
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 04 March 2020
Sec. Antigen Presenting Cell Biology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.00376
 Harriet A. Purvis1*†
Harriet A. Purvis1*† Fiona Clarke1Anna B. Montgomery1
Fiona Clarke1Anna B. Montgomery1 Chloe Colas1
Chloe Colas1 Jack A. Bibby1Georgina H. Cornish1†Xuezhi Dai2,3,4
Jack A. Bibby1Georgina H. Cornish1†Xuezhi Dai2,3,4 Diana Dudziak5David J. Rawlings2,3,4
Diana Dudziak5David J. Rawlings2,3,4 Rose Zamoyska6
Rose Zamoyska6 Pierre Guermonprez1,7‡
Pierre Guermonprez1,7‡ Andrew P. Cope1*‡
Andrew P. Cope1*‡Dendritic cells (DCs) are specialized antigen presenting cells that instruct T cell responses through sensing environmental and inflammatory danger signals. Maintaining the homeostasis of the multiple functionally distinct conventional dendritic cells (cDC) subsets that exist in vivo is crucial for regulating immune responses, with changes in numbers sufficient to break immune tolerance. Using Ptpn22−/− mice we demonstrate that the phosphatase PTPN22 is a highly selective, negative regulator of cDC2 homeostasis, preventing excessive population expansion from as early as 3 weeks of age. Mechanistically, PTPN22 mediates cDC2 homeostasis in a cell intrinsic manner by restricting cDC2 proliferation. A single nucleotide polymorphism, PTPN22R620W, is one of the strongest genetic risk factors for multiple autoantibody associated human autoimmune diseases. We demonstrate that cDC2 are also expanded in mice carrying the orthologous PTPN22619W mutation. As a consequence, cDC2 dependent CD4+ T cell proliferation and T follicular helper cell responses are increased. Collectively, our data demonstrate that PTPN22 controls cDC2 homeostasis, which in turn ensures appropriate cDC2-dependent T cell responses under antigenic challenge. Our findings provide a link between perturbations in DC development and susceptibility to a broad spectrum of PTPN22R620W associated human autoimmune diseases.
Dendritic cells (DCs) are specialized antigen presenting cells that sense danger signals and instruct T cell responses (1). Distinct subsets of DCs exist in vivo, broadly divided into conventional (cDC) and plasmacytoid (pDC) subsets. In mice, cDCs (CD11c+MHCII+) are sub-divided into functionally distinct phenotypes defined as cDC1 (CD8+IRF8+XCR1+Clec9a+CD24+) and cDC2 (IRF4+CD11b+SIRPα+) whilst in humans the equivalent DC subsets are defined by expression of CD8+IRF8+XCR1+Clec9a+CD141+ (cDC1) and IRF4+CD1c+ (cDC2) (1–4). Functionally, cDC1 cross-present exogenous antigens to activate CD8+ T cells and can promote IL-12 dependent Th1 responses (1, 5–7). In comparison, cDC2s are potent activators of CD4+ T cells (8). Under polarizing inflammatory conditions, cDC2 induce Th2 responses in the lung (9, 10), drive Th17 responses through IL-23 secretion (11–17), and initiate SIRPα dependent induction of T follicular helper cells (TFH) and germinal center (GC) formation (18).
Maintenance of cDC homeostasis is crucial for regulating immune responses, with deregulation resulting in infection and autoimmunity (19–22). This control of cDC homeostasis is mediated by a number of factors that drive cDC differentiation, proliferation, and survival or apoptosis (4). Differentiation of cDCs is initiated within the bone marrow where common DC precursors (CDP) transition to an intermediate preDC developmental stage (including pre-cDC1 and pre-cDC2s) (23–28), with terminal differentiation into cDC1 and cDC2 subsets occurring in the periphery (29); cDC1 and cDC2s are then dependent on Flt3L for their development and proliferation (29). Furthermore, local signals transduced through NOTCH2 (12, 15) or LTβR (30, 31) contribute to cDC homeostasis within specific tissue niches. Indeed, LTβR signaling is particularly important for inducing cDC2 proliferation within secondary lymphoid organs (SLOs) (30, 31).
PTPN22 encodes a tyrosine phosphatase that negatively regulates immune receptor activation. It functions by dephosphorylating Src and Syk family kinases operating proximal to immune-receptors such as TCR, BCR, and LFA-1 (32–36). PTPN22 also operates in a phosphatase independent manner, directly binding to TRAF3 in myeloid cells and promoting type 1 interferon dependent TRAF3 ubiquitination (37). Regarding its contribution to disease, a C1858T single nucleotide polymorphism within PTPN22 (encoding R620W) is one of the strongest genetic risk factors outside the HLA for the development of multiple autoimmune diseases, including rheumatoid arthritis, type I diabetes, and lupus (38). Investigations into the functional effects of this variant have demonstrated that PTPN22R620W confers a missense mutation in the P1 domain of the PTPN22 PEST region, resulting in reduced binding to the negative regulatory tyrosine kinase Csk, and TRAF3 (37, 39). However, the consequence of PTPN22R620W on immune function remains unclear, appearing to depend on the cellular context and signaling pathway under investigation. Indeed, both gain- and loss-of-phosphatase function effects of PTPN22R620W have been described (40).
Using Ptpn22 mutant mice, we describe PTPN22 as key mediator in the restriction of cDC2 populations. Perturbation of cDC2 homeostasis is phenocopied in mice carrying the human autoimmune associated variant, translating to accentuated cDC2-driven T cell responses upon antigenic challenge. Based on these data, we propose that disruption of cDC homeostasis by PTPN22R620W genetic polymorphism contributes to the breeching of immune tolerance during the earliest phase of autoimmunity.
Ptpn22−/− mice and Ptpn22R619W mutant mice were backcrossed >10 generations to the C57BL/6 strain, their generation is described in Dai et al. (33) and Brownlie et al. (41). Mice were age and sex-matched within each individual experiment and were used at either 2–4 months or as otherwise indicated. Ptpn22fl/+ mice were bred with PC3-Cre mice and backcrossed to C57BL/6 mice for four generations and were used between 8 and 12 weeks of age. OT-II TCR transgenic CD45.1, CD45.1, and CD45.1/2 transgenic mice were used between 8 and 16 weeks of age. Where indicated, tissue was obtained, shipped on ice and processed within 24 h from mice bred and maintained at Edinburgh University under U.K. Home Office approved guidelines. Mice were age and sex-matched within each individual experiment and were used at either 2 or 6 months of age. Unless otherwise stated mice were maintained under specific pathogen free (SPF) conditions at King's College London Facility according to UK Home Office approved protocols.
Spleens and LNs were injected with RPMI containing Liberase-TL (0.1 mg/ml; Roche) and DNase 1 (0.1 mg/ml; Sigma), and incubated at 37°C 5% CO2 for 30 min. EDTA (10 mM) was added for the final 5 min of the 30-min incubation. Spleen single cell suspensions were RBC lysed (Biolegend). Blood obtained by cardiac puncture was incubated at room temperature 1 h and serum separated following centrifugation. Bone marrow was flushed from the femurs and tibias of WT and Ptpn22−/− mice, RBC lysed and pelleted. Cell suspensions were prepared from the small intestine after removal of Peyer's patches and fat. Intestines were opened longitudinally, washed of fecal contents, cut into 5 cm pieces and incubated in HBSS medium (Life Technologies) with 2 mM EDTA for 20 min at 37°C with rotation. Tissue pieces were washed in HBSS medium, minced and incubated in HBSS medium + 2% FBS with collagenase VIII (100 U/ml, Sigma, C2139) and DNAse1 (20 μg/ml) at 37°C for 40 min with rotation. Cell suspensions was passed through a 40 μm filter and pelleted at 350 g 15 min 4C. Cells were then stained for flow cytometry.
Cells from all tissues were resuspended in PBS and live cells counted by trypan blue discrimination.
CD45.1 or CD45.1/2 mice were hematopoietically-lethally irradiated by exposure to 9Gy for 6 min. Six hours later, bone marrow cells (2.5–5 × 106 cells in 100 μl) were i.v transferred into irradiated recipients. Chimeric mice were analyzed 8 weeks after bone marrow transfer (unless otherwise indicated). As a control for complete replacement of recipient bone marrow, CD45.1+ recipients received 100% CD45.2+ C57BL/6 bone morrow.
Total CD4+ T cells from the lymph nodes (LN) and spleens of 8–16-week old WT OT-II mice were isolated using CD4+ MACS negative selection kit according to manufacturer's instructions (Miltenyi Biotech). Purity of CD4+ T cells was determined by flow cytometry (routinely 90–95%). CD4+ T cells isolated from CD45.1 WT OT-II mice were labeled with cell trace violet (CTV; 2 μM; Invitrogen) at 2 × 107 cells/ml in PBS for 20 min at 37°C, and excess CTV quenched in complete medium for 20 m at 37°C. Recipient mice received 0.5–1 × 106 CTV+CD4+OT-II T cells resuspended in 100 μL PBS i.v. The following day T cell recipient mice were immunized i.v with 33D1-ovalbumin (200 μg/mouse) (42) in the presence or absence of sheep RBC (SRBC; Antibodies-online.com; 20 × 107 cells/mouse). After 3 days spleens were assessed by flow cytometry for CTV dilution and CXCR5+ PD1+ TFH within live, singlet, CD45.1+, CD4+, Vα2Vβ5+ cell gate.
CD4+ T cells isolated from OT-II mice were labeled with cell trace violet (CTV) (2 μM; Invitrogen) at 2 × 107 cells/ml in PBS for 20 min at 37°C, and excess CTV quenched in complete medium for 20 min at 37°C. T cells were co-cultured with isolated DC at a 2:1 T cell to cDC ratio 2 x 106:1 x 106 cells/ml in round bottom 96-well plates in the presence or absence of 33D1-ovalbumin (10 μg/ml) or anti-DEC205-ovalbumin (10 μg/ml) and cells were co-cultured for 6 days at 37°C 5% CO2. At day 6 cells were stained with fixable viability dye eFluor-506 (eBioscience), anti-CD3ε and anti-CD4. CTV dilution gated on live, singlet, CD3+CD4+ T cells was assessed by flow cytometry.
Three days prior to analysis mice were i.p injected daily with BrDU (10 mg/kg) and maintained ad libitum on drinking water containing BrdU (0.5 mg/ml).
Bone marrow was RBC lysed and cells seeded at 1 × 106 cells/ml in 6-well tissue culture plates in RPMI-1640 supplemented with glutamax, 10% heat-inactivated FBS, β-mercaptoethanol (5 0 μM), penicillin/streptomycin (100 μg/ml), and HEPES 1 mM (Sigma) and Flt3L (200 ng/ml; Biolegend). Flt3L-BMDCs were cultured for 8 days at 37°C and 5% CO2. At day 8 non-adherent and adherent Flt3L-BMDC were harvested with trypsin-EDTA (Sigma) incubated for 2 min at room temperature. Cells were washed and live cells counted by trypan blue discrimination and seeded at 1 × 106 cells/ml on 96-well flat bottom plates for 48 h in the presence or absence of anti-LTβR (2 μg/ml; 3C8; AdipoGen).
Fluorochrome or biotin-conjugated antibodies were used to stain single cell suspensions for flow cytometry. Fc receptors were blocked with anti-mouse CD16/CD32 (93; Biolegend) and dead cell exclusion was performed using Fixable Viability Dye (eBioscience). FACS buffer was made of PBS with 1% bovine serum albumin (Sigma) and 2 mM EDTA (Sigma). For BrDU staining following surface staining, splenocytes were fixed and permeabilised using APC-BrDU Flow Kit (BD Pharmingen). Cells were acquired using BD Fortessa or FACSCanto II flow cytometers. Performed in the Biomedical Research Center Flow Core Facility (Guy's and St Thomas' NHS Foundation Trust and King's College London). Flow cytometry gates were determined by fluorescence minus one controls. Flow cytometry analysis was performed using FlowJo software (TreeStar; 10.5.3).
CD3ε (145-2C11), CD4 (GK1.5), CD8a (53-6.7), CD11b (M1/70), CD11c (N418), CD24 (M1/69), CD45R/B220 (RA3-6B2), CD45.1 (A20), CD45.2 (104), CD86 (GL-1), CD103 (2E7), CD172a (SIRP alpha; P84), CD279 (PD1; 29F.1A12), CXCR5 (L138D7), I-Ab (AF6-120.1), TCR Vα2 (B20.1), TCRVβ5 (MR9-4), CD16/32 (93), Ki67 (16A8), Ly-6G/Ly-6C (RB6-8C5), Ly-6G (1A8), (Ly6C (AL-21), Ter-119 (TER-119), NK-1.1 (PK136), CD19 (6D5), DCIR2 (33D1), cKit (2B8), Flt3 (A2-F10), LTβR (5G11) that were bought from Biolegend, eBioscience, or BD.
Spleens were harvested into RPMI and 10% FBS and dried prior to being frozen at −80°C in OCT. Ten micrometers sections were generated and mounted onto slides using a Leica cryostat. Sections were fixed with 4% PFA for 15 min and washed with PBS. Sections were blocked for 30 min in blocking buffer (PBS + 2% FBS, rat serum (1 in 200) and anti-CD16/CD32 (1 in 200) for 30 min at room temperature. Sections were stained for 1 h at room temperature with B220-FITC and 33D1-Alexa-647 in PBS 2% FBS. Slides were washed 3 times with PBS prior to mounting in fluorescence mounting media (DAKO). Images were collected using an Olympus IX83 inverted microscope and image processing was performed using Fiji.
Total RNA was extracted from FACS isolated cDC2 using TRIzol reagent. Equal amounts of mRNA (determined by nanodrop; ThermoScientific) were reverse transcribed to produce cDNA using first strand cDNA synthesis using random hexamers. Gene expression was measured by SYBR Green quantitative real-time PCR using primers: Bcl2 forward, TGAGTACCTGAACCGGCATCT, Bcl2 reverse, GCATCCCAGCCTCCGTTAT; Bim forward, GGCCCCTACCTCCCTACA, Bim reverse, GGGGTTTGTGTTGATTTGTCA; Trim2 forward, TTTCCATAATCACTCTGTCAAGGT, Trim2 reverse, CCATTGGAGCCAAACTTCA; Gapdh forward, ACCACAGTCCATGCCATCAC Gapdh reverse, TCCACCACCCTGTTGCTGTA. Reactions were run using ABI Prism 7700 Sequence Detection System (Applied Biosystems). Ct values were determined with SDS software (Applied Biosystems) and gene expression levels were determined according to the dCt method (relative abundance = 2(−dct) and normalized to GAPDH housekeeper).
Blood obtained by cardiac puncture was incubated at room temperature 1 h and serum separated following centrifugation. Serum Flt3 Ligand was determined by Mouse/Rat Quantikine ELISA (R&D Systems) according to manufacturer's protocol and detected using Victor 1420 multilabel counter (Perkin Elmer).
GraphPad Prism software was used for statistical analysis by unpaired or paired T-test. P < 0.05 were considered significant; NS = not significant, *p < 0.05, ** p < 0.01, ***p < 0.001, ****p < 0.0001.
Ptpn22−/− mice have a well-characterized age dependent increase in effector/memory T lymphocytes and spontaneous GC production. However, the effect of PTPN22 on myeloid lineages is not understood. To address this, we examined if myeloid cell lineages were altered in mice lacking PTPN22. We detected a similar frequency of monocytes, macrophages, and neutrophils (Supplementary Figures 1A–C) within the spleens of WT and Ptpn22−/− mice. In contrast, analysis of the splenic cDC compartment revealed that both the proportion and number of cDCs was increased in Ptpn22−/− mice compared to WT (Figures 1A,B). Further phenotyping of cDC1 and cDC2 subsets revealed that the proportion of cDC2 was increased in Ptpn22−/− mice, whereas the proportion of cDC1 was decreased (Figures 1A,C). By comparing the number of splenic cDC1s and cDC2s within WT and Ptpn22−/− mice we found that changes in cDC subset proportions were due to selective expansion of the cDC2 subset and not loss of cDC1 cells (Figure 1D). Indeed, expansion of splenic cDC2s occurred within the bona fide DCIR2(33D1)+ESAM+CD4+CCR2− cDC2 subset, whereas numbers of the “monocyte-like” DCIR2(33D1)−ESAM−CD4−CCR2+/− DCs were similar (Figures 1E,F and Supplementary Figures 1D–F). Analyzing the kinetics of cDC2 expansion demonstrated that perturbation of cDC2 homeostasis could be detected as early as 3 weeks (Figures 1G,H), increasing further as the mice age (Supplementary Figure 1G). We confirmed these findings in WT and Ptpn22−/− mice bred and maintained in an independent animal facility suggesting that cDC2 expansion was unlikely to be due to facility-associated environmental factors (Supplementary Figures 1H,I).
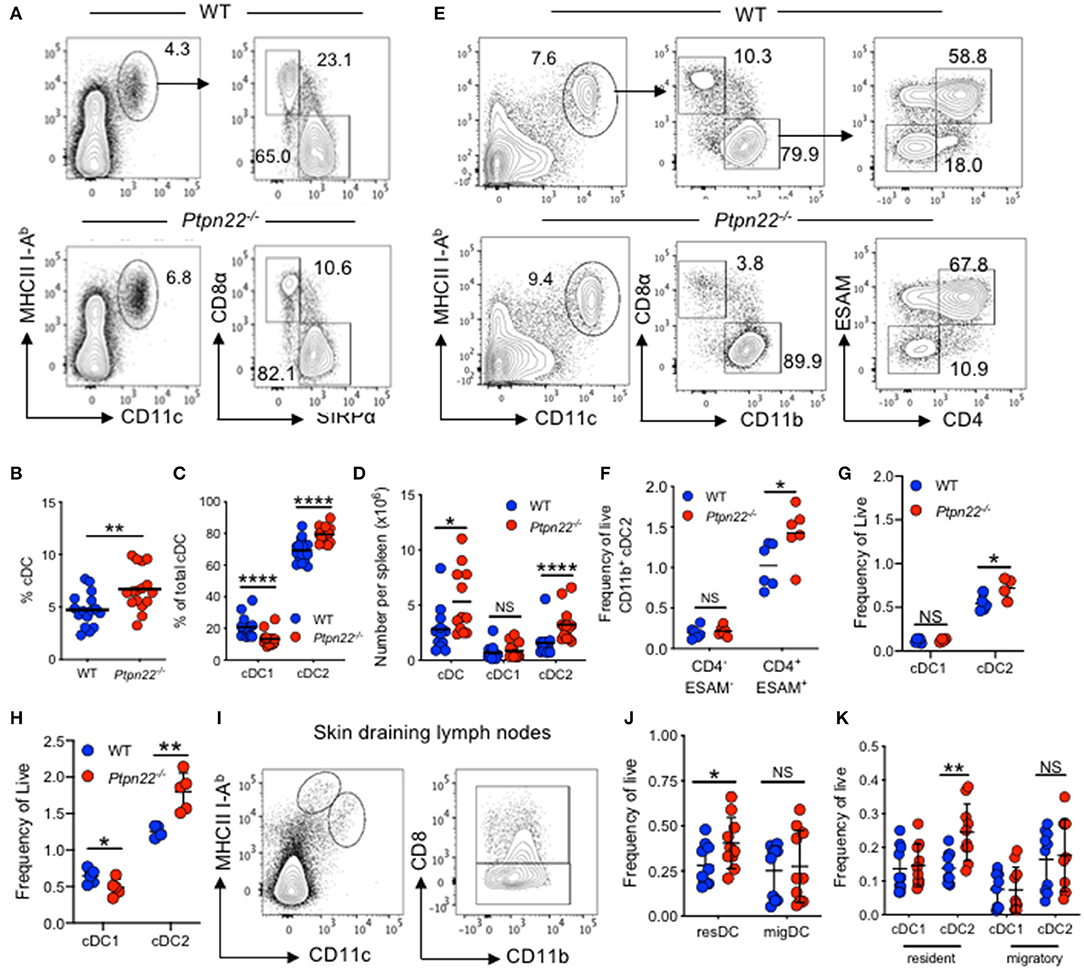
Figure 1. PTPN22 negatively regulates ESAMHI cDC2 homeostasis. (A–D) Spleens of 2–4-months age matched wild type (WT) and Ptpn22−/− mice were evaluated for cDC subsets by flow cytometry, gated on: live, singlet, lin− (CD3, CD19, B220, Ly6C/G, NK1.1, Ter119), CD11c+MHCcII I-Ab+ and then CD8+ vs. SIRPα+ (A) Representative flow cytometry plot analysis of cDC subsets. (B) The proportion of CD11c+I-Ab+ cDC, (C) the proportion of CD8+ cDC1 and SIRPα+ cDC2s, (D) the number of cDC, cDC1 and cDC2 per spleen. N = 12–15 mice per genotype from >3 independent experiments. (E,F) Spleens of 2–4 months age matched wild type (WT) and Ptpn22−/− mice were evaluated for cDC subsets by flow cytometry, gated on: live, singlet, lin− (CD3, CD19, B220, Ly6C/G, NK1.1, Ter119), CD11c+MHCcII I-Ab+, CD8−CD11b+, ESAM vs. CD4. Representative flow cytometry plot analysis of cDC subsets (E) and the frequency of CD11b +DC2 ESAM+/− CD4+/− subsets per spleen (F). N = 6 mice/genotype from two independent experiments. (G) Splenic cDC1 and cDC2 within pre-wean (3 weeks) and (H) post wean (4 weeks) WT and Ptpn22−/− mice. N = 4 mice/genotype. (I–K) Lymph node resident and migratory cDC subsets within 2–4-months age matched WT and Ptpn22−/− mice. Determined by flow cytometry gating on: singlet, live, lin− CD11c+ MHCcII I-AbInt (resident DC) or CD11c+ MHCcII I-AbHigh (migratory DC), and then CD8α (cDC1) vs. CD11b+ (cDC2). (I) Representative flow cytometry plots. (J) Frequency of resident and migratory cDC and (K) frequency of resident and migratory cDC1 and cDC2; N = 10 mice/genotype from 3 independent experiments. Each point represents an individual mouse; bars represent mean, NS, not significant; *p < 0.05, **p < 0.01, ****p < 0.0001, determined by unpaired T-test.
To examine if cDC homeostasis was altered in other lymphoid tissues, we examined cDC populations within skin draining lymph nodes (sdLN), and found that Ptpn22−/− mice displayed expanded resident, but not migratory cDC2. Once again similar numbers of cDC1 were observed within both migratory and resident cDC populations (Figures 1I–K). A similar phenotype was documented within the mesenteric lymph nodes, wherein resident CD11b+ CD103− cDC2 were expanded, while CD103+ resident cDCs and all migratory (IAbhigh) cDC subsets assessed were unaltered (Supplementary Figures 1J-L). Furthermore, in line with Ptpn22 not regulating migratory cDC2, cDC2 positioning within the spleen bridging channels appeared similar between WT and Ptpn22−/− mice (Supplementary Figure 1M). Together, we conclude that PTPN22 regulates resident DC2 homeostasis within secondary lymphoid organs (SLOs).
Given its broad expression in multiple hematopoietic lineages, PTPN22 has the potential to control cDC2 homeostasis through DC intrinsic or DC extrinsic mechanisms. To investigate this, we generated Ptpn22−/− CD45.2: WT CD45.1 mixed bone marrow chimeras and found that CD45.2 Ptpn22−/− bone marrow out-competed WT CD45.1 in the generation of cDC2 (Figures 2A–D), whereas no change was observed in the ratio of Lin+ cells (Supplementary Figure 2A). Consistent with our previous observations, generation of cDC1 was unaffected by genotype, further supporting a role for PTPN22 as a selective, DC intrinsic regulator of cDC2 homeostasis. PTPN22 regulates T cell homeostasis (43), raising the possibility of an indirect cDC2 phenotype driven by enhanced T cell activation in PTPN22:WT chimeras. Therefore, we examined if lineage specific deletion of Ptpn22 within the T cell compartment would have an impact on cDC2 populations. We detected no differences in cDC2 expansion in either mice with T cell restricted Ptpn22−/− (Supplementary Figure 2B) or between chimeras harboring PTPN22 sufficient or deficient T cells (Figure 2E), indicating that deficiency of Ptpn22 exclusively in T cells was not sufficient to perturb cDC homeostasis.
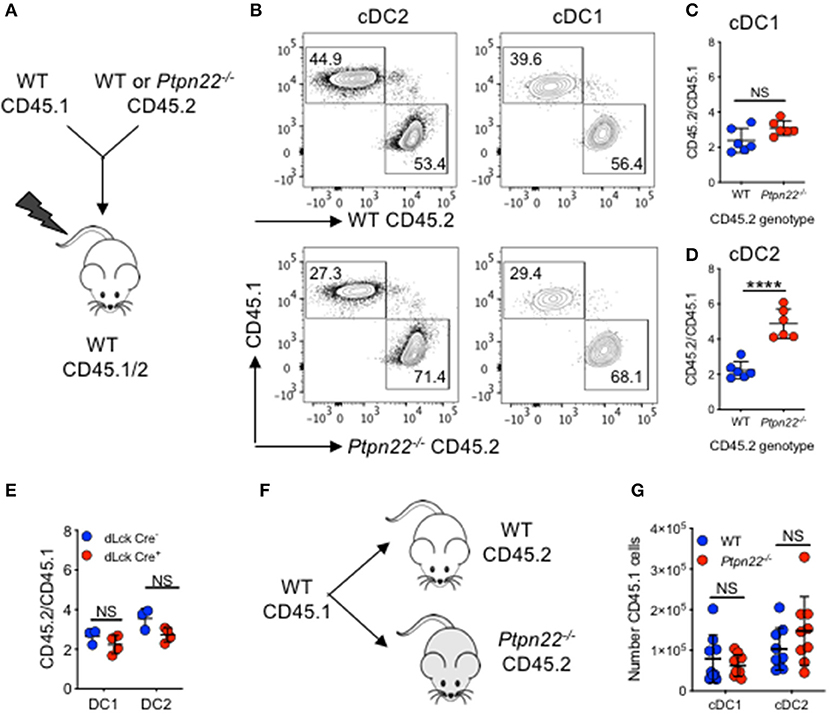
Figure 2. PTPN22 regulates cDC2 homeostasis in a DC intrinsic manner. (A–D) Lethally irradiated CD45.1/2 recipient mice received a 1:1 ratio of WT CD45.1: WT or Ptpn22−/− CD45.2 bone marrow (i.v). After 8 weeks spleens of recipient mice were evaluated for cDC subsets and the ratio of CD45.1:CD45.2 within each subset was determined by flow cytometry gating on: live, singlet, lin− CD11c+, MHCcII I-Ab+ and then CD8+ vs. SIRPα+ CD45.1+ vs. CD45.2+. (A) Experiment schematic, (B) representative flow cytometry staining gated on either cDC2 (left) or cDC1 (right) subsets. (C,D) The ratio of CD45.1:CD45.2 within cDC1 and cDC2 subsets calculated relative to the input ratio. N = 5–6 mice/genotype, one experiment of two. (E) Lethally irradiated wild type (WT) CD45.1/2 mice received a 1:1 ratio of WT CD45.1: dLckCre− or dLckCre+ (Ptpn22−/−) CD45.2 bone marrow (i.v). After 8 weeks spleens of recipient CD45.1/2 mice were evaluated for cDC subsets and the ratio of CD45.1:CD45.2 within each subset was determined by flow cytometry relative to the input ratio, N = 3–4 mice/genotype. (F) WT CD45.1 bone marrow was transferred i.v into WT or Ptpn22−/− CD45.2 recipient mice and after 6 days the spleens of recipient mice were evaluated for the number of CD45.1 cDC1 and cDC2 by flow cytometry. (F) Schematic of experiment (G) N = 9 mice/genotype, two independent experiments. Each point represents an individual mouse; bars represent mean and standard deviation, NS, not significant; ****p < 0.0001 determined by unpaired T-test.
Finally, we determined if the Ptpn22−/− environment (via an indirect effect on stroma), contributed to cDC2 expansion. WT CD45.1 bone marrow was transferred into non-irradiated WT and Ptpn22−/− CD45.2 mice. After 6 days we observed no difference in CD45.1 cDC2 numbers developing within either WT or Ptpn22−/− mice (Figures 2F,G). This result is consistent with previous reports establishing that PTPN22 expression is restricted to hematopoietic cell lineages (44, 45). Together, these data suggest that PTPN22 regulates cDC2 homeostasis via a cDC intrinsic mechanism.
Homeostasis of cDC in SLOs is controlled by multiple factors including differentiation of bone marrow precursors in response to Flt3L, duration of cDC survival, proliferation of incoming precursor cDCs, and the turnover of a small subset of cDCs within SLOs (1). We therefore aimed to examine how PTPN22 might regulate cDC2 development. Firstly, we observed no PTPN22 dependent difference in bone marrow or splenic common DC precursors (CDP) or preDC cells, indicating that PTPN22 operated post pre-cDC development (Supplementary Figures 3A–C). Secondly, as cDC1 and cDC2 are dependent on Flt3L for their differentiation we tested if PTPN22 controls Flt3L dependent cDC2. We observed similar Flt3R expression on ex vivo WT and Ptpn22−/− cDC1 and cDC2 (Supplementary Figures 3D–E), as well as similar concentrations of serum Flt3L in vivo (Supplementary Figure 3F). To compare Flt3L dependent cDC2 development, we cultured WT and Ptpn22−/− bone marrow in vitro with Flt3L. However, no significant changes in cDC2 development were observed (Figure 3A). We then assessed if PTPN22 altered cDC2 survival by comparing the expression of survival genes Bcl2, Bim, and Trim2 in FACS sorted cDC2. Once again we observed no differences between WT and Ptpn22−/− cDC2 (Supplementary Figure 3G). Furthermore, no differences were observed between splenic WT and Ptpn22−/− cDC2 acquiring an apoptotic phenotype (Annexin V+) as a consequence of a 24-h culture in vitro (Supplementary Figure 3H). Based on these data, we reasoned that differences in cell survival were unlikely to be a major mechanism mediating cDC2 expansion in Ptpn22−/− mice.
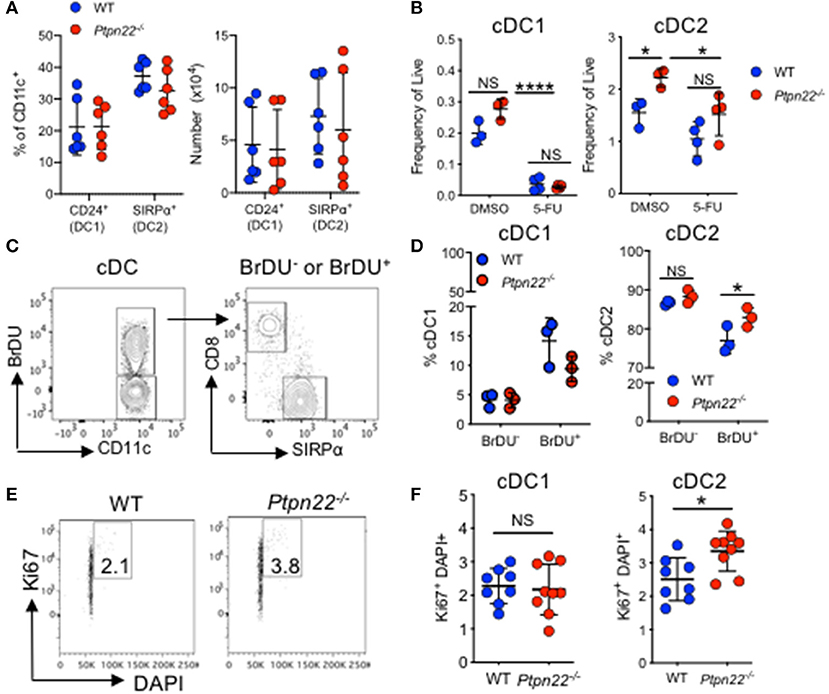
Figure 3. PTPN22 regulates DC2 proliferation. (A) Bone marrow from wild type (WT) or Ptpn22−/− mice cultured in the presence of Flt3L for 8 days (Flt3L-BMDC). At day 8 the proportion and number of CD24+ cDC1 and SIRPα+ cDC2 were determined by flow cytometry. N = 6 mice per genotype from 6 independent experiments. (B) The frequency of live splenic cDC1 and cDC2 from WT and Ptpn22−/− measured 3 days after i.v immunization with 5-flurouracil or DMSO control. N = 3–4 mice per group. (C,D) The percentage of splenic cDC1 and cDC2 within BrDU− and BrDU+ populations within BrDU treated WT and Ptpn22−/− mice. (C) Representative flow plots of analysis, quantified in (D). N = 3 mice per genotype. (E,F) Ki67 and DAPI expression within splenic cDC1 and cDC2 subsets from WT and Ptpn22−/− spleens. (E) Representative flow plot analysis and quantified in (F). N = 8 mice per genotype. (A,B,D,F) Each point represents an individual mouse; bars represent mean and standard deviation. NS = not significant, (A–F) *p < 0.05, determined by unpaired T-test. NS = not significant, *p < 0.05.
Having excluded a role for PTPN22 in regulating DC precursor development, Flt3L dependent differentiation or cDC2 survival, we next addressed if PTPN22 might control cDC2 proliferation. To test this hypothesis, we examined the in vivo effects of 5-flurouracil (5-FU), a pyrimidine analog that inhibits cell proliferation (46). Previous reports have demonstrated that 3 days post-5-FU administration DC populations are significantly reduced within the spleen, indicative of rapid turnover of DCs in vivo (47). The difference in cDC2 expansion between WT and Ptpn22−/− mice was abrogated 3 days after treatment with 5-FU, demonstrating that PTPN22 mediated expansion of cDC2 is indeed dependent on proliferation (Figure 3B). Consistent with our data using 5-FU, administration of thymidine analog BrDU, which incorporates into proliferating cells, demonstrated that the Ptpn22−/− cDC2 population was significantly expanded within the BrDU+ (proliferating), but not the BrDU− (non-proliferating) population (Figures 3C,D). Furthermore, Ki67 and DAPI staining confirmed enhanced proportions of cDC2 undergoing cell cycling (Figures 3E,F and Supplementary Figure 3I). Likewise, cell cycle analysis of competitive bone marrow chimeras 3 weeks post transfer also demonstrated a significant increase in the proportion of cycling Ptpn22−/− cDC2 when compared to WT controls (Supplementary Figure 3J). Taken together, these data support the notion that PTPN22 controls cDC2 homeostasis by restricting cDC2 proliferation.
The human PTPN22R620W polymorphism is one of the highest-ranking genetic risk factors for the development of multiple autoimmune diseases outside MHC loci (38, 48). Given that PTPN22 regulates the expansion of cDC2s, we set out to examine whether the autoimmune associated variant was capable of mediating similar effects. To address this, we enumerated splenic cDC subsets in mice expressing the R620W ortholog, Ptpn22619W. In comparison to Ptpn22619R, mice carrying Ptpn22619W also displayed expansion of splenic cDC2s, which, like Ptpn22−/− mice, occurred specifically within the ESAM+CD4+ DC2 subset (Figures 4A,B). Furthermore, the magnitude of cDC2 expansion was similar to Ptpn22−/− mice when compared to WT (1.5-fold vs. 2-fold respectively). This demonstrated that the autoimmune associated PTPN22 variant is also capable of regulating cDC2 homeostasis, operating as a loss-of-function mutant in this context.
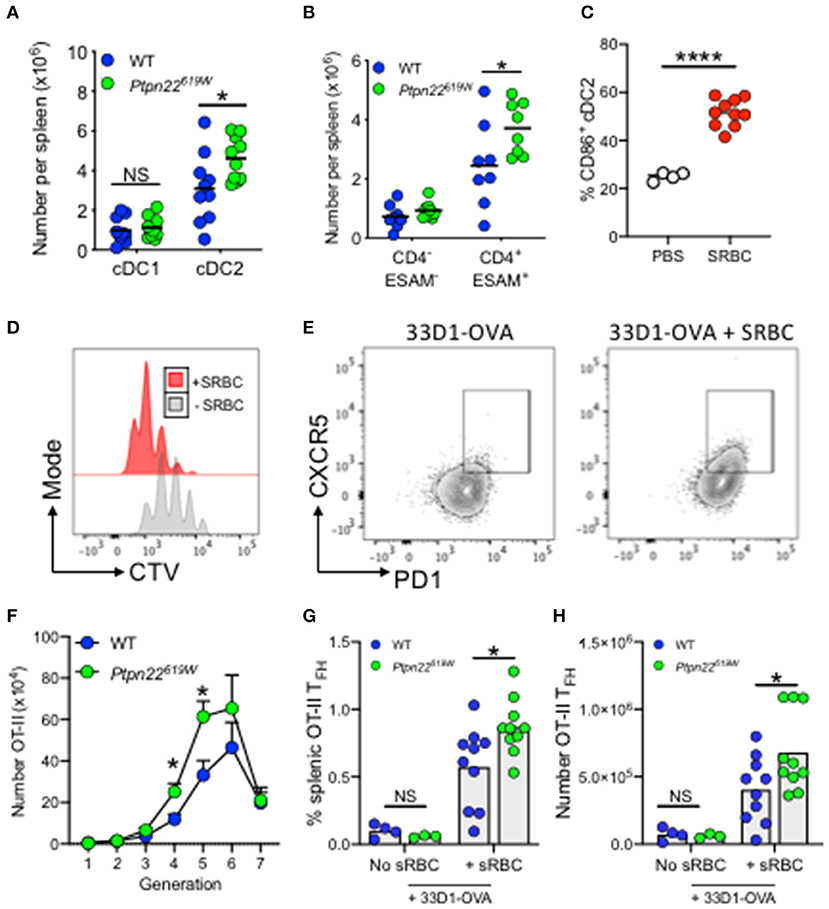
Figure 4. Ptpn22619W conferred cDC2 expansion enhances T cell proliferation and TFH. (A,B) Spleens of 2–4 months age matched WT and Ptpn22619W mice were evaluated for cDC subsets by flow cytometry (A) number of cDC1 and cDC2 per spleen (B) number of ESAM− vs. ESAM+ cDC2 per spleen. (A,B) N = 10 mice per genotype. (C) Mice were immunized i.v. with PBS or SRBC and after 4 h splenic cDC2 were assessed for cell surface CD86 expression by flow cytometry. N = 4 or 10 mice/group. (D–H) CD45.1+CD4+ OT-II T cells were transferred i.v into recipient mice. The following day mice received i.v 33D1-ovalbumin in the presence or absence of sheep RBC (SRBC). After 3 days CD45.1+ CD4+ Vα2Vβ5+ T cells were evaluated for CTV dilution and CXCR5+PD-1+ T follicular helper cell (TFH) by flow cytometry. Representative plots of CTV dilution (D) and TFH induction (E) in the presence or absence of SRBC. (F–H) OT-II proliferation and TFH induction within WT and Ptpn22619W recipient mice determined by flow cytometry. (F) The number of proliferating CD45.1+ CD4+ Vα2Vβ5+ OT-II T cells. (G) The frequency of CD45.1+ Vα2Vβ5+CD4+PD-1+CXCR5+ TFH per spleen. (H) The number of CD45.1+ Vα2Vβ5+CD4+PD-1+CXCR5+ TFH per spleen. (F–H) N = 10 mice/genotype. Each point represents an individual mouse, bars represent mean; error bars represent s.e.m. (A–H) *p < 0.05, ****p < 0.0001 determined by unpaired T-test.
PTPN22R620W is a risk allele associated with multiple autoantibody associated autoimmune diseases. Splenic cDC2 are essential initiators of TFH differentiation, leading to GC formation, and high-affinity antibody production (18, 49). Interestingly, with age Ptpn22−/− and Ptpn22619W mice develop spontaneous GC, and enhanced serum IgG levels (33, 43, 50). Accordingly, we addressed whether expansion of splenic cDC2 in Ptpn22619W mice was sufficient to alter cDC2 dependent T cell activation and TFH induction in vivo. To evaluate cDC2 dependent antigen specific responses in vivo we administered 33D1-OVA conjugates to selectively target the cDC2 subset (Supplementary Figure 4) (8). WT and Ptpn22619W mice received CD45.1 OT-II CD4+ T cells and were immunized with 33D1-OVA in the presence of SRBCs to promote cDC2 activation (Figure 4C), and potentiate OT-II proliferation and TFH responses (Figures 4D,E) (18, 49). When compared to WT recipients, Ptpn22619W cDC2 expansion was sufficient to enhance OT-II proliferation in vivo (Figure 4F). Furthermore, we observed that the proportion and number of splenic OT-II TFH that develop within Ptpn22619W recipients was significantly enhanced compared to WT following 33D1-OVA/SRBC immunization (Figures 4G,H). Our previous investigations demonstrate that Ptpn22 is dispensable for antigen uptake and presentation (51). Furthermore, the in vitro data presented here indicate that differences in OT-II proliferation are due to altered cDC2 homeostasis (specifically the number of cDC2 cells), rather than a cell intrinsic difference in Ptpn22 variant cDC2. When total, unmanipulated splenic cDC are FACS isolated (preserving the cDC2 expansion observed in vivo) and co-cultured with OT-II in the presence of 33D1-OVA, Ptpn22−/− are able to potentiate OT-II T cell proliferation (Supplementary Figure 4B). Conversely, when cDC2 are FACS isolated (normalizing cDC2 numbers) and cultured in the same manner, WT and Ptpn22−/− cDC2 are capable of inducing OT-II proliferation to the same extent. Furthermore, no difference in OT-II proliferation is observed when co-cultured with FACS isolated cDC1 in the presence of DEC205-OVA (Supplementary Figure 4D). Together, these data reveal that Ptpn22619W mediated cDC2 expansion is sufficient to deregulate T cell activation in response to non-self-antigens, and is likely to contribute to the promotion of TFH responses in vivo alongside previously reported T cell and B cell intrinsic effects (33, 50).
Here, we provide evidence that the autoimmune disease-associated phosphatase PTPN22 is a regulator of cDC2 homeostasis. Loss-of-function mutants of PTPN22 result in cDC2 expansion through mechanisms that are DC intrinsic, enhancing cDC2 proliferation. Thus, PTPN22 appears to be a selective regulator of cDC2 homeostasis. Furthermore, cDC2 expansion conferred by the autoimmune associated Ptpn22619W variant resulted in aberrant cDC2 dependent TFH induction in vivo. Our data therefore uncover a novel mechanism by which TFH expansion, first reported in Ptpn22 deficient mice, may be underpinned by a specific cDC2 phenotype.
The precise mechanisms by which Ptpn22 mediates selective expansion of cDC2 remains to be determined. However, we now know that Ptpn22 deficiency mediates the expansion of ESAM+CD4+ cDC2 (Figure 1E), a cDC2 subset that is known to be dependent on LTβR for their proliferation (15). Furthermore, splenic Ptpn22−/− cDC2 were more proliferative ex vivo (Figures 3B–F). Consistent with our data, Ltβr−/− splenic cDC2s are less proliferative, resulting in decreased cDC2 numbers (31). Indeed, our preliminary experiments indicate that Ptpn22 may control LTβR mediated cDC2 proliferation. LTβR agonist treatment of Ptpn22−/− Flt3L-BMDC increased cDC2 numbers compared to WT (Supplementary Figures 5A,B), whilst no difference in cell surface LTβR expression was observed on ex vivo or in vitro generated Flt3L BMDC (Supplementary Figures 5C–F). In addition, phenotypes described in Relb−/− mice, further support the hypothesis that PTPN22 may regulate LTβR dependent cDC2 proliferation. LTβR activates canonical pathway and non-canonical NFκB signaling, resulting in RelB translocation (52). Within Relb−/− mice, there is a severe reduction in splenic and LN resident (but not migratory) cDC2, from as early as 3 weeks of age, whereas cDC1 numbers are unaffected (53), and Flt3L dependent DC development is unaffected by Relb−/−. In addition, despite lamina propria DC being dependent on Notch2 and LTβR signaling, Relb−/− does not affect CD11b+ DC subsets at this location (15, 53). In keeping with these reports, we observed no difference in lamina propria CD11b+ DC subsets between WT and Ptpn22−/− mice (Supplementary Figure 6). Therefore, in both Relb−/− and Ptpn22−/− mice the same cDC2 populations are disrupted, being decreased in Relb−/− and increased in Ptpn22−/−, with both occurring within a 3-weeks time frame. In contrast, due to differences in the specific cDC2 phenotypes reported, our data do not support a role for PTPN22 in regulating IRF4 (54), KLF4 (10), or NOTCH2 (12, 15) dependent cDC2 development. As such, our data are consistent with a model whereby PTPN22 may function to negatively regulate LTβR signaling, limiting RelB translocation to control cDC2 homeostasis.
One question that our data raise, is why and how Ptpn22 selectively regulates cDC2 homeostasis? One explanation might be the differential expression of Ptpn22 in DC subsets, which is substantially higher in cDC2 than cDC1 or pDC (ImmGen); implying that the effects of Ptpn22 deficiency are likely to be greatest in cells with the highest expression. An alternate explanation is that Ptpn22 regulates signaling pathways specifically required for cDC2 but not cDC1 development. In line with this, our data demonstrate no defect in pathways required by both cDC1 and cDC2, since both precursor cDC development (Supplementary Figures 3A–C) and Flt3L responsiveness (Supplementary Figure 3A) remain intact in Ptpn22−/− mice. Conversely, proliferation was enhanced specifically in Ptpn22−/- cDC2 population (Figure 3), indicating that Ptpn22 may operate to restrain cDC2 proliferation in response to signals required for cDC2 turnover. One factor known to mediate the proliferation of a small subset of cDC2 is LTβR, and Ptpn22 may be involved in regulating this proliferative signal in vivo. Alternatively, Ptpn22 may regulate the signaling of an as yet to be identified pathway that is also required for cDC2 proliferation. Further detailed investigation is required to uncover the mechanistic basis for the pathway(s) regulated by Ptpn22 in this context.
PTPN22R620W is one of the strongest autoimmune disease associated genetic risk factors. We demonstrate that cDC2 homeostasis is disrupted in mice that express the ortholog of the human autoimmune associated variant (Figures 4A,B). To our knowledge, this is the first description of cDC homeostasis being regulated by an autoimmune associated genetic risk allele. Our data therefore provide a link between genetic and environmental risk and the breakdown of immune tolerance that leads to autoimmunity. Changes to cDC homeostasis have been described within autoimmune diseases for which PTPN22R620W is a risk factor. In type 1 diabetes the effector phase of murine type I diabetes is characterized by cDC2 expansion (55). Furthermore, in humans with rheumatoid arthritis (RA), cDC2 are decreased within the blood, but expanded within the lymph nodes (56), and the RA synovium is enriched with RelB+ DCs (57). Together, suggesting an association between the enhanced presence of cDC2s within SLOs and tissue and the risk of developing autoimmune disease. Therefore, a failure to maintain cDC homeostasis, as conferred by the PTPN22 risk allele, may be a factor altering the downstream immune responses that ultimately lead to autoimmunity.
Although we accept that the difference between WT and Ptpn22 variant cDC2 populations may appear modest, over the lifetime of a human, these changes could have significant functional impact over the decades that precede autoimmune disease onset. In the context of non-self cDC2-targeted antigen, Ptpn22619W dependent expansion of cDC2 was sufficient to enhance T cell proliferation and TFH expansion following GC promoting SRBC stimulation (Figures 4F,G). Activation of ESAM+ cDC2 is one of the earliest events in splenic GC formation leading to high-affinity antibody production (18, 49). With aging, PTPN22 mutant mice develop many hallmarks of autoimmunity including increased effector T cells, activated B cells and higher immunoglobulin and autoantibody titres (33, 43). Furthermore, humans carrying the PTPN22620W variant have an increased risk of developing autoimmune diseases that are almost exclusively associated with autoantibody production (38, 48). PTPN22 has been reported to regulate TFH (50) cells, GC formation, and antibody production (33) in part via T cell and B cell intrinsic effects. Although our data do not address self-reactivity, they do indicate that changes conferred by Ptpn22R619W altering cDC2 homeostasis, could, alongside T and B intrinsic effects, also contribute to perturb the GC reaction over time. Age is an important risk factor for autoimmune disease onset due to declining immune competence and impaired immune tolerance check points (58). In keeping with this concept, cDC2 expansion was potentiated in aged Ptpn22−/− mice (Supplementary Figure 1E) and as such changes to the balance of activating: inhibitory cells in vivo could alter cDC2 dependent responses that trigger the breaking of immune tolerance. Future work will be required to confirm the link between early expansion of cDC2 and autoimmunity.
In summary, our findings uncover PTPN22 as a selective regulatory checkpoint required to maintain cDC2 homeostasis, and suggest that early perturbation of DC homeostasis may be a trigger for the onset of autoimmunity.
All datasets generated for this study are included in the article/Supplementary Material.
The animal study was reviewed and approved by Animal Welfare and Ethical Review Body (AWERB) King's College London.
HP designed research, performed experiments, analyzed data, and wrote the manuscript. FC, CC, AM, and GC performed experiments and analyzed data. JB analyzed data and contributed to writing the manuscript. XD, DR, and RZ developed and contributed vital mouse models. DD developed and provided vital 33D1 and DEC-205-OVA reagents. PG and AC conceived and funded the project, contributed to data analysis, and wrote the manuscript. All authors reviewed the manuscript.
This research was supported by Versus Arthritis grants 20218 (awarded to HP and AC), 20525 (awarded to PG, RZ, and AC), BBSRC grant BB/M029735/1 (awarded to PG), Cancer Research UK grant C57672/A22369 (awarded to PG), Worldwide Cancer Research grant 18-0422 (awarded to PG), Wellcome Trust Investigator Award 096669AIA (awarded to RZ), German Research Foundation CRC11181-A7, DU548/5-1 (awarded to DD) and NIH: DP3-DK097672 and DP3-DK111802 (to DR). Additional support was provided by the Children's Guild Association Endowed Chair in Pediatric Immunology and the Benaroya Family Gift Fund (to DR). This work was also supported by infrastructure funded by the National Institute for Health Research (NIHR) BioResource Clinical Research facility and Biomedical Research Center based at Guy's and St. Thomas' NHS Foundation Trust and King's College London (reference: guysbrc-2012-17). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Cristina Sanchez Blanco, Tamlyn Peel, Wing Wu, and Esperanza Perucha for their helpful discussions and technical help.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00376/full#supplementary-material
1. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. (2013) 31:563–604. doi: 10.1146/annurev-immunol-020711-074950
2. Tamura T, Tailor P, Yamaoka K, Kong HJ, Tsujimura H, O'Shea JJ, et al. IFN regulatory factor-4 and−8 govern dendritic cell subset development and their functional diversity. J Immunol. (2005) 174:2573–81. doi: 10.4049/jimmunol.174.5.2573
3. Collin M, Bigley V. Human dendritic cell subsets: an update. Immunology. (2018) 154:3–20. doi: 10.1111/imm.12888
4. Dress RJ, Wong AY, Ginhoux F. Homeostatic control of dendritic cell numbers and differentiation. Immunol Cell Biol. (2018) 96:463–76. doi: 10.1111/imcb.12028
5. Maldonado-López R, De Smedt T, Michel P, Godfroid J, Pajak B, Heirman C, et al. CD8α+ and CD8α- subclasses of dendritic cells direct the development of distinct T helper cells in vivo. J Exp Med. (1999) 189:587–92. doi: 10.1084/jem.189.3.587
6. den Haan JM, Lehar SM, Bevan MJ. CD8(+) but not CD8(-) dendritic cells cross-prime cytotoxic T cells in vivo. J Exp Med. (2000) 192:1685–96. doi: 10.1084/jem.192.12.1685
7. Mashayekhi M, Sandau MM, Dunay IR, Frickel EM, Khan A, Goldszmid RS, et al. CD8α(+) dendritic cells are the critical source of interleukin-12 that controls acute infection by Toxoplasma gondii tachyzoites. Immunity. (2011) 35:249–59. doi: 10.1016/j.immuni.2011.08.008
8. Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, et al. Differential antigen processing by dendritic cell subsets in vivo. Science. (2007) 315:107–11. doi: 10.1126/science.1136080
9. Williams JW, Tjota MY, Clay BS, Vander Lugt B, Bandukwala HS, Hrusch CL, et al. Transcription factor IRF4 drives dendritic cells to promote Th2 differentiation. Nat Commun. (2013) 4:2990. doi: 10.1038/ncomms3990
10. Tussiwand R, Everts B, Grajales-Reyes GE, Kretzer NM, Iwata A, Bagaitkar J, et al. Klf4 expression in conventional dendritic cells is required for T helper 2 cell responses. Immunity. (2015) 42:916–28. doi: 10.1016/j.immuni.2015.04.017
11. Denning TL, Norris BA, Medina-Contreras O, Manicassamy S, Geem D, Madan R, et al. Functional specializations of intestinal dendritic cell and macrophage subsets that control Th17 and regulatory T cell responses are dependent on the T cell/APC ratio, source of mouse strain, and regional localization. J Immunol. (2011) 187:733–47. doi: 10.4049/jimmunol.1002701
12. Lewis KL, Caton ML, Bogunovic M, Greter M, Grajkowska LT, Ng D, et al. Notch2 receptor signaling controls functional differentiation of dendritic cells in the spleen and intestine. Immunity. (2011) 35:780–91. doi: 10.1016/j.immuni.2011.08.013
13. Kinnebrew MA, Buffie CG, Diehl GE, Zenewicz LA, Leiner I, Hohl TM, et al. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity. (2012) 36:276–87. doi: 10.1016/j.immuni.2011.12.011
14. Persson EK, Uronen-Hansson H, Semmrich M, Rivollier A, Hägerbrand K, Marsal J, et al. IRF4 transcription-factor-dependent CD103(+)CD11b(+) dendritic cells drive mucosal T helper 17 cell differentiation. Immunity. (2013) 38:958–69. doi: 10.1016/j.immuni.2013.03.009
15. Satpathy AT, Briseño CG, Lee JS, Ng D, Manieri NA, Kc W, et al. Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nat Immunol. (2013) 14:937–48. doi: 10.1038/ni.2679
16. Schlitzer A, McGovern N, Teo P, Zelante T, Atarashi K, Low D, et al. IRF4 Transcription factor-dependent CD11b+dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity. (2013) 38:970–83. doi: 10.1016/j.immuni.2013.04.011
17. Schreiber HA, Loschko J, Karssemeijer RA, Escolano A, Meredith MM, Mucida D, et al. Intestinal monocytes and macrophages are required for T cell polarization in response to Citrobacter rodentium. J Exp Med. (2013) 210:2025–39. doi: 10.1084/jem.20130903
18. Yi T, Li J, Chen H, Wu J, An J, Xu Y, et al. Splenic dendritic cells survey red blood cells for missing self-CD47 to trigger adaptive immune responses. Immunity. (2015) 43:764–75. doi: 10.1016/j.immuni.2015.08.021
19. Ashany D, Savir A, Bhardwaj N, Elkon KB. Dendritic cells are resistant to apoptosis through the Fas (CD95/APO-1) pathway. J Immunol. (1999) 163:5303–11.
20. Birnberg T, Bar-On L, Sapoznikov A, Caton ML, Cervantes-Barragán L, Makia D, et al. Lack of conventional dendritic cells is compatible with normal development and T cell homeostasis, but causes myeloid proliferative syndrome. Immunity. (2008) 29:986–97. doi: 10.1016/j.immuni.2008.10.012
21. Ohnmacht C, Pullner A, King SBS, Drexler I, Meier S, Brocker T, et al. Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity. J Exp Med. (2009) 206:549–59. doi: 10.1084/jem.20082394
22. Cai Y, Yang C, Yu X, Qian J, Dai M, Wang Y, et al. Deficiency of β-arrestin 2 in dendritic cells contributes to autoimmune diseases. J Immunol. (2019) 202:407–20. doi: 10.4049/jimmunol.1800261
23. Naik SH, Sathe P, Park H-Y, Metcalf D, Proietto AI, Dakic A, et al. Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat Immunol. (2007) 8:1217–26. doi: 10.1038/ni1522
24. Onai N, Obata-Onai A, Schmid MA, Ohteki T, Jarrossay D, Manz MG. Identification of clonogenic common Flt3+M-CSFR+ plasmacytoid and conventional dendritic cell progenitors in mouse bone marrow. Nat Immunol. (2007) 8:1207–16. doi: 10.1038/ni1518
25. Liu K, Victora GD, Schwickert TA, Guermonprez P, Meredith MM, Yao K, et al. In vivo analysis of dendritic cell development and homeostasis Kang. Science. (2009) 324:392–7. doi: 10.1126/science.1170540
26. Sathe P, Metcalf D, Vremec D, Naik SH, Langdon WY, Huntington ND, et al. Lymphoid tissue and plasmacytoid dendritic cells and macrophages do not share a common macrophage-dendritic cell-restricted progenitor. Immunity. (2014) 41:104–15. doi: 10.1016/j.immuni.2014.05.020
27. Swiecki M, Wang Y, Riboldi E, Kim AHJ, Dzutsev A, Gilfillan S, et al. Cell depletion in mice that express diphtheria toxin receptor under the control of SiglecH encompasses more than plasmacytoid dendritic cells. J Immunol. (2014) 192:4409–16. doi: 10.4049/jimmunol.1303135
28. Schlitzer A, Sivakamasundari V, Chen J, Sumatoh HRB, Schreuder J, Lum J, et al. Identification of cDC1- and cDC2-committed DC progenitors reveals early lineage priming at the common DC progenitor stage in the bone marrow. Nat. Immunol. (2015) 16:718–28. doi: 10.1038/ni.3200
29. Maraskovsky E, Daro E, Roux E, Teepe M, Maliszewski CR, Hoek J, et al. In vivo generation of human dendritic cell subsets by Flt3 ligand. Blood. (2000) 96:878–84. doi: 10.1182/blood.V96.3.878.015k15_878_884
30. Luther SA, Lopez T, Bai W, Hanahan D, Cyster JG. BLC expression in pancreatic islets causes B cell recruitment and lymphotoxin-dependent lymphoid neogenesis. Immunity. (2000) 12:471–81. doi: 10.1016/S1074-7613(00)80199-5
31. Kabashima K, Banks TA, Ansel KM, Lu TT, Ware CF, Cyster JG. Intrinsic lymphotoxin-beta receptor requirement for homeostasis of lymphoid tissue dendritic cells. Immunity. (2005) 22:439–50. doi: 10.1016/j.immuni.2005.02.007
32. Rieck M, Arechiga A, Onengut-Gumuscu S, Greenbaum C, Concannon P, Buckner JH. Genetic variation in PTPN22 corresponds to altered function of T and B lymphocytes. J Immunol. (2007) 179:4704–10. doi: 10.4049/jimmunol.179.7.4704
33. Dai X, James RG, Habib T, Singh S, Jackson S, Khim S, et al. A disease-associated PTPN22 variant promotes systemic autoimmunity in murine models. J Clin Invest. (2013) 123:2024–36. doi: 10.1172/JCI66963
34. Salmond RJ, Brownlie RJ, Morrison VL, Zamoyska R. The tyrosine phosphatase PTPN22 discriminates weak self peptides from strong agonist TCR signals. Nat Immunol. (2014) 15:875–83. doi: 10.1038/ni.2958
35. Burn GL, Cornish GH, Potrzebowska K, Samuelsson M, Griffié J, Minoughan S, et al. Superresolution imaging of the cytoplasmic phosphatase PTPN22 links integrin-mediated T cell adhesion with autoimmunity. Sci Signal. (2016) 9:ra99. doi: 10.1126/scisignal.aaf2195
36. Sanchez-Blanco C, Clarke F, Cornish GH, Depoil D, Thompson SJ, Dai X, et al. Protein tyrosine phosphatase PTPN22 regulates LFA-1 dependent Th1 responses. J Autoimmun. (2018) 94:45–55. doi: 10.1016/j.jaut.2018.07.008
37. Wang Y, Shaked I, Stanford SM, Zhou W, Curtsinger JM, Mikulski Z, et al. The autoimmunity-associated gene PTPN22 potentiates toll-like receptor-driven, type 1 interferon-dependent immunity. Immunity. (2013) 39:111–22. doi: 10.1016/j.immuni.2013.06.013
38. Burn GL, Svensson L, Sanchez-Blanco C, Saini M, Cope AP. Why is PTPN22 a good candidate susceptibility gene for autoimmune disease? FEBS Lett. (2011) 585:3689–98. doi: 10.1016/j.febslet.2011.04.032
39. Fiorillo E, Orrú V, Stanford SM, Liu Y, Salek M, Rapini N, et al. Autoimmune-associated PTPN22 R620W variation reduces phosphorylation of lymphoid phosphatase on an inhibitory tyrosine residue. J Biol Chem. (2010) 285:26506–18. doi: 10.1074/jbc.M110.111104
40. Rawlings DJ, Dai X, Buckner JH. The role of PTPN22 risk variant in the development of autoimmunity: finding common ground between mouse and human. J Immunol. (2015) 194:2977–84. doi: 10.4049/jimmunol.1403034
41. Brownlie RJ, Miosge LA, Vassilakos D, Svensson LM, Cope A, Zamoyska R. Lack of the phosphatase PTPN22 increases adhesion of murine regulatory T cells to improve their immunosuppressive function. Sci Signal. (2012) 5:ra87. doi: 10.1126/scisignal.2003365
42. Neubert K, Lehmann CHK, Heger L, Baranska A, Staedtler AM, Buchholz VR, et al. Antigen delivery to CD11c+CD8- dendritic cells induces protective immune responses against experimental melanoma in mice in vivo. J Immunol. (2014) 192:5830–8. doi: 10.4049/jimmunol.1300975
43. Hasegawa K, Martin F, Huang G, Tumas D, Diehl L, Chan AC. PEST domain-enriched tyrosine phosphatase (PEP) regulation of effector/memory T cells. Science. (2004) 303:685–9. doi: 10.1126/science.1092138
44. Cohen S, Dadi H, Shaoul E, Sharfe N, Roifman CM. Cloning and characterization of a lymphoid-specific, inducible human protein tyrosine phosphatase, Lyp. Blood. (1999) 93:2013–24. doi: 10.1182/blood.V93.6.2013.406k25_2013_2024
45. He R-J, Yu Z-H, Zhang R-Y, Zhang Z-Y. Protein tyrosine phosphatases as potential therapeutic targets. Acta Pharmacol Sin. (2014) 35:1227–46. doi: 10.1038/aps.2014.80
46. Longley DB, Harkin DP, Johnston PG. 5-Fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. (2003) 3:330–8. doi: 10.1038/nrc1074
47. Zhan Y, Chow KV, Soo P, Xu Z, Brady JL, Lawlor KE, et al. Plasmacytoid dendritic cells are short-lived: reappraising the influence of migration, genetic factors and activation on estimation of lifespan. Sci Rep. (2016) 6:25060. doi: 10.1038/srep25060
48. Stanford SM, Bottini N. PTPN22: the archetypal non-HLA autoimmunity gene. Nat Rev Rheumatol. (2014) 10:602–11. doi: 10.1038/nrrheum.2014.109
49. Briseño CG, Satpathy AT, Davidson JT, Ferris ST, Durai V, Bagadia P, et al. Notch2-dependent DC2s mediate splenic germinal center responses. Proc Natl Acad Sci USA. (2018) 115:10726–31. doi: 10.1073/pnas.1809925115
50. Maine CJ, Marquardt K, Cheung J, Sherman LA. PTPN22 controls the germinal center by influencing the numbers and activity of T follicular helper cells. J Immunol. (2014) 192:1415–24. doi: 10.4049/jimmunol.1302418
51. Clarke F, Jordan CK, Gutiérrez-Martinez E, Bibby JA, Sanchez-Blanco C, Cornish GH, et al. Protein tyrosine phosphatase PTPN22 is dispensable for dendritic cell antigen processing and promotion of T-cell activation by dendritic cells. PLoS ONE. (2017) 12:e0186625. doi: 10.1371/journal.pone.0186625
52. Bista P, Zeng W, Ryan S, Bailly V, Browning JL, Lukashev ME. TRAF3 controls activation of the canonical and alternative NFkappaB by the lymphotoxin beta receptor. J Biol Chem. (2010) 285:12971–8. doi: 10.1074/jbc.M109.076091
53. Briseño CG, Gargaro M, Durai V, Davidson JT, Theisen DJ, Anderson DA, et al. Deficiency of transcription factor RelB perturbs myeloid and DC development by hematopoietic-extrinsic mechanisms. Proc Natl Acad Sci USA. (2017) 114:3957–62. doi: 10.1073/pnas.1619863114
54. Suzuki S, Honma K, Matsuyama T, Suzuki K, Toriyama K, Akitoyo I, et al. Critical roles of interferon regulatory factor 4 in CD11bhighCD8alpha- dendritic cell development. Proc Natl Acad Sci USA. (2004) 101:8981–6. doi: 10.1073/pnas.0402139101
55. Price JD, Tarbell KV. The role of dendritic cell subsets and innate immunity in the pathogenesis of Type 1 diabetes and other autoimmune diseases. Front Immunol. (2015) 6:288. doi: 10.3389/fimmu.2015.00288
56. Ramwadhdoebe TH, van Baarsen LGM, Berger FH, Maas M, Gerlag DM, Tak PP, et al. A8.34 CD1C + dendritic cells are overrepresented in lymph nodes of early arthritis patients and related to B cell responses. Ann Rheum Dis. (2014) 73:A90.1–A90. doi: 10.1136/annrheumdis-2013-205124.208
57. Pettit AR, MacDonald KPA, O'Sullivan B, Thomas R. Differentiated dendritic cells expressing nuclear RelB are predominantly located in rheumatoid synovial tissue perivascular mononuclear cell aggregates. Arthritis Rheum. (2000) 43:791. doi: 10.1002/1529-0131(200004)43:4<791::AID-ANR9>3.0.CO;2-E
Keywords: dendritic cell, PTPN22, homeostasis, T follicular helper cell, proliferation, autoimmunity, polymorphism, cDC2
Citation: Purvis HA, Clarke F, Montgomery AB, Colas C, Bibby JA, Cornish GH, Dai X, Dudziak D, Rawlings DJ, Zamoyska R, Guermonprez P and Cope AP (2020) Phosphatase PTPN22 Regulates Dendritic Cell Homeostasis and cDC2 Dependent T Cell Responses. Front. Immunol. 11:376. doi: 10.3389/fimmu.2020.00376
Received: 27 September 2019; Accepted: 17 February 2020;
Published: 04 March 2020.
Edited by:
Elodie Segura, Institut Curie, FranceReviewed by:
Natalio Garbi, University of Bonn, GermanyCopyright © 2020 Purvis, Clarke, Montgomery, Colas, Bibby, Cornish, Dai, Dudziak, Rawlings, Zamoyska, Guermonprez and Cope. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Harriet A. Purvis, aGFycmlldC5wdXJ2aXNAa2NsLmFjLnVr; aGFycmlldC5wdXJ2aXNAY3JpY2suYWMudWs=; Andrew P. Cope, YW5kcmV3LmNvcGVAa2NsLmFjLnVr
†Present address: Harriet A. Purvis, Immunity and Cancer, The Francis Crick Institute, London, United Kingdom
Georgina H. Cornish, Retroviral Immunology, The Francis Crick Institute, London, United Kingdom
‡These authors have contributed equally to this work and share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.