 Stefan H. E. Kaufmann
Stefan H. E. Kaufmann- 1Max Planck Institute for Infection Biology, Berlin, Germany
- 2Hagler Institute for Advanced Study, Texas A&M University, College Station, TX, United States
Tuberculosis (TB) remains a major health threat. Although a vaccine has been available for almost 100 years termed Bacille Calmette-Guérin (BCG), it is insufficient and better vaccines are urgently needed. This treatise describes first the basic immunology and pathology of TB with an emphasis on the role of T lymphocytes. Better understanding of the immune response to Mycobacterium tuberculosis (Mtb) serves as blueprint for rational design of TB vaccines. Then, disease epidemiology and the benefits and failures of BCG vaccination will be presented. Next, types of novel vaccine candidates are being discussed. These include: (i) antigen/adjuvant subunit vaccines; (ii) viral vectored vaccines; and (III) whole cell mycobacterial vaccines which come as live recombinant vaccines or as dead whole cell or multi-component vaccines. Subsequently, the major endpoints of clinical trials as well as administration schemes are being described. Major endpoints for clinical trials are prevention of infection (PoI), prevention of disease (PoD), and prevention of recurrence (PoR). Vaccines can be administered either pre-exposure or post-exposure with Mtb. A central part of this treatise is the description of the viable BCG-based vaccine, VPM1002, currently undergoing phase III clinical trial assessment. Finally, new approaches which could facilitate design of refined next generation TB vaccines will be discussed.
“ Commit to advancing research for basic science, public health research and the development of innovative products and approaches, …, without which ending the tuberculosis epidemic will be impossible, including towards delivering, as soon as possible, new, safe, effective, equitable, affordable, available vaccines, …” Resolution adopted by General Assembly of the United Nations from the High Level Meeting on the fight against TB, 2018 (1).
Introduction
The only tuberculosis (TB) vaccine in use until today, Bacille Calmette Guérin (BCG), was introduced in 1921 after intensive research & development (R&D) for more than a decade (2). It was not the first tryout to immunize against TB. The very first attempt was made by Robert Koch who used a subunit-adjuvant formulation (3). Subsequently, several other approaches were tested including killed mycobacterial vaccines and live non-tuberculous mycobacterial strains. Yet, these all failed and the only vaccine with proven safety and efficacy until today remains BCG. In fact, today BCG is the most widely used vaccine, which has been given more than 4 billion times. BCG was developed to protect newborns at high risk of TB (2). This mission has been accomplished at least partially since BCG was proven to protect against severe extra pulmonary, but less against pulmonary TB in infants (4–6). Yet, even today infant TB takes a worrisome toll in TB endemic countries with high coverage of BCG immunization (7–9). Later, BCG was also tested as a vaccine against pulmonary TB in adolescents and adults, but this ambitious target was not reached and no vaccine has ever succeeded in reliably protecting against pulmonary TB, the most prevalent form of the disease, in any age group. A better vaccine is urgently needed since Mycobacterium tuberculosis (Mtb), the cause of TB, remains on top of the infamous list of deadly infectious agents (10). In 2018, 10 million individuals fell ill with this disease and 1.5 million died (11) (Figure 1). The early 21st century has witnessed increasing R&D efforts for novel TB vaccines (12–19). These include subunit-adjuvant formulations comprising fusion proteins of Mtb, viral vectored vaccines expressing one or more antigens of Mtb, killed mycobacterial vaccines and viable attenuated mycobacterial vaccines.
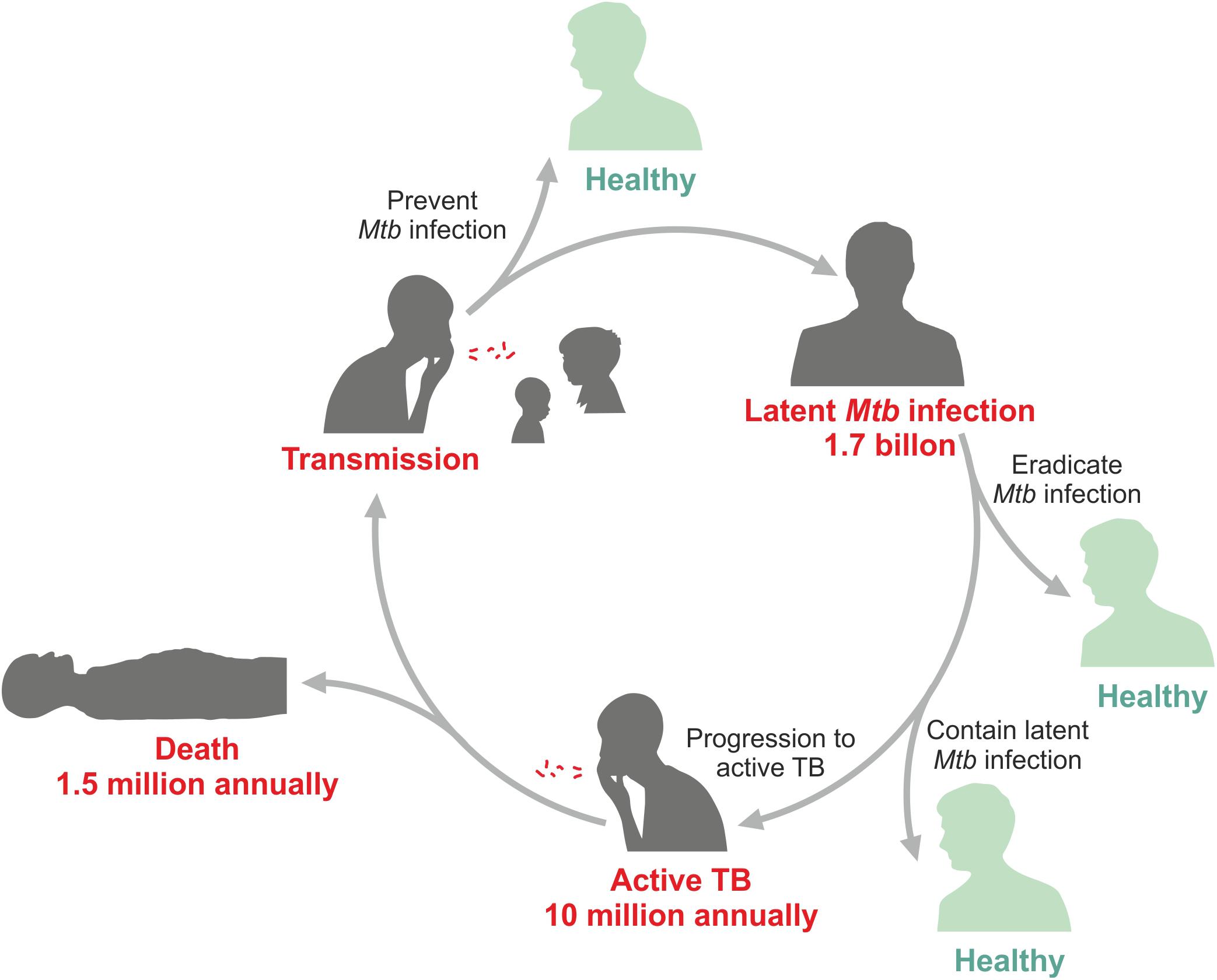
Figure 1. Epidemiologic data for tuberculosis (TB).
Immunopathology of Tuberculosis
Tuberculosis is a chronic infectious disease caused by the intracellular pathogen Mtb (20). This acid-fast bacillus is shielded by a unique lipoid-rich cell wall containing various wax-like substances and glycolipids which contribute to resistance against immune attack. Mtb is generally transmitted by aerosols in which it enters alveoli in lower lung lobes. Once the pathogen has been engulfed by alveolar phagocytes, it ends up in a phagosome, where it keeps the local pH neutral (21). Moreover, Mtb is capable of egressing into the cytosol (22). These and other mechanisms facilitate resistance of Mtb to professional phagocytes including polymorphonuclear neutrophilic granulocytes (in short neutrophils) and mononuclear phagocytes (tissue macrophages and monocytes) (23–27). Resting tissue macrophages generally fail to eliminate Mtb and serve as its retreat due to their long lifespan. Blood monocytes are slightly more aggressive but fail to achieve sterile elimination of Mtb. Neutrophils are highly aggressive phagocytes with the potential to harm Mtb. Due to their short lifespan, they generally will not succeed in completely eliminating Mtb and they do not serve as a harbor, in which Mtb can persist. Once activated by cytokines, notably Interferon-γ (IFN-γ), mononuclear phagocytes increase their anti-bacterial capacities and pose a more serious threat to Mtb although they generally fail to eradicate it completely. The innate immune response mediated by professional phagocytes serves as a first barrier for Mtb. Recent evidence suggests that epigenetic changes induced by Mtb in professional phagocytes leads to trained immunity. Such trained immunity could play a role in early defense against repeated Mtb infections (28, 29). However, thus far compelling evidence for this notion is still incomplete.
In addition, subtypes of dendritic cells (DC) can engulf Mtb (30, 31). They likely translocate Mtb into the lung parenchyma, where the formation of a granuloma is initiated.
Granuloma formation is strongly regulated by T lymphocytes originally stimulated in the draining lymph nodes to which DC harboring Mtb navigate (25, 32). T lymphocytes orchestrate formation of solid granulomas which are primarily composed of macrophages, DCs, and T and B lymphocytes. Within these granulomas Mtb is contained and the infected individual remains healthy and develops latent TB infection (LTBI) (24, 33, 34). CD4 T cells have been proven to be central to acquired resistance against and containment of Mtb (19, 25). According to the cytokines, these CD4 T cells secrete, they can be categorized into TH1, TH2, and TH17 cells. TH1 cells are preferentially stimulated during Mtb infection and are of major importance for defense. They produce cytokines such as IFN-γ, interleukin-2 (IL-2) and tumor necrosis-α (TNF-α). TH2 cells are only weakly induced. They are often considered harmful in TB since they induce inappropriate effector mechanisms. Their major cytokines are IL-4, IL-5, IL-10, and IL-13. However, evidence has been provided that TH2 cytokines, at least in part, can contribute to tissue healing. TH17 cells induce rapid proinflammatory responses by secreting IL-17. They are stimulated during Mtb infection and evidence has been published that they participate in protection against TB, notably at early stages of infection. The role of CD8 T cells in protection and containment – although less profound – is also widely accepted. CD8 T cells often produce cytokines of TH1 type and in addition express cytolytic activity (19, 25, 26). Contribution of cytolytic mechanisms to killing of Mtb has been demonstrated (35). The role of other lymphoid cells including innate lymphoid cells (iLC), NK T cells, mucosa associated immune T cells (MAIT), γδ T lymphocytes, and B lymphocytes is a matter of ongoing discussion (32, 36–45). B lymphocytes could participate in immunity against TB via two mechanisms: First, as regulatory B lymphocytes and second as antibody producing plasma cells. Evidence for regulatory B lymphocytes in immunity against TB is scarce (46, 47). A role for distinct antibody isotypes in defense against TB has been provided (36, 42, 45). Perhaps these antibodies modulate professional phagocytes through their binding to distinct Fc receptors. Convincing evidence has been generated that γδ T cells contribute to early immune defense by secreting IL-17 (38). The iLC can be categorized into iLC-1, iLC-2 and iLC-3 according to their cytokine secretion pattern (40). Cytokines produced by iLC-1 are of TH1 type, iLC-2 cytokines are of TH2 and iLC-3 cytokines are of TH17 type. The iLC-1 and iLC-3 probably contribute to resistance to Mtb and the iLC-2 to healing of lesions (37). During chronic infection, canonical CD4 and CD8 T lymphocytes develop into memory T cells which can be grouped into effector memory T cells (TEM), central memory T cells (TCM), and tissue resident memory T cells (TRM) (48). Although the role of the different memory T cells in protection against Mtb is incompletely understood, evidence for a particular role of TRM and TCM in protection against Mtb has been provided (49, 50). It is likely that different types of memory T cells participate in protective immunity at different stages of infection.
During LTBI, Mtb is contained in solid granuolomas (24, 33, 51). LTBI transforms into active TB disease when granulomas become necrotic and then caseous. This happens in about 5% of individuals with LTBI within the first 2 years and in another 5% at later time points. Thus, only ca. 10% of the 1.7 billion individuals with LTBI develop active TB disease (52). Progression to active disease is due to weakening of the immune response via several incompletely understood mechanisms. It is likely that myeloid-derived suppressor cells and regulatory T lymphocytes participate in dampening of protective immunity (53, 54). These cells produce inhibitory cytokines including IL-4, IL-10, and transforming growth factor-β (TGF-β). Moreover, excessive checkpoint control through inhibitory surface molecules including PD-1/PDL-1 and CTLA-4/B7 co-receptor interactions is likely involved (55, 56).
Notably, progression to active TB from LTBI must be viewed as a continuum rather than a discrete step from one to another stage (33, 57, 58). Mtb is transmitted from a TB patient to a healthy individual in a metabolically active and replicative stage. Hence, the host first encounters highly active Mtb (24). During LTBI, Mtb changes from a metabolically active and replicative stage into a dormant stage in which its activities are markedly downregulated. Once progression to active TB has ensued, Mtb wakes up and becomes active again.
At the early stage of infection, it is possible that Mtb is rapidly eradicated before stable LTBI develops, but the proportion of individuals who become transiently infected, sometimes accompanied by a short episode of clinical symptoms remains unclear (51, 57, 59). Recent evidence suggests LTBI is succeeded by incipient TB, in which the host remains healthy, but becomes alerted and Mtb regains its metabolic and replicative activities (59–62). Subsequently, subclinical TB evolves in which first signs of pathology occur although clinically the patient appears healthy. Signs of host vigilance and pathology can be detected by sensitive gene expression and metabolic profiling (26, 60–62). Given that most, if not all, cases of subclinical TB progress to active TB disease which can be clinically diagnosed, it is possible to predict disease by sensitive profiling by means of transcriptomics and metabolomics (60–63). Note that the different stages are not discrete and that in a single patient areas reflecting LTBI (solid granulomas containing dormant Mtb), incipient TB (solid to necrotic granulomas in which Mtb regains its metabolic and replicative activity), subclinical TB (further increase in pathology due to transition of some solid granulomas to necrotic ones and eventually first signs of caseation) and active TB (all three forms of granulomas present with a preponderance toward caseation and cavitation) can coexist. Accordingly, different stages of granulomas ranging from solid form to caseation and cavitation coexist, as well (58). Obviously, the coexistence of different pathologies and different Mtb activities render TB immunopathology highly complex.
BOX 1. Major vaccine candidates in clinical trials.
Different types of TB vaccines have entered the clinical trial pipeline. These are: viral vectored protein antigens of Mtb, fusion protein antigens of Mtb in adjuvants, killed whole mycobacterial cell vaccines, and recombinant viable mycobacterial vaccines. The viral vectored and the adjuvanted protein vaccines are subunit vaccines, which are generally considered to boost a prime with BCG. The viable TB vaccines are considered for BCG replacement or for boosting previous BCG prime. Killed whole cell vaccines are sometimes considered for booster vaccination and more often for TB therapy in adjunct to chemotherapy.
• Viral vectored vaccines include MVA85A, a modified vaccinia Ankara (MVA) vaccine expressing antigen Ag85A of Mtb. First phase IIb efficacy trials with this vaccine in neonates and in adults failed to provide protection (102, 103). More recently, the vaccine has been tested for safety and immunogenicity after aerosol application (104, 105). Other viral vectored vaccines include replication deficient adenovirus vectors expressing antigen Ag85A and a replication deficient H1N1 influenza vector expressing antigen Ag85A and ESAT-6. Novel prime boost schedules are also being tested including adenovirus vectors for prime and MVA vector for boost expressing antigen Ag85A.
Major viral vectored candidates undergoing clinical testing are:
Ad5Ag85A (phase I), a replication-deficient adenovirus (Ad) 5 vector expressing Antigen 85A (106, 107).
ChAdOx1.85A + MVA85A (phase I), a prime/boost regimen comprising prime with a chimpanzee Adenovirus (ChAd) expressing Antigen 85A (ChAdOx1.85A) followed by a boost with modified Vaccinia Ankara virus (MVA) expressing Antigen 85A (108).
TB-FLU-04L (phase IIa), a replication-deficient H1N1 influenza virus strain expressing Antigen 85A and ESAT-6 (109).
• Protein adjuvant formulations undergoing clinical testing include:
Hybrid 1 (H1, phase I completed) comprising either IC31 or CAF01 as adjuvant and a fusion protein of Antigen 85B and ESAT-6 as antigen (110, 111).
H4 (phase II completed) and H56 (phase IIb) formulated in IC31 as adjuvant and fusion proteins of Antigen 85B and TB10.4 (H4) or Antigen 85B, ESAT-6 and Rv2660c (H56) (73, 112–114).
ID93 (phase IIa) composed of GLA-SE as adjuvant and a fusion protein of 4 antigens, namely Rv2608, Rv3619, Rv3620 and Rv1813 (115, 116).
M72 (phase IIb completed) composed of AS01E as adjuvant and a fusion protein of 2 antigens, Rv1196, and Rv0125. M72 has completed a phase IIb trial revealing its partial protective efficacy (for further details see text) (65, 66, 117).
• Compositions of adjuvants:
IC31, cationic peptides plus TLR-9 agonist;
CAF01, cationic liposome vehicle plus immunomodulatory glycolipid;
GLA-SE, Squalen oil-in-water emulsion plus TLR-4 agonist;
AS01E, liposomes with monophosphoryl lipid A plus saponin QS21.
• Viable vaccines undergoing clinical testing are:
MTBVAC (phase IIa completed), a genetically attenuated Mtb vaccine (118, 119).
VPM1002 (several phase III trials), a rBCG vaccine (for further details see text) (84, 85).
• Killed whole cell vaccines include:
DAR-901 (killed M. obuense) which had already completed a phase III trial under a different name (120–123) and is now under re-evaluation (phase I trial completed) (124).
MIP (phase III) based on killed M. indicus pranii organisms (125–127).
M. vaccae (phase III) based on killed M. vaccae (128–132).
RUTI (phase IIa) a purified killed vaccine of Mtb fragments (133–135).
• Therapeutic vaccines: The above vaccine trials assess outcome of preventive vaccination. Several candidates are also tested as therapeutic vaccines either for TB treatment in adjunct to canonical chemotherapy or for PoR of TB patients who were cured from TB by canonical chemotherapy but may undergo recurrence (136).
Therapeutic vaccines in clinical trials include:
H56:IC31 (phase I), a subunit protein formulation;
ID93:GLA-SE (phase I), a subunit protein formulation;
RUTI (phase IIa), a purified killed vaccine of Mtb fragments;
TB-FLU-04L (phase IIa), a viral vectored vaccine;
MIP (phase III completed), a killed M. indicus pranii preparation;
M. vaccae (phase III completed), a killed M. vaccae preparation;
VPM1002 (phase III), a live rBCG vaccine.
Current Status of Tuberculosis Epidemiology and the Tuberculosis Vaccine Pipeline
According to the latest TB report of the World Health Organization (WHO), 10 million individuals developed active TB disease and 1.5 million died of TB in 2018 (11). Globally 1.7 million individuals are Mtb infected (LTBI, incipient TB, subclinical TB) (52). Thus, the goal of the WHO to eliminate TB over the next decades requires much better intervention measures and notably a highly efficacious vaccine (10). BCG fails to protect against pulmonary TB, which is not only the most prevalent form of disease but also the major source of transmission. This has led to several attempts to design novel vaccination regimens (18). Numerous vaccine candidates have entered clinical trials and first promising results have been obtained (see below). Current vaccine candidates undergoing clinical testing are viral vectored vaccines expressing a few Mtb antigens, adjuvanted subunit vaccines typically comprising fusion proteins representing two to four Mtb antigens, killed whole cell vaccines and viable whole cell vaccines. Further details can be found in Box 1. The vaccine candidates are tested in different clinical situations. These are:
(i) Prevention of Infection (PoI): This clinical endpoint can be applied for pre-exposure vaccination, i.e. vaccination of individuals who have not yet encountered Mtb. The most important target group for PoI are neonates. The WHO has prioritized a vaccine to lower the risk of Mtb infection (11).
(ii) Prevention of Disease (PoD): It is obvious that PoI will result in PoD. The major target population for PoD, however, are individuals with LTBI. Cutting the risk of TB disease in individuals with LTBI has also been prioritized by the WHO (11).
(iii) Prevention of Recurrence (PoR): In high endemic areas, ca. 10% of TB patients who had been cured by canonical drug treatment undergo recurrence, either due to reinfection or relapse (64).
(iv) Therapeutic Vaccination in Adjunct to Canonical Drug Treatment: Such a vaccination regimen gains increasing importance for patients with multi or extensively drug-resistant TB (MDR / XDR-TB) (16). An estimated half million of active TB patients suffer from MDR-TB and 50,000 to 100,000 individuals from XDR-TB (1). Vaccines for PoR are sometimes considered as therapeutic vaccines, as well.
This review will focus on vaccines that prevent active TB either through PoI, PoD, or PoR.
Prevention of Disease by the Subunit Vaccine M72 in a Phase IIb Clinical Trial
The M72 vaccine candidate developed by GlaxoSmithKline has successfully completed a phase IIb clinical trial (65, 66). Participants of this study were HIV– adults with LTBI who had been immunized with BCG as infants. Hence, the study was a post-exposure booster immunization with a subunit vaccine with PoD as clinical endpoint. The clinical endpoint was determined after 2 years of follow-up as pulmonary TB in absence of HIV infection (66). The study revealed ca. 50% protection over placebo control. The follow-up study confirmed the efficacy after the third year (65). This is the first vaccine trial to provide evidence for PoD in human TB. A positive control with BCG was not included in this study. It is hoped that global gene expression profiling and immunologic data will provide information about potential mechanisms underlying PoD induced by this vaccine. The vaccine comprises two TB antigens formulated in a potent adjuvant, AS01E (see Box 1). This adjuvant had been developed as part of the adjuvant system (AS) series and is also used in the shingles vaccines, Shingrix, and the malaria vaccine, Mosquirix (67). Availability of AS01E is limited and production cost is high. It has to be seen whether and how these limitations affect supply of this vaccine for broad-scale immunization programs. Satisfactory supply of vaccines for poverty-related diseases including TB and malaria strongly depends on an affordable price (68).
Promising Prevention of Disease Data in Non-Human Primates (NHP) by a Viral Vectored Tuberculosis Vaccine Candidate
Cytomegalovirus (CMV) based vaccines have been studied in a number of infectious diseases (69). Notably, in a simian immunodeficiency virus (SIV) model of rhesus macaques, CMV vectored vaccines expressing SIV antigens have shown profound protection mediated by CD4 and CD8 T lymphocytes (69). These T cells have been characterized as effector TEM cells and transitional effector memory T cells. Based on these findings, a TB vaccine candidate was designed which is based on a CMV vector expressing 6 or 9 Mtb antigens (70). This vector has been tested for PoD in rhesus macaques and was shown to induce profound protection against TB disease (70). Importantly, in a proportion of animals, evidence for sterile eradication of Mtb by this CMV-vectored TB vaccine was obtained. As expected, the vaccine induced profound CD4 and CD8 T cell responses as well as marked IFN-γ and TNF secretion. In contrast, antibody responses were not induced significantly. The protective CD8 T cell population was not only restricted by MHC I, but also by MHC-E or MHC II. BCG administered intradermally also induced protection, albeit weaker. Intriguingly, prime with BCG and boost with the CMV-based TB vaccine reverted the strong protective effect of the CMV vaccine to levels of protection induced by BCG. Gene expression profiling of vaccinated animals indicated a role for neutrophils in protection induced by the CMV vectored TB vaccine. In conclusion, despite certain disadvantages of CMV-vectored vaccines in general, the CMV-based TB vaccine represents a promising candidate which deserves further investigation. Obviously, the nullifying effect of BCG prime on protective efficacy induced by the CMV TB vaccine boost needs particular attention. Neonatal BCG immunization is done routinely in high TB endemic areas as part of the expanded program of immunization (EPI) recommended by WHO. Hence a new vaccine that provides no added value for BCG-immunized individuals will face major issues before it can be further developed. Similarly, a recent study revealed that in NHP boosting BCG with M72 or H56 (see Box 1) vaccines failed to enhance protection induced by BCG (71).
Recent Findings With the Canonical Bacille Calmette-GuÉRin Vaccine
Two recent studies on BCG immunization have revealed marked impact of the vaccination regimen (72, 73). In the first study, NHP were immunized with BCG intravenously (72). Earlier research in the 1970s had already provided compelling evidence that intravenous immunization with live BCG induces superior protection against TB as compared to other routes of administration in NHP with evidence for sterile eradication of Mtb (74, 75). Thus, in one study 3/3 animals were markedly protected against TB as measured by hematogenous spread, lymphadenopathy and lung involvement (74). On the other hand, profound splenomegaly was reported after intravenous administration of live BCG. Probably this significant adverse event was the major reason that such studies were not followed up. Only very recently this approach was investigated in greater depth. It was shown that intravenous immunization of NHP with BCG induced more profound protection than intradermal or aerogenic vaccination (72). Indeed, from a proportion of animals receiving BCG by the intravenous route no Mtb could be recovered. This study also included a series of highly sophisticated immunologic and pathologic analyses. It was found that antigen responses of CD4 and CD8 T lymphocytes were induced substantially by intravenous immunization prior to Mtb challenge, whereas γ/δ T cells and MAIT cells were, similarly, activated as in groups receiving other routes of immunization. The T cell response was mostly of TH1 type with some contribution of TH17 type. On the negative side, splenomegaly was observed after intravenous immunization with a ca. twofold enlargement of spleens compared to controls. However, splenomegaly was transient and 6 months after BCG immunization no differences were observed in spleen size across the different experimental groups including intravenous administration. Six months after immunization, animals were challenged with a low dose of Mtb. Positron emission tomography – computed tomography (PET/CT) scans revealed fewer granulomas in the intravenously immunized animals compared to controls. These findings provide proof of concept that BCG immunization can induce profound, in some cases sterile, protection in NHP. It needs to be seen how far the splenomegaly observed will be prohibitive for clinical studies in humans.
The second recent study tested the outcome of BCG booster vaccination in Mtb unexposed adults (73). Booster vaccination with BCG had been performed previously although generally it was not endorsed because of the potential risk of adverse events. This assumption was largely based on anecdotal reports describing occasional adverse events after repeated BCG immunization in individuals with LTBI and frequent severe events in TB patients. Principally, BCG revaccination in Mtb uninfected individuals does not cause major side effects and in the recent formal clinical trial, BCG revaccination of Mtb unexposed individuals demonstrated partial prevention of stable Mtb infection (73). More precisely, exposure was determined indirectly via an IFN-γ release assay (IGRA) which determines IFN-γ secretion by canonical T cells after in vitro restimulation with Mtb specific antigens (76–78). This assay is mostly based on CD4 T cell responses with some contribution of CD8 T cells. Whilst initial IGRA conversion did not differ between BCG immunized and untreated study participants, sustained IGRA conversion was significantly reduced by ca. 45% in BCG immunized study participants over controls (73). These findings can be interpreted to mean that stable Mtb infection is prevented by BCG revaccination although in fact it is based on reduced T cell responses as measured by IGRA. It remains to be established more precisely whether prevention of sustained IGRA conversion directly translates into long-term PoI and consequently PoD. Previous observational studies had evaluated PoD by BCG revaccination based on epidemiologic data. Generally, they did not find significant differences between controls and BCG revaccinated individuals (79–81).
These two studies provide strong evidence that the outcome of BCG vaccination is markedly influenced by the kind of administration, notably route of immunization (intravenous) and type of vaccine schedule (pre-exposure revaccination). In conclusion, the BCG vaccine still provides room for improvement.
VPM1002
One of the most advanced TB vaccines, VPM1002, was improved by genetic modification (82). VPM1002 is a recombinant BCG (rBCG) which expresses listeriolysin from Listeria monocytogenes and is devoid of urease C (83). Development of this vaccine had started in the 1990s with the aim to improve BCG by endowing it with the capacity to stimulate a broader, more efficacious T cell response.
VPM1002 has successfully completed phase I and phase IIa clinical trials proving its safety and immunogenicity in adults and neonates (84, 85). A phase II clinical trial in HIV exposed and unexposed neonates has been completed and awaits unblinding (NCT 02391415). A phase III clinical trial in HIV exposed and unexposed neonates is being prepared and expected to start in 2020. This trial has been designed as pre-exposure BCG replacement for infants with PoI as clinical endpoint. In this clinical trial, termed priMe, neonates will be immunized with VPM1002 or BCG as comparator at several sites in Sub-Saharan Africa. A phase III clinical trial with VPM1002 assessing PoR is currently ongoing in India (NCT 03152903). For this trial, patients with TB who had been cured by drug treatment are being recruited. An estimated 10 % of these individuals will develop active TB disease due to reinfection or relapse within 1 year after completion of drug treatment. The clinical trial therefore will reveal whether vaccination with VPM1002 given 3 months after completion of drug treatment can prevent recurrence. A phase III household contact trial has been launched in July 2019 by the Indian Council for Medical Research (ICMR), in which VPM1002 and another vaccine candidate (MIP, see Box 1) will be assessed for PoD in household contacts of patients with active pulmonary TB disease. In addition, VPM1002 is also being assessed as therapeutic agent against non-muscle invasive bladder cancer as substitute for BCG (NCT 02371447). The canonical TB vaccine BCG is the preferred immunomodulatory medicine for treatment of bladder cancer and the current clinical trial assesses whether VPM1002 is safer than and at least equally efficient as BCG against recurrence of bladder cancer. In conclusion, a century after the introduction of the original vaccine BCG, there is hope for a revival of an improved BCG-based TB vaccine. A rationally revamped BCG could contribute to the solution of the TB crisis.
How Does the Intracellular Behavior Differ Between VPM1002, Bacille Calmette-Guérin, and Mtb?
Both BCG and Mtb reside in phagosomes, which are arrested at an early stage by neutralization of the phagosomal pH to prevent its acidification (21). Consequently, phagolysosome fusion is diminished. Yet, BCG is degraded in the phagosome whereas Mtb survives in phagocytes for prolonged periods of time. Only recently, virulence mechanisms of Mtb absent from BCG have been elucidated. Although several sub-strains of BCG exist, it is now clear that the critical step which occurred during attenuation of the parental Mycobacterium bovis strain was the loss of the region of difference (RD) 1 which encodes a number of gene products mediated through the ESX/type VII secretion system and capable of perturbating the phagosomal membrane (86). Membrane perturbation by the RD-1 gene products of Mtb leads to inflammasome activation, apoptosis and autophagy (Figure 2). The signaling cascades involve nod-like receptor protein 3 (NLRP-3) and absent in melanoma 2 (AIM-2), responsible for IL-1 and IL-18 processing from their respective precursor molecules by the inflammasome as well as STING responsible for autophagy and type I IFN dependent responses (87). STING senses cyclic guanosine monophosphate-adenosine monophosphate (cGAMP) derived from double-stranded DNA of Mtb via the enzyme cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS). All these sequelae are caused by Mtb but not or less so by BCG.
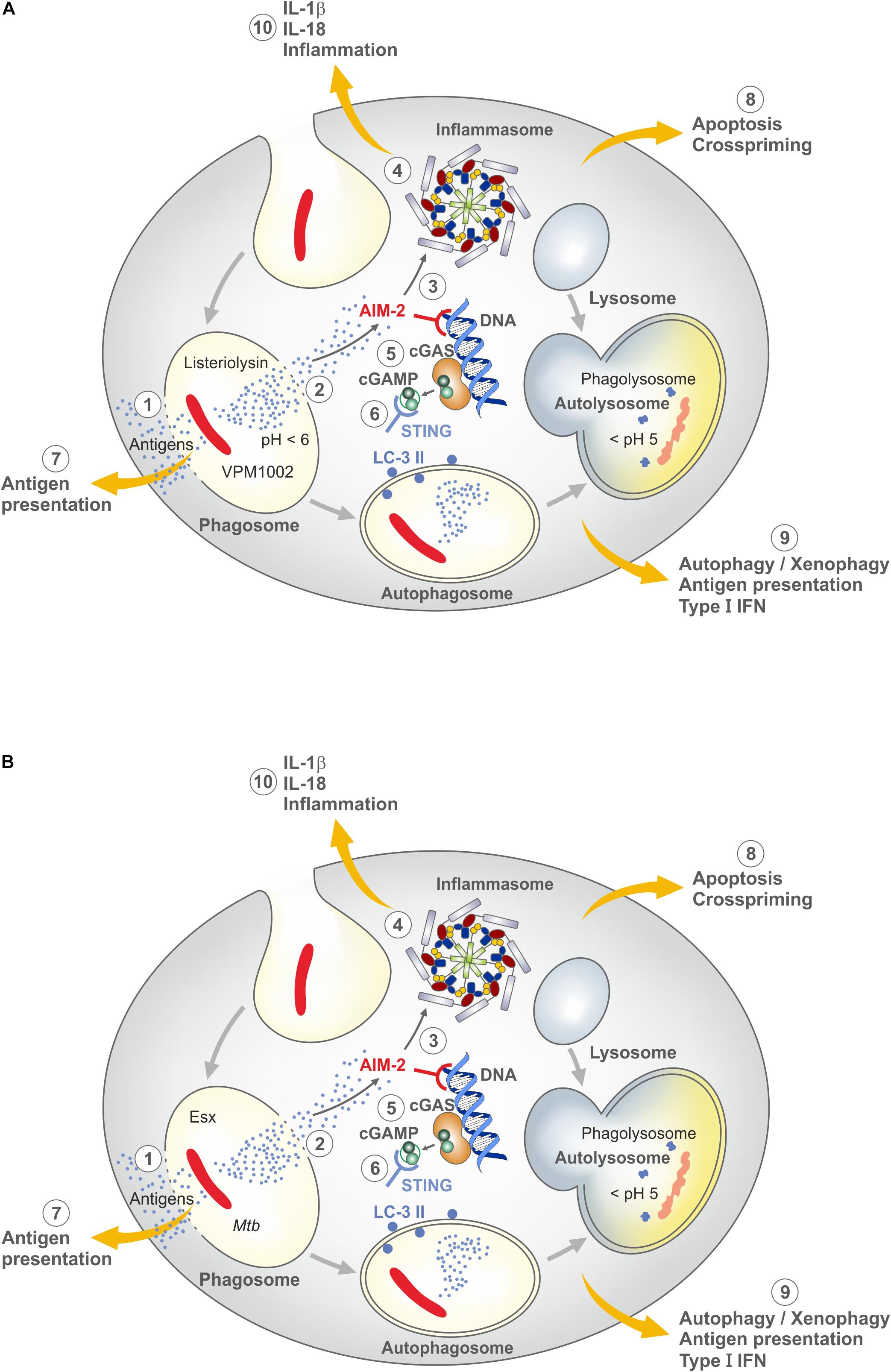
Figure 2. Major mechanisms underlying induction of the host immune response by VPM1002 and M. tuberculosis (Mtb) (for further details see text). (A) VPM1002. VPM1002 (rBCGΔureC::Hly) expresses listeriolysin and lacks urease C activity. Following phagocytosis, VPM1002 ends up in a phagosome. Principally phagosomes become acidic after uptake of particles, but BCG and Mtb actively keep the phagosomal pH neutral. Due to the absence of ureaseC in VPM1002, acidification takes place. This facilitates perturbation of the phagosomal membrane by biologically active listeriolysin. (1) Membrane perturbation allows egress of antigens into the cytosol for processing through the MHC class I pathway. (2) Perturbation can lead to apoptosis. (3) Double-strand DNA released into the cytosol is sensed by absence in melanoma 2 (AIM2). (4) AIM2 activates the inflammasome to generate IL-1β and IL-18. (5) Cyclic GMP-AMP synthase (cGAS) is formed which is then transformed into cyclic guanosine monophosphate-adenosine monophosphate (cGAMP). (6) The latter molecule is sensed by stimulator of IFN genes (STING) which induces autophagy and type I IFN responses. (7) Antigen egress into the cytosol allows stimulation of CD8 T cells in addition to CD4 T cells. (8) Apoptosis promotes crosspriming. (9) Autophagy accelerates elimination of VPM1002 and improves antigen presentation and T cell stimulation. (10) IL-1β and IL-18 induce an inflammatory response. Through these mechanisms, VPM1002 induces an immune response with more depth and breadth than parental BCG (B) Mtb. The genome of Mtb comprises the region of difference 1 (RD-1) which encodes numerous virulence factors which are absent in BCG. Notably genes for Esx dependent mechanisms cause perturbation of phagosomal membranes, very similar to VPM1002. For further details see (A). Because the RD-1 encoded gene products are not degraded after their egress into the cytosol, pathologic consequences prevail. Moreover, RD-1 encoded gene products are not controlled by pH. Hence, inbuilt safety mechanisms of VPM1002 are absent from Mtb (see also Figure 3).
For the design of VPM1002, BCG was equipped with listeriolysin from L. monocytogenes which facilitates perturbation of the phagosomal membrane thereby inducing stronger T cell responses (83). Listeriolysin is a thiol-activated perforin, which perforates cholesterol containing membranes at an acidic pH (88–90). This pH restriction generally prevents listeriolysin activity in the extracellular milieu with neutral pH, e.g. blood and interstitial space. It is however, achieved during natural infection of phagocytes with L. monocytogenes which allows secretion of biologically active listeriolysin. Because BCG neutralizes the phagosomal compartment, acidification is not achieved. For this reason, the urease C encoding gene was deleted in VPM1002 (83). This enzyme is responsible for ammonia production and thereby participates in neutralization of the phagosome where BCG resides (21). Accordingly, VPM1002 lacking urease C favors phagosomal acidification and thereby secretion of biologically active listeriolysin (Figure 2). Once listeriolysin has reached the cytosol, it is rapidly degraded. This is due to the amino acid sequence proline-glutamate-serine-threonine (PEST) in the listeriolysin amino acid sequence which promotes its ubiquitination (Figure 3) (88–90). This represents an inbuilt safety mechanism, which restricts listeriolysin-activity to the perturbation of the membrane of the phagosome, where VPM1002 resides and prevents further potentially detrimental effects on cell membranes. The RD-1 encoded machinery of Mtb is not endowed with such a safety mechanism.
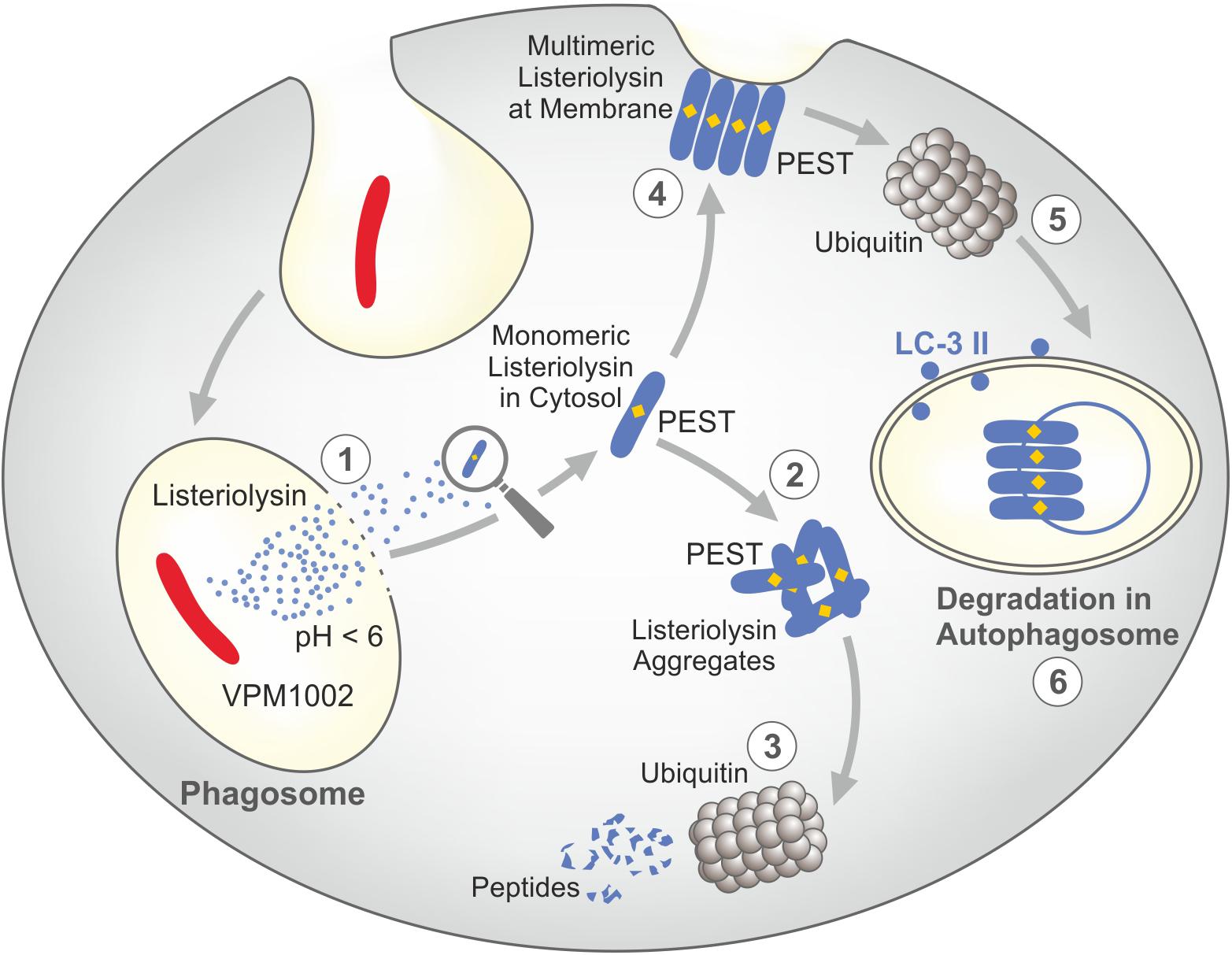
Figure 3. Safety mechanisms of listeriolysin render VPM1002 less virulent than parental BCG. Listeriolysin contains a PEST-like sequence which promotes its degradation. (1) Only at acidic pH, listeriolysin is biologically active and hence perturbates the phagosomal membrane. (2) In the cytosol, monomeric listeriolysin aggregates. (3) Aggregated listeriolysin is degraded by ubiquitin resulting in inactive peptides. (4) Multimeric listeriolysin complexes are formed at the plasma membrane. (5) These complexes are translocated into autophagosomes by ubiquitin. (6) These listeriolysin complexes are inactivated in the phagosome. PEST = Proline (P), Gutalate (E), Serine (S), and Threonine (T). Modified from (88–90).
Similar to the RD-1 machinery in Mtb, listeriolysin-mediated perturbation of the phagosomal membrane by VPM1002 results in inflammasome activation through AIM-2 (91). Hence, IL-1 and IL-18 are processed from their respective precursors. These proinflammatory cytokines create a milieu favorable for activation of TH1 and TH17 cells. Listeriolysin also facilitates autophagy via AIM-2 and STING by promoting sensing of double-strand mycobacterial DNA derived from VPM1002 via cGAS and cGAMP after its egress into the cytosol (91). In addition, membrane perturbation by listeriolysin causes apoptosis, which leads to cross priming of T cells (92). Together these mechanisms improve vaccine efficacy of VPM1002 as compared to canonical BCG (82, 93). Moreover, VPM1002 was shown to be safer than BCG in preclinical studies (83). In experimental models improved stimulation of both CD4 and CD8 T cells has been demonstrated (92) as well as more profound activation of TH17 cells in addition to TH1 cells (94). In addition, central memory T cells were more strongly stimulated by VPM1002 as compared to canonical BCG (50). Finally, VPM1002 was shown to stimulate higher serum levels of specific antibodies both in animal models and in human (50, 84, 85). In conclusion, VPM1002 stimulates an immune response of more depth and breadth and at the same time expresses lower virulence as compared to BCG (82).
Learning From Individuals Resistant to Stable Mtb Infection and Those Capable of Eradicating Mtb After Stable Infection
Individuals with LTBI are generally identified by tuberculin skin test (TST) or IGRA (Figure 4) (76–78). Accordingly, identification of the 1.7 billion individuals on this globe with LTBI is based on measurements of T cell responses against Mtb antigens. These antigens are relatively undefined mixtures of Mtb proteins (purified protein derivative, PPD) in the case of TST and well defined Mtb proteins and/or peptides in the case of IGRA.
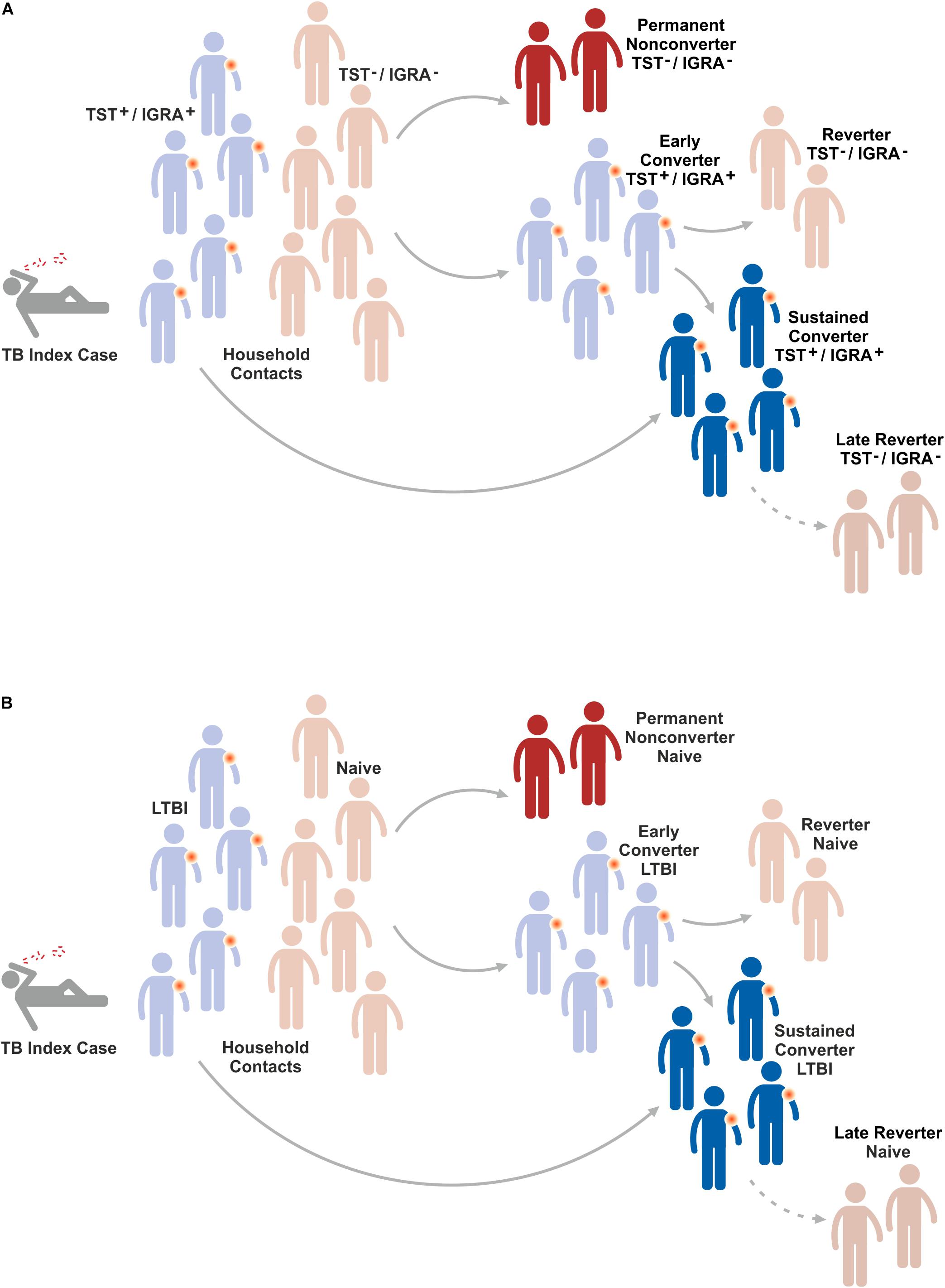
Figure 4. Fate of household contacts of a TB index case. Household contacts of a TB index case are either already latently TB infected (LTBI) or do not show evidence for immunity against Mtb infection. After sustained contact with a TB index case, the majority of naïve individuals will rapidly convert to LTBI because they mount an immune response against Mtb infection. Most of these early converters will remain LTBI and hence become sustained converters. A small proportion of early converters reverts to naïve, i.e. devoid of a measurable immune response to Mtb infection. Some naïve individuals will remain permanent non-converters, i.e. they do not change their status of absent immunity indicating absence of Mtb infection. Finally, some individuals with LTBI will revert to naïve, i.e. they lose their detectable immune response to Mtb indicating elimination of Mtb. The mechanisms underlying these conversions/reversions remain elusive. (A) Indicates response in TST/IGRA and (B) depicts resulting conclusions on conversion/reversion (for further details see text).
Several lines of evidence suggest that a distinct population of individuals remain Mtb uninfected despite their close and prolonged contact with patients with active pulmonary TB who continuously expel Mtb (95–98). This notion is based on the finding that such individuals do not convert when tested by TST or IGRA. Assuming that the lack of the canonical immune response determined by TST and/or IGRA reflects absence of Mtb infection, the following scenario arises (Figure 3): Initially, household contacts of a patient with active pulmonary TB fall into two groups; those who are already LTBI because of previous contact and hence are TST+/IGRA+, and naïve individuals who are TST–/IGRA–. Due to the intensive contact, most of the naïve individuals will convert to TST+/IGRA+ and most of them remain TST+/IGRA+ over longer periods of time, if not lifelong. However, a small group may revert to TST–/IGRA– indicating that they are capable of eradicating Mtb before they become permanently infected. The recent BCG revaccination trial on PoI (73) described above did not reveal significant differences between BCG-immunized and control groups in early conversion to IGRA+. Yet, a 45% reduction in sustained IGRA+ (determined at later time points) was observed in the BCG immunized group as compared to controls without BCG immunization. Moreover, observational studies have identified a distinct group of permanent non-converters (TST–/IGRA–) generally in the order of 20% (95–98).
Obviously, the described effects could also be due to technical reasons and the TST–/IGRA– group could be infected with Mtb but missed by TST/IGRA because these individuals develop a protective immune response which is not detected by TST and IGRA. Underlying mechanisms could include antibodies, MAIT cells, γδ T cells, NK cells and NKT cells (32, 36–45). Furthermore, it remains unclear whether all TST+/IGRA+ individuals are indeed Mtb infected or whether at least a subgroup has succeeded in eliminating Mtb but remains TST+/IGRA+ because of a strong memory T cell response which persists in absence of Mtb antigens. Principally, immunology defines memory as a state of immunity in absence of nominal antigen(s).
Another interesting group may arise years after primary Mtb infection. Whilst many individuals with LTBI remain TST+/IGRA+ livelong, some individuals revert to TST–/IGRA–. It is likely that in these individuals, reversion from TST+/IGRA+ to TST–/IGRA– reflects sterile eradication of Mtb. Yet, it cannot be excluded formally that these individuals remain Mtb infected and control infection by unknown immune mechanisms not detected by TST/IGRA such as antibodies and unconventional T cells. In any case, the permanent non-converters and the late reverters are highly interesting study groups which provide the opportunity to gain deeper insights into the mechanisms of protection against Mtb. TB vaccines which prevent stable infection with Mtb and thereby prevent LTBI and active TB disease would be highly desirable. The specific mechanisms underlying permanent non-conversion and late reversion could be elucidated by determining transcriptomic, metabolomic and immunologic markers and signatures which distinguish permanent non-converters and late reverters from sustained and livelong reverters, respectively (26, 99).
Outlook and Future
Over the last decade, the TB vaccine pipeline has significantly progressed. First, a number of vaccines is ready for clinical efficacy testing for PoI, PoD, or PoR (see Box 1). This implies that several vaccine candidates have already proven their safety and immunogenicity. Second, several positive signals arose from clinical trials over the last years including proof of concept that a subunit vaccine empowered by a strong adjuvant can partially protect against active TB when given post-exposure to individuals with LTBI (65, 66). Third, BCG revaccination of Mtb unexposed individuals has provided indirect evidence for partial prevention of sustained Mtb infection (73). Obviously, major issues remain to be solved. These include: First, BCG revaccination outcome was determined by IGRA which measures canonical T-cell immune responses rather than Mtb infection per se (see above). This raises the question whether BCG indeed prevented Mtb infection or whether infection occurred but was controlled by alternative immune mechanisms such as antibodies and/or unconventional T cells. Second, the M72 clinical trial did not include BCG vaccination as a positive control. Third, studies in NHP revealed that different subunit vaccine constructs including M72, H56, and the CMV vectored TB vaccine failed to increase protection when given as booster on BCG prime (70, 71). In sum, there is well justified hope for better vaccines; but it remains difficult to predict when and to which degree TB can be controlled by improved vaccination strategies.
Nassim Nicholas Taleb is best known for describing the Black Swan Concept which basically includes the notions (100): (i) rare and improbable events do occur more frequently than we assume; (ii) these extreme events can have enormous consequences; (iii) experts generally provide explanations post-hoc which were not plausible ex-ante. This concept was aptly illustrated by the financial stock market crisis in 2008 when numerous stock owners who had gradually accumulated a financial depot went bankrupt through a single event. The most illustrative description for the Black Swan concept is the life of a Thanksgiving Day turkey which is taken care of very well over the first 1000 days by feeding it with most nourishing food. An observer (including the turkey if it could do so) could conclude that the quality of life of this animal increases constantly. Yet, on day 1001, the butcher kills the animal unexpectedly in preparation for Thanksgiving Day. Obviously, this is an extreme event with a major impact on the animal. This scenario can also be turned upside down into a positive direction, i.e. that an unexpected and improbable event turns into something markedly better (an event which would perhaps be better described by the term Pink Swan). With respect to TB vaccine design, continuous funding into R&D (from basic research to preclinical and clinical development) will increase our knowledge about the underlying mechanisms of protection against TB and how this information can be harnessed for TB vaccine design (Figure 5).
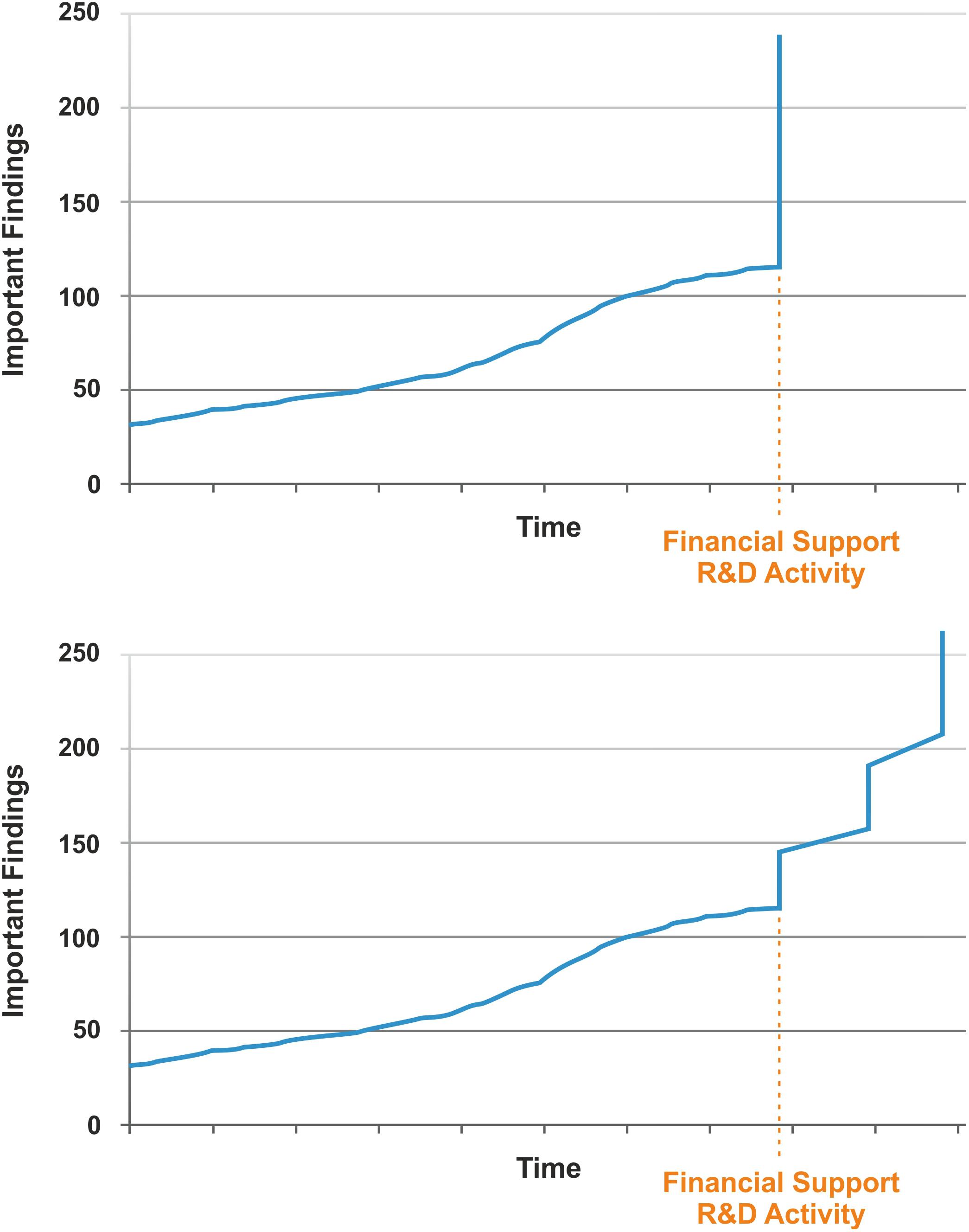
Figure 5. Possible scenarios of TB vaccine development given that adequate financial funding is provided for research & development (R&D). Upper, single step event; Lower, multistep event.
For long periods of time research crawls, but every now and then it jumps. By increasing funding, a fertile soil can be prepared for R&D on better vaccines. Maybe this leads to an extreme event (a single jump), resulting in a novel vaccine that fits all purposes. More likely a couple of smaller, yet significant events will occur which ultimately lead to TB vaccines for different purposes. The type of vaccine and the time when it will be ready for clinical licensing remain unclear as expected for a Black/Pink Swan. Yet, increased funding for R&D will favor chances of success. Undoubtedly, this cannot be accomplished free of cost; yet ultimately it will save cost by reducing the enormous expenses caused by the TB endemic. After all, annual cost for treating active TB disease globally has been estimated to be in the range of 2 billion US dollars and the total burden of TB on the global economy in the order of 100 billion US dollars (101).
It is worth to conclude with a citation from the resolution adopted by the General Assembly of the United Nations from the High Level Meeting on the fight against TB in 2018 (1):
“Commit to mobilize sufficient and sustainable financing, with the aim of increasing overall global investments to 2 billion dollars, in order to close the estimated 1.3 billion dollar gap in funding annually for tuberculosis research, ensuring that all countries contribute appropriately to research and development ….”
Author Contributions
SK conceived the idea and wrote the manuscript.
Conflict of Interest
SK is co-inventor of the TB vaccine, VPM1002 and co-holder of a patent licensed to Vakzine Projekt Management GmbH, Hanover, Germany and sublicensed to Serum Institute of India Pvt. Ltd., Pune, India. The vaccine is currently undergoing clinical trial testing.
Acknowledgments
The author thanks Diane Schad for superb graphics and Souraya Sibaei for excellent help in preparation of the manuscript.
References
1. United Nations UN General Assembly High-Level Meeting on the Fight Against Tuberculosis. (2018). Available online at: www.who.int/tb/unhlmonTBDeclaration.pdf (accessed September 26, 2018).
2. Calmette A, Guérin C, Boquet A, Négre L. La Vaccination Préventive Contre La Tuberculose Par Le “BCG”. Paris: Masson et Cie (1927).
3. Kaufmann SH, Winau F. From bacteriology to immunology: the dualism of specificity. Nat Immunol. (2005) 6:1063–66.
4. Colditz GA, Berkey CS, Mosteller F, Brewer TF, Wilson ME, Burdick E, et al. The Efficacy of bacillus Calmette-Guerin vaccination of newborns and infants in the prevention of tuberculosis: meta-analyses of the published literature. Pediatrics. (1995) 96(1 Pt 1):29–35.
5. Colditz GA, Brewer TF, Berkey S, Wilson ME, Burdick E, Fineberg HV, et al. Efficacy of BCG vaccine in the prevention of tuberculosis. Meta-analysis of the published literature. JAMA. (1994) 271:698–702. doi: 10.1001/jama.271.9.698
6. Roy A, Eisenhut M, Harris RJ, Rodrigues LC, Sridhar S, Habermann S, et al. Effect of BCG vaccination against Mycobacterium tuberculosis infection in children: systematic review and meta-analysis. Br Med J. (2014) 349:g4643. doi: 10.1136/bmj.g4643
7. Chiang SS, Khan FA, Milstein MB, Tolman AW, Benedetti A, Starke JR, et al. Treatment outcomes of childhood tuberculous meningitis: a systematic review and meta-analysis. Lancet Infect Dis. (2014) 14:947–57. doi: 10.1016/S1473-3099(14)70852-7
8. Hesseling AC, Marais BJ, Gie RP, Schaaf HS, Fine PE, Godfrey-Faussett P, et al. The risk of disseminated Bacille Calmette-Guerin (BCG) disease in HIV-infected children. Vaccine. (2007) 25:14–8.
9. Trunz BB, Fine P, Dye C. Effect of BCG vaccination on childhood tuberculous meningitis and miliary tuberculosis worldwide: a meta-analysis and assessment of cost-effectiveness. Lancet. (2006) 367:1173–80.
10. Abu-Raddad LJ, Sabatelli L, Achterberg JT, Sugimoto JD, Longini IM Jr., Dye C, et al. Epidemiological benefits of more-effective tuberculosis vaccines, drugs, and diagnostics. Proc Natl Acad Sci USA. (2009) 106:13980–85. doi: 10.1073/pnas.0901720106
11. World Health Organization.WHO Global Tuberculosis Report 2019. Geneva: World Health Organization (2019).
12. Andersen P, Kaufmann SH. Novel vaccination strategies against tuberculosis. Cold Spring Harb Perspect Med. (2014) 4:a018523. doi: 10.1101/cshperspect.a018523
13. Andersen P, Scriba TJ. Moving tuberculosis vaccines from theory to practice. Nat Rev Immunol. (2019) 19:550–62. doi: 10.1038/s41577-019-0174-z
14. Ginsberg AM. Designing tuberculosis vaccine efficacy trials – lessons from recent studies. Expert Rev Vaccines. (2019) 18:423–32. doi: 10.1080/14760584.2019.1593143
15. Kaufmann SH. Future vaccination strategies against tuberculosis: thinking outside the box. Immunity. (2010) 33:567–77. doi: 10.1016/j.immuni.2010.09.015
16. Kaufmann SH, Weiner J, von Reyn CF. Novel approaches to tuberculosis vaccine development. Int J Infect Dis. (2017) 56:263–67. doi: 10.1016/j.ijid.2016.10.018
17. Kaufmann SHE. Tuberculosis vaccines: time to think about the next generation. Semin Immunol. (2013) 25:172–81. doi: 10.1016/j.smim.2013.04.006
18. Kaufmann SHE, Dockrell HM, Drager N, Ho MM, McShane H, Neyrolles O, et al. TBVAC2020: advancing tuberculosis vaccines from discovery to clinical development. Front Immunol. (2017) 8:1203. doi: 10.3389/fimmu.2017.01203
19. Ottenhoff TH, Kaufmann SH. Vaccines against tuberculosis: where are we and where do we need to go? PLoS Pathog. (2012) 8:e1002607. doi: 10.1371/journal.ppat.1002607
20. Pai M, Behr MA, Dowdy D, Dheda K, Divangahi M, Boehme CC, et al. Tuberculosis. Nat Rev Dis Primers. (2016) 2:16076. doi: 10.1038/nrdp.2016.76
21. Gordon AH, Hart PD, Young MR. Ammonia inhibits phagosome-lysosome fusion in macrophages. Nature. (1980) 286:79–80. doi: 10.1038/286079a0
22. van der Wel N, Hava D, Houben D, Fluitsma D, van Zon M, Pierson J, et al. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell. (2007) 129:1287–98.
23. Dorhoi A, Kaufmann SH. Versatile myeloid cell subsets contribute to tuberculosis-associated inflammation. Eur J Immunol. (2015) 45:2191–202. doi: 10.1002/eji.201545493
24. Gengenbacher M, Kaufmann SHE. Mycobacterium tuberculosis: success through dormancy. FEMS Microbiol Rev. (2012) 36:514–32. doi: 10.1111/j.1574-6976.2012.00331.x
25. O’Garra A, Redford PS, McNab FW, Bloom CI, Wilkinson RJ, Berry MP. The immune response in tuberculosis. Annu Rev Immunol. (2013) 31:475–527. doi: 10.1146/annurev-immunol-032712-095939
26. Ottenhoff TH, Ellner JJ, Kaufmann SH. Ten challenges for TB biomarkers. Tuberculosis. (2012) 92 (Suppl. 1):S17–20. doi: 10.1016/S1472-9792(12)70007-0
27. Schorey JS, Schlesinger LS. Innate immune responses to tuberculosis. Microbiol Spectr. (2016) 4:TBTB2-0010-2016.
28. Aaby P, Benn CS. Stopping live vaccines after disease eradication may increase mortality. Vaccine. (2019) 38:10–4. doi: 10.1016/j.vaccine.2019.10.034
29. Netea MG, Schlitzer A, Placek K, Joosten LAB, Schultze JL. Innate and adaptive immune memory: an evolutionary continuum in the host’s response to pathogens. Cell Host Microbe. (2019) 25:13–26. doi: 10.1016/j.chom.2018.12.006
30. Lozza L, Farinacci M, Fae K, Bechtle M, Staber M, Dorhoi A, et al. Crosstalk between human DC subsets promotes antibacterial activity and CD8+ T-cell stimulation in response to Bacille Calmette-Guerin. Eur J Immunol. (2014) 44:80–92. doi: 10.1002/eji.201343797
31. Randall TD. Pulmonary dendritic cells: thinking globally, acting locally. J Exp Med. (2010) 207:451–4. doi: 10.1084/jem.20100059
32. Behar SM, Carpenter SM, Booty MG, Barber DL, Jayaraman P. Orchestration of pulmonary T cell immunity during Mycobacterium tuberculosis infection: immunity interruptus. Semin Immunol. (2014) 26:559–77. doi: 10.1016/j.smim.2014.09.003
33. Barry CE III, Boshoff HI, Dartois V, Dick T, Ehrt S, Flynn J, et al. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat Rev Microbiol. (2009) 7:845–55. doi: 10.1038/nrmicro2236
34. Ulrichs T, Kaufmann SH. New insights into the function of granulomas in human tuberculosis. J Pathol. (2006) 208:261–9.
35. Stenger S, Hanson DA, Teitelbaum R, Dewan P, Niazi KR, Froelich CJ, et al. An antimicrobial activity of cytolytic T cells mediated by granulysin. Science. (1998) 282:121–5.
36. Achkar JM, Casadevall A. Antibody-mediated immunity against tuberculosis: implications for vaccine development. Cell Host Microbe. (2013) 13:250–62. doi: 10.1016/j.chom.2013.02.009
37. Ardain A, Domingo-Gonzalez R, Das S, Kazer SW, Howard NC, Singh A, et al. Group 3 innate lymphoid cells mediate early protective immunity against tuberculosis. Nature. (2019) 570:528–32. doi: 10.1038/s41586-019-1276-2
38. Coulter F, Parrish A, Manning D, Kampmann B, Mendy J, Garand M, et al. IL-17 production from T helper 17, mucosal-associated invariant T, and γδ cells in tuberculosis infection and disease. Front Immunol. (2017) 8:1252.
39. Downey AM, Kaplonek P, Seeberger PHMAIT. MAIT cells as attractive vaccine targets. FEBS Lett. (2019) 593:1627–40. doi: 10.1002/1873-3468.13488
40. Klose CS, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol. (2016) 17:765–74. doi: 10.1038/ni.3489
41. Li H, Javid B. Antibodies and tuberculosis: finally coming of age? Nat Rev Immunol. (2018) 18:591–6. doi: 10.1038/s41577-018-0028-0
42. Lu LL, Chung AW, Rosebrock TR, Ghebremichael M, Yu WH, Grace PS, et al. A functional role for antibodies in tuberculosis. Cell. (2016) 167:433–43.e14. doi: 10.1016/j.cell.2016.08.072
43. Lu LL, Smith MT, Yu KKQ, Luedemann C, Suscovich TJ, Grace PS, et al. IFN-gamma-independent immune markers of Mycobacterium tuberculosis exposure. Nat Med. (2019) 25:977–87. doi: 10.1038/s41591-019-0441-3
44. Roy Chowdhury R, Vallania F, Yang Q, Lopez Angel CJ, Darboe F, Penn-Nicholson A, et al. A multi-cohort study of the immune factors associated with M. tuberculosis infection outcomes. Nature. (2018) 560:644–8.
45. Zimmermann N, Thormann V, Hu B, Kohler AB, Imai-Matsushima A, Locht C, et al. Human isotype-dependent inhibitory antibody responses against Mycobacterium tuberculosis. EMBO Mol Med. (2016) 8:1325–39. doi: 10.15252/emmm.201606330
46. Benard A, Sakwa I, Schierloh P, Colom A, Mercier I, Tailleux L, et al. B cells producing Type I IFN modulate macrophage polarization in tuberculosis. Am J Respir Crit Care Med. (2018) 197:801–13. doi: 10.1164/rccm.201707-1475OC
47. Cerqueira C, Manfroi B, Fillatreau S. IL-10-producing regulatory B cells and plasmocytes: molecular mechanisms and disease relevance. Semin Immunol. (2019) 44:101323. doi: 10.1016/j.smim.2019.101323
48. Masopust D, Soerens AG. Tissue-resident T cells and other resident leukocytes. Annu Rev Immunol. (2019) 37:521–46. doi: 10.1146/annurev-immunol-042617-053214
49. Perdomo C, Zedler U, Kuhl AA, Lozza L, Saikali P, Sander LE, et al. Mucosal BCG vaccination induces protective lung-resident memory T cell populations against tuberculosis. mBio. (2016) 7:e01686-16. doi: 10.1128/mBio.01686-16
50. Vogelzang A, Perdomo C, Zedler U, Kuhlmann S, Hurwitz R, Gengenbacher M, et al. Central memory CD4+ T cells are responsible for the recombinant Bacillus Calmette-Guerin Δurec::hly vaccine’s superior protection against tuberculosis. J Infect Dis. (2014) 210:1928–37.
51. Simmons JD, Stein CM, Seshadri C, Campo M, Alter G, Fortune S, et al. Immunological mechanisms of human resistance to persistent Mycobacterium tuberculosis infection. Nat Rev Immunol. (2018) 18:575–89. doi: 10.1038/s41577-018-0025-3
52. Houben RM, Dodd PJ. The global burden of latent tuberculosis infection: a re-estimation using mathematical modelling. PLoS Med. (2016) 13:e1002152. doi: 10.1371/journal.pmed.1002152
53. Brighenti S, Ordway DJ. Regulation of immunity to tuberculosis. Microbiol Spectr. (2016) 4:TBTB2-0006-2016. doi: 10.1128/microbiolspec.TBTB2-0006-2016
54. Dorhoi A, Du Plessis N. Monocytic myeloid-derived suppressor cells in chronic infections. Front Immunol. (2017) 8:1895. doi: 10.3389/fimmu.2017.01895
55. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. (2018) 8:1069–86. doi: 10.1158/2159-8290.CD-18-0367
56. Barber DL, Sakai S, Kudchadkar RR, Fling SP, Day TA, Vergara JA, et al. Tuberculosis following PD-1 blockade for cancer immunotherapy. Sci Transl Med. (2019) 11:eaat2702. doi: 10.1126/scitranslmed.aat2702
57. Dodd CE, Schlesinger LS. New concepts in understanding latent tuberculosis. Curr Opin Infect Dis. (2017) 30:316–21. doi: 10.1097/QCO.0000000000000367
58. Lin PL, Ford CB, Coleman MT, Myers A, Gawande R, Ioerger T, et al. Sterilization of granulomas is common in active and latent tuberculosis despite within-host variability in bacterial killing. Nat Med. (2014) 20:75–9. doi: 10.1038/nm.3412
59. Drain PK, Bajema KL, Dowdy D, Dheda K, Naidoo K, Schumacher SG, et al. Incipient and subclinical tuberculosis: a clinical review of early stages and progression of infection. Clin Microbiol Rev. (2018) 31:e00021-18. doi: 10.1128/CMR.00021-18
60. Suliman S, Thompson E, Sutherland J, Weiner Rd J, Ota MOC, Shankar S, et al. Four-gene pan-African blood signature predicts progression to tuberculosis. Am J Respir Crit Care Med. (2018) 197:1198–208. doi: 10.1371/journal.pmed.1002781
61. Weiner J III, Maertzdorf J, Sutherland JS, Duffy FJ, Thompson E, Suliman S, et al. Metabolite changes in blood predict the onset of tuberculosis. Nat Commun. (2018) 9:5208. doi: 10.1038/s41467-018-07635-7
62. Zak DE, Penn-Nicholson A, Scriba TJ, Thompson E, Suliman S, Amon LM, et al. A blood RNA signature for tuberculosis disease risk: a prospective cohort study. Lancet. (2016) 387:2312–22. doi: 10.1016/S0140-6736(15)01316-1
63. Weiner J III, Kaufmann SH. Recent advances towards tuberculosis control: vaccines and biomarkers. J Intern Med. (2014) 275:467–80. doi: 10.1111/joim.12212
64. Rosser A, Marx FM, Pareek M. Recurrent tuberculosis in the pre-elimination era. Int J Tuberc Lung Dis. (2018) 22:139–50. doi: 10.5588/ijtld.17.0590
65. Tait DR, Hatherill M, Van Der Meeren O, Ginsberg AM, Van Brakel E, Salaun B, et al. Final analysis of a trial of M72/AS01E vaccine to prevent tuberculosis. N Engl J Med. (2019) 381:2429–39. doi: 10.1056/NEJMoa1909953
66. Van Der Meeren O, Hatherill M, Nduba V, Wilkinson R, Muyoyeta M, Van Brakel E, et al. Phase 2b controlled trial of M72/AS01E vaccine to prevent tuberculosis. N Engl J Med. (2018) 379:1621–34. doi: 10.1056/NEJMoa1803484
67. Del Giudice G, Rappuoli R, Didierlaurent AM. Correlates of adjuvanticity: a review on adjuvants in licensed vaccines. Semin Immunol. (2018) 39:14–21. doi: 10.1016/j.smim.2018.05.001
68. Kaufmann SHE. Highly affordable vaccines are critical for our continued efforts to reduce global childhood mortality. Hum Vaccin Immunother. (2019) 15:2660–5. doi: 10.1080/21645515.2019.1605817
69. Picker LJ, Hansen SG, Lifson JD. New paradigms for HIV/AIDS vaccine development. Annu Rev Med. (2012) 63:95–111. doi: 10.1146/annurev-med-042010-085643
70. Hansen SG, Zak DE, Xu G, Ford JC, Marshall EE, Malouli D, et al. Prevention of tuberculosis in rhesus macaques by a cytomegalovirus-based vaccine. Nat Med. (2018) 24:130–43. doi: 10.1038/nm.4473
71. Darrah PA, DiFazio RM, Maiello P, Gideon HP, Myers AJ, Rodgers MA, et al. Boosting BCG with proteins or rAD5 does not enhance protection against tuberculosis in rhesus macaques. NPJ Vaccines. (2019) 4:21.
72. Darrah PA, Zeppa JJ, Hackney JA, Wadsworth IIMH, Hughes TK, Pokkali S, et al. Prevention of tuberculosis in macaques after intravenous BCG immunization. Nature. (2020) 577:95–102. doi: 10.1038/s41586-019-1817-8
73. Nemes E, Geldenhuys H, Rozot V, Rutkowski KT, Ratangee F, Bilek N, et al. Prevention of M. tuberculosis infection with H4:IC31 vaccine or BCG revaccination. N Engl J Med. (2018) 379:138–49. doi: 10.1056/NEJMoa1714021
74. Anacker RL, Brehmer W, Barclay WR, Leif WR, Ribi E, Simmons JH, et al. Superiority of intravenously administered BCG and BCG cell walls in protecting rhesus monkeys (Macaca Mulatta) against airborne tuberculosis. Z Immunitatsforsch Exp Klin Immunol. (1972) 143:363–76.
75. Barclay WR, Anacker RL, Brehmer W, Leif W, Ribi E. Aerosol-induced tuberculosis in subhuman primates and the course of the disease after intravenous BCG vaccination. Infect Immun. (1970) 2:574–82.
76. Andrews JR, Hatherill M, Mahomed H, Hanekom WA, Campo M, Hawn TR, et al. The dynamics of quantiferon-TB gold in-tube conversion and reversion in a cohort of South African adolescents. Am J Respir Crit Care Med. (2015) 191:584–91. doi: 10.1164/rccm.201409-1704OC
77. Diel R, Goletti D, Ferrara G, Bothamley G, Cirillo D, Kampmann B, et al. Interferon-gamma release assays for the diagnosis of latent Mycobacterium tuberculosis infection: a systematic review and meta-analysis. Eur Respir J. (2011) 37:88–99. doi: 10.1183/09031936.00115110
78. Pai M, Zwerling A, Menzies D. Systematic review: T-cell-based assays for the diagnosis of latent tuberculosis infection: an update. Ann Intern Med. (2008) 149:177–84.
79. Barreto ML, Pereira SM, Pilger D, Cruz AA, Cunha SS, Sant’Anna C, et al. Evidence of an effect of BCG revaccination on incidence of tuberculosis in school-aged children in Brazil: second report of the BCG-revac cluster-randomised trial. Vaccine. (2011) 29:4875–77. doi: 10.1016/j.vaccine.2011.05.023
80. Karonga Prevention Trial Group. Randomised controlled trial of single BCG, repeated BCG, or combined BCG and killed Mycobacterium leprae vaccine for prevention of leprosy and tuberculosis in Malawi. Lancet. (1996) 348:17–24. doi: 10.1016/s0140-6736(96)02166-6
81. Rodrigues LC, Pereira SM, Cunha SS, Genser B, Ichihara MY, de Brito SC, et al. Effect of BCG revaccination on incidence of tuberculosis in school-aged children in Brazil: the BCG-revac cluster-randomised trial. Lancet. (2005) 366:1290–5.
82. Nieuwenhuizen NE, Kulkarni PS, Shaligram U, Cotton MF, Rentsch CA, Eisele B, et al. The recombinant Bacille Calmette-Guerin vaccine VPM1002: ready for clinical efficacy testing. Front Immunol. (2017) 8:1147. doi: 10.3389/fimmu.2017.01147
83. Grode L, Seiler P, Baumann S, Hess J, Brinkmann V, Nasser Eddine A, et al. Increased vaccine efficacy against tuberculosis of recombinant Mycobacterium bovis Bacille Calmette-Guérin mutants that secrete listeriolysin. J Clin Invest. (2005) 115:2472–9.
84. Grode L, Ganoza CA, Brohm C, Weiner IIIJ, Eisele B, Kaufmann SH. Safety and immunogenicity of the recombinant BCG vaccine VPM1002 in a phase 1 open-label randomized clinical trial. Vaccine. (2013) 31:1340–8. doi: 10.1016/j.vaccine.2012.12.053
85. Loxton AG, Knaul JK, Grode L, Gutschmidt A, Meller C, Eisele B, et al. Safety and immunogenicity of the recombinant Mycobacterium bovis BCG vaccine VPM1002 in HIV-unexposed newborn infants in South Africa. Clin Vaccine Immunol. (2017) 24:e00439-16. doi: 10.1128/CVI.00439-16
86. Vaziri F, Brosch R. ESX/Type VII secretion systems-an important way out for mycobacterial proteins. Microbiol Spectr. (2019) 7:PSIB-0029-2019. doi: 10.1128/microbiolspec.PSIB-0029-2019
87. Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J, et al. Mycobacterium tuberculosis differentially activates cGAS- and inflammasome-dependent intracellular immune responses through ESX-1. Cell Host Microbe. (2015) 17:799–810. doi: 10.1016/j.chom.2015.05.003
88. Chen C, Nguyen BN, Mitchell G, Margolis SR, Ma D, Portnoy DA. The listeriolysin O pest-like sequence Co-opts AP-2-mediated endocytosis to prevent plasma membrane damage during listeria infection. Cell Host Microbe. (2018) 23:786–95.e5. doi: 10.1016/j.chom.2018.05.006
89. Decatur AL, Portnoy DAA. A pest-like sequence in listeriolysin O essential for Listeria monocytogenes pathogenicity. Science. (2000) 290:992–5.
90. Nguyen BN, Peterson BN, Portnoy DA. Listeriolysin O: a phagosome-specific cytolysin revisited. Cell Microbiol. (2019) 21:e12988. doi: 10.1111/cmi.12988
91. Saiga H, Nieuwenhuizen N, Gengenbacher M, Koehler AB, Schuerer S, Moura-Alves P, et al. The recombinant BCG Δurec::hly vaccine targets the AIM2 inflammasome to induce autophagy and inflammation. J Infect Dis. (2015) 211:1831–41. doi: 10.1093/infdis/jiu675
92. Farinacci M, Weber S, Kaufmann SH. The recombinant tuberculosis vaccine rBCG Δurec::hly(+) induces apoptotic vesicles for improved priming of CD4(+) and CD8(+) T cells. Vaccine. (2012) 30:7608–14.
93. Winau F, Weber S, Sad S, de Diego J, Hoops SL, Breiden B, et al. Apoptotic vesicles crossprime CD8 T cells and protect against tuberculosis. Immunity. (2006) 24:105–17.
94. Desel C, Dorhoi A, Bandermann S, Grode L, Eisele B, Kaufmann SH. Recombinant BCG Δurec hly+ induces superior protection over parental BCG by stimulating a balanced combination of Type 1 and Type 17 cytokine responses. J Infect Dis. (2011) 204:1573–84.
95. Badger TL, Spink WW. First-infection type of tuberculosis in adults. N Engl J Med. (1937) 217:424–31.
96. Dickie HA. Tuberculosis in student nurses and medical students at the University of Wisconsin. Ann Intern Med. (1950) 33:941–59.
97. Houk VN, Baker JH, Sorensen K, Kent DC. The epidemiology of tuberculosis infection in a closed environment. Arch Environ Health. (1968) 16:26–35.
98. Kaipilyawar V, Salgame P. Infection resisters: targets of new research for uncovering natural protective immunity against Mycobacterium tuberculosis. F1000Research. (2019) 8:F1000 Faculty Rev-1698. doi: 10.12688/f1000research.19805.1
99. Kaufmann SH, Evans TG, Hanekom WA. Tuberculosis vaccines: time for a global strategy. Sci Transl Med. (2015) 7:276fs8. doi: 10.1126/scitranslmed.aaa4730
100. Taleb NN. The Black Swan. The Impact of the Highly Improbable. 2nd ed. New York, NY: Penguin Random House LLC (2010).
101. APPG The Price of a Pandemic. Counting the Cost of MDR-TB. London: All-Party Parliamentary Group on Global Tuberculosis (2015).
102. Ndiaye BP, Thienemann F, Ota M, Landry BS, Camara M, Dieye S, et al. Safety, immunogenicity, and efficacy of the candidate tuberculosis vaccine MVA85a in healthy adults infected with HIV-1: a randomised, placebo-controlled, Phase 2 trial. Lancet Respir Med. (2015) 3:190–200. doi: 10.1016/S2213-2600(15)00037-5
103. Tameris MD, Hatherill M, Landry BS, Scriba TJ, Snowden MA, Lockhart S, et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: a randomised, placebo-controlled Phase 2b trial. Lancet. (2013) 381:1021–8.
104. Manjaly Thomas Z-R, Satti I, Marshall JL, Harris SA, Lopez Ramon R, Hamidi A, et al. Alternate aerosol and systemic immunisation with a recombinant viral vector for tuberculosis, MVA85A: a Phase I randomised controlled trial. PLoS Med. (2019) 16:e1002790. doi: 10.1371/journal.pmed.1002790
105. Satti I, Meyer J, Harris SA, Manjaly Thomas ZR, Griffiths K, Antrobus RD, et al. Safety and immunogenicity of a candidate tuberculosis vaccine MVA85a delivered by aerosol in BCG-vaccinated healthy adults: a Phase 1, double-blind, randomised controlled trial. Lancet Infect Dis. (2014) 14:939–46. doi: 10.1016/S1473-3099(14)70845-X
106. Smaill F, Jeyanathan M, Smieja M, Medina MF, Thanthrige-Don N, Zganiacz A, et al. A human Type 5 adenovirus-based tuberculosis vaccine induces robust T cell responses in humans despite preexisting anti-adenovirus immunity. Sci Transl Med. (2013) 5:205ra134. doi: 10.1126/scitranslmed.3006843
107. Smaill F, Xing Z. Human Type 5 adenovirus-based tuberculosis vaccine: is the respiratory route of delivery the future? Expert Rev Vaccines. (2014) 13:927–30. doi: 10.1586/14760584.2014.929947
108. Stylianou E, Griffiths KL, Poyntz HC, Harrington-Kandt R, Dicks MD, Stockdale L, et al. Improvement of BCG protective efficacy with a novel chimpanzee adenovirus and a modified vaccinia ankara virus both expressing Ag85A. Vaccine. (2015) 33:6800–8. doi: 10.1016/j.vaccine.2015.10.017
109. Sergeeva MV, Pulkina AA, Vasiliev KA, Romanovskaya-Romanko EA, Komissarov A, Kuchur OA, et al. Safety and immunogenicity of cold-adapted recombinant influenza vector expressing ESAT-6 and Ag85A antigens of M. tuberculosis. Vopr Virusol. (2017) 62:266–72.
110. van Dissel JT, Arend SM, Prins C, Bang P, Tingskov PN, Lingnau K, et al. Ag85B-ESAT-6 adjuvanted with IC31 promotes strong and long-lived Mycobacterium tuberculosis specific T cell responses in naive human volunteers. Vaccine. (2010) 28:3571–81. doi: 10.1016/j.vaccine.2010.02.094
111. van Dissel JT, Joosten SA, Hoff ST, Soonawala D, Prins C, Hokey DA, et al. A novel liposomal adjuvant system, CAF01, promotes long-lived Mycobacterium tuberculosis-specific T-cell responses in human. Vaccine. (2014) 32:7098–107. doi: 10.1016/j.vaccine.2014.10.036
112. Geldenhuys H, Mearns H, Miles DJ, Tameris M, Hokey D, Shi Z, et al. The tuberculosis vaccine H4:IC31 is safe and induces a persistent polyfunctional CD4 T cell response in South African adults: a randomized controlled trial. Vaccine. (2015) 33:3592–9. doi: 10.1016/j.vaccine.2015.05.036
113. Luabeya AK, Kagina BM, Tameris MD, Geldenhuys H, Hoff ST, Shi Z, et al. First-in-human trial of the post-exposure tuberculosis vaccine H56:IC31 in Mycobacterium tuberculosis infected and non-infected healthy adults. Vaccine. (2015) 33:4130–40. doi: 10.1016/j.vaccine.2015.06.051
114. Suliman S, Luabeya AKK, Geldenhuys H, Tameris M, Hoff ST, Shi Z, et al. Dose optimization of H56:IC31 vaccine for tuberculosis-endemic populations. a double-blind, placebo-controlled, dose-selection trial. Am J Respir Crit Care Med. (2019) 199:220–31. doi: 10.1164/rccm.201802-0366OC
115. Coler RN, Day TA, Ellis R, Piazza FM, Beckmann AM, Vergara J, et al. The TLR-4 agonist adjuvant, GLA-SE, improves magnitude and quality of immune responses elicited by the ID93 tuberculosis vaccine: first-in-human trial. NPJ Vaccines. (2018) 3:34. doi: 10.1038/s41541-018-0057-5
116. Penn-Nicholson A, Tameris M, Smit E, Day TA, Musvosvi M, Jayashankar L, et al. Safety and immunogenicity of the novel tuberculosis vaccine ID93 + GLA-SE in BCG-vaccinated healthy adults in South Africa: a randomised, double-blind, placebo-controlled Phase 1 trial. Lancet Respir Med. (2018) 6:287–98. doi: 10.1016/S2213-2600(18)30077-8
117. Leroux-Roels I, Forgus S, De Boever F, Clement F, Demoitie MA, Mettens P, et al. Improved CD4(+) T cell responses to Mycobacterium tuberculosis in PPD-negative adults by M72/AS01 as compared to the M72/AS02 and Mtb72F/AS02 tuberculosis candidate vaccine formulations: a randomized trial. Vaccine. (2013) 31:2196–206. doi: 10.1016/j.vaccine.2012.05.035
118. Spertini F, Audran R, Chakour R, Karoui O, Steiner-Monard V, Thierry AC, et al. Safety of human immunisation with a live-attenuated Mycobacterium tuberculosis vaccine: a randomised, double-blind, controlled Phase I trial. Lancet Respir Med. (2015) 3:953–62. doi: 10.1016/S2213-2600(15)00435-X
119. Tameris M, Mearns H, Penn-Nicholson A, Gregg Y, Bilek N, Mabwe S, et al. Live-attenuated Mycobacterium tuberculosis vaccine MTBVAC versus BCG in adults and neonates: a randomised controlled, double-blind dose-escalation trial. Lancet Respir Med. (2019) 7:757–70. doi: 10.1016/S2213-2600(19)30251-6
120. Johnson JL, Kamya RM, Okwera A, Loughlin AM, Nyole S, Hom DL, et al. Randomized controlled trial of Mycobacterium vaccae immunotherapy in non-human immunodeficiency virus-infected Ugandan adults with newly diagnosed pulmonary tuberculosis. the Uganda-Case Western Reserve University research collaboration. J Infect Dis (2000) 181:1304–12.
121. Lahey T, Arbeit RD, Bakari M, Horsburgh CR, Matee M, Waddell R, et al. Immunogenicity of a protective whole cell mycobacterial vaccine in HIV-infected adults: a Phase III study in Tanzania. Vaccine. (2010) 28:7652–8. doi: 10.1016/j.vaccine.2010.09.041
122. von Reyn CF, Mtei L, Arbeit RD, Waddell R, Cole B, Mackenzie T, et al. Prevention of tuberculosis in Bacille Calmette-Guerin-primed, HIV-infected adults boosted with an inactivated whole-cell mycobacterial vaccine. AIDS. (2010) 24:675–85. doi: 10.1097/QAD.0b013e3283350f1b
123. Vuola JM, Ristola MA, Cole B, Jarviluoma A, Tvaroha S, Ronkko T, et al. Immunogenicity of an inactivated mycobacterial vaccine for the prevention of HIV-associated tuberculosis: a randomized, controlled trial. AIDS. (2003) 17:2351–5.
124. von Reyn CF, Lahey T, Arbeit RD, Landry B, Kailani L, Adams LV, et al. Safety and immunogenicity of an inactivated whole cell tuberculosis vaccine booster in adults primed with BCG: a randomized, controlled trial of DAR-901. PLoS One. (2017) 12:e0175215. doi: 10.1371/journal.pone.0175215
125. Gupta A, Ahmad FJ, Ahmad F, Gupta UD, Natarajan M, Katoch V, et al. Efficacy of Mycobacterium indicus pranii immunotherapy as an adjunct to chemotherapy for tuberculosis and underlying immune responses in the lung. PLoS One. (2012) 7:e39215. doi: 10.1371/journal.pone.0039215
126. Mayosi BM, Ntsekhe M, Bosch J, Pandie S, Jung H, Gumedze F, et al. Prednisolone and Mycobacterium indicus pranii in tuberculous pericarditis. N Engl J Med. (2014) 371:1121–30. doi: 10.1056/NEJMoa1407380
127. Sharma SK, Katoch K, Sarin R, Balambal R, Kumar Jain N, Patel N, et al. Efficacy and safety of Mycobacterium indicus pranii as an adjunct therapy in category II pulmonary tuberculosis in a randomized trial. Sci Rep. (2017) 7:3354. doi: 10.1038/s41598-017-03514-1
128. Butov DA, Efremenko YV, Prihoda ND, Zaitzeva SI, Yurchenko LV, Sokolenko NI, et al. Randomized, placebo-controlled Phase II trial of heat-killed Mycobacterium vaccae (Immodulon Batch) formulated as an oral pill (V7). Immunotherapy. (2013) 5:1047–54. doi: 10.2217/imt.13.110
129. de Bruyn G, Garner P. Mycobacterium vaccae immunotherapy for treating tuberculosis. Cochrane Database Syst Rev. (2003) 2003:CD001166. doi: 10.1002/14651858.CD001166
130. Efremenko YV, Butov DA, Prihoda ND, Zaitzeva SI, Yurchenko LV, Sokolenko NI, et al. Randomized, placebo-controlled Phase II trial of heat-killed Mycobacterium vaccae (Longcom Batch) formulated as an oral pill (V7). Hum Vaccin Immunother. (2013) 9:1852–6. doi: 10.4161/hv.25280
131. Weng H, Huang J-Y, Meng X-Y, Li S, Zhang G-Q. Adjunctive therapy of Mycobacterium vaccae vaccine in the treatment of multidrug-resistant tuberculosis: a systematic review and meta-analysis. Biomed Rep. (2016) 4:595–600. doi: 10.3892/br.2016.624
132. Yang XY, Chen QF, Li YP, Wu SM. Mycobacterium vaccae as adjuvant therapy to anti-tuberculosis chemotherapy in never-treated tuberculosis patients: a meta-analysis. PLoS One. (2011) 6:e23826. doi: 10.1371/journal.pone.0023826
133. Cardona PJ. RUTI: a new chance to shorten the treatment of latent tuberculosis infection. Tuberculosis (Edinb). (2006) 86:273–89.
134. Nell AS, D’Lom E, Bouic P, Sabate M, Bosser R, Picas J, et al. Safety, tolerability, and immunogenicity of the novel antituberculous vaccine ruti: randomized, placebo-controlled Phase II clinical trial in patients with latent tuberculosis infection. PLoS One. (2014) 9:e89612. doi: 10.1371/journal.pone.0089612
135. Vilaplana C, Montane E, Pinto S, Barriocanal AM, Domenech G, Torres F, et al. Double-blind, randomized, placebo-controlled Phase I clinical trial of the therapeutical antituberculous vaccine RUTI. Vaccine. (2010) 28:1106–16. doi: 10.1016/j.vaccine.2009.09.134
Keywords: tuberculosis, vaccine, Bacille Calmette-Guérin, subunit, biomarker, macrophage, T lymphocyte, clinical trial
Citation: Kaufmann SHE (2020) Vaccination Against Tuberculosis: Revamping BCG by Molecular Genetics Guided by Immunology. Front. Immunol. 11:316. doi: 10.3389/fimmu.2020.00316
Received: 13 December 2019; Accepted: 07 February 2020;
Published: 27 February 2020.
Edited by:
Ursula Wiedermann, Medical University of Vienna, AustriaReviewed by:
Arun Kumar, Coalition for Epidemic Preparedness Innovations (CEPI), NorwayNitesh K. Kunda, St. John’s University, United States
Copyright © 2020 Kaufmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefan H. E. Kaufmann, S2F1Zm1hbm5AbXBpaWItYmVybGluLm1wZy5kZQ==