 Nicole Meyer
Nicole Meyer Ana Claudia Zenclussen
Ana Claudia Zenclussen- Experimental Obstetrics and Gynecology, Medical Faculty, Otto-von-Guericke University, Magdeburg, Germany
Sufficient uterine remodeling is essential for fetal survival and development. Pathologies related to poor remodeling have a negative impact on maternal and fetal health even years after birth. Research of the last decades yielded excellent studies demonstrating the key role of immune cells in the remodeling processes. This review summarizes the current knowledge about the relevance of immune cells for uterine remodeling during pregnancy and further discusses immunomodulatory effects of man-made endocrine disrupting chemicals on immune cells.
Introduction
In the last years it has become clear that a healthy intrauterine environment builds the fundament of adult health (1). The efficient remodeling of tissue and vasculature within the uterus during the menstrual cycle and pregnancy is a basic requirement for a healthy intrauterine environment (2, 3). Several studies of the last decades have revealed the relevance of maternal immune cells, specifically uterine natural killer cells (uNKs), macrophages, and T cells but also fetal trophoblast cells for the remodeling process during pregnancy (4). Research within the field has experienced significant progress in recent years and provide new insights. Specifically, there is an increased knowledge about the importance of uterine mast cells (uMCs) for the vascular remodeling process during pregnancy. Equally important, a negative influence of man-made endocrine disrupting chemicals (EDCs) on reproductive health emerge within the literature in recent years, leading to further relevant health questions that need answers, opening new challenging research fields.
Cyclic Endometrial Remodeling
The endometrium of women in reproductive age undergoes cyclic tissue remodeling each month. The menstrual cycle aims to prepare the endometrium for implantation. The activity of the ovarian steroid hormones estrogen and progesterone but also various matrix metalloproteinases (MMPs) and their inhibitors, named tissue inhibitors of MMP (TIMP), regulate the endometrial changes and tissue breakdown during menstruation (5).
The endometrium consists of two layers, the functional layer that is shed with each menstruation, and the basalis layer containing progenitor cells, able to regenerate the functional layer. The proliferative phase of the menstrual cycle is maintained by the hormonal influence of estrogen. Epithelial and stromal cells undergo mitosis and growth, glands increase in length and the extracellular matrix (ECM) expands. Endometrium thickness increases from around 4.5 to 10 mm. The secretory phase of the menstrual cycle is mainly maintained by the effects of progesterone. High levels of progesterone, secreted by the corpus luteum, are responsible for endometrial receptivity after ovulation. Progesterone inhibits the endometrial epithelial mitosis and proliferation, induces the appearance of vacuoles, stromal fibroblast changes, and alterations in ECM that are necessary for the receptivity of the endometrium and subsequently, for the blastocyst attachment (6).
If the fertilization of the oocyte does not take place, the corpus luteum degrades, and the declining progesterone levels cause a local inflammatory response in the endometrium, involving edema and infiltration of specialized maternal immune cells into the stroma (3), predominantly uNKs, macrophages, uMCs, neutrophils, dendritic cells (DCs), and T cells (7). The presence of chemokines in this milieu is important for leukocyte recruitment (8). Macrophages and neutrophils that are recruited to the site represent the microbial protection mechanism while the epithelial barrier is disrupted. MC proteases transform MMP precursors into their active form (9). MMPs in turn degrade the ECM and destruct the tissue. The final result is shedding of the endometrial functional layer, and thereby two-third of the endometrium, during the menstrual phase of each cycle (6). As antigen-presenting cells, DCs and macrophages clear the cellular debris from the uterine cavity. Regulatory T cells (Tregs) control all these processes and maintain the immune balance to avoid an exacerbated inflammatory response (9). Disturbances in endometrial immune cell number or function have been found to contribute to heavy menstrual bleeding or endometriosis (7). Menstruation occurs in human, primates, elephants, and fruit bats. Non-menstruating species show a considerable remodeling and reabsorption of the endometrium (5). A subsequently regeneration, including tissue and vascular repair, growth, and angiogenesis facilitates the receptivity of the endometrium for implantation in the next cycle (10). Also here immune cells play a key role by releasing regulatory molecules stimulating the endometrial repair mechanisms (7).
Uterine Remodeling During Healthy Pregnancy
In non-menstruating mammals, decidualization begins with the implantation process. In contrast, in menstruating species, decidualization occurs prior to implantation and is postulated to be a mechanism to protect the mother from the invasiveness of embryonic trophoblasts. A successful implantation process is followed by several tissue and vascular adaptions. The most important tissue adaption in this regard is the formation and growth of a new transient organ, the placenta. Maternal blood is delivered to the intervillous space of the placenta via the aorta, the uterine artery, the arcuate artery, radial arteries, and spiral arteries (SA), listing from large to small vessels. In response to the altered hemodynamic demands resulting from an increased uterine blood flow during pregnancy, there is the need of a physiological remodeling of the uterine vasculature. The remodeling process starts in the smaller vessels, the SAs, proximal to the sites of placentation and proceeds to the larger, upstream vessels (11).
Many studies focused and still focus on the remodeling of SAs. The helically wounded arteries build the last branch of the uterine artery. SAs transport maternal blood to the intervillous space of the placenta, where the blood enters in direct contact with fetal tissue, for an effective exchange of nutrients and gases (12). During pregnancy, the thick-walled, high resistance vessels transform into thin-walled low resistance vessels by losing several vascular smooth muscle cell (VSMC) layers of the arterial wall (13, 14).
VSMCs are aligned in a circumference in the medial layer of the arterial wall. For maintaining the vascular tone, VSMCs usually acquire a quiescent, contractile phenotype. The contractile phenotype is characterized by high expression of contractility markers and low proliferative or migratory activity. An enormous plasticity enables VSMCs to change their morphology during pregnancy and consequently their functionality changes as well. Expression patterns change leading to increased proliferation, migration, and synthetic capacity (15). These parameters, together with a low expression of contractility markers, are characteristic for the synthetic phenotype of VSMCs. VSMCs can change their expression pattern due to vascular injury or changing hemodynamic demands (16) in response to various stimuli, ligand-receptors interactions, and environmental signals (17). The ECM compounds collagen, elastin, and proteoglycans facilitate a contractile VSMC phenotype. In contrast, high presence of fibronectin favors the shift into a synthetic VSMC phenotype (18). A phenotype switch from the contractile to synthetic VSMCs is associated with a changed protein and receptor expression that modify the binding specificity to the ECM, and an increased VSMC migration (18, 19), that in turn is important for an efficient SA remodeling process (20, 21).
A propervascular remodeling is important for fetal survival, development, and growth for the following reasons. Firstly, an enlarged arterial diameter reduces the velocity of the blood stream (14) and prevents disturbance of the sensitive fetal villi containing fetal blood capillaries. Secondly, due to the loss of muscular VSMC layers, SAs completely lose their contractile ability, preventing an interruption or reduction of the blood stream to the placenta that would be incompatible with fetal survival (22, 23).
Both, maternal and fetal cells contribute to the uterine remodeling process. In preparation for the remodeling process, maternal immune cells release factors that induce the degradation the ECM and directly or indirectly induce apoptosis or a phenotypic switch of VSMCs leading to higher migration (19, 21). Subsequently, fetal trophoblast can invade into the SAs and replace VSMCs.
Immune Cell Function in Uterine Remodeling During Pregnancy
Interaction of Stromal Cells, Immune Cells, and Trophoblasts
The feto-maternal interface consist of decidual stromal cells, immune cells, and trophoblasts. Differentiation of stromal cells within the decidualization process is a pre-requirement for a successful implantation (24).
Stromal cells express integrins, important for the interaction of the blastocyst with the decidua during the implantation process (25). Additionally stromal cells produce chemokines that attract immune cells to the uterus. Interesting examples are Mcp-1, RANTES, or CXCL8 that attract monocytes and macrophages (26, 27), CXCL10 and CXCL11 that attract uNKs (28), and IL-15 and CXCL12 that act as chemoattractants for pNKs (29, 30). Carlino et al. showed that the migratory ability of NKs from pregnant women was higher compared to those of non-pregnant women. The process could be enhanced by progesterone, that upregulated chemokine production by stromal cells (30). Decidual stromal cells are localized near SAs and support their remodeling by the secretion of MMP 2, 7, and 9. These factors are able to promote the disruption of VSMS layers (31) prior to the invasion of extravillous trophoblasts (EVTs). Several studies let assume an association between stromal cell dysfunction and reproductive disorders like endometriosis (32), implantation failure (33) or recurrent pregnancy loss (34).
EVTs are important key players in the vascular remodeling during pregnancy. EVTs arise from the cytotrophoblast in the anchoring villi of the placenta and invade into the maternal SA, thereby replacing VSMCs and the endothelium. By using a three-dimensional bioprinted placenta model, Kuo et al. performed impressive trophoblast-endothelium interaction studies. They reported that the co-culture of trophoblasts with endothelial cells significantly reduced the outgrowth and network formation of endothelial cells and induced endothelial cell apoptosis, shown by a significant upregulation of apoptosis marker (35). Trophoblast invasion is temporally and spatially regulated by autocrine (trophoblastic) and paracrine (uterine) factors as well as cell-to-cell and cell-to-matrix interactions (36). Trophoblast viability, migration and proliferation capacity as well as the expression of pro-apoptotic molecules like Fas-Ligand and tumor necrosis factor (TNF)-related apoptosis-inducing ligand have been shown to support SA remodeling (37–39). Also, the proteinase activity of EVTs influences their invasion capacity. Many studies demonstrate the importance of trophoblast cells for vascular remodeling by showing an association between insufficient trophoblast invasion leading to inadequate vascular remodeling and pregnancy complications, such as preeclampsia (PE) and intrauterine growth restriction (IUGR) (40, 41).
Interestingly, van der Heijden et al. postulated that the conceptus does not contribute to the initiation of uterine artery remodeling. They observed that the cellular processes of the remodeling, including artery lumen and cross-sectional area enlargement, reduced smoothelin expression, and increased VSMC proliferation is comparable in pseudopregnant and pregnant C57BL/6 mice (42). A limitation of this study is that the invasion of the trophoblasts is not as aggressive in mice as it is in humans and as consequence; SA remodeling is not fully comparable. In contrast to this particular study, other studies show that trophoblasts interact with maternal immune cells and this co-operation contributes to vascular changes. For example, the interaction of trophoblast HLA-G with uNKs impacts the maturation, proliferation, and mediator secretion of uNKs (43, 44) that in turn promote uterine vascular remodeling. Mediators of uNKs include chemokines, cytokines, proangiogenic factors, but also growth promoting factors like pleiotropin and osteoglycin. Hauk et al. reported with mouse models that the trophoblast-derived neuropeptide vasoactive intestinal peptide (VIP) is critical for MMP9 expression, migration and invasion capacities and that VIP-deficiency is associated with reduced Treg cell numbers at the feto-maternal interface (45). Further, the syncytiotrophoblast is reportedly able to communicate with maternal immune cells by secreting extracellular vesicles into the maternal circulation. These vesicles interact with monocytes, granulocytes, T cells and uNKs, influence their function, activation, and maturation. Extracellular vesicles from pre-eclamptic women influence immune cells differently when compared to extracellular vesicles from normal pregnant woman. For example, they fail to activate Tregs (46) and this might negatively influence the vascular remodeling during pregnancy.
Uterine Natural Killer Cells
In humans, CD56bright CD16− uNKs can be found in the uterus at the beginning of each menstrual cycle as small agranular cells. Within the cycle, they grow and build numerous mediator-filled granules. Two days before menstruation, apoptosis of uNKs starts. In mice there is no change in the number of CD3− CD122+ PAS+ uNKs during the murine estrous cycle, as they appear firstly after fertilization. During pregnancy in both, humans and mice, uNKs reach 70 % of all lymphocytes at the feto-maternal interface. This fact let to the assumption that they may play an important role in pregnancy. Cell numbers peak at midgestation, decline afterwards and are absent at term. In contrast to peripheral NKs (pNKs), uNKs show no cytotoxic activity and produce high amounts of cytokines, chemokines, and growth factors, for example transforming growth factor (TGF)-β, vascular endothelial growth factor (VEGF) and interferon (IFN)-γ (47, 48).
The differentiation, proliferation, activation, and survival of uNKs is regulated by sex hormones (49, 50) and the cytokine IL-15 (51–53). In addition, inhibitory or activating receptors regulate the function of uNKs. Many recent excellent works revealed the participation and importance of uNKs for the regulation of trophoblast invasion and SA remodeling (54–56). It is assumed that uNKs initiate SA remodeling before colonization by EVTs. In addition, uNKs communicate with trophoblast cells. This is possibly an explanation for the massive enrichment of uNKs near trophoblast cells. Trophoblasts express the non-classical HLA-E/G/C instead of the classical HLA-A/B as ligands for the inhibitory receptors of uNKs (57). As a result of the binding, uNKs may tolerate trophoblasts and support growth and migration of the fetal cells. uNKs may belong to the recently described innate lymphoid cell (ILC) family (58, 59) and more studies are needed to dissect which exact phenotype within this big family is pregnancy-protective. Furthermore, functional studies are needed to fully understand their participation in pregnancy-associated processes.
Mice that lack uNKs show impaired SA remodeling, characterized by thick walls and small vessel lumens, in contrast to control mice (60–63). Interestingly, many studies show that uNKs absence and associated abnormal SA remodeling does not affect progeny growth (61, 64, 65). In contrast, Fu et al. showed that the CD49a+Eomes+ subset of NK cells supports fetal growth in mice by the secretion of growth-promoting factors including pleiotrophin, osteoglycin and osteopontin. Absence of these cells resulted in fetal IUGR. Interestingly, significantly decreased percentages of CD49a+Eomes+ NKs as well as reduced levels of pleiotrophin, osteoglycin and osteopontin were found in first trimester decidua samples of patients suffering from recurrent spontaneous abortion compared to healthy donors (44).
Placental development is negatively affected in rodents that lack uNKs. An increase (61) or decrease (62, 63) in placental weight as well as markedly structural changes (65) were observed in recently published studies. Reasons for the different observations can derive from the fact that different mouse models were employed and different read outs were used as result. Ashkar et al. suggested that uNK-derived IFN-γ mainly contribute to uterine vascular modification at the feto-maternal interface in mice. The authors observed that females deficient for uNKs could initiate SA remodeling after receiving bone marrow from IFN-γ sufficient mice. Mice reconstituted with bone morrow from IFN-γ−/− mice restored uNKs, but were not sufficient to initiate vascular remodeling (66). Human uNKs are able to secrete IFN-γ as well (67). IFN-γ derived by uNKs but also macrophage-derived TNF-α or IL-1β enhance IP-10 (CXCL10) and I-TAC (CXCL11) expression of decidual cells. In turn, these chemokines recruit CXCR3-expressing uNKs. The described regulation process is of clinical relevance, as its dysregulation is used for the prediction of preeclampsia, a disease that is characterized by an incomplete SA remodeling and reduced utero-placental blood flow that is associated with maternal hypertension (68). uNK-derived IFN-γ hinders an aberrant decidual cell MMP1, 3, and 9 expression and prevents thereby the occurrence of pre-eclampsia (69).
Choudhury et al. demonstrated that EVT-conditioned medium activates endothelial cells to secrete CCL14 and CXCL6 that in turn induce the chemotaxis of uNKs and macrophages. Both cell types express receptors for the mentioned chemokines (70). Based on these facts it is tempting to speculate that the crosstalk between fetal EVTs and maternal endothelial and immune cells including uNKs, supports the SA remodeling process that in turn ensures optimal fetal development.
Macrophages
Macrophages reside within the decidua throughout pregnancy. M2 (alternatively activated)-like macrophages are more abundant than M1 (classically activated)-like macrophages in decidual tissue (71). Compared to M1, the M2 phenotype has a stronger pro-angiogenic potential due to higher expression of angiogenic factors (72, 73). In addition, human first trimester decidual macrophages express genes that are relevant for immunomodulation (74), like high levels of the anti-inflammatory IL-10 (75, 76). Additionally, it has been shown in an in vitro model of SA remodeling that human macrophages, isolated from early decidua, express genes for tissue remodeling and induce ECM breakdown of the ECM proteins laminin and fibronectin (77). Other experiments using first trimester placentation sites or 3D co-culture models proved an enrichment of macrophages in close proximity of invasive EVTs (78). Nevertheless, Lash et al. used invasion assays to show that early pregnancy macrophages do not to influence EVT invasion. Additionally, other authors utilized the chorionic plate artery (CPA) model and show that early decidual macrophages do not alter VSMC organization (77).
Apoptosis is an important process during uterine tissue remodeling and the invasion of the developing embryo during pregnancy. Clearance of the resulting apoptotic cells and cell debris presents a crucial event during uterine remodeling, as it is important to maintain tissue homeostasis and to protect the fetus (79). It has been shown that macrophages are important for the effective clearance of cell debris and apoptotic cells (77, 80) likewise trophoblasts, VSMCs, and endothelial cells. Uterine epithelial cells, that surround the blastocyst, undergo apoptosis during embryo implantation and need to be cleared by macrophages. Abrahams et al. postulated that this may explain the proximity of macrophages to EVTs at implantation sites (79).
Compared to those present at term pregnancies, decidual M2-like macrophages are reduced in preterm pregnancies and undergo an M1-like polarization during spontaneous term and preterm labor (71). Moreover, peripheral blood and placental/decidual tissue of preeclampic woman showed imbalanced IL-10 levels (81–85), that could be responsible for a defective M2 polarization of macrophages.
Regulatory T Cells
It is has been proposed by us and many other authors, Tregs are important to induce and maintain immune tolerance toward the semi-allogeneic fetus (86–89). Additionally, several studies provided an association between diminished Treg frequency or disturbed activity and pregnancy complications (90–93).
An interesting recent study from Care et al. revealed that Tregs may also influence maternal vascular function. The authors observed dysregulated hemodynamics of the uterine artery after specific Treg depletion, shown by increased resistance and pulsatility indices as well as enhanced amount of active vasoconstrictors that derived in increased mean arterial pressure (94). In another study it has been shown that an conserved non-coding sequence 1 (CNS1)-dependent mechanism of extrathymic Treg cell differentiation is important for an effective SA remodeling (95). Besides, it was shown that the adoptive transfer of Tregs could reduce uterine perfusion pressure in a rat model of PE by decreasing levels of inflammatory mediators and reactive oxygen species (96). Other studies support the positive role of Tregs for vascular remodeling, and suggest that the mechanism rely on the ability of Tregs to modulate other decidual leucocytes like mast cells (MCs). Concretely, it was found that the transfer of Tregs into abortion-prone mice normalized early pregnancy angiogenesis that was associated with promoting the expansion of uterine mast cells (uMCs) (97). Altogether, it is tempting to speculate that the participation of Tregs and probably of other adaptive cells rely on their interaction with other cell types. An interesting study from 2017 points out that CD8+ T cell reconstitution in recombinase 1-deficient mice (Rag−/−, without T and B cells) before mating abrogates the resistance differences in vasculature that normally persist postpartum (98). Thus, adaptive immune cells overtake unsuspected roles during pregnancy that go beyond their classical function. The next challenge is to understand their participation using experimental models that have translational value.
Uterine Mast Cells (uMCs)
uMCs are abundant in the reproductive tract of rodents and humans (99–101). They are subjected to the hormonal influence of progesterone and estrogen as they express hormone receptors (102). Numbers of uMCs oscillate within the cycle, being the highest at the receptive phase, and increasing during pregnancy (103).
It is well-established that MCs induce myometrium contractions that are important for the induction of birth (100, 104, 105). With the help of different MC-deficient mouse models (Kit mutation-dependent KitW−sh/W−sh and Kit-independent Cpa3-Cre as well as Mcpt5-Cre+ R-DTA mice) it has been shown in the last years that MCs are also crucial for SA remodeling and fetal well-being (60, 63, 94, 100,103, 193). Interestingly, MC-deficient and NK-deficient mice show a comparable pregnancy phenotype, namely impaired SA remodeling. In our hands, the main difference between mouse models for NK- and MC-deficiency relies on the fetal weight outcome with the progeny of NK-deficient mice having normal weight and the progeny of MC- or MC/NK-deficient mice being growth restricted (64).
Poor fetal supply, often induced by insufficient vascular remodeling, can be recognized via ultrasound by an absent or reversed end diastolic flow and high resistance index of the umbilical artery. Those signs for neonatal IUGR could be found in MC/NK-deficient mice, that were growth restricted from midgestation onwards (106, 107). Interestingly, it was demonstrated that the absence of NKs was accompanied by an increased presence of MCs and vice versa (61) leading to the hypothesis that both cell populations work together to ensure a correct vascular modification. MCs are also able to communicate and influence the behavior of other immune cells like neutrophils, DCs, monocytes, macrophages, B and T cells (108–111). Whether the interplay of MCs with the mentioned cell types is relevant for pregnancy needs to be addressed in future studies.
The impressive consequence of MC absence in mouse models raises the question about the putative mediators involved in the remodeling processes at the feto-maternal interface. MC mediators are stored in cytoplasmic granules that can be released immediately upon stimulation. It has been shown that histamine supports ovulation and blastocyst implantation (112). Also chymases seems to play an important role for processes at the feto-maternal interface. The mouse chymase mast cell protease (Mcpt) 5 is expressed by both, uMCs and uNKs (64). In vitro, Mcpt5 mediated the apoptosis of VSMCs (64), an important feature of SA remodeling. Absence of Mcpt5+ cells in Mcpt5-Cre+ R-DTA mice derived in un-remodeled SAs and growth restricted progeny. Similarly, uMCs can be found at the human feto-maternal interface (113). MC-derived α-chymase CMA1 (the human Mcpt5 homolog) stimulated ex vivo the migration of human trophoblasts, a pre-requisite for an efficient SA remodeling process (64).
Chymases are able to convert pro-peptides of MMP2 and MMP9 into their active forms (114). MMPs are important regulators of trophoblast invasion and play an important role during SA remodeling (36). Further, chymases have been shown to degrade the ECM compounds (115). An extensive degradation of ECM compounds led to a loss of matrix survival signals and in turn to VSMCs apoptosis that favor SA remodeling. Additionally, chymases can directly inhibit the growth (116) or induce apoptosis (117) of VSMCs. Chymases are key enzymes of the renin-angiotensin-aldosteron-system (RAAS) that convert angiotensin (Ang) I to AngII independent of the angiotensin converting enzyme (118). Next to the systemic RAAS there exist tissue-specific local RAASs e.g., one in the placenta and another one in the decidua. A high local AngII concentration in the maternal part of the placenta and the resulting maternal-fetal AngII gradient contribute to trophoblast migration and invasion in early pregnancy (119). Additionally, utero-placental AngII is associated with an increased migration of VSMCs (120, 121), a process that is an important step effective SA remodeling (20). Another mediator that is necessary for MC function is the glycogen-binding protein galectin-1. KitW−sh/W−sh mice have a reproductive impaired phenotype, including insufficient SA remodeling. The systemic reconstitution of mice with bone marrow-derived MCs from wildtype, but not Lgals1−/− mice could normalize the remodeling of the arteries and contribute to fetal well-being (103), demonstrating a positive role for galectin-1 for MC function. We anticipate that future studies will contribute to an increasing understanding of the role of single MC mediators for pregnancy success. It is suggested that increased numbers or exacerbated activation of MCs is associated with human pregnancy pathologies like recurrent pregnancy losses (122) and PE (123, 124). The mechanisms underlying this phenomenon are not explored. That an absence of MCs derives in pregnancy pathologies, and an increased number of MCs is associated with pregnancy pathologies again supports the concept of perfectly regulated cellular processes as modulators of pregnancy.
PE and IUGR—Adverse Consequences of Insufficient Uterine Remodeling
Insufficient remodeling of maternal uterine or vascular tissue reportedly leads to severe pregnancy disorders including miscarriage, preterm birth, PE or IUGR (125, 126). These pregnancy diseases dramatically impair maternal and fetal health or survival.
PE affects ~2–8 % of pregnancies worldwide. The multi-system disease is one of the most dangerous complication for both, mother and child with a high morbidity and mortality risk (127). Hallmarks are maternal hypertension that first manifested in pregnancy (≥140/90 mm Hg on at least 2 occasions, 6 h apart) and proteinuria (≥300 mg or greater in a 24 h urine, after 20 weeks of gestation) (128). Although the symptoms of the disease manifest in the second half of pregnancy, the pathogenesis is established during the first trimester, concretely during the time when SA remodeling takes place. Indeed, it has been shown that PE is associated with abnormal trophoblast invasion and inadequate SA remodeling during the first trimester (129, 130). This is thought to be a consequence of imbalanced immune cell numbers or activity. For example increased Th17 and impaired Treg activity, altered activation of macrophages, DCs, T cells, B cells, or NKs (131) were appointed as cause of impaired SA remodeling. To date, delivering the baby and removing of the placenta is the only effective treatment for PE. Here, a decision for the right time point of inducing the birth based on maternal health condition and fetal maturity must be taken.
Insufficient vascular remodeling can be associated with fetal IUGR (22). IUGR is defined as the failure of the neonate to reach its genetically determined growth potential and a weight under the 90th percentile compared to age-matched babies. IUGR occurs as consequence or independent of PE and results from inadequate blood supply due to impaired maternal SA remodeling (22). Besides, it can manifest due to an inadequate nutrition supply resulting from a failure of different placental transporter expression (132, 133). IUGR is associated with an increased risk of intrauterine death and a programming of diseases later in life such as hypertension, heart diseases, stroke, overweight, diabetes, metabolic syndrome or osteoporosis (134, 135).
Additionally to the participation of immune cells in SA remodeling and hence, a role of a dysregulated immune response in abnormal SA remodeling, research of the last years emphasized the negative effects of man-made environmental substances in SA remodeling and pregnancy complications (Figure 1).
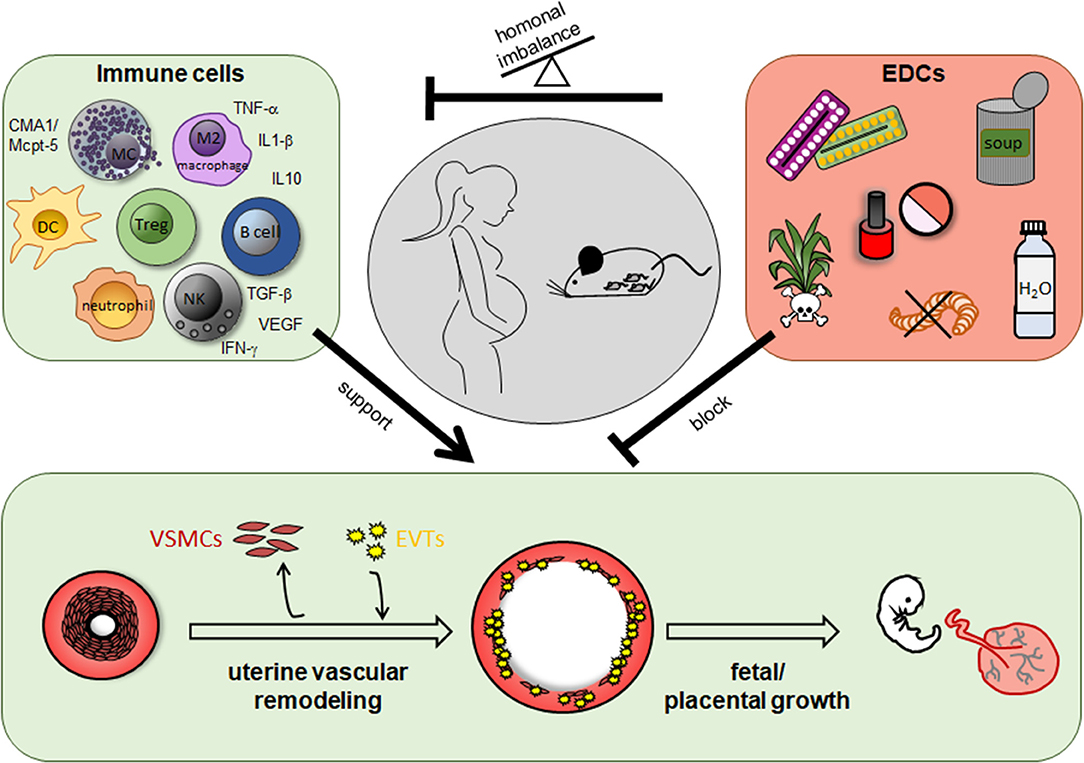
Figure 1. Role of immune cells and endocrine disrupting chemicals (EDCs) for uterine vascular remodeling during pregnancy. In humans and rodents including mice immune cells like uterine natural killer cells, mast cells, dendritic cells, T cells, especially regulatory T cells, B cells, macrophages and their secreted mediators support uterine vascular remodeling at the feto-maternal interface. EDCs are found in herbicides and pesticides as well as in many daily used products like cosmetics, plastic containers and bottles, food cans, and pharmacological drugs like contraceptive pills and many others. EDCs can impair uterine remodeling processes directly or indirectly due to a hormonal imbalance and in turn an altered immune cell number or function.
Endocrine Disrupting Chemicals
The endocrine system is a network of glands and organs that produce and secrete hormones necessary for physiological processes including respiration, metabolism, reproduction, movement, sexual development, and growth. A finely modulated hormonal balance is indispensable for many pregnancy-related processes including time-dependent orchestrating of immune cells that contribute to vascular remodeling, as described earlier in the review. Hormonal imbalance of any kind can be associated with serious pregnancy complications like miscarriage (136, 137), PE (138, 139), preterm birth (140), or IUGR (141).
A large number of man-made or natural compounds, referred as endocrine disrupting chemicals (EDC), disturbs the endocrine system by interfering with biosynthesis, secretion, transport, metabolism, binding action, or elimination of hormones (defined by US Environmental Protection Agency). EDCs are able to bind, activate or inhibit hormone receptors (142) and can change hormone receptor expression levels in tissue (143).
Synthetic EDCs represent a heterogeneous class of molecules that includes plastics like bisphenol A (BPA) and plasticizers named phthalates, substances added to plastics to make them more flexible, durable, and transparent. Also pesticides like dichlorodiphenyltrichloroethane (DDT), fungicides, pharmaceutical agents e.g., diethylstilbestrol (DES), synthetic chemicals like polychlorinated biphenyls (PCBs), polybrominated biphenyls (PBBs), or dioxins, but also heavy metals among others belong to the group of EDCs (144). Due to their widespread usage, EDCs accumulate ubiquitously in our environment and enter the human body via different routes. These routes include inhalation, dermal via direct skin contact, or oral via food or beverages but also contaminated drinking water. Several results from animal studies and human epidemiological studies implicate EDCs as a significant treat to health and thus, a huge concern to public health. Adverse human health effects include metabolic, cardiovascular and behavioral disorders, but also hormone-related cancers (145–147). Also, reproductive failure in human and wildlife (148, 149) has been observed. Especially, people who are highly exposed to chemicals, pesticides, or fungicides at work are at particularly high risk for developing endocrine or reproductive abnormalities (144).
A challenge when working with EDCs is that these substances do not follow the typical dose-response dynamics, known for most chemicals in toxicological studies. In contrast, EDCs often show low dose effects or non-monotonic dose-response curves (150), defined as a non-linear relationship between dose and effect, like U-shaped or inverted-U shaped curves. Therefore, it is difficult to predict the response of an endocrine acting substance. Several EDCs are very stable and have a long half-life. This stability is beneficial for their industrial use, but raises the harmful effects from the use of the products for the environment and human health.
BPA
Most studies that will be mentioned and discussed in the following part of this review, deal with BPA. BPA is one of the most produced and most studied EDC worldwide. The chemical is an organic compound consisting of two phenolic rings connected by a carbon carrying two methyl groups. It belongs to a subgroup of EDCs, the xenoestrogens, that exhibit estrogen-like properties by their ability to bind estrogen receptor (ER) α, ERβ, and the G-protein–coupled estrogen receptors (151–153). As many immune cells express ERs (154) they are highly receptive for the influence of xenoestrogens including BPA. Since the middle of the twentieth century, BPA is mainly used in the manufacturing of polycarbonate plastics and epoxy resins, both of which are used in a variety of daily used applications these days. For example hygiene and cleaning products, electronic devices, medical and dental devices, children toys, paints and coatings, cloth, food containers, plastic bottles, sport protection equipment, and even dental sealants. Under certain conditions BPA is released from the products. Especially high temperatures, e.g., placing plastic food containers in the microwave or the dishwasher, increase BPA release from the products (39). Also, when BPA is used as an additive e.g., for the coating of thermal paper or floor covering, BPA is not chemically bound and easily released from the products (155). BPA ending up in indoor air, dust, soil, wastewater, contact surfaces, food, and drinking water from which it enters the human body.
BPA is under scientific examination since many years. Based on the alarming findings, policies started to enforce restrictions on the manufacturing and use of BPA. Nevertheless, BPA is still present in uncountable daily used products. In 2017, BPA was identified as substances of very high concern (SVHC) under REACH (Registration, Evaluation and Authorisation of Chemicals) and is classificated today as a CMR (carcinogenic, mutagenic or toxic to reproduction) substance of category 1B, meaning BPA is a “presumed human reproductive toxicant based on animal studies.” Companies increasingly advertise for their BPA-free products (Websites European chemicals agency, Umweltbundesamt). What the consumers do not know is, that BPA is replaced by other bisphenols like bisphenol F or S that show comparable or even worse harmful developmental effects and are able to cross the placenta (156, 157). It was recently shown that the exposure of the population to BPA substitutes is almost ubiquitous (158).
Although the numbers of studies is increasing in the last years, the underlying mechanisms as to how exactly EDCs impair health are far from being understood and further research is urgently needed. Next, we will review recent findings concerning the influence of EDCs on reproductive parameters with special emphasis to the question if EDCs are able to influence vascular remodeling, placentation, or immune cell function.
Effects of EDCs on Uterine Remodeling and Immune Cells
Exposure to EDCs in adults can have adverse health effects. During early development, when hormones play an important role, organisms are particularly sensitive to EDC exposure. Several EDCs including BPA are able to pass the placenta barrier and accumulate in placental tissue (159). Next to placental tissue, EDCs have been detected in the urine, cord blood, plasma, amniotic fluid, and breast milk of pregnant women and their developing fetuses (158, 160, 161). This represents a health problem for the mother and in more extent for the unborn, as many enzymes important for degrading dangerous substances are not expressed until birth.
EDCs and Immune Cells
As discussed earlier in this review, a proper development and function of immune cells is indispensable for sufficient uterine remodeling processes during pregnancy. Based on that, we will give an insight into the current findings of the diverse influences of EDCs on immune cells in the following. We will mainly focus on the effects of immune cell phenotypes and functions, important for a successful pregnancy.
In mice, it has been shown that BPA exposure lead to phenotypic changes within distinct immune cell populations (162). Depending on the concentration and the route of administration, BPA decrease TNF-α and nitric oxide secretion of activated macrophages (163–165). Interestingly, BPA did not influence macrophage viability, but decreased the adherence ability of rat peritoneal macrophages (166). Taking into account that adhesion is the first step in the macrophages phagocytic process and that phagocytosis is important for the uterine remodeling process during pregnancy, as mentioned earlier in this review, BPA could in theory inhibit the remodeling process indirectly via macrophage inhibition.
Exposure of mice to BPA via drinking water for 4 weeks induced the production of the Th1 type cytokine IFN-γ and suppressed Th2 type IL-4 expression of CD4+ T cells (167). In contrast another study show an increase of Th2 polarization by an enhanced IL-4 production in antigen-activated T lymphocytes due to BPA exposure (168, 169). Further, Yoshino et al. demonstrated that prenatal fetal exposure to BPA upregulated Th1 but also Th2 responses in adulthood after immunization with hen egg lysozyme. This may explain the dramatic increase in allergic diseases over the last decades. Additionally, male but also female mice prenatally exposed to BPA had markedly increased numbers of splenic CD3+CD4+ and CD3+CD8+ cells (155). Number of Tregs was reduced in mice exposed to BPA either prenatally or in adulthood (169, 170). As discussed earlier, an imbalanced amount of immune cells can cause serious health consequences. Besides, also an imbalanced activity of immune cells due to decreased or increased receptor expression can have detrimental consequences for an organism. A study from 2012 point out that BPA, among other bisphenols, up-regulated HLA-class II, CD11c and CD86 in mouse bone-marrow-derived DCs (171). Further, an increase antibody production by B cells in mice was observed after BPA and DES exposure in mice (172, 173). O'Brien et al. show that the short-term exposure of both, BPA but also estradiol, at levels relevant to human exposure, enhances histamine release by primary bone marrow-derived MCs of mice. This constitutes yet another explanation for the increasing allergy prevalence. BPA additionally enhanced the release of cysteinyl leukotrienes. This was not ER-mediated (174). In contrast, the structure of the specific paraben, used as preservatives in cosmetic, medicines and food, determines if the EDC enhance, inhibit, or do not have any influence on the histamine release of rat peritoneal MCs (175). During the embryo stage of zebrafish, the EDCs 17β-estradiol, 17-α-ethynyestradiol (EE2), permethrin, atrazine, and nonylphenol significantly change innate immune-related gene transcription. This was shown by altered mRNA levels of TNF-α, IFN-γ, IL-1β, IL-8, CXCL-Clc, and CC-chemokine but also genes related to reactive oxygen species (176).
Synthetic EDCs also affect the development and the function of human immune cell populations (177). BPA exposure in human significantly increased in the proliferation of PBMC and modulated their cytokine production leading to a decrease in IL-10 and IL-13 expression. Additionally, BPA altered the phenotype of myeloid DCs by an increased CD1a, but decreased HLA-DR and CD86 expression (178). In contrast, it has been shown that BPA among other bisphenols decreased the expression of CD1a, CD80, CD86, and CD83 but increased the numbers of HLA-DR positive monocyte-derived DCs (179). In response to phthalates and the common EDCs nonylphenol and 4-octylphenol there was a modulation of DC cytokine expression (171, 180). Both of the latter EDCs but also BPA as well as bisphenol B and F interfere additionally with the differentiation of DCs (171).
Di-ethylhexyl-phthalate, dibutyl-phthalate, 4-tert-octylphenol, and BPA interfere with the TNF-α, IL-1β, and IL-8 cytokine secretion (181) and markedly reduce the phagocytosis ability of a human macrophage cell line (182) in an estrogen-mediated manner (181).
Contradictory results of studies may be explained the usage of very different concentrations or different administration routes or time points of the EDCs. Pharmacokinetic studies showed that the route of administration strongly influence the rate of metabolism of EDCs including BPA (183, 184) that in turn determine their concentrations in blood and tissues. For planning animal studies it should be kept in mind, that human beings are mainly exposed orally to BPA.
EDCs and Uterine Remodeling During Pregnancy
A correct placentation is important to maintain maternal and fetal health not only throughout pregnancy, but also impacts the rest of their life. If EDCs interfere with the highly complex mechanisms of placental development or function and thereby impact pregnancy outcome, is under research.
In women that undergo in vitro fertilization (IVF), there is an inverse relationship between BPA urinary concentrations and estradiol levels as well as the total number of oocytes retrieved per cycle (185, 186). In addition, high urinary concentrations of BPA, parabens, and most phthalate metabolites were associated with lower probabilities of implantation, clinical pregnancy, and live birth after IVF (187). Phthalate exposure impacts human placental function by significantly modulating the expression of 93 critical placental genes like the epidermal growth factor receptor through methylation (188). Another study showed that high urinary concentrations of specific phthalate metabolites (mono(2-ethylhexyl) phthalate [MEHP], mono(2-ethyl-5-oxohexyl) phthalate [MEOHP], mono-n-butyl phthalate [MnBP), monoisobutyl phthalate [MiBP], and monobenzyl phthalate [MBzP]) were associated with a lower expression of the target genes reflecting trophoblast differentiation (189) that are in turn important for an efficient vascular remodeling during pregnancy. Moreover, pesticides disturb the hormonal network and the function of trophoblast cells as shown by reduced cell viability and altered hormone secretion and steroidogenesis gene expression in an ER-dependent manner (190).
Although there are many interesting studies about the effect of EDCs for human pregnancy, it is difficult to extrapolate the effect of single EDCs, as human are exposed to many EDCs at the same time. Studies using combined EDC exposure are needed in the future to fully understand the consequences on health.
In mice, short-term oral BPA-exposure (50 μg/kg bw/day) during early pregnancy provoked IUGR in more than half of the offspring from gd12 onwards as documented by ultrasound and fetal weight determination. Although velocity parameters of the uterine artery were normal, SA remodeling was impaired in BPA-exposed mice. This was shown by the fact that many VSMCs remain in the vessels walls in BPA-treated mice. Additionally, the SAs had increased wall thicknesses and increased wall-to-lumen ratios compared to control mice, both signs for insufficient remodeled SAs (191). Similar results were obtained by Ye et al. The authors demonstrated an abnormal vessel remodeling shown by increased retention of VSMCs and reduced vessel areas at the junctional zone of the placenta leading to PE-like features including hypertension. Additionally, they found a decreased expression of MMP2 and 9and an enhanced expression of TIMP1 and−2 in BPA-treated mice that might be the reason for an impaired invasion of the trophoblast cells leading to insufficient remodeled vessels (192). Indeed, in vitro it was shown that BPA impairs trophoblast invasion (192). In these mouse studies it was not clarified if BPA influenced other cells next to trophoblasts. Whereas, uNK and uMCs numbers were not affected (191), a possible influence of the EDC on the activity of the immune cells was not analyzed and is an interesting question for further research. Morphological changes in the placentas of BPA-treated mice, including a smaller labyrinthine zone, narrow intervillous spaces, and degenerative changes in the trophoblastic giant cells and spongiotrophoblast layers were reported. This was associated with a decreased number and weight of embryos (193).
Next to BPA, other EDCs have been analyzed and the results obtained strongly suggest a disturbance of uterine vascular remodeling by EDCs. PCB-exposure of the mink let to degenerated placentas, characterized by vascular lesions in the labyrinthine zones, degeneration of endothelial and trophoblast cells, and hemorrhage. This was associated with fetal growth restriction and death (194). EE2, that is massively used as a compound in contraceptive pills, accumulate in the environment, open waters and enters the human food chain. A recent study shows that oral application of EE2 in mice during early pregnancy leads to reproductive impairments. In a high concentration (5 μg/kg bodyweight (bw)/day) EE2 leads to death of all fetuses in 80 % of the animals at midpregnancy. In contrast, a lower concentration (5 ng/kg bw/day) did not affect fetal survival but was clearly associated with impaired SA remodeling and abnormal increased fetal and placental growth (195). Thus, the impact to fetal and placental growth is different depending on the nature of the EDC.
As earlier mentioned in this review, most EDCs follow non-monotonic dose-response curves. This fact makes it difficult to draw conclusions from EDCs research results. Nevertheless, there is evidence that EDCs do not only exert a direct negative effect on vascular remodeling during pregnancy, but also an indirect effect by their ability to alter immune cell function. Many effects followed by EDC exposure, especially BPA, were not only dose-, but also sex-specific (196). The interesting field of phenotypic gender differences by EDC exposure is still underrepresented and opens potentially new interesting research fields. Also, the epigenetic effect of EDCs is an interesting topic for more detailed research and is not contemplated in this review.
Taken together, the results of numerous research studies clearly demonstrate that EDCs greatly impact the immune system and uterine remodeling during pregnancy that may have harmful effects for the mother and even more for the developing fetus. It is important to mention that most studies investigate the health effects of single EDCs exposure, even though exposures do occur as chemical mixtures of EDCs that most probably impact health quite different and unpredictable. Avoiding unnecessary EDCs-containing products, especially during the critical time of fetal development during pregnancy, may help avoiding harmful health outcomes.
Conclusions
The present review aimed to summarize long-term established and brand-new research results concerning the indispensable role of innate immune cells for an effective uterine remodeling process. Additionally, the manuscript visualizes serious short- and long-term health consequences for mothers and children followed by an ineffective uterine remodeling process. An advanced understanding of mechanisms and disruptors of uterine remodeling helps to identify factors involved in physiological or pathological pregnancies. The long-term goal of research is to develop novel therapeutic options against pregnancy disorders, to elucidate and reduce potential negative effects on unborn life, and to improve in-vitro-fertilization techniques.
Moreover, the review focused on the reproductive health consequences of EDCs that accumulate non-stop in our environment and are impossible to eliminate or neutralize. We are convinced that research has an impact on the understanding of diseases and the potential to improve health care worldwide.
Author Contributions
NM reviewed the literature and wrote the manuscript. AZ reviewed the manuscript.
Funding
The work was funded by grants from the Deutsche Forschungsgemeinschaft (DFG) to NM ME 5360/1-1.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Calkins K, Devaskar SU. Fetal origins of adult disease. Curr Probl Pediatr Adolesc Health Care. (2011) 41:158–76. doi: 10.1016/j.cppeds.2011.01.001
2. Cartwright JE, Fraser R, Leslie K, Wallace AE, James JL. Remodelling at the maternal-fetal interface: relevance to human pregnancy disorders. Reproduction. (2010) 140:803–13. doi: 10.1530/REP-10-0294
3. Maybin JA, Critchley HO. Menstrual physiology: implications for endometrial pathology and beyond. Hum Reprod Update. (2015) 21:748–61. doi: 10.1093/humupd/dmv038
4. Zenclussen AC, Hammerling GJ. Cellular regulation of the uterine microenvironment that enables embryo implantation. Front Immunol. (2015) 6:321. doi: 10.3389/fimmu.2015.00321
5. Grzechocinska B, Dabrowski F, Cyganek A, Panek G, Wielgos M. The role of metalloproteinases in endometrial remodelling during menstrual cycle. Ginekol Pol. (2017) 88:337–42. doi: 10.5603/GP.a2017.0063
7. Berbic M, Fraser IS. Immunology of normal and abnormal menstruation. Womens Health. Springer Verlag (2013) 9:387–95. doi: 10.2217/WHE.13.32
8. Jones RL, Hannan NJ, Kaitu'u TJ, Zhang J, Salamonsen LA. Identification of chemokines important for leukocyte recruitment to the human endometrium at the times of embryo implantation and menstruation. J Clin Endocrinol Metab. (2004) 89:6155–67. doi: 10.1210/jc.2004-0507
9. Berbic M, Ng CH, Fraser IS. Inflammation and endometrial bleeding. Climacteric. (2014) 17(Suppl.2):47–53. doi: 10.3109/13697137.2014.963964
10. Jabbour HN, Kelly RW, Fraser HM, Critchley HO. Endocrine regulation of menstruation. Endocr Rev. (2006) 27:17–46. doi: 10.1210/er.2004-0021
11. Osol G, Mandala M. Maternal uterine vascular remodeling during pregnancy. Physiology. (2009) 24:58–71. doi: 10.1152/physiol.00033.2008
12. Boss AL, Chamley LW, James JL. Placental formation in early pregnancy: how is the centre of the placenta made?. Hum Reprod Update. (2018) 24:750–60. doi: 10.1093/humupd/dmy030
13. Burke SD, Barrette VF, Gravel J, Carter AL, Hatta J, Zhang Z, et al. Uterine NK cells, spiral artery modification and the regulation of blood pressure during mouse pregnancy. Am J Reprod Immunol. (2010) 63:472–81. doi: 10.1111/j.1600-0897.2010.00818.x
14. Whitley GS, Cartwright JE. Cellular and molecular regulation of spiral artery remodelling: lessons from the cardiovascular field. Placenta. (2010) 31:465–74. doi: 10.1016/j.placenta.2010.03.002
15. Cecchettini A, Rocchiccioli S, Boccardi C, Citti L. Vascular smooth-muscle-cell activation: proteomics point of view. Int Rev Cell Mol Biol. (2011) 288:43–99. doi: 10.1016/B978-0-12-386041-5.00002-9
16. Wanjare M, Kuo F, Gerecht S. Derivation and maturation of synthetic and contractile vascular smooth muscle cells from human pluripotent stem cells. Cardiovasc Res. (2013) 97:321–30. doi: 10.1093/cvr/cvs315
17. Lacolley P, Regnault V, Nicoletti A, Li Z, Michel JB. The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc Res. (2012) 95:194–204. doi: 10.1093/cvr/cvs135
18. Rensen SS, Doevendans PA, van Eys GJ. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth Heart J. (2007) 15:100–8. doi: 10.1007/BF03085963
19. Louis SF, Zahradka P. Vascular smooth muscle cell motility: from migration to invasion. Exp Clin Cardiol. (2010) 15:e75–85.
20. Bulmer JN, Innes BA, Levey J, Robson SC, Lash GE. The role of vascular smooth muscle cell apoptosis and migration during uterine spiral artery remodeling in normal human pregnancy. FASEB J. (2012) 26:2975–85. doi: 10.1096/fj.12-203679
21. Liu W, Liu X, Luo M, Liu X, Luo Q, Tao H, et al. dNK derived IFN-gamma mediates VSMC migration and apoptosis via the induction of LncRNA MEG3: a role in utero-vascular transformation. Placenta. (2017) 50:32–9. doi: 10.1016/j.placenta.2016.12.023
22. Kaufmann P, Black S, Huppertz B. Endovascular trophoblast invasion: implications for the pathogenesis of intrauterine growth retardation and preeclampsia. Biol Reprod. (2003) 69:1–7. doi: 10.1095/biolreprod.102.014977
23. Whitley GS, Cartwright JE. Trophoblast-mediated spiral artery remodelling: a role for apoptosis. J Anat. (2009) 215:21–6. doi: 10.1111/j.1469-7580.2008.01039.x
24. Gellersen B, Brosens JJ. Cyclic decidualization of the human endometrium in reproductive health and failure. Endocr Rev. (2014) 35:851–905. doi: 10.1210/er.2014-1045
25. Vinketova K, Mourdjeva M, Oreshkova T. Human decidual stromal cells as a component of the implantation niche and a modulator of maternal immunity. J Pregnancy. (2016) 2016:8689436. doi: 10.1155/2016/8689436
26. Pollard JW, Lin EY, Zhu L. Complexity in uterine macrophage responses to cytokines in mice. Biol Reprod. (1998) 58:1469–75. doi: 10.1095/biolreprod58.6.1469
27. Huang SJ, Schatz F, Masch R, Rahman M, Buchwalder L, Niven-Fairchild T, et al. Regulation of chemokine production in response to pro-inflammatory cytokines in first trimester decidual cells. J Reprod Immunol. (2006) 72:60–73. doi: 10.1016/j.jri.2006.03.002
28. Sentman CL, Meadows SK, Wira CR, Eriksson M. Recruitment of uterine NK cells: induction of CXC chemokine ligands 10 and 11 in human endometrium by estradiol and progesterone. J Immunol. (2004) 173:6760–6. doi: 10.4049/jimmunol.173.11.6760
29. Kitaya K, Yamaguchi T, Honjo H. Central role of interleukin-15 in postovulatory recruitment of peripheral blood CD16(-) natural killer cells into human endometrium. J Clin Endocrinol Metab. (2005) 90:2932–40. doi: 10.1210/jc.2004-2447
30. Carlino C, Stabile H, Morrone S, Bulla R, Soriani A, Agostinis C, et al. Recruitment of circulating NK cells through decidual tissues: a possible mechanism controlling NK cell accumulation in the uterus during early pregnancy. Blood. (2008) 111:3108–15. doi: 10.1182/blood-2007-08-105965
31. Magatti M, Stefani FR, Papait A, Cargnoni A, Masserdotti A, Silini AR, et al. Perinatal mesenchymal stromal cells and their possible contribution to fetal-maternal tolerance. Cells. (2019) 8:1401. doi: 10.3390/cells8111401
32. Klemmt PA, Carver JG, Kennedy SH, Koninckx P, Mardon HJ. Stromal cells from endometriotic lesions and endometrium from women with endometriosis have reduced decidualization capacity. Fertil Steril. (2006) 85:564–72. doi: 10.1016/j.fertnstert.2005.08.046
33. Giuliani E, Parkin KL, Lessey BA, Young S, Fazleabas AT. Characterization of uterine NK cells in women with infertility or recurrent pregnancy loss and associated endometriosis. Am J Reprod Immunol. (2014) 72:262–9. doi: 10.1111/aji.12259
34. Salker M, Teklenburg G, Molokhia M, Lavery S, Trew G, Aojanepong T, et al. Natural selection of human embryos: impaired decidualization of endometrium disables embryo-maternal interactions and causes recurrent pregnancy loss. PLoS ONE. (2010) 5:e10287. doi: 10.1371/journal.pone.0010287
35. Kuo CY, Shevchuk M, Opfermann J, Guo T, Santoro M, Fisher J, et al. Trophoblast-endothelium signaling involves angiogenesis and apoptosis in a dynamic bioprinted placenta model. Biotechnol Bioeng. (2019) 116:181–92. doi: 10.1002/bit.26850
36. Harris LK. Review: trophoblast-vascular cell interactions in early pregnancy: how to remodel a vessel. Placenta. (2010) 31(Suppl.):S93–8. doi: 10.1016/j.placenta.2009.12.012
37. Keogh RJ, Harris LK, Freeman A, Baker PN, Aplin JD, Whitley GS, et al. Fetal-derived trophoblast use the apoptotic cytokine tumor necrosis factor-α-related apoptosis-inducing ligand to induce smooth muscle cell death. Circ Res. (2007) 100:834–41. doi: 10.1161/01.RES.0000261352.81736.37
38. Harris LK, Keogh RJ, Wareing M, Baker PN, Cartwright JE, Aplin JD, et al. Invasive trophoblasts stimulate vascular smooth muscle cell apoptosis by a fas ligand-dependent mechanism. Am J Pathol. (2006) 169:1863–74. doi: 10.2353/ajpath.2006.060265
39. Ashton SV, Whitley GS, Dash PR, Wareing M, Crocker IP, Baker PN, et al. Uterine spiral artery remodeling involves endothelial apoptosis induced by extravillous trophoblasts through Fas/FasL interactions. Arterioscler Thromb Vasc Biol. (2005) 25:102–8. doi: 10.1161/01.ATV.0000148547.70187.89
40. James JL, Whitley GS, Cartwright JE. Pre-eclampsia: fitting together the placental, immune and cardiovascular pieces. J Pathol. (2010) 221:363–78. doi: 10.1002/path.2719
41. Khong TY, De Wolf F, Robertson WB, Brosens I. Inadequate maternal vascular response to placentation in pregnancies complicated by pre-eclampsia and by small-for-gestational age infants. Br J Obstet Gynaecol. (1986) 93:1049–59. doi: 10.1111/j.1471-0528.1986.tb07830.x
42. van der Heijden OW, Essers YP, Spaanderman ME, De Mey J, van Eys GJ, Peeters LL. Uterine artery remodeling in pseudopregnancy is comparable to that in early pregnancy. Biol Reprod. (2005) 73:1289–93. doi: 10.1095/biolreprod.105.044438
43. Stieglitz F, Celik AA, von Kaisenberg C, Camps MA, Blasczyk R, Bade-Doding C. The microstructure in the placenta is influenced by the functional diversity of HLA-G allelic variants. Immunogenetics. (2019) 71:455–63. doi: 10.1007/s00251-019-01121-0
44. Fu B, Zhou Y, Ni X, Tong X, Xu X, Dong Z, et al. Natural Killer cells promote fetal development through the secretion of growth-promoting factors. Immunity. (2017) 47:1100–13.e6. doi: 10.1016/j.immuni.2017.11.018
45. Hauk V, Vota D, Gallino L, Calo G, Paparini D, Merech F, et al. Trophoblast VIP deficiency entails immune homeostasis loss and adverse pregnancy outcome in mice. FASEB J. (2019) 33:1801–10. doi: 10.1096/fj.201800592RR
46. Gohner C, Plosch T, Faas MM. Immune-modulatory effects of syncytiotrophoblast extracellular vesicles in pregnancy and preeclampsia. Placenta. (2017) 60(Suppl. 1):S41–51. doi: 10.1016/j.placenta.2017.06.004
47. Croy BA, van den Heuvel MJ, Borzychowski AM, Tayade C. Uterine natural killer cells: a specialized differentiation regulated by ovarian hormones. Immunol Rev. (2006) 214:161–85. doi: 10.1111/j.1600-065X.2006.00447.x
48. Vacca P, Moretta L, Moretta A, Mingari MC. Origin, phenotype and function of human natural killer cells in pregnancy. Trends Immunol. (2011) 32:517–23. doi: 10.1016/j.it.2011.06.013
49. Beagley KW, Gockel CM. Regulation of innate and adaptive immunity by the female sex hormones oestradiol and progesterone. FEMS Immunol Med Microbiol. (2003) 38:13–22. doi: 10.1016/S0928-8244(03)00202-5
50. Gibson DA, Greaves E, Critchley HO, Saunders PT. Estrogen-dependent regulation of human uterine natural killer cells promotes vascular remodelling via secretion of CCL2. Hum Reprod. (2015) 30:1290–301. doi: 10.1093/humrep/dev067
51. Ledee-Bataille N, Bonnet-Chea K, Hosny G, Dubanchet S, Frydman R, Chaouat G. Role of the endometrial tripod interleukin-18,−15, and−12 in inadequate uterine receptivity in patients with a history of repeated in vitro fertilization-embryo transfer failure. Fertil Steril. (2005) 83:598–605. doi: 10.1016/j.fertnstert.2004.11.021
52. Cooper MA, Bush JE, Fehniger TA, VanDeusen J, Waite RE, Liu Y, et al. In vivo evidence for a dependence on interleukin 15 for survival of natural killer cells. Blood. (2002) 100:3633–8. doi: 10.1182/blood-2001-12-0293
53. Beuneu H, Deguine J, Breart B, Mandelboim O, Di Santo JP, et al. Dynamic behavior of NK cells during activation in lymph nodes. Blood. (2009) 114:3227–34. doi: 10.1182/blood-2009-06-228759
54. Croy BA, He H, Esadeg S, Wei Q, McCartney D, Zhang J, et al. Uterine natural killer cells: insights into their cellular and molecular biology from mouse modelling. Reproduction. (2003) 126:149–60. doi: 10.1530/rep.0.1260149
55. Croy BA, Chen Z, Hofmann AP, Lord EM, Sedlacek AL, Gerber SA. Imaging of vascular development in early mouse decidua and its association with leukocytes and trophoblasts. Biol Reprod. (2012) 87:125. doi: 10.1095/biolreprod.112.102830
56. Ratsep MT, Felker AM, Kay VR, Tolusso L, Hofmann AP, Croy. BA. Uterine natural killer cells: supervisors of vasculature construction in early decidua basalis. Reproduction. (2015) 149:R91–102. doi: 10.1530/REP-14-0271
57. King A, Hiby SE, Gardner L, Joseph S, Bowen J, Verma S, et al. Recognition of trophoblast HLA class I molecules by decidual NK cell receptors–a review. Placenta. (2000) 21(Suppl.A):S81–5. doi: 10.1053/plac.1999.0520
58. Sojka DK, Yang L, Yokoyama WM. Uterine natural killer cells. Front Immunol. (2019) 10:960. doi: 10.3389/fimmu.2019.00960
59. Vacca P, Chiossone L, Mingari MC, Moretta L. Heterogeneity of NK cells and other innate lymphoid cells in human and murine decidua. Front Immunol. (2019) 10:170. doi: 10.3389/fimmu.2019.00170
60. Ashkar AA, Black GP, Wei Q, He H, Liang L, Head JR, Croy BA. Assessment of requirements for IL-15 and IFN regulatory factors in uterine NK cell differentiation and function during pregnancy. J Immunol. (2003) 171:2937–44. doi: 10.4049/jimmunol.171.6.2937
61. Meyer N, Woidacki K, Maurer M, Zenclussen AC. Safeguarding of fetal growth by mast cells and natural killer cells: deficiency of one is counterbalanced by the other. Front Immunol. (2017) 8:711. doi: 10.3389/fimmu.2017.00711
62. Greenwood JD, Minhas K, di Santo JP, Makita M, Kiso Y, Croy BA. Ultrastructural studies of implantation sites from mice deficient in uterine natural killer cells. Placenta. (2000) 21:693–702. doi: 10.1053/plac.2000.0556
63. Guimond MJ, Wang B, Croy BA. Engraftment of bone marrow from severe combined immunodeficient (SCID) mice reverses the reproductive deficits in natural killer cell-deficient tg epsilon 26 mice. J Exp Med. (1998) 187:217–23. doi: 10.1084/jem.187.2.217
64. Meyer N, Woidacki K, Knofler M, Meinhardt G, Nowak D, Velicky P, et al. Chymase-producing cells of the innate immune system are required for decidual vascular remodeling and fetal growth. Sci Rep. (2017) 7:45106. doi: 10.1038/srep45106
65. Renaud SJ, Scott RL, Chakraborty D, Rumi MA, Soares MJ. Natural Killer-cell deficiency alters placental development in rats. Biol Reprod. (2017) 96:145–58. doi: 10.1095/biolreprod.116.142752
66. Ashkar AA, Di Santo JP, Croy BA. Interferon gamma contributes to initiation of uterine vascular modification, decidual integrity, and uterine natural killer cell maturation during normal murine pregnancy. J Exp Med. (2000) 192:259–70. doi: 10.1084/jem.192.2.259
67. Chen CP, Piao L, Chen X, Yu J, Masch R, Schatz F, et al. Expression of interferon gamma by decidual cells and natural killer cells at the human implantation site: implications for preeclampsia, spontaneous abortion, and intrauterine growth restriction. Reprod Sci. (2015) 22:1461–7. doi: 10.1177/1933719115585148
68. Lockwood CJ, Huang SJ, Chen CP, Huang Y, Xu S, Faramarzi O, et al. Decidual cell regulation of natural killer cell-recruiting chemokines: implications for the pathogenesis and prediction of preeclampsia. Am J Pathol. (2013) 183:841–56. doi: 10.1016/j.ajpath.2013.05.029
69. Lockwood CJ, Basar M, Kayisli UA, Guzeloglu-Kayisli O, Murk W, Wang N, et al. Interferon-gamma protects first-trimester decidual cells against aberrant matrix metalloproteinases 1. 3, and 9 expression in preeclampsia. Am J Pathol. (2014) 184:2549–59. doi: 10.1016/j.ajpath.2014.05.025
70. Choudhury RH, Dunk CE, Lye SJ, Aplin J, Harris LK, Jones RL. Extravillous trophoblast and endothelial cell crosstalk mediates leukocyte infiltration to the early remodeling decidual spiral arteriole wall. J Immunol. (2017) 198:4115–28. doi: 10.4049/jimmunol.1601175
71. Xu Y, Romero R, Miller D, Kadam L, Mial TN, Plazyo O, et al. An M1-like macrophage polarization in decidual tissue during spontaneous preterm labor that is attenuated by rosiglitazone treatment. J Immunol. (2016) 196:2476–91. doi: 10.4049/jimmunol.1502055
72. Zhao H, Kalish FS, Wong RJ, Stevenson DK. Hypoxia regulates placental angiogenesis via alternatively activated macrophages. Am J Reprod Immunol. (2018) 80:e12989. doi: 10.1111/aji.12989
73. Jetten N, Verbruggen S, Gijbels MJ, Post MJ, De Winther MP, Donners MM. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis. (2014) 17:109–18. doi: 10.1007/s10456-013-9381-6
74. Gustafsson C, Mjosberg J, Matussek A, Geffers R, Matthiesen L, Berg G, et al. Gene expression profiling of human decidual macrophages: evidence for immunosuppressive phenotype. PLoS ONE. (2008) 3:e2078. doi: 10.1371/journal.pone.0002078
75. Heikkinen J, Mottonen M, Komi J, Alanen A, Lassila O. Phenotypic characterization of human decidual macrophages. Clin Exp Immunol. (2003) 131:498–505. doi: 10.1046/j.1365-2249.2003.02092.x
76. Svensson J, Jenmalm MC, Matussek A, Geffers R, Berg G, Ernerudh J. Macrophages at the fetal-maternal interface express markers of alternative activation and are induced by M-CSF and IL-10. J Immunol. (2011) 187:3671–82. doi: 10.4049/jimmunol.1100130
77. Lash GE, Pitman H, Morgan HL, Innes BA, Agwu CN, Bulmer JN. Decidual macrophages: key regulators of vascular remodeling in human pregnancy. J Leukoc Biol. (2016) 100:315–25. doi: 10.1189/jlb.1A0815-351R
78. Helige C, Ahammer H, Moser G, Hammer A, Dohr G, Huppertz B, et al. Distribution of decidual natural killer cells and macrophages in the neighbourhood of the trophoblast invasion front: a quantitative evaluation. Hum Reprod. (2014) 29:8–17. doi: 10.1093/humrep/det353
79. Abrahams VM, Kim YM, Straszewski SL, Romero R, Mor G. Macrophages and apoptotic cell clearance during pregnancy. Am J Reprod Immunol. (2004) 51:275–82. doi: 10.1111/j.1600-0897.2004.00156.x
80. Sonora C, Mourglia-Ettlin G, Calo G, Hauk V, Ramhorst R, Hernandez A, et al. Anti-tissue transglutaminase antibody inhibits apoptotic cell clearance by macrophages in pregnant NOD mice. J Reprod Immunol. (2014) 103:59–66. doi: 10.1016/j.jri.2013.11.001
81. Madazli R, Aydin S, Uludag S, Vildan O, Tolun N. Maternal plasma levels of cytokines in normal and preeclamptic pregnancies and their relationship with diastolic blood pressure and fibronectin levels. Acta Obstet Gynecol Scand. (2003) 82:797–802. doi: 10.1034/j.1600-0412.2003.00206.x
82. Schonkeren D, van der Hoorn ML, Khedoe P, Swings G, van Beelen E, Claas F, et al. Differential distribution and phenotype of decidual macrophages in preeclamptic versus control pregnancies. Am J Pathol. (2011) 178:709–17. doi: 10.1016/j.ajpath.2010.10.011
83. Sharma A, Satyam A, Sharma JB. Leptin, IL-10 and inflammatory markers (TNF-α, IL-6, and IL-8) in pre-eclamptic, normotensive pregnant and healthy non-pregnant women. Am J Reprod Immunol. (2007) 58:21–30. doi: 10.1111/j.1600-0897.2007.00486.x
84. Zak P, Soucek M. Correlation of tumor necrosis factor alpha, interleukin 6 and interleukin 10 with blood pressure, risk of preeclampsia and low birth weight in gestational diabetes. Physiol Res. (2019) 68:395–408. doi: 10.33549/physiolres.934002
85. Hennessy A, Pilmore HL, Simmons LA, Painter DM. A deficiency of placental IL-10 in preeclampsia. J Immunol. (1999) 163:3491–5.
86. Zenclussen AC, Gerlof K, Zenclussen ML, Sollwedel A, Bertoja AZ, Ritter T, et al. Abnormal T-Cell reactivity against paternal antigens in spontaneous abortion. Am J Pathol. (2005). 166:811–22. doi: 10.1016/S0002-9440(10)62302-4
87. Svensson M, Maroof A, Ato M, Kaye PM. Stromal cells direct local differentation of regulatory dendritic cells. Immunity. (2004). 21:805–16. doi: 10.1016/j.immuni.2004.10.012
88. Schumacher A, Zenclussen AC. Regulatory T cells: regulators of life. Am J Reprod Immunol. (2014) 72:158–70. doi: 10.1111/aji.12238
89. Svensson-Arvelund J, Mehta RB, Lindau R, Mirrasekhian E, Rodriguez-Martinez H, Berg GE, et al. The human fetal placenta promotes tolerance against the semiallogeneic fetus by inducing regulatory T cells and homeostatic M2 macrophages. J Immunol. (2015) 194:1534–44. doi: 10.4049/jimmunol.1401536
90. Boij R, Mjosberg J, Svensson-Arvelund J, Hjorth M, Berg G, Matthiesen L, et al. Regulatory T-cell subpopulations in severe or early-onset preeclampsia. Am J Reprod Immunol. (2015) 74:368–78. doi: 10.1111/aji.12410
91. Sasaki Y, Sakai M, Miyazaki S, Higuma S, Shiozaki A, Saito S. Decidual and peripheral blood CD4+CD25+ regulatory T cells in early pregnancy subjects and spontaneous abortion cases. Mol Hum Reprod. (2004) 10:347–53. doi: 10.1093/molehr/gah044
92. Zare M, Namavar Jahromi B, Gharesi-Fard B. Analysis of the frequencies and functions of CD4(+)CD25(+)CD127(low/neg), CD4(+)HLA-G(+), and CD8(+)HLA-G(+) regulatory T cells in pre-eclampsia. J Reprod Immunol. (2019) 133:43–51. doi: 10.1016/j.jri.2019.06.002
93. Hafeez NA, Fouda Mel T, Abdel Gawad ER, Assar T, Mansour AI. The role of regulatory T cells in preeclampsia. Egypt J Immunol. (2014) 21:45–55.
94. Care AS, Bourque SL, Morton JS, Hjartarson E, Robertson SA, Davidge ST. Reduction in regulatory T cells in early pregnancy causes uterine artery dysfunction in mice. Hypertension. (2018) 72:177–87. doi: 10.1161/HYPERTENSIONAHA.118.10858
95. Samstein RM, Josefowicz SZ, Arvey A, Treuting PM, Rudensky AY. Extrathymic generation of regulatory T cells in placental mammals mitigates maternal-fetal conflict. Cell. (2012) 150:29–38. doi: 10.1016/j.cell.2012.05.031
96. Cornelius DC, Amaral LM, Harmon A, Wallace K, Thomas A, Campbell N, et al. An increased population of regulatory T cells improves the pathophysiology of placental ischemia in a rat model of preeclampsia. Am J Physiol Regul Integr Comp Physiol. (2015) 309:R884–91. doi: 10.1152/ajpregu.00154.2015
97. Woidacki K, Meyer N, Schumacher A, Goldschmidt A, Maurer M, Zenclussen AC. Transfer of regulatory T cells into abortion-prone mice promotes the expansion of uterine mast cells and normalizes early pregnancy angiogenesis. Sci Rep. (2015) 5:13938. doi: 10.1038/srep13938
98. Bonney EA, Howard A, Krebs K, Begin K, Veilleux K, Gokina NI. Impact of immune deficiency on remodeling of maternal resistance vasculature 4 weeks postpartum in mice. Reprod Sci. (2017) 24:514–25. doi: 10.1177/1933719116678691
99. Drudy L, Sheppard BL, Bonnar J. The ultrastructure of mast cells in the uterus throughout the normal menstrual cycle and the postmenopause. J Anat. (1991) 175:51–63. doi: 10.1016/0378-5122(91)90169-Q
100. Bytautiene E, Vedernikov YP, Saade GR, Romero R, Garfield RE. IgE-independent mast cell activation augments contractility of non-pregnant and pregnant guinea pig myometrium. Int Arch Allergy Immunol. (2008) 147:140–46. doi: 10.1159/000135701
101. Schmerse F, Woidacki K, Riek-Burchardt M, Reichardt P, Roers A, Tadokoro C, et al. In vivo visualization of uterine mast cells by two-photon microscopy. Reproduction. (2014) 147:781–8. doi: 10.1530/REP-13-0570
102. Jensen F, Woudwyk M, Teles A, Woidacki K, Taran F, Costa S, et al. Estradiol and progesterone regulate the migration of mast cells from the periphery to the uterus and induce their maturation and degranulation. PLoS ONE. (2010) 5:e14409. doi: 10.1371/journal.pone.0014409
103. Woidacki K, Popovic M, Metz M, Schumacher A, Linzke N, Teles A, et al. Mast cells rescue implantation defects caused by c-kit deficiency. Cell Death Dis. (2013) 4:e462. doi: 10.1038/cddis.2012.214
104. Rudolph MI, Bardisa L, Cruz MA, Reinicke K. Mast cells mediators evoke contractility and potentiate each other in mouse uterine horns. Gen Pharmacol. (1992) 23:833–6. doi: 10.1016/0306-3623(92)90233-A
105. Rudolph MI, Cabanillas A, Gomez P, Garcia MA, Villan L. On the mechanism of action of ethodin in inducing myometrium contractions. Gen Pharmacol. (1997) 28:381–5. doi: 10.1016/S0306-3623(96)00288-1
106. Meyer N, Schuler T, Zenclussen AC. Simultaneous ablation of uterine natural killer cells and uterine mast cells in mice leads to poor vascularization and abnormal doppler measurements that compromise fetal well-being. Front Immunol. (2017) 8:1913. doi: 10.3389/fimmu.2017.01913
107. Meyer N, Schuler T, Zenclussen AC. High frequency ultrasound for the analysis of fetal and placental development in vivo. J Vis Exp. (2018) 8:141. doi: 10.3791/58616.
108. De Filippo K, Dudeck A, Hasenberg M, Nye E, van Rooijen N, Hartmann K, et al. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood. (2013) 121:4930–7. doi: 10.1182/blood-2013-02-486217
109. Dudeck J, Ghouse SM, Lehmann CH, Hoppe A, Schubert N, Nedospasov SA, et al. Mast-cell-derived TNF amplifies CD8(+) dendritic cell functionality and CD8(+) T cell priming. Cell Rep. (2015) 13:399–411. doi: 10.1016/j.celrep.2015.08.078
110. Galli SJ, Grimbaldeston M, Tsai M. Immunomodulatory mast cells: negative, as well as positive, regulators of immunity. Nat Rev Immunol. (2008) 8:478–86. doi: 10.1038/nri2327
111. Lu LF, Lind EF, Gondek DC, Bennett KA, Gleeson MW, Pino-Lagos K, et al. Mast cells are essential intermediaries in regulatory T-cell tolerance. Nature. (2006) 442:997–1002. doi: 10.1038/nature05010
112. Bloxham PA. From ovulation to blastocyst attachment. A review of early embryonic life in the mouse. Folia Vet Lat. (1976) 6:319–34.
113. Mezouar S, Ben Amara A, Vitte J, Mege JL. Isolation of human placental mast cells. Curr Protoc Cell Biol. (2018) 80:e52. doi: 10.1002/cpcb.52
114. Tchougounova E, Pejler G, Abrink M. The chymase, mouse mast cell protease 4, constitutes the major chymotrypsin-like activity in peritoneum and ear tissue. a role for mouse mast cell protease 4 in thrombin regulation and fibronectin turnover. J Exp Med. (2003) 198:423–31. doi: 10.1084/jem.20030671
115. Leskinen, M. J., Lindstedt KA, Wang Y, Kovanen PT. Mast cell chymase induces smooth muscle cell apoptosis by a mechanism involving fibronectin degradation and disruption of focal adhesions. Arterioscler Thromb Vasc Biol. (2003) 23:238–43. doi: 10.1161/01.ATV.0000051405.68811.4D
116. Wang Y, Shiota N, Leskinen MJ, Lindstedt KA, Kovanen PT. Mast cell chymase inhibits smooth muscle cell growth and collagen expression in vitro: transforming growth factor-beta1-dependent and -independent effects. Arterioscler Thromb Vasc Biol. (2001) 21:1928–33. doi: 10.1161/hq1201.100227
117. den Dekker WK, Tempel D, Bot I, Biessen EA, Joosten L, Netea MG, et al. (2012). Mast cells induce vascular smooth muscle cell apoptosis via a toll-like receptor 4 activation pathway. Arterioscler Thromb Vasc Biol. 32:1960–9. doi: 10.1161/ATVBAHA.112.250605
118. Urata H, Kinoshita A, Misono KS, Bumpus FM, Husain A. Identification of a highly specific chymase as the major angiotensin II-forming enzyme in the human heart. J Biol Chem. (1990) 265:22348–57.
119. Hering L, Herse F, Geusens N, Verlohren S, Wenzel K, Staff A, et al. Effects of circulating and local utero-placental angiotensin II in rat pregnancy. Hypertension. (2010) 56:311–8. doi: 10.1161/HYPERTENSIONAHA.110.150961
120. Yaghini FA, Song CY, Lavrentyev EN, Ghafoor HU, Fang XR, Estes AM, et al. Angiotensin II-induced vascular smooth muscle cell migration and growth are mediated by cytochrome P450 1B1-dependent superoxide generation. Hypertension. (2010) 55:1461–7. doi: 10.1161/HYPERTENSIONAHA.110.150029
121. Meng Y, Chen C, Liu Y, Tian C, Li HH. Angiotensin II regulates dendritic cells through activation of NF-κB /p65, ERK1/2, and STAT1 Pathways. Cell Physiol Biochem. (2017) 42:1550–8. doi: 10.1159/000479272
122. Derbala Y, Elazzamy H, Bilal M, Reed R, Salazar Garcia MD, Skariah A, et al. Mast cell-induced immunopathology in recurrent pregnancy losses. Am J Reprod Immunol. (2019) 82:e13128. doi: 10.1111/aji.13128
123. Purcell WM. Human placental mast cells: a role in pre-eclampsia?. Med Hypotheses. (1992) 39:281–3. doi: 10.1016/0306-9877(92)90123-T
124. Szewczyk G, Pyzlak M, Klimkiewicz J, Smiertka W, Miedzinska-Maciejewska M, Szukiewicz D. Mast cells and histamine: do they influence placental vascular network and development in preeclampsia?. Mediators Inflamm. (2012) 2012:307189. doi: 10.1155/2012/307189
125. Lyall F, Robson SC, Bulmer JN. Spiral artery remodeling and trophoblast invasion in preeclampsia and fetal growth restriction: relationship to clinical outcome. Hypertension. (2013) 62:1046–54. doi: 10.1161/HYPERTENSIONAHA.113.01892
126. Ball E, Bulmer JN, Ayis S, Lyall F, Robson SC. Late sporadic miscarriage is associated with abnormalities in spiral artery transformation and trophoblast invasion. J Pathol. (2006) 208:535–42. doi: 10.1002/path.1927
127. Duley L. The global impact of pre-eclampsia and eclampsia. Semin Perinatol. (2009) 33:130–7. doi: 10.1053/j.semperi.2009.02.010
128. Garovic VD, August P. Preeclampsia and the future risk of hypertension: the pregnant evidence. Curr Hypertens Rep. (2013) 15:114–21. doi: 10.1007/s11906-013-0329-4
129. Brosens IA, Robertson WB, Dixon HG. The role of the spiral arteries in the pathogenesis of preeclampsia. Obstet Gynecol Annu. (1972) 1:177–91.
130. Pijnenborg R, Anthony J, Davey DA, Rees A, Tiltman A, Vercruysse L, et al. Placental bed spiral arteries in the hypertensive disorders of pregnancy. Br J Obstet Gynaecol. (1991) 98:648–55. doi: 10.1111/j.1471-0528.1991.tb13450.x
131. Jafri S, Ormiston ML. Immune regulation of systemic hypertension, pulmonary arterial hypertension, and preeclampsia: shared disease mechanisms and translational opportunities. Am J Physiol Regul Integr Comp Physiol. (2017) 313:R693–705. doi: 10.1152/ajpregu.00259.2017
132. Huang X, Anderle P, Hostettler L, Baumann MU, Surbek DV, Ontsouka EC, et al. Identification of placental nutrient transporters associated with intrauterine growth restriction and pre-eclampsia. BMC Genomics. (2018) 19:173. doi: 10.1186/s12864-018-4518-z
133. Chen YY, Gupta MB, Grattton R, Powell TL, Jansson T. Down-regulation of placental folate transporters in intrauterine growth restriction. J Nutr Biochem. (2018) 59:136–41. doi: 10.1016/j.jnutbio.2018.06.003
134. Godfrey KM, Barker DJ. Fetal nutrition and adult disease. Am J Clin Nutr. (2000) 71:1344–52. doi: 10.1093/ajcn/71.5.1344s
135. Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. (2008) 359:61–73. doi: 10.1056/NEJMra0708473
136. Zhang Y, Wang H, Pan X, Teng W, Shan Z. Patients with subclinical hypothyroidism before 20 weeks of pregnancy have a higher risk of miscarriage: a systematic review and meta-analysis. PLoS ONE. (2017) 12:e0175708. doi: 10.1371/journal.pone.0175708
137. Pluchino N, Drakopoulos P, Wenger JM, Petignat P, Streuli I, Genazzani AR. Hormonal causes of recurrent pregnancy loss (RPL). Hormones. (2014) 13:314–22. doi: 10.14310/horm.2002.1505
138. Berkane N, Liere P, Oudinet JP, Hertig A, Lefevre G, Pluchino N, et al. From pregnancy to preeclampsia: a key role for estrogens. Endocr Rev. (2017) 38:123–44. doi: 10.1210/er.2016-1065
139. Cantonwine DE, McElrath TF, Trabert B, Xu X, Sampson J, Roberts JM, et al. Estrogen metabolism pathways in preeclampsia and normal pregnancy. Steroids. (2019) 144:8–14. doi: 10.1016/j.steroids.2019.01.005
140. Korevaar TIM, Derakhshan A, Taylor PN, Meima M, Chen L, Bliddal S, et al. Association of thyroid function test abnormalities and thyroid autoimmunity with preterm birth: a systematic review and meta-analysis. JAMA. (2019) 322:632–41. doi: 10.1001/jama.2019.10931
141. Baud O, Berkane N. Hormonal changes associated with intra-uterine growth restriction: impact on the developing brain and future neurodevelopment. Front Endocrinol. (2019) 10:179. doi: 10.3389/fendo.2019.00179
142. Balaguer P, Delfosse V, Grimaldi M, Bourguet W. Structural and functional evidences for the interactions between nuclear hormone receptors and endocrine disruptors at low doses. C R Biol. (2017) 340:414–20. doi: 10.1016/j.crvi.2017.08.002
143. Feng Y, Zhang P, Zhang Z, Shi J, Jiao Z, Shao B. Endocrine disrupting effects of triclosan on the placenta in pregnant rats. PLoS ONE. (2016) 11:e0154758. doi: 10.1371/journal.pone.0154758
144. Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, Hauser R, Prins GS, Soto AM, et al. Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr Rev. (2009) 30:293–342. doi: 10.1210/er.2009-0002
145. Petrakis D, Vassilopoulou L, Mamoulakis C, Psycharakis C, Anifantaki A, Sifakis S, et al. Endocrine disruptors leading to obesity and related diseases. Int J Environ Res Public Health. (2017) 14:e1282. doi: 10.3390/ijerph14101282
146. Heindel JJ, Blumberg B, Cave M, Machtinger R, Mantovani A, Mendez MA, et al. Metabolism disrupting chemicals and metabolic disorders. Reprod Toxicol. (2017) 68:3–33. doi: 10.1016/j.reprotox.2016.10.001
147. Papalou O, Kandaraki EA, Papadakis G, Diamanti-Kandarakis E. Endocrine disrupting chemicals: an occult mediator of metabolic disease. Front Endocrinol. (2019) 10:112. doi: 10.3389/fendo.2019.00112
148. Rasier G, Toppari J, Parent AS, Bourguignon JP. Female sexual maturation and reproduction after prepubertal exposure to estrogens and endocrine disrupting chemicals: a review of rodent and human data. Mol Cell Endocrinol. (2006) 254–5:187–201. doi: 10.1016/j.mce.2006.04.002
149. Adeel M, Song X, Wang Y, Francis D, Yang Y. Environmental impact of estrogens on human, animal and plant life: a critical review. Environ Int. (2017) 99:107–19. doi: 10.1016/j.envint.2016.12.010
150. Vandenberg LN, Colborn T, Hayes TB, Heindel JJ, Jacobs DR Jr, Lee DH, et al. (2012). Hormones and endocrine-disrupting chemicals: low-dose effects and non-monotonic dose responses. Endocr Rev. 33:378–455. doi: 10.1210/er.2011-1050
151. Kadokawa H, Pandey K, Onalenna K, Nahar A. Reconsidering the roles of endogenous estrogens and xenoestrogens: the membrane estradiol receptor G protein-coupled receptor 30 (GPR30) mediates the effects of various estrogens. J Reprod Dev. (2018) 64:203–8. doi: 10.1262/jrd.2017-153
152. La Rosa P, Pellegrini M, Totta P, Acconcia F, Marino M. Xenoestrogens alter estrogen receptor (ER) α intracellular levels. PLoS ONE. (2014) 9:e88961. doi: 10.1371/journal.pone.0088961
153. Pennie WD, Aldridge TC, Brooks AN. Differential activation by xenoestrogens of ER alpha and ER beta when linked to different response elements. J Endocrinol. (1998) 158:R11–4. doi: 10.1677/joe.0.158r011
154. Kovats S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell Immunol. (2015) 294:63–9. doi: 10.1016/j.cellimm.2015.01.018
155. Yoshino S, Yamaki K, Li X, Sai T, Yanagisawa R, Takano H, et al. Prenatal exposure to bisphenol A up-regulates immune responses, including T helper 1 and T helper 2 responses, in mice. Immunology. (2004) 112:489–95. doi: 10.1111/j.1365-2567.2004.01900.x
156. Liu J, Li J, Wu Y, Zhao Y, Luo F, Li S, et al. Bisphenol A metabolites and Bisphenol S in paired maternal and cord serum. Environ Sci Technol. (2017) 51:2456–63. doi: 10.1021/acs.est.6b05718
157. Mu X, Huang Y, Li X, Lei Y, Teng M, Li X, et al. Developmental effects and estrogenicity of Bisphenol A alternatives in a zebrafish embryo model. Environ Sci Technol. (2018) 52:3222–31. doi: 10.1021/acs.est.7b06255
158. Lehmler HJ, Liu B, Gadogbe M, Bao W. Exposure to Bisphenol A, Bisphenol F, and Bisphenol S in adults, U. S., and children: The National Health and Nutrition Examination Survey 2013–2014. ACS Omega. (2018) 3:6523–32. doi: 10.1021/acsomega.8b00824
159. Yang C, Song G, Lim W. A mechanism for the effect of endocrine disrupting chemicals on placentation. Chemosphere. (2019) 231:326–36. doi: 10.1016/j.chemosphere.2019.05.133
160. Li LX, Chen L, Meng XZ, Chen BH, Chen SQ, Zhao Y, et al. Exposure levels of environmental endocrine disruptors in mother-newborn pairs in China and their placental transfer characteristics. PLoS ONE. (2013) 8:e62526. doi: 10.1371/journal.pone.0062526
161. Covaci A, Jorens P, Jacquemyn Y, Schepens P. Distribution of PCBs and organochlorine pesticides in umbilical cord and maternal serum. Sci Total Environ. (2002) 298:45–53. doi: 10.1016/S0048-9697(02)00167-5
162. Rogers JA, Metz L, Yong VW. Review: endocrine disrupting chemicals and immune responses: a focus on bisphenol-A and its potential mechanisms. Mol Immunol. (2013) 53:421–30. doi: 10.1016/j.molimm.2012.09.013
163. Kim JY, Jeong HG. Down-regulation of inducible nitric oxide synthase and tumor necrosis factor-alpha expression by Bisphenol A via nuclear factor-kappaB inactivation in macrophages. Cancer Lett. (2003) 196:69–76. doi: 10.1016/S0304-3835(03)00219-2
164. Yoshitake J, Kato K, Yoshioka D, Sueishi Y, Sawa T, Akaike T, et al. Suppression of NO production and 8-nitroguanosine formation by phenol-containing endocrine-disrupting chemicals in LPS-stimulated macrophages: involvement of estrogen receptor-dependent or -independent pathways. Nitric Oxide. (2008) 18:223–8. doi: 10.1016/j.niox.2008.01.003
165. Byun JA, Heo Y, Kim YO, Pyo MY. Bisphenol A-induced downregulation of murine macrophage activities in vitro and ex vivo. Environ Toxicol Pharmacol. (2005) 19:19–24. doi: 10.1016/j.etap.2004.02.006
166. Segura JJ, Jimenez-Rubio A, Pulgar R, Olea N, Guerrero JM, Calvo JR. In vitro effect of the resin component Bisphenol A on substrate adherence capacity of macrophages. J Endod. (1999) 25:341–4. doi: 10.1016/S0099-2399(06)81168-4
167. Youn JY, Park HY, Lee JW, Jung I, Choi KH, Kim K, et al. Evaluation of the immune response following exposure of mice to Bisphenol A: induction of Th1 cytokine and prolactin by BPA exposure in the mouse spleen cells. Arch Pharm Res. (2002) 25:946–53. doi: 10.1007/BF02977018
168. Lee MH, Chung SW, Kang BY, Park J, Lee CH, Hwang SY, et al. Enhanced interleukin-4 production in CD4+ T cells and elevated immunoglobulin E levels in antigen-primed mice by bisphenol A and nonylphenol, endocrine disruptors: involvement of nuclear factor-AT and Ca2+. Immunology. (2003) 109:76–86. doi: 10.1046/j.1365-2567.2003.01631.x
169. Yan H, Takamoto M, Sugane K. Exposure to Bisphenol A prenatally or in adulthood promotes T(H)2 cytokine production associated with reduction of CD4CD25 regulatory T cells. Environ Health Perspect. (2008) 116:514–9. doi: 10.1289/ehp.10829
170. Ohshima Y, Yamada A, Tokuriki S, Yasutomi M, Omata N, Mayumi M. Transmaternal exposure to bisphenol a modulates the development of oral tolerance. Pediatr Res. (2007) 62:60–4. doi: 10.1203/PDR.0b013e3180674dae
171. Pisapia L, Del Pozzo G, Barba P, Caputo L, Mita L, Viggiano E, et al. Effects of some endocrine disruptors on cell cycle progression and murine dendritic cell differentiation. Gen Comp Endocrinol. (2012) 178:54–63. doi: 10.1016/j.ygcen.2012.04.005
172. Goto M, Takano-Ishikawa Y, Ono H, Yoshida M, Yamaki K, Shinmoto H. Orally administered bisphenol A disturbed antigen specific immunoresponses in the naive condition. Biosci Biotechnol Biochem. (2007) 71:2136–43. doi: 10.1271/bbb.70004
173. Yurino H, Ishikawa S, Sato T, Akadegawa K, Ito T, Ueha S, et al. Endocrine disruptors (environmental estrogens) enhance autoantibody production by B1 cells. Toxicol Sci. (2004) 81:139–47. doi: 10.1093/toxsci/kfh179
174. O'Brien E, Dolinoy DC, Mancuso P. Bisphenol A at concentrations relevant to human exposure enhances histamine and cysteinyl leukotriene release from bone marrow-derived mast cells. J Immunotoxicol. (2014) 11:84–9. doi: 10.3109/1547691X.2013.800925
175. Uramaru N, Inoue T, Watanabe Y, Shigematsu H, Ohta S, Kitamura S. Structure-activity relationship of a series of 17 parabens and related compounds for histamine release in rat peritoneal mast cells and skin allergic reaction in guinea pigs. J Toxicol Sci. (2014) 39:83–90. doi: 10.2131/jts.39.83
176. Jin Y, Chen R, Liu W, Fu Z. Effect of endocrine disrupting chemicals on the transcription of genes related to the innate immune system in the early developmental stage of zebrafish (Danio rerio). Fish Shellfish Immunol. (2010) 28:854–61. doi: 10.1016/j.fsi.2010.02.009
177. Nowak K, Jablonska E, Ratajczak-Wrona W. Immunomodulatory effects of synthetic endocrine disrupting chemicals on the development and functions of human immune cells. Environ Int. (2019) 125:350–64. doi: 10.1016/j.envint.2019.01.078
178. Camarca A, Gianfrani C, Ariemma F, Cimmino I, Bruzzese D, Scerbo R, et al. Human peripheral blood mononuclear cell function and dendritic cell differentiation are affected by bisphenol-A exposure. PLoS ONE. (2016) 11:e0161122. doi: 10.1371/journal.pone.0161122
179. Svajger U, Dolenc MS, Jeras M. In vitro impact of bisphenols BPA, BPF, BPAF, and 17β-estradiol (E2) on human monocyte-derived dendritic cell generation, maturation and function. Int Immunopharmacol. (2016) 34:146–54. doi: 10.1016/j.intimp.2016.02.030
180. Hung CH, Yang SN, Kuo PL, Chu Y, Chang HW, Wei WJ, et al. Modulation of cytokine expression in human myeloid dendritic cells by environmental endocrine-disrupting chemicals involves epigenetic regulation. Environ. Health Perspect. (2010) 118:67–72. doi: 10.1289/ehp.0901011
181. Couleau N, Falla J, Beillerot A, Battaglia E, D'Innocenzo M, Plancon S, et al. Effects of endocrine disruptor compounds, alone or in combination, on human macrophage-like THP-1 cell response. PLoS ONE. (2015) 10:e0131428. doi: 10.1371/journal.pone.0131428
182. Ustyugova IV, Frost LL, Van Dyke K, Brundage KM, Schafer R, Barnett JB, et al. 3,4-dichloropropionaniline suppresses normal macrophage function. Toxicol Sci. (2007) 97:364–74. doi: 10.1093/toxsci/kfm048
183. Pottenger LH, Domoradzki JY, Markham DA, Hansen SC, Cagen SZ, Waechter JM Jr. The relative bioavailability and metabolism of bisphenol A in rats is dependent upon the route of administration. Toxicol Sci. (2000) 54:3–18. doi: 10.1093/toxsci/54.1.3
184. Richter CA, Birnbaum LS, Farabollini F, Newbold RR, Rubin BS, Talsness CE, et al. In vivo effects of bisphenol A in laboratory rodent studies. Reprod Toxicol. (2007) 24:199–224. doi: 10.1016/j.reprotox.2007.06.004
185. Ehrlich S, Williams PL, Missmer SA, Flaws JA, Ye X, Calafat AM, et al. Urinary bisphenol A concentrations and early reproductive health outcomes among women undergoing IVF. Hum Reprod. (2012) 27:3583–92. doi: 10.1093/humrep/des328
186. Mok-Lin E, Ehrlich S, Williams PL, Petrozza J, Wright D, Calafat AM, et al. Urinary bisphenol A concentrations and ovarian response among women undergoing IVF. Int J Androl. (2010) 33:385–93. doi: 10.1111/j.1365-2605.2009.01014.x
187. Minguez-Alarcon L, Messerlian C, Bellavia A, Gaskins AJ, Chiu Y, Ford JB, et al. Urinary concentrations of bisphenol A, parabens and phthalate metabolite mixtures in relation to reproductive success among women undergoing in vitro fertilization. Environ Int. (2019) 126:355–62. doi: 10.1016/j.envint.2019.02.025
188. Grindler NM, Vanderlinden L, Karthikraj R, Kannan K, Teal S, Polotsky A, et al. Exposure to phthalate, an endocrine disrupting chemical, alters the first trimester placental methylome and transcriptome in women. Sci Rep. (2018) 8:6086. doi: 10.1038/s41598-018-24505-w
189. Adibi JJ, Whyatt RM, Hauser R, Bhat HK, Davis BJ, Calafat AM, et al. Transcriptional biomarkers of steroidogenesis and trophoblast differentiation in the placenta in relation to prenatal phthalate exposure. Environ Health Perspect. (2010) 118:291–6. doi: 10.1289/ehp.0900788
190. Zhao M, Zhang Y, Zhuang S, Zhang Q, Lu C, Liu W. Disruption of the hormonal network and the enantioselectivity of bifenthrin in trophoblast: maternal-fetal health risk of chiral pesticides. Environ Sci Technol. (2014) 48:8109–16. doi: 10.1021/es501903b
191. Muller JE, Meyer N, Santamaria CG, Schumacher A, Luque E, Zenclussen ML, et al. Bisphenol A exposure during early pregnancy impairs uterine spiral artery remodeling and provokes intrauterine growth restriction in mice. Sci Rep. (2018) 8:9196. doi: 10.1038/s41598-018-27575-y
192. Ye Y, Tang Y, Xiong Y, Feng L, Li X. Bisphenol A exposure alters placentation and causes preeclampsia-like features in pregnant mice involved in reprogramming of DNA methylation of WNT2. FASEB J. (2019) 33:2732–42. doi: 10.1096/fj.201800934RRR
193. Tachibana T, Wakimoto Y, Nakamuta N, Phichitraslip T, Wakitani S, Kusakabe K, et al. Effects of bisphenol A (BPA) on placentation and survival of the neonates in mice. J Reprod Dev. (2007) 53:509–14. doi: 10.1262/jrd.18171
194. Backlin BM, Persson E, Jones CJ, Dantzer V. Polychlorinated biphenyl (PCB) exposure produces placental vascular and trophoblastic lesions in the mink (Mustela vison): a light and electron microscopic study. APMIS. (1998) 106:785–99. doi: 10.1111/j.1699-0463.1998.tb00225.x
195. Meyer N, Santamaria CG, Muller JE, Schumacher A, Rodriguez HA, Zenclussen AC. Exposure to 17α-ethinyl estradiol during early pregnancy affects fetal growth and survival in mice. Environ Pollut. (2019) 251:493–501. doi: 10.1016/j.envpol.2019.04.144
Keywords: uterine remodeling, immune cells, pregnancy, pregnancy pathologies, endocrine disrupting chemicals, fetal development, maternal health
Citation: Meyer N and Zenclussen AC (2020) Immune Cells in the Uterine Remodeling: Are They the Target of Endocrine Disrupting Chemicals? Front. Immunol. 11:246. doi: 10.3389/fimmu.2020.00246
Received: 07 October 2019; Accepted: 30 January 2020;
Published: 19 February 2020.
Edited by:
Irun R. Cohen, Weizmann Institute of Science, IsraelReviewed by:
Umit A. Kayisli, University of South Florida, United StatesZhibin Chen, University of Miami, United States
Copyright © 2020 Meyer and Zenclussen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicole Meyer, bmljb2xlLm1leWVyQG1lZC5vdmd1LmRl