 Charles Caër
Charles Caër Mary Jo Wick
Mary Jo Wick- Department of Microbiology and Immunology, Institute of Biomedicine, University of Gothenburg, Gothenburg, Sweden
Inflammatory bowel disease (IBD), including Crohn's disease and ulcerative colitis, is a complex immune-mediated disease of the gastrointestinal tract that increases morbidity and negatively influences the quality of life. Intestinal mononuclear phagocytes (MNPs) have a crucial role in maintaining epithelial barrier integrity while controlling pathogen invasion by activating an appropriate immune response. However, in genetically predisposed individuals, uncontrolled immune activation to intestinal flora is thought to underlie the chronic mucosal inflammation that can ultimately result in IBD. Thus, MNPs are involved in fine-tuning mucosal immune system responsiveness and have a critical role in maintaining homeostasis or, potentially, the emergence of IBD. MNPs include monocytes, macrophages and dendritic cells, which are functionally diverse but highly complementary. Despite their crucial role in maintaining intestinal homeostasis, specific functions of human MNP subsets are poorly understood, especially during diseases such as IBD. Here we review the current understanding of MNP ontogeny, as well as the recently identified human intestinal MNP subsets, and discuss their role in health and IBD.
Introduction
Crohn's disease and ulcerative colitis (UC) are chronic inflammatory disorders of the digestive tract that comprise the term inflammatory bowel disease (IBD) (1, 2). These diseases are complex, severe, and chronic public health problems for which the incidence and prevalence are increasing worldwide (1, 2). Prevalence rates are highest in westernized countries, but ethnic and geographical differences are beginning to fade due to globalization (3, 4). The onset and pathophysiology of IBD are not fully understood, but the current concept is that uncontrolled immune reactivity against intestinal microorganisms combined with environmental factors in genetically predisposed individuals underlie pathogenesis (5–10). Infiltration of pro-inflammatory immune cells into the intestinal mucosa is induced; this releases cytokines and chemokines, creating a vicious circle and perpetuating tissue damage (11). Moreover, IBD is characterized by intestinal microbiota dysbiosis, with a reduction in both bacterial quantity and diversity (10, 12, 13). In some patients, mucosal inflammation is linked to these alterations and to bacteria-derived factors (14–16). However, whether changes in intestinal microbiota is a cause or a consequence of IBD is currently not known (8, 17–19). Finally, the disease course is characterized by repeated cycles of remission and relapse, adding further complexity to disease pathogenesis.
Crohn's disease can involve any part of the digestive tract, but predominantly the terminal ileum, while UC involves only the large intestine, mainly the rectum (1, 2). Generally, the onset of IBD is in young individuals, often 20–30 years old, and most IBD patients have a normal life expectancy thanks to existing treatments. However, despite very low mortality from IBD, morbidity remains a significant problem, and conventional medication involves escalating drug regimens with concomitant side effects. Moreover, IBD is not curable and increases the risk for lymphoma, biliary cancer, and colorectal cancer (20, 21). A significant number of IBD patients do not respond to treatments and must instead undergo surgery to relieve symptoms, often multiple times. Surgery is not only a major procedure for patients, but can also result in postoperative complications and infection, and negatively influences the quality of life (1, 2, 22). Regarding immunomodulators, some, albeit relatively few, targets have been identified; however, there is unfortunately a loss of treatment efficacy over time (1, 2, 11). Moreover, immunoregulation is altered during disease course and flare-ups, which affects treatment timing and efficacy (23). Thus, there is a great need to develop new targeted immunotherapies and, importantly, to identify methods to screen patients for likeliness to respond to a given therapy prior to starting treatment (24–26). To achieve these goals, it is important to further our understanding of IBD immunopathogenesis in humans.
Mononuclear phagocytes (MNPs) consist of multiple specialized innate immune cell types, including monocytes, macrophages (Mfs), and dendritic cells (DCs) (27–29). These cells are central to eliminating pathogens by their ability to sense, internalize and digest microbes and present antigens to T cells to drive adaptive immunity. They also secrete chemokines and cytokines, resulting in the migration and activation of immune cells (30–33). Importantly, both DCs and Mfs collaborate to maintain intestinal tolerance against food antigens and commensal bacteria through the induction and maintenance of regulatory T cells (Tregs) (34–40). Thus, MNPs have critical roles throughout the body in maintaining homeostasis and health. However, inappropriate activation of MNPs can induce sustained inflammation and tissue damage resulting in autoimmune and chronic inflammatory diseases such as IBD (41–47).
Therefore, defining how MNPs control immune homeostasis in the healthy human gastrointestinal tract, and their contribution to the aberrant immunoregulation that results in disease, is critical to improving treatments for IBD patients (7, 48–50).
This review focuses on the current understanding of MNP subset ontogeny as well as their role in the human intestine during health and IBD. Deciphering human intestinal MNP subset characteristics and understanding their roles in tipping the balance from intestinal health to IBD will provide insight for the development of new therapies to reset aberrant cellular functions that drive the chronic inflammation of IBD.
Ontogeny, Location, and Characterization of MNP Populations
Monocytes and Macrophages
Origin
Monocytes are produced in the bone marrow from common monocyte progenitors that derived from common myeloid progenitors (51–54) (Figure 1). They represent 2–8% of leukocytes in the peripheral blood of healthy individuals and constitute a versatile and dynamic cell population, composed of three major subsets: classical, intermediate and non-classical monocytes (55–57) (Figure 1). Classical monocytes circulate for only a day in the bloodstream and transmigrate to peripheral organs where a majority of them differentiate into tissue-specific resident Mfs and DCs following exposure to growth factors, cytokines, and microbial products in the local microenvironment (57, 58). However, a decade of data from mouse models provides evidence that most tissue-resident Mfs are seeded before birth, derived from erythro-myeloid progenitors in the yolk sac during embryonic development; they acquire tissue-specific characteristics through the microenvironment and possess self-renewal capacity (53, 59–67) (Figure 1). Interestingly, recent articles observed equivalent development and characteristics of tissue-resident Mfs in humans (68–70). There are two exceptions, dermal Mfs (53, 71) and intestinal Mfs, in both mice (53, 72–74) and in humans (75), which are continuously reconstituted by blood classical monocytes. Therefore, each organ has its own unique combination of embryonic and adult-derived Mf populations that change throughout life according to immune responses and tissue repair (53, 69, 75–77).
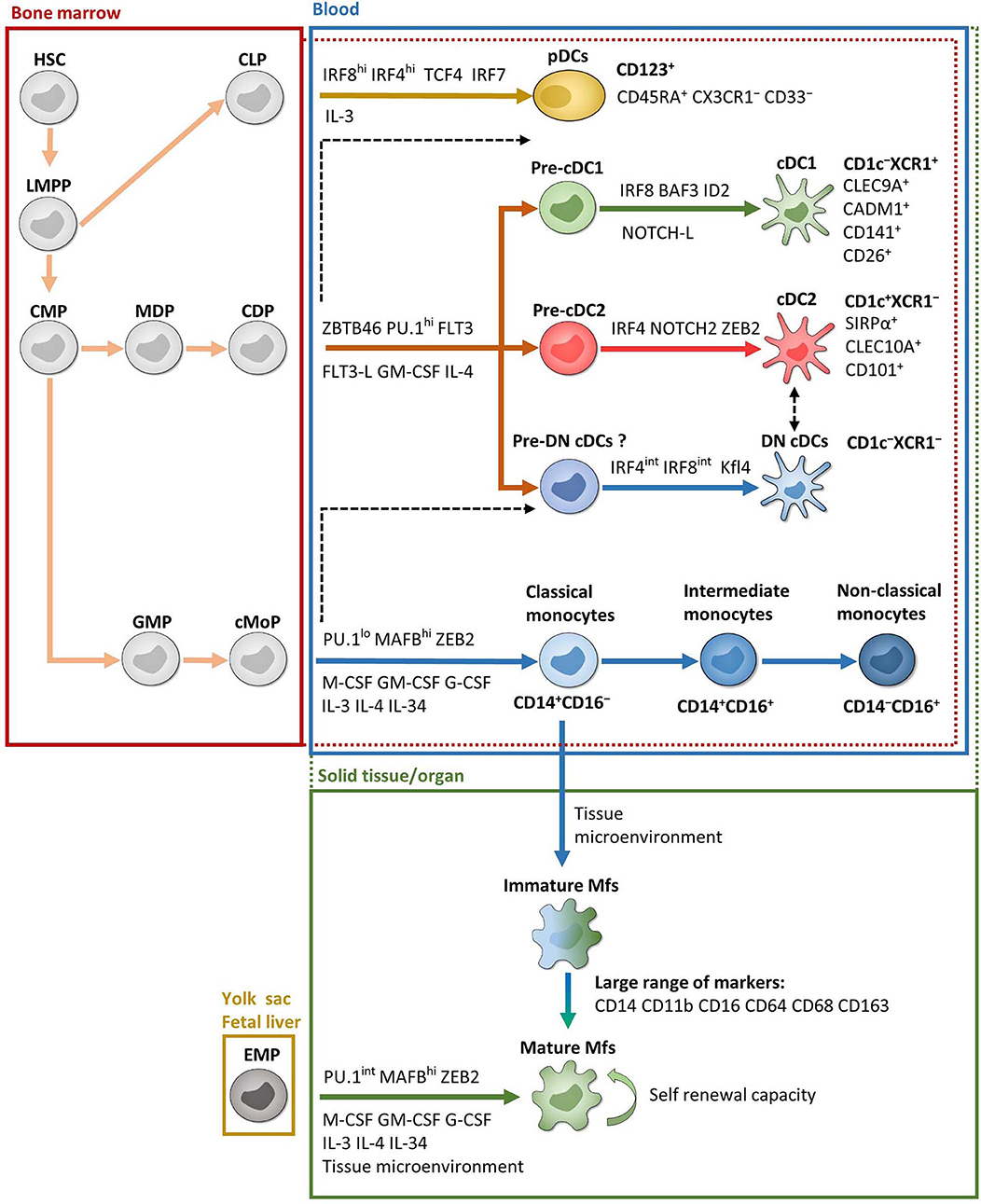
Figure 1. Ontogeny and development of MNPs. Except EMP and pDC ontogenies, which are exclusively from mice, most of the data are from humans. Red and green dotted lines represent possible overlaps between compartments. For example, pre-cDCs are present in both bone marrow and blood and cDCs are present in both blood and solid tissues. Black dashed lines represent possible developmental processes still under debate. CDP, common DC progenitor; CLP, common lymphoid progenitor; cMoP, common monocyte progenitor; CMP, common myeloid progenitor; EMP, erythro-myeloid progenitor; GMP, granulocyte-macrophage progenitor; HSC, hematopoietic stem cell; LMPP, lymphoid-primed multipotent progenitor; MDP, macrophage-DC progenitor. See the main text for other acronyms.
Development
In mouse, development of monocytes and Mfs from progenitors depends on essential transcription factors such as PU.1, MAFB, ZEB2, and macrophage colony-stimulating factor (M-CSF) (65, 78–81) (Figure 1). Several other growth factors and interleukins also play a significant role for their maintenance and homeostasis. This includes granulocyte macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), IL-3, IL-4, and IL-34 (41, 82–85). Importantly, a specific combination of transcription factors is required to maintain the tissue-specific identity of Mfs (81, 86). In humans, even if it is more difficult to study cell development, the same transcription factors and growth factors seem to be involved in monocyte and Mf development such as PU.1, MAFB, and M-CSF (87, 88) (Figure 1).
Location and Characterization
As stated above, the blood monocyte population has traditionally been divided into three subsets: CD14+CD16− classical monocytes, which represent 85% of the monocyte pool, CD14+CD16+ intermediate monocytes and CD14−CD16+ non-classical monocytes (55–57) (Figure 1). Each of these subsets possess specific extravasation and cell fate properties and are implicated in distinct functions and diseases (51, 57, 89, 90). Recently, using single-cell RNA sequencing (scRNA-Seq), Villani et al. have observed four blood monocyte subsets as a result of the high heterogeneity of intermediate monocytes (91). Nevertheless, the frequency of these cells is very low and supplementary studies are needed to fully understand their biological relevance. Moreover, two recent studies showed that one of these four subsets was a contamination by NK cells (92, 93).
Regarding human Mfs, the main phenotypic markers used to characterize them are CD14, CD11b, CD16, CD64, CD68, and CD163 depending on the tissue analyzed (Figure 1). Indeed, Mfs are tissue-specific populations, such as alveolar Mfs in lung or Kupffer cells in liver, which acquire and maintain identities according to their local microenvironment (32, 66, 81, 94–96). Moreover, each organ comprises Mf subsets with distinct phenotypes and functions according to their origin, fate and location. Finally, Mfs are essential to maintain tissue homeostasis, clear apoptotic cells, provide immune system regulation, perform tissue remodeling and repair, as well as defend against pathogens (32, 77, 84, 97–101). Characterization of human intestinal Mf subsets is discussed in detail below.
Dendritic Cells
Origin
The DC population is divided into three major subsets according to their ontogeny and functions: conventional DC1 (cDC1), conventional DC2 (cDC2) and plasmacytoid DCs (pDCs) (55, 102). Typically, DCs derive from bone marrow common DC progenitors, which diverge into pre-cDCs and pDCs (103) (Figure 1). Pre-cDCs undergo maturation in the blood, resulting in cDC1 and cDC2, where they have a short lifespan, and some of which transmigrate to organs (103) (Figure 1). Nonetheless, recent conceptual models in hematopoiesis have shaken up DC ontogeny, as reviewed in (51, 102). Indeed, even if earlier studies have shown that pre-cDCs could be programmed to become cDC1 or cDC2 at several steps of their development (104–107), it is now thought that each bone marrow progenitor follows a predestined pathway according to lineage priming that occurs at early stages in development (51, 108–110). Thus, each phenotypically defined population contains cells primed by related yet distinct developmental pathways that share a common transient phenotype. This has been shown both in mouse models (108), as well as in humans (109, 110). In addition, Rodrigues et al. identified two distinct mature pDC subsets in mouse models (111). One pDC subset is derived from common DC progenitors and the other is derived from common lymphoid progenitors and represents the majority of mature pDCs (111) (Figure 1). However, it is currently unknown whether these two subsets of mature pDCs also exist in humans. Moreover, Dress et al. have recently claimed that all pDCs are derived from common lymphoid progenitors and could be called “plasmacytoid innate lymphoid cells” (112). Thus, although the recent development of scRNA-Seq analysis has led to better understanding of DC subset origins, much work remains, especially in humans.
Development
In mouse, DC development is dependent on transcription factors ZBTB46 and PU.1, as well as FLT3 and its ligand, GM-CSF and IL-4 (80, 113–115) (Figure 1). Further development of each DC subset then involves specific transcription factors such as interferon regulatory factor (IRF) 4 and IRF8. More precisely, pDCs require both IRF4 and IRF8 while cDC1 and cDC2 require IRF8 and IRF4, respectively (102, 103, 116–119). Other factors are involved in DC subset development and sustention including BATF3 and ID2 for cDC1, NOTCH2 and ZEB2 for cDC2, and TCF4 (also known as E2-2) and IRF7 for pDCs (43, 45, 111, 120–124) (Figure 1). In humans, the same factors are involved in DC development, particularly PU.1, FLT3 and GM-CSF (87, 106, 107, 125–127), as well as DC subset development via IRF4, IRF8, BATF3, ID2, TCF4, and IRF4 (103, 115). Recently, two studies have shown that adding NOTCH ligands to FLT3L in bone marrow precursor cultures increased the yield of cDC1 and that these NOTCH-cDC1 were transcriptionally and functionally closer to in vivo cDC1 (127–129) (Figure 1). However, transcription factor dependency differs considerably between tissues, and the question remains whether this specificity is enforced at the precursor level in the bone marrow or if microenvironmental cues in the organs are the primary regulators of the final steps in DC development (124, 130). This phenomenon seems to be tissue-specific (131). Indeed, Heidkamp et al. showed that DC subsets in lymphohematopoietic organs, i.e., spleen, thymus and blood, are strongly defined by ontogeny rather than by signals from the microenvironment, while it is the opposite in DC subsets from lung or skin (131).
Location and Characterization
First, among PBMCs, DCs are identified as CD14−CD16− cells among MNPs, i.e., CD45+Lin−(CD3/CD19/CD56)HLA-DR+ cells (132, 133). Then among DCs, cDCs are CD11cint−hi while pDCs are CD11c− (91).
The cDC2 subset is characterized by CD1c and SIRPα among cDCs (91, 118, 131, 134, 135) (Figure 1). CD1c is a glycoprotein involved in the presentation of lipid antigens while SIRPα is an inhibitory receptor, mainly expressed by myeloid cells (136). While all SIRPα+ cDCs comprise IRF4+IRF8− bona fide cDC2 in mouse, two populations of SIRPα+ cDCs have been detected in humans: a population of bona fide cDC2 with a CD1c+IRF4+IRF8−phenotype and a population of CD1c− cDCs showing the typical IRF4intIRF8int expression observed in the monocyte-macrophage population (118). Therefore, CD1c is required to define human bona fide cDC2 (Figure 1). In mice, cDC2 are specialized in CD4+ naïve T cell polarization in LNs (137, 138). On the contrary, in humans, cDC2 do not have an enhanced capacity to prime CD4+ T cells compared to cDC1 (139, 140).
The cDC1 subset was first described as CD141+ cells among cDCs (55, 141, 142). However, although CD141 is associated with cDC1, it is also expressed by other blood MNP subsets, including pDCs (91). Moreover, several human tissues contain a CD141+CD1c+ double-positive population (143, 144), which has been associated with either cDC2 (135) or cDC1 (91). This makes the subset identity of this double-positive population unclear. Fortunately, transcriptional profiling identified new markers that better define cDC1 and can be used for subset confirmation. Such markers include CLEC9A (also called DNGR-1), CADM1, CD26, and CD13 (91, 118, 134, 135, 145–147) (Figure 1). XCR1, a receptor for XCL1 and XCL2 chemokines, can also be used and is conserved in many species (91, 118, 134, 148, 149) (Figure 1). Actually, XCR1+ cDCs seem to be the “final form” of cDC1 subset development (127). Indeed, Balan et al. showed that the blood CADM1+CD141+CLEC9A+XCR1− DC fraction proliferates and acquires XCR1 expression during culture, suggesting that these cells are the immediate precursors of the XCR1+ cDC1 (127). Moreover, lack of expression of monocyte-macrophage and cDC2 markers such as CD14, CD1c, CD11b and SIRPα is also important to thoroughly identify the cDC1 population. Finally, as some cDC1 have intermediate CD11c expression, caution needs to be used to include all cDC1 by gating cDCs as CD11cint−hi cells (102, 135, 143). Functionally, cDC1 are involved in CD8+ T cell priming through antigen cross-presentation as well as in CD4+ Th1 and Treg polarization (150, 151). They also seem optimal for the generation of tissue-resident memory T cells, but not for circulating memory T cells, during viral infection, at least in mouse models (152). Thus, the cDC1 population constitutes an interesting DC subset for the design of immunotherapeutic treatments against intracellular pathogens or cancer cells. However, in humans, cross-presentation is also done by cDC2, monocyte-derived cDCs and monocyte-derived Mfs (140, 153–156). Nevertheless, it has been demonstrated that only cDC1 have the capacity to cross-present antigens from necrotic cells (157). Unfortunately, the human cDC1 subset is rare in blood and tissues (135), making it difficult to study them ex vivo. Thus, the division of labor between cDC1 and cDC2 subsets is still not fully understood (158), but their physical location in the LNs could contribute to differences in T cell activation, as recently reviewed (159, 160).
There is also a cDC population double negative for CD1c and XCR1 among cDCs (called DN cDCs hereafter), which could be a third bona fide cDC subset or a monocyte-derived cell type (Figure 1). This population is also present in several organs (118, 135); however, little is known about their functions or their involvement in diseases. Thus, further investigation is needed to fully characterize this DN cDC population.
Finally, it has recently been shown that the traditional gating strategy characterizing human pDCs, which is CD123+ cells among CD14−CD11c− MNPs, also includes pre-cDCs (91, 103). Thus, additional markers such as CD45RA, CX3CR1, and CD33 are required to analyze bona fide pDCs (103) (Figure 1). Concerning their functionally, pDCs are mostly involved in antiviral responses through the secretion of type I IFNs (102, 122, 161).
Monocyte-Derived Cells
The fate of monocyte-derived cells is an area of active research and contains issues that are actively debated (51, 130, 162, 163). Indeed, it is now clear that blood DCs and blood monocytes arise from bone marrow precursors (Figure 1). In tissues, Mfs can arise from both embryogenic precursors and blood monocytes while DCs can arise from blood pre-DCs, blood DCs, tissue pre-DCs or even blood monocytes (Figure 1). Thus, the origin of tissue Mfs and DCs are multiple and complex and also depend on the tissue type as well the inflammatory and wound healing status (35, 58, 164, 165). Therefore, phenotype and functions of these cells during tissue homeostasis and their alterations during disease are not fully understood.
In human tissues, Segura and colleagues have suggested that HLA-DR+CD14+CD1c+ monocyte-derived cells, which increase during inflammation, are inflammatory DCs (162, 166, 167). These cells display a typical DC morphology and possess hallmark DC functions, such as the ability to stimulate naïve T cells (162). However, these CD14+CD1c+ monocyte-derived cells also expressed markers found on Mfs, including CD64 (166, 167). In addition, Schrøder et al. have recently shown that a fraction of CD14+ monocytes already expressed CD1c in blood (168). These CD14+CD1c+ cells characterized by Schrøder et al. possess hallmarks of monocytes such as CCR2 expression, TNF induction after LPS treatment and lower efficiency to promote naïve T cell proliferation compared to blood CD14−CD1c+ cDCs (168). Together, these data support that tissue CD14+CD1c+ monocyte-derived cells could represent a highly plastic Mf subset, which shared some capacities with cDCs, rather being than a bona fide cDC subset (130, 169). These data underscore that nomenclature within the MNP compartment should be based on ontogeny rather than phenotypic characterization. Thus, labeling a cell as a “DC” should be restricted to cells derived from dedicated precursors, pre-DCs. Consequently, CD14+CD1c+ cells with DC-like functions should be referred to as monocyte-derived cells rather than CD14+ DCs (163).
To note, a new twist has come from a recent publication by Ginhoux's lab, where data indicate that human inflammatory CD14+ DC3, a subset of blood cDC2, are not monocyte-derived cells, but are FLT3L responsive and rather belong to DC lineage (92). In addition, the most inflammatory CD14+ DC3 subset, namely CD163+CD14+ DC3, increase in the blood of patients with systemic lupus erythematosus (92). Therefore, the use of powerful single-cell techniques will likely continue to add to the depth and breadth, as well as the complexity, of the seemingly ever-expanding human MNP family.
Nevertheless, monocyte-derived cells, which have high plasticity, and bona fide cDCs are synergistic close collaborators that complement each other in time and space and work toward the same goal—the clearance of pathogens without inducing an immunopathological response (130). Furthermore, DC heterogeneity is highly variable among individuals (133), and surface markers, TLR repertoire and functions of MNP subsets are tissue-specific, as discussed above. Therefore, the continuing delineation of MNP subsets underscores the ongoing need to determine the specific functions of these cells to better understand the development and propagation of diseases such as IBD.
Human Intestinal Homeostasis and Disruption During IBD
Homeostasis
In addition to the skin, the intestine is one of the major interfaces with the external environment; it is in contact with pathogens as well as commensal microbiota and food antigens (Figure 2). To maintain homeostasis, this bodily niche thus requires a balance between immune tolerance and immune responses against pathogens (170–182). The intestinal epithelium, mainly composed of a single-cell layer of enterocytes, forms a critical continuous physical barrier with tight junctions connecting adjacent cells and regulates selective permeability for luminal content (Figure 2) (183, 184). In addition to this physical barrier, stem cells located at the base of intestinal crypts (185) continuously give rise to several other epithelial cell types that are involved in specialized functions to maintain homeostasis (186, 187). This includes Paneth cells (188) and goblet cells (189) that secrete antimicrobial peptides and mucins, respectively (Figure 2). The small intestine has a single mucus layer while the colon has an inner mucus layer, lacking bacteria, and an outer mucus layer, which forms a habitat for numerous microorganisms (189) (Figure 2). Despite these systems, luminal antigens can cross the epithelial barrier using one or more routes, such as microfold cells in Peyer's patches, as recently reviewed (50, 190, 191) (Figure 2). Subsequently, antigens come in contact with immune cells, including MNPs, in secondary and tertiary lymphoid organs in the lamina propria (LP) (192, 193). After internalization by MNPs, processed antigens are presented to lymphocytes to induce oral tolerance (173, 191, 194–196) and thus establish homeostatic interaction with dietary factors and intestinal microbiota (37–40, 197). To this end, in addition to their interaction in solitary intestinal lymphoid tissues and Peyer's patches, cDCs are able to migrate to mesenteric lymph nodes (mLNs) through afferent lymphatic vessels to polarize naïve T cells (198) (Figure 2). In contrast, Mfs lack active migratory properties and rather contribute to amplifying T cell responses in the LP. Additionally, intestinal Mfs maintain tissue homeostasis by removing apoptotic and dead cells, remodeling the epithelium and secreting cytokines to sustain Treg functions (32, 35, 38, 42, 45, 71, 98, 199–204). These active regulatory processes, as well as deletion and anergy of T cells, have been implicated in maintaining oral tolerance (191, 205–207). Finally, in response to microbial sensing, cDCs also favor class switching of IgM and IgG to IgA on B cells (208, 209). This is essential for gut homeostasis as IgA transcytoses across the epithelial cell layer to restrain interaction between microorganisms and epithelial cells (210) (Figure 2). To conclude, MNPs control intestinal homeostasis by maintaining immune tolerance to diet- and commensal-antigens while sustaining the capacity to trigger immune responses against pathogens (38, 42, 194, 201, 211). Ideally, these immune responses are a self-limiting process that leads to a complete resolution of inflammation and rapid return to tissue homeostasis. Unfortunately, repeated and aberrant activation of the immune system can result in a chronic inflammatory microenvironment leading to IBD.
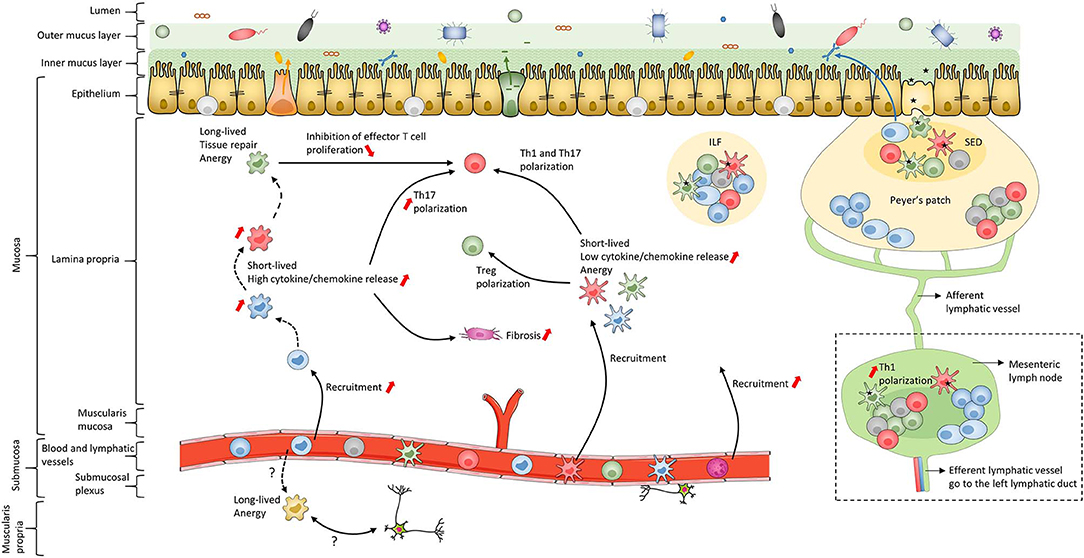
Figure 2. Schematic of human intestinal mucosa and submucosa structure. The schematic is an overall representation of intestinal organization. For example, only the small intestine has Peyer's patches while only the colon has both inner and outer mucus layers composed of firm and loose mucus, respectively. In addition, this schematic does not represent the villi or the crypts of the intestinal mucosa. The color coding of macrophage subsets and dendritic cell subsets matches that in Figures 1, 3, 4 and 6 to allow continuity in the Figures. Black dashed lines represent the maturation waterfall of macrophages. Questions marks represent remaining unresolved issues regarding differentiation and function of the Mf4 subset. Red arrows show processes that increase or decrease during IBD. ILF, isolated lymphoid follicle; SED, subepithelial dome.
Disruption of Intestinal Homeostasis Through MNPs During IBD
Several defects in intestinal homeostasis have been linked to IBD. These include immune responses against commensal bacteria, epithelial barrier dysfunction, diminution of nutrient absorption, alterations in tissue oxygenation and in autophagy, which induce immune cell recruitment such as MNPs (25, 184, 187, 191, 212–217). In addition, expression of genes associated with variation in Crohn's disease prognosis can be mapped to MNPs (218). Thus, MNPs have a key role in cellular signaling pathways that modulate tolerance vs. chronic inflammation during IBD, as described in detail below (Figure 2).
Human Intestinal Mucosa MNP Populations During Homeostasis and IBD
First, we summarize MNP subset characteristics and their functions during homeostasis, as assessed in tissue from control individuals, and thereafter we describe MNP alterations during IBD. To note, several layers add complexity to this field, such as various surface markers and gating strategies used in different studies, low number of cells available to perform functional analyzes, and differences between intestinal regions such as the ileum and colon (219, 220). Finally, a caveat to samples used as “controls” is that they are typically from colorectal cancer patients or obese patients.
Macrophages in Intestinal Mucosa During Homeostasis
Phenotype and Frequency of Intestinal Mucosa Macrophages
The first step to characterize intestinal Mfs by flow cytometry is gating on MNPs, i.e., CD45+Lin−(CD3/CD19/CD56)HLA-DRint−hiCD14neg−hi cells (Figures 3A, 4). Then, among MNPs, CD14 (Figures 3A, 4), CD64 and CD163 are used to distinguish Mfs from DCs (72, 75, 221–225). To note, various studies have shown that CD64 alone was not sufficient to distinguish intestinal Mfs from DCs, as some cDC2 are CD64+ (72, 135, 220, 224). In control intestinal mucosa, several studies showed that Mfs, i.e., CD14+ cells, represent ~0.2 and 0.5% of LP cells (Figure 5A), and 20 and 40% of MNPs (Figure 5B), in ileum and in colon, respectively (132, 225–227). This suggests that the frequency of Mfs is higher in colon than in ileum. However, Granot et al. showed the contrary by analyzing CD14+ cells among CD45+ cells in organ donors (135), underscoring once again the complexity of working with human tissues.
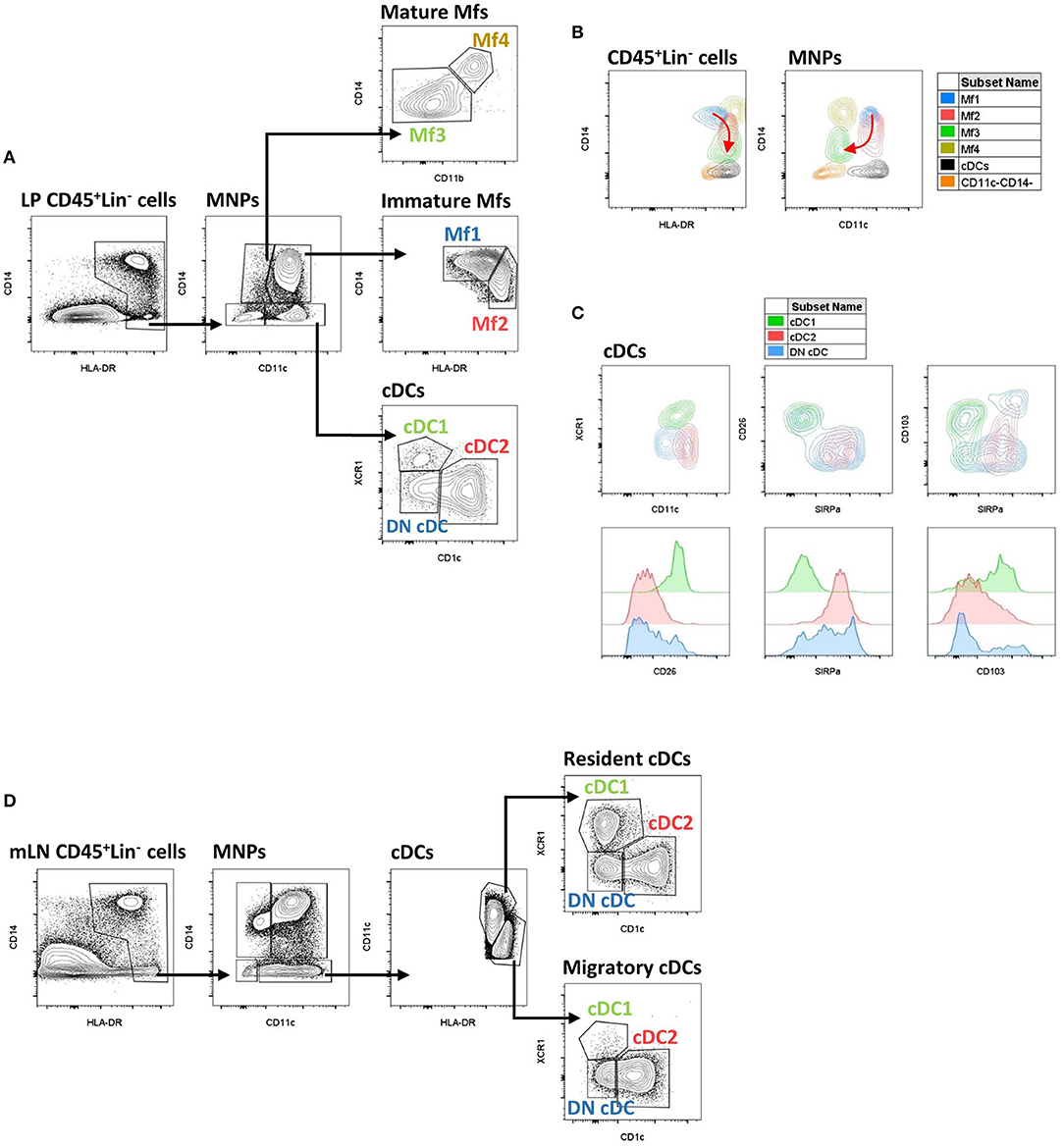
Figure 3. Gating strategy and phenotypic characteristics of human intestinal mucosa MNP subsets and human mLN cDC subsets. (A) Gating strategy to analyze human intestinal mucosa MNP subsets from lamina propria cells. This example is from ileum of a Crohn's disease patient. Lineage is composed of CD3, CD19, and CD56. (B) Expression level of HLA-DR, CD14, and CD11c on human intestinal MNP subsets. The red arrow indicates the Mf maturation waterfall from Mfl to Mf3. (A,B) Based on reference (75). (C) Expression level of CD11c, SIRPα, CD26, CD103, and XCR1 on human intestinal cDC subsets. (D) Gating strategy to analyze human cDC subsets from mesenteric lymph node cells. This example is from mLN of a ulcerative colitis patient. Lineage is composed of CD3, CD19, and CD56. LP, lamina propria; mLN, mesenteric lymph node.
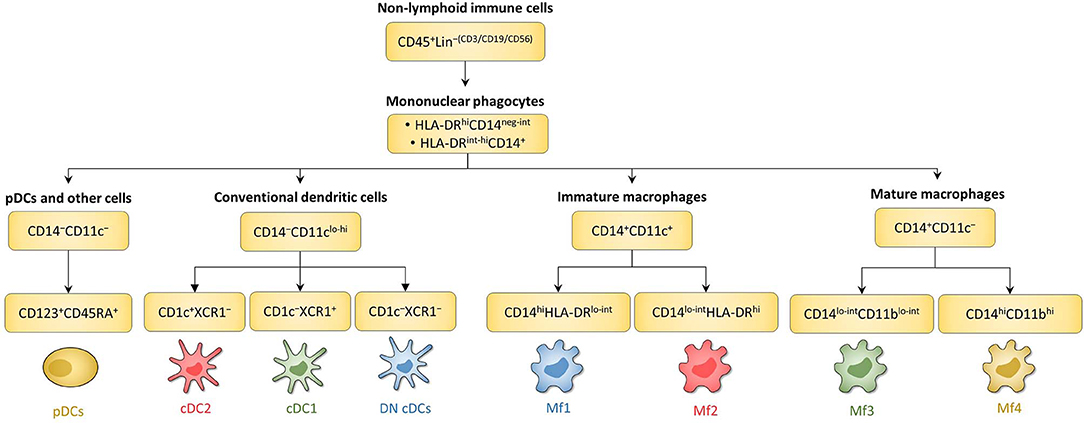
Figure 4. Schematic overview of human intestinal MNP subsets. Main surface markers to identify human intestinal MNP subsets using the gating strategy in Figure 3. This schematic is not intended to show the ontogenic or developmental relationship between the human intestinal MNP subsets.
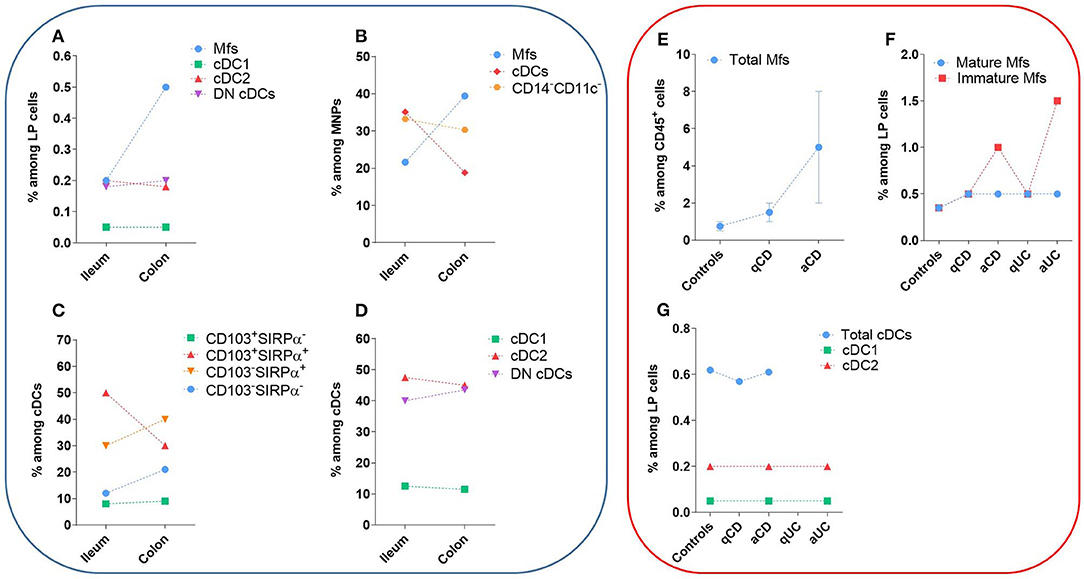
Figure 5. Frequency of human intestinal MNP subsets in homeostasis and during IBD. These values do not take in account the number of patients or the standard error of the mean from each study. The dashed lines are only to clarify the difference between the type of tissue and do not represent paired values. (A–D) Frequency of human intestinal MNP subsets in homeostasis. (E–G) Comparison of human intestinal MNP subset frequencies in homeostasis and during IBD. a, active lesion areas; q, quiescent lesion areas. References: (A) (132, 225); (B) (226, 227); (C) (75, 134, 220); (D) (132); (E) (223, 228); (F) (132, 225, 228); (G) (132, 229).
Concerning Mf subsets, it is now well-defined that the human intestinal Mf population comprises a continuum of blood monocyte-derived cells differentiating first into a newly recruited monocyte population (called immature Mfs hereafter) and then into a mature Mf population, similar to the situation in mouse intestine (42, 72, 73, 75, 221). Based on flow cytometry expression of HLA-DR, CD14, CD11c and CD11b, Bujko et al. recently described four Mf subsets (75). More precisely, they showed that both immature and mature Mf populations are composed of two subsets, Mf1 and Mf2, and Mf3 and Mf4, respectively (75) (Figures 3A,B, 4, 6). These four Mf subsets are comparable with those described in human ileum by Bain et al. using HLA-DR, CD14, CD11c, CD163, and CD209 (221) (Figure 6). The Mf maturation waterfall from Mf1 to Mf3 includes a decreased expression of a set of blood monocyte markers such as CD11c and CCR2, as well as an increased expression of CD163 and CD209 (75) (Figures 3B, 6). The Mf4 population is primarily located deep in the mucosa and in the densely innervated submucosa and muscularis propria/externa (75) (Figures 2, 6). Thus, the Mf4 population might be specialized in nervous system interactions, as described in mice (195, 230–233). However, further investigation is needed to better understand the origin and function of these cells in the human intestine.
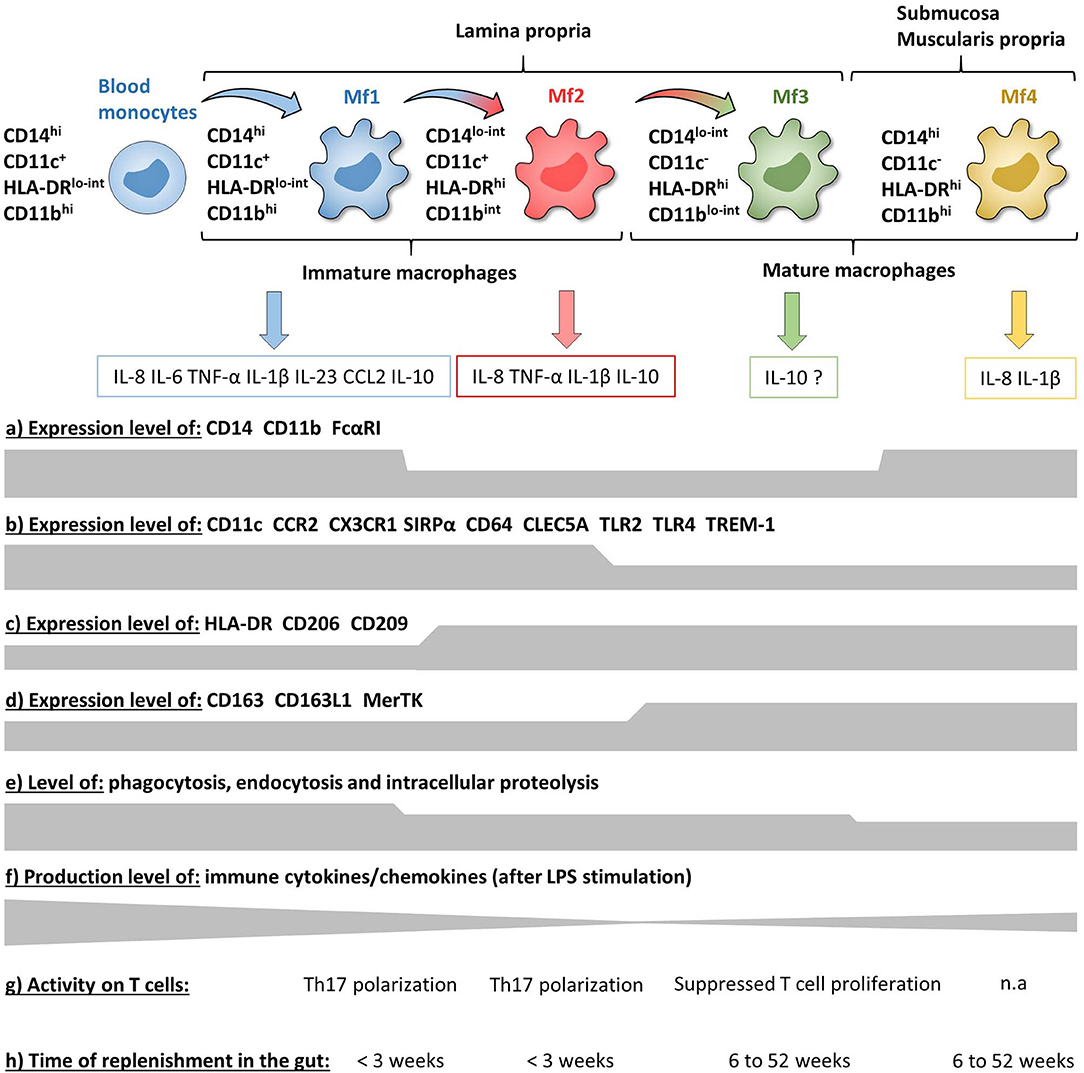
Figure 6. Phenotypic and functional characteristics of human intestinal mucosa Mf subsets during homeostasis. (a–d) Expression level of major surface markers on blood monocytes and intestinal mucosa Mf subsets assessed by flow cytometry (intensity level from blood monocytes to Mf4 subset). (e–g) Functional characteristics of blood monocytes and intestinal mucosa Mf subsets assessed ex vivo (intensity level from blood monocytes to Mf4 subset). (h) Time of replenishment of intestinal mucosa Mf subsets assessed in duodenum-pancreas transplanted patients. n.a, not analyzed.
As previously shown by Jahnsen's group (75, 168, 234), Bernardo et al. confirmed that immature Mfs are characterized as CD11c+ Mfs and expressed monocyte-associated markers such as CCR2 and CX3CR1, while mature Mfs are characterized as CD11c− Mfs (225) (Figure 6). However, they did not find a difference of CD206 and CD163 expression between Mf populations (225), which are markers typically associated with mature Mfs (75, 221) (Figure 6). Regarding frequency, mature Mfs were the most abundant Mf population in duodenum but decreased from duodenum to colon while the opposite was observed for the frequency of immature Mfs (75, 225).
To summarize, circulating classical monocytes enter in the intestinal mucosa and differentiate in situ, first into transient immature Mfs and then to mature Mfs. The Mf maturation waterfall is based on stepwise acquisition of a set of markers related to bacteria clearance concomitant with loss of inflammatory markers (Figure 6).
Function of Intestinal Mucosa Macrophages
Intestinal mucosal Mfs are strategically located in the subepithelial area where they regulate lumen-derived commensal microbe penetrance through their capacities of phagocytosis and degradation. Thus, intestinal mucosal Mfs are involved in pathogen clearance and immune regulation by maintaining tolerance to commensal microbiota and food antigens as well as tissue repair (42, 195, 235–237). The three intestinal mucosal Mf subsets described by Bujko et al. are highly proficient at endocytosis, antigen uptake, and intracellular degradation of proteins, although less than blood monocytes, while the submucosal Mf4 population is weakly competent in these capacities (75) (Figure 6). Compared with the other intestinal Mf subsets, unstimulated and LPS stimulated Mf1 release significantly higher amounts of numerous chemokines and cytokines (75, 225) (Figure 6). To note, the frequency of blood monocyte-derived CD11c− Mfs, related to intestinal mature Mf3, increased in the presence of mucosa-conditioned medium from control intestine (225). In addition, Maheshwari et al. showed that blood monocyte-derived Mfs developed LPS tolerance on exposure to mucosa-conditioned medium, mainly provided by TGF-β2 (238). This suggests that intestinal mature CD11c− Mfs are generated from newly recruited monocytes through the intestinal microenvironment, which promotes an anti-inflammatory and anergic state during the Mf maturation waterfall. This would result in mature Mfs that are less responsive against food antigens and commensal microbiota. Indeed, several studies have shown that, contrary to monocytes and most other tissue-resident Mfs, intestinal Mfs lack surface expression of TLR2, TLR4 and FcαRI, and are consequently down-regulated for LPS- and IgA-mediated activities (75, 239, 240) (Figure 6). Moreover, mature Mfs reduce the release of proinflammatory molecules together with desensitization to TLR ligands, which is a functional feature of anergy (75, 239, 241, 242). This anergic status is thought to be driven by several micro-environmental factors, as recently reviewed (237), such as TGF-β, which induces downregulation of the MyD88 pathway in blood monocytes and results in a hyporesponsive mature Mf-like phenotype (242, 243). Confirming this, Kelly et al. recently showed that human intestinal Mfs highly expressed integrin αvβ8, which down-regulated TNF production via TGF-β activation, at least in blood monocytes (244). Some earlier studies also support the hyporesponsive nature of intestinal Mfs. Indeed, more than 10 years ago, using an unconventional Mf definition, i.e., CD13+CD14−CD33−CD64−CD16− cells, Smith et al. showed that even if jejunum Mfs had strong basal phagocytic and bactericidal activities they were anergic (239, 242, 245). More precisely, they did not secrete cytokines and chemokines, except low amounts of IL-8, with or without LPS stimulation (239, 242) and did not activate the NF-kB pathway (243). However, in contrast, several recent studies showed that intestinal Mfs, defined more conventionally as CD14+ cells, express TLRs and secrete pro-inflammatory molecules with or without TLR stimulation (75, 226, 227, 246). Indeed, intestinal Mfs from controls produced few cytokines in the absence of stimulation while secretion of IL-12p40, IL-23, TNF, IL-6, and IL-10 were induced by commensal bacteria (246). More specifically, Mfs were the major producers of IFN-γ, TNF, IL-6, TL1A, and IL-23 among LP cells activated or not by commensal bacteria (246, 247). Furthermore, Mf-derived IL-23 induced IFN-γ and TNF release from LP cells (246) as well as IL-22 secretion by innate lymphoid cells (248).
Human intestinal Mfs also express a receptor called TREM-1 (triggering receptor expressed on myeloid cells 1), which amplifies inflammatory responses upon engagement by thus far poorly understood ligands (249–251). TREM-1 activation leads to the secretion of pro-inflammatory cytokines and chemokines such as TNF, IL-6, IL-1β, IL-8 and CCL2, and synergizes with TLR activation (251–254). Our group and another showed that intestinal mature Mfs expressed less TREM-1 than immature Mfs and blood monocytes (253, 255), which has been corroborated by a recent study using scRNA-Seq (228) (Figure 6). The decrease of TREM-1 expression during Mf maturation seems to be due to the intestinal anti-inflammatory microenvironment, as monocytes cultured in the presence of IL-10 and TGF-β have reduced TREM-1 expression (253).
Takeda's group described three human intestinal Mf subsets as CD163lo, CD163hiCD160lo, and CD163hiCD160hi (226, 256). They showed that CD163lo Mfs, which seem related to Mf1, highly secreted pro-inflammatory cytokines, while the CD163hiCD160hi Mf subset, which seem related to mature Mfs, secreted IL-10 (226, 256). These data corroborate those from Bernardo et al. (225). On the contrary, Bujko et al. showed that, in addition to their high pro-inflammatory cytokine release, immature Mfs, mainly Mf1, secreted more IL-10 than mature Mfs (75). Thus, which subset of intestinal Mfs is the main producer of IL-10 is still an open question (Figure 6). Nevertheless, regarding T cell induction, the CD163lo Mfs (immature Mf1-like) induced naïve CD4+ T cell polarization into Th17 cells while CD163hiCD160hi Mfs (mature Mf3-like) suppressed effector T cell proliferation (226, 256) (Figures 2, 6). However, none of these Mf subsets induced naïve CD4+ T cell polarization into Th1 or Tregs (226, 256). This is consistent with data from Matsuno et al. who showed that Mfs could polarize naïve CD4+ T cells into Th17, but not into Th1 or Tregs (227). To note, intestinal Mfs do not express CCR7 (135, 222, 246), suggesting that they cannot migrate to mLNs to interact with naïve T cells in situ. Therefore, given that human intestinal T cells are almost entirely memory T cells (257–259), the biological significance of the ability of intestinal Mfs to regulate naïve CD4+ T cells as shown ex vivo is not clear.
Finally, Bujko et al. have also analyzed the replenishment of the four Mf subsets from pancreatico-duodenal transplantation patients (75). Three weeks after surgery almost all donor Mf1 and Mf2 were replaced by recipient Mfs, while only 20% of Mf3 and Mf4 were replaced 6 weeks after transplantation. However, after 1 year, all donor Mfs were replaced by recipient monocytes. These data elegantly showed that human intestinal Mf compartment is fully replenish through blood monocytes in a maximum of 1 year, and consists of transient immature Mfs (Mf1 and Mf2 subsets) that die or differentiate into long-lived mature Mfs (Mf3 and Mf4 subsets) (75) (Figure 6). This is in contradiction with mouse data, where it has been recently shown that there is a self-maintaining Mf population that persists throughout adulthood and is essential for intestinal homeostasis (232, 260).
To summarize, during homeostasis, circulating classical monocytes constantly replenish the intestinal Mf population, first constituting functionally plastic immature Mfs, which is a very appropriate way to respond rapidly and aggressively to pathogens through phagocytosis and cytokine secretion. Then, under the influence of the intestinal microenvironment, maturation steps generate long-lived anergic resident mature Mfs, which maintain oral tolerance and tissue homeostasis to prevent chronic inflammatory responses (Figure 2). The exact mechanisms involved in the Mf maturation waterfall remain to be fully elucidated in order to understand alterations during intestinal diseases such as IBD.
Conventional Dendritic Cells in Intestinal Mucosa During Homeostasis
Before venturing deeper into the specifics of human intestinal cDC subsets, it is worth taking a moment to say that definition of these subsets is difficult. Indeed, based on data from mouse models where the integrins CD103 and CD11b are widely used to identify four intestinal cDC subsets (45, 261), many human studies use CD103 along with SIRPα (also called CD172a), as the human equivalent of CD11b in mice, to likewise identify four intestinal cDC subsets (45, 261). It has been shown that CD103+SIRPα− were usually related to cDC1, CD103+SIRPα+ and CD103− SIRPα+ were related to cDC2 while CD103−SIRPα− cDCs are poorly studied (45, 75, 134, 220, 224). However, these markers do not robustly define cDC subsets in humans (132) and recent analyzes use other markers such as CLECL9A/CAMD1/CD26 and CD1c to characterize them (118, 135) (Figure 3C). Thus, the discussion below is divided to reflect these different ways to define human cDC subsets.
Frequency of Intestinal Mucosa cDC Subsets Defined Using CD103 and SIRPα
The frequency of CD103+SIRPα+ cells among cDCs predominated in small intestine but decreased in colon, while the contrary was true for CD103−SIRPα+ and CD103−SIRPα− cells (75, 134, 220) (Figure 5C). Furthermore, the frequency of total CD103+ cDCs, as well as the frequency of CD103+ cells among each cDC subset, decreased in colon relative to ileum (132, 227). Thus, CD103-expressing cDCs decreased in colon relative to small intestine regardless of the subset analyzed. Importantly, the mechanisms regulating CD103 expression on cDCs are poorly known, suggesting that other markers could be more robust to defined human intestinal cDC subsets.
Phenotype and Frequency of Intestinal Mucosa cDC Subsets Using Markers Other Than CD103 and SIRPα
First, among MNPs, intestinal cDCs are characterized as CD14−CD11cint−hi cells (Figures 3A, 4) (75, 132, 135, 224). Within this population, cDC1, cDC2, and DN DCs can be identified based on two major surface markers, CD1c and XCR1. Precisely, cDC2 are defined as CD1c+XCR1−, cDC1 as CD1c−XCR1+, and DN cDCs as CD1c−XCR1− (Figures 3A, 4). In addition, these three cDC subsets expressed other specific markers as shown in Figure 1 (132, 134, 135, 224). DN cDCs seemed more heterogeneous with at least three other subsets based on SIRPα and CD26 expression (118, 134, 135) (Figure 3C). Regarding their abundance in control intestinal mucosa, cDCs represent 0.3–0.6% of colonic LP cells, which corresponds to a median of 114 cDCs per mg of tissue (229, 262). More precisely, our group showed that cDC1, cDC2, and DN cDCs represent around 0.05, 0.2, and 0.2%, respectively, of LP cells in both ileum and colon (132) (Figure 5A). However, among MNPs, the total cDC population seemed to decrease from ileum to colon (226) (Figure 5B). Finally, among cDCs, cDC1, cDC2, and DN cDCs represented around 10–15, 40–50, and 35–50%, respectively, both in ileum and colon (132) (Figure 5D).
To note, as described above in the blood, intestinal pDCs are present among the CD14−CD11c− population and can be identified as CD123+CD45RA+ cells (135) (Figure 4). However, even if pDC frequency seems to increase by 10-fold from jejunum to colon, i.e., from 0.001 to 0.01% of CD45+ cells (135), human intestinal pDCs represent a very low amount of cells (135, 263), and will not be discussed further in this review.
Function of “Total” Intestinal Mucosa cDCs
In control intestinal mucosa, the frequency of cytokine-producing cDCs was absent/low for IL-6, IL-12, IL-22, and IL-23 and intermediate for IL-10, TGF-β, TNF, and IL-1β (220, 264). In addition, intestinal cDCs secreted almost no cytokines without stimulation and did not respond to TLR ligands ex vivo (75, 226). Supporting this, cDCs seemed immature as judged by their low expression of CD80, CD83, CD86, and TLRs (220, 226, 229, 264, 265). Mann et al. showed that functional differences could exist between colonic and ileal cDCs, such as induction the gut-homing receptor CCR9 (220), which makes the comparison of studies even more complex.
With respect to T cell induction, Ogino et al. showed that colonic cDCs induced naïve CD4+ T cell polarization into Th1 but not Th17 cells (226) (Table 1). In contrast, Mann et al. showed that colonic cDCs induced high production of several cytokines such as IL-10, TGF-β, IL-17, IFN-γ, and IL-22 by naïve CD4+ T cells (265), at least by dividing CFSElo T cells (Table 1). To note, Fenton et al. recently showed that intestinal cDCs that highly express integrin αVβ8, such as cDC2, but not cDC1, might induce higher Treg polarization through TGF-β secretion (266). Nevertheless, given that there are few naïve CD4+ T cells in human intestinal mucosa (257–259), it is more relevant to study naïve CD4+ T cell polarization with cDCs from mLNs, as discussed below.
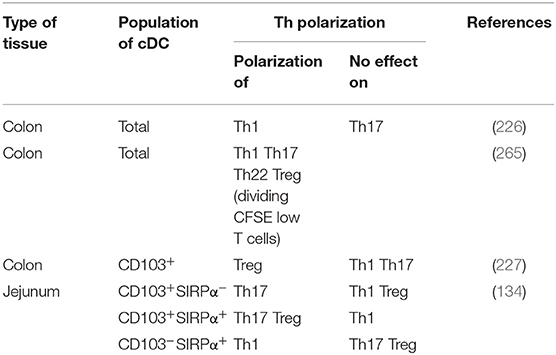
Table 1. Th polarization by human intestinal cDCs.
Function of Intestinal Mucosa cDC Subsets Defined Using CD103 and SIRPα
Watchmaker et al. showed that CD103+SIRPα−, CD103+SIRPα+, and CD103−SIRPα+ in jejunum expressed a low level of CD80, an intermediate level of CD83 and CD86, while a high level of CD40 (134). In addition, they expressed CCR7, suggesting that these three cDC subsets may migrate to mLNs (134, 220). Furthermore, these cDC subsets induced the mucosa-associated integrin α4β7 and the gut-homing receptor CCR9 on naïve CD4+ T cells (134), suggesting that they support T cell homing to intestinal tissue. Matsuno et al. showed that colonic CD103+ cDCs induced Tregs, but neither Th1 nor Th17 (227) (Table 1). In contrast, Watchmaker et al. showed that both CD103+SIRPα+ cDCs and CD103+SIRPα− cDCs induce Th17, while CD103+SIRPα+ cDCs induce Tregs and CD103−SIRPα+ cDCs induce Th1, at least with cDCs from jejunum of three obese subjects (134) (Table 1).
To note, Richter et al. recently described a monocyte-derived cell population in the cDC compartment, identified as HLA-DR+CD14−/loCD11c+SIRPα+, which are mainly cDC2 (224). Indeed, some SIRPα+ cDCs expressing calprotectin and low level of CD14 were enriched in monocyte gene signatures, were morphologically similar to monocytes and did not express FLT3 receptor (224). Moreover, they exhibited a higher capacity for antigen processing, yet an inferior potential for migration and priming of naïve T cells compared to SIRPα+FLT3+calprotectin− cDCs (224). These data suggest that CD14loCD11c+SIRPα+FLT3−calprotectin+ cDCs, although mimicking a typical cDC phenotype, are more related to the monocyte lineage than to bona fide cDCs, highlighting once again the diversity and complexity of human intestinal MNP subsets. Finally, using pancreatico-duodenal transplantation patients, Richter et al. showed all intestinal cDCs were replaced by recipient cells 6 weeks after transplantation, suggesting that cDCs lack self-renewal capacity and long-life phenotype in the human intestine (224).
To summarize, the different gating strategies used and the low number of cDCs complicates getting a clear picture of subset-specific function. However, it has been shown that human intestinal cDCs are replenished by blood-derived cells in a few weeks, are poor cytokine-producing cells and play a role of sampling antigens to activate T cell proliferation in mLNs, which is consistent with the well-characterized overall function of cDCs (Figure 2). Nevertheless, additional studies of human intestinal cDCs are warranted to understand which and how cDC subsets maintain tissue homeostasis and initiating effective immunity without driving disease pathogenesis.
Macrophages in Intestinal Mucosa During IBD
Phenotype and Frequency of Intestinal Mucosa Macrophages
It is now clear that the frequency and number of Mfs among intestinal LP cells is increased in IBD patients compared to controls, especially in active lesion areas (132, 221, 223, 225, 226, 228, 246, 263, 267–272) (Figure 5E). More specifically, the augmentation of intestinal Mfs is due to an increase in the frequency of immature Mfs among LP cells, despite that somewhat different surface markers and combinations thereof have been used to characterized them (i.e., HLA-DR or CD11c levels; scRNA-seq) (132, 221, 223, 225, 226, 228, 256, 263, 270, 272–275) (Figure 5F). This accumulation seems to be due to the inflammatory intestinal microenvironment of IBD patients, which boosts the recruitment of classical monocytes through mechanisms involving CCL2, IL-8, and TGF-β signaling (223, 276). These newly recruited monocytes are maintained in the immature pro-inflammatory state, which in turn amplify intestinal chronic inflammation (11, 277). Chapuy et al. showed that the frequency of immature Mfs, but not mature Mfs, is positively correlated with endoscopic score of disease severity in Crohn's patients (228). To note, age, gender, age at diagnosis, disease location and disease behavior, as well treatment history, did not influence the increased frequency of immature Mfs in Crohn's patients (228).
Some studies have addressed the interesting question of the effect of immunotherapy in IBD patients on the intestinal Mf compartment. For example, the frequency of immature Mfs decreased slightly in IBD patients after 5 weeks of anti-TNF adalimumab treatment (278). Specifically, this effect appears to be limited to patients in remission, at least after 14 weeks of infliximab therapy (279). Moreover, Vos et al. showed that the frequency of mature Mfs increased in IBD patients after 4 weeks of anti-TNF infliximab treatment (280). On the contrary, the two aforementioned studies did not find a change in the frequency of mature Mfs (278, 279). In addition, the frequency of immature Mfs decreased and the frequency of mature Mfs increased in IBD patients after 14 weeks of anti-α4β7 vedolizumab treatment, specifically in patients in remission (279). Even if these three studies have not used the same markers to define Mf subsets, their data go in the same direction. That is, reduced immature Mf and/or increased mature Mf frequencies accompany disease quiescence after immunotherapy. This suggests that the restoration of homeostatic Mf composition is resulting from and/or is necessary during the remission of IBD patients after immunotherapy.
A fundamental question that remains is which components of the intestinal microenvironment drive Mf maturation during homeostasis, and how this process is affected during IBD. One candidate could be GM-CSF. Indeed, it has been shown that blood monocytes differentiate to immature Mf-like cells through GM-CSF ex vivo (281, 282), a factor that increases in Crohn's disease and UC, particularly in active lesion areas (283). Moreover, GM-CSF can act in concert with IFN-γ and TNF to reprogram blood monocytes into Mfs with an inflammatory profile (284). However, GM-CSF has also been shown to induce blood monocytes with a tissue repair and anti-inflammatory profile, which can dampen intestinal inflammation in mouse models (285). This mechanism could contribute to the benefit of GM-CSF therapy observed in some Crohn's patients (286). Therefore, either the absence or chronic production of GM-CSF can result in high susceptibility to intestinal pathology, demonstrating the importance of its balanced production to maintain homeostasis. However, the regulation of Mf function goes far beyond a single factor. Indeed, it is difficult to imagine the scenario when numerous immunomodulating factors such as IFN-γ, TNF, IL-1β, IL-6, IL-36, TGF-β, and IL-10 are present in various combinations and amounts, as is the case in vivo (11). Clearly, this area of research warrants further investigation.
Function of Intestinal Mucosa Macrophages
In addition to their accumulation, intestinal Mfs produced more pro-inflammatory cytokines, such as TNF, IL-23, IL-1β and IL-6, in basal conditions as well as after TLR stimulation, in UC patients and even more in Crohn's patients compared to controls (226, 228, 246, 256, 271, 287). These pro-inflammatory cytokines can promote and/or perpetuate a pathologic environment (11). For example, Takayama et al. showed that Mfs from Crohn's patients induced IFN-γ secretion by NK cells via IL-23 release and cell-cell contact (288). Moreover, factors in LP-conditioned medium from Crohn's patients, including IFN-γ, induced inflammatory monocyte differentiation and IL-23 secretion by these cells, leading to a vicious circle perpetuating inflammation (246). Importantly, Mfs from Crohn's patients also expressed higher levels of both IL-10 and latent TGF-β, which have anti-inflammatory effects (226, 256). Nevertheless, Kelly et al. recently showed that integrin αvβ8 expression, which regulates immune tolerance via TGF-β activation as discussed above, was highly reduced on Mfs from IBD patients (244). This suggests that, even if Mfs from Crohn's patients expressed more latent TGF-β, less TGF-β was in the active form (244).
Additional mechanisms that amply inflammation may also contribute to IBD pathogenesis. For example, the frequency and the number of TREM-1+ Mfs, which are mainly immature Mfs, are increased in IBD patients, especially in active lesion areas (254, 255). As described above, TREM-1 is an inflammation-amplifying receptor expressed on myeloid cells; it is involved in immune responses triggered by bacteria (249, 250), yet its role in IBD is poorly understood. Our group showed that an anti-TREM-1 antagonist antibody dampened secretion of several pro-inflammatory cytokines and chemokines by LP cells from highly inflamed intestinal mucosa of IBD patients (255), supporting a potential role of TREM-1 in perpetuating intestinal inflammation in IBD patients.
Other disease-associated changes in Mf function may promote IBD. For example, it has been suggested that intestinal Mfs have bacterial clearance impairment in IBD patients (289), and in patients manifesting a Crohn's disease phenotype, i.e., Niemann–Pick disease type C1, mainly through dysfunctional autophagy (290). Moreover, they appear to contribute to intestinal barrier dysfunction. Indeed, Mfs from inflamed Crohn's disease tissue induced less IL-22 secretion by innate lymphoid cells than those from quiescent area (248), and blood monocytes reduced epithelial barrier efficiency in vitro by altering the structure and function of tight junctions (271). Finally, intestinal Mfs from IBD patients had increased ROS production (291), which can also contribute to epithelial injury (292).
Another role of intestinal Mfs is their involvement in tissue repair and fibrosis (99, 293). To note, at least 10% of Crohn's patients have an intestinal fibrostenosis phenotype at the time of diagnosis (294). In addition, fibrotic complications, such as strictures, occur in ~20–30% of Crohn's patients 10 years after diagnosis (294). In UC patients, the degree of fibrosis is proportional to the degree of inflammation, even if the fibrosis-associated strictures are less prevalent than in Crohn's patients (294). Despite the problem of fibrosis in IBD (295, 296), the mechanism by which Mfs contribute to fibrosis-associated pathology is poorly understood. Several Crohn's-associated susceptibility loci, including some related to MNP-associated inflammation such as NOD2, ATG16L1, IL-12B, IL-23R, and CX3CR1, are predictors of fibrostenosis (294). In addition, Scheibe et al. showed an increase number of intestinal IL-36α+ Mfs in IBD patients, which correlated with the degree of inflammation (297) and the accumulation of αSMA+ myofibroblasts (298). More precisely, the number of intestinal IL-36α+ Mfs increase in the colon of Crohn's patients with stenosis (298). Functionally, IL-36 acted directly on human mesenchymal cells to elicit a profibrotic transcriptional program (298), suggesting that the increase of IL-36α+ Mfs could induce intestinal fibrosis during chronic inflammation in IBD patients (298–300). Corroborating this, Martin et al. recently showed in situ that immature Mfs were always in the close vicinity of activated fibroblasts in intestinal mucosa of Crohn's patients (263). In particular, immature Mfs, as well as cDC2, induced intestinal inflammation through fibroblast activation via oncostatin M/OSMR signaling, which increased in IBD patients and predicted anti-TNF therapy response (275, 301). However, even if OSM induced IL-11 expression by activated fibroblast (275), which is known to be a major fibrotic component (302), whether OSM promotes intestinal fibrosis in IBD patients remains to be determined.
Regarding T cell activation, intestinal Mfs from Crohn's patients induced naïve CD4+ T cell proliferation as well as integrin β7 and CCR9 expression in the same range as those from controls (287). However, Barman et al. showed that mature Mfs from UC patients were unable to suppress effector T cell proliferation compared to those from controls (256) (Figure 2). In addition, intestinal Mfs from Crohn's patients induced more Th1 and Th17 polarization from naïve CD4+ T cells (226, 287) (Figure 2). This seems to be due to immature Mf accumulation within the total Mf population in Crohn's patients. Indeed, Chapuy et al. have recently shown that immature Mfs from IBD patients, but seemingly not mature Mfs, induce Th17 cells, as well pathologic IFN-y+IL-17+ T cells (303), from autologous colonic CD4+ T cells mainly through their production of IL-1β (228, 272). Corroborating this, Martin et al. have shown that, while initial steps of lymphocyte aggregate formation depend on DCs, immature Mfs likely participate in T cell activation in situ (263).
In summary, data support that there is a large influx of inflammatory immature Mfs that drive inflammation and tissue damage in IBD (Figure 2). Moreover, although mature Mfs seem to maintain their anti-inflammatory and tissue repair functions in IBD, their relative abundance is reduced during inflammation as immature Mfs dominate. However, it is still unclear if the disruption of blood monocyte differentiation into mature Mfs reflects a loss of intrinsic maturation cues that normally program recruited monocytes toward cells with tolerogenic properties or if the chronic inflammatory microenvironment generates new factors that actively overhaul this homeostatic process. Thus, development of new therapies to restore the Mf maturation process and/or neutralize factors that drive monocyte recruitment may be beneficial for treating IBD.
Conventional Dendritic Cells in Intestinal Mucosa During IBD
Phenotype and Frequency of Intestinal Mucosa cDC Subsets
Several studies found no difference in the number, frequency or maturation state (with respect to CD80, CD83, and CD86 levels) of total intestinal cDCs, as well cDC1 and cDC2 subsets, in active lesion areas of IBD patients compared to quiescent lesion areas as well compared to controls (132, 229, 263, 269, 278) (Figure 5G). These data contrast a single study showing increased cDC2 among LP cells of IBD patients (262). To note, using scRNA-seq, Martin et al. have recently defined four cDC subsets in ileum from Crohn's patients, namely cDC1, cDC2, monocyte-derived DC-like cells and activated cDCs (263). Activated cDCs expressed CCR7 and PD-L1, as well as the lymphocyte-attracting chemokines CCL17, CCL22, and CCL19, and was the only cDC subset increased in inflamed lesions compared to uninflamed lesions (263).
Given the caveat that CD103 expression does not robustly define functionally distinct subsets of human intestinal cDCs, as discussed above, it has been shown that cDCs expressing CD103 are decreased among LP cells, as well as among MNPs, in both Crohn's and UC patients (132, 227, 278). In addition, the frequency of CD103+ cells among cDC1, cDC2 and DN cDCs subsets was lower in active IBD intestinal tissue compared to quiescent tissue, and even more so compared to controls (132). However, mechanisms underlying reduced human intestinal CD103+ cDC frequency in IBD patients are not known. It could be due to inflammation-induced cell death, downregulation of CD103 expression and/or emigration of CD103+ cDCs from intestinal LP. Thus, further investigation to understand CD103 regulation in human intestinal cDCs, its role on cDC function and its possible impairment during IBD are needed.
Function of Intestinal Mucosa cDC Subsets
Several studies showed that intestinal cDCs have a higher inflammatory state in UC patients, and even more so in Crohn's patients, compared to controls. This is supported by the increased frequency of CD40+, TLR2+, TLR4+, IL-12+ and IL-6+ cDCs, but not IL-10+ cDCs in patients' tissues (262, 264, 265, 304). Consistent with this, LPS-stimulated intestinal cDCs from Crohn's and UC patients secreted more TNF and IL-8 compared to those from controls (262). In addition, the frequency of IL-6+ cDCs and TLR4+ cDCs were associated with the Crohn's Disease Activity Index (304). To note, even if the frequency of CD103+ cDCs decreased in UC compared to controls, they were more inflammatory and induced less Tregs but more Th1, Th2, and Th17 polarization of naïve CD4+ T cells (227).
As for intestinal Mfs, the factors that trigger the inflammatory state of cDCs in the intestine of IBD patients are not fully understood. Wu et al. showed that TNF and IFN-γ reduced miR-10a expression in DCs from IBD patients, resulting in enhanced IL-12/23p40 and NOD2 expression as well as Th1 and Th17 polarization (305). The microbiome composition may also influence DC function, as suggested by Ng et al. (304). Indeed, in this study, the frequency of IL-12p40+ DCs positively correlated with Bacteroides and the frequency of IL-6+ DCs negatively correlated with F. prausnitzii, which are considered detrimental and beneficial, respectively, during IBD (304). Thus, although a cause/effect relationship between dysbiosis and altered DC function is not established, these data raise the possibility that, during IBD, intestinal dysbiosis drives higher production of pro-inflammatory cytokines by intestinal cDCs which, in turn, overcomes their regulatory properties and tips the balance toward inflammation (304).
Regarding T cell induction, intestinal cDCs from UC patients induced less T cell proliferation and the same amount of integrin β7 but more CCR9 on naïve CD4+ T cells compared to those from controls (265). Moreover, the dividing CFSElo CD4+ T cells produced less IFN-y and IL-22, similar IL-10, TGF-β and IL-17, but more IL-4 when co-cultured with cDCs from UC patients compared to cDCs from controls (265). To note, some of these alterations have been restored to control levels in the presence of Lactobacillus casei Shirota, a bacterium found in the commensal microbiota and used as a probiotic (265). This again suggests a role of microbiota in intestinal cDC regulation. Interestingly, Martin et al. showed that activated cDCs, which expressed lymphocyte-attracting chemokines, formed dense aggregates with lymphocytes (263). Moreover, both activated cDCs and Ki-67+ cycling lymphocytes were enriched in Crohn's patients with a “high inflammatory signature,” suggesting a role for activated DCs in the recruitment, local activation, expansion, and spatial organization of adaptive immune responses in inflamed lesions of Crohn's patients (263). Finally, Fenton et al. showed that the frequency of αVβ8+ cDC2, which are thought to induce Tregs, doubled in Crohn's patients compared to controls (266). However, intestinal Treg frequency among CD4+ T cells decreased in Crohn's patients (306). Nevertheless, given recent evidence that enhancing the ability of intestinal T cells to sense active TGF-β is effective in inducing remission in some Crohn's patients (307), boosting the αVβ8–TGF-β pathway may be an attractive complementary therapeutic approach to weaken inflammatory T cell responses (266).
Retinoic Acid Influence on Human Intestinal Mucosa MNP Subsets
A specific factor derived from dietary vitamin A1, retinoic acid (RA), has been shown to be a regulator of intestinal MNP functions (308). More precisely, RA release by cDCs has been related to Treg polarization, especially in mouse models (309), but it can be pro- or anti-inflammatory depending on the local microenvironment (310). In humans, intestinal Mfs and cDC subsets had RALDH activity and expressed ALDH1A1, ALDH1A2, and/or RDH10 suggesting that they possess the complete enzymatic machinery to generate RA from vitamin A1 (132, 134, 222, 287). Moreover, they induced the gut-homing α4β7 and CCR9 on naïve CD4+ T cells in a RA-dependent manner (222, 287). To note, there was no difference between ileal or colonic cDCs, or between cDC subsets, concerning RALDH activity and induction of α4β7 and CCR9 on naïve CD4+ T cells (132, 134, 222). This contrasts data from mice (309, 311).
Regarding RALDH activity by intestinal MNPs during IBD, there are conflicting data (132, 222, 287). For example, Sanders et al. showed that intestinal Mfs, and both CD103+ and CD103− cDC subsets, from Crohn's patients had higher RALDH activity compared to controls (222). In contrast, Magnusson et al. showed that RALDH activity in intestinal Mfs, cDC1, and cDC2 subsets decreased in UC patients compared to controls, and the same trend was observed in Crohn's patients although not reaching statistical significant (132). In addition, there are also discrepancies regarding ALDH1A1, ALDH1A2, and/or RDH10 gene expression between these three studies. For example, Kamada et al. observed the same level of RALDH2 expression but a decrease of RDH10 expression by intestinal Mfs from Crohn's patients compared to those from controls (287). Furthermore, they showed that intestinal Mfs from Crohn's patients induced more Th17 polarization from naïve CD4+ T cells compared to those from controls and that RA can act as a suppressor of this Th17 polarization (287). Therefore, they suggested that an RA-dependent Th17 polarization suppressive pathway was impaired in intestinal Mfs from Crohn's disease patients (287). However, others found no difference, or even an increase, in ALDH1A1, ALDH1A2, and/or RDH10 gene expression in MNP subsets from Crohn's patients compared to controls (132, 222). Nevertheless, none of these three studies measured RA itself nor assessed other factors that regulate RA availability. Additionally, given that intestinal T cells are almost entirely memory T cells (257, 258), the biological significance of the ability of RA-producing intestinal MNPs to regulate naïve CD4+ T cells as shown ex vivo is not clear. To conclude, it is more relevant to analyze this property in mLN MNP subsets, especially cDCs, as described below.
To summarize, in most tissues, exposure to microbial components is sufficient to induce inflammatory cDCs, while in the intestine additional signals are required due to the necessity to maintain homeostasis in this microbe-rich environment. Thus, inflammation-dampening mechanisms in cDCs must be overcome to enable them to drive inflammation in the intestine. Even if the additional signals are not defined, they likely increase during IBD and lead to pro-inflammatory cytokine production by cDCs (Figure 2). However, it is unclear if these inflammatory/activated cDCs arise from modulation of local cDC populations, recruitment of blood cDCs, differentiation of monocyte-derived cDCs, or a combination of these scenarios. Moreover, the non-redundant roles of the cDC1 and cDC2 subsets during IBD, such as mLN migration capacity and Th polarization, are poorly understood. Therefore, better understanding of intestinal cDC dysregulation is required to target these cells as a means to treat IBD.
Human Mesenteric Lymph Node MNP Populations During Homeostasis and IBD
Macrophages in Mesenteric Lymph Nodes
Since Mfs are primarily involved in non-lymphoid tissues, there is little data available on Mfs from mLNs, especially in humans. Granot et al. showed that the Mf population represents 0.8% of CD45+ cells in mLNs from control individuals (135). In IBD patients, the frequency of Mfs in mLNs increases and they produced more IL-1β and TNF compared to those from controls (268, 312, 313). Chapuy et al. recently showed that mLN Mfs had high frequency of cells positive for inflammatory cytokines compared to mLN SIRPα+ DCs, which contain a mix of cDC2 and pDCs (313). However, these mLN Mfs poorly activated naïve T cells and did not contribute to Th17 plasticity toward Th1 and Th1/Th17 profiles compared to mLN SIRPα+ DCs (313). Finally, mLN Mfs highly expressed genes involved in phagocytosis and in leucocyte chemotaxis (313). These data corroborated with those from mice, where it has been shown that mLN Mfs poorly activate naïve T cells and were involved in clearing apoptotic cells and promoting B cell activation (314, 315). To note, using CyTOF, Chapuy et al. described 7 mLN Mf subsets in IBD patients (313), but their location, function and disease involvement remain to be deciphered.
Conventional Dendritic Cells in Mesenteric Lymph Nodes During Homeostasis
In mLNs, as in the other draining LNs, there are two cDC subpopulations: resident immature cDCs derived directly from the blood (called resident cDCs hereafter) and migratory mature cDCs derived from intestinal mucosa (called migratory cDCs hereafter) (135, 316, 317). Both populations are CD45+Lin−(CD3/CD19/CD56)HLA-DR+CD11c+CD14− but differ in the level of HLA-DR and CD11c expression. That is, resident cDCs are HLA-DRintCD11chi while migratory cDCs are HLA-DRhiCD11cint (Figure 3D). In contrast to lymphoid organs lacking afferent lymphatic vessels, such as the spleen and tonsils, where resident cDCs represent the vast majority (~98%) of the cDC fraction, resident cDCs represent 50 to 90% of total cDCs in draining LNs (135, 143).
Phenotype and Frequency of mLN cDCs
Both resident and migratory cDCs have been identified in mLNs of control individuals, but the percentage of each differs between studies (132, 135, 318) (Table 2). Indeed, Magnusson et al. observed a 7:1 migratory: resident cDC ratio in ileal mLNs (132), while Sakuraba et al. reported a 1:1 ratio in colonic mLNs (318) (Table 2). Recently Granot et al. found a 1:5 ratio in mLNs from unspecified intestinal regions (135) (Table 2). These discrepancies could be due to several reasons such as flow cytometry gating strategies or the “type” of control patients (Table 2). Thus, so far, available data do not allow a clear picture regarding the migratory:resident cDC ratio in mLNs during homeostasis. Regarding cDC subsets, Granot et al. found that cDC1 and cDC2 represent 0.05 and 0.1% of CD45+ cells in mLNs, respectively (135). More precisely, they observed a 3:1 cDC2/cDC1 ratio among resident cDCs while a 20:1 cDC2/cDC1 ratio among migratory cDCs (135) (Table 2). Magnusson et al. also showed a higher cDC2/cDC1 ratio among migratory cDCs compared to resident cDCs (132), even if it was not in the same range as that of Granot et al. (Table 2). Nevertheless, it seems that cDC2 represent the major subset of migratory cDCs in human mLNs (Figure 3D). However, migratory cDC1 may still go unnoticed by flow cytometry analysis if they downregulated their markers upon migration or/and die shortly after reaching mLNs (316). Thus, it remains to be determined if there is an imbalance between cDC2 and cDC1 migration per se to the mLNs, or if the proportional differences in mLNs simply reflects the relative frequencies that already exist in the intestinal mucosa.
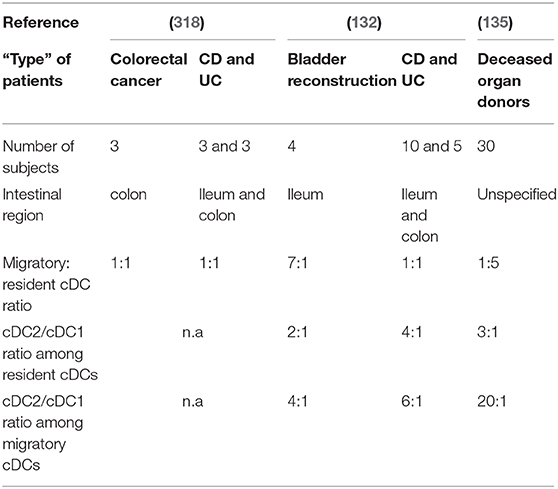
Table 2. Characteristics of human mLN cDCs during homeostasis and IBD.
Function of mLN cDCs
First, contrary to dogma, Sakuraba et al. showed that resident and migratory cDCs induce the same release of IFN-γ, IL-4, and IL-10 by naïve CD4+ T cells (318). Regarding functional assays on cDC subsets, Jaensson et al. showed that CD103+ cDCs, which represented around 30% of cDCs in mLNs, were more mature than CD103− cDCs, as judged by expression of CD40 and CD83 (311). In addition, CD103+ cDCs seem more inclined to immunosuppressive effects and induce more naïve CD4+ T cell polarization toward Tregs (319, 320). However, concerning RA activity, which is considered to be involved in Treg polarization in intestinal mucosa as discussed above, Sato et al. showed that neither human mLN CD103+ cDCs nor CD103− cDCs have ex vivo ALDH activity at basal state (321). This contrasts data from mice (309, 322). In addition, after ex vivo stimulation with GM-CSF, RA, and Vitamin D3, CD103− cDCs had more ALDH activity than CD103+ cDCs (321). Thus, as it has been shown in mouse skin-draining LN cDCs (323), CD103 expression does not constitute a marker for RA-producing human mLN cDCs. Thus, even if mLN CD103+ cDCs seem more tolerogenic than CD103− cDCs, the mechanisms for this are unknown. Moreover, analyzes need to be performed on cDC subsets delineated with more robust phenotypic markers.
Conventional Dendritic Cells in Mesenteric Lymph Nodes During IBD
Phenotype and Frequency of mLN cDCs
Concerning the migratory: resident cDC ratio in mLNs, Sakuraba et al. reported no difference between controls and IBD patients, which was 1:1 in both groups (318) (Table 2). In contrast, Magnusson et al. showed that this ratio decreased from 7:1 to 1:1 (132) (Table 2). Regarding cDC subsets, it has been shown that the cDC2/cDC1 ratio, among both resident and migratory cDCs, tends to increase in mLNs of IBD patients (132) (Table 2). Additional work is needed to clarify the migratory: resident cDC ratio and the cDC2/cDC1 ratio between controls and IBD patients, as well as to understand the significance of any revealed differences and the mechanisms driving ratio alterations in health vs. IBD.
Function of mLN cDCs
Sakuraba et al. showed that total mLN cDCs from IBD patients release little IL-12, IL-23, and IL-10 with or without LPS treatment ex vivo (318). However, in response to Enterococcus faecalis extract, the release of these cytokines increased in both Crohn's disease and UC patients (318). MLN cDCs from Crohn's patients induced more Th1 but similar Th2, Th17, and Treg polarization from naïve CD4+ T cells compared to those from controls or UC patients (318) (Figure 2, dotted square lower right). Regarding mLN CD103+ and CD103− cDCs, there was no difference in CCR9 and α4β7 induction on CD8+ T cells (311) or in ALDH activity (321) between cells from Crohn's patients compared to those from controls. In summary, much work remains to decipher the function of the cDC network, including the role of bona fide cDC1 and cDC2 subsets identified using robust markers, in mLNs during homeostasis and IBD pathogenesis.
Concluding Remarks and Future Perspectives
What Is the Current Status of Human Intestinal MNP Studies?
Despite the recent advances that have furthered our understanding of human intestinal MNPs, in both homeostasis and IBD, many important questions remain. In particular, intestinal MNP subsets seem to have both overlapping and distinct functional abilities, and unraveling this complexity is definitely a challenge. This is exemplified by the absence of a clear picture of T cell skewing capacities of MNP subsets despite experimental efforts. Part of the difficulty in understanding MNP function may be the spectrum of MNP subsets, which can complicate comparison of data from different research groups and lead to discrepancies (45, 261, 324, 325). Moreover, intestinal MNP regulation is complex and influenced by other immune cells, epithelial cells, and stromal cells (263, 274, 275, 326), as well as microbiota and metabolic components (17, 327, 328). Nevertheless, it is clear that the increase of immature Mfs and activated cDCs play a major role in IBD pathogenesis given their production of inflammatory cytokines/chemokines and their activation of stromal cells promoting their own recruitment and perpetuating an inflammatory cycle that leads to intestinal damage.
Can We Improve Conventional Strategies to Treat IBD?
Therapeutics for IBD that suppress intestinal inflammation by cytokine blockade have been used for some time (329), and ongoing testing of new drugs that target cytokines supports that this treatment avenue will remain viable (11, 330, 331). However, important questions remain. For example, do robust biomarkers exist to predict efficacy prior to initiating a treatment? What are the prospects of developing predictive biomarkers for existing therapies where they are lacking, and for new therapies as they enter the clinic? Will we be able to screen patients and stratify them for appropriate therapies?
The most significant breakthrough thus far in treating IBD, and is perhaps the classic example of suppressing a pro-inflammatory cytokine, is anti-TNF treatment. However, neutralizing TNF by no means helps all patients, as 30% of patients do not respond 1 year after treatment (332). Moreover, most of strategies targeting single effector cytokines in IBD have been disappointing in clinical trials (333), reinforcing the complexity and heterogeneity of IBD. Indeed, as many types of intestinal cells produce a wide range of effector cytokines, it is not surprising that it may be necessary to simultaneously target multiple cytokines to effectively suppress intestinal pathology. This is supported by positive effects of concomitant blockade of IL-12 and IL-23 using a monoclonal antibody against the IL-12p40 subunit (11, 330, 331, 333–339).
In addition to strategies that neutralize pro-inflammatory cytokines, an alternate approach has been to promote anti-inflammatory responses through application of cytokines such as IL-2, IL-10, IL-22, or TGF- β1 (11, 331, 336). Although these have been promising to some extent in animal models, they have not undergone rigorous clinical trials. For example, directly targeting TGF-β1 could be difficult given its multiple functions that distinctly influence the disease; nevertheless, recent clinical studies showed the efficacy of TGF-β signal restoration in IBD (307, 340, 341). Moreover, promoting intestinal repair through IL-22Fc could be another way to reduce burden in IBD patients (342).
One more pillar in IBD therapy is blocking immune cell trafficking molecules, such as α4β7 and MadCAM-1, as a general way to prevent influx of potentially pathogenic cells into the intestine. This approach has shown promise in experimental models but has had limited success in treating IBD patients (279, 331, 343). Nonetheless, etrolizumab anti-β7, which blocks both α4β7/MAdCAM-1 and αEβ7/E-cadherin interaction, is currently under evaluation in a phase III trial (344, 345). Another interesting way could be to do the opposite. Indeed, activating the integrin Mac-1 (CD11b/CD18) with small-molecule agonists inhibited immune cell recruitment by increasing their adhesion to the inflamed endothelium, at least in mice (346, 347). Therefore, development of therapies that target immune cell trafficking in intestine of IBD patients is attractive.
Can Specific MNP Populations Be Targeted to Treat IBD?
Monocytes and Macrophages
Significant data support that newly recruited monocytes in inflamed intestinal mucosa are central to driving the immunopathogenesis of IBD. Thus, developing treatments that dampen the number and/or pathologic function of these cells is an exciting avenue to pursue. Therefore, targeting the CCR2-CCL2/CCL7 axis could be one possibility to block monocyte recruitment to the intestine (263, 348). However, this could also inhibit their recruitment to other tissues, thus inducing side effects. Additionally, an overall lack of intestinal Mfs would likely increase susceptibility to infections as well as suppress benefits of their tissue repair activity (349, 350). The potential risks associated with this type of immunotherapy necessitate careful monitoring programs.
Other approaches are to decrease inflammatory programs in intestinal Mfs. One possibility among other examples reviewed previously (11, 325) could be the inflammation-amplifying TREM-1 receptor expressed by Mfs (249–251). Indeed, it has been shown that TREM-1 plays a role in initial signaling toward an inflammatory state of newly recruited monocytes in a mouse model of small intestine inflammation (351), and inhibition of TREM-1/CLEC5A pathways reduced intestinal inflammation during colitis (254, 352). In humans, TREM-1+ Mfs are increased in IBD patients and its engagement enhanced pro-inflammatory cytokine secretion while a TREM-1 antagonist dampened it (254, 255). Finally, Verstockt et al. have shown that low TREM-1 expression in both whole blood and intestinal mucosa can predict anti-TNF therapy responders (353, 354). Thus, TREM-1 could be a potential biomarker for predicting the effect of anti-TNF therapy and, secondly, blocking TREM-1 could be an attractive target for IBD treatment.
Other ways to improve intestinal homeostasis could be to promote anti-inflammatory and pro-resolving functions in Mfs. This could be achieved, for example, by enhancing negative regulation of TLR signaling and silent clearance of apoptotic cells (33, 355–359), as well as by augmenting IL-10 receptor signaling (360–362). To note, thanks to their high phagocytic capacity, targeting intestinal Mfs could be facilitated by “delivery systems” such as nanomaterials and biomaterials (363), as has been shown in mouse model of organ transplantation (364). Finally, reprogramming of Mf using metabolites could become a promising approach to dampen intestinal inflammation. For instance, the short-chain fatty acid n-butyrate (365, 366) and itaconate (327, 367) induce anti-inflammatory program in Mfs, which could represent an opportunity to treat IBD (368).
Conventional Dendritic Cells
In view of the data from preclinical models of IBD supporting a role of cDCs in intestinal inflammation (11, 45), it is appealing to speculate that selectively targeting cDCs could be a treatment strategy. However, the main function of cDCs is in draining lymph nodes where they influence T cell skewing and imprint tissue homing properties (317). Therapeutic approaches targeting cDC function in IBD would optimally influence cDC function, preferably specifically in mLNs, which is challenging but potentially possible (369). However, intestinal cDCs rapidly respond to environmental cues, including dietary changes and pathogen exposure, suggesting their environmental responsiveness could be exploited to influence their function. They also produce and respond to key cytokines implicated in IBD pathogenesis, and other drug-sensitive pathways that can potentially be exploited to modulate cDC function for therapeutic approaches also exist (11). This could include prevention of cDC activation by blockade of activating and/or survival signals, interference with intracellular signaling pathways, neutralization of effector cytokines they produce, or perturbation of cDC trafficking to target organs, which in the case of IBD is the intestinal mucosa and mLNs. Important questions essentially unexplored are the effect of existing treatments on cDC number and cDC subset ratio, and if these are disease-relevant, as well as cDC function, which likely has implications on tipping the balance from health to disease.
Conclusion
To conclude, advancing our understanding of the extended family of MNPs and dissecting their interactions with other cells comprising the networks that drive IBD is crucial to develop additional strategies to alleviate the chronic inflammation that underlies this debilitating disease. Indeed, recent publications using multi-dimensional analyses revealed cellular networks, including MNP subsets, involved in IBD pathogenesis, providing a platform for future therapeutic development. Finally, better characterization of pathophysiology in subgroups of IBD patients and developing combined immunotherapies for stratified, or possibly personalized, strategies is required to limit disease progression and develop new treatments for patients where current therapies are ineffective.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
The author's research is supported by the Swedish Cancer Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ungaro R, Mehandru S, Allen PB, Peyrin-Biroulet L, Jean-Frédéric C. Ulcerative colitis. Lancet. (2017) 389:1756–70. doi: 10.1016/S0140-6736(16)32126-2
2. Torres J, Mehandru S, Jean-Frédéric C, Peyrin-Biroulet L. Crohn's disease. Lancet. (2017) 389:1741–55. doi: 10.1016/S0140-6736(16)31711-1
3. Kaplan GG, Ng SC. Understanding and preventing the global increase of inflammatory bowel disease. Gastroenterology. (2017) 152:313–21.e2. doi: 10.1053/j.gastro.2016.10.020
4. Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. (2017) 390:2769–78. doi: 10.1016/S0140-6736(17)32448-0
5. Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. (2011) 474:307–17. doi: 10.1038/nature10209
6. Liu TC, Stappenbeck TS. Genetics and pathogenesis of inflammatory bowel disease. Annu Rev Pathol. (2016) 11:127–48. doi: 10.1146/annurev-pathol-012615-044152
7. de Souza HS, Fiocchi C. Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol. (2016) 13:13–27. doi: 10.1038/nrgastro.2015.186
8. Ni J, Wu GD, Albenberg L, Tomov VT. Gut microbiota and IBD: causation or correlation? Nat Rev Gastroenterol Hepatol. (2017) 14:573–84. doi: 10.1038/nrgastro.2017.88
9. Ananthakrishnan AN, Bernstein CN, Iliopoulos D, Macpherson A, Neurath MF, Ali RAR, et al. Environmental triggers in IBD: a review of progress and evidence. Nat Rev Gastroenterol Hepatol. (2018) 15:39–49. doi: 10.1038/nrgastro.2017.136
10. Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology. (2014) 146:1489–99. doi: 10.1053/j.gastro.2014.02.009
11. Friedrich M, Pohin M, Powrie F. Cytokine networks in the pathophysiology of inflammatory bowel disease. Immunity. (2019) 50:992–1006. doi: 10.1016/j.immuni.2019.03.017
12. Frank DN, St. Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA. (2007) 104:13780–5. doi: 10.1073/pnas.0706625104
13. Lepage P, Hasler R, Spehlmann ME, Rehman A, Zvirbliene A, Begun A, et al. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology. (2011) 141:227–36. doi: 10.1053/j.gastro.2011.04.011
14. Gevers D, Kugathasan S, Denson LA, Vazquez-Baeza Y, Van Treuren W, Ren B, et al. The treatment-naive microbiome in new-onset Crohn's disease. Cell Host Microbe. (2014) 15:382–92. doi: 10.1016/j.chom.2014.02.005
15. Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. (2019) 569:655–62. doi: 10.1038/s41586-019-1237-9
16. Franzosa EA, Sirota-Madi A, Avila-Pacheco J, Fornelos N, Haiser HJ, Reinker S, et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol. (2019) 4:293–305. doi: 10.1038/s41564-018-0306-4
17. Schirmer M, Garner A, Vlamakis H, Xavier RJ. Microbial genes and pathways in inflammatory bowel disease. Nat Rev Microbiol. (2019) 17:497–511. doi: 10.1038/s41579-019-0213-6
18. Zuo T, Kamm MA, Colombel JF, Ng SC. Urbanization and the gut microbiota in health and inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. (2018) 15:440–52. doi: 10.1038/s41575-018-0003-z
19. Zhang X, Deeke SA, Ning Z, Starr AE, Butcher J, Li J, et al. Metaproteomics reveals associations between microbiome and intestinal extracellular vesicle proteins in pediatric inflammatory bowel disease. Nat Commun. (2018) 9:2873. doi: 10.1038/s41467-018-05357-4
20. Scharl S, Barthel C, Rossel JB, Biedermann L, Misselwitz B, Schoepfer AM, et al. Malignancies in inflammatory bowel disease: frequency, incidence and risk factors-results from the swiss IBD cohort study. Am J Gastroenterol. (2018) 114:116–26. doi: 10.1038/s41395-018-0360-9
21. Samadder NJ, Valentine JF, Guthery S, Singh H, Bernstein CN, Leighton JA, et al. Family history associates with increased risk of colorectal cancer in patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol. (2018) 17:1807–13.e1. doi: 10.1016/j.cgh.2018.09.038
22. Liang H, Jiang B, Manne S, Lissoos T, Bennett D, Dolin P. Risk factors for postoperative infection after gastrointestinal surgery among adult patients with inflammatory bowel disease: findings from a large observational US cohort study. JGH Open. (2018) 2:182–90. doi: 10.1002/jgh3.12072
23. Kugathasan S, Saubermann LJ, Smith L, Kou D, Itoh J, Binion DG, et al. Mucosal T-cell immunoregulation varies in early and late inflammatory bowel disease. Gut. (2007) 56:1696–705. doi: 10.1136/gut.2006.116467
24. Coskun M, Vermeire S, Nielsen OH. Novel targeted therapies for inflammatory bowel disease. Trends Pharmacol Sci. (2017) 38:127–42. doi: 10.1016/j.tips.2016.10.014
25. Ahluwalia B, Moraes L, Magnusson MK, Ohman L. Immunopathogenesis of inflammatory bowel disease and mechanisms of biological therapies. Scand J Gastroenterol. (2018) 53:379–89. doi: 10.1080/00365521.2018.1447597
26. Biasci D, Lee JC, Noor NM, Pombal DR, Hou M, Lewis N, et al. A blood-based prognostic biomarker in IBD. Gut. (2019) 68:1386–95. doi: 10.1136/gutjnl-2019-318343
27. Gordon S, Pluddemann A. The mononuclear phagocytic system: generation of diversity. Front Immunol. (2019) 10:1893. doi: 10.3389/fimmu.2019.01893
28. van Furth R, Cohn ZA, Hirsch JG, Humphrey JH, Spector WG, Langevoort HL. The mononuclear phagocyte system a new classification of macrophages, monocytes, and their precursor cells. Bull World Health Organ. (1972) 46:845–52.
29. Hume DA. The mononuclear phagocyte system. Curr Opin Immunol. (2006) 18:49–53. doi: 10.1016/j.coi.2005.11.008
30. Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, et al. Immunobiology of dendritic cells. Annu Rev Immunol. (2000) 18:767–811. doi: 10.1146/annurev.immunol.18.1.767
31. Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol. (2015) 16:343–53. doi: 10.1038/ni.3123
32. Gordon S, Pluddemann A. Tissue macrophages: heterogeneity and functions. BMC Biol. (2017) 15:53. doi: 10.1186/s12915-017-0392-4
33. Nagata S. Apoptosis and clearance of apoptotic cells. Annu Rev Immunol. (2018) 36:489–517. doi: 10.1146/annurev-immunol-042617-053010
34. Schulz O, Jaensson E, Persson EK, Liu X, Worbs T, Agace WW, et al. Intestinal CD103+, but not CX3CR1+, antigen sampling cells migrate in lymph and serve classical dendritic cell functions. J Exp Med. (2009) 206:3101–14. doi: 10.1084/jem.20091925
35. Rivollier A, He J, Kole A, Valatas V, Kelsall BL. Inflammation switches the differentiation program of Ly6Chi monocytes from anti-inflammatory macrophages to inflammatory dendritic cells in the colon. J Exp Med. (2012) 209:139–55. doi: 10.1084/jem.20101387
36. Tanoue T, Atarashi K, Honda K. Development and maintenance of intestinal regulatory T cells. Nat Rev Immunol. (2016) 16:295–309. doi: 10.1038/nri.2016.36
37. Esterhazy D, Loschko J, London M, Jove V, Oliveira TY, Mucida D. Classical dendritic cells are required for dietary antigen-mediated induction of peripheral T(reg) cells and tolerance. Nat Immunol. (2016) 17:545–55. doi: 10.1038/ni.3408
38. Kim M, Galan C, Hill AA, Wu WJ, Fehlner-Peach H, Song HW, et al. Critical role for the microbiota in CX3CR1+ intestinal mononuclear phagocyte regulation of intestinal T cell responses. Immunity. (2018) 49:151–63.e5. doi: 10.1016/j.immuni.2018.05.009
39. Loschko J, Schreiber HA, Rieke GJ, Esterhazy D, Meredith MM, Pedicord VA, et al. Absence of MHC class II on cDCs results in microbial-dependent intestinal inflammation. J Exp Med. (2016) 213:517–34. doi: 10.1084/jem.20160062
40. Nutsch K, Chai JN, Ai TL, Russler-Germain E, Feehley T, Nagler CR, et al. Rapid and efficient generation of regulatory T cells to commensal antigens in the periphery. Cell Rep. (2016) 17:206–20. doi: 10.1016/j.celrep.2016.08.092
41. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. (2013) 496:445–55. doi: 10.1038/nature12034
42. Mowat AM, Bain CC. Macrophages in intestinal homeostasis and inflammation. Immunol Rev. (2014) 3:550–64. doi: 10.1111/imr.12192
43. Bekiaris V, Persson EK, Agace WW. Intestinal dendritic cells in the regulation of mucosal immunity. Immunol Rev. (2014) 260:86–101. doi: 10.1111/imr.12194
44. Gren ST, Grip O. Role of monocytes and intestinal macrophages in Crohn's Disease and ulcerative colitis. Inflamm Bowel Dis. (2016) 22:1992–8. doi: 10.1097/MIB.0000000000000824
45. Joeris T, Muller-Luda K, Agace WW, Mowat AM. Diversity and functions of intestinal mononuclear phagocytes. Mucosal Immunol. (2017) 10:845–64. doi: 10.1038/mi.2017.22
46. Baillie JK, Arner E, Daub C, De Hoon M, Itoh M, Kawaji H, et al. Analysis of the human monocyte-derived macrophage transcriptome and response to lipopolysaccharide provides new insights into genetic aetiology of inflammatory bowel disease. PLoS Genet. (2017) 13:e1006641. doi: 10.1371/journal.pgen.1006641
47. Bernardo D, Chaparro M, Gisbert JP. Human intestinal dendritic cells in inflammatory bowel diseases. Mol Nutr Food Res. (2018) 62:e1700931. doi: 10.1002/mnfr.201700931
48. Cader MZ, Kaser A. Recent advances in inflammatory bowel disease: mucosal immune cells in intestinal inflammation. Gut. (2013) 62:1653–64. doi: 10.1136/gutjnl-2012-303955
49. Kmiec Z, Cyman M, Slebioda TJ. Cells of the innate and adaptive immunity and their interactions in inflammatory bowel disease. Adv Med Sci. (2017) 62:1–6. doi: 10.1016/j.advms.2016.09.001
50. Ahluwalia B, Magnusson MK, Ohman L. Mucosal immune system of the gastrointestinal tract: maintaining balance between the good and the bad. Scand J Gastroenterol. (2017) 52:1185–93. doi: 10.1080/00365521.2017.1349173
51. Guilliams M, Mildner A, Yona S. Developmental and functional heterogeneity of monocytes. Immunity. (2018) 49:595–613. doi: 10.1016/j.immuni.2018.10.005
52. Kawamura S, Onai N, Miya F, Sato T, Tsunoda T, Kurabayashi K, et al. Identification of a human clonogenic progenitor with strict monocyte differentiation potential: a counterpart of mouse cMoPs. Immunity. (2017) 46:835–48.e4. doi: 10.1016/j.immuni.2017.04.019
53. Liu Z, Gu Y, Chakarov S, Bleriot C, Kwok I, Chen X, et al. Fate mapping via Ms4a3-Expression history traces monocyte-derived cells. Cell. (2019) 178:1509–25.e19. doi: 10.1016/j.cell.2019.08.009
54. Buenrostro JD, Corces MR, Lareau CA, Wu B, Schep AN, Aryee MJ, et al. Integrated single-cell analysis maps the continuous regulatory landscape of human hematopoietic differentiation. Cell. (2018) 173:1535–48.e16. doi: 10.1016/j.cell.2018.03.074
55. Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, et al. Nomenclature of monocytes and dendritic cells in blood. Blood. (2010) 116:e74–80. doi: 10.1182/blood-2010-02-258558
56. Zawada AM, Rogacev KS, Rotter B, Winter P, Marell RR, Fliser D, et al. SuperSAGE evidence for CD14++CD16+ monocytes as a third monocyte subset. Blood. (2011) 118:e50–61. doi: 10.1182/blood-2011-01-326827
57. Patel AA, Zhang Y, Fullerton JN, Boelen L, Rongvaux A, Maini AA, et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J Exp Med. (2017) 214:1913–23. doi: 10.1084/jem.20170355
58. Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol. (2017) 17:349–62. doi: 10.1038/nri.2017.28
59. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. (2010) 330:841–5. doi: 10.1126/science.1194637
60. Schulz C, Perdiguero EG, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. (2012) 336:86–90. doi: 10.1126/science.1219179
61. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. (2013) 38:79–91. doi: 10.1016/j.immuni.2013.05.008
62. Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol. (2013) 14:986–95. doi: 10.1038/ni.2705
63. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. (2013) 38:792–804. doi: 10.1016/j.immuni.2013.04.004
64. Lavin Y, Mortha A, Rahman A, Merad M. Regulation of macrophage development and function in peripheral tissues. Nat Rev Immunol. (2015) 15:731–44. doi: 10.1038/nri3920
65. Perdiguero EG, Geissmann F. The development and maintenance of resident macrophages. Nat Immunol. (2016) 17:2–8. doi: 10.1038/ni.3341
66. Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity. (2016) 44:439–49. doi: 10.1016/j.immuni.2016.02.024
67. Guilliams M, Scott CL. Does niche competition determine the origin of tissue-resident macrophages? Nat Rev Immunol. (2017) 17:451–60. doi: 10.1038/nri.2017.42
68. Stremmel C, Schuchert R, Wagner F, Thaler R, Weinberger T, Pick R, et al. Yolk sac macrophage progenitors traffic to the embryo during defined stages of development. Nat Commun. (2018) 9:75. doi: 10.1038/s41467-017-02492-2
69. Bajpai G, Schneider C, Wong N, Bredemeyer A, Hulsmans M, Nahrendorf M, et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med. (2018) 24:1234–45. doi: 10.1038/s41591-018-0059-x
70. Eguiluz-Gracia I, Schultz HH, Sikkeland LI, Danilova E, Holm AM, Pronk CJ, et al. Long-term persistence of human donor alveolar macrophages in lung transplant recipients. Thorax. (2016) 71:1006–11. doi: 10.1136/thoraxjnl-2016-208292
71. Tamoutounour S, Guilliams M, Montanana Sanchis F, Liu H, Terhorst D, Malosse C, et al. Origins and functional specialization of macrophages and of conventional and monocyte-derived dendritic cells in mouse skin. Immunity. (2013) 39:925–38. doi: 10.1016/j.immuni.2013.10.004
72. Tamoutounour S, Henri S, Lelouard H, de Bovis B, de Haar C, van der Woude CJ, et al. CD64 distinguishes macrophages from dendritic cells in the gut and reveals the Th1-inducing role of mesenteric lymph node macrophages during colitis. Eur J Immunol. (2012) 42:3150–66. doi: 10.1002/eji.201242847
73. Bain CC, Bravo-Blas A, Scott CL, Perdiguero EG, Geissmann F, Henri S, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol. (2014) 15:929–37. doi: 10.1038/ni.2967
74. Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol. (2016) 17:34–40. doi: 10.1038/ni.3324
75. Bujko A, Atlasy N, Landsverk OJB, Richter L, Yaqub S, Horneland R, et al. Transcriptional and functional profiling defines human small intestinal macrophage subsets. J Exp Med. (2018) 215:441–58. doi: 10.1084/jem.20170057
76. Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. (2014) 41:21–35. doi: 10.1016/j.immuni.2014.06.013
77. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. (2018) 233:6425–40. doi: 10.1002/jcp.26429
78. Zhang DE, Hetherington CJ, Chen HM, Tenen DG. The macrophage transcription factor PU.1 directs tissue-specific expression of the macrophage colony-stimulating factor receptor. Mol Cell Biol. (1994) 14:373–81. doi: 10.1128/MCB.14.1.373
79. Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. (2010) 327:656–61. doi: 10.1126/science.1178331
80. Monticelli S, Natoli G. Transcriptional determination and functional specificity of myeloid cells: making sense of diversity. Nat Rev Immunol. (2017) 17:595–607. doi: 10.1038/nri.2017.51
81. Scott CL, T'Jonck W, Martens L, Todorov H, Sichien D, Soen B, et al. The transcription factor ZEB2 is required to maintain the tissue-specific identities of macrophages. Immunity. (2018) 49:312–25.e5. doi: 10.1016/j.immuni.2018.07.004
82. Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol. (2009) 27:669–92. doi: 10.1146/annurev.immunol.021908.132557
83. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. (2013) 210:1977–92. doi: 10.1084/jem.20131199
84. McGovern N, Schlitzer A, Gunawan M, Jardine L, Shin A, Poyner E, et al. Human dermal CD14+ cells are a transient population of monocyte-derived macrophages. Immunity. (2014) 41:465–77. doi: 10.1016/j.immuni.2014.08.006
85. Kurotaki D, Sasaki H, Tamura T. Transcriptional control of monocyte and macrophage development. Int Immunol. (2017) 29:97–107. doi: 10.1093/intimm/dxx016
86. T'Jonck W, Guilliams M, Bonnardel J. Niche signals and transcription factors involved in tissue-resident macrophage development. Cell Immunol. (2018) 330:43–53. doi: 10.1016/j.cellimm.2018.02.005
87. Bakri Y, Sarrazin S, Mayer UP, Tillmanns S, Nerlov C, Boned A, et al. Balance of MafB and PU.1 specifies alternative macrophage or dendritic cell fate. Blood. (2005) 105:2707–16. doi: 10.1182/blood-2004-04-1448
88. Goudot C, Coillard A, Villani AC, Gueguen P, Cros A, Sarkizova S, et al. Aryl hydrocarbon receptor controls monocyte differentiation into dendritic cells versus macrophages. Immunity. (2017) 47:582–96.e6. doi: 10.1016/j.immuni.2017.08.016
89. Wong KL, Yeap WH, Tai JJ, Ong SM, Dang TM, Wong SC. The three human monocyte subsets: implications for health and disease. Immunol Res. (2012) 53:41–57. doi: 10.1007/s12026-012-8297-3
90. Boyette LB, Macedo C, Hadi K, Elinoff BD, Walters JT, Ramaswami B, et al. Phenotype, function, and differentiation potential of human monocyte subsets. PLoS ONE. (2017) 12:e0176460. doi: 10.1371/journal.pone.0176460
91. Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science. (2017) 356:eaah4573. doi: 10.1126/science.aah4573
92. Dutertre CA, Becht E, Irac SE, Khalilnezhad A, Narang V, Khalilnezhad S, et al. Single-cell analysis of human mononuclear phagocytes reveals subset-defining markers and identifies circulating inflammatory dendritic cells. Immunity. (2019) 51:573–89.e8. doi: 10.1016/j.immuni.2019.08.008
93. Günther P, Cirovic B, Baßler K, Händler K, Becker M, Dutertre CA, et al. A rule-based data-informed cellular consensus map of the human mononuclear phagocyte cell space. bioRxiv 658179; doi: 10.1101/658179
94. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
95. Varol C, Mildner A, Jung S. Macrophages: development and tissue specialization. Annu Rev Immunol. (2015) 33:643–75. doi: 10.1146/annurev-immunol-032414-112220
96. Mass E, Ballesteros I, Farlik M, Halbritter F, Gunther P, Crozet L, et al. Specification of tissue-resident macrophages during organogenesis. Science. (2016) 353:aaf4238. doi: 10.1126/science.aaf4238
97. Das A, Sinha M, Datta S, Abas M, Chaffee S, Sen CK, et al. Monocyte and macrophage plasticity in tissue repair and regeneration. Am J Pathol. (2015) 185:2596–606. doi: 10.1016/j.ajpath.2015.06.001
98. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. (2016) 44:450–62. doi: 10.1016/j.immuni.2016.02.015
99. Vannella KM, Wynn TA. Mechanisms of organ injury and repair by macrophages. Annu Rev Physiol. (2017) 79:593–617. doi: 10.1146/annurev-physiol-022516-034356
100. Eming SA, Wynn TA, Martin P. Inflammation and metabolism in tissue repair and regeneration. Science. (2017) 356:1026–30. doi: 10.1126/science.aam7928
101. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. (2011) 11:723–37. doi: 10.1038/nri3073
102. Collin M, Bigley V. Human dendritic cell subsets: an update. Immunology. (2018) 154:3–20. doi: 10.1111/imm.12888
103. See P, Dutertre CA, Chen J, Gunther P, McGovern N, Irac SE, et al. Mapping the human DC lineage through the integration of high-dimensional techniques. Science. (2017) 356:eaag3009. doi: 10.1126/science.aag3009
104. Grajales-Reyes GE, Iwata A, Albring J, Wu X, Tussiwand R, Kc W, et al. Batf3 maintains autoactivation of Irf8 for commitment of a CD8α+ conventional DC clonogenic progenitor. Nat Immunol. (2015) 16:708–17. doi: 10.1038/ni.3197
105. Schlitzer A, Sivakamasundari V, Chen J, Sumatoh HR, Schreuder J, Lum J, et al. Identification of cDC1- and cDC2-committed DC progenitors reveals early lineage priming at the common DC progenitor stage in the bone marrow. Nat Immunol. (2015) 16:718–28. doi: 10.1038/ni.3200
106. Breton G, Lee J, Zhou YJ, Schreiber JJ, Keler T, Puhr S, et al. Circulating precursors of human CD1c+ and CD141+ dendritic cells. J Exp Med. (2015) 212:401–13. doi: 10.1084/jem.20141441
107. Lee J, Breton G, Oliveira TY, Zhou YJ, Aljoufi A, Puhr S, et al. Restricted dendritic cell and monocyte progenitors in human cord blood and bone marrow. J Exp Med. (2015) 212:385–99. doi: 10.1084/jem.20141442
108. Paul F, Arkin Y, Giladi A, Jaitin DA, Kenigsberg E, Keren-Shaul H, et al. Transcriptional heterogeneity and lineage commitment in myeloid progenitors. Cell. (2015) 163:1663–77. doi: 10.1016/j.cell.2015.11.013
109. Lee J, Zhou YJ, Ma W, Zhang W, Aljoufi A, Luh T, et al. Lineage specification of human dendritic cells is marked by IRF8 expression in hematopoietic stem cells and multipotent progenitors. Nat Immunol. (2017) 18:877–88. doi: 10.1038/ni.3789
110. Velten L, Haas SF, Raffel S, Blaszkiewicz S, Islam S, Hennig BP, et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat Cell Biol. (2017) 19:271–81. doi: 10.1038/ncb3493
111. Rodrigues PF, Alberti-Servera L, Eremin A, Grajales-Reyes GE, Ivanek R, Tussiwand R. Distinct progenitor lineages contribute to the heterogeneity of plasmacytoid dendritic cells. Nat Immunol. (2018) 19:711–22. doi: 10.1038/s41590-018-0136-9
112. Dress RJ, Dutertre CA, Giladi A, Schlitzer A, Low I, Shadan NB, et al. Plasmacytoid dendritic cells develop from Ly6D+ lymphoid progenitors distinct from the myeloid lineage. Nat Immunol. (2019) 20:852–64. doi: 10.1038/s41590-019-0420-3
113. Bogunovic M, Ginhoux F, Helft J, Shang L, Hashimoto D, Greter M, et al. Origin of the lamina propria dendritic cell network. Immunity. (2009) 31:513–25. doi: 10.1016/j.immuni.2009.08.010
114. Varol C, Vallon-Eberhard A, Elinav E, Aychek T, Shapira Y, Luche H, et al. Intestinal lamina propria dendritic cell subsets have different origin and functions. Immunity. (2009) 31:502–12. doi: 10.1016/j.immuni.2009.06.025
115. Murphy TL, Grajales-Reyes GE, Wu X, Tussiwand R, Briseno CG, Iwata A, et al. Transcriptional control of dendritic cell development. Annu Rev Immunol. (2016) 34:93–119. doi: 10.1146/annurev-immunol-032713-120204
116. Persson EK, Uronen-Hansson H, Semmrich M, Rivollier A, Hagerbrand K, Marsal J, et al. IRF4 transcription-factor-dependent CD103+CD11b+ dendritic cells drive mucosal T helper 17 cell differentiation. Immunity. (2013) 38:958–69. doi: 10.1016/j.immuni.2013.03.009
117. Schlitzer A, McGovern N, Teo P, Zelante T, Atarashi K, Low D, et al. IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity. (2013) 38:970–83. doi: 10.1016/j.immuni.2013.04.011
118. Guilliams M, Dutertre C-A, Scott CL, McGovern N, Sichien D, Chakarov S, et al. Unsupervised high-dimensional analysis aligns dendritic cells across tissues and species. Immunity. (2016) 45:669–84. doi: 10.1016/j.immuni.2016.08.015
119. Sichien D, Scott CL, Martens L, Vanderkerken M, Van Gassen S, Plantinga M, et al. IRF8 transcription factor controls survival and function of terminally differentiated conventional and plasmacytoid dendritic cells, respectively. Immunity. (2016) 45:626–40. doi: 10.1016/j.immuni.2016.08.013
120. Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, et al. Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell. (2008) 135:37–48. doi: 10.1016/j.cell.2008.09.016
121. Edelson BT, Kc W, Juang R, Kohyama M, Benoit LA, Klekotka PA, et al. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8α+ conventional dendritic cells. J Exp Med. (2010) 207:823–36. doi: 10.1084/jem.20091627
122. Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol. (2015) 15:471–85. doi: 10.1038/nri3865
123. Luda KM, Joeris T, Persson EK, Rivollier A, Demiri M, Sitnik KM, et al. IRF8 transcription-factor-dependent classical dendritic cells are essential for intestinal T cell homeostasis. Immunity. (2016) 44:860–74. doi: 10.1016/j.immuni.2016.02.008
124. Sichien D, Lambrecht BN, Guilliams M, Scott CL. Development of conventional dendritic cells: from common bone marrow progenitors to multiple subsets in peripheral tissues. Mucosal Immunol. (2017) 10:831–44. doi: 10.1038/mi.2017.8
125. Pulendran B, Banchereau J, Burkeholder S, Kraus E, Guinet E, Chalouni C, et al. Flt3-Ligand and granulocyte colony-stimulating factor mobilize distinct human dendritic cell subsets in vivo. J Immunol. (2000) 165:566–72. doi: 10.4049/jimmunol.165.1.566
126. Maraskovsky E, Daro E, Roux E, Teepe M, Maliszewski CR, Hoek J, et al. In vivo generation of human dendritic cell subsets by Flt3 ligand. Blood. (2000) 96:878–84. doi: 10.1182/blood.V96.3.878.015k15_878_884
127. Balan S, Arnold-Schrauf C, Abbas A, Couespel N, Savoret J, Imperatore F, et al. Large-scale human dendritic cell differentiation revealing notch-dependent lineage bifurcation and heterogeneity. Cell Rep. (2018) 24:1902–15.e6. doi: 10.1016/j.celrep.2018.07.033
128. Guilliams M, Scott CL. 'NOTCHing up' the in vitro production of dendritic cells. Trends Immunol. (2018) 39:765–7. doi: 10.1016/j.it.2018.08.002
129. Kirkling ME, Cytlak U, Lau CM, Lewis KL, Resteu A, Khodadadi-Jamayran A, et al. Notch signaling facilitates in vitro generation of cross-presenting classical dendritic cells. Cell Rep. (2018) 23:3658–72.e6. doi: 10.1016/j.celrep.2018.05.068
130. Schlitzer A, McGovern N, Ginhoux F. Dendritic cells and monocyte-derived cells: two complementary and integrated functional systems. Semin Cell Dev Biol. (2015) 41:9–22. doi: 10.1016/j.semcdb.2015.03.011
131. Heidkamp GF, Sander J, Lehmann CHK, Heger L, Eissing N, Baranska A, et al. Human lymphoid organ dendritic cell identity is predominantly dictated by ontogeny, not tissue microenvironment. Sci Immunol. (2016) 1:eaai7677. doi: 10.1126/sciimmunol.aai7677
132. Magnusson MK, Brynjolfsson SF, Dige A, Uronen-Hansson H, Borjesson LG, Bengtsson JL, et al. Macrophage and dendritic cell subsets in IBD: ALDH+ cells are reduced in colon tissue of patients with ulcerative colitis regardless of inflammation. Mucosal Immunol. (2016) 9:171–82. doi: 10.1038/mi.2015.48
133. Alcantara-Hernandez M, Leylek R, Wagar LE, Engleman EG, Keler T, Marinkovich MP, et al. High-dimensional phenotypic mapping of human dendritic cells reveals interindividual variation and tissue specialization. Immunity. (2017) 47:1037–50.e6. doi: 10.1016/j.immuni.2017.11.001
134. Watchmaker PB, Lahl K, Lee M, Baumjohann D, Morton J, Kim SJ, et al. Comparative transcriptional and functional profiling defines conserved programs of intestinal DC differentiation in humans and mice. Nat Immunol. (2014) 15:98–108. doi: 10.1038/ni.2768
135. Granot T, Senda T, Carpenter DJ, Matsuoka N, Weiner J, Gordon CL, et al. Dendritic cells display subset and tissue-specific maturation dynamics over human life. Immunity. (2017) 46:504–15. doi: 10.1016/j.immuni.2017.02.019
136. Barclay AN, Van den Berg TK. The interaction between signal regulatory protein alpha (SIRPalpha) and CD47: structure, function, and therapeutic target. Annu Rev Immunol. (2014) 32:25–50. doi: 10.1146/annurev-immunol-032713-120142
137. Durai V, Murphy KM. Functions of murine dendritic cells. Immunity. (2016) 45:719–36. doi: 10.1016/j.immuni.2016.10.010
138. Vu Manh TP, Bertho N, Hosmalin A, Schwartz-Cornil I, Dalod M. Investigating evolutionary conservation of dendritic cell subset identity and functions. Front Immunol. (2015) 6:260. doi: 10.3389/fimmu.2015.00260
139. Sittig SP, Bakdash G, Weiden J, Skold AE, Tel J, Figdor CG, et al. A comparative study of the T cell stimulatory and polarizing capacity of human primary blood dendritic cell subsets. Mediators Inflamm. (2016) 2016:3605643. doi: 10.1155/2016/3605643
140. Mittag D, Proietto AI, Loudovaris T, Mannering SI, Vremec D, Shortman K, et al. Human dendritic cell subsets from spleen and blood are similar in phenotype and function but modified by donor health status. J Immunol. (2011) 186:6207–17. doi: 10.4049/jimmunol.1002632
141. Dzionek A, Fuchs A, Schmidt P, Cremer S, Zysk M, Miltenyi S, et al. BDCA-2, BDCA-3, and BDCA-4: three markers for distinct subsets of dendritic cells in human peripheral blood. J Immunol. (2000) 165:6037–46. doi: 10.4049/jimmunol.165.11.6037
142. MacDonald KPA, Munster DJ, Clark GJ, Dzionek A, Schmitz J, Hart DNJ. Characterization of human blood dendritic cell subsets. Blood. (2002) 100:4512–20. doi: 10.1182/blood-2001-11-0097
143. Haniffa M, Shin A, Bigley V, McGovern N, Teo P, See P, et al. Human tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity. (2012) 37:60–73. doi: 10.1016/j.immuni.2012.04.012
144. Chu CC, Ali N, Karagiannis P, Di Meglio P, Skowera A, Napolitano L, et al. Resident CD141 (BDCA3)+ dendritic cells in human skin produce IL-10 and induce regulatory T cells that suppress skin inflammation. J Exp Med. (2012) 209:935–45. doi: 10.1084/jem.20112583
145. Contreras V, Urien C, Guiton R, Alexandre Y, Vu Manh TP, Andrieu T, et al. Existence of CD8α-like dendritic cells with a conserved functional specialization and a common molecular signature in distant mammalian species. J Immunol. (2010) 185:3313–25. doi: 10.4049/jimmunol.1000824
146. Poulin LF, Salio M, Griessinger E, Anjos-Afonso F, Craciun L, Chen JL, et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8α+ dendritic cells. J Exp Med. (2010) 207:1261–71. doi: 10.1084/jem.20092618
147. Poulin LF, Reyal Y, Uronen-Hansson H, Schraml BU, Sancho D, Murphy KM, et al. DNGR-1 is a specific and universal marker of mouse and human Batf3-dependent dendritic cells in lymphoid and nonlymphoid tissues. Blood. (2012) 119:6052–62. doi: 10.1182/blood-2012-01-406967
148. Bachem A, Guttler S, Hartung E, Ebstein F, Schaefer M, Tannert A, et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med. (2010) 207:1273–81. doi: 10.1084/jem.20100348
149. Crozat K, Guiton R, Contreras V, Feuillet V, Dutertre CA, Ventre E, et al. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8α+ dendritic cells. J Exp Med. (2010) 207:1283–92. doi: 10.1084/jem.20100223
150. Gutierrez-Martinez E, Planes R, Anselmi G, Reynolds M, Menezes S, Adiko AC, et al. Cross-Presentation of cell-associated antigens by MHC class i in dendritic cell subsets. Front Immunol. (2015) 6:363. doi: 10.3389/fimmu.2015.00363
151. Blander JM. Regulation of the cell biology of antigen cross-presentation. Annu Rev Immunol. (2018) 36:717–53. doi: 10.1146/annurev-immunol-041015-055523
152. Iborra S, Martinez-Lopez M, Khouili SC, Enamorado M, Cueto FJ, Conde-Garrosa R, et al. Optimal generation of tissue-resident but not circulating memory T cells during viral infection requires crosspriming by DNGR-1+ dendritic cells. Immunity. (2016) 45:847–60. doi: 10.1016/j.immuni.2016.08.019
153. Segura E, Durand M, Amigorena S. Similar antigen cross-presentation capacity and phagocytic functions in all freshly isolated human lymphoid organ-resident dendritic cells. J Exp Med. (2013) 210:1035–47. doi: 10.1084/jem.20121103
154. Nizzoli G, Krietsch J, Weick A, Steinfelder S, Facciotti F, Gruarin P, et al. Human CD1c+ dendritic cells secrete high levels of IL-12 and potently prime cytotoxic T-cell responses. Blood. (2013) 122:932–42. doi: 10.1182/blood-2013-04-495424
155. Tang-Huau TL, Gueguen P, Goudot C, Durand M, Bohec M, Baulande S, et al. Human in vivo-generated monocyte-derived dendritic cells and macrophages cross-present antigens through a vacuolar pathway. Nat Commun. (2018) 9:2570. doi: 10.1038/s41467-018-04985-0
156. Mantegazza AR, Savina A, Vermeulen M, Perez L, Geffner J, Hermine O, et al. NADPH oxidase controls phagosomal pH and antigen cross-presentation in human dendritic cells. Blood. (2008) 112:4712–22. doi: 10.1182/blood-2008-01-134791
157. Jongbloed SL, Kassianos AJ, McDonald KJ, Clark GJ, Ju X, Angel CE, et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med. (2010) 207:1247–60. doi: 10.1084/jem.20092140
158. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. (2013) 31:563–604. doi: 10.1146/annurev-immunol-020711-074950
159. Eisenbarth SC. Dendritic cell subsets in T cell programming: location dictates function. Nat Rev Immunol. (2019) 19:89–103. doi: 10.1038/s41577-018-0088-1
160. Leon B, Lund FE. Compartmentalization of dendritic cell and T-cell interactions in the lymph node: Anatomy of T-cell fate decisions. Immunol Rev. (2019) 289:84–100. doi: 10.1111/imr.12758
161. Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. (2012) 12:125–35. doi: 10.1038/nri3133
162. Tang-Huau TL, Segura E. Human in vivo-differentiated monocyte-derived dendritic cells. Semin Cell Dev Biol. (2018) 86:44–9. doi: 10.1016/j.semcdb.2018.02.018
163. Amigorena S. Dendritic cells on the way to glory. J Immunol. (2018) 200:885–6. doi: 10.4049/jimmunol.1701693
164. Domínguez PM, Ardavín C. Differentiation and function of mouse monocyte-derived dendritic cells in steady state and inflammation. Immunol Rev. (2010) 234:90–104. doi: 10.1111/j.0105-2896.2009.00876.x
165. Sprangers S, de Vries TJ, Everts V. Monocyte heterogeneity: consequences for monocyte-derived immune cells. J Immunol Res. (2016) 2016:1475435. doi: 10.1155/2016/1475435
166. Michea P, Noel F, Zakine E, Czerwinska U, Sirven P, Abouzid O, et al. Adjustment of dendritic cells to the breast-cancer microenvironment is subset specific. Nat Immunol. (2018) 19:885–97. doi: 10.1038/s41590-018-0145-8
167. Segura E, Touzot M, Bohineust A, Cappuccio A, Chiocchia G, Hosmalin A, et al. Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity. (2013) 38:336–48. doi: 10.1016/j.immuni.2012.10.018
168. Schroder M, Melum GR, Landsverk OJ, Bujko A, Yaqub S, Gran E, et al. CD1c-expression by monocytes - implications for the use of commercial CD1c+ dendritic cell isolation kits. PLoS ONE. (2016) 11:e0157387. doi: 10.1371/journal.pone.0157387
169. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol. (2014) 14:571–8. doi: 10.1038/nri3712
170. Mowat AM. Anatomical basis of tolerance and immunity to intestinal antigens. Nat Rev Immunol. (2003) 3:331–41. doi: 10.1038/nri1057
171. Hettinger J, Richards DM, Hansson J, Barra MM, Joschko AC, Krijgsveld J, et al. Origin of monocytes and macrophages in a committed progenitor. Nat Immunol. (2013) 14:821–30. doi: 10.1038/ni.2638
172. Hill DA, Artis D. Intestinal bacteria and the regulation of immune cell homeostasis. Annu Rev Immunol. (2010) 28:623–67. doi: 10.1146/annurev-immunol-030409-101330
173. Pabst O, Mowat AM. Oral tolerance to food protein. Mucosal Immunol. (2012) 5:232–9. doi: 10.1038/mi.2012.4
174. Kamada N, Seo SU, Chen GY, Nunez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol. (2013) 13:321–35. doi: 10.1038/nri3430
175. Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. (2014) 157:121–41. doi: 10.1016/j.cell.2014.03.011
176. Parigi SM, Eldh M, Larssen P, Gabrielsson S, Villablanca EJ. Breast milk and solid food shaping intestinal immunity. Front Immunol. (2015) 6:415. doi: 10.3389/fimmu.2015.00415
177. Rooks MG, Garrett WS. Gut microbiota, metabolites and host immunity. Nat Rev Immunol. (2016) 16:341–52. doi: 10.1038/nri.2016.42
178. Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E. Dysbiosis and the immune system. Nat Rev Immunol. (2017) 17:219–32. doi: 10.1038/nri.2017.7
179. Steensels S, Depoortere I. Chemoreceptors in the Gut. Annu Rev Physiol. (2018) 80:117–41. doi: 10.1146/annurev-physiol-021317-121332
180. Allaire JM, Crowley SM, Law HT, Chang SY, Ko HJ, Vallance BA. The intestinal epithelium: central coordinator of mucosal immunity. Trends Immunol. (2018) 39:677–96. doi: 10.1016/j.it.2018.04.002
181. Olivares-Villagomez D, Van Kaer L. Intestinal intraepithelial lymphocytes: sentinels of the mucosal barrier. Trends Immunol. (2018) 39:264–275. doi: 10.1016/j.it.2017.11.003
182. Lutter L, Hoytema van Konijnenburg DP, Brand EC, Oldenburg B, van Wijk F. The elusive case of human intraepithelial T cells in gut homeostasis and inflammation. Nat Rev Gastroenterol Hepatol. (2018) 15:637–49. doi: 10.1038/s41575-018-0039-0
183. Chelakkot C, Ghim J, Ryu SH. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp Mol Med. (2018) 50:103. doi: 10.1038/s12276-018-0126-x
184. Odenwald MA, Turner JR. The intestinal epithelial barrier: a therapeutic target? Nat Rev Gastroenterol Hepatol. (2017) 14:9–21. doi: 10.1038/nrgastro.2016.169
185. Guiu J, Hannezo E, Yui S, Demharter S, Ulyanchenko S, Maimets M, et al. Tracing the origin of adult intestinal stem cells. Nature. (2019) 570:107–11. doi: 10.1038/s41586-019-1212-5
186. Martens EC, Neumann M, Desai MS. Interactions of commensal and pathogenic microorganisms with the intestinal mucosal barrier. Nat Rev Microbiol. (2018) 16:457–70. doi: 10.1038/s41579-018-0036-x
187. Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. (2014) 14:141–53. doi: 10.1038/nri3608
188. Clevers HC, Bevins CL. Paneth cells: maestros of the small intestinal crypts. Annu Rev Physiol. (2013) 75:289–311. doi: 10.1146/annurev-physiol-030212-183744
189. Johansson ME, Hansson GC. Immunological aspects of intestinal mucus and mucins. Nat Rev Immunol. (2016) 16:639–49. doi: 10.1038/nri.2016.88
190. Tordesillas L, Berin MC. Mechanisms of oral tolerance. Clin Rev Allergy Immunol. (2018) 55:107–17. doi: 10.1007/s12016-018-8680-5
191. Mowat AM. To respond or not to respond - a personal perspective of intestinal tolerance. Nat Rev Immunol. (2018) 18:405–15. doi: 10.1038/s41577-018-0002-x
192. Da Silva C, Wagner C, Bonnardel J, Gorvel JP, Lelouard H. The peyer's patch mononuclear phagocyte system at steady state and during infection. Front Immunol. (2017) 8:1254. doi: 10.3389/fimmu.2017.01254
193. Buettner M, Lochner M. Development and function of secondary and tertiary lymphoid organs in the small intestine and the colon. Front Immunol. (2016) 7:342. doi: 10.3389/fimmu.2016.00342
194. Hadis U, Wahl B, Schulz O, Hardtke-Wolenski M, Schippers A, Wagner N, et al. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity. (2011) 34:237–46. doi: 10.1016/j.immuni.2011.01.016
195. Muller PA, Koscso B, Rajani GM, Stevanovic K, Berres ML, Hashimoto D, et al. Crosstalk between muscularis macrophages and enteric neurons regulates gastrointestinal motility. Cell. (2014) 158:300–13. doi: 10.1016/j.cell.2014.08.002
196. Chinthrajah RS, Hernandez JD, Boyd SD, Galli SJ, Nadeau KC. Molecular and cellular mechanisms of food allergy and food tolerance. J Allergy Clin Immunol. (2016) 137:984–97. doi: 10.1016/j.jaci.2016.02.004
197. Belkaid Y, Harrison OJ. Homeostatic immunity and the microbiota. Immunity. (2017) 46:562–76. doi: 10.1016/j.immuni.2017.04.008
198. Gaudino SJ, Kumar P. Cross-talk between antigen presenting cells and t cells impacts intestinal homeostasis, bacterial infections, and tumorigenesis. Front Immunol. (2019) 10:360. doi: 10.3389/fimmu.2019.00360
199. Zigmond E, Varol C, Farache J, Elmaliah E, Satpathy AT, Friedlander G, et al. Ly6Chi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity. (2012) 37:1076–90. doi: 10.1016/j.immuni.2012.08.026
200. Zigmond E, Bernshtein B, Friedlander G, Walker CR, Yona S, Kim KW, et al. Macrophage-restricted interleukin-10 receptor deficiency, but not IL-10 deficiency, causes severe spontaneous colitis. Immunity. (2014) 40:720–33. doi: 10.1016/j.immuni.2014.03.012
201. Cerovic V, Bain CC, Mowat AM, Milling SW. Intestinal macrophages and dendritic cells: what's the difference? Trends Immunol. (2014) 35:270–7. doi: 10.1016/j.it.2014.04.003
202. Schett G, Neurath MF. Resolution of chronic inflammatory disease: universal and tissue-specific concepts. Nat Commun. (2018) 9:3261. doi: 10.1038/s41467-018-05800-6
203. Ortega-Gomez A, Perretti M, Soehnlein O. Resolution of inflammation: an integrated view. EMBO Mol Med. (2013) 5:661–74. doi: 10.1002/emmm.201202382
204. Sugimoto MA, Vago JP, Perretti M, Teixeira MM. Mediators of the resolution of the inflammatory response. Trends Immunol. (2019) 40:212–27. doi: 10.1016/j.it.2019.01.007
205. Sun M, He C, Cong Y, Liu Z. Regulatory immune cells in regulation of intestinal inflammatory response to microbiota. Mucosal Immunol. (2015) 8:969–78. doi: 10.1038/mi.2015.49
206. Wawrzyniak M, O'Mahony L, Akdis M. Role of regulatory cells in oral tolerance. Allergy Asthma Immunol Res. (2017) 9:107–15. doi: 10.4168/aair.2017.9.2.107
207. Luu M, Steinhoff U, Visekruna A. Functional heterogeneity of gut-resident regulatory T cells. Clin Transl Immunol. (2017) 6:e156. doi: 10.1038/cti.2017.39
208. Litinskiy MB, Nardelli B, Hilbert DM, He B, Schaffer A, Casali P, et al. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol. (2002) 3:822–9. doi: 10.1038/ni829
209. Castro-Dopico T, Clatworthy MR. IgG and fcγ receptors in intestinal immunity and inflammation. Front Immunol. (2019) 10:805. doi: 10.3389/fimmu.2019.00805
210. Macpherson AJ, Yilmaz B, Limenitakis JP, Ganal-Vonarburg SC. IgA function in relation to the intestinal microbiota. Annu Rev Immunol. (2018) 36:359–81. doi: 10.1146/annurev-immunol-042617-053238
211. Leonardi I, Li X, Semon A, Li D, Doron I, Putzel G, et al. CX3CR1+ mononuclear phagocytes control immunity to intestinal fungi. Science. (2018) 359:232–6. doi: 10.1126/science.aao1503
212. Okumura R, Takeda K. Roles of intestinal epithelial cells in the maintenance of gut homeostasis. Exp Mol Med. (2017) 49:e338. doi: 10.1038/emm.2017.20
213. Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. (2011) 474:298–306. doi: 10.1038/nature10208
214. Johansson ME, Sjövall H, Hansson GC. The gastrointestinal mucus system in health and disease. Nat Rev Gastroenterol Hepatol. (2013) 10:352–61. doi: 10.1038/nrgastro.2013.35
215. Ramakrishnan SK, Shah YM. Role of Intestinal HIF-2α in Health and Disease. Annu Rev Physiol. (2016) 78:301–25. doi: 10.1146/annurev-physiol-021115-105202
216. Colgan SP, Campbell EL, Kominsky DJ. Hypoxia and Mucosal Inflammation. Annu Rev Pathol. (2016) 11:77–100. doi: 10.1146/annurev-pathol-012615-044231
217. VanDussen KL, Stojmirovic A, Li K, Liu TC, Kimes PK, Muegge BD, et al. Abnormal small intestinal epithelial microvilli in patients with Crohn's disease. Gastroenterology. (2018) 155:815–28. doi: 10.1053/j.gastro.2018.05.028
218. Lee JC, Biasci D, Roberts R, Gearry RB, Mansfield JC, Ahmad T, et al. Genome-wide association study identifies distinct genetic contributions to prognosis and susceptibility in Crohn's disease. Nat Genet. (2017) 49:262–8. doi: 10.1038/ng.3755
219. Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol. (2014) 14:667–85. doi: 10.1038/nri3738
220. Mann ER, Bernardo D, English NR, Landy J, Al-Hassi HO, Peake ST, et al. Compartment-specific immunity in the human gut: properties and functions of dendritic cells in the colon versus the ileum. Gut. (2016) 65:256–70. doi: 10.1136/gutjnl-2014-307916
221. Bain CC, Scott CL, Uronen-Hansson H, Gudjonsson S, Jansson O, Grip O, et al. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. (2013) 6:498–510. doi: 10.1038/mi.2012.89
222. Sanders TJ, McCarthy NE, Giles EM, Davidson KL, Haltalli ML, Hazell S, et al. Increased production of retinoic acid by intestinal macrophages contributes to their inflammatory phenotype in patients with Crohn's disease. Gastroenterology. (2014) 146:1278–88.e1–2. doi: 10.1053/j.gastro.2014.01.057
223. Thiesen S, Janciauskiene S, Uronen-Hansson H, Agace W, Hogerkorp CM, Spee P, et al. CD14hiHLA-DRdim macrophages, with a resemblance to classical blood monocytes, dominate inflamed mucosa in Crohn's disease. J Leukoc Biol. (2014) 95:531–41. doi: 10.1189/jlb.0113021
224. Richter L, Landsverk OJB, Atlasy N, Bujko A, Yaqub S, Horneland R, et al. Transcriptional profiling reveals monocyte-related macrophages phenotypically resembling DC in human intestine. Mucosal Immunol. (2018) 11:1512–23. doi: 10.1038/s41385-018-0060-1
225. Bernardo D, Marin AC, Fernandez-Tome S, Montalban-Arques A, Carrasco A, Tristan E, et al. Human intestinal pro-inflammatory CD11chighCCR2+CX3CR1+ macrophages, but not their tolerogenic CD11c−CCR2−CX3CR1− counterparts, are expanded in inflammatory bowel disease. Mucosal Immunol. (2018) 11:1114–26. doi: 10.1038/s41385-018-0030-7
226. Ogino T, Nishimura J, Barman S, Kayama H, Uematsu S, Okuzaki D, et al. Increased Th17-inducing activity of CD14+ CD163low myeloid cells in intestinal lamina propria of patients with Crohn's disease. Gastroenterology. (2013) 145:1380–91.e1. doi: 10.1053/j.gastro.2013.08.049
227. Matsuno H, Kayama H, Nishimura J, Sekido Y, Osawa H, Barman S, et al. CD103+ dendritic cell function is altered in the colons of patients with ulcerative colitis. Inflamm Bowel Dis. (2017) 23:1524–34. doi: 10.1097/MIB.0000000000001204
228. Chapuy L, Bsat M, Sarkizova S, Rubio M, Therrien A, Wassef E, et al. Two distinct colonic CD14+ subsets characterized by single-cell RNA profiling in Crohn's disease. Mucosal Immunol. (2019) 12:703–19. doi: 10.1038/s41385-018-0126-0
229. Bell SJ, Rigby R, English N, Mann SD, Knight SC, Kamm MA, et al. Migration and maturation of human colonic dendritic cells. J Immunol. (2001) 166:4958–67. doi: 10.4049/jimmunol.166.8.4958
230. Meroni E, Stakenborg N, Viola MF, Boeckxstaens GE. Intestinal macrophages and their interaction with the enteric nervous system in health and inflammatory bowel disease. Acta Physiol. (2019) 225:e13163. doi: 10.1111/apha.13163
231. Gabanyi I, Muller PA, Feighery L, Oliveira TY, Costa-Pinto FA, Mucida D. Neuro-immune interactions drive tissue programming in intestinal macrophages. Cell. (2016) 164:378–91. doi: 10.1016/j.cell.2015.12.023
232. De Schepper S, Verheijden S, Aguilera-Lizarraga J, Viola MF, Boesmans W, Stakenborg N, et al. Self-maintaining gut macrophages are essential for intestinal homeostasis. Cell. (2018) 175:400–15.e13. doi: 10.1016/j.cell.2018.07.048
233. Wehner S, Engel DR. Resident macrophages in the healthy and inflamed intestinal muscularis externa. Pflugers Arch. (2017) 469:541–52. doi: 10.1007/s00424-017-1948-4
234. Beitnes AC, Raki M, Brottveit M, Lundin KE, Jahnsen FL, Sollid LM. Rapid accumulation of CD14+CD11c+ dendritic cells in gut mucosa of celiac disease after in vivo gluten challenge. PLoS ONE. (2012) 7:e33556. doi: 10.1371/journal.pone.0033556
235. Cipriani G, Gibbons SJ, Kashyap PC, Farrugia G. Intrinsic gastrointestinal macrophages: their phenotype and role in gastrointestinal motility. Cell Mol Gastroenterol Hepatol. (2016) 2:120–30.e1. doi: 10.1016/j.jcmgh.2016.01.003
236. Grainger JR, Konkel JE, Zangerle-Murray T, Shaw TN. Macrophages in gastrointestinal homeostasis and inflammation. Pflugers Arch. (2017) 469:527–39. doi: 10.1007/s00424-017-1958-2
237. Bain CC, Schridde A. Origin, differentiation, and function of intestinal macrophages. Front Immunol. (2018) 9:2733. doi: 10.3389/fimmu.2018.02733
238. Maheshwari A, Kelly DR, Nicola T, Ambalavanan N, Jain SK, Murphy-Ullrich J, et al. TGF-β2 suppresses macrophage cytokine production and mucosal inflammatory responses in the developing intestine. Gastroenterology. (2011) 140:242–53. doi: 10.1053/j.gastro.2010.09.043
239. Smith PD, Smythies LE, Mosteller-Barnum M, Sibley DA, Russell MW, Merger M, et al. Intestinal macrophages lack CD14 and CD89 and consequently are down-regulated for LPS- and IgA-mediated activities. J Immunol. (2001) 167:2651–6. doi: 10.4049/jimmunol.167.5.2651
240. Hausmann M, Kiessling S, Mestermann S, Webb G, Spöttl T, Andus T, et al. Toll-like receptors 2 and 4 are up-regulated during intestinal inflammation. Gastroenterology. (2002) 122:1987–2000. doi: 10.1053/gast.2002.33662
241. Rugtveit J, Nilsen EM, Bakka A, Carlsen H, Brandtzaeg P, Scott H. Cytokine profiles differ in newly recruited and resident subsets of mucosal macrophages from inflammatory bowel disease. Gastroenterology. (1997) 112:1493–505. doi: 10.1016/S0016-5085(97)70030-1
242. Smythies LE, Sellers M, Clements RH, Mosteller-Barnum M, Meng G, Benjamin WH, et al. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J Clin Invest. (2005) 115:66–75. doi: 10.1172/JCI200519229
243. Smythies LE, Shen R, Bimczok D, Novak L, Clements RH, Eckhoff DE, et al. Inflammation anergy in human intestinal macrophages is due to Smad-induced IkappaBalpha expression and NF-kappaB inactivation. J Biol Chem. (2010) 285:19593–604. doi: 10.1074/jbc.M109.069955
244. Kelly A, Gunaltay S, McEntee CP, Shuttleworth EE, Smedley C, Houston SA, et al. Human monocytes and macrophages regulate immune tolerance via integrin αvβ8-mediated TGFβ activation. J Exp Med. (2018) 215:2725–36. doi: 10.1084/jem.20171491
245. Smith PD, Smythies LE, Shen R, Greenwell-Wild T, Gliozzi M, Wahl SM. Intestinal macrophages and response to microbial encroachment. Mucosal Immunol. (2011) 4:31–42. doi: 10.1038/mi.2010.66
246. Kamada N, Hisamatsu T, Okamoto S, Chinen H, Kobayashi T, Sato T, et al. Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-gamma axis. J Clin Invest. (2008) 118:2269–80. doi: 10.1172/JCI34610
247. Kamada N, Hisamatsu T, Honda H, Kobayashi T, Chinen H, Takayama T, et al. TL1A produced by lamina propria macrophages induces Th1 and Th17 immune responses in cooperation with IL-23 in patients with Crohn's disease. Inflamm Bowel Dis. (2010) 16:568–75. doi: 10.1002/ibd.21124
248. Mizuno S, Mikami Y, Kamada N, Handa T, Hayashi A, Sato T, et al. Cross-talk between RORγt+ innate lymphoid cells and intestinal macrophages induces mucosal IL-22 production in Crohn's disease. Inflamm Bowel Dis. (2014) 20:1426–34. doi: 10.1097/MIB.0000000000000105
249. Klesney-Tait J, Turnbull IR, Colonna M. The TREM receptor family and signal integration. Nat Immunol. (2006) 7:1266–73. doi: 10.1038/ni1411
250. Tammaro A, Derive M, Gibot S, Leemans JC, Florquin S, Dessing MC. TREM-1 and its potential ligands in non-infectious diseases: from biology to clinical perspectives. Pharmacol Ther. (2017) 177:81–95. doi: 10.1016/j.pharmthera.2017.02.043
251. Read CB, Kuijper JL, Hjorth SA, Heipel MD, Tang X, Fleetwood AJ, et al. Cutting edge: identification of neutrophil PGLYRP1 as a ligand for TREM-1. J Immunol. (2015) 194:1417–21. doi: 10.4049/jimmunol.1402303
252. Bouchon A, Dietrich J, Colonna M. Cutting edge: inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J Immunol. (2000) 164:4991–5. doi: 10.4049/jimmunol.164.10.4991
253. Schenk M, Bouchon A, Birrer S, Colonna M, Mueller C. Macrophages expressing triggering receptor expressed on myeloid cells-1 are underrepresented in the human intestine. J Immunol. (2004) 174:517–24. doi: 10.4049/jimmunol.174.1.517
254. Schenk M, Bouchon A, Seibold F, Mueller C. TREM-1–expressing intestinal macrophages crucially amplify chronic inflammation in experimental colitis and inflammatory bowel diseases. J Clin Invest. (2007) 117:3097–106. doi: 10.1172/JCI30602
255. Brynjolfsson SF, Magnusson MK, Kong PL, Jensen T, Kuijper JL, Hakansson K, et al. An antibody against triggering receptor expressed on myeloid cells 1 (TREM-1) dampens proinflammatory cytokine secretion by lamina propria cells from patients with IBD. Inflamm Bowel Dis. (2016) 22:1803–11. doi: 10.1097/MIB.0000000000000822
256. Barman S, Kayama H, Okuzaki D, Ogino T, Osawa H, Matsuno H, et al. Identification of a human intestinal myeloid cell subset that regulates gut homeostasis. Int Immunol. (2016) 28:533–45. doi: 10.1093/intimm/dxw034
257. Sathaliyawala T, Kubota M, Yudanin N, Turner D, Camp P, Thome JJ, et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity. (2013) 38:187–97. doi: 10.1016/j.immuni.2012.09.020
258. Kumar BV, Connors TJ, Farber DL. Human T cell development, localization, and function throughout life. Immunity. (2018) 48:202–13. doi: 10.1016/j.immuni.2018.01.007
259. Bartolome-Casado R, Landsverk OJB, Chauhan SK, Richter L, Phung D, Greiff V, et al. Resident memory CD8 T cells persist for years in human small intestine. J Exp Med. (2019) 216:2412–26. doi: 10.1084/jem.20190414
260. Shaw TN, Houston SA, Wemyss K, Bridgeman HM, Barbera TA, Zangerle-Murray T, et al. Tissue-resident macrophages in the intestine are long lived and defined by Tim-4 and CD4 expression. J Exp Med. (2018) 215:1507–18. doi: 10.1084/jem.20180019
261. Stagg AJ. Intestinal dendritic cells in health and Gut inflammation. Front Immunol. (2018) 9:2883. doi: 10.3389/fimmu.2018.02883
262. Baumgart DC, Thomas S, Przesdzing I, Metzke D, Bielecki C, Lehmann SM, et al. Exaggerated inflammatory response of primary human myeloid dendritic cells to lipopolysaccharide in patients with inflammatory bowel disease. Clin Exp Immunol. (2009) 157:423–36. doi: 10.1111/j.1365-2249.2009.03981.x
263. Martin JC, Chang C, Boschetti G, Ungaro R, Giri M, Grout JA, et al. Single-cell analysis of Crohn's disease lesions identifies a pathogenic cellular module associated with resistance to anti-TNF therapy. Cell. (2019) 178:1493–508.e20. doi: 10.1016/j.cell.2019.08.008
264. Hart AL, Al-Hassi HO, Rigby RJ, Bell SJ, Emmanuel AV, Knight SC, et al. Characteristics of intestinal dendritic cells in inflammatory bowel diseases. Gastroenterology. (2005) 129:50–65. doi: 10.1053/j.gastro.2005.05.013
265. Mann ER, Bernardo D, Ng SC, Rigby RJ, Al-Hassi HO, Landy J, et al. Human gut dendritic cells drive aberrant gut-specific T-cell responses in ulcerative colitis, characterized by increased IL-4 production and loss of IL-22 and IFNγ. Inflamm Bowel Dis. (2014) 20:2299–307. doi: 10.1097/MIB.0000000000000223
266. Fenton TM, Kelly A, Shuttleworth EE, Smedley C, Atakilit A, Powrie F, et al. Inflammatory cues enhance TGFβ activation by distinct subsets of human intestinal dendritic cells via integrin αvβ8. Mucosal Immunol. (2017) 10:624–34. doi: 10.1038/mi.2016.94
267. Lampinen M, Waddell A, Ahrens R, Carlson M, Hogan SP. CD14+CD33+ myeloid cell-CCL11-eosinophil signature in ulcerative colitis. J Leukoc Biol. (2013) 94:1061–70. doi: 10.1189/jlb.1212640
268. Bsat M, Chapuy L, Baba N, Rubio M, Panzini B, Wassef R, et al. Differential accumulation and function of proinflammatory 6-sulfo LacNAc dendritic cells in lymph node and colon of Crohn's versus ulcerative colitis patients. J Leukoc Biol. (2015) 98:671–81. doi: 10.1189/jlb.5A1014-509RR
269. Chuang LS, Villaverde N, Hui KY, Mortha A, Rahman A, Levine AP, et al. A frameshift in CSF2RB predominant among ashkenazi jews increases risk for Crohn's disease and reduces monocyte signaling via GM-CSF. Gastroenterology. (2016) 151:710–23.e2. doi: 10.1053/j.gastro.2016.06.045
270. Gonzalez-Dominguez E, Samaniego R, Flores-Sevilla JL, Campos-Campos SF, Gomez-Campos G, Salas A, et al. CD163L1 and CLEC5A discriminate subsets of human resident and inflammatory macrophages in vivo. J Leukoc Biol. (2015) 98:453–66. doi: 10.1189/jlb.3HI1114-531R
271. Lissner D, Schumann M, Batra A, Kredel LI, Kuhl AA, Erben U, et al. Monocyte and M1 macrophage-induced barrier defect contributes to chronic intestinal inflammation in IBD. Inflamm Bowel Dis. (2015) 21:1297–305. doi: 10.1097/MIB.0000000000000384
272. Chapuy L, Bsat M, Rubio M, Sarkizova S, Therrien A, Bouin M, et al. IL-12 and mucosal CD14+ monocyte-like cells induce IL-8 in colonic memory CD4+ T cells of patients with ulcerative colitis but not Crohn's disease. J Crohns Colitis. (2019) 14:79–95. doi: 10.1093/ecco-jcc/jjz115
273. Jones GR, Bain CC, Fenton TM, Kelly A, Brown SL, Ivens AC, et al. Dynamics of colon monocyte and macrophage activation during colitis. Front Immunol. (2018) 9:2764. doi: 10.3389/fimmu.2018.02764
274. Huang B, Chen Z, Geng L, Wang J, Liang H, Cao Y, et al. Mucosal profiling of pediatric-onset colitis and IBD reveals common pathogenics and therapeutic pathways. Cell. (2019) 179:1160–76.e24. doi: 10.1016/j.cell.2019.10.027
275. Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, et al. Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell. (2019) 178:714–30.e22. doi: 10.1016/j.cell.2019.06.029
276. Smythies LE, Maheshwari A, Clements R, Eckhoff D, Novak L, Vu HL, et al. Mucosal IL-8 and TGF-beta recruit blood monocytes: evidence for cross-talk between the lamina propria stroma and myeloid cells. J Leukoc Biol. (2006) 80:492–9. doi: 10.1189/jlb.1005566
277. Grimm MC, Pullman WE, Bennett GM, Sullivan PJ, Pavli P, Doe WF. Direct evidence of monocyte recruitment to inflammatory bowel disease mucosa. J Gastroenterol Hepatol. (1995) 10:387–95. doi: 10.1111/j.1440-1746.1995.tb01589.x
278. Dige A, Magnusson MK, Ohman L, Hvas CL, Kelsen J, Wick MJ, et al. Reduced numbers of mucosal DRint macrophages and increased numbers of CD103+ dendritic cells during anti-TNF-α treatment in patients with Crohn's disease. Scand J Gastroenterol. (2016) 51:692–9. doi: 10.3109/00365521.2015.1134649
279. Zeissig S, Rosati E, Dowds CM, Aden K, Bethge J, Schulte B, et al. Vedolizumab is associated with changes in innate rather than adaptive immunity in patients with inflammatory bowel disease. Gut. (2019) 68:25–39. doi: 10.1136/gutjnl-2018-316023
280. Vos AC, Wildenberg ME, Arijs I, Duijvestein M, Verhaar AP, de Hertogh G, et al. Regulatory macrophages induced by infliximab are involved in healing in vivo and in vitro. Inflamm Bowel Dis. (2012) 18:401–8. doi: 10.1002/ibd.21818
281. Lacey DC, Achuthan A, Fleetwood AJ, Dinh H, Roiniotis J, Scholz GM, et al. Defining GM-CSF- and macrophage-CSF-dependent macrophage responses by in vitro models. J Immunol. (2012) 188:5752–65. doi: 10.4049/jimmunol.1103426
282. Ohradanova-Repic A, Machacek C, Fischer MB, Stockinger H. Differentiation of human monocytes and derived subsets of macrophages and dendritic cells by the HLDA10 monoclonal antibody panel. Clin Transl Immunol. (2016) 5:e55. doi: 10.1038/cti.2015.39
283. Pearson C, Thornton EE, McKenzie B, Schaupp AL, Huskens N, Griseri T, et al. ILC3 GM-CSF production and mobilisation orchestrate acute intestinal inflammation. Elife. (2016) 5:e10066. doi: 10.7554/eLife.10066
284. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. (2014) 40:274–88. doi: 10.1016/j.immuni.2014.01.006
285. Dabritz J, Weinhage T, Varga G, Wirth T, Walscheid K, Brockhausen A, et al. Reprogramming of monocytes by GM-CSF contributes to regulatory immune functions during intestinal inflammation. J Immunol. (2015) 194:2424–38. doi: 10.4049/jimmunol.1401482
286. Roth L, MacDonald JK, McDonald JW, Chande N. Sargramostim (GM-CSF) for induction of remission in Crohn's disease: a cochrane inflammatory bowel disease and functional bowel disorders systematic review of randomized trials. Inflamm Bowel Dis. (2012) 18:1333–9. doi: 10.1002/ibd.22973
287. Kamada N, Hisamatsu T, Honda H, Kobayashi T, Chinen H, Kitazume MT, et al. Human CD14+ macrophages in intestinal lamina propria exhibit potent antigen-presenting ability. J Immunol. (2009) 183:1724–31. doi: 10.4049/jimmunol.0804369
288. Takayama T, Kamada N, Chinen H, Okamoto S, Kitazume MT, Chang J, et al. Imbalance of NKp44+NKp46− and NKp44−NKp46+ natural killer cells in the intestinal mucosa of patients with Crohn's disease. Gastroenterology. (2010) 139:882–92:892.e1–3. doi: 10.1053/j.gastro.2010.05.040
289. Smith AM, Rahman FZ, Hayee B, Graham SJ, Marks DJ, Sewell GW, et al. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn's disease. J Exp Med. (2009) 206:1883–97. doi: 10.1084/jem.20091233
290. Schwerd T, Pandey S, Yang HT, Bagola K, Jameson E, Jung J, et al. Impaired antibacterial autophagy links granulomatous intestinal inflammation in Niemann-Pick disease type C1 and XIAP deficiency with NOD2 variants in Crohn's disease. Gut. (2017) 66:1060–73. doi: 10.1136/gutjnl-2015-310382
291. Aden K, Tran F, Ito G, Sheibani-Tezerji R, Lipinski S, Kuiper JW, et al. ATG16L1 orchestrates interleukin-22 signaling in the intestinal epithelium via cGAS-STING. J Exp Med. (2018) 215:2868–86. doi: 10.1084/jem.20171029
292. Nathan C, Cunningham-Bussel A. Beyond oxidative stress: an immunologist's guide to reactive oxygen species. Nat Rev Immunol. (2013) 13:349–61. doi: 10.1038/nri3423
293. Karin M, Clevers H. Reparative inflammation takes charge of tissue regeneration. Nature. (2016) 529:307–15. doi: 10.1038/nature17039
294. Rieder F, Fiocchi C, Rogler G. Mechanisms, management, and treatment of fibrosis in patients with inflammatory bowel diseases. Gastroenterology. (2017) 152:340–50.e6. doi: 10.1053/j.gastro.2016.09.047
295. Bettenworth D, Bokemeyer A, Baker M, Mao R, Parker CE, Nguyen T, et al. Anti-Fibrotic research: assessment of Crohn's disease-associated small bowel strictures and fibrosis on cross-sectional imaging: a systematic review. Gut. (2019) 68:1115–26. doi: 10.1136/gutjnl-2018-318081
296. Lenti MV, Di Sabatino A. Intestinal fibrosis. Mol Aspects Med. (2019) 65:100–9. doi: 10.1016/j.mam.2018.10.003
297. Scheibe K, Backert I, Wirtz S, Hueber A, Schett G, Vieth M, et al. IL-36R signalling activates intestinal epithelial cells and fibroblasts and promotes mucosal healing in vivo. Gut. (2017) 66:823–38. doi: 10.1136/gutjnl-2015-310374
298. Scheibe K, Kersten C, Schmied A, Vieth M, Primbs T, Carle B, et al. Inhibiting Interleukin 36 receptor signaling reduces fibrosis in mice with chronic intestinal inflammation. Gastroenterology. (2019) 156:1082–97.e11. doi: 10.1053/j.gastro.2018.11.029
299. Bettenworth D, Rieder F. Pathogenesis of intestinal fibrosis in inflammatory bowel disease and perspectives for therapeutic implication. Dig Dis. (2017) 35:25–31. doi: 10.1159/000449079
300. Salvador P, Macias-Ceja DC, Gisbert-Ferrandiz L, Hernandez C, Bernardo D, Alos R, et al. CD16+ macrophages mediate fibrosis in inflammatory bowel disease. J Crohns Colitis. (2018) 12:589–99. doi: 10.1093/ecco-jcc/jjx185
301. West NR, Hegazy AN, Owens BMJ, Bullers SJ, Linggi B, Buonocore S, et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat Med. (2017) 23:579–89. doi: 10.1038/nm.4307
302. West NR. Coordination of immune-stroma crosstalk by IL-6 family cytokines. Front Immunol. (2019) 10:1093. doi: 10.3389/fimmu.2019.01093
303. Ramesh R, Kozhaya L, McKevitt K, Djuretic IM, Carlson TJ, Quintero MA, et al. Pro-inflammatory human Th17 cells selectively express P-glycoprotein and are refractory to glucocorticoids. J Exp Med. (2014) 211:89–104. doi: 10.1084/jem.20130301
304. Ng SC, Benjamin JL, McCarthy NE, Hedin CR, Koutsoumpas A, Plamondon S, et al. Relationship between human intestinal dendritic cells, gut microbiota, and disease activity in Crohn's disease. Inflamm Bowel Dis. (2011) 17:2027–37. doi: 10.1002/ibd.21590
305. Wu W, He C, Liu C, Cao AT, Xue X, Evans-Marin HL, et al. miR-10a inhibits dendritic cell activation and Th1/Th17 cell immune responses in IBD. Gut. (2015) 64:1755–64. doi: 10.1136/gutjnl-2014-307980
306. Zhou L, Chu C, Teng F, Bessman NJ, Goc J, Santosa EK, et al. Innate lymphoid cells support regulatory T cells in the intestine through interleukin-2. Nature. (2019) 568:405–9. doi: 10.1038/s41586-019-1082-x
307. Monteleone G, Neurath MF, Ardizzone S, Di Sabatino A, Fantini MC, Castiglione F, et al. Mongersen, an oral SMAD7 antisense oligonucleotide, and Crohn's disease. N Engl J Med. (2015) 372:1104–13. doi: 10.1056/NEJMoa1407250
308. Manicassamy S, Pulendran B. Retinoic acid-dependent regulation of immune responses by dendritic cells and macrophages. Semin Immunol. (2009) 21:22–7. doi: 10.1016/j.smim.2008.07.007
309. Agace WW, Persson EK. How vitamin A metabolizing dendritic cells are generated in the gut mucosa. Trends Immunol. (2012) 33:42–8. doi: 10.1016/j.it.2011.10.001
310. Erkelens MN, Mebius RE. Retinoic acid and immune homeostasis: a balancing act. Trends Immunol. (2017) 38:168–80. doi: 10.1016/j.it.2016.12.006
311. Jaensson E, Uronen-Hansson H, Pabst O, Eksteen B, Tian J, Coombes JL, et al. Small intestinal CD103+ dendritic cells display unique functional properties that are conserved between mice and humans. J Exp Med. (2008) 205:2139–49. doi: 10.1084/jem.20080414
312. Baba N, Van VQ, Wakahara K, Rubio M, Fortin G, Panzini B, et al. CD47 fusion protein targets CD172a+ cells in Crohn's disease and dampens the production of IL-1beta and TNF. J Exp Med. (2013) 210:1251–63. doi: 10.1084/jem.20122037
313. Chapuy L, Bsat M, Rubio M, Harvey F, Motta V, Schwenter F, et al. Transcriptomic analysis and high dimensional phenotypic mapping of mononuclear phagocytes in mesenteric lymph nodes reveal differences between ulcerative colitis and Crohn's disease. J Crohns Colitis. (2019) jjz156. doi: 10.1093/ecco-jcc/jjz156
314. Baratin M, Simon L, Jorquera A, Ghigo C, Dembele D, Nowak J, et al. T cell zone resident macrophages silently dispose of apoptotic cells in the lymph node. Immunity. (2017) 47:349–62.e5. doi: 10.1016/j.immuni.2017.07.019
315. Bellomo A, Gentek R, Bajenoff M, Baratin M. Lymph node macrophages: scavengers, immune sentinels and trophic effectors. Cell Immunol. (2018) 330:168–74. doi: 10.1016/j.cellimm.2018.01.010
316. Segura E, Soumelis V. Of human DC migrants and residents. Immunity. (2017) 46:342–4. doi: 10.1016/j.immuni.2017.03.006
317. Worbs T, Hammerschmidt SI, Forster R. Dendritic cell migration in health and disease. Nat Rev Immunol. (2017) 17:30–48. doi: 10.1038/nri.2016.116
318. Sakuraba A, Sato T, Kamada N, Kitazume M, Sugita A, Hibi T. Th1/Th17 immune response is induced by mesenteric lymph node dendritic cells in Crohn's disease. Gastroenterology. (2009) 137:1736–45. doi: 10.1053/j.gastro.2009.07.049
319. Matteoli G, Mazzini E, Iliev ID, Mileti E, Fallarino F, Puccetti P, et al. Gut CD103+ dendritic cells express indoleamine 2,3-dioxygenase which influences T regulatory/T effector cell balance and oral tolerance induction. Gut. (2010) 59:595–604. doi: 10.1136/gut.2009.185108
320. Iliev ID, Spadoni I, Mileti E, Matteoli G, Sonzogni A, Sampietro GM, et al. Human intestinal epithelial cells promote the differentiation of tolerogenic dendritic cells. Gut. (2009) 58:1481–9. doi: 10.1136/gut.2008.175166
321. Sato T, Kitawaki T, Fujita H, Iwata M, Iyoda T, Inaba K, et al. Human CD1c+ myeloid dendritic cells acquire a high level of retinoic acid-producing capacity in response to vitamin D(3). J Immunol. (2013) 191:3152–60. doi: 10.4049/jimmunol.1203517
322. del Rio ML, Bernhardt G, Rodriguez-Barbosa JI, Förster R. Development and functional specialization of CD103+ dendritic cells. Immunol Rev. (2010) 234:268–81. doi: 10.1111/j.0105-2896.2009.00874.x
323. Guilliams M, Crozat K, Henri S, Tamoutounour S, Grenot P, Devilard E, et al. Skin-draining lymph nodes contain dermis-derived CD103− dendritic cells that constitutively produce retinoic acid and induce Foxp3+ regulatory T cells. Blood. (2010) 115:1958–68. doi: 10.1182/blood-2009-09-245274
324. Roszer T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflamm. (2015) 2015:816460. doi: 10.1155/2015/816460
325. Na YR, Stakenborg M, Seok SH, Matteoli G. Macrophages in intestinal inflammation and resolution: a potential therapeutic target in IBD. Nat Rev Gastroenterol Hepatol. (2019) 16:531–43. doi: 10.1038/s41575-019-0172-4
326. Kinchen J, Chen HH, Parikh K, Antanaviciute A, Jagielowicz M, Fawkner-Corbett D, et al. Structural remodeling of the human colonic mesenchyme in inflammatory bowel disease. Cell. (2018) 175:372–86.e17. doi: 10.1016/j.cell.2018.08.067
327. Russell DG, Huang L, VanderVen BC. Immunometabolism at the interface between macrophages and pathogens. Nat Rev Immunol. (2019) 19:291–304. doi: 10.1038/s41577-019-0124-9
328. Wang G, Huang S, Wang Y, Cai S, Yu H, Liu H, et al. Bridging intestinal immunity and gut microbiota by metabolites. Cell Mol Life Sci. (2019) 76:3917–37. doi: 10.1007/s00018-019-03190-6
329. Abraham C, Dulai PS, Vermeire S, Sandborn WJ. Lessons learned from trials targeting cytokine pathways in patients with inflammatory bowel diseases. Gastroenterology. (2017) 152:374–88.e4. doi: 10.1053/j.gastro.2016.10.018
330. Neurath MF. Current and emerging therapeutic targets for IBD. Nat Rev Gastroenterol Hepatol. (2017) 14:269–78. doi: 10.1038/nrgastro.2016.208
331. Neurath MF. Targeting immune cell circuits and trafficking in inflammatory bowel disease. Nat Immunol. (2019) 20:970–9. doi: 10.1038/s41590-019-0415-0
332. Shivaji UN, Sharratt CL, Thomas T, Smith SCL, Iacucci M, Moran GW, et al. Review article: managing the adverse events caused by anti-TNF therapy in inflammatory bowel disease. Aliment Pharmacol Ther. (2019) 49:664–80. doi: 10.1111/apt.15097
333. Baker KF, Isaacs JD. Novel therapies for immune-mediated inflammatory diseases: what can we learn from their use in rheumatoid arthritis, spondyloarthritis, systemic lupus erythematosus, psoriasis, Crohn's disease and ulcerative colitis? Ann Rheum Dis. (2018) 77:175–87. doi: 10.1136/annrheumdis-2017-211555
334. Feagan BG, Sandborn WJ, Gasink C, Jacobstein D, Lang Y, Friedman JR, et al. Ustekinumab as induction and maintenance therapy for Crohn's disease. N Engl J Med. (2016) 375:1946–60. doi: 10.1056/NEJMoa1602773
335. Teng MW, Bowman EP, McElwee JJ, Smyth MJ, Casanova JL, Cooper AM, et al. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat Med. (2015) 21:719–29. doi: 10.1038/nm.3895
336. Uhlig HH, Powrie F. Translating immunology into therapeutic concepts for inflammatory bowel disease. Annu Rev Immunol. (2018) 36:755–81. doi: 10.1146/annurev-immunol-042617-053055
337. Bloemendaal FM, Koelink PJ, van Schie KA, Rispens T, Peters CP, Buskens CJ, et al. TNF-anti-TNF immune complexes Inhibit IL-12/IL-23 Secretion by Inflammatory Macrophages via an Fc-dependent mechanism. J Crohns Colitis. (2018) 12:1122–30. doi: 10.1093/ecco-jcc/jjy075
338. Pagnini C, Pizarro TT, Cominelli F. Novel pharmacological therapy in inflammatory bowel diseases: beyond anti-tumor necrosis factor. Front Pharmacol. (2019) 10:671. doi: 10.3389/fphar.2019.00671
339. Moschen AR, Tilg H, Raine T. IL-12, IL-23 and IL-17 in IBD: immunobiology and therapeutic targeting. Nat Rev Gastroenterol Hepatol. (2018) 16:185–96. doi: 10.1038/s41575-018-0084-8
340. Ihara S, Hirata Y, Koike K. TGF-β in inflammatory bowel disease: a key regulator of immune cells, epithelium, and the intestinal microbiota. J Gastroenterol. (2017) 52:777–87. doi: 10.1007/s00535-017-1350-1
341. Feagan BG, Sands BE, Rossiter G, Li X, Usiskin K, Zhan X, et al. Effects of mongersen (GED-0301) on endoscopic and clinical outcomes in patients with active Crohn's disease. Gastroenterology. (2018) 154:61–4.e6. doi: 10.1053/j.gastro.2017.08.035
342. Rothenberg ME, Wang Y, Lekkerkerker A, Danilenko DM, Maciuca R, Erickson R, et al. Randomized phase I healthy volunteer study of UTTR1147A (IL-22Fc): a potential therapy for epithelial injury. Clin Pharmacol Ther. (2019) 105:177–89. doi: 10.1002/cpt.1164
343. Zundler S, Becker E, Schulze LL, Neurath MF. Immune cell trafficking and retention in inflammatory bowel disease: mechanistic insights and therapeutic advances. Gut. (2019) 68:1688–700. doi: 10.1136/gutjnl-2018-317977
344. Tang MT, Keir ME, Erickson R, Stefanich EG, Fuh FK, Ramirez-Montagut T, et al. Review article: nonclinical and clinical pharmacology, pharmacokinetics and pharmacodynamics of etrolizumab, an anti-β7 integrin therapy for inflammatory bowel disease. Aliment Pharmacol Ther. (2018) 47:1440–52. doi: 10.1111/apt.14631
345. Smids C, Horjus Talabur Horje CS, van Wijk F, van Lochem EG. The complexity of alpha E beta 7 blockade in inflammatory bowel diseases. J Crohns Colitis. (2017) 11:500–8. doi: 10.1093/ecco-jcc/jjw163
346. Maiguel D, Faridi MH, Wei C, Kuwano Y, Balla KM, Hernandez D. Small molecule-mediated activation of the integrin CD11bCD18 reduces inflammatory disease. Sci Signal. (2011) 4:ra57. doi: 10.1126/scisignal.2001811
347. Bermudez A. P142 Constitutively active Mac-1 (CD11b) ameliorates inflammation in mouse DSS colitis model. Inflamm Bowel Dis. (2019). doi: 10.1053/j.gastro.2019.01.221
348. Atreya R, Neurath MF. Chemokines in inflammatory bowel diseases. Dig Dis. (2010) 28:386–94. doi: 10.1159/000320392
349. Farro G, Stakenborg M, Gomez-Pinilla PJ, Labeeuw E, Goverse G, Di Giovangiulio M, et al. CCR2-dependent monocyte-derived macrophages resolve inflammation and restore gut motility in postoperative ileus. Gut. (2017) 66:2098–109. doi: 10.1136/gutjnl-2016-313144
350. Watanabe S, Alexander M, Misharin AV, Budinger GRS. The role of macrophages in the resolution of inflammation. J Clin Invest. (2019) 129:2619–28. doi: 10.1172/JCI124615
351. Desalegn G, Pabst O. Inflammation triggers immediate rather than progressive changes in monocyte differentiation in the small intestine. Nat Commun. (2019) 10:3229. doi: 10.1038/s41467-019-11148-2
352. Biagioli M, Mencarelli A, Carino A, Cipriani S, Marchiano S, Fiorucci C, et al. Genetic and pharmacological dissection of the role of spleen tyrosine kinase (Syk) in intestinal inflammation and immune dysfunction in inflammatory bowel diseases. Inflamm Bowel Dis. (2017) 24:123–35. doi: 10.1093/ibd/izx031
353. Verstockt B, Verstockt S, Dehairs J, Ballet V, Blevi H, Wollants WJ, et al. Low TREM1 expression in whole blood predicts anti-TNF response in inflammatory bowel disease. EBioMed. (2019) 40:733–42. doi: 10.1016/j.ebiom.2019.01.027
354. Verstockt B, Verstockt S, Blevi H, Cleynen I, de Bruyn M, Van Assche G, et al. TREM-1, the ideal predictive biomarker for endoscopic healing in anti-TNF-treated Crohn's disease patients? Gut. (2018) 68:1531–3. doi: 10.1136/gutjnl-2018-316845
355. Hamidzadeh K, Christensen SM, Dalby E, Chandrasekaran P, Mosser DM. Macrophages and the recovery from acute and chronic inflammation. Annu Rev Physiol. (2017) 79:567–92. doi: 10.1146/annurev-physiol-022516-034348
356. Gobbetti T, Dalli J, Colas RA, Federici Canova D, Aursnes M, Bonnet D, et al. Protectin D1n-3 DPA and resolvin D5n-3 DPA are effectors of intestinal protection. Proc Natl Acad Sci USA. (2017) 114:3963–8. doi: 10.1073/pnas.1617290114
357. Fullerton JN, Gilroy DW. Resolution of inflammation: a new therapeutic frontier. Nat Rev Drug Discov. (2016) 15:551–67. doi: 10.1038/nrd.2016.39
358. Gordon S, Pluddemann A. Macrophage clearance of apoptotic cells: a critical assessment. Front Immunol. (2018) 9:127. doi: 10.3389/fimmu.2018.00127
359. A-Gonzalez N, Quintana JA, Garcia-Silva S, Mazariegos M, González de la Aleja A, Nicolás-Ávila JA, et al. Phagocytosis imprints heterogeneity in tissue-resident macrophages. J Exp Med. (2017) 214:1281–96. doi: 10.1084/jem.20161375
360. Shouval DS, Biswas A, Goettel JA, McCann K, Conaway E, Redhu NS, et al. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity. (2014) 40:706–19. doi: 10.1016/j.immuni.2014.03.011
361. Shouval DS, Biswas A, Kang YH, Griffith AE, Konnikova L, Mascanfroni ID, et al. Interleukin 1β mediates intestinal inflammation in mice and patients with interleukin 10 receptor deficiency. Gastroenterology. (2016) 151:1100–4. doi: 10.1053/j.gastro.2016.08.055
362. Shouval DS, Konnikova L, Griffith AE, Wall SM, Biswas A, Werner L, et al. Enhanced TH17 responses in patients with IL10 Receptor Deficiency and Infantile-onset IBD. Inflamm Bowel Dis. (2017) 23:1950–61. doi: 10.1097/MIB.0000000000001270
363. Sylvestre M, Crane CA, Pun SH. Progress on modulating tumor-associated macrophages with biomaterials. Adv Mater. (2019) 27:e1902007. doi: 10.1002/adma.201902007
364. Braza MS, van Leent MMT, Lameijer M, Sanchez-Gaytan BL, Arts RJW, Perez-Medina C, et al. Inhibiting inflammation with myeloid cell-specific nanobiologics promotes organ transplant acceptance. Immunity. (2018) 49:819–28.e6. doi: 10.1016/j.immuni.2018.09.008
365. Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci USA. (2014) 111:2247–52. doi: 10.1073/pnas.1322269111
366. Schulthess J, Pandey S, Capitani M, Rue-Albrecht KC, Arnold I, Franchini F, et al. The short chain fatty acid butyrate imprints an antimicrobial program in macrophages. Immunity. (2019) 50:432–45.e7. doi: 10.1016/j.immuni.2018.12.018
367. O'Neill LAJ, Artyomov MN. The poster child of metabolic reprogramming in macrophage function. Nat Rev Immunol. (2019) 19:273–81. doi: 10.1038/s41577-019-0128-5
368. Parada Venegas D, De la Fuente MK, Landskron G, Gonzalez MJ, Quera R, Dijkstra G, et al. Short chain fatty acids (SCFAs)-mediated Gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front Immunol. (2019) 10:277. doi: 10.3389/fimmu.2019.01486
Keywords: crohn's disease, ulcerative colitis, intestine, macrophages, dendritic cells
Citation: Caër C and Wick MJ (2020) Human Intestinal Mononuclear Phagocytes in Health and Inflammatory Bowel Disease. Front. Immunol. 11:410. doi: 10.3389/fimmu.2020.00410
Received: 25 November 2019; Accepted: 21 February 2020;
Published: 18 March 2020.
Edited by:
Eric Cox, Ghent University, BelgiumCopyright © 2020 Caër and Wick. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mary Jo Wick, bWFyeS1qby53aWNrQGltbXVuby5ndS5zZQ==
†These authors have contributed equally to this work