaroli V, Fagerberg B, Petranovic D, et al. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat Commun. (2012) 3:1245. doi: 10.1038/ncomms2266
76. Pluznick J. A novel SCFA receptor, the microbiota, and blood pressure regulation. Gut Microbes. (2014) 5:202–7. doi: 10.4161/gmic.27492
Keywords: Treg, pregnancy, autoimmunity, cancer, cardiovascular disease, microbiota, evolution
Citation: Martini E, Giugliano S, Rescigno M and Kallikourdis M (2020) Regulatory T Cells Beyond Autoimmunity: From Pregnancy to Cancer and Cardiovascular Disease. Front. Immunol. 11:509. doi: 10.3389/fimmu.2020.00509
Received: 22 October 2019; Accepted: 05 March 2020;
Published: 31 March 2020.
Edited by:
Margarita Dominguez-Villar, Imperial College London, United KingdomReviewed by:
Thomas Vauvert Hviid, Zealand University Hospital, DenmarkCopyright © 2020 Martini, Giugliano, Rescigno and Kallikourdis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marinos Kallikourdis, bWFyaW5vcy5rYWxsaWtvdXJkaXNAaHVtYW5pdGFzcmVzZWFyY2guaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.