
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 31 March 2020
Sec. Multiple Sclerosis and Neuroimmunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.00456
This article is part of the Research Topic Role of Inflammation in Neurodegenerative Diseases View all 33 articles
 Harald Hampel1*
Harald Hampel1* Filippo Caraci2,3
Filippo Caraci2,3 A. Claudio Cuello4,5,6,7
A. Claudio Cuello4,5,6,7 Giuseppe Caruso3
Giuseppe Caruso3 Robert Nisticò8,9
Robert Nisticò8,9 Massimo Corbo10
Massimo Corbo10 Filippo Baldacci1,11,12,13
Filippo Baldacci1,11,12,13 Nicola Toschi14,15,16Francesco Garaci14,17Patrizia A. Chiesa1,11,12Steven R. Verdooner18
Nicola Toschi14,15,16Francesco Garaci14,17Patrizia A. Chiesa1,11,12Steven R. Verdooner18 Leyla Akman-Anderson18
Leyla Akman-Anderson18 Félix Hernández19,20Jesús Ávila19,20Enzo Emanuele21
Félix Hernández19,20Jesús Ávila19,20Enzo Emanuele21 Pedro L. Valenzuela22
Pedro L. Valenzuela22 Alejandro Lucía23,24,25Mark Watling26
Alejandro Lucía23,24,25Mark Watling26 Bruno P. Imbimbo27
Bruno P. Imbimbo27 Andrea Vergallo1†
Andrea Vergallo1† Simone Lista1,11,12† On behalf of the Alzheimer Precision Medicine Initiative (APMI)
Simone Lista1,11,12† On behalf of the Alzheimer Precision Medicine Initiative (APMI)Neuroinflammation commences decades before Alzheimer's disease (AD) clinical onset and represents one of the earliest pathomechanistic alterations throughout the AD continuum. Large-scale genome-wide association studies point out several genetic variants—TREM2, CD33, PILRA, CR1, MS4A, CLU, ABCA7, EPHA1, and HLA-DRB5-HLA-DRB1—potentially linked to neuroinflammation. Most of these genes are involved in proinflammatory intracellular signaling, cytokines/interleukins/cell turnover, synaptic activity, lipid metabolism, and vesicle trafficking. Proteomic studies indicate that a plethora of interconnected aberrant molecular pathways, set off and perpetuated by TNF-α, TGF-β, IL-1β, and the receptor protein TREM2, are involved in neuroinflammation. Microglia and astrocytes are key cellular drivers and regulators of neuroinflammation. Under physiological conditions, they are important for neurotransmission and synaptic homeostasis. In AD, there is a turning point throughout its pathophysiological evolution where glial cells sustain an overexpressed inflammatory response that synergizes with amyloid-β and tau accumulation, and drives synaptotoxicity and neurodegeneration in a self-reinforcing manner. Despite a strong therapeutic rationale, previous clinical trials investigating compounds with anti-inflammatory properties, including non-steroidal anti-inflammatory drugs (NSAIDs), did not achieve primary efficacy endpoints. It is conceivable that study design issues, including the lack of diagnostic accuracy and biomarkers for target population identification and proof of mechanism, may partially explain the negative outcomes. However, a recent meta-analysis indicates a potential biological effect of NSAIDs. In this regard, candidate fluid biomarkers of neuroinflammation are under analytical/clinical validation, i.e., TREM2, IL-1β, MCP-1, IL-6, TNF-α receptor complexes, TGF-β, and YKL-40. PET radio-ligands are investigated to accomplish in vivo and longitudinal regional exploration of neuroinflammation. Biomarkers tracking different molecular pathways (body fluid matrixes) along with brain neuroinflammatory endophenotypes (neuroimaging markers), can untangle temporal–spatial dynamics between neuroinflammation and other AD pathophysiological mechanisms. Robust biomarker–drug codevelopment pipelines are expected to enrich large-scale clinical trials testing new-generation compounds active, directly or indirectly, on neuroinflammatory targets and displaying putative disease-modifying effects: novel NSAIDs, AL002 (anti-TREM2 antibody), anti-Aβ protofibrils (BAN2401), and AL003 (anti-CD33 antibody). As a next step, taking advantage of breakthrough and multimodal techniques coupled with a systems biology approach is the path to pursue for developing individualized therapeutic strategies targeting neuroinflammation under the framework of precision medicine.
Alzheimer's disease (AD) is the most commoncause of neurodegenerative dementia. According to current estimates, 17% of people aged 75–84 years in the United States have AD, and the disease costs the country US$236 billion per year. The prevalence is projected to triple by 2050 to >15 million, with annual costs of >$700 billion (1). There is an urgent need for developing pharmacological treatments with a disease-modifying effect to halt the disease at its earliest preclinical stage where brain and cognitive functions can still be preserved (2, 3). Indeed, drugs currently available on the pharmaceutical market (i.e., acetylcholinesterase inhibitors and non-competitive N-methyl-D-aspartate antagonists) have been approved for a symptomatic effect only and for the dementia stage of AD (4).
The acknowledged pathophysiological hallmarks—(I) extracellular deposition of amyloid beta (Aβ), (II) intracellular aggregates of tau proteins, ultimately called neurofibrillary tangles (NFT), and (III) neurodegeneration—have been integrated in research diagnostic criteria (5–8).
The hypothesis-free biomarker-guided “A/T/N” classification scheme was introduced to categorize subjects based on core AD hallmarks (9). The A/T/N scheme is anticipated to provide consistent recruitment of individuals and target engagement among various different sites in AD clinical trials. Even though the A/T/N classification scheme provides crucial pathophysiological insights, it offers a partial depiction of the spectrum of pathomechanistic modifications of AD (10, 11).
The increasing animal and in-human evidence for the upstream role that neuroinflammation may play in AD has posed several conceptual therapeutic concerns and open up new avenues for preventing AD cognitive decline.
The pathophysiological mechanisms of multifactorial and polygenic AD are not limited to the neuronal tissue; they are related to cerebral immunological responses (12). Indeed, brains of patients with AD and other neurodegenerative diseases (ND) show chronic inflammation (13). Neuroinflammation is as an innate immunological response of the nervous system that comprises microglia, astrocytes, cytokines, and chemokines, which play a central role in an early phase of AD pathogenesis (12, 14). The key contribution of inflammation in the AD pathophysiology has been hypothesized more than 20 years ago (12, 15–17). Recent studies demonstrate that this early disease-aggravating central nervous system (CNS) inflammation starts decades before the appearance of severe cognitive decay or AD (18–20). Along this line, different longitudinal studies show that inflammation and microglial activation occur years before AD onset (21–23). Furthermore, there is a strong link between neuroinflammation and amyloid and tau accumulation in the human brain (23–26).
The acknowledged cell mediators of inflammatory mechanisms in AD are microglia and astrocytes (12). In general, these cells play a substantial role in neural transmission and synapse remodeling, as they facilitate the removal of non-essential synapses by eradicating inadequate connections (27, 28). Thus, the efficiency of neuronal transmission is increased.
Synapses exhibit a quad-partite arrangement that consists of an axon terminal, a dendritic spine put in direct communication with a microglial and an astrocytic process (29). Astrocytes and microglia—the brain-resident macrophages—play a key role in neural circuit development and synaptic homeodynamics during adulthood. Astrocytes are essential for supporting synaptogenesis (axonal and dendritic spines sprouting) and regulating synaptic robustness (30–32). Astrocytes also contribute to the spatiotemporal integration of several synaptic signals and regulate the synaptic transmission (33, 34). Microglial cells play a key role in the immune surveillance of the presynaptic microenvironment and also for the synaptic remodeling toward axonal and dendritic terminals pruning by reshaping proteolytic and phagocytic processes. Microglial cells are able to recruit astroglia, or they can be recruited by the latter (30–32, 35). They are thought to drive the well-known age-related regional synaptic vulnerability, as recently reported (36). Indeed, an age-related ultrastructural and functional shift of microglia cells is associated with increased synaptic susceptibility and neurodegeneration (35).
Therefore, astrocytes and microglia express physiological properties essential for synaptic transmission, the accurate modulation of neural and synaptic plasticity, and both synaptic adaptation and homeostasis (30–32).
In summary, it is well-established that microglia and astrocytes take part in aberrant molecular pathways that, ultimately, reflect AD pathomechanistic alterations, i.e., brain proteinopathies, synaptic failure, loss of brain plasticity, neuroinflammation, axonal damage, and neurodegeneration (37–41).
Microglial cells, arising from the mesodermal (myeloid) lineage (42), are the main category of macrophages in the CNS parenchyma. They express a large assortment of receptors that recognize exogenous or endogenous CNS insults and initiate an immune response. Besides their typical immune cell role, microglial cells protect the brain by stimulating phagocytic clearance and providing trophic sustenance to preserve cerebral homeostasis and support tissue repair. When circumstances related to loss of homeostasis or tissue alterations occur, then many dynamic microglial mechanisms are triggered, leading to the “activated state” of microglia (43). These encompass cellular morphology modifications, changes in the secretory profile of molecular mediators, and increased proliferative responses (44). A persistent homeodynamic imbalance, such as brain accumulation of Aβ, can trigger a step further in activation, referred to as “priming” (37). Priming of microglia is directed by alterations in their microenvironment and the release of molecules guiding their proliferation. Priming makes microglia inclined to secondary inflammatory stimulating factors, which can then elicit amplified inflammatory reactions (37).
Activated microglia is a typical pathophysiological feature of AD and other ND (12, 43, 45). Two main types of microglia cells are present in the brain, “resting” (or “quiescent”) and “active” microglia. In particular, there is evidence for the high degree of heterogeneity of microglial activation in the CNS, which can be categorized into two opposite activation phenotypes: M1 and M2 (43, 46, 47). According to the phenotype activated, microglia can generate either cytotoxic or neuroprotective effects (46). The M1 or “proinflammatory” phenotype (classically activated) displays proinflammatory cytokines and nitric oxide. It decreases the release of neurotrophic factors, thus exacerbating inflammation and cytotoxicity (43). In contrast, the M2 or “anti-inflammatory” phenotype (alternatively activated) displays anti-inflammatory cytokines, increased expression of neurotrophic factors, and several other signals involved in downregulation, protection, or repair processes in response to inflammation (43). Preliminary evidence from experimental studies suggests that the phenotypic transformation of the activated M1/M2 functional states (“phenotypic switching”) (48, 49) can be determined by both the stage and the severity of the disease. In preclinical models, M1 microglia seems to prevail at the injury site, at the end stage of disease, and once inflammation resolution and repair processes of M2 microglia are diminished (46).
In light of the increasing evidence that the modality by which microglia is activated is a continuum between proinflammatory (M1) and anti-inflammatory (M2) phenotypes, the M1/M2 “dichotomy” (or “polarization” scheme) is still disputed. Actually, it seems possible that the global process of microglia activation represents a much larger heterogeneous spectrum of very dissimilar responses (43).
Experimental models of AD demonstrate that microglia cluster around plaques, likely via chemotactic mechanisms, and may contribute both in Aβ (39, 44) clearance and in limiting the growth and further accumulation plaques (39, 44). Moreover, the dysregulation of microglia activity, including dystrophic microglia, may be either a trigger, or a worsening factor, or both, of the seeding of aberrant protein aggregates in the brain (39, 44).
In AD, during inflammation, there is a transition from the resting to the active functional state of microglia that, at a general level, might be the consequence of stress or depressive-like behavior (50). At a molecular level, inflammation is promoted by the presence of Aβ aggregates, including oligomers and fibrils (51–54). Indeed, microglia can bind to soluble Aβ oligomers and insoluble Aβ fibrils through cell surface receptors, including the class A1 scavenger receptor (SCARA1), cell surface cluster of differentiation (CD) markers (CD36, CD14, and CD47), the α6β1 integrin, and the Toll-like receptors (TLRs) (55–58). A key point within the scientific debate is represented by a recent evidence indicating that microglia displays either beneficial or harmful effects throughout the beginning and advancement of AD (45). This is strictly related to the nature of the major activities: (I) clearance of Aβ or (II) release of proinflammatory mediators. In early AD pathogenesis, Aβ oligomers and fibrils gather in the extracellular space and elicit a pathological cascade resulting in neuronal apoptosis and depletion. Microglia eliminate Aβ peptides and dying/dead cells through phagocytosis (59, 60). Besides clearance of Aβ oligomers and fibrils, microglia surrounds plaques and fibrils likely creating a physical barrier that can prevent their spreading and toxicity (61). Aβ clearance is also stimulated by the release of numerous proteases participating in Aβ degradation (62). In spite of the advantageous actions of early activation of microglia cells, their chronic activation by Aβ is detrimental and induces protracted inflammation and disproportionate Aβ deposition, thus rushing neurodegeneration (Figure 1). During AD pathogenesis, the production and release of proinflammatory cytokines and other detrimental components are intensified. In addition, the typical phagocytic action of microglia is decreased. Moreover, the microglial-dependent release of apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) modulates the diffusion of the pathology within and between cerebral areas (63). Extracellular vesicles—constituted by microvesicles and exosomes and released by reactive microglia—play a role in AD pathogenesis (64) (Figure 1). Finally, microglial cells are able to regulate AD pathogenesis via active interaction with neurons, astrocytes, and oligodendrocytes. Indeed, activated microglial cells induce altered astrocytes via proinflammatory cytokines (Figure 1). These astrocytes can rush and aggravate neuronal and oligodendrocytes death (65).
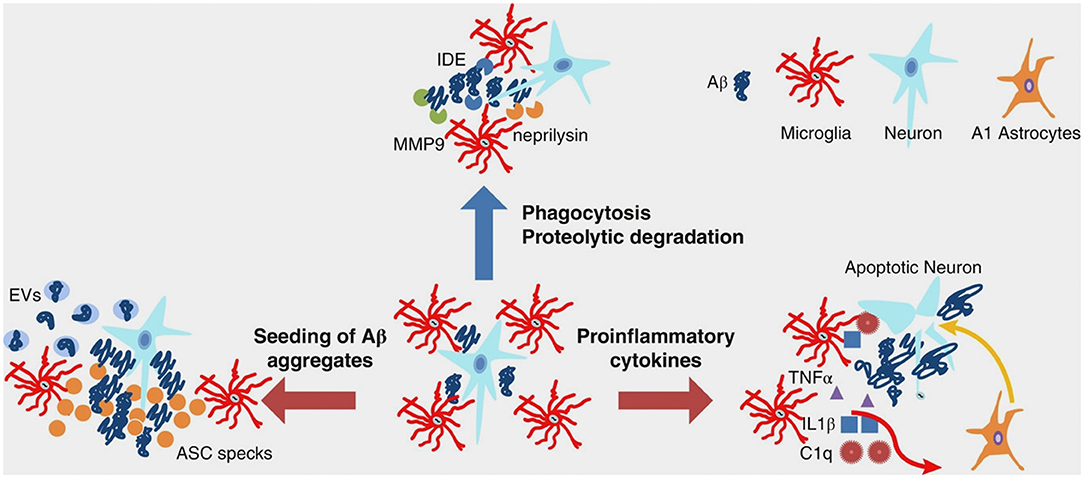
Figure 1. Multifaceted functions of microglia during Aβ pathology. In healthy brain and early stages of AD, microglia clear small aggregates of Aβ peptides by phagocytosis and by secreting proteolytic enzymes, such as IDE, neprilysin, and MMP9. During advanced AD, microglia exacerbate AD pathology by releasing proinflammatory cytokines that induce neuronal cell death as well as A1 astrocytes, which, in turn, affect neuronal survival. Moreover, during advanced AD, microglia-derived ASC specks and EVs promote seeding of Aβ aggregates. Aβ, amyloid beta; AD, Alzheimer's disease; ASC, apoptosis-associated speck-like protein containing a CARD; C1q, complement component 1q; EVs, extracellular vesicles; IDE, insulin degrading enzyme; IL-1β, interleukin-1 beta; MMP-9, metalloprotease-9; TNF-α, tumor necrosis factor-alpha. From Wang and Colonna (45). Copyright© 2019, Society for Leukocyte Biology. Reprinted with permission from Wiley.
The still open question is to understand the specific contributions of neuronal and glial cells in the early phase of inflammation in preclinical AD. Aβ1−42 oligomers have a major role in synaptic depletion and gradual cognitive deterioration (66, 67). They induce neuroinflammation and neurodegeneration by stimulating the microglia to produce and release proinflammatory cytokines (14, 68) and also by interfering with the synthesis of anti-inflammatory cytokines, for instance the transforming growth factor-beta 1 (TGF-β1) (Figure 2) (69–71). This early proinflammatory process is characterized by neuronal and microglia-derived cytokines and chemokines as well as by mobilization of microglia toward Aβ-burdened neurons (Figure 2) (19, 72). In addition to Aβ, extracellular non-phosphorylated tau, rather than hyperphosphorylated tau (p-tau), activates the p38 mitogen-activated protein kinase (MAPK) pathway, eliciting a proinflammatory reaction (73).
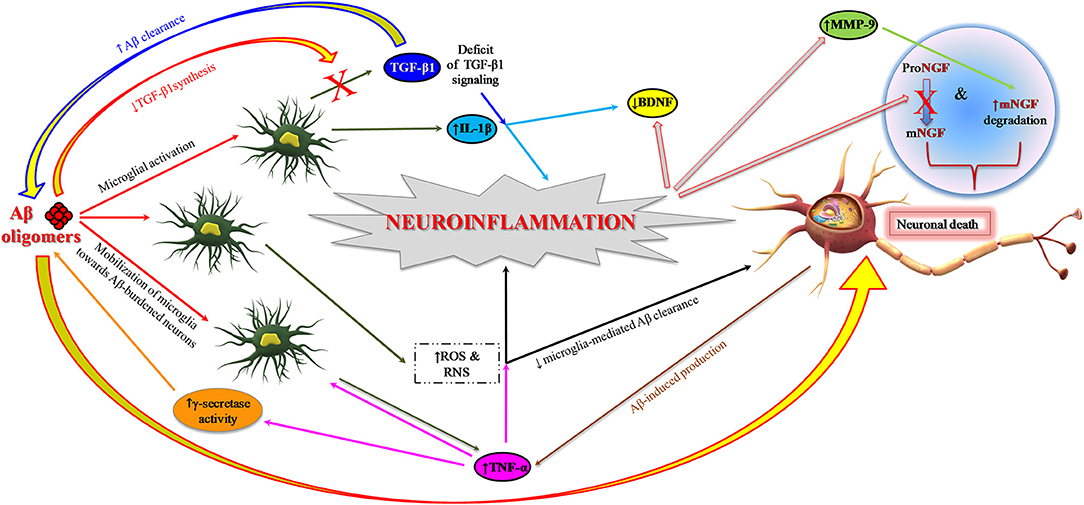
Figure 2. Role of neuroinflammation in AD pathogenesis: impairment of neurotrophin signaling. Aβ1−42 oligomers promote neuroinflammation and neuronal death in AD brain by eliciting the release of proinflammatory cytokines (IL-1β and TNF-α) from microglia and also interfering with the synthesis of anti-inflammatory cytokines such as TGF-β1. TNF-α inhibits microglia phagocytosis of Aβ and stimulates γ-secretase activity, thus facilitating Aβ accumulation and microglia-mediated neuroinflammation. Proinflammatory microglial activities promote neuronal death also through the formation of ROS and RNS. Neuroinflammatory phenomena can finally contribute to the pathogenesis of AD by impairing neurotrophin signaling function: (I) reducing the synthesis of BDNF and TGF-β1 and (II) causing an impairment of NGF metabolic pathway characterized by a reduced conversion of proNGF to biologically active mNGF and by an increased degradation of mNGF promoted by MMP-9. Aβ, amyloid beta; Aβ1−42, 42-amino acid-long amyloid beta peptide; BDNF, brain-derived neurotrophic factor; IL-1β, interleukin-1 beta; MMP-9, metalloprotease-9; NGF, nerve growth factor; mNGF, mature nerve growth factor; proNGF, precursor of the nerve growth factor; RNS, reactive nitrogen species; ROS, reactive oxygen species; TGF-β, transforming growth factor-beta; TNF-α, tumor necrosis factor-alpha.
Astrocytes, differently from microglia and similarly to neurons and oligodendrocytes, arise from the neuroectoderm (74). These cells promotes synaptogenesis (axonal and dendritic spines sprouting), regulates the synaptic strength, take part in the spatial–temporal integration of multiple synaptic processes, and modulate the neurotransmission. Hence, astrocytes execute a variety of physiological activities, in both developing and adult brain, that are essential for synaptic plasticity and a solid and organized cognitive activity (74). Of note, astrocytes modulate Ca2+-dependent signaling pathways that are crucial for hippocampal synaptic function and plasticity (75, 76). Indeed, depending on the fluctuations of intracellular Ca2+ concentrations, they release gliotransmitters, such as glutamate, D-serine, and ATP, which have feedback actions on neurons (77). Moreover, each astrocyte wraps several neurons, thus interacting with hundreds of neuronal dendrites (78) and connecting with up to two million synapses in the human cortex (79). This kind of interconnectedness indicates that each astrocyte creates a hub to facilitate the integration of the information (74). Moreover, remodeling of astrocytes promotes neuroprotection and recovery of injured neural tissue (80, 81). Along with microglia activation, hypertrophic reactive astrocytes gather around Aβ plaques as reported in human postmortem studies (82) as well as in animal models (83). Like microglia, astrocytes are also activated by tissue injury, infection, and inflammation (84). In AD, after exposure to Aβ, astrocytes release various proinflammatory molecules, such as cytokines, interleukins (ILs), complement components (85–87), nitric oxide, and other cytotoxic compounds, ultimately amplifying the neuroinflammatory response.
Human neuropathological studies conducted on AD brains report the presence of cytoplasmic inclusions of non-fibrillar Aβ in astrocytes, supposed to reflect a phagocytic engulfment from extracellular Aβ deposits (86). In addition, rodent models of AD indicate the ability of astrocytes to uptake and clear Aβ in subjects bearing cerebral fibrillar aggregates and diffuse plaques (16, 17, 33, 86). Conversely, the shutdown of astrocyte-mediated homeodynamics is associated with increased Aβ plaque burden and synaptic terminals dystrophy (68). This enhanced phagocytic activity may represent a compensatory mechanism to incipient increased Aβ accumulation to neutralize its induced toxicity.
Genome-wide association studies (GWAS) allowed the detection of more than 40 susceptibility gene variants associated with a bigger risk of developing late-onset AD (88). These results include genes associated with immune reaction (in particular, ABCA7, CD33, CLU, CR1, EPHA1, HLA-DRB5-HLA-DRB1, and MS4A). The relevance of neuroinflammation is further sustained by recent large-scale GWAS showing that the risk of developing late-onset AD is substantially more elevated in individuals with rare variants of microglial immunoreceptors: TREM2, encoding the triggering receptor expressed on myeloid cells 2 protein (89); CD33 (transmembrane receptor CD33), expressed on cells of myeloid lineage (90, 91); and PILRA (paired immunoglobulin-like type 2 receptor alpha) (92).
The receptor protein TREM2 enhances the rate of phagocytosis in microglia and macrophages, modulates inflammatory signaling, and controls myeloid cell number, proliferation, and survival (89). Recent studies show that triggering TREM2 receptor in microglial cells is closely associated with the pathogenesis of AD (93). TREM2 modulates microglial functions (e.g., stimulates the production of inflammatory cytokines) in response to Aβ plaques and tau tangles (94, 95). TREM2 absence enhances amyloid pathology, during early AD; however, this is exacerbated at later stages due to the loss of phagocytic Aβ clearance (94). TREM2 variants cause AD by decreasing the Aβ phagocytic ability of microglia and through the dysregulation of the proinflammatory response of these immune cells (96). Interestingly, the analysis of the existing single-cell transcriptome datasets for human neurons highlights the association of microglia with late-onset AD (97). In addition, the study of regulatory networks of genes showing differential expression in AD brains indicates that immune- and microglia-specific gene modules primarily contribute to AD pathophysiology (98). Finally, Tanzi and colleagues, after exploring the potential role of the cross-talk between CD33 and TREM2 in both neuroinflammation and the cause of AD, propose that TREM2 is working downstream of CD33 to modulate the neuroinflammatory process (99).
Besides the above-mentioned role of Aβ and tau in triggering neuroinflammation, it is assumed that the presence of extracellular tau plays a role in the transition from resting to active microglia. In the resting microglia, the protein fractalkine (CX3CL1), secreted by healthy neurons, binds to the cell receptor (CX3CR1) present in the microglia allowing the maintenance of microglia in the resting state. Tau pathology is shown to be associated with neuroinflammatory processes. On the other hand, microglia could be involved in tau propagation in tauopathies. In this scenario, microglial CX3CR1 acts like a receptor for extracellular tau, since the absence of CX3CR1 impairs the internalization of tau microglia (100). Thus, extracellular tau can compete with CX3CL1 for a common receptor. Microglia cells lacking CX3CR1 are deficient in neuronal CX3CL1 signaling and are not in the resting state. As a result, these active microglial cells could secrete some compounds, such as cytokines, potentially affecting neuronal functions like adult neurogenesis. The absence of the microglial CX3CR1 impairs the synaptic integration of adult born hippocampal granule neurons (101). Mice lacking CX3CR1 show modifications in both microglia and neurons of some cerebral areas, like dentate gyrus. Adult-newborn neurons, in CX3CR1–/– mice, show a deficient synaptic integration in the neuronal network and exhibit a diminished amount of dendritic spines. These display some morphological alterations, since mice lacking CX3CR1 protein have a hyperactive, anxiolytic-like, and depressive-like phenotype (101).
Mainly, all the previous remarks are observed in mouse models, but little is known about the consequences of changes in microglia in humans. Interestingly, CX3CL1 concentrations are reduced in the cerebrospinal fluid (CSF) of AD patients compared to control subjects, thus suggesting that variations in CX3CL1 levels might represent a new target to use in inflammation and AD (102). Two recent different publications describe the consequences, in humans, of having homozygous mutations in the colony-stimulating factor 1 receptor (CSF-1R) gene expressing a cell receptor essential for the development and maintenance of microglia. The consequences are the presence of abnormalities not only in brain structures, like corpus callosum, but also in bones that, in some cases, are overly dense and malformed (103, 104). In the future, it will be interesting to explore possible changes in adult neurogenesis at the dentate gyrus in the autopsy, in the cases of patients with biallelic CSF-1R mutations.
Neuroinflammatory pathways and microglial cells activation are associated with neuronal ectopic cell cycle activation (105). In particular, microglial activation induced by Aβ oligomers promotes neuronal ectopic cell cycle events (CCEs) via the tumor necrosis factor-alpha (TNF-α) and the c-Jun kinase (JNK) signaling pathways. Hence, administering of non-steroidal anti-inflammatory drugs (NSAIDs) in AD transgenic mice precludes both microglial activation and stimulation of CCE (105, 106). Two analyses report the capability of ibuprofen to alter the advancement of mild AD (107). However, forthcoming AD clinical trials show no effectiveness in mild dementia individuals, probably because these drugs are administered in a late phase of CNS inflammation. Indeed, recent studies designate initial CNS inflammation as an encouraging target to prevent the advancement of the pathology (19).
Today, it is widely accepted that oxidative stress is strongly associated with the inflammation observed in AD (108). In fact, neuroinflammatory processes can act both as cause and as effect of chronic oxidative stress (Figure 2). In this context, microglia play a pivotal role. Proinflammatory microglial activities may be detrimental in AD due to reactive oxygen and nitrogen intermediate species—ROS and RNS, respectively—leading to oxidative stress-induced neuronal death, which could be further exacerbated by chronic stress (109, 110). Cumulative evidence suggests that microglial inflammation-induced oxidative stress in AD is amplified. In contrast, microglial-mediated clearance mechanisms are not functional (43, 110).
TNF-α exerts a key role in this early proinflammatory process observed in preclinical AD as emerges from preclinical studies in animal models of AD (111–114) as well as from human longitudinal studies (21, 113–115). TNF-α is chronically released during the course of AD pathology, likely by activated microglia, neurons, and astrocytes stimulated by increased levels of extracellular Aβ (111). Aβ oligomeric forms activate microglia with anomalous TNF-α-mediated pathways in mouse models (68). Such an atypical stimulation of cerebral innate immunity is responsible for reduced serotonergic tonus, a primary event in depression due to Aβ, a prodromal symptom of AD (70). On the other hand, TNF-α can stimulate γ-secretase activity, which results in an increased synthesis of Aβ peptides and a further increase in TNF-α release (113, 116). It is hypothesized that this auto-amplified loop in the AD brain can contribute to the maintenance of excessive levels of TNF-α, which could then stimulate Aβ synthesis and neuronal loss, also inhibiting microglia phagocytosis of Aβ (Figure 2) (113, 117). Finally, TNF-α significantly contributes to promote insulin resistance and the following cognitive decline in AD (118, 119). Aβ oligomeric forms prompt peripheral glucose intolerance in mice by activating TNF-α signaling in the hypothalamus (120). Multiple studies detected elevated TNF-α levels in both mild cognitive impairment (MCI) and AD (21, 113). Interestingly, Down syndrome cases with preclinical AD show significant links among augmented levels of plasma TNF-α, Aβ accumulation, and the following cognitive deterioration in the subsequent years (115).
TNF-α exerts its activity by binding two distinct high-affinity receptors (TNF-Rs) placed at the cell surface: TNF-RI, ubiquitously expressed apart from erythrocytes, and TNF-RII, whose expression is limited to myeloid cells, endothelial cells, oligodendrocytes, microglia, astrocytes, and subpopulations of neurons (113). The concentrations of the soluble forms of the TNF receptors (sTNF-RI and sTNF-RII) are typically unaltered in CSF and blood of AD patients compared to controls (21). However, both TNF-α and TNF-RI concentrations are increased in postmortem brains of early-stage AD patients (113). MCI subjects present controversial data; longitudinal studies report associations between TNF-R concentrations and the risk of conversion from MCI to AD (21). Notably, the TNF-α receptor complex and its functional proteins are assumed to play a crucial role since they link neuroinflammatory pathways to amyloid deposition process in a chronically damaging and self-perpetuating way (21).
A strong neurobiological link is also found in the AD brain between the deficit of anti-inflammatory cytokines, such as TGF-β1, and the early proinflammatory process observed in preclinical AD (70). TGF-β1 is a neurotrophic factor whose deficit exerts a key role in AD. A selective impairment of TGF-β1 pathway is present in early AD, both in the AD brain (121, 122) and in AD animal models (71, 123, 124). This deficit seems to critically contribute to neuroinflammation in AD brain. TGF-β1 displays both anti-inflammatory and neuroprotective actions (123, 125) and stimulates Aβ clearance by microglia (126). Furthermore, it exhibits a primary role in synaptic plasticity and memory creation processes, thus supporting the path from early to late long-term potentiation (LTP) (127). We should reconsider the relevance of TGF-β1 in neuroinflammation resulting from microglia activation, contributing to reactivate the neuronal cell cycle in the AD brain (128). According to this scenario, the reactivation of the neuronal cell cycle might be assisted by the disruption of Smad-dependent TGF-β1 pathways. Overall, these studies suggest the potential contribution of the deficit of Smad-dependent TGF-β1 pathway to neuroinflammation and cognitive impairment (70).
Moreover, neuroinflammatory phenomena might impair neurotrophin signaling (Figure 2) and interfere with brain-derived neurotrophic factor (BDNF)-induced neuroprotection (129–131).
Finally, neuroinflammation can exert a primary function in AD pathophysiology by interfering with nerve growth factor (NGF) maturation and function. NGF is a neurotrophic factor essential for the survival and homeostasis of basal forebrain cholinergic neurons whose selective degeneration critically contributes to cognitive decline in AD patients (132, 133). Studies in transgenic animal models of AD indicate that the proinflammatory process—initiated before plaque deposition and promoted by soluble Aβ oligomers—leads to an impairment of NGF metabolic pathway characterized by a reduced conversion of the precursor proNGF to the mature NGF (mNGF) as well as by an increased deprivation of mNGF (18, 132, 134). Neuroinflammatory processes promote an overactivation of metalloprotease-9 (MMP-9), as observed in the brains of Down syndrome patients (132), MCI subjects, and AD patients (135). Increased MMP-9 activity would then facilitate the degradation of mNGF, finally compromising mNGF activity in sustaining the trophic dependence of the cholinergic neurons (132). Notably, a strong correlation is present in Down syndrome cases showing preclinical AD among the plasma TNF-α increase, a deficit in NGF maturation (with grown concentrations of proNGF), and an increased degree of cognitive impairment (115). This study substantiates the key contribution of inflammatory markers (i.e., TNF-α) in combination with plasma Aβ1−42 levels and increased proNGF levels to better predict the worsening of “latent” AD pathology with the consequential cognitive decline in Down syndrome patients (115). The discovery of an imbalance in the metabolic pathway controlling NGF maturation and degradation in Down syndrome/AD patients provides a platform for the identification of novel biomarker candidates as well for the development of disease-modifying drugs. Therefore, drug discovery processes should be directed in the future to develop new drugs that are able to interfere with early CNS inflammation and, at the same time, rescue neurotrophin signaling (e.g., BDNF, NGF, TGF-β1) in the AD brain.
Among the different mediators of inflammation explored, TNF-α mediates proinflammatory processes in various ND including AD (136). In normal conditions, TNF-α from glial cells modulates homeostatic activity-dependent regulation of synaptic connectivity (137). On the other hand, this cytokine mediates the disrupting effects of Aβ on LTP in experimental AD. Accordingly, mutant mice lacking TNF receptor type 1 exhibit normal LTP following Aβ application and similar results are obtained with the use of anti-TNF agents including the monoclonal antibody infliximab and thalidomide, which also inhibits TNF-α production (138). Generally, several studies indicate that blocking the TNF-α pathway in AD models is associated with: (I) improvement in memory decline, as tested in different behavioral tests evaluating cognitive function; (II) reduction in immunohistochemical and histopathological markers like formation of Aβ plaques and NFT; and (III) reduction in the number of microglial cells in the AD brain (139).
Similarly to TNF-α, also the proinflammatory cytokine IL-1β mediates the synaptotoxic effects of Aβ peptide (140). Indeed, the interleukin-1 receptor antagonist (IL-1Ra) is able to reverse synaptic plasticity alteration triggered by the administration of the 40-amino acid-long Aβ peptide (Aβ1−40) (141). However, the role of ILs in AD pathogenesis is far more complex since some exert proinflammatory while others exert anti-inflammatory actions. In this frame, it is worth mentioning IL-12 and IL-23 which are increased in CSF in both AD and MCI (142, 143). Notably, genetic ablation of IL-12 and IL-23 or therapeutic approaches directed against IL-12 and IL-23 signal reduce the AD-like pathology, including histopathological and behavioral changes, making them attractive targets for the treatment of AD (144). On the other hand, IL-10 seems to play a protective role since delivery of this cytokine via adeno-associated virus leads to markedly decreased microgliosis and astrogliosis as well as reversed cognitive impairment in transgenic AD mice (145), although the use of a different adeno-associated virus approach generates a different outcome (146).
There is a growing interest on the role of complement and microglia in AD pathology (147). Microglia cells have prominent functions in complement-mediated synaptic pruning, in the postnatal period (148, 149). It is hypothesized that an inappropriate reactivation of this mechanism later in life could result in synapse loss, thus facilitating the progression of ND (150). In this frame, C1q, which mediates the toxic effects of Aβ oligomers on LTP, is increased in synaptic connections before plaque deposition, and inhibition of C1q, C3, or the microglial complement receptor CR3 diminishes phagocytic microglia, resulting in protection against synapse loss (151).
Investigations conducted in transgenic AD mice also address the effects of NSAIDs on amyloid load and inflammation (152). These studies suggest that NSAIDS not only exert neuroprotection through the suppression of inflammatory events but also reduce early amyloid pathology by mechanisms that remain unclear (153). Of note, two selective cyclooxygenase-2 (COX-2) inhibitors are found to be effective in rescuing LTP impairment by synthetic soluble Aβ1−42, whereas the same effect is not achieved with the cyclooxygenase-1 (COX-1) inhibitor piroxicam (154). Similarly, ibuprofen prevents early memory decline in AD model, and this effect is associated with activation of hippocampal plasticity-related genes (155). Overall, these studies indicate that NSAIDs exert neuroprotection and prevent memory decline through the modulation of multiple neuronal pathways (156).
Most of the failed AD clinical trials—including trials investigating anti-inflammatory compounds—did not assess any biological in vivo identification of AD-related pathomechanistic alterations, thus preventing proof of mechanisms (157) and including a percentage of subject displaying non-AD pathophysiology (158). Therefore, robust biomarkers–drug codevelopment pipelines are strongly recommended for next-generation clinical trials (159).
Modifications of the concentrations of several cytokines (160–164) and other inflammatory biomarkers associated with either microglia—e.g., soluble TREM2 (sTREM2), monocyte chemoattractant protein-1 (MCP-1), and YKL-40 (165–168)—or astroglia, e.g., YKL-40 (161), are extensively investigated in AD patients. These alterations, potentially, reflect the inflammatory mechanisms within the CNS coupled with the neurodegenerative pathways (11, 166). A recent meta-analysis reports higher concentration of YKL-40, sTREM2, MCP-1, and TGF-β in the CSF of AD patients compared to controls (160). In particular, robust evidence from several studies focus on CSF YKL-40 that shows a fair classificatory capability in differentiating between AD individuals and controls as well as in predicting the progression from the asymptomatic to later prodromal and dementia stages (166–168). However, its function in differentiating subjects with AD and other dementia remains controversial since neuroinflammation seems to be associated with neurodegeneration tout court and not with specific neurodegenerative pathways (12, 169).
The clinical meaning of inflammatory biomarkers in blood needs to be elucidated, as they might represent low-invasive and low-cost screening tools of cerebral inflammatory activity during the early asymptomatic stages of AD (170–172). The main issue concerning the peripheral measurements of inflammatory biomarkers is that they may not directly reflect brain neuroinflammation (163). Nonetheless, IL-6 and IL-1β concentrations are significantly higher in AD compared to cognitively normal controls in four meta-analysis (160–163). IL-1β is a key molecule participating in the inflammatory response, cell proliferation, differentiation, and apoptosis. Some evidence suggest that IL-1β is produced and secreted by microglia cells in response to Aβ deposition, thus resulting in chronic neuroinflammation and, eventually, neuronal disruption, dysfunction, and neurodegeneration (173, 174). A negative correlation between CSF concentrations of this cytokine and cognitive scores has also been described in AD (175). IL-6 levels are associated with the severity of cognitive decline as assessed by Mini-Mental State Examination (MMSE) scores (161). Notably, peripheral IL-6 concentrations positively correlate with the cerebral ventricular volumes (176) and with matched CSF samples (177) in AD. The peripheral modifications of IL-6 levels could begin in the prodromal phase of AD; indeed, a recent meta-analysis highlights the greater IL-6 concentrations in MCI subjects compared to controls (160). In line with these findings, a longitudinal study reports the association of elevated plasma IL-6 levels with a greater risk of cognitive decay, at 2-year clinical follow-up. Other cytokines emerging as candidate peripheral inflammatory biomarkers are IL-2, IL-12, IL-18, and TGF-β (160–163).
Overall, these studies have several biases to consider. First, the risk of misdiagnosis is high since AD and MCI diagnoses are mainly clinical based in the majority of the studies lacking the necessary biomarker information [e.g., cerebral amyloid-positron emission tomography (PET) uptake or CSF Aβ1−42 measurements]. This means that at least 20–25% of the AD patients and MCI subjects enrolled in the previous studies do not have cerebral amyloid deposition (6). Moreover, these studies are cross-sectional without an appropriate follow-up, and this could lead to incorrect MCI diagnosis. Indeed, the clinical picture of MCI is heterogeneous not only with a 10–15% annual rate of developing AD (178) but also with a consistent proportion of individuals who recover, remain stable, or develop ND other than AD (179). In addition, the MCI classification (e.g., amnestic or non-amnestic), which significantly impacts clinical outcome (8, 179, 180), is inadequately specified in most of the studies. Furthermore, data regarding comorbidities—such as cerebrovascular diseases, coronary diseases, atrial fibrillation, periodontitis, and diabetes or concomitant drugs (e.g., non-steroidal inflammatory medications, corticosteroids, statins) that can significantly modify peripheral inflammatory biomarkers—have been rarely reported. For instance, persistent higher plasma levels of IL-1β and IL-6 are observed in relation to cardiovascular diseases as well as atherosclerosis (181, 182). Other potential biases include technical issues: detection methods (e.g., ELISA kits) for inflammatory biomarkers in biological fluids are consistently different among studies as well as sample handling approaches [e.g., measurements on different fluids matrix (plasma or serum) and storage protocols].
In conclusion, neuroinflammation is certainly a relevant pathophysiological mechanism of neurodegeneration in AD. However, we still lack reliable inflammatory biomarkers to be used in a screening context of use. In essence, sTREM2, MCP-1, IL-6, TGF-β, and, particularly, YKL-40 are interesting novel inflammatory CSF biomarkers, but they cannot be proposed in detecting the early asymptomatic phases of AD, as it would be altered with disease-modifying treatments. Prospective observational studies enrolling large cohorts of participants with accurate clinical and biomarker-based characterizations are needed to identify potentially effective inflammatory blood-based biomarkers of AD.
There are several genetic association studies highlighting a key role of neuroinflammation in AD by demonstrating the occurrence of specific genetic variations related to immune response in patients with ND including AD (183). As a direct consequence, the possibility of tracking the regional evolution of neuroinflammation and imaging non-invasively the neuroinflammatory process in AD patients opens up exciting novel opportunities to monitor disease progression and, eventually, to explore immune-therapeutic strategies to prevent or decelerate disease progression.
It is interesting to note that it could be possible to assess the neuroinflammatory status by conventional [18F]-fluorodeoxyglucose (FDG)-PET, provided that the whole uptake curve is studied (184). Neuroinflammation can be measured more specifically using targeted radio-ligands for PET imaging—that allow accomplishing the regional in vivo exploration of neuroinflammation—like [11C]-PK11195 (185). A number of studies show alterations in [11C]-PK11195 binding in AD and several other ND (186–189), Parkinson's disease (190), and progressive supranuclear palsy (189, 191), and the distributions of [11C]-PK11195 found in these studies are akin to the well-known distribution of neurodegeneration (e.g., posterior cortical regions in AD). However, one should also note that translocator protein (TSPO) gene polymorphisms can greatly affect binding affinity (192), and TSPO expression is not circumscribed to activated microglia, which can also occur on astrocytes, or endothelial cells (193). In this context, a number of novel TSPO-specific PET radiotracers are currently available, both carbon-11 (i.e., [11C]-PK11195, [11C]-PBR28) and fluoroine-18 labeled [e.g., [18F]-GE-180, [18F]-DPA-714, and [18F]-PBR06], typically used in preclinical investigations (194, 195). In addition, a very recent tracer named [18F]-FEPPA is able to provide a high potential for TSPO-PET in humans (192).
Interestingly, microglia activation is only one (albeit important) part of the chain of events that eventually lead to neuroinflammation and that can potentially be imaged with even more specific tracers. For example, protein misfolding, aggregation, and accumulation may trigger glial response and, therefore, neurotoxicity. To date, the causal relationships between neuroinflammation and other pathogenetic mechanisms of AD is not elucidated yet. PET radiotracers can represent a suitable tool for untangling these dynamics along the roadmap of discovering new targets for anti-inflammatory disease-modifying strategies. In this context, a number of specific PET tracers can target protein aggregates in the brain. For example, the [11C]-Pittsburgh compound-B ([11C]-PIB) is able to bind Aβ fibers (186, 190). Aβ can also be imaged through, e.g., (18F)-labeled derivatives like [18F]-Florbetaben, [18F]-Florbetapir, and [18F]-Flutemetamol (196). In addition, hyperphosphorylation and abnormal aggregation of tau, which is crucial to neuronal activity, can be imaged using definite tracers, using specific ligands: T807, Flortaucipir as well as the phenyl/pyridinyl-butadienyl-benzothiazole/benzothiazolium derivative PBB3 (197, 198). Additionally, tracers [18F]-FA and [18F]-EFA (analogs of 2-fluoroacetate, which can be utilized to inhibit glial cell metabolism) are able to selectively enter the metabolic compartment (199) and may, therefore, be promising candidates for evaluating glial metabolism when thinking of astrocytic response.
Finally, there are other molecular targets that can offer a more exhaustive depiction of in vivo neuroinflammation (200). For example, the cyclooxygenase (COX) enzyme is involved in both inflammation and generation of proinflammatory mediators. In this context, COX-1 radioligands, like [11C]-KTP-Me, show promising results in AD animal models (201). In addition, the cannabinoid receptor type 2 (CB2R) is subject to upregulation in activated microglia in various ND (202), possibly in conjunction with a neuroprotective effect (203), and postmortem studies emphasize the potential of compounds like [11C]-RS-016, which show high specific binding (204). This is emphasizing the role of CB2R as an additional potential target for PET imaging of neuroinflammation, in humans. Further encouraging targets examined in preclinical examinations are the purinergic receptor P2X7 ([11C]-GSK1482160) (205) and the adenosine receptor A2AR (i.e., [11C]-TMSX).
NSAIDs have long been hypothesized to play a protective role in AD. This assumption is reinforced by several cohort analyses. A recent meta-analysis including 16 investigations demonstrate that present or previous utilization of NSAIDs is linked to a decreased relative risk of AD (0.81; 95% confidence interval, 0.70–0.94) (206). Despite the observational epidemiological data suggesting a protective effect of NSAIDs and the evidence for a biologically plausible role for anti-inflammatory treatment, all placebo-controlled trials of a wide range of anti-inflammatory agents (NSAIDs, corticosteroids, and others) in both mild-to-moderate AD patients (Table 1) and MCI subjects (Table 2) are negative. Studies in cognitively normal subjects at risk of developing AD are also negative (Table 3). The first, large primary prevention study of naproxen and celecoxib [Alzheimer's Disease Anti-inflammatory Prevention Trial (ADAPT)] has been prematurely interrupted for cardiovascular safety concerns after the enrollment of 2, 528 subjects in the study and their treatment for a median time of 2 years. The study is not able to support the hypothesis that either drugs could postpone AD beginning in adults with a family history of dementia (227). A subsequent 2-year, primary prevention trial [Impact of Naproxen Treatment in Pre-symptomatic Alzheimer's Disease (INTREPAD)] has been used to compare the effects of naproxen and placebo on the Alzheimer Progression Score (APS) in 195 cognitively normal older persons with a positive family history of AD (226). Over time, the APS scores progressively increase to a similar extent in both study groups, thus suggesting that naproxen does not provide any benefit over placebo in slowing the progression of presymptomatic AD.
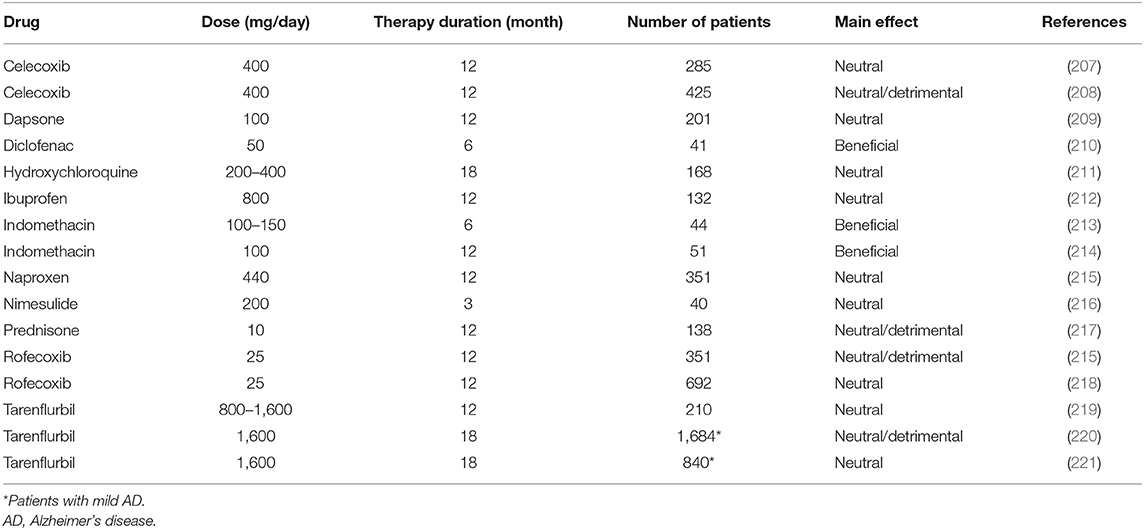
Table 1. Double-blind, randomized, placebo-controlled trials using anti-inflammatory drugs in mild-to-moderate AD patients.

Table 2. Double-blind, randomized, placebo-controlled trials using NSAIDs in MCI individuals.

Table 3. Double-blind, randomized, placebo-controlled primary prevention trials using NSAIDs in AD.
In spite of emerging epidemiological evidence, all large, longstanding, randomized, placebo-controlled investigations aiming at attenuating cerebral inflammation in AD display negative outcomes. The fact that anti-inflammatory therapies are not able to safeguard patients with overt dementia has been debated. Actually, a trial recruiting MCI individuals highlights that rofecoxib could rush the conversion to AD (223). Moreover, a primary prevention study involving celecoxib and naproxen in cognitively healthy elderly individuals showing family history of AD has been terminated in advance due to the existence of negative or harmful effects generated by the drugs (225, 228). Additional longstanding, controlled studies examining anti-inflammatory drugs, including tarenflurbil in mild AD patients (220), prednisone (217), and celecoxib (208) in mild-to-moderate AD patients, report the presence of detrimental consequences vs. placebo. NSAIDs negative and/or harmful effects, documented in AD, MCI, as well as in the stages preceding AD, are apparently in conflict with epidemiological data indicating diminished AD incidence after sustained treatment with NSAIDs. This is potentially related to the different impact of the disease stages on NSAIDs exposure. In this context, two different inflammatory responses in the AD pathophysiological process are assumed to exist: (I) one, at the early preclinical stage, with a predominantly proinflammatory component that is amenable to therapy; (II) another, at a later clinical stage, with predominantly innate/adaptive immune reactions not responsive to anti-inflammatory therapy (19). During the early inflammation stage, neurons stimulated by Aβ initiate the inflammatory process, and then, they induce intermediate microglia cells activation and their recruitment around Aβ-burdened neurons. Both neurons and microglia elicit a process exacerbating the disease characterized by the release of proinflammatory mediators (cytokines and chemokines) (Figure 3). The inflammatory immune response of the late plaque-associated stage involves different processes, including full microglial activation, microgliosis, and CNS invasion by peripheral monocytes. Both microglia and monocytes participate in phagocytic activities to eradicate toxic Aβ oligomers and, probably, cellular debris (19). This assumption is in line with data from the Rotterdam (229), the Cache County (230), and the US Veterans (231) observational studies. The above-mentioned analyses emphasize the lack of protection following 2-year NSAID exposure before dementia onset. In case the timing of exposure defines whether NSAID administration is beneficial or harmful, then the negative results of the previously mentioned studies (ADAPT and INTREPAD) are not unexpected, given that the timing of exposure of the participants to NSAIDs was restricted (2 years). On this basis, NSAIDs might be useful for AD prevention when their administration occurs years before the usual onset age; however, when used later in life, they might increase the risk of disease. We cannot exclude the possibility that (I) the majority of the advantageous effects of NSAIDs, documented from epidemiological studies, may originate from different types of bias (232), and (II) actually, there is no established impact of NSAIDs on AD prevention or treatment (233).
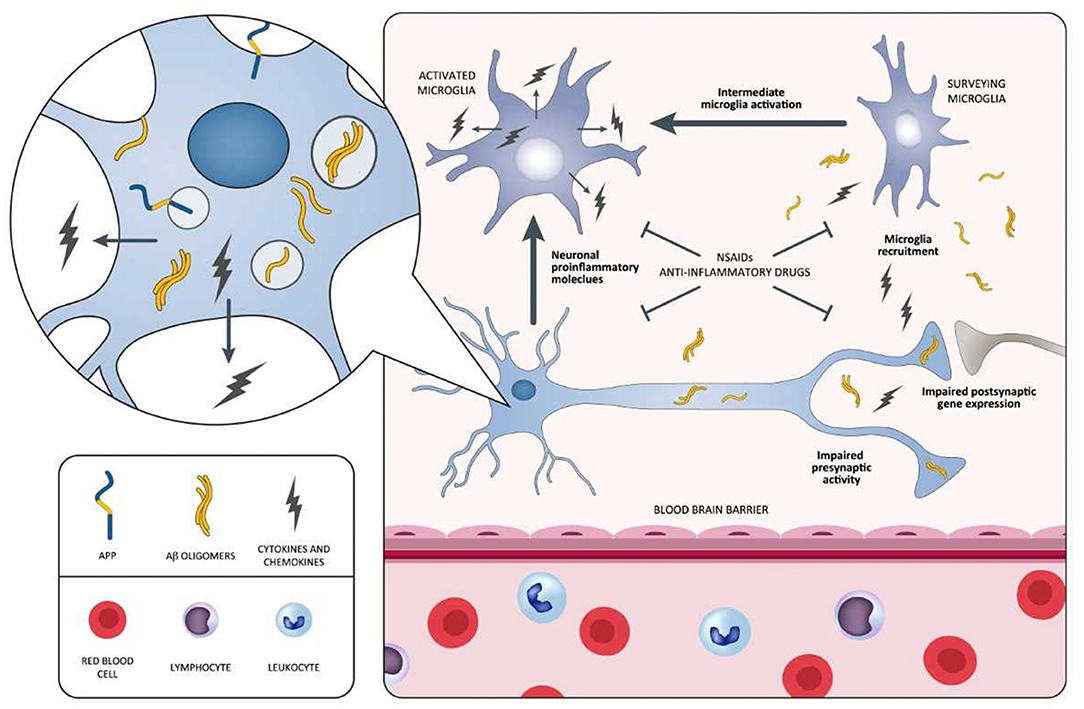
Figure 3. Schematic representation of neuroinflammatory process occurring during the early stages of the AD pathology and potential points of attack of NSAIDs and anti-inflammatory drugs. In this process, neurons surrounded by Aβ oligomers release proinflammatory cytokines triggering the intermediate activation of microglia and their mobilization toward Aβ-burdened neurons. Both Aβ-burdened neurons and activated microglia are responsible for a disease-aggravating process in which the release of proinflammatory cytokines and chemokines predominates. NSAIDs and anti-inflammatory drugs may be potentially effective during this early inflammatory phase, antagonizing the aggravating activity of proinflammatory mediators. Selective agents stabilizing microglia may also be effective in attenuating the inflammatory process. Aβ, amyloid beta; AD, Alzheimer's disease; NSAIDs, non-steroidal anti-inflammatory drugs.
Alector/Abbvie claim the generation of a monoclonal antibody (AL002) that binds and activates TREM2. AL002 entered its first phase 1 trial in 51 healthy adults and 16 AD patients (234). Alector is also starting its first trial of the anti-CD33 antibody, AL003. The microglial receptor CD33 opposes the effects of TREM2 signaling and may present a more amenable target because it would be inhibited rather than activated. In the first phase of the trial, 42 healthy adults will receive a single treatment of either placebo or one of seven different AL003 doses. The second, multiple-dose phase will enroll 12 AD patients, two of whom will receive placebo (234).
Of note, other drugs targeting neuroinflammation to treat AD are being developed and underwent clinical testing. XPro1595 is currently undergoing phase 1b clinical trials. Other examples are GC021109 and NP001. XPro1595 is a variant of TNF-α that forms heterotrimers with native soluble TNF-α and prevents its interaction with the type 1 TNF-α receptors (235). Unlike other non-selective TNF-α inhibitors, XPro1595 does not suppress innate immunity or myelination mediated by type 2 receptors (236). Differently from etanercept, long-term treatment with XPro1595 does not suppress hippocampal neurogenesis, learning, and memory in adult mice (237). In 5xFAD mice, twice-weekly subcutaneous administration of XPro1595 for 2 months reduced brain amyloid deposition and immune cell infiltration, and improved synaptic function (238). In young TgCRND8 mice, continuous subcutaneous infusion of XPro1595 for 1 month prevented brain amyloid deposition and normalized hippocampal neuron synaptic function (239). In 3xTg mice, intracranial administration of XPro1595 reduced amyloid pathology (240). In aged wild-type rats, intracranial infusions of XPro1595 for 6 weeks reduced microglia activation and improved synaptic function and cognition (241). A 12-week, open-label, phase 1b study of XPro1595 (weekly injections of 0.03, 1.0, or 3.0 mg/kg) is ongoing in 18 mild-to-moderate AD patients (NCT03943264). Participants were requested to have a positive amyloid test and evidence of peripheral inflammation [elevated blood C-reactive protein (CRP)]. Biomarkers of neuroinflammation in blood and CSF (CRP, TNF-α), IL-1β, and IL-6 are being measured.
GC 021109 targets microglial cells by binding the P2Y6 receptor, a metabotropic G-protein-coupled receptor, whose natural ligand is adenosine diphosphate, a metabolite of ATP. Astrocytes release ATP in response to the presence of Aβ aggregates and P2Y6 signaling is thought to be involved in shifting the phenotype of microglia, which tend to surround amyloid plaques, from patrolling to phagocytic (242). GC 021109 has been reported in the biotech press to stimulate both microglial phagocytosis and inhibit microglial release of proinflammatory cytokines such as IL-12; however, this information is not published in the peer-reviewed literature. A phase 1a study in 44 healthy volunteers has been carried out in 2015 (NCT02254369), and a 4-week, phase 1b study in 39 mild-to-moderate AD was completed in 2016 (NCT02386306). However, no results were reported.
NP001 is a pH-adjusted intravenous formulation of purified sodium chlorite. Within monocytes/macrophages, chlorite is converted into taurine chloramine that downregulates the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) expression and inhibits production of proinflammatory cytokine IL-1β. These mechanisms of downregulation transform inflammatory monocytes/macrophages from a proinflammatory to a basal phagocytic state. NP001 has been tested in patients with amyotrophic lateral sclerosis (243). A small study planned to be carried out in 14 mild-to-moderate AD patients (NCT03179501) was interrupted in 2018 for poor enrollment.
Profiling molecular pathways related to ND is expected to reveal novel pathways for therapeutic agents. In this context, inflammation represents a primarily involved pathway (12, 19). Interestingly, a meta-analysis including 175 studies reports changes in several inflammatory biomarkers (IL-6, CRP, and TNF-α) in AD (161). Another meta-analysis including nine longitudinal studies shows a protective effect by NSAIDs against AD progress (244). Changes in the concentrations of blood (serum) inflammatory proteins—including IL-6, CRP, and TNF-α–define a serum-based proteomic signature potentially useful for AD diagnosis (245–247). Hence, according to the literature, anti-inflammatory compounds might be employed as therapeutic agents in AD and others ND. In this regard, a novel model based on PM for targeted NSAIDs therapy to specific AD patients is recently proposed by O'Bryant and colleagues. In particular, they determine whether a blood proteomic companion diagnostic (CDx) is able to predict response to NSAID treatment (248). The analysis of the proteome in plasma samples from the Alzheimer's Disease Cooperative Studies (ADCS) anti-inflammatory clinical trial, including 1-year administration of rofecoxib (25 mg once daily), naproxen (220 mg twice daily), or placebo (N = 351) (215)—indicates that an overall NSAID-general CDx is accurate in detecting treatment response with 87% accuracy. Drug-specific companion diagnostics—Rofecoxib-CDx and Naproxen-CDx—generate a very high degree of accuracy in both rofecoxib (98%) and naproxen (97%) arms (234). This is a relevant example of direct evidence for a precision medicine-based model to address AD treatment via the creation of CDx-driven therapeutics.
In the last 20 years, a rising body of experimental studies has indicated that soluble Aβ protofibrils are more synaptotoxic than insoluble Aβ plaque cores. For instance, the former display higher rates of synapse structure impairment, including LTP, than plaques (249–251). Of note, solubilization of Aβ plaque cores is strictly related to the release of smaller Aβ species, such as dimers, and downstream increase in synaptotoxicity (252). Therefore, it is argued that prefibrillar Aβ1−42 assemblies, rather than monomers or dimers, are the proximate mediators of Aβ toxicity (253).
With regard to CNS immune resident cells, growing evidence indicates that small soluble Aβ1−42 protofibrils are the main trigger of microglial activation. Indeed, experimental models of AD indicate that both microglia and astrocytes display not only a high sensitivity to Aβ structure (16, 17, 33, 68, 86) in the internalization process but also emphasize their greater affinity for soluble Aβ protofibrils than mature insoluble fibrils (254, 255). In this context, it is reported that small soluble Aβ1−42 protofibrils, rather than fibrils, can induce microglial activation, as reflected by increased cerebral levels of TNF-α (255).
The murine monoclonal antibody mAb158 displays a 1, 000-fold higher selectivity for protofibrils [N-terminal (1–16) of the Aβ sequence] vs. monomers and 10–15 times more efficient binding to protofibrils vs. fibrils (256, 257).
Several studies, employing mAb158, suggest that astrocytic Aβ uptake depends on size and/or composition of Aβ aggregates, since astrocytes, if possible, engulf oligomeric Aβ over its fibrillar aggregation states (258).
Recent trials conducted in mice models of AD demonstrate that the antibody significantly slows down Aβ accumulation in astrocytes reducing the downstream Aβ-induced neuronal toxicity (256). The authors argue that their results provide a strong evidence for astrocytes to play a key mechanistic role in anti-Aβ immunotherapy.
The phase 2 preliminary results regarding the humanized IgG1 monoclonal version of mAb158 [BAN2401 (Biogen, Eisai Co., Ltd./BioArctic Neuroscience AB)]1 (259) show that BAN2401 significantly reduces Aβ-PET standardized uptake value ratio (SUVr) as well as CSF neurogranin, p-tau, and neurofilament light chain protein levels over a 18-month clinical trial (260, 261).
Acute, unaccustomed exercise (i.e., of an unusual duration and/or intensity) can increase oxidative stress and act as a proinflammatory stimulus (262, 263). However, this response is attenuated when exercise is performed regularly, with strong evidence actually supporting that “chronic” exercise upregulates an endogenous systemic anti-inflammatory response (16).
Large cohort studies indicate that higher levels of physical activity are inversely associated with inflammatory biomarkers, for instance CRP (264, 265). There is meta-analytical evidence that regular physical exercise can reduce inflammation-related biomarkers (e.g., CRP, TNF-α) in middle-aged and older adults (266, 267), these benefits being also present in individuals with cognitive impairment (268). Animal research indicates that the anti-inflammatory effects of exercise can also reach the brain tissue. Physical exercise training results in an enhanced anti-inflammatory status—as reflected by an increased expression of anti-inflammatory cytokines including IL-10β coupled with the decrease in proinflammatory cytokines (including TNF-α)—at the hippocampus level in a rat model of AD (269). Chronic exercise also promotes a conversion of the microglia from the proinflammatory (M1) to the anti-inflammatory (M2) phenotype in different rodent models of disease, including AD (269–272).
Although the mechanisms underlying exercise anti-inflammatory effects remain to be clearly elucidated, several pathways are currently proposed. Notably, contracting muscles act as an endocrine organ by releasing myokines (i.e., cytokines and other small peptides) to the bloodstream, which, in turn, induce numerous health benefits (such as a decrease in inflammation) at the multisystemic level, including the brain (273, 274). Muscle-derived IL-6 promotes the systemic production of anti-inflammatory cytokines (IL-1Ra, IL-10) and downregulates the expression of proinflammatory cytokines (TNF-α, IL-1β) (275). Other proposed mechanisms include exercise-induced reductions in adiposity (which, especially visceral fat, contribute to systemic inflammation), on the one hand, and increases in vagal tone, on the other hand, through the cholinergic anti-inflammatory pathway, an evolutionarily ancient circuit that modulates immune responses and the progression of inflammatory diseases (276, 277). In conclusion, given the documented relevance of inflammation in most ND (169), there is strong biological rationale to support that exercise might serve as a coadjuvant therapeutic strategy against such conditions.
The official launch of the US Precision Medicine Initiative (PMI), in 2015 (https://obamawhitehouse.archives.gov/precision-medicine), by the US President Obama followed by the National Institutes of Health (NIH) development of the US PMI Cohort Program (PMI-CP) (278) and the creation of the US “All of Us Research Program” (available at https://allofus.nih.gov/) are contributing to make precision medicine one of the key topics in biomedical research, worldwide. These facts support the evolution of Medicine from the outdated “one-size-fits-all” paradigm—according to which treatments are conceived for the “average patient”—to the search for comprehensive and accurate stratification of individuals and future individually tailored therapeutic modalities and targeted therapies (279). Indeed, genetic and biological heterogeneity among individuals sharing the same clinical features (so-called clinical syndrome) is highly frequent in polygenic, multifactorial diseases with complex and non-linear pathophysiological dynamics, such as cancer and AD. In this regard, it is acknowledged that the adaptive and innate immune systems are characterized by enormous individual heterogeneity that accounts for the subject-specific response to vaccines and other immunomodulatory therapies (280–282). As a result, some drugs, regularly administered, can be of benefit only to a restricted subset of patients; other drugs might even have detrimental effects to some definite ethnic groups (283). Hence, the identification of the molecular/cellular and environmental factors indicating the presence and the type of reaction of a single AD patient to a specific therapy is crucial (284). The shift to individualized therapies and targeted treatments needs exploratory, unbiased, high-throughput, integrative, large-scale analyses of the features of the cohort's individuals with the disease (279, 284, 285). Cohorts stratified according to different multimodal-throughput technological platforms (“omic” sciences)—via systems biology (285, 286)—and different neuroimaging modalities—via systems neurophysiology (285)—can be assimilated in the disease modeling to stratify and predict AD patient subgroups (279, 285). Both systems biology and neurophysiology enable to perform a holistic, systemic exploration of complex interactions in biological systems, thus allowing an overview of cells/groups of cells, tissues, organs, organisms, and populations at multiple scales. High-throughput, integrative approaches permit to recover exhaustive biological information, supported by advanced powerful bioinformatics. This will enable the inclusive integration of both multiomic and clinical data to attain fast and significant interpretation. Precision medicine capitalizes on these theoretical and technological advancements (287). Particularly, the integration of the “omics” and the development of the “multiomic” disciplines—such as proteogenomics, whereby the involved technologies are next-generation sequencing and mass spectrometry—seem to be able to offer a substantial support for accurate phenotype prediction, individualized patient management, and precision medicine (288, 289). Establishing precision medicine needs the implementation of a network of integrated disciplines and methods including the “omic” sciences, neuroimaging modalities, cognitive examinations, and clinical features. All these congregate toward many domains investigated using the systems theory approach (290). This allows the development of models explaining all systems levels—explored via systems biology and systems neurophysiology—and the different categories and scales of spatiotemporal data describing the complexity and clinical heterogeneity of any polygenic disease belonging to any medical fields, from oncology, to immunology (284) (Figure 4), to neurology (285, 291, 292). Precision medicine aims at ameliorating the efficacy of prevention strategies and therapies using customized treatments tailored on the individual's “biological make-up” (285, 291, 292), based on the “P4 Medicine” (P4M) framework (293). To safeguard the rapid and full expansion of precision medicine in AD, the international Alzheimer Precision Medicine Initiative (APMI) and its own Cohort Program (APMI-CP) (available at https://www.apmiscience.com/)—thematicall associated with the US PMI and the US “All of Us Research Program”—are currently established and operational (279). In this connection, a therapeutic plan based on immune/inflammation modulation for a subset of AD and associated dementias is currently ongoing within the “Korean AD Research Platform Initiative Based on Immune-Inflammatory Biomarkers” (K-ARPI) (294).
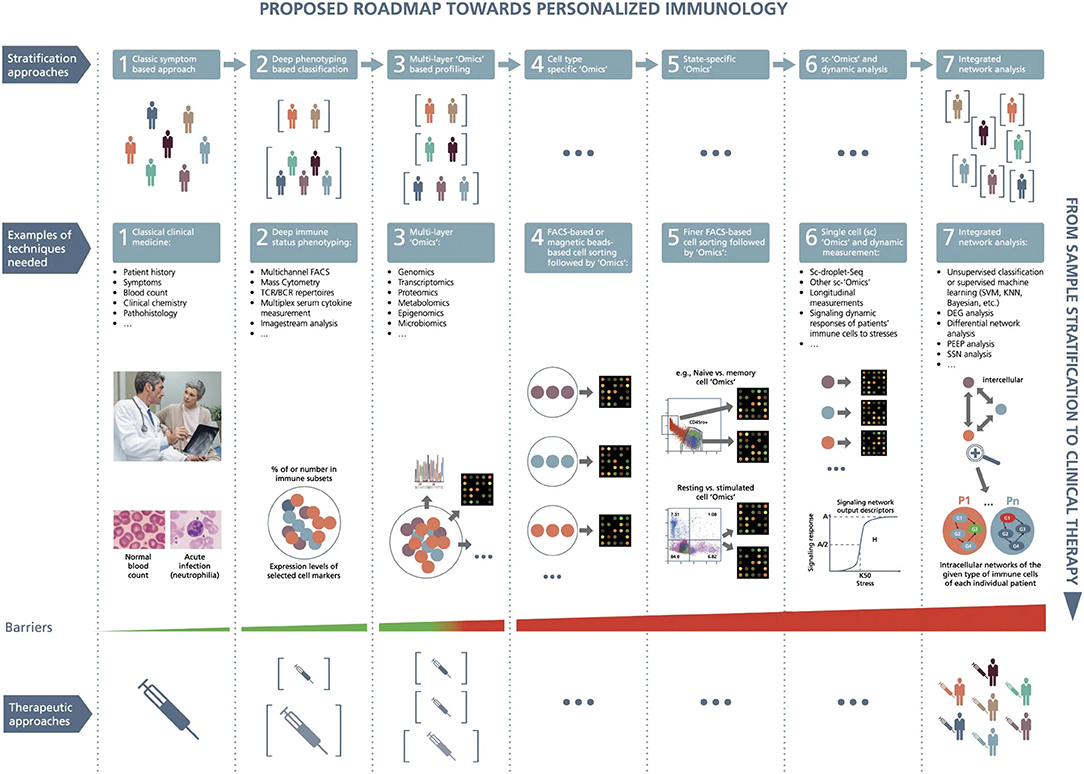
Figure 4. A roadmap proposed toward personalized immunology. There exist both horizontal and vertical roadmaps toward personalized immunology. Vertically, to translate sample stratification to clinical therapies, it is necessary to utilize the state-of-the-art “Omics” analysis and network integration approaches to stratify patients into subgroups and then implement personalized therapeutic approaches to treat individual patients, which needs to overcome various types of barriers at different steps. Horizontally, it might be necessary to go through at least seven steps to enable personalized immunotherapies: (1) classic symptom-based approach, (2) deep phenotyping approach, (3) multilayer “Omics”-based profiling, (4) cell-type-specific “Omics,” (5) state-specific “Omics,” (6) single-cell “Omics” and dynamic response analysis of immune cells, and (7) integrated network analysis. Under the first layer (the so-called stratification layer), different colors of patients indicate individual patients with different cellular and/or molecular profiles, while brackets represent patient subgroups; under the second layer (the so-called technique layers), different small circles with distinct colors indicate different immune cells, while big circles represent patient (sub)groups; under the technique layers, the snapshot of microarray representing either microarray-based or RNA-seq-based transcriptome analysis; under the third layer (the so-called therapeutic layer), the syringes with different colors or tonalities indicate different therapeutic approaches; P1,…, Pn at step 7 designate different patients; G1, G2, G3, and G4 represent different genes, the arrows between them representing regulatory relationships. DEG, differential expression gene; FACS, fluorescence-activated cell sorting; KNN, K-nearest neighbors; PEEP, personalized expression perturbation profile; sc, single-cell; SSN, sample-specific network; SVM, support vector machine; TCR/BCR, T-cell receptor/B-cell receptor. From Delhalle et al. (284). Copyright© 2018, Springer Nature. Reprinted with permission from Creative Commons CC BY. S.
Systems theory/biology-based studies are needed to untangle the spatiotemporal dynamics of neuroinflammation and its related subcomponents. Biomarkers simultaneously tracking different molecular pathways (body fluid matrixes) along with brain neuroinflammation endophenotypes (neuroimaging markers) can untangle key temporal–spatial dynamics among glia, neuroinflammation, and other AD pathophysiological mechanisms. Implementing this approach will be necessary to fill the gap in the understanding of whether neuroinflammation represents a direct pathophysiological or compensatory mechanism or both, along the AD continuum. According to this assumption, a new pathway (mechanism)-based pharmacological model—intended to establish effective and functional biomarker-guided targeted and tailored treatments for preventive and neuroinflammation-freezing strategies—needs to be developed.
HH, AV, and SL designed the concept of the manuscript, provided supervision and assisted with the writing and content of the manuscript. AV and SL assisted in the preparation of the figures. All authors provided their contribution in writing the manuscript, critically reviewing the completed manuscript, and approved the submitted version of the manuscript.
This research benefited from the support of the Program PHOENIX led by the Sorbonne University Foundation and sponsored by la Fondation pour la Recherche sur Alzheimer. HH is an employee of Eisai Inc. During his previous work (until April 2019), he was supported by the AXA Research Fund, the Fondation partenariale Sorbonne Université and the Fondation pour la Recherche sur Alzheimer, Paris, France.
HH is an employee of Eisai Inc. and serves as Senior Associate Editor for the Journal Alzheimer's & Dementia; during the past 3 years he had received lecture fees from Servier, Biogen, and Roche; research grants from Pfizer, Avid, and MSD Avenir (paid to the institution); travel funding from Eisai, Functional Neuromodulation, Axovant, Eli Lilly and company, Takeda and Zinfandel, GE-Healthcare, and Oryzon Genomics; consultancy fees from Qynapse, Jung Diagnostics, Cytox Ltd., Axovant, Anavex, Takeda and Zinfandel, GE Healthcare, Oryzon Genomics, and Functional Neuromodulation; and participated in scientific advisory boards of Functional Neuromodulation, Axovant, Eisai, Eli Lilly and company, Cytox Ltd., GE Healthcare, Takeda and Zinfandel, Oryzon Genomics, and Roche Diagnostics. He is coinventor in the following patents as a scientific expert and has received no royalties: In Vitro Multiparameter Determination Method for the Diagnosis and Early Diagnosis of Neurodegenerative Disorders Patent Number: 8916388; In Vitro Procedure for Diagnosis and Early Diagnosis of Neurodegenerative Diseases Patent Number: 8298784; Neurodegenerative Markers for Psychiatric Conditions Publication Number: 20120196300; In Vitro Multiparameter Determination Method for the Diagnosis and Early Diagnosis of Neurodegenerative Disorders Publication Number: 20100062463; In Vitro Method for the Diagnosis and Early Diagnosis of Neurodegenerative Disorders Publication Number: 20100035286; In Vitro Procedure for Diagnosis and Early Diagnosis of Neurodegenerative Diseases Publication Number: 20090263822. In Vitro Method for the Diagnosis of Neurodegenerative Diseases Patent Number: 7547553; CSF Diagnostic In Vitro Method for Diagnosis of Dementias and Neuroinflammatory Diseases Publication Number: 20080206797; In Vitro Method for the Diagnosis of Neurodegenerative Diseases Publication Number: 20080199966; Neurodegenerative Markers for Psychiatric Conditions Publication Number: 20080131921. SV is an officer and director of NeuroVision. LA-A is an employee of NeuroVision. EE is the unique owner of 2E Science, a for-profit private scientific company. Neither EE nor 2E Science have any commercial interest or financial tie in relation with this article. MW is employed by the company TranScrip Partners, Reading, United Kingdom. He is a clinical consultant of Chiesi Farmaceutici. AV is an employee of Eisai Inc. and received lecture honoraria from Roche, MagQu LLC, and Servier. SL received lecture honoraria from Roche and Servier.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Contributors to the Alzheimer Precision Medicine Initiative Working Group (APMI-WG): Mohammad Afshar (France), Lisi Flores Aguilar (Canada), Leyla Akman-Anderson (USA), Joaquín Arenas (Spain), Jesús Ávila (Spain), Claudio Babiloni (Italy), Filippo Baldacci (Italy), Richard Batrla (Switzerland), Norbert Benda (Germany), Keith L. Black (USA), Arun L. W. Bokde (Ireland), Ubaldo Bonuccelli (Italy), Karl Broich (Germany), Francesco Cacciola (Italy), Filippo Caraci (Italy), Giuseppe Caruso (Italy), Juan Castrillo† (Spain), Enrica Cavedo (France), Roberto Ceravolo (Italy), Patrizia A. Chiesa (France), Massimo Corbo (Italy), Jean-Christophe Corvol (France), Augusto Claudio Cuello (Canada), Jeffrey L. Cummings (USA), Herman Depypere (Belgium), Bruno Dubois (France), Andrea Duggento (Italy), Enzo Emanuele (Italy), Valentina Escott-Price (UK), Howard Federoff (USA), Maria Teresa Ferretti (Switzerland), Massimo Fiandaca (USA), Richard A. Frank (USA), Francesco Garaci (Italy), Hugo Geerts (USA), Ezio Giacobini (Switzerland), Filippo S. Giorgi (Italy), Edward J. Goetzl (USA), Manuela Graziani (Italy), Marion Haberkamp (Germany), Marie-Odile Habert (France), Britta Hänisch (Germany), Harald Hampel (USA), Karl Herholz (UK), Felix Hernandez (Spain), Bruno P. Imbimbo (Italy), Dimitrios Kapogiannis (USA), Eric Karran (USA), Steven J. Kiddle (UK), Seung H. Kim (South Korea), Yosef Koronyo (USA), Maya Koronyo-Hamaoui (USA), Todd Langevin (USA), Stéphane Lehéricy (France), Pablo Lemercier (France), Simone Lista (France), Francisco Llavero (Spain), Jean Lorenceau (France), Alejandro Lucia (Spain), Dalila Mango (Italy), Mark Mapstone (USA), Christian Neri (France), Robert Nisticò (Italy), Sid E. O'Bryant (USA), Giovanni Palermo (Italy), George Perry (USA), Craig Ritchie (Scotland), Simone Rossi (Italy), Amira Saidi (Italy), Emiliano Santarnecchi (Italy), Lon S. Schneider (USA), Olaf Sporns (USA), Nicola Toschi (Italy), Pedro L. Valenzuela (Spain), Bruno Vellas (France) Steven R. Verdooner (USA), Andrea Vergallo (USA), Nicolas Villain (France), Kelly Virecoulon Giudici (France), Mark Watling (UK), Lindsay A. Welikovitch (Canada), Janet Woodcock (USA), Erfan Younesi (Luxembourg), José L. Zugaza (Spain).
[11C]-PIB, [11C]-Pittsburgh compound-B; [18F]-fluorodeoxyglucose-PET, [18F]-fluorodeoxyglucose-positron emission tomography; Aβ, amyloid beta; Aβ1−40, 40-amino acid-long amyloid beta peptide; Aβ1−42, 42-amino acid-long amyloid beta peptide; AD, Alzheimer's disease; ADAPT, Alzheimer's disease anti-inflammatory prevention trial; ADCS, Alzheimer's disease cooperative studies; APMI, Alzheimer precision medicine initiative; APMI-CP, Alzheimer precision medicine initiative cohort program; APS, Alzheimer progression score; ASC, apoptosis-associated speck-like protein containing a caspase recruitment domain; BDNF, brain-derived neurotrophic factor; CB2R, cannabinoid receptor type 2; CCE, cell cycle events; CD, cell surface cluster of differentiation; CDx, companion diagnostic; CNS, central nervous system; COX, cyclooxygenase; COX-1, cyclooxygenase-1; COX-2, cyclooxygenase-2; CRP, C-reactive protein; CSF, cerebrospinal fluid; CSF-1R, colony stimulating factor 1 receptor; GWAS, genome-wide association studies; ILs, interleukins; IL-1Ra, interleukin-1 receptor antagonist; INTREPAD, impact of naproxen treatment in presymptomatic Alzheimer's disease; JNK, c-Jun Kinase; K-ARPI, Korean AD research platform initiative based on immune-inflammatory biomarkers; LTP, long-term potentiation; MAPK, p38 mitogen-activated protein kinase; MCI, mild cognitive impairment; MCP-1, monocyte chemoattractant protein-1; MMP-9, metalloprotease-9; MMSE, Mini-Mental State Examination; mNGF, mature nerve growth factor; Nap1, Nck-associated protein 1; ND, neurodegenerative diseases; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NFT, neurofibrillary tangles; NGF, nerve growth factor; NIH, National Institutes of Health; NSAIDs, non-steroidal anti-inflammatory drugs; P4M, predictive, preventive, personalized and participatory medicine; PET, positron emission tomography; PMI, US precision medicine initiative; PMI-CP, US PMI cohort program; proNGF, precursor of the nerve growth factor; p-tau, hyperphosphorylated tau; RNS, reactive nitrogen species; ROS, reactive oxygen species; SCARA1, class A1 scavenger receptor; SphK1, sphingosine kinase 1; sTREM2, soluble TREM2; TGF-β1, transforming growth factor-beta1; TLRs, Toll-like receptors; TNF-α, tumor necrosis factor-alpha; TNF-Rs, TNF receptors; TNF-RI, TNF receptor I; TNF-RII, TNF receptor II; TREM2, triggering receptor expressed on myeloid cells 2; TSPO, translocator protein.
1. ^https://clinicaltrials.gov/ct2/show/NCT01230853 (A Randomized, Double-blind, Placebo-controlled, Combined Single Ascending Dose and Multiple Ascending Dose Study to Assess Safety, Tolerability, Immunogenicity, Pharmacodynamic Response, and Pharmacokinetics of Intravenous Infusions of BAN2401 in Subjects With Mild to Moderate Alzheimer's Disease).
1. Association A. 2017 Alzheimer's disease facts and figures. Alzheimer's Dement. (2017) 13:325–73. doi: 10.1016/j.jalz.2017.02.001
2. Panza F, Lozupone M, Logroscino G, Imbimbo BP. A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat Rev Neurol. (2019) 15:73–88. doi: 10.1038/s41582-018-0116-6
3. Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. (2018) 14:535–62. doi: 10.1016/j.jalz.2018.02.018
4. Hampel H, Mesulam M-M, Cuello AC, Farlow MR, Giacobini E, Grossberg GT, et al. The cholinergic system in the pathophysiology and treatment of Alzheimer's disease. Brain. (2018) 141:1917–33. doi: 10.1093/brain/awy132
5. Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the national institute on Aging-Alzheimer's association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. (2011) 7:280–92. doi: 10.1016/j.jalz.2011.03.003
6. Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K, et al. Advancing research diagnostic criteria for Alzheimer's disease: the IWG-2 criteria. Lancet Neurol. (2014) 13:614–29. doi: 10.1016/S1474-4422(14)70090-0
7. Mckhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH, et al. The diagnosis of dementia due to Alzheimer's disease : recommendations from the national institute on Aging-Alzheimer's association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's Dement. (2011) 7:263–9. doi: 10.1016/j.jalz.2011.03.005
8. Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the national institute on Aging-Alzheimer's association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's Dement. (2011) 7:270–9. doi: 10.1016/j.jalz.2011.03.008
9. Jack CR, Bennett DA, Blennow K, Carrillo MC, Feldman HH, Frisoni GB, et al. A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology. (2016) 87:539–47. doi: 10.1212/WNL.0000000000002923
10. Hampel H, O'Bryant SE, Molinuevo JL, Zetterberg H, Masters CL, Lista S, et al. Blood-based biomarkers for Alzheimer disease: mapping the road to the clinic. Nat Rev Neurol. (2018) 14:639–52. doi: 10.1038/s41582-018-0079-7
11. Molinuevo JL, Ayton S, Batrla R, Bednar MM, Bittner T, Cummings J, et al. Current state of Alzheimer's fluid biomarkers. Acta Neuropathol. (2018) 136:821–53. doi: 10.1007/s00401-018-1932-x
12. Heneka MT, Carson MJ, Khoury J El, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. (2015) 14:388–405. doi: 10.1016/S1474-4422(15)70016-5
13. Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. (2000) 21:383–421. doi: 10.1016/S0197-4580(00)00124-X
14. Businaro R, Corsi M, Asprino R, Di Lorenzo C, Laskin D, Corbo RM, et al. Modulation of inflammation as a way of delaying Alzheimer's Disease progression: the diet's role. Curr Alzheimer Res. (2018) 15:363–80. doi: 10.2174/1567205014666170829100100
15. Rogers J, Webster S, Lue LF, Brachova L, Civin WH, Emmerling M, et al. Inflammation and Alzheimer's disease pathogenesis. Neurobiol Aging. (1996) 17:681–6. doi: 10.1016/0197-4580(96)00115-7
16. McGeer PL, McGeer EG. The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Brain Res Rev. (1995) 21:195–218. doi: 10.1016/0165-0173(95)00011-9
17. Eikelenboom P, Zhan S-S, van Gool WA, Allsop D. Inflammatory mechanisms in Alzheimer's disease. Trends Pharmacol Sci. (1994) 15:447–50. doi: 10.1016/0165-6147(94)90057-4
18. Iulita MF, Ganesh A, Pentz R, Flores Aguilar L, Gubert P, Ducatenzeiler A, et al. Identification and preliminary validation of a plasma profile associated with cognitive decline in dementia and at-risk individuals: a retrospective cohort analysis. J Alzheimer's Dis. (2019) 67:327–41. doi: 10.3233/JAD-180970
19. Cuello AC. Early and late CNS Inflammation in Alzheimer's disease: two extremes of a continuum? Trends Pharmacol Sci. (2017) 38:956–66. doi: 10.1016/j.tips.2017.07.005
20. Rogers J. Principles for central nervous system inflammation research: a call for a consortium approach. Alzheimer's Dement. (2018) 14:1553–9. doi: 10.1016/j.jalz.2018.01.008
21. Brosseron F, Krauthausen M, Kummer M, Heneka MT. Body fluid cytokine levels in mild cognitive impairment and Alzheimer's disease: a comparative overview. Mol Neurobiol. (2014) 50:534–44. doi: 10.1007/s12035-014-8657-1
22. Morgan AR, Touchard S, Leckey C, O'Hagan C, Nevado-Holgado AJ, Barkhof F, et al. Inflammatory biomarkers in Alzheimer's disease plasma. Alzheimer's Dement. (2019) 15:776–87. doi: 10.1016/j.jalz.2019.03.007
23. Nordengen K, Kirsebom B-E, Henjum K, Selnes P, Gísladóttir B, Wettergreen M, et al. Glial activation and inflammation along the Alzheimer's disease continuum. J Neuroinflammation. (2019) 16:46. doi: 10.1186/s12974-019-1399-2
24. Fan Z, Brooks DJ, Okello A, Edison P. An early and late peak in microglial activation in Alzheimer's disease trajectory. Brain. (2017) 140:aww349. doi: 10.1093/brain/aww349
25. Edison P, Brooks DJ. Role of neuroinflammation in the trajectory of Alzheimer's disease and in vivo quantification using PET. J Alzheimer's Dis. (2018) 64:S339–51. doi: 10.3233/JAD-179929
26. Parbo P, Ismail R, Hansen K V, Amidi A, Mårup FH, Gottrup H, et al. Brain inflammation accompanies amyloid in the majority of mild cognitive impairment cases due to Alzheimer's disease. Brain. (2017) 140:2002–11. doi: 10.1093/brain/awx120
27. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell. (2007) 131:1164–78. doi: 10.1016/j.cell.2007.10.036
28. Henstridge CM, Tzioras M, Paolicelli RC. Glial contribution to excitatory and inhibitory synapse loss in neurodegeneration. Front Cell Neurosci. (2019) 13:63. doi: 10.3389/fncel.2019.00063
29. Schafer DP, Lehrman EK, Stevens B. The “quad-partite” synapse: microglia-synapse interactions in the developing and mature CNS. Glia. (2013) 61:24–36. doi: 10.1002/glia.22389
30. Cohen J, Torres C. Astrocyte senescence: evidence and significance. Aging Cell. (2019) 18:e12937. doi: 10.1111/acel.12937
31. Stojiljkovic MR, Ain Q, Bondeva T, Heller R, Schmeer C, Witte OW. Phenotypic and functional differences between senescent and aged murine microglia. Neurobiol Aging. (2019) 74:56–69. doi: 10.1016/j.neurobiolaging.2018.10.007
32. Arranz AM, De Strooper B. The role of astroglia in Alzheimer's disease: pathophysiology and clinical implications. Lancet Neurol. (2019) 18:406–14. doi: 10.1016/S1474-4422(18)30490-3
33. Gomez-Arboledas A, Davila JC, Sanchez-Mejias E, Navarro V, Nuñez-Diaz C, Sanchez-Varo R, et al. Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer's disease. Glia. (2018) 66:637–53. doi: 10.1002/glia.23270
34. Hansen D V, Hanson JE, Sheng M. Microglia in Alzheimer's disease. J Cell Biol. (2018) 217:459–72. doi: 10.1083/jcb.201709069
35. Harry GJ. Microglia during development and aging. Pharmacol Ther. (2013) 139:313–26. doi: 10.1016/j.pharmthera.2013.04.013
36. Graham LC, Naldrett MJ, Kohama SG, Smith C, Lamont DJ, McColl BW, et al. Regional molecular mapping of primate synapses during normal healthy aging. Cell Rep. (2019) 27:1018–26.e4. doi: 10.1016/j.celrep.2019.03.096
37. Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol. (2014) 10:217–24. doi: 10.1038/nrneurol.2014.38
38. De Strooper B, Karran E. The cellular phase of Alzheimer's disease. Cell. (2016) 164:603–15. doi: 10.1016/j.cell.2015.12.056
39. Edwards FA. A unifying hypothesis for Alzheimer's disease: from Plaques to neurodegeneration. Trends Neurosci. (2019) 42:310–22. doi: 10.1016/j.tins.2019.03.003
40. Rajendran L, Paolicelli RC. Microglia-mediated synapse loss in Alzheimer's disease. J Neurosci. (2018) 38:2911–9. doi: 10.1523/JNEUROSCI.1136-17.2017
41. Arnold SE, Louneva N, Cao K, Wang L-S, Han L-Y, Wolk DA, et al. Cellular, synaptic, and biochemical features of resilient cognition in Alzheimer's disease. Neurobiol Aging. (2013) 34:157–68. doi: 10.1016/j.neurobiolaging.2012.03.004
42. Chan WY, Kohsaka S, Rezaie P. The origin and cell lineage of microglia—New concepts. Brain Res Rev. (2007) 53:344–54. doi: 10.1016/j.brainresrev.2006.11.002
43. Sarlus H, Heneka MT. Microglia in Alzheimer's disease. J Clin Invest. (2017) 127:3240–9. doi: 10.1172/JCI90606
44. Streit WJ, Xue Q-S, Tischer J, Bechmann I. Microglial pathology. Acta Neuropathol Commun. (2014) 2:142. doi: 10.1186/s40478-014-0142-6
45. Wang S, Colonna M. Microglia in Alzheimer's disease: a target for immunotherapy. J Leukoc Biol. (2019) 106:JLB.MR0818-319R. doi: 10.1002/JLB.MR0818-319R
46. Tang Y, Le W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol. (2016) 53:1181–194. doi: 10.1007/s12035-014-9070-5
47. Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. (2016) 19:987–91. doi: 10.1038/nn.4338
48. Stratoulias V, Venero JL, Tremblay MÈ, Joseph B. Microglial subtypes: diversity within the microglial community. EMBO J. (2019) 38:e101997. doi: 10.15252/embj.2019101997
49. Simon E, Obst J, Gomez-Nicola D. The evolving dialogue of microglia and neurons in Alzheimer's disease: microglia as necessary transducers of pathology. Neuroscience. (2019) 405:24–34. doi: 10.1016/j.neuroscience.2018.01.059
50. Wang Y, Zhu T, Wang M, Zhang F, Zhang G, Zhao J, et al. Icariin attenuates M1 activation of microglia and Aβ plaque accumulation in the hippocampus and prefrontal cortex by up-regulating PPARγ in restraint/isolation-stressed APP/PS1 Mice. Front Neurosci. (2019) 13:291. doi: 10.3389/fnins.2019.00291
51. Forloni G, Balducci C. Alzheimer's disease, oligomers, and inflammation. J Alzheimers Dis. (2018) 62:1261–76. doi: 10.3233/JAD-170819
52. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. (2010) 140:918–34. doi: 10.1016/j.cell.2010.02.016
53. Cameron B, Landreth GE. Inflammation, microglia, and Alzheimer's disease. Neurobiol Dis. (2010) 37:503–9. doi: 10.1016/j.nbd.2009.10.006
54. Yates SL, Kocsis-Angle J, Embury P, Brunden KR. Inflammatory responses to amyloid fibrils. Methods Enzymol. (1999) 309:723–33. doi: 10.1016/S0076-6879(99)09048-5
55. Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J Neurosci. (2003) 23:2665–74. doi: 10.1523/JNEUROSCI.23-07-02665.2003
56. Liu Y, Walter S, Stagi M, Cherny D, Letiembre M, Schulz-Schaeffer W, et al. LPS receptor (CD14): a receptor for phagocytosis of Alzheimer's amyloid peptide. Brain. (2005) 128:1778–89. doi: 10.1093/brain/awh531
57. Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer's disease amyloid β-protein via a scavenger receptor. Neuron. (1996) 17:553–65. doi: 10.1016/S0896-6273(00)80187-7
58. Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, et al. CD36 ligands promote sterile inflammation through assembly of a toll-like receptor 4 and 6 heterodimer. Nat Immunol. (2010) 11:155–61. doi: 10.1038/ni.1836
59. Simard AR, Soulet D, Gowing G, Julien J-P, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. (2006) 49:489–502. doi: 10.1016/j.neuron.2006.01.022
60. Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, et al. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. (2008) 28:4283–92. doi: 10.1523/JNEUROSCI.4814-07.2008
61. Condello C, Yuan P, Schain A, Grutzendler J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat Commun. (2015) 6:6176. doi: 10.1038/ncomms7176
62. Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective -amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. (2008) 28:8354–60. doi: 10.1523/JNEUROSCI.0616-08.2008
63. Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer's disease. Nature. (2017) 552:355–61. doi: 10.1038/nature25158
64. Joshi P, Turola E, Ruiz A, Bergami A, Libera DD, Benussi L, et al. Microglia convert aggregated amyloid-β into neurotoxic forms through the shedding of microvesicles. Cell Death Differ. (2014) 21:582–93. doi: 10.1038/cdd.2013.180
65. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. (2017) 541:481–7. doi: 10.1038/nature21029
66. Ferretti MT, Cuello AC. Does a pro-inflammatory process precede Alzheimer's disease and mild cognitive impairment? Curr Alzheimer Res. (2011) 8:164–74. doi: 10.2174/156720511795255982
67. Klein WL. Synaptotoxic amyloid-β oligomers: a molecular basis for the cause, diagnosis, and treatment of alzheimer's disease? J Alzheimer's Dis. (2012) 33:S49–65. doi: 10.3233/JAD-2012-129039
68. Ledo JH, Azevedo EP, Beckman D, Ribeiro FC, Santos LE, Razolli DS, et al. Cross talk between brain innate immunity and serotonin signaling underlies depressive-like behavior induced by Alzheimer's amyloid- oligomers in mice. J Neurosci. (2016) 36:12106–16. doi: 10.1523/JNEUROSCI.1269-16.2016
69. Caruso G, Fresta C, Musso N, Giambirtone M, Grasso M, Spampinato S, et al. Carnosine prevents Aβ-induced oxidative stress and inflammation in microglial cells: a key role of TGF-β1. Cells. (2019) 8:64. doi: 10.3390/cells8010064
70. Caraci F, Spampinato SF, Morgese MG, Tascedda F, Salluzzo MG, Giambirtone MC, et al. Neurobiological links between depression and AD: the role of TGF-β1 signaling as a new pharmacological target. Pharmacol Res. (2018) 130:374–84. doi: 10.1016/j.phrs.2018.02.007
71. Torrisi SA, Geraci F, Tropea MR, Grasso M, Caruso G, Fidilio A, et al. Fluoxetine and vortioxetine reverse depressive-like phenotype and memory deficits induced by Aβ1-42 oligomers in mice: a key role of transforming growth factor-β1. Front Pharmacol. (2019) 10:693. doi: 10.3389/fphar.2019.00693
72. Hanzel CE, Pichet-Binette A, Pimentel LSB, Iulita MF, Allard S, Ducatenzeiler A, et al. Neuronal driven pre-plaque inflammation in a transgenic rat model of Alzheimer's disease. Neurobiol Aging. (2014) 35:2249–62. doi: 10.1016/j.neurobiolaging.2014.03.026
73. Perea JR, Ávila J, Bolós M. Dephosphorylated rather than hyperphosphorylated tau triggers a pro-inflammatory profile in microglia through the p38 MAPK pathway. Exp Neurol. (2018) 310:14–21. doi: 10.1016/j.expneurol.2018.08.007
74. Jensen CJ, Massie A, De Keyser J. Immune players in the CNS: the astrocyte. J Neuroimmune Pharmacol. (2013) 8:824–39. doi: 10.1007/s11481-013-9480-6
75. Ben Achour S, Pont-Lezica L, Béchade C, Pascual O. Is astrocyte calcium signaling relevant for synaptic plasticity? Neuron Glia Biol. (2010) 6:147–55. doi: 10.1017/S1740925X10000207
76. Vincent AJ, Gasperini R, Foa L, Small DH. Astrocytes in Alzheimer's disease: emerging roles in calcium dysregulation and synaptic plasticity. J Alzheimers Dis. (2010) 22:699–714. doi: 10.3233/JAD-2010-101089
77. Halassa MM, Fellin T, Haydon PG. The tripartite synapse: roles for gliotransmission in health and disease. Trends Mol Med. (2007) 13:54–63. doi: 10.1016/j.molmed.2006.12.005
78. Halassa MM, Fellin T, Takano H, Dong J-H, Haydon PG. Synaptic islands defined by the territory of a single astrocyte. J Neurosci. (2007) 27:6473–7. doi: 10.1523/JNEUROSCI.1419-07.2007
79. Oberheim NA, Wang X, Goldman S, Nedergaard M. Astrocytic complexity distinguishes the human brain. Trends Neurosci. (2006) 29:547–53. doi: 10.1016/j.tins.2006.08.004
80. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. (2010) 119:7–35. doi: 10.1007/s00401-009-0619-8
81. Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. (2009) 32:638–47. doi: 10.1016/j.tins.2009.08.002
82. Medeiros R, LaFerla FM. Astrocytes: conductors of the Alzheimer disease neuroinflammatory symphony. Exp Neurol. (2013) 239:133–8. doi: 10.1016/j.expneurol.2012.10.007
83. Olabarria M, Noristani HN, Verkhratsky A, Rodríguez JJ. Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer's disease. Glia. (2010) 58:831–8. doi: 10.1002/glia.20967
84. Sekar S, McDonald J, Cuyugan L, Aldrich J, Kurdoglu A, Adkins J, et al. Alzheimer's disease is associated with altered expression of genes involved in immune response and mitochondrial processes in astrocytes. Neurobiol Aging. (2015) 36:583–91. doi: 10.1016/j.neurobiolaging.2014.09.027
85. Yamaguchi H, Sugihara S, Ogawa A, Saido TC, Ihara Y. Diffuse plaques associated with astroglial amyloid β protein, possibly showing a disappearing stage of senile plaques. Acta Neuropathol. (1998) 95:217–22. doi: 10.1007/s004010050790
86. Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, et al. Adult mouse astrocytes degrade amyloid-β in vitro and in situ. Nat Med. (2003) 9:453–7. doi: 10.1038/nm838
87. Chao CC, Hu S, Sheng WS, Bu D, Bukrinsky MI, Peterson PK. Cytokine-stimulated astrocytes damage human neurons via a nitric oxide mechanism. Glia. (1996) 16:276–84. doi: 10.1002/(SICI)1098-1136(199603)16:3<276::AID-GLIA10>3.0.CO;2-X
88. Kamboh MI. A brief synopsis on the genetics of Alzheimer's disease. Curr Genet Med Rep. (2018) 6:133–5. doi: 10.1007/s40142-018-0155-8
89. Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer's sisease. N Engl J Med. (2013) 368:117–27. doi: 10.1056/NEJMoa1211851
90. Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, et al. Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. (2013) 78:631–43. doi: 10.1016/j.neuron.2013.04.014
91. Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, et al. CD33 Alzheimer's disease locus: altered monocyte function and amyloid biology. Nat Neurosci. (2013) 16:848–50. doi: 10.1038/nn.3435
92. Rathore N, Ramani SR, Pantua H, Payandeh J, Bhangale T, Wuster A, et al. Paired immunoglobulin-like type 2 receptor alpha G78R variant alters ligand binding and confers protection to Alzheimer's disease. PLoS Genet. (2018) 14:e1007427. doi: 10.1371/journal.pgen.1007427
93. Li J-T, Zhang Y. TREM2 regulates innate immunity in Alzheimer's disease. J Neuroinflammation. (2018) 15:107. doi: 10.1186/s12974-018-1148-y
94. Zheng H, Cheng B, Li Y, Li X, Chen X, Zhang Y. TREM2 in Alzheimer's disease: microglial survival and energy metabolism. Front Aging Neurosci. (2018) 10:395. doi: 10.3389/fnagi.2018.00395
95. Zhong L, Chen X-F, Wang T, Wang Z, Liao C, Wang Z, et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J Exp Med. (2017) 214:jem.20160844. doi: 10.1084/jem.20160844
96. Hickman SE, El Khoury J. TREM2 and the neuroimmunology of Alzheimer's disease. Biochem Pharmacol. (2014) 88:495–8. doi: 10.1016/j.bcp.2013.11.021
97. Calderon D, Bhaskar A, Knowles DA, Golan D, Raj T, Fu AQ, et al. Inferring relevant cell types for complex traits by using single-cell gene expression. Am J Hum Genet. (2017) 101:686–99. doi: 10.1016/j.ajhg.2017.09.009
98. Zhang B, Gaiteri C, Bodea L-G, Wang Z, McElwee J, Podtelezhnikov AA, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's Disease. Cell. (2013) 153:707–20. doi: 10.1016/j.cell.2013.03.030
99. Griciuc A, Patel S, Federico AN, Choi SH, Innes BJ, Oram MK, et al. TREM2 acts downstream of CD33 in modulating microglial pathology in Alzheimer's disease. Neuron. (2019) 103:820–35.e7. doi: 10.1016/j.neuron.2019.06.010
100. Bolós M, Llorens-Martín M, Perea JR, Jurado-Arjona J, Rábano A, Hernández F, et al. Absence of CX3CR1 impairs the internalization of tau by microglia. Mol Neurodegener. (2017) 12:59. doi: 10.1186/s13024-017-0200-1
101. Bolós M, Perea JR, Terreros-Roncal J, Pallas-Bazarra N, Jurado-Arjona J, Ávila J, Llorens-Martín M. Absence of microglial CX3CR1 impairs the synaptic integration of adult-born hippocampal granule neurons. Brain Behav Immun. (2018) 68:76–89. doi: 10.1016/j.bbi.2017.10.002
102. Perea JR, Lleó A, Alcolea D, Fortea J, Ávila J, Bolós M. Decreased CX3CL1 levels in the cerebrospinal fluid of patients with Alzheimer's disease. Front Neurosci. (2018) 12:609. doi: 10.3389/fnins.2018.00609
103. Oosterhof N, Chang IJ, Karimiani EG, Kuil LE, Jensen DM, Daza R, et al. Homozygous mutations in CSF1R cause a pediatric-onset leukoencephalopathy and can result in congenital absence of microglia. Am J Hum Genet. (2019) 104:936–47. doi: 10.1016/j.ajhg.2019.03.010
104. Guo L, Bertola DR, Takanohashi A, Saito A, Segawa Y, Yokota T, et al. Bi-allelic CSF1R mutations cause skeletal dysplasia of dysosteosclerosis-pyle disease spectrum and degenerative encephalopathy with brain malformation. Am J Hum Genet. (2019) 104:925–35. doi: 10.1016/j.ajhg.2019.03.004
105. Bhaskar K, Maphis N, Xu G, Varvel NH, Kokiko-Cochran ON, Weick JP, et al. Microglial derived tumor necrosis factor-α drives Alzheimer's disease-related neuronal cell cycle events. Neurobiol Dis. (2014) 62:273–85. doi: 10.1016/j.nbd.2013.10.007
106. Varvel NH, Bhaskar K, Kounnas MZ, Wagner SL, Yang Y, Lamb BT, et al. NSAIDs prevent, but do not reverse, neuronal cell cycle reentry in a mouse model of Alzheimer disease. J Clin Invest. (2009) 119:3692–702. doi: 10.1172/JCI39716
107. Babiloni C, Frisoni GB, Del Percio C, Zanetti O, Bonomini C, Cassetta E, et al. Ibuprofen treatment modifies cortical sources of EEG rhythms in mild Alzheimer's disease. Clin Neurophysiol. (2009) 120:709–18. doi: 10.1016/j.clinph.2009.02.005
108. Mhatre M, Floyd RA, Hensley K. Oxidative stress and neuroinflammation in Alzheimer's disease and amyotrophic lateral sclerosis: common links and potential therapeutic targets. J Alzheimers Dis. (2004) 6:147–57. doi: 10.3233/JAD-2004-6206
109. Bisht K, Sharma K, Tremblay M-È. Chronic stress as a risk factor for Alzheimer's disease: roles of microglia-mediated synaptic remodeling, inflammation, and oxidative stress. Neurobiol Stress. (2018) 9:9–21. doi: 10.1016/j.ynstr.2018.05.003
110. Caruso G, Caraci F, Jolivet RB. Pivotal role of carnosine in the modulation of brain cells activity: multimodal mechanism of action and therapeutic potential in neurodegenerative disorders. Prog Neurobiol. (2019) 175:35–53. doi: 10.1016/j.pneurobio.2018.12.004
111. McAlpine FE, Tansey MG. Neuroinflammation and tumor necrosis factor signaling in the pathophysiology of Alzheimer's disease. J Inflamm Res. (2008) 1:29–39. doi: 10.2147/JIR.S4397
112. Cantarella G, Di Benedetto G, Puzzo D, Privitera L, Loreto C, Saccone S, et al. Neutralization of TNFSF10 ameliorates functional outcome in a murine model of Alzheimer's disease. Brain. (2015) 138:203–16. doi: 10.1093/brain/awu318
113. Decourt B, Lahiri D, Sabbagh M. Targeting tumor necrosis factor alpha for Alzheimer's disease. Curr Alzheimer Res. (2016) 14:412–25. doi: 10.2174/1567205013666160930110551
114. Ekert JO, Gould RL, Reynolds G, Howard RJ. TNF alpha inhibitors in Alzheimer's disease: a systematic review. Int J Geriatr Psychiatry. (2018) 33:688–94. doi: 10.1002/gps.4871
115. Iulita MF, Caraci F, Cuello AC. A link between nerve growth factor metabolic deregulation and amyloid-β-driven inflammation in down syndrome. CNS Neurol Disord Drug Targets. (2016) 15:434–47. doi: 10.2174/1871527315666160321104916
116. Liao Y-F, Wang B-J, Cheng H-T, Kuo L-H, Wolfe MS. Tumor necrosis factor-α, interleukin-1β, and interferon-γ stimulate γ-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J Biol Chem. (2004) 279:49523–32. doi: 10.1074/jbc.M402034200
117. Koenigsknecht-Talboo J. Microglial phagocytosis induced by fibrillar -amyloid and IgGs are differentially regulated by proinflammatory cytokines. J Neurosci. (2005) 25:8240–9. doi: 10.1523/JNEUROSCI.1808-05.2005
118. Clark I, Atwood C, Bowen R, Paz-Filho G, Vissel B. Tumor necrosis factor-induced cerebral insulin resistance in Alzheimer's disease links numerous treatment rationales. Pharmacol Rev. (2012) 64:1004–26. doi: 10.1124/pr.112.005850
119. Lourenco M V, Clarke JR, Frozza RL, Bomfim TR, Forny-Germano L, Batista AF, et al. TNF-α mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer's β-Amyloid oligomers in mice and monkeys. Cell Metab. (2013) 18:831–43. doi: 10.1016/j.cmet.2013.11.002
120. Clarke JR, Lyra e Silva NM, Figueiredo CP, Frozza RL, Ledo JH, Beckman D, et al. Alzheimer-associated Aβ oligomers impact the central nervous system to induce peripheral metabolic deregulation. EMBO Mol Med. (2015) 7:190–210. doi: 10.15252/emmm.201404183
121. Ueberham U, Ueberham E, Gruschka H, Arendt T. Altered subcellular location of phosphorylated smads in Alzheimer's disease. Eur J Neurosci. (2006) 24:2327–34. doi: 10.1111/j.1460-9568.2006.05109.x
122. Tesseur I, Zou K, Esposito L, Bard F, Berber E, Can J Van, et al. Deficiency in neuronal TGF-β signaling promotes neurodegeneration and Alzheimer's pathology. J Clin Invest. (2006) 116:3060–9. doi: 10.1172/JCI27341
123. Caraci F, Spampinato S, Sortino MA, Bosco P, Battaglia G, Bruno V, et al. Dysfunction of TGF-β1 signaling in Alzheimer's disease: perspectives for neuroprotection. Cell Tissue Res. (2012) 347:291–301. doi: 10.1007/s00441-011-1230-6
124. Liu C, Liu Q, Song L, Gu Y, Jie J, Bai X, et al. Dab2 attenuates brain injury in APP/PS1 mice via targeting transforming growth factor-beta/SMAD signaling. Neural Regen Res. (2014) 9:41–50. doi: 10.4103/1673-5374.125328
125. Chen J-H, Ke K-F, Lu J-H, Qiu Y-H, Peng Y-P. Protection of TGF-β1 against Neuroinflammation and Neurodegeneration in Aβ1–42-Induced Alzheimer's Disease Model Rats. PLoS ONE. (2015) 10:e0116549. doi: 10.1371/journal.pone.0116549
126. Tichauer JE, von Bernhardi R. Transforming growth factor-β stimulates β amyloid uptake by microglia through Smad3-dependent mechanisms. J Neurosci Res. (2012) 90:1970–80. doi: 10.1002/jnr.23082
127. Caraci F, Gulisano W, Guida CA, Impellizzeri AAR, Drago F, Puzzo D, et al. A key role for TGF-β1 in hippocampal synaptic plasticity and memory. Sci Rep. (2015) 5:11252. doi: 10.1038/srep11252
128. Herrup K. The involvement of cell cycle events in the pathogenesis of Alzheimer's disease. Alzheimers Res Ther. (2010) 2:13. doi: 10.1186/alzrt37
129. Tong L, Balazs R, Soiampornkul R, Thangnipon W, Cotman CW. Interleukin-1β impairs brain derived neurotrophic factor-induced signal transduction. Neurobiol Aging. (2008) 29:1380–93. doi: 10.1016/j.neurobiolaging.2007.02.027
130. Iulita MF, Bistué Millón MB, Pentz R, Aguilar LF, Do Carmo S, Allard S, et al. Differential deregulation of NGF and BDNF neurotrophins in a transgenic rat model of Alzheimer's disease. Neurobiol Dis. (2017) 108:307–23. doi: 10.1016/j.nbd.2017.08.019
131. Song J-H, Yu J-T, Tan L. Brain-Derived neurotrophic factor in Alzheimer's disease: risk, mechanisms, and therapy. Mol Neurobiol. (2015) 52:1477–93. doi: 10.1007/s12035-014-8958-4
132. Iulita MF, Do Carmo S, Ower AK, Fortress AM, Flores Aguilar L, Hanna M, et al. Nerve growth factor metabolic dysfunction in down's syndrome brains. Brain. (2014) 137:860–72. doi: 10.1093/brain/awt372
133. Cuello AC, Pentz R, Hall H. The brain NGF metabolic pathway in health and in Alzheimer's pathology. Front Neurosci. (2019) 13:62. doi: 10.3389/fnins.2019.00062
134. Bruno MA, Leon WC, Fragoso G, Mushynski WE, Almazan G, Cuello AC. Amyloid β-Induced nerve growth factor dysmetabolism in Alzheimer disease. J Neuropathol Exp Neurol. (2009) 68:857–69. doi: 10.1097/NEN.0b013e3181aed9e6
135. Bruno MA, Mufson EJ, Wuu J, Cuello AC. Increased matrix metalloproteinase 9 activity in mild cognitive impairment. J Neuropathol Exp Neurol. (2009) 68:1309–18. doi: 10.1097/NEN.0b013e3181c22569
136. Cheng X, Shen Y, Li R. Targeting TNF: a therapeutic strategy for Alzheimer's disease. Drug Discov Today. (2014) 19:1822–7. doi: 10.1016/j.drudis.2014.06.029
137. Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-α. Nature. (2006) 440:1054–9. doi: 10.1038/nature04671
138. Wang Q, Wu J, Rowan MJ, Anwyl R. β-amyloid inhibition of long-term potentiation is mediated via tumor necrosis factor. Eur J Neurosci. (2005) 22:2827–32. doi: 10.1111/j.1460-9568.2005.04457.x
139. Shamim D, Laskowski M. Inhibition of inflammation mediated through the tumor necrosis factor α biochemical pathway can lead to favorable outcomes in Alzheimer disease. J Cent Nerv Syst Dis. (2017) 9:117957351772251. doi: 10.1177/1179573517722512
140. Mrak R. Interleukin-1, neuroinflammation, and Alzheimer's disease. Neurobiol Aging. (2001) 22:903–8. doi: 10.1016/S0197-4580(01)00287-1
141. Schmid AW, Lynch MA, Herron CE. The effects of IL-1 receptor antagonist on beta amyloid mediated depression of LTP in the rat CA1 in vivo. Hippocampus. (2009) 19:670–6. doi: 10.1002/hipo.20542
142. vom Berg J, Prokop S, Miller KR, Obst J, Kälin RE, Lopategui-Cabezas I, et al. Inhibition of IL-12/IL-23 signaling reduces Alzheimer's disease–like pathology and cognitive decline. Nat Med. (2012) 18:1812–9. doi: 10.1038/nm.2965
143. Guerreiro RJ, Santana I, Brás JM, Santiago B, Paiva A, Oliveira C. Peripheral inflammatory cytokines as biomarkers in Alzheimer's disease and mild cognitive impairment. Neurodegener Dis. (2007) 4:406–12. doi: 10.1159/000107700
144. Griffin WST. Neuroinflammatory cytokine signaling and Alzheimer's disease. N Engl J Med. (2013) 368:770–1. doi: 10.1056/NEJMcibr1214546
145. Kiyota T, Ingraham KL, Swan RJ, Jacobsen MT, Andrews SJ, Ikezu T. AAV serotype 2/1-mediated gene delivery of anti-inflammatory interleukin-10 enhances neurogenesis and cognitive function in APP+PS1 mice. Gene Ther. (2012) 19:724–33. doi: 10.1038/gt.2011.126
146. Chakrabarty P, Li A, Ceballos-Diaz C, Eddy JA, Funk CC, Moore B, et al. IL-10 Alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron. (2015) 85:519–33. doi: 10.1016/j.neuron.2014.11.020
147. Wyss-Coray T, Yan F, Lin AH-T, Lambris JD, Alexander JJ, Quigg RJ, et al. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer's mice. Proc Natl Acad Sci USA. (2002) 99:10837–42. doi: 10.1073/pnas.162350199
148. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. (2011) 333:1456–8. doi: 10.1126/science.1202529
149. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. (2012) 74:691–705. doi: 10.1016/j.neuron.2012.03.026
150. Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. (2012) 35:369–89. doi: 10.1146/annurev-neuro-061010-113810
151. Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. (2016) 352:712–6. doi: 10.1126/science.aad8373
152. McGeer PL, McGeer EG. NSAIDs and Alzheimer disease: epidemiological, animal model and clinical studies. Neurobiol Aging. (2007) 28:639–47. doi: 10.1016/j.neurobiolaging.2006.03.013
153. Cole GM, Frautschy SA. Mechanisms of action of non-steroidal anti-inflammatory drugs for the prevention of Alzheimer's disease. CNS Neurol Disord Drug Targets. (2010) 9:140–8. doi: 10.2174/187152710791011991
154. Kotilinek LA, Westerman MA, Wang Q, Panizzon K, Lim GP, Simonyi A, et al. Cyclooxygenase-2 inhibition improves amyloid-beta-mediated suppression of memory and synaptic plasticity. Brain. (2008) 131:651–64. doi: 10.1093/brain/awn008
155. Woodling NS, Colas D, Wang Q, Minhas P, Panchal M, Liang X, et al. Cyclooxygenase inhibition targets neurons to prevent early behavioural decline in Alzheimer's disease model mice. Brain. (2016) 139:2063–81. doi: 10.1093/brain/aww117
156. Malkki H. NSAIDs protect neurons and preserve memory in a mouse model of AD. Nat Rev Neurol. (2016) 12:370–1. doi: 10.1038/nrneurol.2016.79
157. Hampel H, Goetzl EJ, Kapogiannis D, Lista S, Vergallo A. Biomarker-drug and liquid biopsy co-development for disease staging and targeted therapy: cornerstones for Alzheimer's precision medicine and pharmacology. Front Pharmacol. (2019) 10:310. doi: 10.3389/fphar.2019.00310
158. Nicoll JAR, Buckland GR, Harrison CH, Page A, Harris S, Love S, et al. Persistent neuropathological effects 14 years following amyloid-β immunization in Alzheimer's disease. Brain. (2019) 142:2113–26. doi: 10.1093/brain/awz142
159. Hampel H, Lista S, Neri C, Vergallo A. Time for the systems-level integration of aging: resilience enhancing strategies to prevent Alzheimer's disease. Prog Neurobiol. (2019) 181:101662. doi: 10.1016/j.pneurobio.2019.101662
160. Shen X-N, Niu L-D, Wang Y-J, Cao X-P, Liu Q, Tan L, et al. Inflammatory markers in Alzheimer's disease and mild cognitive impairment: a meta-analysis and systematic review of 170 studies. J Neurol Neurosurg Psychiatry. (2019) 90:590–8. doi: 10.1136/jnnp-2018-319148
161. Lai KSP, Liu CS, Rau A, Lanctôt KL, Köhler CA, Pakosh M, et al. Peripheral inflammatory markers in Alzheimer's disease: a systematic review and meta-analysis of 175 studies. J Neurol Neurosurg Psychiatry. (2017) 88:876–82. doi: 10.1136/jnnp-2017-316201
162. Su C, Zhao K, Xia H, Xu Y. Peripheral inflammatory biomarkers in Alzheimer's disease and mild cognitive impairment: a systematic review and meta-analysis. Psychogeriatrics. (2019) 19:300–9. doi: 10.1111/psyg.12403
163. Swardfager W, Lanctôt K, Rothenburg L, Wong A, Cappell J, Herrmann N. A meta-analysis of cytokines in Alzheimer's disease. Biol Psychiatry. (2010) 68:930–41. doi: 10.1016/j.biopsych.2010.06.012
164. Saleem M, Herrmann N, Swardfager W, Eisen R, Lanctôt KL. Inflammatory markers in mild cognitive impairment: a meta-Analysis. J Alzheimer's Dis. (2015) 47:669–79. doi: 10.3233/JAD-150042
165. Olsson B, Lautner R, Andreasson U, Öhrfelt A, Portelius E, Bjerke M, Hölttä M, et al. CSF and blood biomarkers for the diagnosis of Alzheimer's disease: a systematic review and meta-analysis. Lancet Neurol. (2016) 15:673–84. doi: 10.1016/S1474-4422(16)00070-3
166. Dhiman K, Blennow K, Zetterberg H, Martins RN, Gupta VB. Cerebrospinal fluid biomarkers for understanding multiple aspects of Alzheimer's disease pathogenesis. Cell Mol Life Sci. (2019) 76:1833–63. doi: 10.1007/s00018-019-03040-5
167. Baldacci F, Lista S, Cavedo E, Bonuccelli U, Hampel H. Diagnostic function of the neuroinflammatory biomarker YKL-40 in Alzheimer's disease and other neurodegenerative diseases. Expert Rev Proteomics. (2017) 14:285–99. doi: 10.1080/14789450.2017.1304217
168. Baldacci F, Lista S, Palermo G, Giorgi FS, Vergallo A, Hampel H. The neuroinflammatory biomarker YKL-40 for neurodegenerative diseases: advances in development. Expert Rev Proteomics. (2019) 16:593–600. doi: 10.1080/14789450.2019.1628643
169. Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. (2015) 16:358–72. doi: 10.1038/nrn3880
170. Hampel H, Vergallo A, Afshar M, Akman-Anderson L, Arenas J, Benda N, et al. Blood-based systems biology biomarkers for next-generation clinical trials in Alzheimer's disease? Dialogues Clin Neurosci. (2019) 21:177–91. doi: 10.31887/DCNS.2019.21.2/hhampel
171. Hampel H, Vergallo A, Giorgi FS, Kim SH, Depypere H, Graziani M, et al. Precision medicine and drug development in Alzheimer's disease: the importance of sexual dimorphism and patient stratification. Front Neuroendocrinol. (2018) 50:31–51. doi: 10.1016/j.yfrne.2018.06.001
172. Baldacci F, Lista S, O'Bryant SE, Ceravolo R, Toschi N, Hampel H. Blood-based biomarker screening with agnostic biological definitions for an accurate diagnosis within the dimensional spectrum of neurodegenerative diseases. In: Perneczky R, editor. Methods in Molecular Biology. New York, NY: Humana Press Inc (2016). p. 139–155. doi: 10.1007/978-1-4939-7704-8_9
173. Tarkowski E, Liljeroth A-M, Minthon L, Tarkowski A, Wallin A, Blennow K. Cerebral pattern of pro- and anti-inflammatory cytokines in dementias. Brain Res Bull. (2003) 61:255–60. doi: 10.1016/S0361-9230(03)00088-1
174. Teixeira AL, Reis HJ, Coelho FM, Carneiro DS, Teixeira MM, Vieira LB, et al. All-or-Nothing type biphasic cytokine production of human lymphocytes after exposure to Alzheimer's β-Amyloid Peptide. Biol Psychiatry. (2008) 64:891–5. doi: 10.1016/j.biopsych.2008.07.019
175. Hesse R, Wahler A, Gummert P, Kirschmer S, Otto M, Tumani H, et al. Decreased IL-8 levels in CSF and serum of AD patients and negative correlation of MMSE and IL-1β. BMC Neurol. (2016) 16:185. doi: 10.1186/s12883-016-0707-z
176. Leung R, Proitsi P, Simmons A, Lunnon K, Güntert A, Kronenberg D, et al. Inflammatory Proteins in Plasma Are Associated with Severity of Alzheimer's Disease. PLoS ONE. (2013) 8:e64971. doi: 10.1371/journal.pone.0064971
177. Sun Y-X, Minthon L, Wallmark A, Warkentin S, Blennow K, Janciauskiene S. Inflammatory Markers in Matched Plasma and Cerebrospinal Fluid from Patients with Alzheimer's Disease. Dement Geriatr Cogn Disord. (2003) 16:136–44. doi: 10.1159/000071001
178. Bermejo P, Martín-Aragón S, Benedí J, Susín C, Felici E, Gil P, et al. Differences of peripheral inflammatory markers between mild cognitive impairment and Alzheimer's disease. Immunol Lett. (2008) 117:198–202. doi: 10.1016/j.imlet.2008.02.002
179. Winblad B, Palmer K, Kivipelto M, Jelic V, Fratiglioni L, Wahlund L-O, et al. Mild cognitive impairment–beyond controversies, toward a consensus: report of the international working group on mild cognitive impairment. J Intern Med. (2004) 256:240–6. doi: 10.1111/j.1365-2796.2004.01380.x
180. Dubois B, Feldman HH, Jacova C, Cummings JL, DeKosky ST, Barberger-Gateau P, et al. Revising the definition of Alzheimer's disease: a new lexicon. Lancet Neurol. (2010) 9:1118–27. doi: 10.1016/S1474-4422(10)70223-4
182. Ikeda U, Ito T, Shimada K. Interleukin-6 and acute coronary syndrome. Clin Cardiol. (2001) 24:701–4. doi: 10.1002/clc.4960241103
183. Villegas-Llerena C, Phillips A, Garcia-Reitboeck P, Hardy J, Pocock JM. Microglial genes regulating neuroinflammation in the progression of Alzheimer's disease. Curr Opin Neurobiol. (2016) 36:74–81. doi: 10.1016/j.conb.2015.10.004
184. Yang Z, Zan Y, Zheng X, Hai W, Chen K, Huang Q, et al. Dynamic FDG-PET imaging to differentiate malignancies from inflammation in subcutaneous and in situ mouse model for Non-Small Cell Lung Carcinoma (NSCLC). PLoS ONE. (2015) 10:e0139089. doi: 10.1371/journal.pone.0139089
185. Cosenza-Nashat M, Zhao M-L, Suh H-S, Morgan J, Natividad R, Morgello S, et al. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol Appl Neurobiol. (2009) 35:306–28. doi: 10.1111/j.1365-2990.2008.01006.x
186. Edison P, Archer HA, Gerhard A, Hinz R, Pavese N, Turkheimer FE, et al. Microglia, amyloid, and cognition in Alzheimer's disease: an [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis. (2008) 32:412–9. doi: 10.1016/j.nbd.2008.08.001
187. Schuitemaker A, Kropholler MA, Boellaard R, van der Flier WM, Kloet RW, van der Doef TF, et al. Microglial activation in Alzheimer's disease: an (R)-[11C]PK11195 positron emission tomography study. Neurobiol Aging. (2013) 34:128–36. doi: 10.1016/j.neurobiolaging.2012.04.021
188. Stefaniak J, O'Brien J. Imaging of neuroinflammation in dementia: a review. J Neurol Neurosurg Psychiatry. (2015) 87:jnnp-2015-311336. doi: 10.1136/jnnp-2015-311336
189. Passamonti L, Rodríguez PV, Hong YT, Allinson KSJ, Bevan-Jones WR, Williamson D, et al. [11C]PK11195 binding in Alzheimer disease and progressive supranuclear palsy. Neurology. (2018) 90:e1989–96. doi: 10.1212/WNL.0000000000005610
190. Fan Z, Aman Y, Ahmed I, Chetelat G, Landeau B, Ray Chaudhuri K, et al. Influence of microglial activation on neuronal function in Alzheimer's and parkinson's disease dementia. Alzheimer's Dement. (2015) 11:608–21.e7. doi: 10.1016/j.jalz.2014.06.016
191. Fernández-Botrán R, Ahmed Z, Crespo FA, Gatenbee C, Gonzalez J, Dickson DW, et al. Cytokine expression and microglial activation in progressive supranuclear palsy. Parkinsonism Relat Disord. (2011) 17:683–8. doi: 10.1016/j.parkreldis.2011.06.007
192. Owen DRJ, Gunn RN, Rabiner EA, Bennacef I, Fujita M, Kreisl WC, et al. Mixed-affinity binding in humans with 18-kDa translocator protein ligands. J Nucl Med. (2011) 52:24–32. doi: 10.2967/jnumed.110.079459
193. Lavisse S, Guillermier M, Herard A-S, Petit F, Delahaye M, Van Camp N, et al. Reactive astrocytes overexpress TSPO and are detected by TSPO positron emission tomography imaging. J Neurosci. (2012) 32:10809–18. doi: 10.1523/JNEUROSCI.1487-12.2012
194. Airas L, Dickens AM, Elo P, Marjamaki P, Johansson J, Eskola O, et al. In Vivo pet imaging demonstrates diminished microglial activation after fingolimod treatment in an animal model of multiple sclerosis. J Nucl Med. (2015) 56:305–10. doi: 10.2967/jnumed.114.149955
195. Rusjan PM, Wilson AA, Bloomfield PM, Vitcu I, Meyer JH, Houle S, et al. Quantitation of translocator protein binding in human brain with the novel radioligand [ 18 F]-FEPPA and positron emission tomography. J Cereb Blood Flow Metab. (2011) 31:1807–16. doi: 10.1038/jcbfm.2011.55
196. Morris E, Chalkidou A, Hammers A, Peacock J, Summers J, Keevil S. Diagnostic accuracy of 18F amyloid PET tracers for the diagnosis of Alzheimer's disease: a systematic review and meta-analysis. Eur J Nucl Med Mol Imaging. (2016) 43:374–85. doi: 10.1007/s00259-015-3228-x
197. Saint-Aubert L, Lemoine L, Chiotis K, Leuzy A, Rodriguez-Vieitez E, Nordberg A. Tau PET imaging: present and future directions. Mol Neurodegener. (2017) 12:19. doi: 10.1186/s13024-017-0162-3
198. Passamonti L, Vázquez RP, Hong YT, Allinson KSJ, Williamson D, Borchert RJ, et al. 18 F-AV-1451 positron emission tomography in Alzheimer's disease and progressive supranuclear palsy. Brain. (2017) 140:aww340. doi: 10.1093/brain/aww340
199. Marik J, Ogasawara A, Martin-McNulty B, Ross J, Flores JE, Gill HS, et al. PET of glial metabolism using 2-18F-Fluoroacetate. J Nucl Med. (2009) 50:982–90. doi: 10.2967/jnumed.108.057356
200. Tronel C, Largeau B, Santiago Ribeiro M, Guilloteau D, Dupont A-C, Arlicot N. Molecular targets for PET imaging of activated microglia: the current situation and future expectations. Int J Mol Sci. (2017) 18:802. doi: 10.3390/ijms18040802
201. Shukuri M, Mawatari A, Ohno M, Suzuki M, Doi H, Watanabe Y, et al. Detection of cyclooxygenase-1 in activated microglia during amyloid plaque progression: PET studies in alzheimers disease model mice. J Nucl Med. (2016) 57:291–6. doi: 10.2967/jnumed.115.166116
202. Benito C, Tolón RM, Pazos MR, Núñez E, Castillo AI, Romero J. Cannabinoid CB 2 receptors in human brain inflammation. Br J Pharmacol. (2008) 153:277–85. doi: 10.1038/sj.bjp.0707505
203. Palazuelos J, Aguado T, Pazos MR, Julien B, Carrasco C, Resel E, et al. Microglial CB2 cannabinoid receptors are neuroprotective in Huntington's disease excitotoxicity. Brain. (2009) 132:3152–64. doi: 10.1093/brain/awp239
204. Slavik R, Herde AM, Bieri D, Weber M, Schibli R, Krämer SD, et al. Synthesis, radiolabeling and evaluation of novel 4-oxo-quinoline derivatives as PET tracers for imaging cannabinoid type 2 receptor. Eur J Med Chem. (2015) 92:554–64. doi: 10.1016/j.ejmech.2015.01.028
205. Territo PR, Meyer JA, Peters JS, Riley AA, McCarthy BP, Gao M, et al. Characterization of 11 C-GSK1482160 for targeting the P2X7 receptor as a biomarker for neuroinflammation. J Nucl Med. (2017) 58:458–65. doi: 10.2967/jnumed.116.181354
206. Zhang C, Wang Y, Wang D, Zhang J, Zhang F. NSAID exposure and risk of Alzheimer's disease: an updated meta-analysis from cohort studies. Front Aging Neurosci. (2018) 10:83. doi: 10.3389/fnagi.2018.00083
207. Sainati SM, Ingram DM, Talwalker S, Geis G. Results of a double-blind, randomized, placebo-controlled study of celecoxib in the treatment of progression of Alzheimer's disease. In: Sixth International Stockholm/Springfield Symposium on Advances in Alzheimer Therapy. Sweden: Stockholm (2000).
208. Soininen H, West C, Robbins J, Niculescu L. Long-term efficacy and safety of celecoxib in Alzheimer's Disease. Dement Geriatr Cogn Disord. (2007) 23:8–21. doi: 10.1159/000096588
209. Bain A. Alzheimer disease: dapsone phase 2 trial results reported. Immune Network Ltd. Press release, October 15, 2002
210. Scharf S, Mander A, Ugoni A, Vajda F, Christophidis NP. A double-blind, placebo-controlled trial of diclofenac/misoprostol in Alzheimer's disease. Neurology. (1999) 53:197–201. doi: 10.1212/WNL.53.1.197
211. Van Gool WA, Weinstein HC, Scheltens P, Walstra GJ. Effect of hydroxychloroquine on progression of dementia in early Alzheimer's disease: an 18-month randomised, double-blind, placebo-controlled study. Lancet. (2001) 358:455–60. doi: 10.1016/S0140-6736(01)05623-9
212. Pasqualetti P, Bonomini C, Dal Forno G, Paulon L, Sinforiani E, Marra C, et al. A randomized controlled study on effects of ibuprofen on cognitive progression of Alzheimer's disease. Aging Clin Exp Res. (2009) 21:102–10. doi: 10.1007/BF03325217
213. Rogers J, Kirby LC, Hempelman SR, Berry DL, Mcgeer PL, Kaszniak AW, et al. Clinical trial of indomethacin in Alzheimer's disease. Neurology. (1993) 43:1609–11. doi: 10.1212/WNL.43.8.1609
214. de Jong D, Jansen R, Hoefnagels W, Jellesma-Eggenkamp M, Verbeek M, Borm G, et al. No effect of one-year treatment with indomethacin on Alzheimer's disease progression: a randomized controlled trial. PLoS ONE. (2008) 3:e1475. doi: 10.1371/journal.pone.0001475
215. Aisen PS, Schafer KA, Grundman M, Pfeiffer E, Sano M, Davis KL, et al. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression. JAMA. (2003) 289:2819. doi: 10.1001/jama.289.21.2819
216. Aisen PS, Schmeidler J, Pasinetti GM. Randomized pilot study of nimesulide treatment in Alzheimer's disease. Neurology. (2002) 58:1050–4. doi: 10.1212/WNL.58.7.1050
217. Aisen PS, Davis KL, Berg JD, Schafer K, Campbell K, Thomas RG, et al. A randomized controlled trial of prednisone in Alzheimer's disease. Neurology. (2000) 54:588. doi: 10.1212/WNL.54.3.588
218. Reines SA, Block GA, Morris JC, Liu G, Nessly ML, Lines CR, et al. Rofecoxib: no effect on Alzheimer's disease in a 1-year, randomized, blinded, controlled study. Neurology. (2004) 62:66–71. doi: 10.1212/WNL.62.1.66
219. Wilcock GK, Black SE, Hendrix SB, Zavitz KH, Swabb EA, Laughlin MA. Tarenflurbil Phase II Study investigators. Efficacy and safety of tarenflurbil in mild to moderate Alzheimer's disease: a randomised phase II trial. Lancet Neurol. (2008) 7:483–93. doi: 10.1016/S1474-4422(08)70090-5
220. Green RC. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild alzheimer disease a randomized controlled trial. JAMA. (2009) 302:2557–64. doi: 10.1001/jama.2009.1866
221. Wilcock GK, Black SE, Balch AH, Amato DA, Beelen AP, Schneider LS, et al. Safety and efficacy of tarenflurbil in subjects with mild Alzheimer's disease: results from an 18-month international multi-center Phase 3 trial. Alzheimers Dement. (2009) 5(Suppl. 1):P86. doi: 10.1016/j.jalz.2009.05.220
222. Small GW, Siddarth P, Silverman DH, Ercoli LM, Miller KJ, Lavretsky H, et al. Cognitive and cerebral metabolic effects of celecoxib versus placebo in people with age-related memory loss: randomized controlled study. Am J Geriatr Psychiatry. (2008) 16:999–1009. doi: 10.1097/JGP.0b013e31818cd3a4
223. Thal LJ, Ferris SH, Kirby L, Block GA, Lines CR, Yuen E, et al. A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology. (2005) 30:1204–15. doi: 10.1038/sj.npp.1300690
224. Gómez-Isla T, Blesa R, Boada M, Clarimón J, Del Ser T, Domenech G. A randomized, double-blind, placebo controlled-trial of triflusal in mild cognitive impairment: the TRIMCI study. Alzheimer Dis Assoc Disord. (2008) 22:21–9. doi: 10.1097/WAD.0b013e3181611024
225. Lyketsos CG, Breitner JCS, Green RC, Martin BK, Meinert C, Piantadosi S, et al. Naproxen and celecoxib do not prevent AD in early results from a randomized controlled trial. Neurology. (2007) 68:1800–8. doi: 10.1212/01.wnl.0000260269.93245.d2
226. Meyer P-F, Tremblay-Mercier J, Leoutsakos J, Madjar C, Lafaille-Maignan M-É, Savard M, et al. INTREPAD: a randomized trial of naproxen to slow progress of presymptomatic Alzheimer disease. Neurology. (2019) 92:e2070–80. doi: 10.1212/WNL.0000000000007232
227. Alzheimer's Disease Anti-inflammatory Prevention Trial Research Group. Results of a follow-up study to the randomized Alzheimer's Disease Anti-Inflammatory Prevention Trial (ADAPT). Alzheimers Dement. (2013) 9:714–23. doi: 10.1016/j.jalz.2012.11.012
228. Martin BK, Szekely C, Brandt J, Piantadosi S, Breitner JCS, Craft S, et al. Cognitive function over time in the Alzheimer's Disease Anti-inflammatory Prevention Trial (ADAPT). Arch Neurol. (2008) 65:896–905. doi: 10.1001/archneur.2008.65.7.nct70006
229. Szekely CA, Thorne JE, Zandi PP, Ek M, Messias E, Breitner JCS, et al. Nonsteroidal Anti-inflammatory drugs for the prevention of Alzheimer's disease: a systematic review. Neuroepidemiology. (2004) 23:159–69. doi: 10.1159/000078501
230. in 't Veld BA, Ruitenberg A, Hofman A, Launer LJ, van Duijn CM, Stijnen T, et al. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's Disease. N Engl J Med. (2001) 345:1515–21. doi: 10.1056/NEJMoa010178
231. Vlad SC, Miller DR, Kowall NW, Felson DT. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology. (2008) 70:1672–7. doi: 10.1212/01.wnl.0000311269.57716.63
232. de Craen AJM. Meta-Analysis of nonsteroidal antiinflammatory drug use and risk of dementia. Am J Epidemiol. (2005) 161:114–20. doi: 10.1093/aje/kwi029
233. Wichmann MA, Cruickshanks KJ, Carlsson CM, Chappell R, Fischer ME, Klein BEK, et al. NSAID use and incident cognitive impairment in a population-based cohort. Alzheimer Dis Assoc Disord. (2016) 30:105–12. doi: 10.1097/WAD.0000000000000098
234. Rogers MB. Antibodies against microglial receptors TREM2 and CD33 head to trials | ALZFORUM. Alzforum (2019) Available at: https://www.alzforum.org/news/conference-coverage/antibodies-against-microglial-receptors-trem2-and-cd33-head-trials (accessed January 30, 2020).
235. Steed PM, Tansey MG, Zalevsky J, Zhukovsky EA, Desjarlais JR, Szymkowski DE, et al. Inactivation of TNF signaling by rationally designed dominant-negative TNF variants. Science. (2003) 301:1895–8. doi: 10.1126/science.1081297
236. Zalevsky J, Secher T, Ezhevsky SA, Janot L, Steed PM, O'Brien C, et al. Dominant-negative inhibitors of soluble TNF attenuate experimental arthritis without suppressing innate immunity to infection. J Immunol. (2007) 179:1872–83. doi: 10.4049/jimmunol.179.3.1872
237. Yli-Karjanmaa M, Larsen KS, Fenger CD, Kristensen LK, Martin NA, Jensen PT, et al. TNF deficiency causes alterations in the spatial organization of neurogenic zones and alters the number of microglia and neurons in the cerebral cortex. Brain Behav Immun. (2019) 82:279–97. doi: 10.1016/j.bbi.2019.08.195
238. MacPherson KP, Sompol P, Kannarkat GT, Chang J, Sniffen L, Wildner ME, et al. Peripheral administration of the soluble TNF inhibitor XPro1595 modifies brain immune cell profiles, decreases beta-amyloid plaque load, and rescues impaired long-term potentiation in 5xFAD mice. Neurobiol Dis. (2017) 102:81–95. doi: 10.1016/j.nbd.2017.02.010
239. Cavanagh C, Tse YC, Nguyen HB, Krantic S, Breitner JC, Quirion R, et al. Inhibiting tumor necrosis factor-α before amyloidosis prevents synaptic deficits in an Alzheimer's disease model. Neurobiol Aging. (2016) 47:41–9. doi: 10.1016/j.neurobiolaging.2016.07.009
240. McAlpine FE, Lee JK, Harms AS, Ruhn KA, Blurton-Jones M, Hong J, et al. Inhibition of soluble TNF signaling in a mouse model of Alzheimer's disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol Dis. (2009) 34:163–77. doi: 10.1016/j.nbd.2009.01.006
241. Sama DM, Mohmmad Abdul H, Furman JL, Artiushin IA, Szymkowski DE, Scheff SW, et al. Inhibition of soluble tumor necrosis factor ameliorates synaptic alterations and Ca2+ dysregulation in aged rats. PLoS ONE. (2012) 7:e38170. doi: 10.1371/journal.pone.0038170
242. Li HQ, Chen C, Dou Y, Wu HJ, Liu YJ, Lou HF, et al. P2Y4 receptor-mediated pinocytosis contributes to amyloid beta-induced self-uptake by microglia. Mol Cell Biol. (2013) 33:4282–93. doi: 10.1128/MCB.00544-13
243. Miller RG, Block G, Katz JS, Barohn RJ, Gopalakrishnan V, Cudkowicz M, et al. Randomized phase 2 trial of NP001-a novel immune regulator: Safetyearly efficacy in ALS. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e100. doi: 10.1212/NXI.0000000000000100
244. Etminan M. Effect of non-steroidal anti-inflammatory drugs on risk of Alzheimer's disease: systematic review and meta-analysis of observational studies. BMJ. (2003) 327:128. doi: 10.1136/bmj.327.7407.128
245. O'Bryant SE. A serum protein–based algorithm for the detection of Alzheimer Disease. Arch Neurol. (2010) 67:1077–81. doi: 10.1001/archneurol.2010.215
246. O'Bryant SE, Xiao G, Barber R, Huebinger R, Wilhelmsen K, Edwards M, et al. A blood-based screening tool for Alzheimer's disease that spans serum and plasma: findings from TARC and ADNI. PLoS ONE. (2011) 6:e28092. doi: 10.1371/journal.pone.0028092
247. O'Bryant SE, Xiao G, Zhang F, Edwards M, German DC, Yin X, et al. Validation of a serum screen for Alzheimer's disease across assay platforms, species, and tissues. J Alzheimer's Dis. (2014) 42:1325–35. doi: 10.3233/JAD-141041
248. O'Bryant SE, Zhang F, Johnson LA, Hall J, Edwards M, Grammas P, et al. A precision medicine model for targeted NSAID therapy in Alzheimer's Disease. J Alzheimer's Dis. (2018) 66:97–104. doi: 10.3233/JAD-180619
249. Walsh DM1, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. (2002) 416:535–9. doi: 10.1038/416535a
250. Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, et al. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. (1999) 155:853–62. doi: 10.1016/S0002-9440(10)65184-X
251. McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, et al. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. (1999) 46:860–6. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m
252. Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. (2008) 14:837–42. doi: 10.1038/nm1782
253. O'Nuallain B, Freir DB, Nicoll AJ, Risse E, Ferguson N, Herron CE, et al. Amyloid beta-protein dimers rapidly form stable synaptotoxic protofibrils. J Neurosci. (2010) 30:14411–9. doi: 10.1523/JNEUROSCI.3537-10.2010
254. Gouwens LK, Makoni NJ, Rogers VA, Nichols MR. Amyloid-β42 protofibrils are internalized by microglia more extensively than monomers. Brain Res. (2016) 1648:485–95. doi: 10.1016/j.brainres.2016.08.016
255. Paranjape GS, Gouwens LK, Osborn DC, Nichols MR. Isolated Amyloid-β(1–42) Protofibrils, but not isolated fibrils, are robust stimulators of microglia. ACS Chem Neurosci. (2012) 3:302–11. doi: 10.1021/cn2001238
256. Söllvander S, Nikitidou E, Gallasch L, Zyśk M, Söderberg L, Sehlin D, et al. The Aβ protofibril selective antibody mAb158 prevents accumulation of Aβ in astrocytes and rescues neurons from Aβ-induced cell death. J Neuroinflammation. (2018) 15:98. doi: 10.1186/s12974-018-1134-4
257. Englund H, Sehlin D, Johansson AS, Nilsson LN, Gellerfors P, Paulie S, et al. Sensitive ELISA detection of amyloid-beta protofibrils in biological samples. J Neurochem. (2007) 103:334–45. doi: 10.1111/j.1471-4159.2007.04759.x
258. Nielsen HM, Mulder SD, Beliën JAM, Musters RJP, Eikelenboom P, Veerhuis R, et al. Astrocytic A beta 1-42 uptake is determined by A beta-aggregation state and the presence of amyloid-associated proteins. Glia. (2010) 58:1235–46. doi: 10.1002/glia.21004
259. Logovinsky V, Satlin A, Lai R, Swanson C, Kaplow J, Osswald G, et al. Safety and tolerability of BAN2401–a clinical study in Alzheimer's disease with a protofibril selective Aβ antibody. Alzheimers Res Ther. (2016) 8:14. doi: 10.1186/s13195-016-0181-2
260. Fagan T, Strobel G. BAN2401 Removes Brain Amyloid, Possibly Slows Cognitive Decline. AlzForum. (2016). Available online at: https://www.alzforum.org/news/conference-coverage/ban2401-removes-brain-amyloid-possibly-slows-cognitive-decline (accessed March 1, 2020).
261. ClinicalBiomarker Updates from BAN2401 Study 201 in Early AD. Presented at the 11thClinical Trials on Alzheimer's Disease (CTAD) Conference October 24–27. (2018). Available online at: https://www.eisai.com/ir/library/presentations/pdf/e4523_181025.pdf
262. Powers SK, Talbert EE, Adhihetty PJ. Reactive oxygen and nitrogen species as intracellular signals in skeletal muscle. J Physiol. (2011) 589:2129–38. doi: 10.1113/jphysiol.2010.201327
263. Suzuki K. Cytokine response to exercise and its modulation. Antioxidants. (2018) 7:17. doi: 10.3390/antiox7010017
264. Geffken DF, Cushman M, Burke GL, Polak JF, Sakkinen PA, Tracy RP. Association between physical activity and markers of inflammation in a healthy elderly population. Am J Epidemiol. (2001) 153:242–50. doi: 10.1093/aje/153.3.242
265. Wannamethee SG. physical activity and hemostatic and inflammatory variables in elderly men. Circulation. (2002) 105:1785–90. doi: 10.1161/hc1502.107117
266. Monteiro-Junior RS, de Tarso Maciel-Pinheiro P, da Matta Mello Portugal E, da Silva Figueiredo LF, Terra R, Carneiro LSF, et al. Effect of exercise on inflammatory profile of older persons: systematic review and meta-analyses. J Phys Act Heal. (2018) 15:64–71. doi: 10.1123/jpah.2016-0735
267. Sobol NA, Hoffmann K, Frederiksen KS, Vogel A, Vestergaard K, Brændgaard H, et al. Effect of aerobic exercise on physical performance in patients with Alzheimer's disease. Alzheimer's Dement. (2016) 12:1207–15. doi: 10.1016/j.jalz.2016.05.004
268. Nascimento CMC, Cominetti MR, Pereira JR, Andrade LP, Garuffi M, Kerr DS, et al. Regular multimodal aerobic exercise reduces pro-inflammatory cytokines and improves BDNF peripheral levels and executive functions in elderly MCI individuals with different BDNF Val66Met genotypes. Alzheimer's Dement. (2015) 11:P323. doi: 10.1016/j.jalz.2015.07.465
269. Lu Y, Dong Y, Tucker D, Wang R, Ahmed ME, Brann D, et al. Treadmill exercise exerts neuroprotection and regulates microglial polarization and oxidative stress in a streptozotocin-induced rat model of sporadic Alzheimer's disease. J Alzheimer's Dis. (2017) 56:1469–84. doi: 10.3233/JAD-160869
270. He X, Liu D, Zhang Q, Liang F, Dai G, Zeng J, et al. Voluntary exercise promotes glymphatic clearance of amyloid beta and reduces the activation of astrocytes and microglia in aged mice. Front Mol Neurosci. (2017) 10:144. doi: 10.3389/fnmol.2017.00144
271. Jiang T, Zhang L, Pan X, Zheng H, Chen X, Li L, et al. Physical exercise improves cognitive function together with microglia phenotype modulation and remyelination in chronic cerebral hypoperfusion. Front Cell Neurosci. (2017) 11:404. doi: 10.3389/fncel.2017.00404
272. Kohman RA, Bhattacharya TK, Wojcik E, Rhodes JS. Exercise reduces activation of microglia isolated from hippocampus and brain of aged mice. J Neuroinflammation. (2013) 10:885. doi: 10.1186/1742-2094-10-114
273. Delezie J, Handschin C. Endocrine crosstalk between skeletal muscle and the brain. Front Neurol. (2018) 9:698. doi: 10.3389/fneur.2018.00698
274. Fiuza-Luces C, Garatachea N, Berger NA, Lucia A. Exercise is the real polypill. Physiology. (2013) 28:330–58. doi: 10.1152/physiol.00019.2013
275. Gleeson M, Bishop NC, Stensel DJ, Lindley MR, Mastana SS, Nimmo MA. The anti-inflammatory effects of exercise: mechanisms and implications for the prevention and treatment of disease. Nat Rev Immunol. (2011) 11:607–15. doi: 10.1038/nri3041
276. Woods JA, Wilund KR, Martin SA, Kistler BM. Exercise, inflammation and aging. Aging Dis. (2012) 3:130–40.
278. Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. (2015) 372:793–5. doi: 10.1056/NEJMp1500523
279. Hampel H, Vergallo A, Perry G, Lista S. The Alzheimer precision medicine initiative (APMI). J Alzheimer's Dis. (2019) 68:1–25. doi: 10.3233/JAD-181121
280. Ye CJ, Feng T, Kwon H-K, Raj T, Wilson MT, Asinovski N, et al. Intersection of population variation and autoimmunity genetics in human T cell activation. Science. (2014) 345:1254665. doi: 10.1126/science.1254665
281. Duffy D, Rouilly V, Libri V, Hasan M, Beitz B, David M, et al. Functional analysis via standardized whole-blood stimulation systems defines the boundaries of a healthy immune response to complex stimuli. Immunity. (2014) 40:436–50. doi: 10.1016/j.immuni.2014.03.002
282. Willis JCD, Lord GM. Immune biomarkers: the promises and pitfalls of personalized medicine. Nat Rev Immunol. (2015) 15:323–9. doi: 10.1038/nri3820
283. Schork NJ. Personalized medicine: time for one-person trials. Nature. (2015) 520:609–11. doi: 10.1038/520609a
284. Delhalle S, Bode SFN, Balling R, Ollert M, He FQ. A roadmap toward personalized immunology. NPJ Syst Biol Appl. (2018) 4:9. doi: 10.1038/s41540-017-0045-9
285. Hampel H, Toschi N, Babiloni C, Baldacci F, Black KL, Bokde ALW, et al. Revolution of Alzheimer Precision Neurology. Passageway of Systems Biology and Neurophysiology. J Alzheimer's Dis. (2018) 64:S47–105. doi: 10.3233/JAD-179932
286. Castrillo JI, Lista S, Hampel H, Ritchie CW. Systems biology methods for alzheimer's disease research toward molecular signatures, subtypes, and stages and precision medicine: application in cohort studies and trials. In: Perneczky R, editor. Methods in Molecular Biology. New York, NY: Humana Press Inc (2016). p. 31–66. doi: 10.1007/978-1-4939-7704-8_3
287. Tebani A, Afonso C, Marret S, Bekri S. Omics-Based strategies in precision medicine: toward a paradigm shift in inborn errors of metabolism investigations. Int J Mol Sci. (2016) 17:1555. doi: 10.3390/ijms17091555
288. Dimitrakopoulos L, Prassas I, Diamandis EP, Charames GS. Onco-proteogenomics: Multi-omics level data integration for accurate phenotype prediction. Crit Rev Clin Lab Sci. (2017) 54:414–32. doi: 10.1080/10408363.2017.1384446
289. Ang MY, Low TY, Lee PY, Wan Mohamad Nazarie WF, Guryev V, Jamal R. Proteogenomics: from next-generation sequencing (NGS) and mass spectrometry-based proteomics to precision medicine. Clin Chim Acta. (2019) 498:38–46. doi: 10.1016/j.cca.2019.08.010
290. Lista S, Khachaturian ZS, Rujescu D, Garaci F, Dubois B, Hampel H. Application of systems theory in longitudinal studies on the origin and progression of Alzheimer's Disease. In: Castrillo JI, Oliver SG, editors. Methods in Molecular Biology. New York, NY: Humana Press (2016). p. 49–67. doi: 10.1007/978-1-4939-2627-5_2
291. Hampel H, O'Bryant SE, Castrillo JI, Ritchie C, Rojkova K, Broich K, et al. PRECISION MEDICINE - the golden gate for detection, treatment and prevention of Alzheimer's Disease. J Prev Alzheimer's Dis. (2016) 3:243–59. doi: 10.14283/jpad.2016.112
292. Hampel H, Vergallo A, Aguilar LF, Benda N, Broich K, Cuello AC, et al. Precision pharmacology for Alzheimer's disease. Pharmacol Res. (2018). 130:331–65. doi: 10.1016/j.phrs.2018.02.014
293. Hood L, Flores M. A personal view on systems medicine and the emergence of proactive P4 medicine: predictive, preventive, personalized and participatory. N Biotechnol. (2012) 29:613–24. doi: 10.1016/j.nbt.2012.03.004
Keywords: Alzheimer's disease, neuroinflammation, microglia, neuroinflammatory pathways, biomarkers, anti-inflammatory therapy, systems biology, precision medicine
Citation: Hampel H, Caraci F, Cuello AC, Caruso G, Nisticò R, Corbo M, Baldacci F, Toschi N, Garaci F, Chiesa PA, Verdooner SR, Akman-Anderson L, Hernández F, Ávila J, Emanuele E, Valenzuela PL, Lucía A, Watling M, Imbimbo BP, Vergallo A and Lista S (2020) A Path Toward Precision Medicine for Neuroinflammatory Mechanisms in Alzheimer's Disease. Front. Immunol. 11:456. doi: 10.3389/fimmu.2020.00456
Received: 02 November 2019; Accepted: 27 February 2020;
Published: 31 March 2020.
Edited by:
Sally Ann Frautschy, University of California, Los Angeles, United StatesReviewed by:
Maria Grazia Cifone, University of L'Aquila, ItalyCopyright © 2020 Hampel, Caraci, Cuello, Caruso, Nisticò, Corbo, Baldacci, Toschi, Garaci, Chiesa, Verdooner, Akman-Anderson, Hernández, Ávila, Emanuele, Valenzuela, Lucía, Watling, Imbimbo, Vergallo and Lista. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Harald Hampel, aGFyYWxkLmhhbXBlbEBtZWQudW5pLW11ZW5jaGVuLmRl
†These authors have contributed equally to this work and share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.