 Rachael M. Zemek1
Rachael M. Zemek1 Wee Loong Chin2,3,4
Wee Loong Chin2,3,4 Anna K. Nowak2,3,4Michael J. Millward3,4Richard A. Lake2,5
Anna K. Nowak2,3,4Michael J. Millward3,4Richard A. Lake2,5 W. Joost Lesterhuis1,2,5*
W. Joost Lesterhuis1,2,5*- 1Telethon Kids Institute, University of Western Australia, West Perth, WA, Australia
- 2National Centre for Asbestos Related Diseases, Nedlands, WA, Australia
- 3Medical School, University of Western Australia, Crawley, WA, Australia
- 4Department of Medical Oncology, Sir Charles Gairdner Hospital, Nedlands, WA, Australia
- 5School of Biomedical Sciences, University of Western Australia, Crawley, WA, Australia
Immune checkpoint blockade (ICB) has revolutionized cancer treatment, providing remarkable clinical responses in some patients. However, the majority of patients do not respond. It is therefore crucial both to identify predictive biomarkers of response and to increase the response rates to immune checkpoint therapy. In this review we explore the current literature about the predictive characteristics of the tumor microenvironment and discuss therapeutic approaches that aim to change this toward a milieu that is conducive to response. We propose a personalized biomarker-based adaptive approach to immunotherapy, whereby a sensitizing therapy is tailored to the patient's specific tumor microenvironment, followed by on-treatment verification of a change in the targeted biomarker, followed by immune checkpoint therapy. By incorporating detailed knowledge of the immunological tumor microenvironment, we may be able to sensitize currently non-responsive tumors to respond to immune checkpoint therapy.
Introduction
Therapeutic approaches that inhibit negative regulatory immune checkpoints or stimulate activating immune checkpoints have shown great success in preclinical models and clinical trials (1, 2). In particular, antibodies that block cytotoxic T lymphocyte associated protein 4 (CTLA4), and programmed death 1 (PD-1) or its ligand PD-L1 have demonstrated unprecedented therapeutic efficacy in metastatic melanoma, non-small cell lung cancer, mismatch repair deficient cancers, and several other cancer types (3, 4). In these cancer types, significant response rates and survival benefits are seen, with a proportion of durable complete regressions, allowing the word “cure” to enter the oncologists' vocabulary (5). However, despite some positive outcomes, survival gains are modest in most cancers, and even in the most responsive cancers, many patients do not experience clinical benefit (6). This heterogeneity in response has led to a search for predictive biomarkers that could identify whether a patient will or will not respond to immune checkpoint blockade (ICB). In addition, although the targets for these antibodies are known, the down-stream biological consequences of therapeutic target engagement—both systemic and in the tumor microenvironment (TME)—are incompletely understood (6). Hence, although there is a clear need to improve the therapeutic efficacy of ICB, most clinical trials testing combination therapies are empirical, and often based on scant biological or preclinical data (7, 8).
In mouse cancer models using subcutaneous tumors derived from clonal cell lines we also observe a dichotomy in response to ICB; some mice experience complete response to treatment, while in others, their tumor continues to progress, despite having been treated under identical environmental and experimental conditions, including equal tumor burden and presumably identical (neo-)antigen expression (9–13). It is noteworthy that the probability of response in these mice can generally be increased by treating with ICB earlier, at a smaller tumor size (9). Together, these data suggest that each mouse has the capacity to respond, but that even potentially sensitive tumors may not respond if the pre-treatment conditions are not optimal. Furthermore, some murine tumor models never respond to ICB. Are these tumors intrinsically resistant, or do they lack functional TME attributes that could be therapeutically induced? Promisingly, some recent studies have been able to render otherwise resistant models sensitive to subsequent ICB (9, 14).
Despite known associations between pre-treatment TME characteristics and response [see references (15) and (16) for comprehensive reviews of predictive biomarkers], strategies to induce a responsive phenotype and thus sensitize cancers to ICB are only beginning to be developed. Systemic immunity and the local immune response at the effector site are obviously linked, and indeed several studies, both in animal models and patients, have shown that a degree of systemic immunity is required for tumors to respond to ICB (17, 18). Here, we focus on factors within the TME: we summarize and contextualize recent studies characterizing the features of an ICB responsive, contrasting with a non-responsive, TME (Figure 1), and discuss selected therapeutic interventions designed to modulate that environment toward a responsive phenotype (Figure 2).
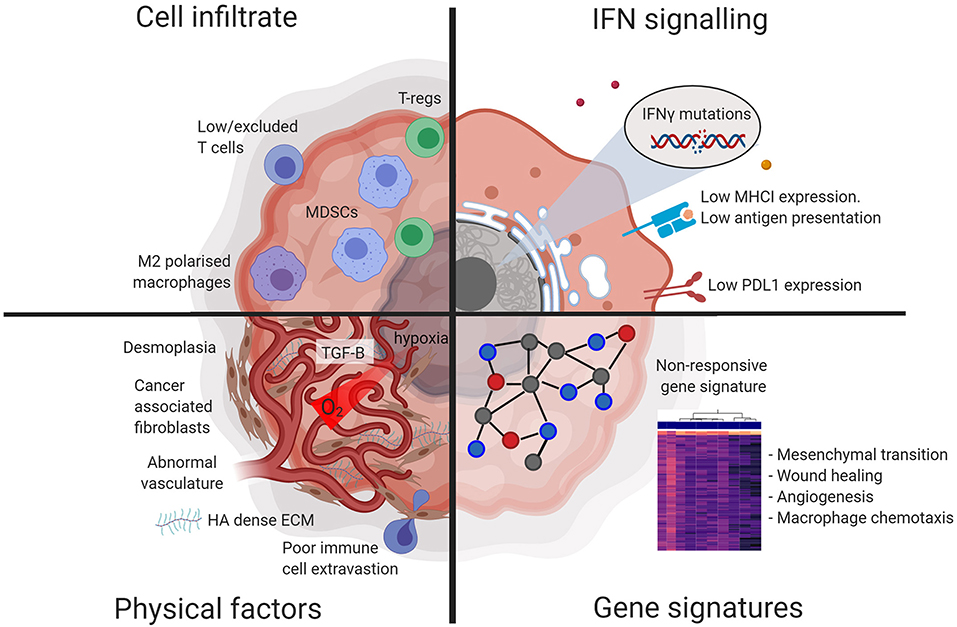
Figure 1. Diagram illustrating factors characteristic of a non-responsive tumor microenvironment. Tumors that are non-responsive to checkpoint blockade display resistance at the physical, cellular, protein, and gene expression level. Figures made in ©BioRender-biorender.com.
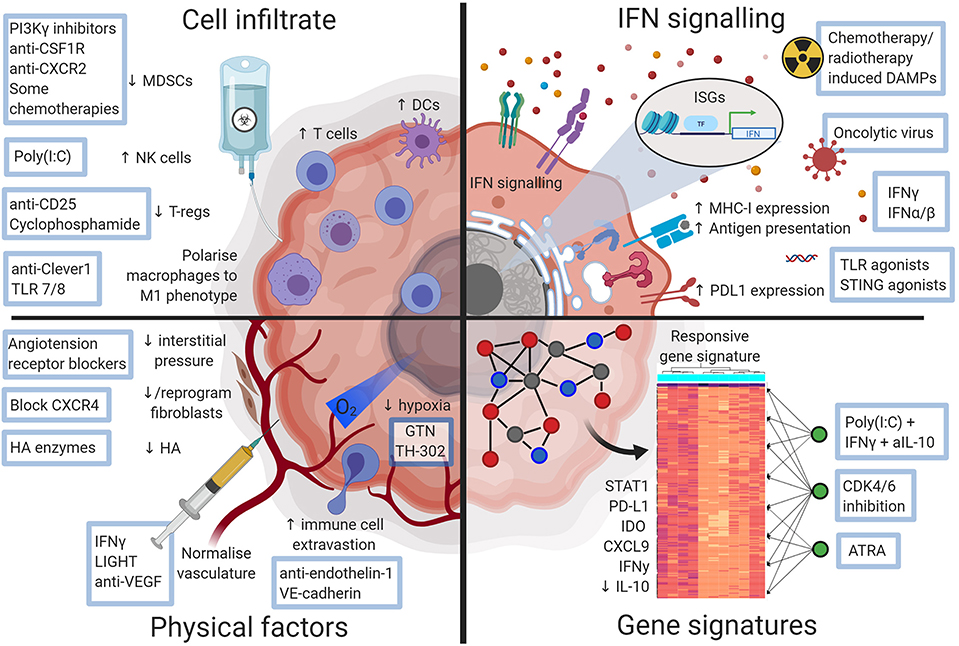
Figure 2. Diagram illustrating ways a checkpoint blockade favorable tumor microenvironment may be therapeutically attained. Approaches that could alter the non-responsive tumor microenvironment and sensitize tumors to checkpoint blockade. Figures made in ©BioRender-biorender.com.
Turning “Cold” Tumors “Hot”—Cells in the Responsive TME
Recently, the concept of hot vs. cold tumors has become widely accepted in immuno-oncology, where “hot” denotes tumors that contain more than a defined threshold of inflammatory cells, while “cold” tumors do not. Other terms characterizing response-associated cellular TMEs include describing cold tumors as either “immune desert” (absence of immune cells) or “infiltrated-excluded” (tumors with only peripheral invasion of immune cells); and classifying “hot” tumors into those with tertiary lymphoid structures, or “infiltration-inflamed” tumors (inflammatory myeloid cells and activated CD8+ T cells) (19, 20). Alternatively, more comprehensive characterizations incorporate several different aspects of T cell immunity resulting in more subcategories and allowing a scoring metric to be applied (21, 22). A potential caveat of these approaches is that innate immune cells are underrepresented. Accumulating evidence indicates that innate immune cells play a role in the responsive TME and could be exploited therapeutically to improve outcomes. Relevant cells include macrophages (23–25), dendritic cells (DCs) (26) and natural killer (NK) cells (9, 26). We will briefly discuss several cell types in the TME associated with response.
T Cells
Experiments in murine cancer models have consistently demonstrated the importance of T cells for the efficacy of ICB (27–29). In humans, secondary resistance to ICB has also been associated with T cell deficits, including mutations in cancer cells associated with decreased sensitivity to T cell-mediated killing, or reduced antigen presentation to T cells (30, 31). Baseline numbers of tumor-infiltrating lymphocytes (TILs) have been found to correlate with response to anti-PD-1 alone, but not with combination ICB therapy (32–34). CD8+ T cells have been shown to be the anti-tumor effector cells, and sensitivity to ICB was enhanced in tumors enriched for CD8+ T cells reactive to clonal neoantigens (35). The differential role of CD8+ vs. CD4+ T cells is less clear, with conflicting outcomes in different tumor models (28, 29). The interpretation of these results is difficult because CD4 depletion in mice not only depletes T effector cells, but also regulatory T cells (T-regs) (36). It should be noted that anti-mouse PD-1 antibodies used in murine models are often raised in rats, and repeated dosing will result in anti-antibody formation. In addition, there are important differences in IgG isotype and their affinity for Fc receptors (37, 38), which may partly explain the difference in efficacy between murine and human studies.
Pre-treatment with selected chemotherapeutics has been shown to enhance T cell responses in the experimental setting (39). For example, oxaliplatin plus cyclophosphamide treatment of lung adenocarcinomas increased the ratio of CD8+ T cells vs. T-regs, increased the presence of tumor-specific CD8+ T cells, and resulted in enhanced expression of PD-1 and PD-L1 with subsequent improved responsiveness to ICB (40). Similarly, gemcitabine only synergized with CD40 directed immunotherapy when mice were pre-treated with gemcitabine, but it was not effective when given concurrently or after immunotherapy (41).
T-regs express high levels of CTLA4, and antibodies blocking CTLA4 can deplete T-regs in the TME in murine models, dependent on Fc subclass and host Fc receptor (42–44). T-reg depletion has also been hypothesized as a key mechanism of action for anti-CTLA4 treatment in humans (45, 46), however clinical data does not support this (47), which may be due to the different isotype and Fc portion of the human antibody. Indeed, there is an association between pre-treatment Foxp3+ T-reg infiltration, followed by a subsequent increase in TILs 3 weeks into treatment in melanoma biopsies which was associated with response to the CTLA4-targeting antibody ipilimumab (48). Whether this is a causal relationship, or a bystander effect of enhanced inflammation remains to be established, as infiltration of the TME with T-regs usually coexists with an inflammatory response that also includes CD8+ T cells, macrophages and granulocytes (49, 50). Conversely, T-regs and T effector cells express similar levels of PD-1 (51), and anti-PD-1 antibodies do not deplete cells expressing the target, but the Fc portion can modulate myeloid cell activity (37). Depletion of T-regs, nevertheless, may enhance the efficacy of ICB. Depletion of T-reg using an Fc-optimized anti-CD25 antibody prior to anti-PD-1 treatment resulted in a greater response rate in a murine model, indicating that depleting T-regs prior to ICB may be viable sensitization strategy (52). An intriguing approach turned T-regs into interferon (IFN)γ-producing T effector cells by targeting the CARMA1-BCL10-MALT1 signalosome complex. This resulted in an inflammatory TME with increased expression of MHC class II by macrophages and MHC class I and PD-L1 by tumor cells, facilitating increased T cell mediated tumor lysis (53). Another strategy to target Treg is pharmacological inhibition using the allosteric MALT1 inhibitor mepazine (previously used in psychiatric diseases). Combination therapy with mepazine and PD-1 blockade resulted in an additive anti-tumor effect (53). However, as yet there are no clinically available therapeutic agents which deplete human T-reg.
Macrophages
Macrophages may also play a role in response to ICB, although both tumor-promoting and tumoricidal effects have been noted. Macrophages are enriched in anti-PD-1 resistant human non-small cell lung cancer (NSCLC) (24). Conversely, macrophages have also been noted in the regression bed of neoadjuvant treated NSCLC patients with complete response (25). These seemingly opposing effects may be partially explained by the heterogeneity of macrophages, which can change polarization from a pro-tumorigenic M2 phenotype to a more tumoricidal M1 phenotype, a phenomenon which could be exploited therapeutically. Viitala et al. identified common lymphatic endothelial and vascular endothelial receptor-1 (Clever-1) as a driver of M2 polarization. Treatment of Lewis lung cancer-bearing mice with an antibody targeting Clever-1 concomitant with PD-1 blockade provided a modest additive anti-tumor effect (54). Similarly, Rodell et al. used Toll-like receptor 7/8 agonist-loaded nanoparticles to polarize macrophages toward an M1 phenotype, resulting in single-agent anti-tumor efficacy, and additive effects in combination with PD-1 blockade in mice bearing MC38 or B16 tumors (55). Notably, these studies used concomitant treatments and did not explore sensitization strategies using sequential scheduling of treatment to alter the TME prior to ICB.
Myeloid Derived Suppressor Cells
Myeloid-derived suppressor cells (MDSCs) have been linked to a reduced efficacy of several immune-based cancer therapies (56–60), and therefore can also be targeted to enhance ICB efficacy. In murine cancer models, the gamma isoform of phosphoinositide 3-kinase (PI3Kγ), which is highly expressed in myeloid cells, can be targeted with a selective inhibitor that reprograms MDSCs and improves responses to antibodies targeting CTLA4, PD-1, or both (61). Selective PI3Kγ inhibitors are currently being evaluated in clinical trials (62). Similarly, blocking CSF1R prevents MDSCs from exerting immunosuppressive effects, enhances anti-tumor T-cell responses, and improves response to checkpoint blockade in several murine models to ICB (63–65). Other approaches include blocking the chemokine receptor CXCR2 to prevent MDSC recruitment into the tumor, which sensitizes a mouse model of rhabdomyosarcoma to anti-PD-1 (66), and using the repurposed drug ibrutinib which inhibits MDSCs and sensitizes murine breast cancer models to anti-PD-L1 (67, 68).
Natural Killer Cells
There has been increasing interest in the role of NK cells in anti-tumor immunity, and the potential to modulate their function therapeutically. In human nasopharyngeal cancer, the presence of functionally exhausted NK cells predicted worse outcomes, and reversing NK cell exhaustion in vitro restored anti-tumor effects (69). Furthermore, a high number of intra-tumoral NK cells in patient melanoma samples at various stages of treatment predicted responsiveness to anti-PD-1. NK cell associated genes were correlated with expression of Flt3lg in the Cancer Genome Atlas melanoma dataset, suggesting a DC stimulatory role for NK cells (26). Although NK cells may not be required for the direct anti-tumor effects driven by ICB, they appear to play a role in supporting an immune-favorable TME. NK cells are required for the accumulation of conventional type I dendritic cells (cDC1) into tumors in mouse models, which are crucial for T cell anti-tumor immunity (70). In addition, we recently identified higher numbers of activated NK cells in the pre-treatment TME of responsive tumors in mouse models treated with anti-CTLA4/anti-PD-L1 compared with those that did not respond. This observation was validated in data from patients treated with anti-PD-L1 (9, 71). Depletion of NK cells prior to ICB abrogated the response, confirming a role of NK cells in the priming of the TME, potentially through local IFNγ production (9). Pre-treatment with poly(I:C), IFNγ and anti-IL-10 increased NK infiltration and sensitized four different tumor models to subsequent ICB. This sensitizing effect was similarly abrogated when NK cells were depleted, despite intratumoral IFNγ administration, suggesting that the effect of NK cells was not restricted to local IFNγ production (9).
In conclusion, the concept of hot vs. cold continues to be useful to explain some differential sensitivity to ICB. However, as more granular information emerges about the functionality of infiltrating cells in the TME and how this is associated with response, this binary description may miss some nuances. For example, in a homogenous background, using tumors derived from clonal cancer cell lines in inbred mice, we found that the composition of cellular infiltrates between responsive and non-responsive tumors was largely identical for CD8+ T cells, macrophages, DCs and granulocytes (9). However, cells in the TME of responding mice had a more activated phenotype as measured by MHCI, PD-L1 and activated regulatory networks, compared to non-responding mice (discussed in more detail below) (9). Specific characteristics of infiltrating cells, such as cellular phenotype and activation state, may define the sensitive or resistant nature of the TME more than cell lineage or origin. The examples above show that therapeutic approaches that change these phenotypic and functional characteristics are able to sensitize tumors to ICB (Figure 2).
Sensitizing the TME Through Enhanced IFN Signaling
Interferons have been associated with both responsiveness and resistance to immunotherapy (72). Expression of IFNγ and IFNγ-inducible genes, such as IDO and CXCL9, are positive biomarkers of response to ICB (73). Upregulation of IFNγ can promote T cell responses, as well as upregulate MHC class I molecules on tumor cells, increasing their sensitivity to cytotoxic T cells (40, 74, 75). At the same time, IFNγ upregulates PD-L1 on cancer cells, leading to the possibility of T cell exhaustion. Targeted activation of the type I IFN system (IFN α and β) renders resistant immune-cell poor tumors sensitive to ICB (76). Some chemotherapies also upregulate type I IFNs, attracting T cells to the TME (77). Combination anti-CTLA4 and anti-PD-1 work synergistically by increasing IFNγ signaling, which in turn increases IL-7 signaling, resulting in superior tumor eradication (78). Inducing IFN signaling may therefore be exploited to increase response to ICB. However, prolonged activation of type I IFN can induce resistance to ICB through stimulation of nitric oxide synthase 2 (NOS2), resulting in increased infiltration of Treg and myeloid cells (79). The finding that IFN-pathways can drive resistance has been reported by Benci et al. (80). Using a B16 cell line that progressed after ICB, they found that resistance was due to enhanced IFNγ/PD-L1 signaling in the tumor cells. Specific blockade of IFNγ signaling in tumor cells resulted in increased sensitivity to PD-1 blockade in an NK/ILC1-dependent manner. This is mirrored by the observation that genetic deletion of the type I IFN pathway in cancer cells increases responses to ICB (81). Interestingly, by inhibiting tumor derived IFNγ and decreasing the immune stimulated genes in tumor cells, IFNγ production by T cells was increased, promoting tumor cell killing (82). In addition, prolonged IFN signaling can drive clonal selection leading to recurrence after an initial response to ICB. In the clinical setting, defects in the pathways involved in interferon-receptor signaling favored outgrowth and sequential progression in melanoma patients treated with anti-PD-1(83, 84). Together, these data indicate that stimulation of the IFN system is a balance between priming the TME to subsequent ICB or rendering it resistant. The spatial and temporal characteristics of the IFN response likely play a role in this outcome.
Cytosolic Sensor Activation
Interferons can be upregulated via several mechanisms. Activation of cytosolic sensors by pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) results in a robust IFN response. These DAMPs can be released as a consequence of treatment with some chemotherapeutics or radiotherapy, resulting in increased production of type I and II IFNs in the TME (85, 86). Interestingly, a post-hoc analysis of the KEYNOTE-001 trial of NSCLC patients treated with anti-PD-1 suggested that patients who had received radiation prior to anti-PD-1 had significantly longer survival than those who did not (87).
The stimulator of interferon genes (STING) sensor can be activated by free DNA, which is released after radiotherapy (88), or by direct injection of cyclic dinucleotides (89), resulting in strong immune activation which can overcome tumor immune suppression (88, 90). Similarly, Toll-like receptors (TLRs) such as TLR3 and TLR9 can be activated by poly(I:C) and CpG, respectively, which can result in a potent inflammatory response when injected into tumors, and can induce long-lasting CD8+ T cell-dependent anti-tumor responses (76, 91). Activation of these cytosolic sensors results in type I IFN production, which as described above can control anti-tumor immunity (76, 92). There is experimental evidence that induction of IFN in the TME via activation of Toll-like receptors (TLR) increases the effectiveness of ICB. Peri-tumoral injection of TLR9 ligand CpG increased sensitivity to anti-PD-1 in murine models of bladder cancer (93). Similarly, several studies identified that TLR3 ligand poly(I:C) improved response rates to ICB in murine models of melanoma, lung, and colon cancer (76, 94). Importantly, these beneficial effects of poly(I:C) are schedule-dependent. A short course of treatment with a Poly(I:C)-based therapeutic combination prior to ICB sensitized several tumor models to anti-CTLA4/anti-PD-L1. However, treatment with ICB first, followed by poly(I:C) was not additive over ICB alone, emphasizing the importance of temporal aspects of an effective anti-tumor response, and by extension, of scheduling drugs in combination immunotherapy (9). There are ongoing clinical trials combining polyICLC [a stable derivative of poly(I:C)] with ICB (clinicaltrial.gov numbers: NCT02834052, NCT03721679, NCT02643303). Treatment with immunostimulatory molecules prior to ICB is a rational strategy to impose a sensitive phenotype onto tumors, however, they must be applied directly into the tumor, which may not always be feasible. A novel small molecule STING agonist has recently been developed for systemic use, which may overcome this limitation (95).
Oncolytic Virotherapy
Oncolytic virotherapy is another strategy which has been used to induce IFN. Combining oncolytic virotherapy and CTLA4 blockade resulted in rejection of distant (non-virally injected) tumors in the poorly immunogenic B16 melanoma model, which was dependent on NK cells and type I IFN (96). Oncolytic virotherapy was also able to sensitize a model of triple-negative breast cancer to ICB (97). In the clinical setting, intravenous oncolytic Orthoreovirus increased T cell infiltration in primary and metastatic brain cancer and up-regulated IFN-regulated gene expression and PD-L1 expression, creating a favorable TME for subsequent ICB therapy (98). Another clinical trial using an attenuated herpes simplex virus type 1, followed by anti-PD-1 therapy for melanoma patients resulted in a 33% complete response rate, with increased CD8+ T cells, and increased PD-L1 protein and IFNγ gene expression in responders. Baseline CD8+ T cell infiltration or a baseline IFNγ signature was not associated with response (99). Oncolytic viral therapy is therefore a potential future option to skew the TME toward an ICB responsive phenotype, provided that at least some of the tumor is accessible for local injection.
Altering Physical Factors of Tumors to Improve Response
Recent research has highlighted that, besides biological, and chemical cues from the microenvironment, physical cues can also greatly alter cellular behavior of cancer cells. Abnormal vasculature and barriers to perfusion, such as high interstitial pressure within tumors can antagonize the effectiveness of ICB by promoting TME-mediated immune suppression. The impaired perfusion capacity of tumor blood vessels helps to create an immune cell unfavorable TME (100, 101). However, these physical characteristics can vary greatly between tumor types. For example, biomarkers used for typical ICB-responsive cancer-types, such as mutational burden, level of tumor-infiltrating T lymphocytes or expression of immune-checkpoints are not predictive for glioblastoma, partly due to the different physical environment (102).
Normalizing Tumor Vasculature and Immune Cell Infiltration
Responsive tumors are characterized as having greater immune cell infiltrate, and the baseline infiltration level of T cells has important predictive and prognostic implications for ICB (103, 104). Although activated T cells are observed in the periphery of non-responsive tumors after ICB, they often fail to infiltrate the tumor itself (105). Tumors exploit many mechanisms to limit immune cell infiltration. Proangiogenic growth factors downregulate adhesion molecule expression, limiting extravasation of immune cells across the tumor endothelium (101). The tumor endothelium can be manipulated through selective blockade of angiogenic factors including VEGF and endothelin-1 (105, 106), or by increasing VE-cadherin expression (107), resulting in increased T-cell infiltration. Combination blockade of VEGFA and angiopoietin-2 normalized tumor blood vessels and increased lymphocyte infiltration, improving outcomes when combined with anti-PD-1 in B16-OVA and MC38 mouse models (108). A small clinical study in 10 patients with metastatic renal cell carcinoma found that pre-treatment with the VEGF blocking drug bevacizumab followed by a combination of bevacizumab and PD-L1 resulted in a 40% response rate, which was high compared to historical controls for either agent alone (109). A unique approach using vascular targeting peptides allowed targeted delivery of the pro-inflammatory cytokine LIGHT to tumor vessels in murine models (110). This approach normalized tumor vasculature and increased intratumoral effector T cells when combined with ICB, resulting in responses in otherwise immunotherapy-resistant tumors.
Abnormal tumor vasculature promotes resistance to ICB via various effects. Abnormal vasculature limits access of ICB antibodies into the TME and reduces oxygen availability, leading to a hypoxic TME. Hypoxia in turn, upregulates several immune checkpoints in the TME, including PD-L1, CD47, VISTA and 4-1BB (CD137), impairing anti-tumor responses. Furthermore, hypoxia facilitates recruitment of MDSCs and enhances their immunosuppressive function (111). The immunosuppressor TGF-β1 is promoted by overexpression of HIF-1α in tumor hypoxia, which fosters exclusion of T lymphocytes (71, 112). Targeting hypoxia-induced immunosuppression improves outcomes from ICB, but there are few studies addressing whether targeting hypoxia directly sensitizes to ICB. In one study, in vitro pre-treatment of B16-OVA tumor cells with GTN, an agonist of nitric oxide, inhibited hypoxia-induced resistance to CTL-mediated lysis (113). In another, combining the hypoxia reducing prodrug (TH-302) with checkpoint blockade in a mouse prostate cancer model significantly reduced the number of tumor infiltrating MDSCs and cured around 80% of mice (114).
Circumventing High Interstitial Pressures in Tumors
Another feature of the TME is “solid stress,” defined as a growth-induced increase of physical pressure, commonly exacerbated by overproduction of hyaluronic acid (HA). This leads to high interstitial pressure and vessel collapse which may result in the exclusion of immune cells (115). Treatment with pegvorhyaluronidase alfa, which enzymatically degrades HA, has been tested to ameliorate solid stress, increasing both infiltration of immune cells and intratumoral uptake of anti-PD-L1 antibodies. Pre-treatment with pegvorhyaluronidase alfa 24 h prior to anti-PD-L1 resulted in significant growth inhibition in an anti-PD-L1-resistant breast cancer model which was genetically engineered to express hyaluronan synthase 3 (116). Desmoplasia, the growth of fibrous or connective tissue produced by cancer-associated fibroblasts, can also contribute to solid stress. A dense fibrotic stroma is associated with poor prognosis and is a common feature of immunotherapy resistant tumors such as pancreatic and breast cancer. One approach to decrease tumor interstitial pressure is to reprogram cancer associated fibroblasts to a quiescent state using angiotensin receptor blockers. Treatment with tumor-targeted angiotensin receptor blockers increased tumor perfusion, reduced immunosuppression, and enhanced the efficacy of anti-PD-1 treatment (117). Fibroblast function is also modulated by hypoxia, which induces signaling by CXCL12 via CXCR4, which promotes CAF recruitment, activation, and matrix production. Inhibition of CXCR4 signaling alleviated solid stress and increased the response to anti-CTLA4/anti PD-1 ICB in three metastatic breast cancer models (118).
Altering the Microbiome to Augment Immune Cells Within the TME
The microbiome has been shown to directly impact the TME, as antibiotic-treated or germ-free mice have defective tumor-infiltrating myeloid-derived cells with lower cytokine production and tumor necrosis after CpG-oligonucleotide treatment compared to controls (119). Conversely, commercialization with the bacterial species Bifidobacterium improved dendritic cell function and subsequent tumor-killing capabilities of cytotoxic T cells, resulting in reduced growth of subcutaneous melanoma xenograft models in mice. Additionally, Bifidobacterium administration in combination with anti-PD-L1 nearly abolished tumor growth (120). Other bacterial species such as Bacteroides thetaiotamicron and non-toxigenic B. fragilis have shown to improve antitumor cytotoxic T-cell immunity improving the efficacy of anti-CTLA-4 in multiple cancer mouse models (121). In melanoma patients treated with anti-PD-1, a positive correlation was found between the number of tumor infiltrating CD8+ T cells and the abundance of Faecalibacterium in responders (122). Several studies have found a link between the fecal microbiome and response to ICB (120–125). Fecal transplants from responding patients into mice has shown to improve tumor control, augment T cell responses, and increase the efficacy of anti-PD-L1 therapy (124) or PD-1 therapy (125), showing promise as a potential sensitizing strategy.
Modulating Gene Signatures Toward a Permissive TME
In the context of immune checkpoint blockade, important biological changes in the tumor microenvironment are reflected in changes in gene expression. Gene expression data from pre-treatment melanoma samples revealed immune-related signatures that were highly expressed in patients whose tumors responded to anti-CTLA4 or anti-PD-1 therapy, compared to non-responders (73, 126). In another study using bulk RNAseq data, non-responders to anti-PD-1 had higher pre-treatment expression of genes involved in mesenchymal transition, immunosuppression (including genes associated with wound healing and angiogenesis), and monocyte and macrophage chemotaxis (127). More recently, time-dependent transcriptomic changes have been associated with response to checkpoint blockade. RNA sequencing before and during anti-PD-1 therapy showed that tumor samples from responsive patients displayed upregulation of immune checkpoint genes and activation of response-specific transcriptional networks (128). Taken together, these results highlight the importance of transcriptomic changes in the tumor microenvironment. Moreover, it raises the possibility that manipulating gene expression patterns in the tumor microenvironment will impact treatment with ICB.
As proof of concept of this approach, gene expression changes in non-responsive tumors to ICB were used to inform computational drug discovery. Using single cell data of 33 melanoma patients, Jerby-Arnon et al. identified a transcriptional program expressed in malignant melanoma cells indicating a poor response to checkpoint blockade (129). They linked this poorer response to a less permissive TME, as evidenced by intratumoral niches of T-cell depletion. In a drug screen on 131 cell lines, they found their transcriptional signature to be antagonized by CDK4/6-inhibitors such as abemaciclib and palbociclib. Combining CDK4/6-inhibitors with checkpoint blockade inhibited tumor outgrowth in mouse models of melanoma, validating the approach of using gene signatures and bioinformatics interrogation to identify effective combination therapies (129).
In our own work, we used a systems biology approach to contrast responsive and non-responsive tumors prior to therapy in two murine models, to identify upstream regulators of the gene signature response signature to ICB (6, 9, 10). We found that responsive tumors were characterized by an inflammatory gene expression signature consistent with upregulation of STAT1 and TLR3 signaling, and down-regulation of IL-10 signaling. Therapeutically targeting these drivers using poly(I:C), IFNγ and an anti-IL-10 monoclonal antibody sensitized the TME and significantly increased response to ICB in multiple murine models. The triple combination was superior to any of the single drugs, validating the approach to identify complex therapeutic combinations for tumor sensitization (9). Similarly, by interrogating drug repurposing databases for the response-associated gene expression profile of anti-CTLA4 treated mice, we identified all-trans retinoic acid as a potential drug candidate to improve outcomes to ICB therapy. Treatment with all-trans retinoic acid indeed significantly increased the response rate when combined with anti-CTLA4 over either treatment alone (10). Bioinformatics approaches will facilitate rapid identification of new ways to sensitize the TME.
Discussion
Going Forward: Personalized Biomarker-Based Adaptive Therapy
There are likely to be many different ways to increase the chance that a patient will respond to ICB, and these are likely to differ between individuals. A personalized pathway to improve treatment effectiveness may be possible in the future (Figure 3). We propose that a pre-treatment biopsy will determine the baseline TME. If the patient's tumor has many characteristics associated with a responsive TME, they can start treatment with ICB. If not, a sensitizing therapy could be considered first, based on the genetic and immunological profile of the tumor. If, for example, the biopsy demonstrates many macrophages, but little IFN/STAT1 signaling, therapy aimed to polarize macrophages could be considered; if there are many T-regs in the biopsy, therapy aimed at reducing those could be considered. An on-treatment biopsy shortly after initiation of the sensitization strategy can verify whether the biological endpoint has been achieved. If so, the patient can progress to treatment with ICB. If not, another sensitization strategy could be considered, or another therapy altogether. Although this proposal is attractive, and would prevent patients from undergoing potentially futile therapy with attendant physical and financial toxicity, none of the biomarkers discussed are currently robust enough to justify withholding ICB in patients with an appropriate indication, and none of the sensitization strategies have been clinically validated in prospective trials.
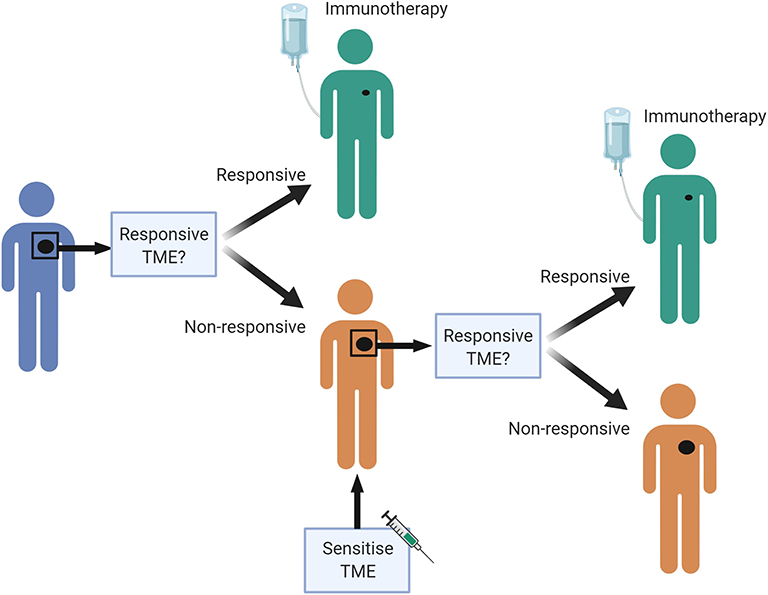
Figure 3. A personalized pre-treatment strategy to optimize outcomes to immune checkpoint blockade. We propose a pre-treatment approach to treating cancer patients, where tumors displaying features of a non-responsive TME can be first sensitized to attain a favorable TME, to improve chances of response to immune checkpoint blockade. Figures made in ©BioRender-biorender.com. Adapted from a figure we published recently in Science Translational Medicine (7).
How can we progress toward personalized immunotherapy and sensitization? Firstly, preclinical studies must include more robust study of scheduling effects. As the anti-tumor immune response is a highly orchestrated program involving many cells from both innate and adaptive immunity, changing over time, it is likely that temporal aspects are important for optimal immunological control, similar to the antiviral response (6, 130). However, as discussed above, most murine cancer studies do not rigorously study scheduling when investigating combination treatments. Secondly, human window of opportunity trials could help screen for drugs that induce a response-associated TME, by making use of the short period between cancer diagnostic biopsy and primary surgery (131). Sequential biopsies in clinical trials can also provide more insight into how drugs change the TME. Lastly, bioinformatics approaches will also need to continue to develop, to improve our understanding of a responsive TME (132).
The future goal of this personalized biomarker-based adaptive treatment approach is to give each patient the best chance of a successful response to immunotherapy.
Author Contributions
RZ and WL conceptualized and wrote the review. WC contributed to sections of the review. RL, AN, and MM provided critical review and editing of the manuscript. RZ designed and generated the figures. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
RZ was funded by the Children's Leukemia and Cancer Research Foundation and the Sock it to Sarcoma! Foundation. WC was funded by a Western Australian Cancer and Palliative Care Network fellowship. WL was funded by an NHMRC RD Wright Fellowship, a Simon Lee Fellowship and a Fellowship from the Cancer Council of Western Australia. The National Centre for Asbestos Related Diseases was supported by NHMRC Centre of Research Excellence grant 1107043.
Conflict of Interest
Patent application pertaining to aspects of discussed work: RZ, RL, and WL (PCT/AU2019/050259, “Method for immunotherapy drug treatment”). AN, MM, RL, WL research funding and consultancy for Douglas Pharmaceuticals. WL research funding from AstraZeneca. AN consultant or advisory boards for Boehringer Ingelheim, Bayer Pharmaceuticals, Roche Pharmaceutics, Merck Sharp Dohme, Pharmabcine, Atara biotherapeutics and Trizell; research funding from AstraZeneca. MM advisory boards for Merck Sharp & Dohme, Bristol-Myers Squibb, Roche, Astra-Zeneca.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Lesterhuis WJ, Haanen JB, Punt CJ. Cancer immunotherapy–revisited. Nat Rev Drug Discov. (2011) 10:591–600. doi: 10.1038/nrd3500
2. Hoos A. Development of immuno-oncology drugs — from CTLA4 to PD1 to the next generations. Nat Rev Drug Discov. (2016) 15:235. doi: 10.1038/nrd.2015.35
3. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. (2014) 371:2189–99. doi: 10.1056/NEJMoa1406498
4. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non–small-cell lung cancer. N Engl J Med. (2015) 372:2018–28. doi: 10.1056/NEJMoa1501824
5. Eggermont AMM, Kroemer G, Zitvogel L. Immunotherapy and the concept of a clinical cure. Eur J Cancer. (2013) 49:2965–7. doi: 10.1016/j.ejca.2013.06.019
6. Lesterhuis WJ, Bosco A, Millward MJ, Small M, Nowak AK, Lake RA. Dynamic versus static biomarkers in cancer immune checkpoint blockade: unravelling complexity. Nat Rev Drug Discov. (2017) 16:264. doi: 10.1038/nrd.2016.233
7. The Lancet Oncology. Calling time on the immunotherapy gold rush. Lancet Oncol. (2017) 18:981. doi: 10.1016/S1470-2045(17)30521-1
8. Nature Medicine. Rationalizing combination therapies. Nat Med. (2017) 23:1113. doi: 10.1038/nm.4426
9. Zemek RM, De Jong E, Chin WL, Schuster IS, Fear VS, Casey TH, et al. Sensitization to immune checkpoint blockade through activation of a STAT1/NK axis in the tumor microenvironment. Sci Transl Med. (2019) 11:eaav7816. doi: 10.1126/scitranslmed.aav7816
10. Lesterhuis WJ, Rinaldi C, Jones A, Rozali EN, Dick IM, Khong A, et al. Network analysis of immunotherapy-induced regressing tumours identifies novel synergistic drug combinations. Sci Rep. (2015) 5:12298. doi: 10.1038/srep12298
11. Sutmuller RP, Van Duivenvoorde LM, Van Elsas A, Schumacher TN, Wildenberg ME, Allison JP, et al. Synergism of cytotoxic T lymphocyte–associated antigen 4 blockade and depletion of CD25+ regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J Exp Med. (2001) 194:823–32. doi: 10.1084/jem.194.6.823
12. Grosso JF, Jure-Kunkel MN. CTLA-4 blockade in tumor models: an overview of preclinical and translational research. Cancer Immunology. (2013) 13:5.
13. van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti–cytotoxic T lymphocyte–associated antigen 4 (Ctla-4) and granulocyte/macrophage colony-stimulating factor (Gm-Csf)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. (1999) 190:355. doi: 10.1084/jem.190.3.355
14. Wilson DR, Sen R, Sunshine JC, Pardoll DM, Green JJ, Kim YJ. Biodegradable STING agonist nanoparticles for enhanced cancer immunotherapy. Nanomedicine. (2018) 14:237–46. doi: 10.1016/j.nano.2017.10.013
15. Shen H, Yang ES-H, Conry M, Fiveash J, Contreras C, Bonner JA, et al. Predictive biomarkers for immune checkpoint blockade and opportunities for combination therapies. Genes Dis. (2019) 6:232–46. doi: 10.1016/j.gendis.2019.06.006
16. Nakamura Y. Biomarkers for immune checkpoint inhibitor-mediated tumor response and adverse events. Front. Med. (2019) 6:127. doi: 10.3389/fmed.2019.00119
17. Zuazo M, Arasanz H, Fernández-Hinojal G, García-Granda MJ, Gato M, Bocanegra A, et al. Functional systemic CD4 immunity is required for clinical responses to PD-L1/PD-1 blockade therapy. EMBO Mol Med. (2019) 11:e10293. doi: 10.15252/emmm.201910293
18. Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, et al. Systemic immunity is required for effective cancer immunotherapy. Cell. (2017) 168:487–502.e15. doi: 10.1016/j.cell.2016.12.022
19. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. (2018) 24:541–50. doi: 10.1038/s41591-018-0014-x
20. Hegde PS, Karanikas V, Evers S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin Cancer Res. (2016) 22:1865. doi: 10.1158/1078-0432.CCR-15-1507
21. Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. (2019) 18:197–218. doi: 10.1038/s41573-018-0007-y
22. Blank CU, Haanen JB, Ribas A, Schumacher TN. The “cancer immunogram”. Science. (2016) 352:658–60. doi: 10.1126/science.aaf2834
23. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature. (2019) 572:392–6. doi: 10.1038/s41586-019-1456-0
24. Duruisseaux M, Martínez-Cardús A, Calleja-Cervantes ME, Moran S, Castro de Moura M, Davalos V, et al. Epigenetic prediction of response to anti-PD-1 treatment in non-small-cell lung cancer: a multicentre, retrospective analysis. Lancet Respirat Med. (2018) 6:771–81. doi: 10.1016/S2213-2600(18)30284-4
25. Cottrell TR, Thompson ED, Forde PM, Stein JE, Duffield AS, Anagnostou V, et al. Pathologic features of response to neoadjuvant anti-PD-1 in resected non-small-cell lung carcinoma: a proposal for quantitative immune-related pathologic response criteria (irPRC). Annals Oncol. (2018) 29:1853–60. doi: 10.1093/annonc/mdy218
26. Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, et al. A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nat Med. (2018) 24:1178–91. doi: 10.1038/s41591-018-0085-8
27. Lesterhuis WJ, Salmons J, Nowak AK, Rozali EN, Khong A, Dick IM, et al. Synergistic effect of CTLA-4 blockade and cancer chemotherapy in the induction of anti-tumor immunity. PLoS ONE. (2013) 8:e61895. doi: 10.1371/journal.pone.0061895
28. Homet Moreno B, Zaretsky JM, Garcia-Diaz A, Tsoi J, Parisi G, Robert L, et al. Response to programmed cell death-1 blockade in a murine melanoma syngeneic model requires costimulation, CD4, and CD8 T Cells. Cancer Immunol Res. (2016) 4:845–57. doi: 10.1158/2326-6066.CIR-16-0060
29. Rigo V, Emionite L, Daga A, Astigiano S, Corrias MV, Quintarelli C, et al. Combined immunotherapy with anti-PDL-1/PD-1 and anti-CD4 antibodies cures syngeneic disseminated neuroblastoma. Sci Rep. (2017) 7:14049. doi: 10.1038/s41598-017-14417-6
30. Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. (2016) 167:397–404.e9. doi: 10.1016/j.cell.2016.08.069
31. Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane J-P, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun. (2017) 8:1136. doi: 10.1038/s41467-017-01062-w
32. Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci USA. (2010) 107:4275–80. doi: 10.1073/pnas.0915174107
33. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. (2013) 369:122–33. doi: 10.1056/NEJMoa1302369
34. Das R, Verma R, Sznol M, Boddupalli CS, Gettinger SN, Kluger H, et al. Combination therapy with anti–CTLA-4 and anti–PD-1 leads to distinct immunologic changes in vivo. J Immunol. (2015) 194:950–9. doi: 10.4049/jimmunol.1401686
35. McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. (2016) 351:1463–9. doi: 10.1126/science.aaf1490
36. Tang H, Wang Y, Chlewicki LK, Zhang Y, Guo J, Liang W, et al. Facilitating T cell infiltration in tumor microenvironment overcomes resistance to PD-L1 blockade. Cancer Cell. (2016) 29:285–96. doi: 10.1016/j.ccell.2016.02.004
37. Dahan R, Sega E, Engelhardt J, Selby M, Korman Alan J, Ravetch Jeffrey V. FcγRs modulate the anti-tumor activity of antibodies targeting the PD-1/PD-L1 axis. Cancer Cell. (2015) 28:285–95. doi: 10.1016/j.ccell.2015.08.004
38. Zhang T, Song X, Xu L, Ma J, Zhang Y, Gong W, et al. The binding of an anti-PD-1 antibody to FcγR? has a profound impact on its biological functions. Cancer Immunol Immunother. (2018) 67:1079–90. doi: 10.1007/s00262-018-2160-x
39. Hodge JW, Garnett CT, Farsaci B, Palena C, Tsang K-Y, Ferrone S, et al. Chemotherapy-induced immunogenic modulation of tumor cells enhances killing by cytotoxic T lymphocytes and is distinct from immunogenic cell death. Int J Cancer. (2013) 133:624–36. doi: 10.1002/ijc.28070
40. Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity. (2016) 44:343–54. doi: 10.1016/j.immuni.2015.11.024
41. Nowak AK, Robinson BWS, Lake RA. Synergy between chemotherapy and immunotherapy in the treatment of established murine solid tumors. Cancer Res. (2003) 63:4490–6.
42. Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, et al. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. (2013) 2013:32–42. doi: 10.1158/2326-6066.CIR-13-0013
43. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti–CTLA-4 therapy against melanoma. J Exp Med. (2013) 210:1695–710. doi: 10.1084/jem.20130579
44. Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, et al. Activating Fc γ receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J Exp Med. (2013) 210:1685–93. doi: 10.1084/jem.20130573
45. Tang F, Du X, Liu M, Zheng P, Liu Y. Anti-CTLA-4 antibodies in cancer immunotherapy: selective depletion of intratumoral regulatory T cells or checkpoint blockade? Cell Biosci. (2018) 8:30. doi: 10.1186/s13578-018-0229-z
46. Du X, Liu M, Su J, Zhang P, Tang F, Ye P, et al. Uncoupling therapeutic from immunotherapy-related adverse effects for safer and effective anti-CTLA-4 antibodies in CTLA4 humanized mice. Cell Res. (2018) 28:433–47. doi: 10.1038/s41422-018-0012-z
47. Sharma A, Subudhi SK, Blando J, Scutti J, Vence L, Wargo J, et al. Anti-CTLA-4 immunotherapy does not deplete FOXP3<sup>+</sup> regulatory T cells (Tregs) in human cancers. Clin Cancer Res. (2019) 25:1233–8. doi: 10.1158/1078-0432.CCR-18-0762
48. Hamid O, Schmidt H, Nissan A, Ridolfi L, Aamdal S, Hansson J, et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J Transl Med. (2011) 9:204. doi: 10.1186/1479-5876-9-204
49. Stromnes IM, Hulbert A, Pierce RH, Greenberg PD, Hingorani SR. T-cell localization, activation, and clonal expansion in human pancreatic ductal adenocarcinoma. Cancer Immunol Res. (2017) 5:978–991. doi: 10.1158/2326-6066.CIR-16-0322
50. Asaka S, Yen T, Wang T, Shih I, Gaillard S. T cell-inflamed phenotype and increased Foxp3 expression in infiltrating T-cells of mismatch-repair deficient endometrial cancers. Modern Pathol. (2019) 32:576–84. doi: 10.1038/s41379-018-0172-x
51. van de Ven R, Niemeijer A-LN, Stam AGM, Hashemi SMS, Slockers CG, Daniels JM, et al. High PD-1 expression on regulatory and effector T-cells in lung cancer draining lymph nodes. ERJ Open Res. (2017) 3:00110–2016. doi: 10.1183/23120541.00110-2016
52. Vargas FA, Furness AJS, Solomon I, Joshi K, Mekkaoui L, Lesko MH, et al. Fc-optimized anti-CD25 depletes tumor-infiltrating regulatory t cells and synergizes with PD-1 blockade to eradicate established tumors. Immunity. (2017) 46:577–86. doi: 10.1016/j.immuni.2017.03.013
53. Di Pilato M, Kim EY, Cadilha BL, Prüßmann JN, Nasrallah MN, Seruggia D, et al. Targeting the CBM complex causes Treg cells to prime tumours for immune checkpoint therapy. Nature. (2019) 570:112–6. doi: 10.1038/s41586-019-1215-2
54. Viitala M, Virtakoivu R, Tadayon S, Rannikko J, Jalkanen S, Hollmén M. Immunotherapeutic blockade of macrophage clever-1 reactivates the CD8<sup>+</sup> T-cell response against immunosuppressive tumors. Clin Cancer Res. (2019) 25:3289. doi: 10.1158/1078-0432.CCR-18-3016
55. Rodell CB, Arlauckas SP, Cuccarese MF, Garris CS, Li R, Ahmed MS, et al. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat Biomed Eng. (2018) 2:578–88. doi: 10.1038/s41551-018-0236-8
56. Santegoets SJ, Stam AG, Lougheed SM, Gall H, Jooss K, Sacks N, et al. Myeloid derived suppressor and dendritic cell subsets are related to clinical outcome in prostate cancer patients treated with prostate GVAX and ipilimumab. J Immunother Cancer. (2014) 2:31. doi: 10.1186/s40425-014-0031-3
57. Gebhardt C, Sevko A, Jiang H, Lichtenberger R, Reith M, Tarnanidis K, et al. Myeloid cells and related chronic inflammatory factors as novel predictive markers in melanoma treatment with ipilimumab. Clin Cancer Res. (2015) 21:5453–9. doi: 10.1158/1078-0432.CCR-15-0676
58. Meyer C, Cagnon L, Costa-Nunes C, Baumgaertner P, Montandon N, Leyvraz L, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother. (2014) 63:247–57. doi: 10.1007/s00262-013-1508-5
59. Kitano S, Postow MA, Cortez C, Rasalan T, Gallardo HF, Panageas K, et al. Myeloid-derived suppressor cell quantity prior to treatment with ipilimumab at 10mg/kg to predict for overall survival in patients with metastatic melanoma. J Clin Oncol. (2012) 30:2518. doi: 10.1200/jco.2012.30.15_suppl.2518
60. Delaunay M, Guibert N, Lusque A, Farella M, Boubekeur N, Gouin S, et al. Baseline circulating myeloid-derived suppressor cells and response to PD-1 inhibitor in non-small cell lung cancer patients. J Clin Oncol. (2018) 36:145. doi: 10.1200/JCO.2018.36.5_suppl.145
61. De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature. (2016) 539:443–7. doi: 10.1038/nature20554
62. Sullivan RJ, Hong DS, Tolcher AW, Patnaik A, Shapiro G, Chmielowski B, et al. Initial results from first-in-human study of IPI-549, a tumor macrophage-targeting agent, combined with nivolumab in advanced solid tumors. J Clin Oncol. (2018) 36(15_suppl):3013. doi: 10.1200/JCO.2018.36.15_suppl.3013
63. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. (2014) 74:5057–69. doi: 10.1158/0008-5472.CAN-13-3723
64. Holmgaard RB, Zamarin D, Lesokhin A, Merghoub T, Wolchok JD. Targeting myeloid-derived suppressor cells with colony stimulating factor-1 receptor blockade can reverse immune resistance to immunotherapy in indoleamine 2,3-dioxygenase-expressing tumors. EBioMed. (2016) 6:50–8. doi: 10.1016/j.ebiom.2016.02.024
65. Mao Y, Eissler N, Blanc KL, Johnsen JI, Kogner P, Kiessling R. Targeting suppressive myeloid cells potentiates checkpoint inhibitors to control spontaneous neuroblastoma. Clin Cancer Res. (2016) 22:3849–59. doi: 10.1158/1078-0432.CCR-15-1912
66. Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Translat Med. (2014) 6:237ra67. doi: 10.1126/scitranslmed.3007974
67. Stiff A, Trikha P, Mundy-Bosse B, McMichael E, Mace TA, Benner B, et al. Nitric oxide production by myeloid-derived suppressor cells plays a role in impairing Fc receptor–mediated natural killer cell function. Clin Cancer Res. (2018) 24:1891–1904. doi: 10.1158/1078-0432.CCR-17-0691
68. Sagiv-Barfi I, Kohrt HEK, Czerwinski DK, Ng PP, Chang BY, Levy R. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc Natl Acad Sci USA. (2015) 112:E966. doi: 10.1073/pnas.1500712112
69. Sun H, Huang Q, Huang M, Wen H, Lin R, Zheng M, et al. Human CD96 correlates to natural killer cell exhaustion and predicts the prognosis of human hepatocellular carcinoma. Hepatology. (2019) 70:168–83. doi: 10.1002/hep.30347
70. Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. (2018) 172:1022–37.e14. doi: 10.1016/j.cell.2018.01.004
71. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. (2018) 554:7693. doi: 10.1038/nature25501
72. Minn AJ. Interferons and the immunogenic effects of cancer therapy. Trends Immunol. (2015) 36:725–37. doi: 10.1016/j.it.2015.09.007
73. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. (2014) 515:563–7. doi: 10.1038/nature14011
74. Hervieu A, Rébé C, Végran F, Chalmin F, Bruchard M, Vabres P, et al. Dacarbazine-mediated upregulation of NKG2D ligands on tumor cells activates NK and CD8 T cells and restrains melanoma growth. J Invest Dermatol. (2013) 133:499–508. doi: 10.1038/jid.2012.273
75. Liu WM, Fowler DW, Smith P, Dalgleish AG. Pre-treatment with chemotherapy can enhance the antigenicity and immunogenicity of tumours by promoting adaptive immune responses. Br J Cancer. (2009) 102:115–23. doi: 10.1038/sj.bjc.6605465
76. Bald T, Landsberg J, Lopez-Ramos D, Renn M, Glodde N, Jansen P, et al. Immune cell–poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov. (2014) 4:674–87. doi: 10.1158/2159-8290.CD-13-0458
77. Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, et al. Cancer cell–autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. (2014) 20:1301–9. doi: 10.1038/nm.3708
78. Shi LZ, Fu T, Guan B, Chen J, Blando JM, Allison JP, et al. Interdependent IL-7 and IFN-γ signalling in T-cell controls tumour eradication by combined α-CTLA-4+α-PD-1 therapy. Nat Commun. (2016) 7:12335. doi: 10.1038/ncomms12335
79. Jacquelot N, Yamazaki T, Roberti MP, Duong CPM, Andrews MC, Verlingue L, et al. Sustained Type I interferon signaling as a mechanism of resistance to PD-1 blockade. Cell Res. (2019) 29:846–61. doi: 10.1038/s41422-019-0224-x
80. Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell. (2016) 167:1540–54.e12. doi: 10.1016/j.cell.2016.11.022
81. Chen J, Cao Y, Markelc B, Kaeppler J, Vermeer JAF, Muschel RJ. Type I IFN protects cancer cells from CD8+ T cell–mediated cytotoxicity after radiation. J Clin Invest. (2019) 129:4224–38. doi: 10.1172/JCI127458
82. Benci JL, Johnson LR, Choa R, Xu Y, Qiu J, Zhou Z, et al. Opposing functions of interferon coordinate adaptive and innate immune responses to cancer immune checkpoint blockade. Cell. (2019) 178:933–48.e14. doi: 10.1016/j.cell.2019.07.019
83. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. (2016) 375:819–29. doi: 10.1056/NEJMoa1604958
84. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. (2017) 7:188–201. doi: 10.1158/2159-8290.CD-16-1223
85. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell. (2015) 28:690–714. doi: 10.1016/j.ccell.2015.10.012
86. Vanpouille-Box C, Formenti SC, Demaria S. Toward precision radiotherapy for use with immune checkpoint blockers. Clin Cancer Res. (2018) 24:259. doi: 10.1158/1078-0432.CCR-16-0037
87. Shaverdian N, Lisberg AE, Bornazyan K, Veruttipong D, Goldman JW, Formenti SC, et al. Previous radiotherapy and the clinical activity and toxicity of pembrolizumab in the treatment of non-small-cell lung cancer: a secondary analysis of the KEYNOTE-001 phase 1 trial. Lancet Oncol. (2017) 18:895–903. doi: 10.1016/S1470-2045(17)30380-7
88. Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. (2014) 41:843–52. doi: 10.1016/j.immuni.2014.10.019
89. Baird JR, Monjazeb AM, Shah O, McGee H, Murphy WJ, Crittenden MR, et al. Stimulating innate immunity to enhance radiation therapy–induced tumor control. Int J Radiat Oncol Biol Phys. (2017) 99:362–73. doi: 10.1016/j.ijrobp.2017.04.014
90. Corrales L, Glickman Laura H, McWhirter Sarah M, Kanne David B, Sivick Kelsey E, Katibah George E, et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. (2015) 11:1018–30. doi: 10.1016/j.celrep.2015.04.031
91. Charlebois R, Allard B, Allard D, Buisseret L, Turcotte M, Pommey S, et al. PolyI: C and CpG synergize with anti-ErbB2 mAb for treatment of breast tumors resistant to immune checkpoint inhibitors. Cancer Res. (2017) 77:312–9. doi: 10.1158/0008-5472.CAN-16-1873
92. Tel J, Sittig SP, Blom RAM, Cruz LJ, Schreibelt G, Figdor CG, et al. Targeting uptake receptors on human plasmacytoid dendritic cells triggers antigen cross-presentation and robust type I IFN secretion. J Immunol. (2013) 191:5005–12. doi: 10.4049/jimmunol.1300787
93. Mangsbo SM, Sandin LC, Anger K, Korman AJ, Loskog A, Tötterman TH. Enhanced tumor eradication by combining CTLA-4 or PD-1 blockade with CpG therapy. J Immunother. (2010) 33:225–35. doi: 10.1097/CJI.0b013e3181c01fcb
94. Nagato T, Lee Y-R, Harabuchi Y, Celis E. Combinatorial immunotherapy of polyinosinic–polycytidylic acid and blockade of programmed death-ligand 1 induce effective CD8 T-cell responses against established tumors. Clin Cancer Res. (2014) 20:1223–34. doi: 10.1158/1078-0432.CCR-13-2781
95. Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang S-Y, et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature. (2018) 564:439–43. doi: 10.1038/s41586-018-0705-y
96. Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. (2014) 6:226ra32. doi: 10.1126/scitranslmed.3008095
97. Bourgeois-Daigneault M-C, Roy DG, Aitken AS, El Sayes N, Martin NT, Varette O, et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci Translat Med. (2018) 10:eaao1641. doi: 10.1126/scitranslmed.aao1641
98. Samson A, Scott KJ, Taggart D, West EJ, Wilson E, Nuovo GJ, et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci Transl Med. (2018) 10:eaam7577. doi: 10.1126/scitranslmed.aam7577
99. Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, et al. Oncolytic virotherapy promotes intratumoral t cell infiltration and improves anti-PD-1 immunotherapy. Cell. (2017) 170:1109–19.e10. doi: 10.1016/j.cell.2017.08.027
100. Huang Y, Goel S, Duda DG, Fukumura D, Jain RK. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. (2013) 73:2943. doi: 10.1158/0008-5472.CAN-12-4354
101. Lanitis E, Irving M, Coukos G. Targeting the tumor vasculature to enhance T cell activity. Curr Opin Immunol. (2015) 33:55–63. doi: 10.1016/j.coi.2015.01.011
102. Garg AD, Vandenberk L, Van Woensel M, Belmans J, Schaaf M, Boon L, et al. Preclinical efficacy of immune-checkpoint monotherapy does not recapitulate corresponding biomarkers-based clinical predictions in glioblastoma. OncoImmunology. (2017) 6:e1295903. doi: 10.1080/2162402X.2017.1295903
103. Daud AI, Loo K, Pauli ML, Sanchez-Rodriguez R, Sandoval PM, Taravati K, et al. Tumor immune profiling predicts response to anti–PD-1 therapy in human melanoma. J Clin Invest. (2016) 126:3447–52. doi: 10.1172/JCI87324
104. Disis M. Immunologic biomarkers as correlates of clinical response to cancer immunotherapy. Cancer Immunol Immunother. (2011) 60:433–42. doi: 10.1007/s00262-010-0960-8
105. Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, et al. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med. (2008) 14:28. doi: 10.1038/nm1699
106. Gremonprez F, Descamps B, Izmer A, Vanhove C, Vanhaecke F, De Wever O, et al. Pretreatment with VEGF(R)-inhibitors reduces interstitial fluid pressure, increases intraperitoneal chemotherapy drug penetration, and impedes tumor growth in a mouse colorectal carcinomatosis model. Oncotarget. (2015) 6:29889–900. doi: 10.18632/oncotarget.5092
107. Zhao Y, Ting KK, Li J, Cogger VC, Chen J, Johansson-Percival A, et al. Targeting vascular endothelial-cadherin in tumor-associated blood vessels promotes T-cell–mediated immunotherapy. Cancer Res. (2017) 77:4434. doi: 10.1158/0008-5472.CAN-16-3129
108. Schmittnaegel M, Rigamonti N, Kadioglu E, Cassará A, Wyser Rmili C, Kiialainen A, et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci Transl Med. (2017) 9:eaak9670. doi: 10.1126/scitranslmed.aak9670
109. Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun. (2016) 7:12624. doi: 10.1038/ncomms12624
110. Johansson-Percival A, He B, Li Z-J, Kjellén A, Russell K, Li J, et al. De novo induction of intratumoral lymphoid structures and vessel normalization enhances immunotherapy in resistant tumors. Nat Immunol. (2017) 18:1207–17. doi: 10.1038/ni.3836
111. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. (2014) 211:781–90. doi: 10.1084/jem.20131916
112. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. (2018) 554:538–43. doi: 10.1038/nature25492
113. Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. (2014) 74:665–74. doi: 10.1158/0008-5472.CAN-13-0992
114. Jayaprakash P, Ai M, Liu A, Budhani P, Bartkowiak T, Sheng J, et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J Clin Invest. (2018) 128:5137–49. doi: 10.1172/JCI96268
115. Stylianopoulos T, Martin JD, Chauhan VP, Jain SR, Diop-Frimpong B, Bardeesy N, et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc Natl Acad Sci USA. (2012) 109:15101–8. doi: 10.1073/pnas.1213353109
116. Clift R, Souratha J, Garrovillo SA, Zimmerman S, Blouw B. Remodeling the tumor microenvironment sensitizes breast tumors to anti-programmed death-ligand 1 immunotherapy. Cancer Res. (2018) 79:4149–59. doi: 10.1158/0008-5472.CAN-18-3060
117. Chauhan VP, Chen IX, Tong R, Ng MR, Martin JD, Naxerova K, et al. Reprogramming the microenvironment with tumor-selective angiotensin blockers enhances cancer immunotherapy. Proc Natl Acad Sci USA. (2019) 116:10674–80. doi: 10.1073/pnas.1819889116
118. Chen IX, Chauhan VP, Posada J, Ng MR, Wu MW, Adstamongkonkul P, et al. Blocking CXCR4 alleviates desmoplasia, increases T-lymphocyte infiltration, and improves immunotherapy in metastatic breast cancer. Proc Natl Acad Sci USA. (2019) 116:4558–66. doi: 10.1073/pnas.1815515116
119. Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. (2013) 342:967–70. doi: 10.1126/science.1240527
120. Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal & Bifidobacterium < promotes antitumor immunity and facilitates anti–PD-L1 efficacy. Science. (2015) 350:1084–9. doi: 10.1126/science.aac4255
121. Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. (2015) 350:1079–84. doi: 10.1126/science.aad1329
122. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science. (2018) 359:97–103. doi: 10.1126/science.aan4236
123. Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Annals Oncol. (2017) 28:1368–79. doi: 10.1093/annonc/mdx108
124. Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre M-L, et al. The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science. (2018) 359:104–8. doi: 10.1126/science.aao3290
125. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science. (2018) 359:91–7. doi: 10.1126/science.aan3706
126. Ji R-R, Chasalow S, Wang L, Hamid O, Schmidt H, Cogswell J, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother. (2012) 61:1019–31. doi: 10.1007/s00262-011-1172-6
127. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. (2016) 165:35–44. doi: 10.1016/j.cell.2016.02.065
128. Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. (2017) 171:934–49.e15. doi: 10.1016/j.cell.2017.09.028
129. Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su M-J, Melms JC, et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell. (2018) 175:984–97.e24. doi: 10.1016/j.cell.2018.09.006
130. Park S-H, Rehermann B. Immune responses to HCV and other hepatitis viruses. Immunity. (2014) 40:13–24. doi: 10.1016/j.immuni.2013.12.010
131. Schmitz S, Duhoux F, Machiels J-P. Window of opportunity studies: do they fulfil our expectations? Cancer Treatment Rev. (2016) 43:50–7. doi: 10.1016/j.ctrv.2015.12.005
Keywords: cancer immunotherapy, immune checkpoints, sensitization, tumor microenvironment, PD-1, CTLA4, biomarkers, systems biology
Citation: Zemek RM, Chin WL, Nowak AK, Millward MJ, Lake RA and Lesterhuis WJ (2020) Sensitizing the Tumor Microenvironment to Immune Checkpoint Therapy. Front. Immunol. 11:223. doi: 10.3389/fimmu.2020.00223
Received: 13 November 2019; Accepted: 28 January 2020;
Published: 18 February 2020.
Edited by:
Umaimainthan Palendira, University of Sydney, AustraliaReviewed by:
David Escors, University College London, United KingdomLewis Zhichang Shi, University of Alabama at Birmingham, United States
Copyright © 2020 Zemek, Chin, Nowak, Millward, Lake and Lesterhuis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: W. Joost Lesterhuis, d2lsbGVtLmxlc3Rlcmh1aXNAdXdhLmVkdS5hdQ==