
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 17 March 2020
Sec. Comparative Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.00425
 Yo Okamura1
Yo Okamura1 Natsuki Morimoto1Daisuke Ikeda2Nanami Mizusawa2Shugo Watabe2Hiroshi Miyanishi3Yuichi Saeki4Haruko Takeyama5Takashi Aoki6
Natsuki Morimoto1Daisuke Ikeda2Nanami Mizusawa2Shugo Watabe2Hiroshi Miyanishi3Yuichi Saeki4Haruko Takeyama5Takashi Aoki6 Masato Kinoshita7
Masato Kinoshita7 Tomoya Kono4
Tomoya Kono4 Masahiro Sakai4
Masahiro Sakai4 Jun-ichi Hikima4*
Jun-ichi Hikima4*In mammals, interleukin (IL)-17A and F are hallmark inflammatory cytokines that play key roles in protection against infection and intestinal mucosal immunity. In the gastrointestinal tract (GI), the induction of antimicrobial peptide (AMP) production via Paneth cells is a fundamental role of IL-17A and F in maintaining homeostasis of the GI microbiome and health. Although mammalian IL-17A and F homologs (referred to as IL-17A/F1-3) have been identified in several fish species, their function in the intestine is poorly understood. Additionally, the fish intestine lacks Paneth cells, and its GI structure is very different from that of mammals. Therefore, the GI microbiome modulatory mechanism via IL-17A/F genes has not been fully elucidated. In this study, Japanese medaka (Oryzias latipes) were used as a teleost model, and IL-17A/F1-knockout (IL-17A/F1-KO) medaka were established using the CRISPR/Cas9 genome editing technique. Furthermore, two IL-17A/F1-deficient medaka strains were generated, including one strain containing a 7-bp deletion (-7) and another with an 11-bp addition (+11). After establishing F2 homozygous KO medaka, transcriptome analysis (RNA-seq) was conducted to elucidate IL-17A/F1-dependent gene induction in the intestine. Results of RNA-seq and real-time PCR (qPCR) demonstrated down-regulation of immune-related genes, including interleukin-1β (IL-1β), complement 1q subunit C (C1qc), transferrin a (Tfa), and G-type lysozyme (LyzG), in IL-17A/F1-KO medaka. Interestingly, protein and lipid digestive enzyme genes, including phospholipase A2, group IB (pla2g1b), and elastase-1-like (CELA1), were also downregulated in the intestines of IL-17A/F1-KO medaka. Furthermore, to reveal the influence of these downregulated genes on the gut microbiome in IL-17A/F1-KO, 16S rRNA-based metagenomic sequencing analysis was conducted to analyze the microbiome constitution. Under a non-exposed state, the intestinal microbiome of IL-17A/F1-KO medaka differed at the phylum level from wild-type, with significantly higher levels of Verrucomicrobia and Planctomycetes. Additionally, at the operational taxonomic unit (OTU) level of the human and fish pathogens, the Enterobacteriaceae Plesiomonas shigelloides was the dominant species in IL-17A/F1-KO medaka. These findings suggest that IL-17A/F1 is involved in the maintenance of a healthy gut microbiome.
In mammals, the relationship between host mucosal immunity in the gastrointestinal tract (GI) and a good microbiome environment has received increased attention with regards to promoting body health (1). The human microbiome is comprised of different types of microorganisms, including bacteria (prokaryotes), archaea, fungi, protists, and viruses, which inhabit the GI, skin, oral cavity, urogenital organ, and nasal fossa, forming different and uniquely diverse microbiomes (2). In the GI, the number of inhabiting microorganisms has been estimated to exceed 1014, with bacteria representing a major player in the GI microbiome (3, 4). GI microbiomes are modulated by various host biochemical factors, such as pH in the stomach, antimicrobial peptides (AMPs) in the small intestines, and mucin (MUC) in the colon (4). Failure to modulate this balance can lead to deterioration of enteritis pathophysiology, such as Crohn's disease and ulcerative colitis (5).
Interleukin 17 (IL-17) is a hallmark inflammatory cytokine produced by the T helper 17 (Th17) subset of CD4+ T cells that plays key roles in protection against infection and intestinal mucosal immunity and is also a critical microbiome regulator in mammals (6). Th17 subset of CD4+ T cells have been reported to accumulate at a high density in the mammalian intestinal tract, and it is known that both transforming growth factor (TGF-β) and IL-6 are essential for Th17 differentiation (7). Furthermore, segmented filamentous bacteria (SFB), belonging to the genus of Clostridium, play important roles in Th17 differentiation (8, 9), where SFB antigen presentation by resident dendritic cells in the intestinal tract has been suggested as a key step in the process (10). Studies on the relationship between the Th17-mediated intestinal immune system and gut microbiome have increased in recent years. It has recently been revealed that IL-17 is produced from other lymphocytes, γδ-T cells, natural killer T (NKT) cells, innate lymphoid cells (ILC3), neutrophils, mast cells, and macrophages (11). Currently, six family members, namely IL-17A, B, C, D, E, and F, have been identified in mammals (12). Among them, IL-17A and F are known as key inflammatory cytokines that modulate the microbiome. IL-17A and F share the highest homology among IL-17 family members, and their gene loci are adjacent to each other on the same chromosome (13). These two IL-17s can bind both the IL-17 receptor A (RA) and receptor C (RC) heterodimer complex. However, IL-17F has a decreased ability to induce inflammation compared to IL-17A, and the reason why remains unknown. Moreover, differences in the roles of IL-17A and IL-17F in pathogenesis have yet to be elucidated (12). Nevertheless, IL-17A and F are known to induce pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α, the chemokines CXCL1, CXCL8, and CCL20, and AMPs, including defensin and calprotectin (11, 14). The mammalian intestinal tract, particularly the small intestine, exhibits a unique AMP production mechanism via Paneth cells. Paneth cells are neutrophil-like, intestinal epithelial cells that contain granules and are localized in crypts of villus tissue, with an ability to produce AMPs such as phospholipase A2, lysozyme, and defensin in the intestine (15). Of these, the human alpha-defensin (HD5) homolog gene in mice, cryptdin (Crp)—a representative AMP secreted from Paneth cells—potently kills pathogens, yet it shows almost no anti-bactericidal activity against commensal bacteria (16). Moreover, Paneth cells transmit signals to adjacent intestinal epithelial stem cells via Wnt and Notch signaling and play an important role in determining the differentiation pathway of each epithelial cell (15). Thus, AMP exhibits anti-bactericidal activity with some selectivity. Therefore, IL-17A and F are considered critical for pathogen elimination and maintenance of the gut microbiome via the intestinal immune system. By contrast, in fish, the relationship between IL-17 and other immune systems and the gut microbiome has hardly been clarified. Therefore, we focused on the importance of the regulatory function of IL-17 on the gut microbiome in maintaining fish health.
In teleosts, the mammalian IL-17A and F homologs, referred to as IL-17A/F1-3, have been identified in many fish species such as zebrafish (Danio rerio), Japanese medaka (Oryzias latipes), Japanese puffer (Takifugu rubripes), channel catfish (Ictalurus punctatus), European seabass (Dicentrarchus labrax), rainbow trout (Oncorhynchus mykiss), and Atlantic salmon (Salmo salar) (17–21). Among the three types of IL-17A/F genes, IL-17A/F1 and IL-17A/F2 are adjacently located on the same chromosome, similar to that of mammalian IL-17A and F, but share low sequence identity. IL-17A/F3 is located on a different chromosome from that of IL-17A/F1 and 2 and shows higher amino acid sequence identity and similarity to IL-17A/F1 than to IL-17A/F2. Moreover, phylogenetic analysis determined that IL-17A/F3 forms a cluster that is closer to IL-17A/F1 than to IL-17A/F2 (20). It has been reported that expression levels of IL-17A/F1, 2, and 3 genes fluctuate at the mRNA level during bacterial infection in multiple fish species. Yersinia ruckeri up-regulates salmonid IL-17A/F1, 2, and 3 in the spleen (22), whereas Aeromonas hydrophila up-regulates large yellow croaker counterparts in the head, kidney, and gill (23). However, details regarding the mechanism of fish IL-17A/F gene regulation of the intestinal tract microbiome are unknown. Furthermore, in the teleost gut, Paneth cells have not been found. In addition, the pH of the teleost GI is closer to neutral compared with that of mammals (24). Based on these differences in the GI physiological structure, we hypothesized that the aquatic environment drives different mechanisms to control the gut microbiome in teleosts compared with land mammals.
In this study, we aimed to understand the role of IL-17A and F in the intestines of teleosts by constructing two IL-17A/F1-mutated strains of Japanese medaka (O. latipes) [IL-17A/F1-knockout(KO)-medaka] using CRISPR-Cas9 genome editing technology. After establishing the mutant strains, transcriptome analysis (RNA-seq) was conducted to investigate IL-17A/F1-dependent gene regulation. Additionally, 16S metagenome analysis of intestinal content was performed. This is the first report using IL-17 gene-mutated strains to extensively analyze the role of IL-17 in the intestines of teleosts.
The Cab strain of Japanese medaka was maintained in several transparent plastic tanks with a water circulating system (26°C) under a 14-h light and 10-h dark cycle. In all experiments, fish that were 3–4 months old and weighing 200–300 mg were used. All animal experiments were conducted according to the relevant national (Act on Welfare and Management of Animals, Ministry of the Environment, Japan) and international guidelines. Ethics approval from the local IACUC was not sought as this law does not mandate protection of fish.
Approximately 0.5 nL of a solution containing crRNA (25 ng/μL), tracrRNA (40 ng/μL), and Cas9 mRNA (100 ng/μL) was injected into one- to two-cell stage embryos with a manipulator (Narishige, Tokyo, Japan). All three crRNAs were designed in exon 2 of the medaka IL-17A/F1 gene (Figure 1A). Prior to designing crRNAs, the medaka Cab IL-17A/F1 mRNA nucleotide sequence was deposited in the GenBank database under accession number LC501388. The three crRNA sequences are listed in Supplementary Table 1. After injection into the embryo, genomic DNA was extracted from randomly selected embryos to confirm gene editing efficiency using the heteroduplex mobility assay (HMA; Supplementary Figure 1). Filial generation 0 (F0) fish, grown from the injected fertilized eggs, were interbred with wild-type medaka (Cab strain) to produce F1 heterozygotes. Among the F1 fish, males and females carrying an identical mutation were mated (in-crossed) to obtain homozygous progeny and/or a mutant line. For detection of the mutated locus in the genome, HMA was performed. Additionally, to confirm crRNA efficiency, primer sets amplifying specific regions, including the crRNA identical regions, were used; locations are listed in Supplementary Table 1. Briefly, genomic DNA was prepared from the embryo or an excised fin by dissolving in 20 μL of a solution containing 0.2 mM EDTA and 25 mM NaOH and incubating at 95°C for 20 min. After incubation, samples were neutralized by the same volume of 40 mM Tris/HCl (pH 8.0). Before collecting the tail fins, the medaka were anesthetized using MS-222 (Sigma-Aldrich, St. Louis, MO, USA). Using this genomic DNA-containing solution as a template, PCR was carried out using KOD FX Neo (Toyobo, Osaka, Japan). PCR amplification was carried out in a total volume of 10 μL containing 5 μL PCR buffer, 2 μL dNTP, 0.2 μL KOD FX Neo, 1 μL gDNA-containing solution, 0.2 μL each of the forward (Ol_IL-17A/F1_F1) and reverse primers (Ol_IL-17A/F1_R1) at a final concentration of 5 pmol, and 1.4 μL distilled water. The PCR program was initially run at 94°C for 2 min, followed by 37 cycles of 94°C for 30 s, 62°C for 30 s, and 72°C for 20 s. The amplicon products were separated by 12% PAGE to compare their migration patterns.
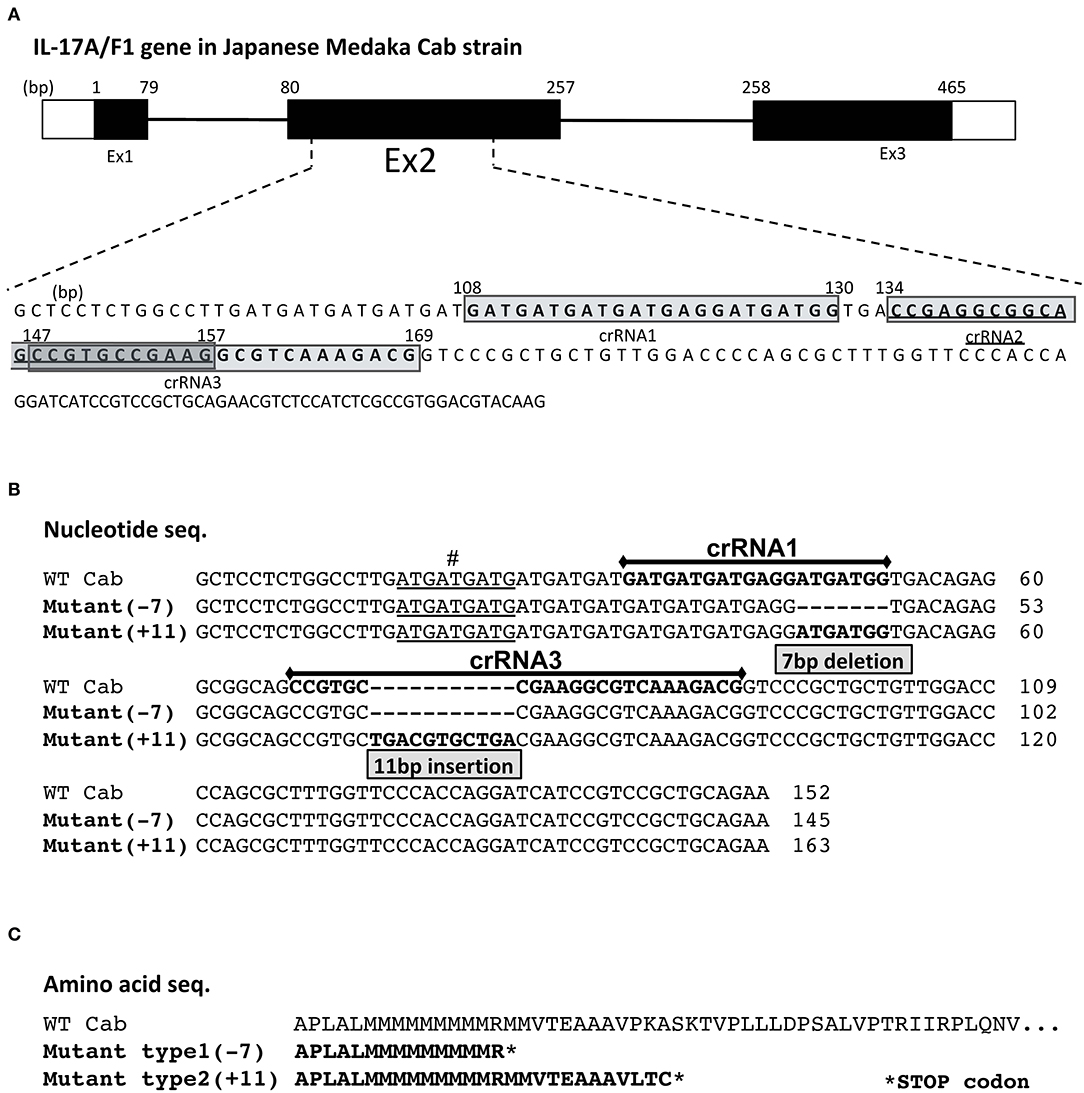
Figure 1. crRNA regions and mutated sequences in the medaka IL-17A/F1 gene. (A) Three crRNA regions in the medaka IL-17A/F1 gene. Three crRNAs were designed in exon 2 of the IL-17A/F1 gene. The mutated nucleotide (B) and amino acid sequences (C) of WT and two KO strains. The mutated regions in F0 founder and F2 homo individuals were confirmed by sequencing. The nine underlined nucleotides “#” indicate different nucleotide sequences of the IL-17A/F1 gene in the medaka Cab strain compared with that of the Hd-rR strain and GenBank-deposited sequence (acc. no. NM_001204785). In (C), both mutant strains contained premature stop codons in the amino acid sequences due to frameshift mutations.
For qPCR analysis, total RNA was extracted from whole intestines of adult Japanese medaka Cab strains using an RNAiso Plus Kit (TaKaRa Bio, Kusatsu, Japan) according to manufacturer's instructions. Total RNA from each medaka was extracted separately (not normalized). Total RNA was quantified with a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). RNA purity was measured by the OD260/OD280 ratio, with a minimum ratio of 1.8 as the quality cut-off value. cDNA was synthesized using 500 ng of total RNA extracted from each sample with the ReverTra Ace qPCR RT Master Mix with gDNA Remover (Toyobo) according to manufacturer's instructions. For RNA-seq, total RNA from five individuals was equally pooled, and KAPA Stranded RNA-Seq Library Preparation Kit (KAPA Biosystems, Wilmington, MA, USA) was used to construct cDNA libraries according to manufacturer's instructions. cDNA libraries were sequenced using the MiSeq Reagent Kit v3 (150 Cycles; Illumina, San Diego, CA, USA).
Processed reads were submitted to the DDBJ Sequence Read Archive (DRA) under accession number DRA008715. Subsequently, the collected reads were mapped to the annotated medaka Hd-rR reference genome (release 85; http://www.ensembl.org/index.html) with the TopHat program (25) and further analyzed by the Cufflinks-Cuffdiff program (26, 27). Transcription expression values were estimated as fragments per kilobase of exon length per million reads (FPKM), and transcripts with a q < 0.05 were considered significantly differentially expressed (26). To normalize individual differences, the RNA-seq samples were pooled from individual fish that were maintained at a constant temperature, and RNA-seq was not replicated. Therefore, the statistical analysis was not reliable, although the Cuffdiff program can calculate q-values without biological replicates. Thus, we focused on genes whose expression levels differed substantially (4-fold or higher) between the WT and IL-17A/F1-KO medaka intestinal tissues. Gene enrichment analysis was carried out with the DAVID program (28). Gene ontology (GO) terms in the biological processes (GOTERM_BP_FAT) and the cellular component (GOTERM_CC_FAT) as well as Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were selected. GO terms were summarized and visualized with the REVIGO program (29).
qPCR was used to analyze differences in expression patterns between WT and KO intestinal tissues. We compared IL-17A/F1 (+11) homo-mutant, WT, and medaka exposed with Edwardsiella piscicida (immersion in 6.9 × 108 CFU/mL E. piscicida for 24 h). Total RNA was extracted from the intestines of medaka (n = 5) from each group, and their respective cDNAs were prepared separately as described in section RNA Extraction and cDNA Synthesis for Real-Time PCR (qPCR) Analysis and Next-Generation Sequencing. Target genes for expression analysis were selected based on RNA-seq results, and the primers used are listed in Supplementary Table 1. qPCR amplification was carried out in triplicate in a total volume of 15 μL containing 7.5 μL Brilliant III Ultra-Fast SYBR® Green QPCR Master Mix (Agilent Technologies, Santa Clara, CA, USA), 1.5 μL cDNA, 1.5 μL (5 pmol) each of the forward and reverse primers, and 3 μL distilled water. The qPCR program was run at 95°C for 15 s and 60°C for 30 s, followed by 40 cycles on a CFX connect TM (Bio-Rad Laboratories, Hercules, CA, USA). A melting curve analysis of the amplified products was performed at the end of each cycle to confirm the specificity of amplification. The medaka β-actin gene served as an internal control to confirm the quality and quantity of the cDNA. Relative expression ratios were calculated based on the comparative threshold cycle (CT) method (2−ΔΔCT method). Briefly, the target gene and internal control Ct values were determined for each sample, after which the average Ct value of the three replicates was used to calculate expression levels relative to β-actin.
Before dissection, the surface of medaka bodies was washed three times with PBS containing 0.2% Tween® 20 (molecular biology grade; Promega, Madison, WI, USA) to avoid outer bacterial contamination. After washing, whole intestinal tissues were extracted, and intestinal tracts were filled with PBS and squeezed to extract intestinal contents. From each individual medaka's intestinal content, bacterial genomic DNA was extracted separately (not normalized) using the QIAamp Fast DNA Stool Mini Kit (Qiagen, Hilden, Germany) according to manufacturer's instructions. The concentration of each DNA sample was set to 5 ng/μL using nuclease-free water for the PCR reaction (not pooled).
The V3-V4 region of 16S rRNA was targeted for PCR amplification, and the first PCR was carried out in a total volume of 20 μL containing 10 μL KAPA HiFi HotStart ReadyMix (2X; Nippon genetics, Tokyo, Japan), 8 μL cDNA, and 1 μL (5 pmol) each of the forward (V3V4f) and reverse primers (V3V4r). Primer sequences for the first and second PCR are listed in Supplementary Table 1. The PCR program was run at 94°C for 2 min, followed by 28 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 30 s. Before reading sequencing analysis, PCR products were examined for target specificity by 2% agarose gel electrophoresis and purified using AMPure XP (Beckman Coulter, Brea, CA, USA). Using purified DNA, the second PCR was carried out in a total volume of 10 μL containing 1 μL 10X Ex Buffer, 0.8 μL dNTPs (each 2.5 mM), 0.5 μL (10 pmol) each of the forward (2ndF) and reverse primers (2ndR), 2.0 μL purified DNA (purified 1st PCR product, maximum 2 ng/μL), 0.1 μL ExTaq HS (TaKaRa Bio), and 5.1 μL DDW. After amplification, PCR products were purified using AMPure XP, and then purified DNA derived from each individual was used as a library. Before sequencing, library qualities were assessed by Fragment Analyzer and dsDNA 915 Reagent Kit (Advanced Analytical Technologies, Santa Clara, CA, USA). Sequencing was performed under the condition of 2 × 300 bp read lengths using MiSeq (Illumina).
After sequencing, we extracted only those sequences whose beginning sequence reads were an exact match of the primers used by the Fastaq barcode splitter of the Fastx toolkit. The raw paired-end readings were then subjected to a quality control procedure using QIIME 2 (30). Qualified reads were clustered to generate operational taxonomic units (OTUs) at the 97% similarity level, and phylogenetics were estimated using Greengenes OTU (version 13.8). Chimeric sequences were removed using the UCHIME algorithm (version 4.1). The processed 16S sequence data from this study have been deposited at the DRA under accession number DRA008844. For diversity tests and statistical analysis, we executed R codes in RStudio v1.1.442 (31) and employed the below-mentioned packages to analyze the data. Data were imported into R using the package “xlsx.” Observed OTU number and Chao 1 were used to reflect community richness, and diversity was assessed using Shannon indices. In the β-diversity test, we performed weighted UniFrac analysis. For statistical analysis, the abundance of phylum, family, and OTU level between WT and KO was assessed by a Student's t-test with subsequent Bonferroni correction. Furthermore, Chao1 and Shannon indices, Kruskal–Wallis test, and Dunn's test with the Benjamini–Hochberg FDR correction using “dunn.test” and “FSA” in R package were used to identify significant differences in the α-diversity indices of the groups. In weighted UniFrac analysis, PERMANOVA using “MASS” and “Vegan” in R package were used to identify significant differences in distance between different groups.
qPCR was used to calculate the plasmid copy number of P. shigelloides in each of the WT and KO groups. The P. shigelloides genomic DNA fragment (GenBank accession number AJ300545) was amplified via PCR using specific primers from the DNA sample extracted for metagenome analysis. PCR products were cloned into the pTAC-2 vector (BioDynamics, Kumamoto, Japan), and plasmid DNA was extracted using the QIAprep Spin Mini-prep Kit (Qiagen) from transformed Escherichia coli (DH5α). Extracted plasmid DNA was diluted from 1.0 × 1010 to 1.0 × 103 copies/μL to create a standard curve. qPCR amplification was carried out in triplicate in a total volume of 15 μL containing 7.5 μL Brilliant III Ultra-Fast SYBR® Green QPCR Master Mix, 5.5 μL DNA, and 1.0 μL (5 pmol) each of the forward and reverse primers. The qPCR program was run at 95°C for 10 s and 60°C for 20 s, followed by 40 cycles on a CFX connect TM.
The Japanese medaka Cab strain was exposed by immersion in fresh water containing E. piscicida [E381 strain (former name E. tarda) isolated from the Nile tilapia (Oreochromis niloticus) kindly gifted by Dr. Tomokazu Takano]. E. piscicida was cultured with heart infusion medium (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) at 28°C for 48 h. After 24-h immersion of medaka in freshwater + E. piscicida, the water in the tanks was changed to new freshwater without E. piscicida. CFU counts in the tank water during bacterial challenge were determined by the plate counting method. The concentration of the E. piscicida in the freshwater was confirmed by plate counting methods (i.e., gene expression experiment by qPCR; 6.9 × 108 CFU/mL and metagenomic experiment; 2.1 × 108 CFU/mL). For both experiments, five fish were selected at 0, 24, and 48 h from WT and IL-17A/F1-KO(+11) groups.
Of the three crRNAs located in exon 2 of IL-17A/F1, crRNA1 and 3 showed a high mutation level (Supplementary Figure 1). After injection into embryos with an RNA mixture containing crRNA, tracRNA, and Cas9 mRNA, two strains of IL-17A/F1-KO medaka were obtained. One strain had a 7-bp deletion (-7) in the region of the crRNA1, and another strain had an 11-bp insertion (+11) in crRNA3 (Figure 1B). For both KO strains, the amino acid sequence of IL-17A/F1 was terminated in the middle of its full-length sequence due to a codon frame shift (Figure 1C). The IL-17A/F1-KO(+11) strain did not show any morphological abnormalities in juvenile and adult fish (Supplementary Figures 2A,B). In F2 homo-mutant individuals, no differences were found in intestinal villus tissue (Supplementary Figure 2C), and no significant changes in body weight were observed between WT and IL-17A/F1-KO medaka (Supplementary Figure 2D).
Of the three IL-17A/F genes in the WT intestine, the expression level of IL-17A/F3 was the highest, followed by IL-17A/F1 and 2, which was statistically significant (Figure 2). Furthermore, no changes were observed in the expression patterns of IL-17A/F1, 2, and 3 in IL-17A/F1-KO(+11) (primer set of IL-17A/F1 for qPCR amplified the extra region of the +11 bp mutation) compared to WT.
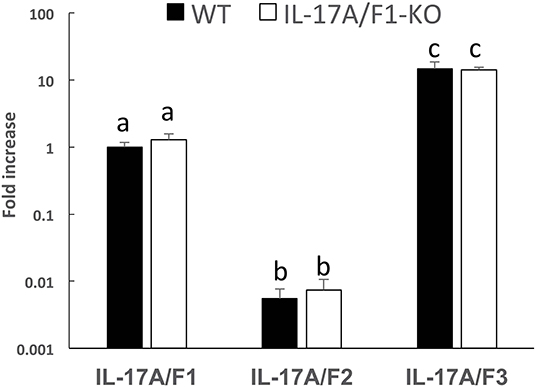
Figure 2. Gene expression analysis of IL-17A/F1, 2, and 3 in WT and KO medaka. No significant changes in the three IL-17A/Fs at the transcriptional level were observed in KO medaka (+11 inserted strain) intestines compared with WT. Comparisons were performed using healthy medaka. Symbols with different letters are significantly different at p < 0.05 with Tukey Kramer's multiple comparison test after a one-way ANOVA. Data are presented as mean ± SD; n = 5 fish' SD depends on the statistical analyses run.
For transcriptome analysis, F2 homo individuals of the IL-17A/F1(-7)-deficient strain were used to compare gene expression in whole intestines with that of the WT. Next-generation sequencing provided the 2993350 and 4443631 raw sequence reads from WT and KO cDNA samples, respectively. RNA-seq revealed that WT and IL-17A/F1-KO medaka had different expression patterns under the non-exposed state (Figure 3A). Furthermore, REVIGO analysis showed down-regulation in several genes that participate in the immune response and lipid catabolism in KO medaka intestines (Figure 3B). Conversely, gene groups related to endosomal transport and muscle structure development were mainly up-regulated (Figure 3C). Table 1 shows a list of genes with a significant down-regulation of ≥4-fold in the IL-17A/F1-KO intestines compared with WT, and includes various immune-related genes, such as AMPs, transferrin a (Tfa), and complement 1q subunit C (C1qc) located on chromosome 18. In addition to these genes, various lipid and protein digestive enzyme genes, including phospholipase A2, group IB (pla2g1b), bile salt-activated lipase-like (CEL), and elastase 1 (CELA1), were down-regulated (≥4-fold) in IL-17A/F1-KO intestines compared with WT. Furthermore, Table 2 lists several genes, including craniofacial development protein 2-like (CFDP2L), mitogen-activated protein kinase 4 (MAP4K4), and ethanolamine-phosphate phospho-lyase (ETNPPL), that were found to be significantly up-regulated (≥4-fold) in IL-17A/F1-KO intestines compared with WT.
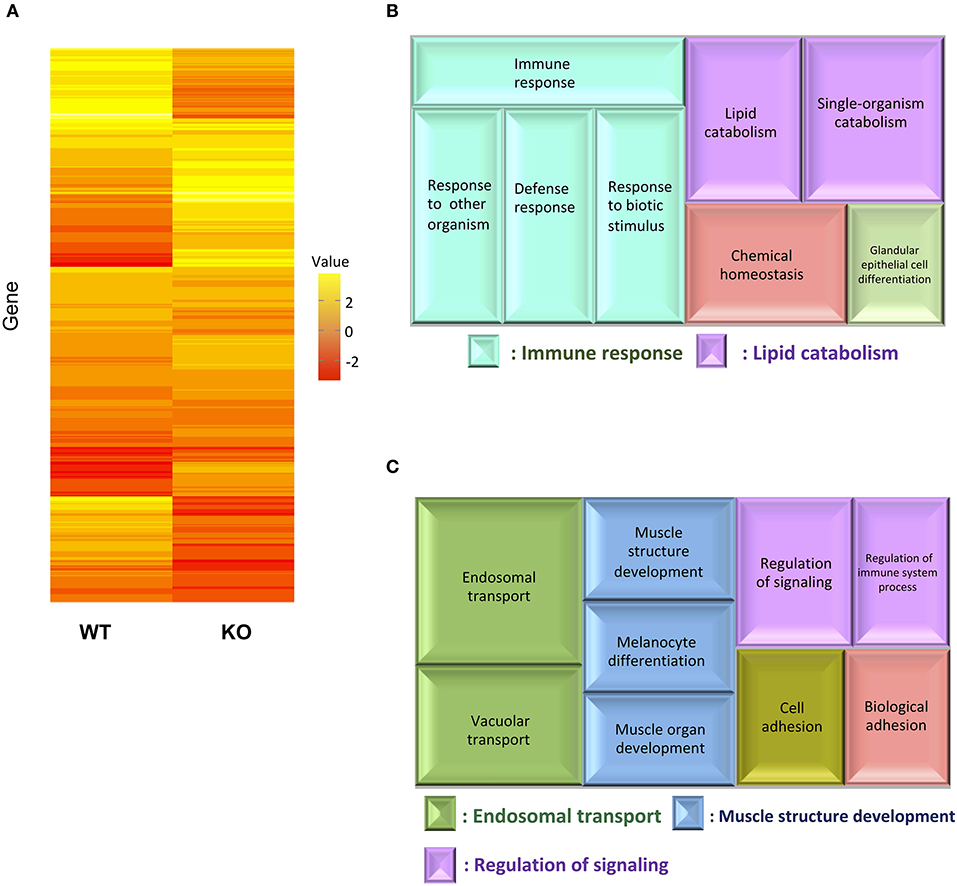
Figure 3. Comparative analyses between WT and IL-17A/F1-KO intestines by RNA-seq. (A) Differences in expression patterns of detected genes. Heatmaps of RNA-seq were generated using unsupervised hierarchical clustering with Euclidian distance metric using 863 genes. The input matrix on rows has been converted to –log10 p-value. (B,C) Treemap view of the summarized lists of GO terms in the biological process category. The size of the color panel reflects the number of relevant genes. Enriched GO terms for genes down-regulated by ≥4-fold in IL-17A/F1-KO (7-bp deletion strain) (B) and upregulated by ≥4-fold in KO(-7) (C).
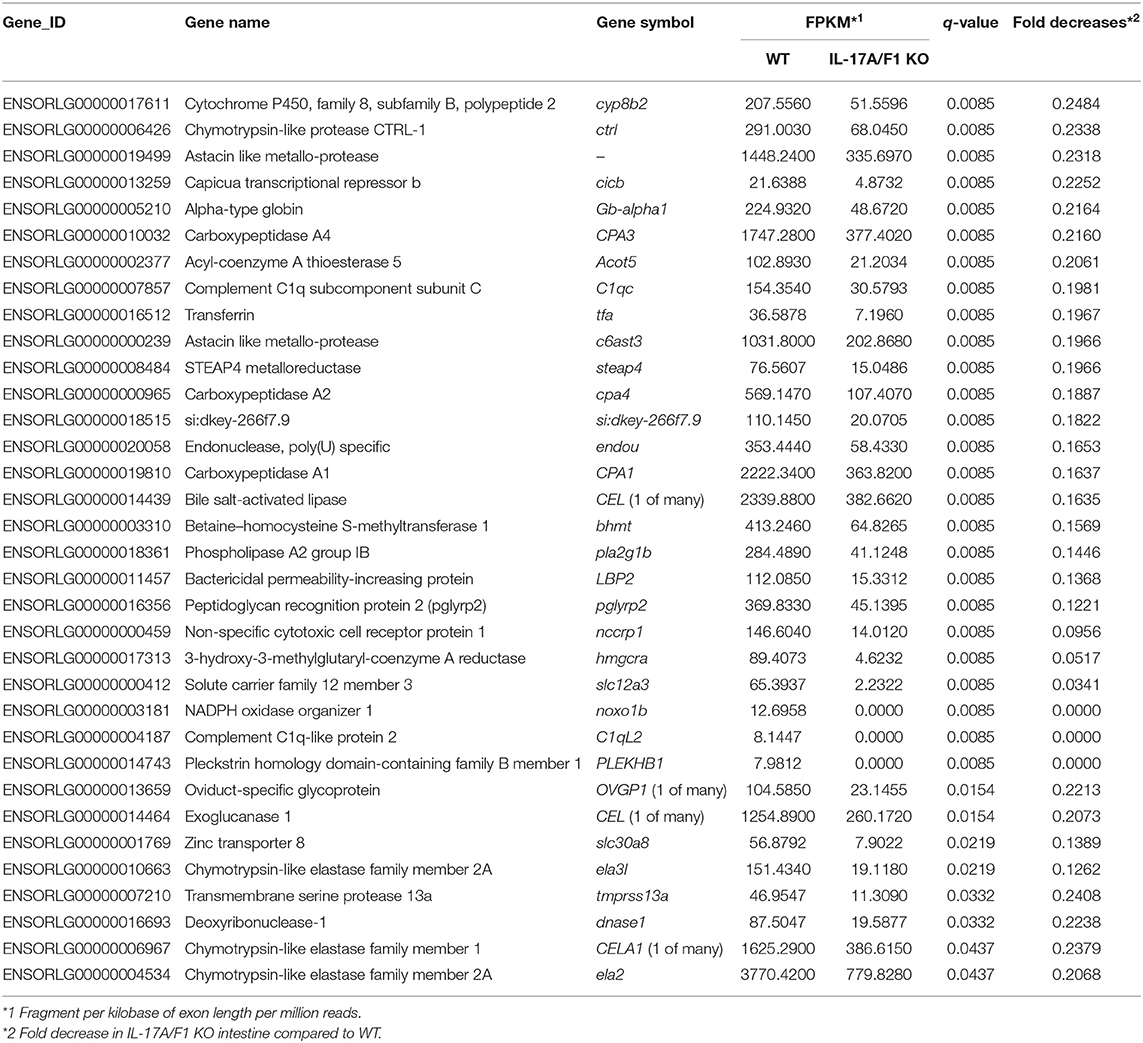
Table 1. List of genes down-regulated (1/4-fold) in IL-17A/F1-KO medaka intestines.
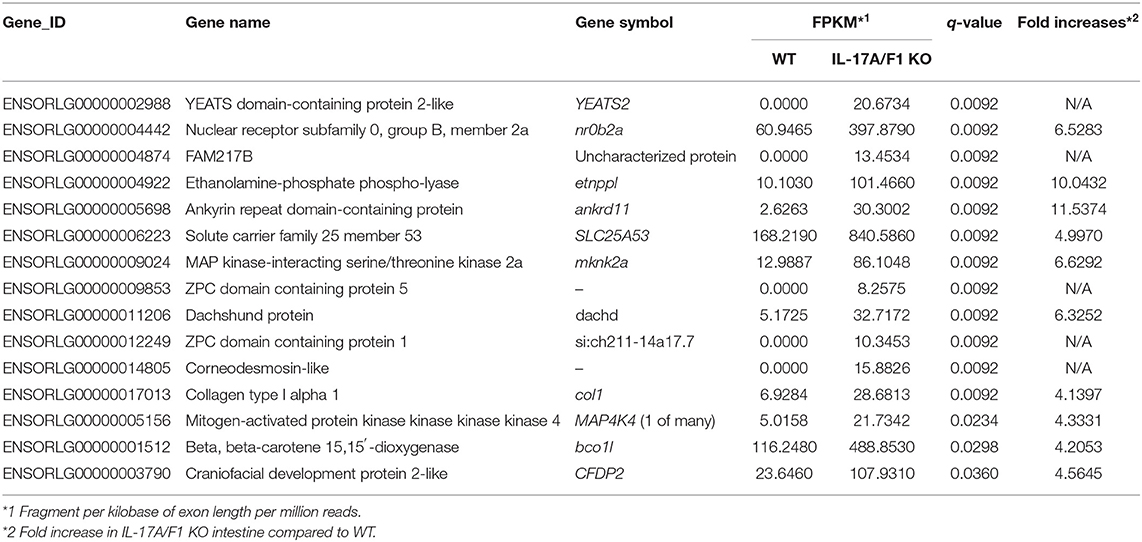
Table 2. List of genes up-regulated (≥4-fold) in IL-17A/F1 KO medaka intestines.
The genes down-regulated in the IL-17A/F1-KO(-7) intestines were also confirmed in the IL-17A/F1-KO(+11) intestines by qPCR. Furthermore, we analyzed healthy and E. piscicida-exposed medaka via qPCR and found that the immune-related genes Tfa and C1qc (Chr. 18) were significantly reduced (Tfa, 0-, 24-, and 48-h exposure; C1q, 0- and 24-h exposure) in the intestines of the 11-bp insertion line (Figures 4A,B). Additionally, the proinflammatory cytokine IL-1β, antimicrobial peptide G type lysozyme (LyzG), and other types of C1q genes [C1qb and C1qc (Chr. 5)] were found to be significantly decreased in the IL-17A/F1-KO(+11) medaka intestines (IL-1β, non-exposed state; LyzG, non-exposed state and 24-h exposure; C1qb, non-exposed state and 48-h exposure; C1qc, non-exposed state; Figures 4C–F). Although these genes were down-regulated in the 7-bp deletion line, it was not statistically significant, as evidenced by RNA-seq (Supplementary Table 2). The expression of lipase pla2g1b and CELA1 was also significantly reduced in the IL-17A/F1-KO(+11) (Figures 4G,H). Nevertheless, several immune-related and digestive enzyme genes showed no significant changes between WT and KO (Supplementary Figure 3).
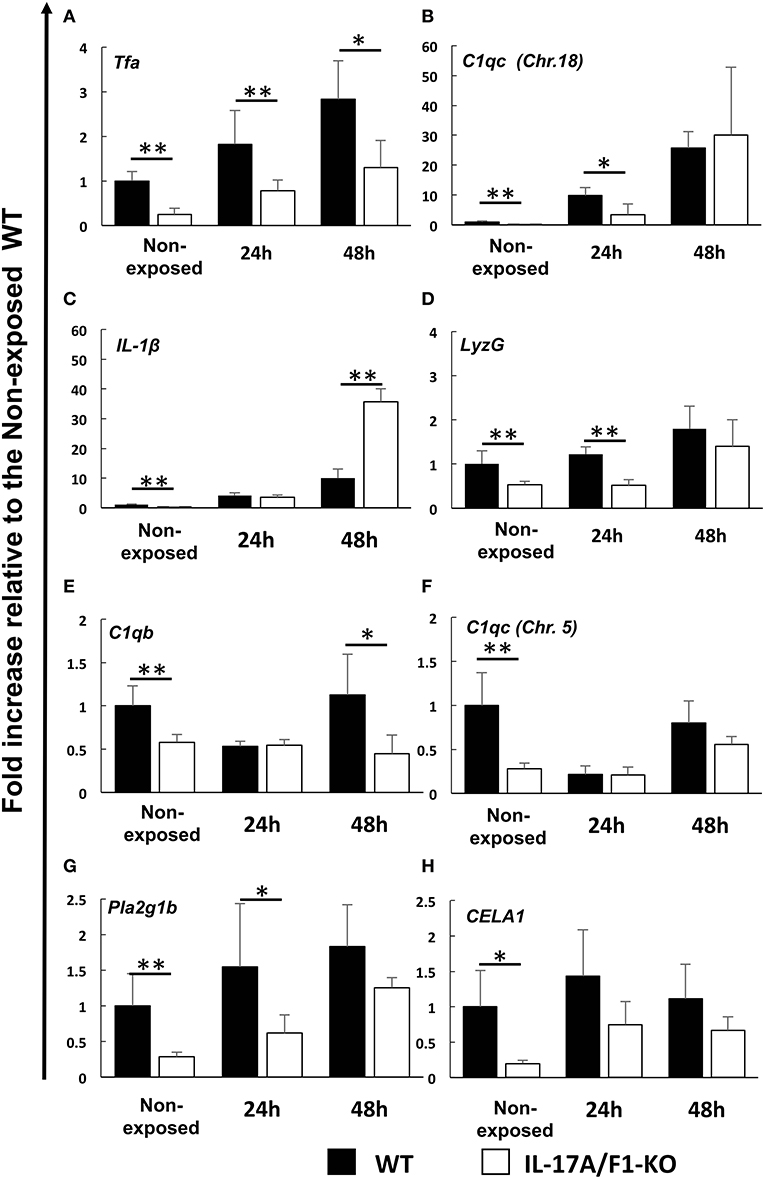
Figure 4. Real-time PCR analysis of the down-regulated immune-related and digestive enzyme genes identified under the healthy condition via RNA-seq. Expression levels of (A) transferrin a, (B) complement C1q subcomponent subunit C (Chr.18), (C) IL-1β, (D) G-type lysozyme, (E) complement C1q subcomponent subunit b, (F) complement C1q subcomponent subunit C (Chr. 5), (G) phospholipase A2 (group IB), and (H) chymotrypsin-like elastase family member 1 were compared between healthy and exposed conditions (24 and 48 h) of immersed WT and IL-17A/F1 (11-bp insertion strain) medaka in E. piscicida-containing tanks (6.9 × 108 CFU/mL). **P < 0.01, *P < 0.05 (Student's t-test) WT medaka group vs. KO(+11) medaka group at each time point. Data are presented as mean ± SD; n = 5 fish' SD depends on the statistical analyses run.
We next analyzed the 16S rRNA V3 and V4 amplicon sequences of the bacterial communities from 30 intestinal content samples. WT and IL-17A/F1-KO were compared under non-exposed and E. piscicida-exposed conditions. In total, 1,340,455 raw sequence reads were obtained. After adapter and quality trimming, 870,792 reads were used for OTU characterization and OTU-based analysis by QIIME 2. Finally, these sequences were clustered into 2065 OTUs at a 97% identity threshold, with OTUs representing 711 different taxa. For representative OTUs in non-exposed samples, the OTU numbers of KO medaka were higher than for WT (P = 0.058). The average number of OTUs was 104 and 137.4 in the WT and KO medaka, respectively. Furthermore, the number of OTUs in the non-exposed WT group was significantly increased compared with that of the two exposed WT groups (non-exposed state, 104.0 OTUs; 24-h exposure, 192.4 OTUs; 48-h exposure, 229 OTUs). However, these significant increases in OTUs after E. piscicida exposure were not observed in the KO medaka (Supplementary Figure 4).
At the phylum level, Proteobacteria (65.90%) was the dominant phylum, followed by Fusobacteria (20.96%) and Bacteroidetes (3.65%). These phyla of representative OTUs accounted for 90.5% of the reads and were commonly found across all 30 intestinal content samples. Other than these dominant phyla, 31 phyla, including Chlamydiae, OD-1, Planctomycetes, TM7, Verrucomicrobia, Cyanobacteria, Actinobacteria, Firmicutes, and Tenericutes, were identified from the representative OTUs (Figure 5A). At the phylum level, KO medaka showed different intestinal bacterial diversity compared to WT under a non-exposed state. Although not significant, Cyanobacteria showed an increased tendency in the KO compared with WT medaka under a non-exposed state (t-test; P = 0.08; Figure 6A). The representative OTU percentages of Verrucomicrobia and Planctomycetes were significantly higher in KO than in WT medaka under a non-exposed state (P < 0.05; Figures 6B,C). After exposure, WT and KO medaka showed variations in phylum proportions. The proportion of Actinobacteria in the WT increased significantly after 48 h, whereas KO medaka did not show a significant increase. Additionally, Actinobacteria was significantly higher in WT than KO after 48 h exposure (Figure 6D). Furthermore, for Firmicutes and OD-1, WT showed a significant increase after 48 h of exposure, whereas KO did not show a significant increase (Figures 6E,F). Conversely, the Chlamydiae phylum was decreased after 48 h of exposure in both WT and KO (Figure 6G).
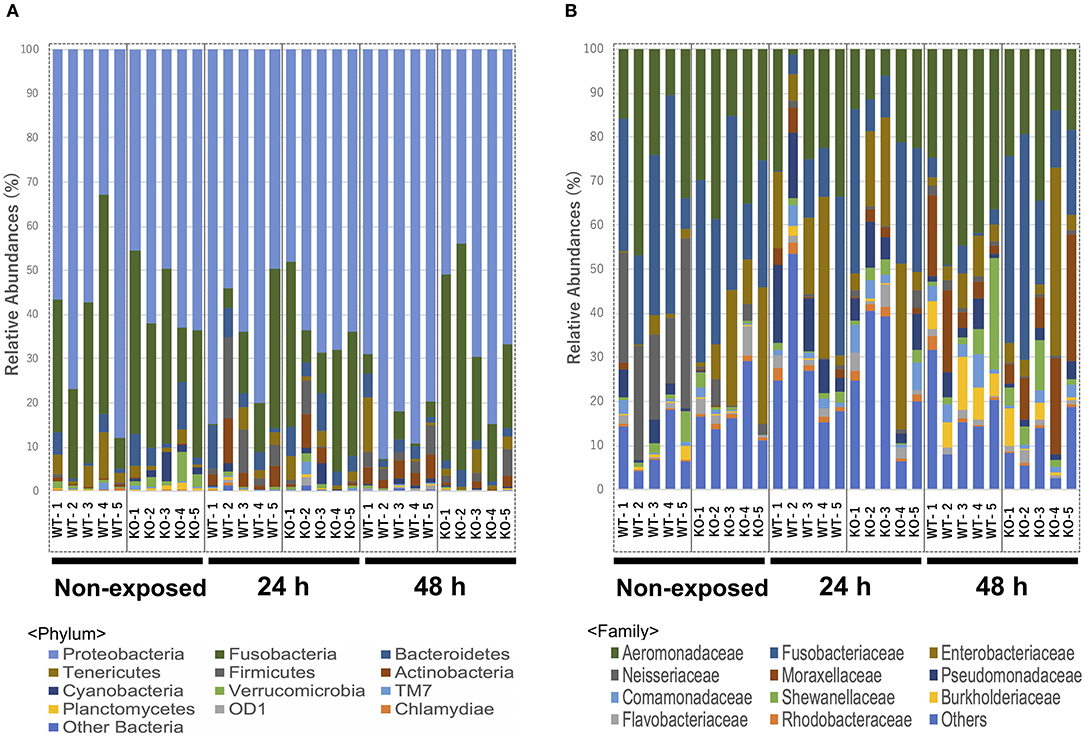
Figure 5. Relative abundance of the intestinal bacterial communities of WT and IL-17A/F1-KO at different taxonomic levels in healthy medaka and medaka immersed for 24 h in E. piscicida-containing tanks (2.1 × 108 CFU/mL). (A) Relative abundance of gut microbiome communities of WT and IL-17A/F1-KO at the phylum level (n = 5). (B) Relative abundance of intestinal bacterial communities of WT and IL-17A/F1-KO at the family level. Data were obtained from one experiment with five individual fish (n = 5).
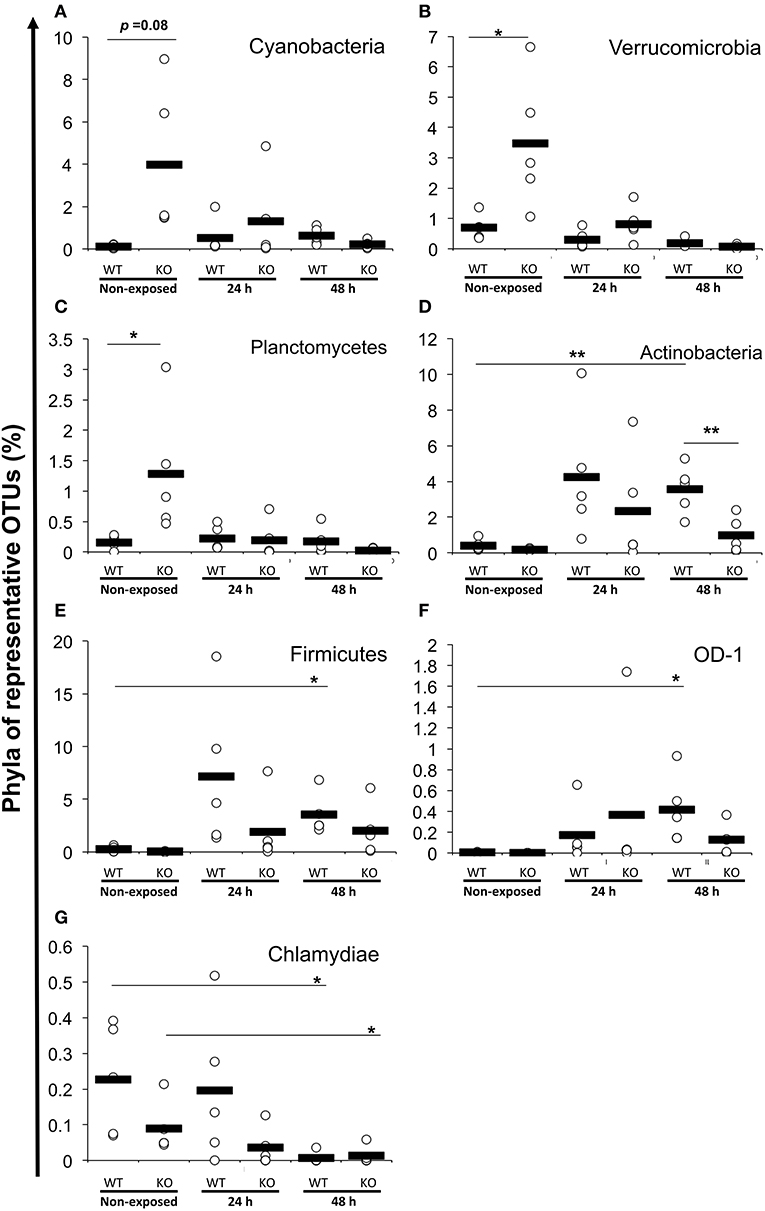
Figure 6. Increased or decreased phylum abundance of the top 12 dominant phylum total reads between WT and KO medaka or against E. piscicida exposure. Abundance of (A) Cyanobacteria, (B) Verrucomicrobia, (C) Planctomycetes, (D) Actinobacteria, (E) Firmicutes, (F) OD-1, and (G) Chlamydiae. **P < 0.01, *P < 0.05 (Student's t-test). Data are from one experiment with five individual fish (n = 5).
At the family level, Aeromonadaceae, Fusobacteriaceae, Enterobacteriaceae, Neisseriaceae, Moraxellaceae, Pseudomonadaceae, Shewanellaceae, Comamonadaceae, Burkholderiaceae, Flavobacteriaceae, and Rhodobacteraceae were dominant among core bacteria, which occupied over 82% of total reads (Figure 5B). Under a non-exposed state, the proportions of Enterobacteriaceae, Rhodobacteraceae, Xanthomonadaceae, Cryomorphaceae, OM60, and Acetobacteraceae were significantly higher in KO than WT groups, whereas Neisseriaceae and Pseudomonadaceae were significantly lower. These significant differences were not observed after 24 h of exposure. However, after 48 h, the WT and KO groups showed significant differences once more in the microbiome at the family level, with increased proportions of Aeromonadaceae, Neisseriaceae, Corynebacteriaceae, Xanthomonadaceae, and Propionibacteriaceae in the WT compared with the KO group. Meanwhile, only Fusobacteriaceae in the KO group was significantly higher than in WT at 48 h (Supplementary Table 3). At the genus level, Edwardsiella, for which E. piscicida belongs, was detected only in E. piscicida immersed groups in both WT and KO groups at 24 and 48 h. However, no significant changes in containing rate were observed between WT and KO (Supplementary Figure 5). Furthermore, at the OTU level, P. shigelloides was significantly higher in KO than WT under a non-exposed state (Table 3), which was confirmed by qPCR (Supplementary Figure 6). Other than P. shigelloides, Cetobacterium somerae level was significantly higher in KO than WT at 48 h. Chitinibacter tainanensis, however, was significantly higher in the WT than the KO group at 0 and 48 h of exposure (Table 3).
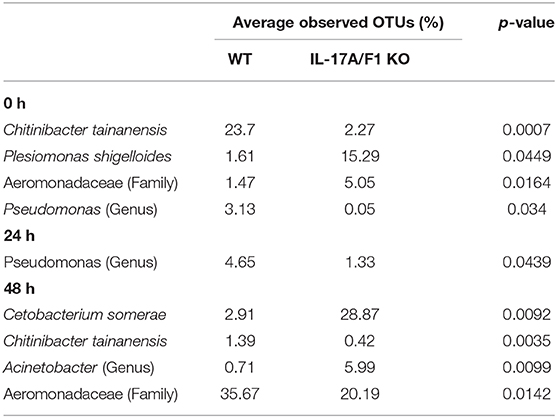
Table 3. Comparison of gut microbiome OTUs in WT and KO medaka intestines.
Shannon and Chao 1 diversity indices were determined for α-diversity analysis of the metagenome data. The Shannon diversity index indicated that bacterial diversity in the WT increased significantly as exposure progressed (non-exposed state vs. 24-h exposure, P < 0.01; non-exposed state vs. 48-h exposure, P < 0.05). However, between the KO groups, no significant changes were observed (Figure 7A). The Chao 1 diversity index showed a significant change (P < 0.05) only in the WT after 48 h exposure when compared to non-exposure (Figure 7B).
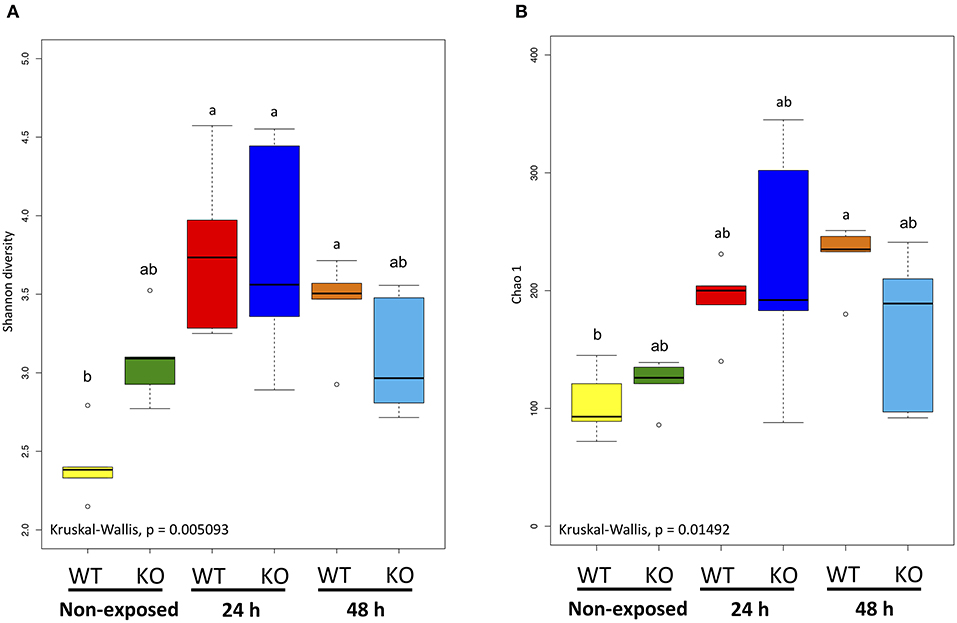
Figure 7. α-Diversity indices of the intestinal bacterial communities of WT and IL-17A/F1-KO medaka. (A) Shannon diversity and (B) Chao 1 indices were determined by R studio. Different letters above the bars indicate significant differences as determined by Dunn's tests. Data are from one experiment with five individual fish (n = 5).
The microbial community composition of the WT and KO as well as each non-exposed and E. piscicida-exposed groups were visualized according to the weighted UniFrac distance-based analysis (with statistical confirmation), which accounts for bacterial abundance rather than presence and absence of taxa. In non-exposed samples, WT and KO groups formed significantly different clusters (P = 0.011). After 24 h of exposure, these clusters became mixed and showed no significant differences. However, after 48 h of exposure, the WT and KO groups formed different clusters again (P = 0.018). Comparisons between the exposed and non-exposed WT groups showed that the non-exposed groups at 24 and 48 h were in statistically different clusters (non-exposed state vs. 24 h, P = 0.011; non-exposed state vs. 48 h, P = 0.007; 24 h vs. 48 h, P = 0.009). However, in the KO groups, no significant differences were observed between 24 and 48 h of exposure (non-exposed vs. 24 h, P = 0.013; non-exposed state vs. 48 h, P = 0.005; 24 h vs. 48 h, P = 0.122; Figure 8).
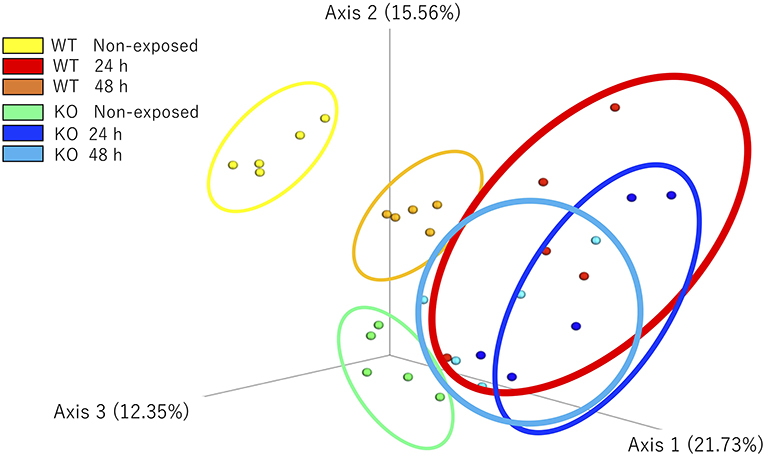
Figure 8. β-Diversity indices of the intestinal bacterial communities of WT and IL-17A/F1-KO medaka. Weighted UniFrac distance-based analyses were performed by R studio. Different letters above the bars indicate significant differences as determined by PERMANOVA. Data are from one experiment with five individual fish (n = 5).
In this study, we established two strains of IL-17A/F1-KO medaka. Medaka IL-17A/F1 and 2 are adjacent on the same chromosome as well as mammalian IL-17A and -F, whereas IL-17A/F3 is located on a different chromosome (20). Gene expression analysis of IL-17A/F1, 2, and 3 in the medaka intestinal tract revealed that IL-17A/F3 levels were the highest, whereas IL-17A/F2 levels were extremely low. This expression pattern of IL-17A/F1, 2, and 3 was previously reported in yellow croaker (Larimichthys polyactis) (23). In contrast, IL-17A/F2 expression in the intestinal tract of salmonids was relatively high (22). Thus, IL-17A/F1, 2, and 3 gene expression levels are different among fish species. In teleost IL-17A/F1, it was reported that yellow croaker IL-17A/F1 has a higher ability to activate the transcriptional activity of NF-κB than IL-17A/F2 and 3 (23). Furthermore, in Japanese pufferfish, IL-17A/F1 showed a more acute up-regulated response compared to IL-17A/F3 (20). Based on these previous studies, we selected medaka IL-17A/F1 as a target gene of genome editing. However, the differences in the functionality of IL-17A/F1, 2, and 3 remain unclear, which warrants future research to increase our understanding of these genes.
In mammalian small intestines, IL-17A and F play important roles in the production of AMPs, such as defensin and calprotectin (11, 14). In the present study, we performed transcriptome analysis (RNA-seq) to compare gene expression pattern between WT and IL-17A/F1-KO intestines to elucidate the role of medaka IL-17A/F1. Expression levels of immune-related genes, such as Tfa and C1qc, and various lipases and proteinases, such as phospholipase A2 and chymotrypsin, were found to be down-regulated in the intestines of IL-17A/F1-KO medaka compared to WT. The induction of these genes has not been reported to depend on IL-17A and F in mammals.
Among the genes with reduced expression in KO individuals, several have been reported to be important in regulating the gut microbiome in vertebrates. Transferrin a is an iron ion transporter and an important factor in homeostasis maintenance, in which the iron ion absorption effect is known to attenuate bacterial proliferation. Transferrin a is also abundant in mucosal tissues and considered to inhibit bacterial survival (32, 33). Moreover, the transferrin gene has been identified in several fish species, including the Japanese medaka, and is known to be induced at the transcriptional level upon bacterial infection stress (34–36). C1q is known to trigger initiation of the classical pathway of the complement system (37). In zebrafish, C1q production via the transcription factor interferon regulatory factor-8 (IRF8) in macrophages modulates the intestinal microbiome (38).
Digestive enzymes are also important for regulation of the intestinal tract microbiome and immune-related genes, such as Tfa and C1q. Members of the chymotrypsin and elastase family are enzymes involved in the degradation of the outer membrane protein OmpA of Gram-negative bacteria (39). Although phospholipase A2 (group IB; pla2g1b) mainly acts as a lipid degrading enzyme, phospholipase A2 (PLA2) produced from intestinal epithelial cells is also important for defense against parasite infection (40). Furthermore, phospholipase A2 (group IIA; pla2g2A), which is classified as the same phospholipase, is known as a marker of Paneth cells. In fact, secreted PLA2 modulates intestinal stem cell and Paneth cell differentiation (41). In addition, expression levels of pla2g2A in IL-17A- or F-KO mouse intestines are reduced compared with those of WT mice, with IL-17F-KO mice showing a particularly marked decrease (42). Thus, the significant down-regulation of several genes, as shown by RNA-seq, appear to affect gut microbiome control in the intestinal tract of IL-17A/F1-KO(-7) medaka. Considering that the expression of IL-17 KO-dependent genes was different from that in mammals, we theorized that the fish intestinal tract may have a unique and specific IL-17A/F1-mediated microbiome control mechanism, which differs from that of the mammalian intestinal tract.
Gene expression analysis by qPCR confirmed that the two IL-17A/F1 mutant strains established in this study showed similar trends in gene down-regulation; Tfa, C1qc, C1qb, IL-1β, lyzG, and the lipase and proteinase genes PLA2 and CELA1 were down-regulated compared to the WT strain. RNA-seq results showed that, although not significant, expression levels of IL-1β, LyzG, and C1q genes [C1qb and C1qc (Chr. 5)] were decreased in the IL-17A/F1 knockout. In mammalian intestines, lysozyme is known as a marker of Paneth cells, which secrete AMPs from the intestinal epithelia layer (43). In this study, RNA-seq indicated that expression levels of both lysozymes, LyzC and lyzG, were reduced. However, LyzC expression was not significantly decreased in the qPCR analysis. It is of note that expression levels of Paneth cell markers (e.g., PLA2 and LyzC) were reduced in the IL-17A/F1-KO medaka strains, although Paneth cells have not yet been identified in fish. Moreover, among the down-regulated genes identified by RNA-seq, IL-1β was the only gene induced via mammalian IL-17 signaling. In the mammalian intestinal tract, IL-17A and F stimulate macrophages to induce IL-1β and promote the aggregation of neutrophils and other lymphocytes (12). In addition, IL-1β is essential for the differentiation of lymphocytes, including Th17 and γδ T cells, which are IL-17-producing cells (44). Moreover, in fish, it has been reported that recombinant IL-17A/F1 of grass carp (Ctenopharyngodon Idella) and IL-17A/F2 of common carp (Cyprinus carpio) induce IL-1β in vitro (45, 46). However, it has not been reported that IL-1β is induced in an IL-17A/F-dependent manner in the fish intestinal tract.
For metagenomic analysis, the IL-17A/F1-KO(+11) strain was used, and changes in the gut microbiome during E. piscicida exposure were analyzed. Between WT and KO groups, there was no remarkable difference in the number of OTUs or diversity. Under a non-exposed state, however, WT and IL-17A/F1-KO medaka exhibited different gut microbiomes. The observed number of OTUs and the α-diversity index increased in the KO strain compared with the WT. Furthermore, the β-diversity test revealed that the two groups were in significantly different clusters. In a previous β-diversity test, IL-17A-KO mice formed a different gut microbiome to WT mice (47), with IL-17F-KO mice showing similar results (42). In addition, mice with knock out of IL-17RA and RC, which are receptors for IL-17A and F, respectively, are highly sensitive to the immune disease, Graft-vs.-host disease (GVDH). Moreover, after co-housing WT mice with IL-17RA- or RC-KO mice, the gut microbiome of WT mice became similar to that of IL-17RA/RC-KO mice, with increased sensitivity to GVHD (48). Based on these findings, the health importance of gut microbiome regulation by IL-17A and F signaling via IL-17RA/RC in mammals is clear. However, this importance has not been shown in fish. Our findings suggest that the IL-17A/F gene is an important inflammatory cytokine for regulating the gut microbiome in fish. For a better understanding of this role of IL-17A/F, it is necessary to simultaneously analyze other IL-17A/F ligands (i.e., IL-17A/F2 and/or 3) and their receptors (i.e., IL-17RA and RC).
Prior to metagenomic analysis, we compared the susceptibility of WT and IL-17A/F1-KO(+11) strains to E. piscicida exposure, but no differences in cumulative mortality after exposure were observed (Supplementary Figure 7). The effect of IL-17A/F1-KO on the gut microbiome may be limited to the control of opportunistic bacteria resident in the intestines. Opportunistic bacteria were examined at the phylum level, and the results revealed Proteobacteria as the most dominant phylum in the intestinal microbiome of medaka, followed by Fusobacteria. By contrast, the gut microbiome of mammals at the phylum level is dominated by Firmicutes and Bacteroides, with only a low percentage of Proteobacteria (49). However, the fish gut microbiome has been reported to be dominated by Proteobacteria and Fusobacteria, which is consistent with our results. In addition, in both freshwater and marine fish species, the predominance of Proteobacteria and Fusobacteria is common (50, 51); however, this is not a simple reflection of the bacteria present in the surrounding aquatic environment. Shrimp, which is also an aquatic organism, has predominant Proteobacteria levels but hardly contains Fusobacterium (52), whereas the whale gut microbiome is similar to that of land mammals (53). Although the percentage of Proteobacteria and Fusobacteria in this study showed no significant change between the WT and KO strains, others types of phyla showed different composition rates between WT and KO. Under a non-exposed state, Cyanobacteria, Verrucomicrobia, and Planctomycetes were increased in the KO gut microbiome compared with the WT. Cyanobacteria are known to play important roles in the activation of host cell signaling by producing their primary metabolites, but the cyanotoxins produced may be harmful to many vertebrate species (54). Meanwhile, at the OTU level, significant changes were observed between WT and KO with regards to several bacterial species. No pathogenicity has been reported for any of these species, except for P. shigelloides. P. shigelloides is an intestinal bacterium that is widely known as a causative agent of food poisoning in humans and is also present in freshwater. In recent years, P. shigelloides infection has been reported to be responsible for the high mortality of several freshwater fish species as it is an opportunistic pathogen (55). Increases in the proportion of P. shigelloides in the KO strains may indicate an increased risk of death due to an opportunistic infection. The effects of P. shigelloides on health and changes in susceptibility to infections other than E. piscicida have not been clarified in this study, warranting future research.
In summary, we established IL-17A/F1-deficient Japanese medaka strains in this study. In the IL-17A/F1-KO medaka intestines, the expression of various immune-related genes as well as proteinase and lipase genes was significantly down-regulated. Furthermore, 16S rRNA-based metagenomic analysis revealed that the WT and KO strains formed different gut microbiomes, with a significantly increased proportion of P. shigelloides—a fish opportunistic pathogen—in the KO strains, suggesting that medaka IL-17A/F1 may have a role in maintaining healthy intestines by modulating the gut microbiome via transcriptional control of the affected genes.
The datasets generated for this study can be found in the DDBJ Sequence Read Archive (DRA) under accession number DRA008715, DRA008844, GenBank accession number AJ300545.
The animal study was reviewed and approved by University of Miyazaki.
YO, TA, MS, and JH designed the research. YO, NMo, DI, NMi, HM, MK, TK, and JH conducted the research. YO, DI, SW, YS, HT, and TK analyzed the data. YO, MS, and JH wrote the paper. All authors reviewed and approved the final manuscript.
This work was supported by a Grant-in-Aid for Scientific Research (KAKENHI grant numbers 17H01486 and 17H03863) from the Japan Society for the Promotion of Science (JSPS), Japan.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Tomokazu Takano (National Research Institute of Aquaculture, Japan Fisheries Research, and Education Agency) for providing E. piscicida (E381 strain). We would like to thank Editage (www.editage.jp) for English language editing.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00425/full#supplementary-material
1. Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. (2007) 449:804–10. doi: 10.1038/nature06244
2. Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. (2012) 486:207–14. doi: 10.1038/nature11234
3. Hillman ET, Lu H, Yao T, Nakatsu CH. Microbial ecology along the gastrointestinal tract. Microbes Environ. (2017) 32:300–13. doi: 10.1264/jsme2.ME17017
4. Lopetuso LR, Scaldaferri F, Franceschi F, Gasbarrini A. The gastrointestinal microbiome - Functional interference between stomach and intestine. Best Pract Res Clin Gastroenterol. (2014) 28:995–1002. doi: 10.1016/j.bpg.2014.10.004
5. Halfvarson J, Brislawn CJ, Lamendella R, Vázquez-Baeza Y, Walters WA, Bramer LM, et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat Microbiol. (2017) 2:17004. doi: 10.1038/nmicrobiol.2017.4
6. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. (2006) 441:235–8. doi: 10.1038/nature04753
7. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+T helper cells. Cell. (2006) 126:1121–33. doi: 10.1016/j.cell.2006.07.035
8. Huber S, Gagliani N, Flavell RA. Life, death, and miracles: Th17 cells in the intestine. Eur J Immunol. (2012) 42:2238–45. doi: 10.1002/eji.201242619
9. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. (2009) 139:485–98. doi: 10.1016/j.cell.2009.09.033
10. Goto Y, Panea C, Nakato G, Cebula A, Lee C, Diez MG, et al. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity. (2014) 40:594–607. doi: 10.1016/j.immuni.2014.03.005
11. Isailovic N, Daigo K, Mantovani A, Selmi C. Interleukin-17 and innate immunity in infections and chronic inflammation. J Autoimmun. (2015) 60:1–11. doi: 10.1016/j.jaut.2015.04.006
12. Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin-17 family members. Immunity. (2011) 34:149–62. doi: 10.1016/j.immuni.2011.02.012
13. Moseley TA, Haudenschild DR, Rose L, Reddi AH. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. (2003) 14:155–74. doi: 10.1016/S1359-6101(03)00002-9
14. Kirkham BW, Kavanaugh A, Reich K. Interleukin-17A: a unique pathway in immune-mediated diseases: psoriasis, psoriatic arthritis and rheumatoid arthritis. Immunology. (2014) 141:133–42. doi: 10.1111/imm.12142
15. Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol. (2011) 9:356–68. doi: 10.1038/nrmicro2546
16. Masuda K, Sakai N, Nakamura K, Yoshioka S, Ayabe T. Bactericidal activity of mouse α-defensin cryptdin-4 predominantly affects noncommensal bacteria. J Innate Immun. (2011) 3:315–26. doi: 10.1159/000322037
17. Kono T, Korenaga H, Sakai M. Genomics of fish IL-17 ligand and receptors: a review. Fish Shellfish Immunol. (2011) 31:635–43. doi: 10.1016/j.fsi.2010.11.028
18. Wang X, Li C, Thongda W, Luo Y, Beck B, Peatman E. Characterization and mucosal responses of interleukin 17 family ligand and receptor genes in channel catfish Ictalurus punctatus. Fish Shellfish Immunol. (2014) 38:47–55. doi: 10.1016/j.fsi.2014.02.020
19. Monte MM, Wang T, Holland JW, Zou J, Secombes CJ. Cloning and characterization of rainbow trout interleukin-17A/F2 (IL-17A/F2) and IL-17 receptor a: expression during infection and bioactivity of recombinant IL-17A/F2. Infect Immun. (2013) 81:340–53. doi: 10.1128/IAI.00599-12
20. Korenaga H, Kono T, Sakai M. Isolation of seven IL-17 family genes from the Japanese pufferfish Takifugu rubripes. Fish Shellfish Immunol. (2010) 28:809–18. doi: 10.1016/j.fsi.2010.01.016
21. Nuñez Ortiz N, Gerdol M, Stocchi V, Marozzi C, Randelli E, Bernini C, et al. T cell transcripts and T cell activities in the gills of the teleost fish sea bass (Dicentrarchus labrax). Dev Comp Immunol. (2014) 47:309–18. doi: 10.1016/j.dci.2014.07.015
22. Wang T, Jiang Y, Wang A, Husain M, Xu Q, Secombes CJ. Identification of the salmonid IL-17A/F1a/b, IL-17A/F2b, IL-17A/F3 and IL-17N genes and analysis of their expression following in vitro stimulation and infection. Immunogenetics. (2015) 67:395–412. doi: 10.1007/s00251-015-0838-1
23. Ding Y, Ao J, Ai C, Chen X. Molecular and functional identification of three interleukin-17A/F (IL-17A/F) homologues in large yellow croaker (Larimichthys crocea). Dev Comp Immunol. (2016) 55:221–32. doi: 10.1016/j.dci.2015.09.010
24. Brugman S. The zebrafish as a model to study intestinal inflammation. Dev Comp Immunol. (2016) 64:82–92. doi: 10.1016/j.dci.2016.02.020
25. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. (2009) 25:1105–11. doi: 10.1093/bioinformatics/btp120
26. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. (2010) 28:511–5. doi: 10.1038/nbt.1621
27. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. (2012) 7:562–78. doi: 10.1038/nprot.2012.016
28. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using David bioinformatics resources. Nat Protoc. (2009) 4:44–57. doi: 10.1038/nprot.2008.211
29. Supek F, Bošnjak M, Škunca N, Šmuc T. Revigo summarizes and visualizes long lists of gene ontology terms. PLoS ONE. (2011) 6:e21800. doi: 10.1371/journal.pone.0021800
30. Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable, and extensible microbiome data science using QIIME. Nat Biotechnol. (2019) 37:852–7. doi: 10.1038/s41587-019-0209-9
31. R Core Team. R: A Language and Environment for Statistical Computing. Vol. 3. Vienna: R Foundation for Statistical Computing (2018). p. 201. Available online at: https://www.R-project.org/
32. Grange PA, Mouricout MA. Transferrin associated with the porcine intestinal mucosa is a receptor specific for K88ab fimbriae of Escherichia coli. Infect Immun. (1996) 64:606–10. doi: 10.1128/IAI.64.2.606-610.1996
33. Andrews SC, Robinson AK, Rodríguez-Quiñones F. Bacterial iron homeostasis. FEMS Microbiol Rev. (2003) 27:215–37. doi: 10.1016/S0168-6445(03)00055-X
34. Mikawa N, Hirono I, Aoki T. Structure of medaka transferrin gene and its 5′-flanking region. Mol Mar Biol Biotechnol. (1996) 5:225–9.
35. Nynca J, Dietrich MA, Adamek M, Steinhagen D, Bilinska B, Hejmej A, et al. Purification, characterization and expression of transferrin from rainbow trout seminal plasma. Comp Biochem Physiol B Biochem Mol Biol. (2017) 208–9:38–46. doi: 10.1016/j.cbpb.2017.04.002
36. Perera NCN, Godahewa GI, Hwang JY, Kwon MG, Hwang SD, Lee J. Molecular, structural, and functional comparison of N lobe and C lobe of the transferrin from rock bream, Oplegnathus fasciatus, with respect to its immune response. Fish Shellfish Immunol. (2017) 68:299–309. doi: 10.1016/j.fsi.2017.07.033
37. Mortensen SA, Sander B, Jensen RK, Pedersen JS, Golas MM, Jensenius JC, et al. Structure and activation of C1, the complex initiating the classical pathway of the complement cascade. Proc Natl Acad Sci USA. (2017) 114:986–91. doi: 10.1073/pnas.1616998114
38. Earley AM, Graves CL, Shiau CE. Critical role for a subset of intestinal macrophages in shaping gut microbiota in adult zebrafish. Cell Rep. (2018) 25:424–36. doi: 10.1016/j.celrep.2018.09.025
39. Belaaouaj AA, Kim KS, Shapiro SD. Degradation of outer membrane protein A in Escherichia coli killing by neutrophil elastase. Science. (2000) 289:1185–8. doi: 10.1126/science.289.5482.1185
40. Entwistle LJ, Pelly VS, Coomes SM, Kannan Y, Perez-Lloret J, Czieso S, et al. epithelial-cell-derived phospholipase A2 group 1B is an endogenous anthelmintic. Cell Host Microbe. (2017) 22:484–93.e5. doi: 10.1016/j.chom.2017.09.006
41. Schewe M, Franken PF, Sacchetti A, Schmitt M, Joosten R, Böttcher R, et al. Secreted phospholipases A2 are intestinal stem cell niche factors with distinct roles in homeostasis, inflammation, and cancer. Cell Stem Cell. (2016) 19:38–51. doi: 10.1016/j.stem.2016.05.023
42. Tang C, Kakuta S, Shimizu K, Kadoki M, Kamiya T, Shimazu T, et al. Suppression of IL-17F, but not of IL-17A, provides protection against colitis by inducing T reg cells through modification of the intestinal microbiota. Nat Immunol. (2018) 19:755–65. doi: 10.1038/s41590-018-0134-y
43. Coutinho HB, da Mota HC, Coutinho VB, Robalinho TI, Furtado AF, Walker E, et al. Absence of lysozyme. (muramidase) in the intestinal Paneth cells of newborn infants with necrotising enterocolitis. J Clin Pathol. (1998) 51:512–4. doi: 10.1136/jcp.51.7.512
44. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KHG. Interleukin-1 and IL-23 induce innate IL-17 production from γδ T cells, amplifying Th17 responses and autoimmunity. Immunity. (2009) 31:331–41. doi: 10.1016/j.immuni.2009.08.001
45. Du L, Feng S, Yin L, Wang X, Zhang A, Yang K, et al. Identification and functional characterization of grass carp IL-17A/F1: an evaluation of the immunoregulatory role of teleost IL-17A/F1. Dev Comp Immunol. (2015) 51:202–11. doi: 10.1016/j.dci.2015.03.014
46. Li H, Yu J, Li J, Tang Y, Yu F, Zhou J, et al. Cloning and characterization of two duplicated interleukin-17A/F2 genes in common carp (Cyprinus carpio L.): transcripts expression and bioactivity of recombinant IL-17A/F2. Fish Shellfish Immunol. (2016) 51:303–12. doi: 10.1016/j.fsi.2016.01.042
47. Bi Y, Li C, Liu L, Zhou J, Li Z, Deng H, et al. IL-17A-dependent gut microbiota is essential for regulating diet-induced disorders in mice. Sci Bull. (2017) 62:1052–63. doi: 10.1016/j.scib.2017.07.011
48. Varelias A, Ormerod KL, Bunting MD, Koyama M, Gartlan KH, Kuns RD, et al. Acute graft-versus-host disease is regulated by an IL-17–sensitive microbiome. Blood. (2017) 129:2172–85. doi: 10.1182/blood-2016-08-732628
49. Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, et al. Enterotypes of the human gut microbiome. Nature. (2011) 473:174–80. doi: 10.1038/nature09944
50. Riiser ES, Haverkamp THA, Borgan Ø, Jakobsen KS, Jentoft S, Star B. A single Vibrionales 16S rRNA oligotype dominates the intestinal microbiome in two geographically separated Atlantic cod populations. Front Microbiol. (2018) 9:1561. doi: 10.3389/fmicb.2018.01561
51. Liu H, Guo X, Gooneratne R, Lai R, Zeng C, Zhan F, et al. The gut microbiome and degradation enzyme activity of wild freshwater fishes influenced by their trophic levels. Sci Rep. (2016) 6:24340. doi: 10.1038/srep24340
52. Gainza O, Ramírez C, Ramos AS, Romero J. Intestinal microbiota of white shrimp Penaeus vannamei under intensive cultivation conditions in Ecuador. Microb Ecol. (2018) 75:562–8. doi: 10.1007/s00248-017-1066-z
53. Erwin PM, Rhodes RG, Kiser KB, Keenan-Bateman TF, McLellan WA, Pabst DA. High diversity and unique composition of gut microbiomes in pygmy (Kogia breviceps) and dwarf (K. sima) sperm whales. Sci Rep. (2017) 7:7205. doi: 10.1038/s41598-017-07425-z
54. Ziková A, Palíková M, Mareš J, Navrátil S, Kopp R. Impacts of dietary cyanobacteria on fish. Acta. Univ Agric Silvic Mendelianae Brun. (2010) 58:277–84. doi: 10.11118/actaun201058040277
Keywords: genome editing, interleukin-17A/F1, Japanese medaka, Oryzias latipes, transcriptome analysis, metagenomics, teleosts
Citation: Okamura Y, Morimoto N, Ikeda D, Mizusawa N, Watabe S, Miyanishi H, Saeki Y, Takeyama H, Aoki T, Kinoshita M, Kono T, Sakai M and Hikima J (2020) Interleukin-17A/F1 Deficiency Reduces Antimicrobial Gene Expression and Contributes to Microbiome Alterations in Intestines of Japanese medaka (Oryzias latipes). Front. Immunol. 11:425. doi: 10.3389/fimmu.2020.00425
Received: 18 November 2019; Accepted: 25 February 2020;
Published: 17 March 2020.
Edited by:
Jonathan P. Rast, Emory University, United StatesReviewed by:
Katherine Buckley, Auburn University, United StatesCopyright © 2020 Okamura, Morimoto, Ikeda, Mizusawa, Watabe, Miyanishi, Saeki, Takeyama, Aoki, Kinoshita, Kono, Sakai and Hikima. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun-ichi Hikima, amhpa2ltYUBjYy5taXlhemFraS11LmFjLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.