 Chloé Michaudel1
Chloé Michaudel1 Florent Bataille1Isabelle Maillet1Louis Fauconnier2
Florent Bataille1Isabelle Maillet1Louis Fauconnier2 Cyril Colas3,4
Cyril Colas3,4 Harry Sokol5Marjolène Straube5
Harry Sokol5Marjolène Straube5 Aurélie Couturier-Maillard1Laure Dumoutier6Jacques van Snick7Valérie F. Quesniaux1Dieudonnée Togbe1,2
Aurélie Couturier-Maillard1Laure Dumoutier6Jacques van Snick7Valérie F. Quesniaux1Dieudonnée Togbe1,2 Bernhard Ryffel1*
Bernhard Ryffel1*- 1Laboratory of Experimental and Molecular Immunology and Neurogenetics, UMR 7355 CNRS-University of Orleans, Orléans, France
- 2ArtImmune SAS, Orléans, France
- 3University of Orléans, CNRS ICOA, UMR7311, F-45067, Orléans, France
- 4CNRS, CBM, UPR4301, University Orléans, Orléans, France
- 5Avenir Team Gut Microbiota and Immunity, Equipe de Recherche Labélisée 1157, Institut National de la Santé et de la Recherche Médicale, Paris, France
- 6Institut de Duve, Université Catholique de Louvain, Brussels, Belgium
- 7Ludwig Institute for Cancer Research, Université Catholique de Louvain, Brussels, Belgium
Airborne ozone exposure causes severe lung injury and inflammation. The aryl hydrocarbon Receptor (AhR) (1), activated in pollutant-induced inflammation, is critical for cytokine production, especially IL-22 and IL-17A. The role of AhR in ozone-induced lung inflammation is unknown. We report here that chronic ozone exposure activates AhR with increased tryptophan and lipoxin A4 production in mice. AhR−/− mice show increased lung inflammation, airway hyperresponsiveness, and tissue remodeling with an increased recruitment of IL-17A and IL-22-expressing cells in comparison to control mice. IL-17A- and IL-22-neutralizing antibodies attenuate lung inflammation in AhR−/− and control mice. Enhanced lung inflammation and recruitment of ILC3, ILC2, and T cells were observed after T cell-specific AhR depletion using the AhRCD4cre-deficient mice. Together, the data demonstrate that ozone exposure activates AhR, which controls lung inflammation, airway hyperresponsiveness, and tissue remodeling via the reduction of IL-22 expression.
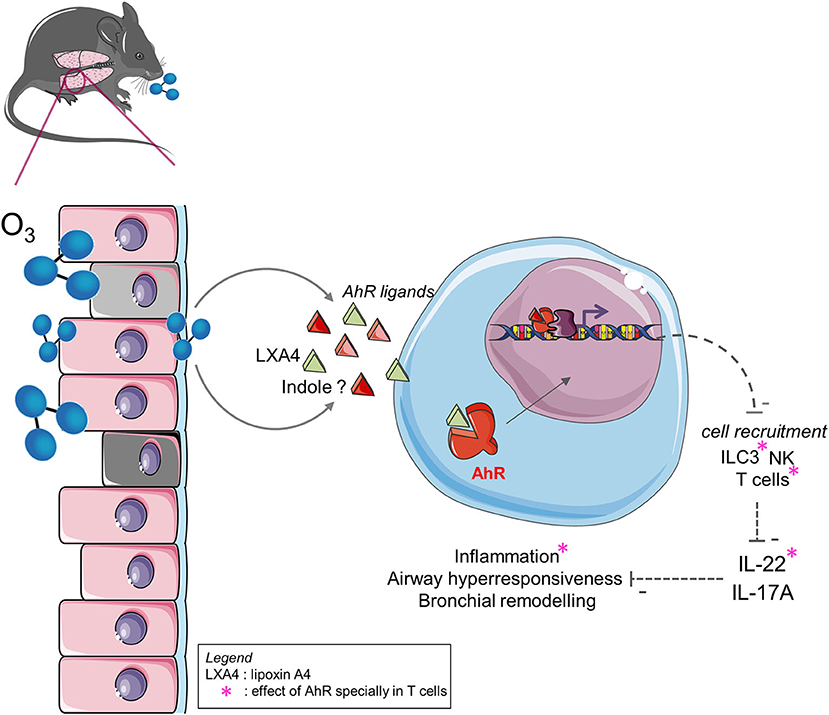
Graphical Abstract. Ozone exposure induces tryptophan metabolites and LXA4 that activates AhR. This activation likely control cell recruitment, TH2 and TH17/22 response, airway hyperresponsiveness and tissue remodeling with IL-22 dependent manner. AhR activation in lymphocytes influences cells recruitment, IL-22 expression and inflammation.
Introduction
Ozone is an abundant air pollutant that causes respiratory inflammation. Peaks of ozone correlate with severe respiratory disease, morbidity, mortality (2, 3), and hospital admissions (4–6). An increase of 10 μg/m3 of ozone exposure for 1 h daily induces an increase of 0.26% in mortality rate (7). The consequences of ozone exposure are especially deleterious for vulnerable populations with asthma or COPD (8, 9) and exacerbate asthma (10, 11). Ozone causes severe lung tissue damage, with inflammation and emphysema, loss of lung function, and airway hyperresponsiveness in human and mice (12–14).
The aryl hydrocarbon receptor (AhR) (1) is broadly expressed in immune cells and non-hematopoietic cells (e.g., epithelial cells). AhR is implicated in pollutant metabolism and/or degradation in response to polycyclic aromatic hydrocarbons. At steady state, AhR is cytoplasmic. In the absence of ligands, AhR resides in the cytoplasm under the control of a chaperone protein complex. Upon ligand binding, the AhR complex translocates into the nucleus, the chaperones are released, and AhR heterodimerizes with AhR Nuclear Translocator (ARNT) (15). This complex binds to Dioxin Response Elements (14) on the DNA and induces gene transcription, including P450 cytochrome and cytokines (e.g., IL-17A, IL-22). Non-canonical AhR signaling pathways have also been reported, either at the genomic level, through association with other transcription factors (e.g., NF-κB), or at the non-genomic level (e.g., through the release of the Src kinase) (16, 17). AhR is known to mediate several aspects of immune response and homeostatic maintenance, and its action is dependent on the context. For example, AhR can interact with NF-κB and STAT1 to inhibit IL-6 production after LPS treatment (18). AhR activation affects different pathways, such as T cell differentiation and antioxidant response. Furthermore, AhR induces ILC3 maintenance in the gut via Cyp1a1 (19) and triggers TH17 cells differentiation, promoting IL-17 and IL-22 production by inhibiting IFN-γ (20). The AhR activity is dependent on context, but also ligands. AhR ligands can be of endogenous (e.g., tryptophan metabolism derivatives from the microbiota) or of exogenous origin (e.g., Benzo[a]pyren, environmental pollutants, and dietary-derived compounds) (21). The response of AhR activation depends on the nature of the ligand. For example, AhR in PBMCs stimulated by TCDD (2,3,7,8-tetrachlorodibenzo-p-dioxine) induces IL-22 and IL-17A production, while stimulation by prostaglandin E2 (PGE2) has the opposite effect (22). AhR's effect on T cell polarization also depends on the nature of the ligand, as it can either promote TH17 or Treg cell differentiation (23). Taken together, the literature reported that AhR could have a principally protective effect, for example during major depressive disorder, multiple sclerosis, rheumatoid arthritis (24), and intestinal disease (16).
IL-17A has a pro-inflammatory role in several models of inflammation, such as asthma or colitis (25, 26). IL-17A could be produced by T cells, iNKT, NK, γδ T cells, and ILC3 and could modulate the production of other pro-inflammatory cytokines (e.g., IL-6 and IL-8), chemokines, and molecules involved in tissue remodeling, such as MMPs (27). During acute ozone exposure, IL-17A is produced by γδ T cells and iNKT (28, 29) and induces neutrophil recruitment and airway hyperresponsiveness. Upon chronic ozone exposure, IL-17A also induces inflammation, neutrophil recruitment, and airway hyperresponsiveness (30, 31). Furthermore, IL-17A induces M2 macrophage polarization and promotes apoptotic cell clearance (32). Similarly to IL-17A, IL-22 is produced by αβ T cells (TH17, TH22), γδ T cells, NK, and ILC3, after stimulation with IL-1β, TGF-β, or IL-23 and the transcription factor RORγt (23, 33, 34). IL-22 is a pro- or anti-inflammatory cytokine, depending on the inflammatory context. IL-22 plays a protective role when produced during epithelial or tissue damage, while IL-17A/IL-22 collaboration promotes IL-22 pro-inflammatory activity (35, 36).
In the current study, we identified an AhR ligand, the lipoxin A4, a tryptophan metabolite, which is produced after chronic exposure to ozone and induces AhR activation in the lung. AhR-deficient mice showed an increased lung inflammation and an increased cytokine production, including IL-17A and IL-22. Consistent with previous observations, we hypothesized that the protective effect of AhR is linked to the indirect repression of IL-17A and IL-22 by the reduction of cell recruitment of NK, T cells, and ILC3. Using T cell-specific AhR-deficient mice (CD4cre AhRf/f), we showed that AhR present in T cells induces the indirect repression of IL-22 and IL-17A by mediation of the recruitment of T cells of IL-22+ or IL-17+ and ILC3 of IL-22+ or IL-17+. Using IL-22−/−, IL-22Rα−/−, and IL-22xIL-17Rα−/− mice, we demonstrate the pro-inflammatory role of IL-22 during chronic ozone-induced inflammation (Graphical Abstract). IL-22 blockade in AhR−/− mice restores WT mouse phenotype. Altogether, these findings suggest that AhR activation during ozone exposure may be beneficial for the host, and thus, AhR agonists or IL-22 blockade may represent potential therapeutic strategies in ozone-induced lung inflammation.
Methods
Mice and Reagents
AhR−/− (from Frank Gonzalez) (37), AhRflox/flox (from Christopher Bradford), AhRCD4cre, IL-22−/− (38), and IL-22 × IL-17Rα−/− (38) mice and littermate controls of 7–9 weeks of age were used for the study. IL22Ra1tm1a(EUCOMM)Wtsi mice were obtained from the International Mouse Phenotyping Consortium via EMMA network. Generation of mice carrying the Il22Ra1tm1c(KOMP)Wtsi and Il22Ra1tm1d(KOMP)Wtsi alleles was achieved by breeding to StellaCre mice expressing Cre (Dppa3tm1(cre)Peli) (39) and/or Flp (ROSA26Fki) recombinases. Colonies were maintained on a C57BL/6 genetic background.
All gene-deficient mice and WT (40) controls (C57BL/6J background) were bred and housed in our specific pathogen-free animal facility at Transgenose Institute (TAMM-CNRS, UPS 44 under agreement D-45-234-6, 2014, Orleans, France). In all experiments, 5–6 female mice per group were used, were maintained in a temperature controlled (23°C) facility with a strict 12 h light/dark cycles, and were given free access to food and water. Animal experiments were performed according to the French Institutional Committee under agreement CLE CCO 2015-1088.
Anti-IL-22 antibody (AM22.1, mouse anti-mouse, 14 μg/mouse, from Laure Dumoutier) and anti-IL-17A antibody (MM17AF3, mouse anti-mouse, 20 μg/mouse, from Jacques van Snick) were administered intranasally, once a week for 6 weeks.
Ozone Induced Airway Inflammation
Mice were exposed to ozone in a plexiglass chamber (EMB 104, EMMS®) at 1.5 ppm for 2 h, two times a week for 6 weeks. Ozone was created by an ozonisator (Ozonisator Ozoniser S 500 mg, Sander®) and a level of 1.5 ppm was controlled by a sensor (ATI 2-wire transmitter, Analytical Technology®). Mice were euthanized by progressive CO2 inhalation, 24 h after last ozone exposure and BAL was collected. After a cardiac perfusion with ISOTON II (acid-free balanced electrolyte solution Beckman Coulter, Krefeld, Germany), lung was collected and sampled for analyses.
Broncho Alveolar Lavage (11)
After ozone exposure, BAL was performed by four lavages of lung with 500 μL of saline solution (NaCl 0.9%) each time, via a cannula introduced into mice trachea. BAL fluids were centrifuged at 300 g for 10 min at 4°C, the supernatants were stored at −20°C for ELISA analysis, and cell pellets were recovered to prepare cytospin (Thermo Scientific, Waltham, USA) on glass slides, followed by a staining with Diff-Quik solution (Merz & Dade A.G., Dudingen, Switzerland). Differential cell counts were performed with at least 400 cells.
DNA Measurement
DNA release was measured to evaluate the tissue damage in the BAL supernatant using the Quant-iT™ PicoGreen™ dsDNA assay kit (ThermoFisher), according to the manufacturer's instructions.
Measurement of Cytokines, Collagen, and Total Proteins
IL-22, IL-17A, MPO, and AREG in BALF were determined by ELISA (R & D systems, Abingdon, UK), while IL-4, IL-5 in lung, and TGF-β1 from BAL were measured by Luminex immunoassay using MagPix reader (Bio-Rad) according to the manufacturer's instructions. Data were analyzed with Bio-Plex Manager software (Bio-Rad). Total protein levels in BALF were analyzed with the Bio-Rad DC Protein Assay. Collagen was measured with Soluble Collagen Assay Sircol™ (biocolor), according to the manufacturer's instructions on BAL and lung supernatants. For the protein measurement on the lung supernatant, the same part of the lung was harvested and gridded in 1 mL of PBS with proteases inhibitor cocktail (Roche). After centrifugation, supernatant was collected and frozen.
AhR Ligand Determination
Lung supernatants were mixed with DPBS up to a total volume of 3 mL. SPE (solid-phase extraction) was then performed on Supelclean LC-8 SPE cartridges (Supelco) conditioned with methanol and water. The sample was loaded then washed with 3 mL of ultrapure water and dried for 15 min under vacuum (10 mmHg) before being eluted with 5 mL of an 80:20 dichloromethane:isopropyl alcohol mixture. The exudate was evaporated to dryness under a nitrogen flow and reconstituted in 200 μL of a 50:50 acetonitrile:water mixture.
LC-HRMS analyses were performed on a maXis Q-TOF mass spectrometer (Bruker, Bremen, Germany) coupled to an Ultimate 3000 RSLC system (Dionex, Germering, Germany). The column was a Kinetex C18 (150 × 2.1 mm) with a particle size of 1.7 μm (Phenomenex, Le Pecq, France), equipped with a C18 SecurityGuard Ultra (2.1 mm) guard filter (Phenomenex, Le Pecq, France). The mobile phase consisted of a gradient of water with 0.1% of formic acid (solvent A) and acetonitrile with 0.08% of formic acid (solvent B) as follows: 0–0.3 min 3% B, 0.3–10 min 3–90% B, 10–10.5 min 90% B, and finally, 10.5–10.6 min 90–3% B, maintained for 1.5 min before each new injection. The column was thermostated at 40°C, and the flow rate was 500 μL/min. The injection volume was 2.5 μL. The mass spectra were acquired with an ESI (electrospray Ionization) source in positive mode in the range of 50–1,650 m/z at a frequency of 1 Hz. The capillary voltage was set at 4.5 kV, the pressure of nebulizing gas was 2.0 bar, and the flow rate of drying gas was 9 L/min, heated at 200°C.
Histology
The left lung tissue was fixed in 4% buffered formaldehyde and paraffin embedded under standard conditions. Tissue sections (3 μm) were stained with Periodic acid Schiff (41). Histological changes were determined by a semi-quantitative severity score (0–3) for inflammatory cell infiltration (Table 1). The slides (one mouse per slide) were blindly examined by two independent investigators with a Nikon microscope (Nikon eclipse 80i, United States). All mice were scored (1 big and 3 small bronchi per mice).
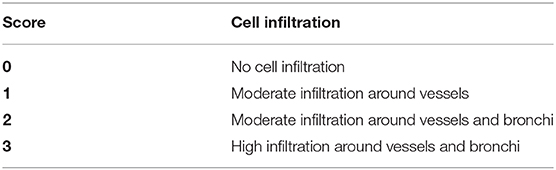
Table 1. Lung inflammation histological scoring.
FACS Analysis
Lungs were collected from mice, minced, digested with DNase (Sigma, 1 mg/mL) and Liberase (Roche, 5 mg/mL) during 1 h at 37°C, and red bloods cells were lysed with Lysing buffer (BD Pharm LyseTM, BD Pharmingen). Stimulation was performed with PMA (50 ng/ml, Sigma) and ionomycin (750 ng/ml, Sigma), for 2 h and 30 min. Cells were incubated with the antibodies for 25 min at 4°C in FACS buffer (PBS, 2% FCS, 2 mM EDTA). For intracellular staining, BD Cytofix/Cytoperm™ was used according to the manufacturer's instructions. Cells were washed with FACS buffer and fixed by lysing buffer 1 × (BD Pharmingen). The antibodies used were against mouse molecules and described in the Table S1. The gating strategy is presented in Supplemental Figure 1 and was conducted as follows: epithelial cells (CD45−EpCam+), NK cells (CD45+NKp46+NK1.1+), interstitial macrophages (CD45+F4/80+CD11c−), alveolar macrophages (CD45+F4/80+CD11c+), neutrophils (CD45+GR1+), CD4+ T cells (CD45+CD3ε+CD4+), γδ T cells (CD45+CD3ε+TCRγδ+), ILC2 (CD45+lin−ICOS+ST2+), and ILC3 (CD45+lin−RORγt+CD127+). The lineage staining used was composed of CD11b, CD3ε, CD45R/B220, Siglec-F, and FcεRIα. Data were acquired (~2 million cells) with a flow cytometer (BD FacsCanto II) and analyzed with FlowJo software (TreeStar, Mountain View, CA).
Ex vivo Stimulation of Lung Cells
Lung cells were collected from mice as described above. Stimulation was performed on 106 cells with PMA (50 ng/ml, Sigma) and ionomycin (750 ng/ml, Sigma) for 24 h. After stimulation, supernatant was harvested and frozen at −20°C.
Immunofluorescence
Lungs were treated with paraformaldehyde (42) perfusion, and tissues were fixed for 3 days in PFA 4% and transferred in sucrose 20% for 2 weeks. Half of right lung was included in tissue-tek® O.C.T (4583, tissue-tek®) and 10 μm frozen tissue were sectioned by using a cryomicrotome (Leica) at −20°C. For immunofluorescence staining, slides were incubated 30 min in citrate buffer at 80°C. Slides were washed with PBS 1× after each step, followed by 45-min incubation in saturation medium (TBS 1× , 1% BSA, 10% SVF, 0.3% triton X100). The primary antibody, an anti-AhR (10 μg/mL, ab2769, Abcam), was incubated overnight at 4°C. After washing, slides were treated with pontamin (0.05%, Chicago sky Blue 6B, Sigma) for 15 min. After washing (TBS 1×), slides were incubated with a secondary antibody, a goat-anti-mouse (4 μg/mL, ab150113, Abcam) during 1 h at room temperature. After washing, slides were incubated with DAPI (1/5,000 dilution) for 10 min, washed, and mounted in mowiol® (Sigma). Lung sections were observed under a fluorescence microscope Leica (Leica®, CTR6000) at 400× magnification for microphotography.
Lung Function Determined by Invasive Plethysmography
Airway hyperresponsiveness was measured by using increasing concentrations of methacholine (25–200 mg/mL) using the FinePointe system (Buxco, DSI) as previously described (43).
AhR/Luciferase Reporter Assay
The H1L1.1c2 cell line was used as described before (16). Control (10% NaCl), supernatant of BAL and lung were used and incubated with the H1L1.1c2 cell line. AhR activity was calculated by subtracting the luminescence of the control (10% NaCl) from the luminescence obtained with the samples multiplied by the cytotoxicity value.
CD4+ T Cells Isolation
CD4+ T cell were isolated from the spleens of WT, AhRCD4cre, and AhR-/- mice with Dynabeads™ Untouched™ Mouse CD4 Cells Kit (ThermoFisher). 6–7 × 106 CD4+ T cells were extracted from each spleen, and RNA was extracted as described below.
RNA Extraction, Reverse Transcription and Quantitative Real-Time PCR
Total RNA was extracted using TRIzol reagent (Sigma) followed by a phenol/chloroform extraction. Total RNA (1 μg) was reverse-transcribed using GoScript™ Reverse Transcription System (Promega). The mRNA levels for the genes of interest were examined by quantitative RT-PCR using the GoTaq® qPCR Master Mix according to the manufacturer's protocol. The primer used were obtained from Qiagen: AhR (QT00174251), Cyp1a1 (QT00105756), and AhRR (QT00161693). Relative levels of mRNA expression were normalized to HPRT1 (QT00166768) mRNA levels using a comparative method (2−ΔΔCt). Non-reverse-transcribed RNA samples and water were included as negative controls.
Statistical Analysis
Data were analyzed using Prism version 6 (Graphpad Software, San Diego, USA). The non-parametric Mann Whitney test or parametric one-way ANOVA tests with multiple Bonferroni's comparison tests were performed. Values are expressed as mean ± SEM. Statistical significance was defined at a p-value **** < 0.0001, *** < 0.001, ** < 0.01, and * < 0.05. Statistical differences between air and ozone mice in the same genotypes were represented by a star (or ns) just on the top of column of ozone mice, other comparisons are represented with bar between the groups compared.
Results
Chronic Ozone Exposure Induces Tryptophan Metabolites Release and AhR Activation
First, we asked whether AhR is activated upon chronic respiratory ozone exposure. Thus, we investigated the expression of associated AhR genes after chronic ozone exposure (twice weekly to 1.5 ppm for 2 h for 6 weeks). We observed an increased mRNA expression of the AhR repressor (AhRR) and P450 cytochrome (Cyp1A1) in the lung homogenate (Figure 1A). Furthermore, an increased number of AhR+ cells in the lung tissue by immunofluorescence (Figure 1B) and by flow cytometry analysis (Figure 1C, Table S1, Supplemental Figures 1, 2) were observed.
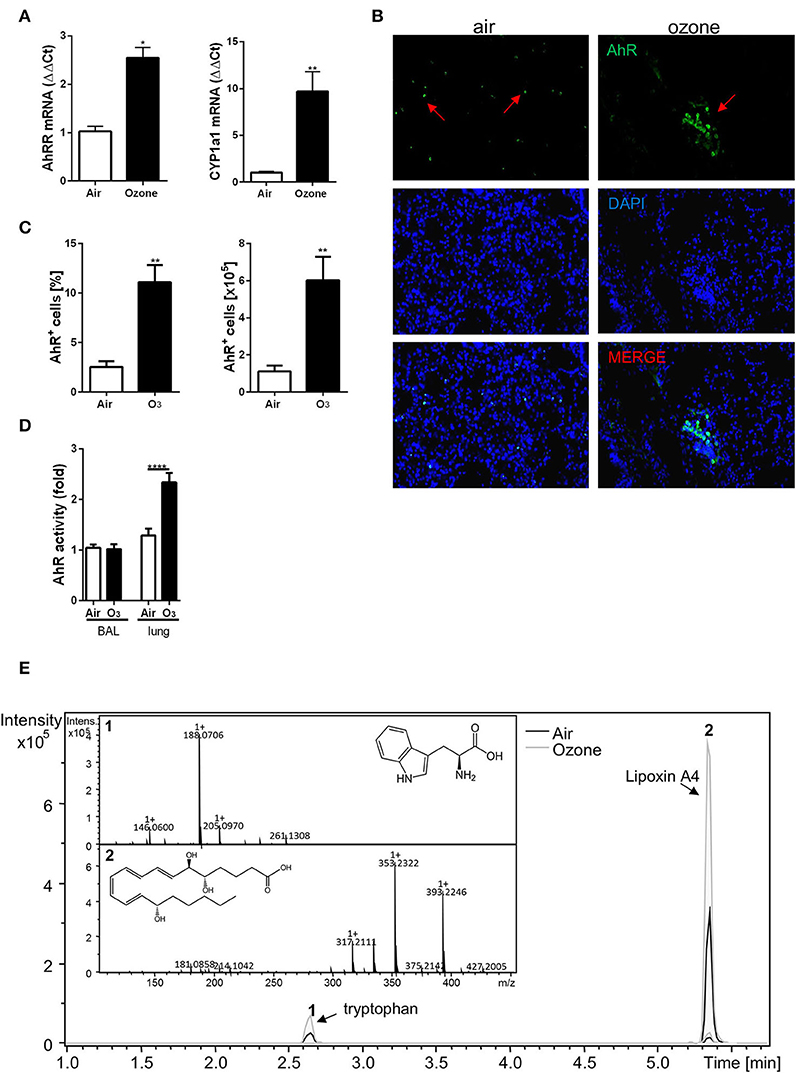
Figure 1. Chronic ozone exposure induces AhR activation. Quantification of AhRR and Cyp1a1 involved in the AhR pathway activation (A). AhR detection in lung, by immunofluorescence staining (magnification 400×; arrows represent AhR+ cells) (B) and FACS (C). AhR activity in the BAL and the lung after 6 weeks ozone exposure (D). Relative abundance of tryptophan and Lipoxin A4 in lung supernatant analyzed by LC-MS (E). The data are representative of one from two independent experiments with n = 5–6 mice per group. Values are expressed as mean ± SEM. Statistical significance was defined at a p-value **** < 0.0001, *** < 0.001, ** < 0.01, and * < 0.05.
Since ozone activates AhR expression, we performed functional tests on the BAL supernatant and lung homogenate from mice exposed to ozone using the H1L1.1c2 luciferase reporter cell line (16) and found an activation of the reporter cell line in the lung homogenate (Figure 1D). These data therefore demonstrate the presence of an AhR-activating moiety in lung supernatant of ozone-exposed mice.
Endogenous metabolites from the host and commensals are involved in AhR activation (15, 16, 22). To identify potential metabolites in our model, lungs from mice exposed to ozone were homogenized, and the supernatants were analyzed by LC-MS. Several known ligands of AhR were assessed (Table S2). Two peaks were observed, the first one corresponding to tryptophan, suggesting an increase of tryptophan metabolism and metabolites that activate AhR, and a second peak corresponding to the lipoxin A4 (LXA4), an anti-inflammatory molecule (44–46) (Figure 1E).
Therefore, ozone exposure induced AhR ligand production in the lung, and we identified LXA4 as a candidate activating AhR.
AhR Regulates Lung Inflammation, Injury, and Airway Hyperreactivity Upon Chronic Ozone Exposure
To investigate the role of AhR in ozone induced lung inflammation model, AhR-deficient mice (AhR−/−) were exposed to ozone (1.5 ppm for 2 h over 6 weeks). Increased cell infiltration in the lung of AhR−/− mice was observed after ozone exposure in comparison to WT mice (Figures 2A,B). Indeed, in the BAL, an increased number of macrophages, neutrophils, and T cells were detected (Figure 2A) with augmented MPO, AREG, and collagen levels (Supplemental Figure 3A). More particularly, increased absolute numbers of interstitial macrophages, ILC2, ILC3, αβ T cells, γδ T cells, and NK were found in the lung of AhR−/− in comparison to WT mice after ozone exposure (Figure 2B, Supplemental Figure 1). Furthermore, the TH2 cytokines and IL-4 and IL-5 levels were higher in the lung from AhR-deficient mice in comparison to WT mice (Figure 2C) with an increased recruitment of IL-5+ ILC2 and/or IL-13+ ILC2 (Supplemental Figure 3B).
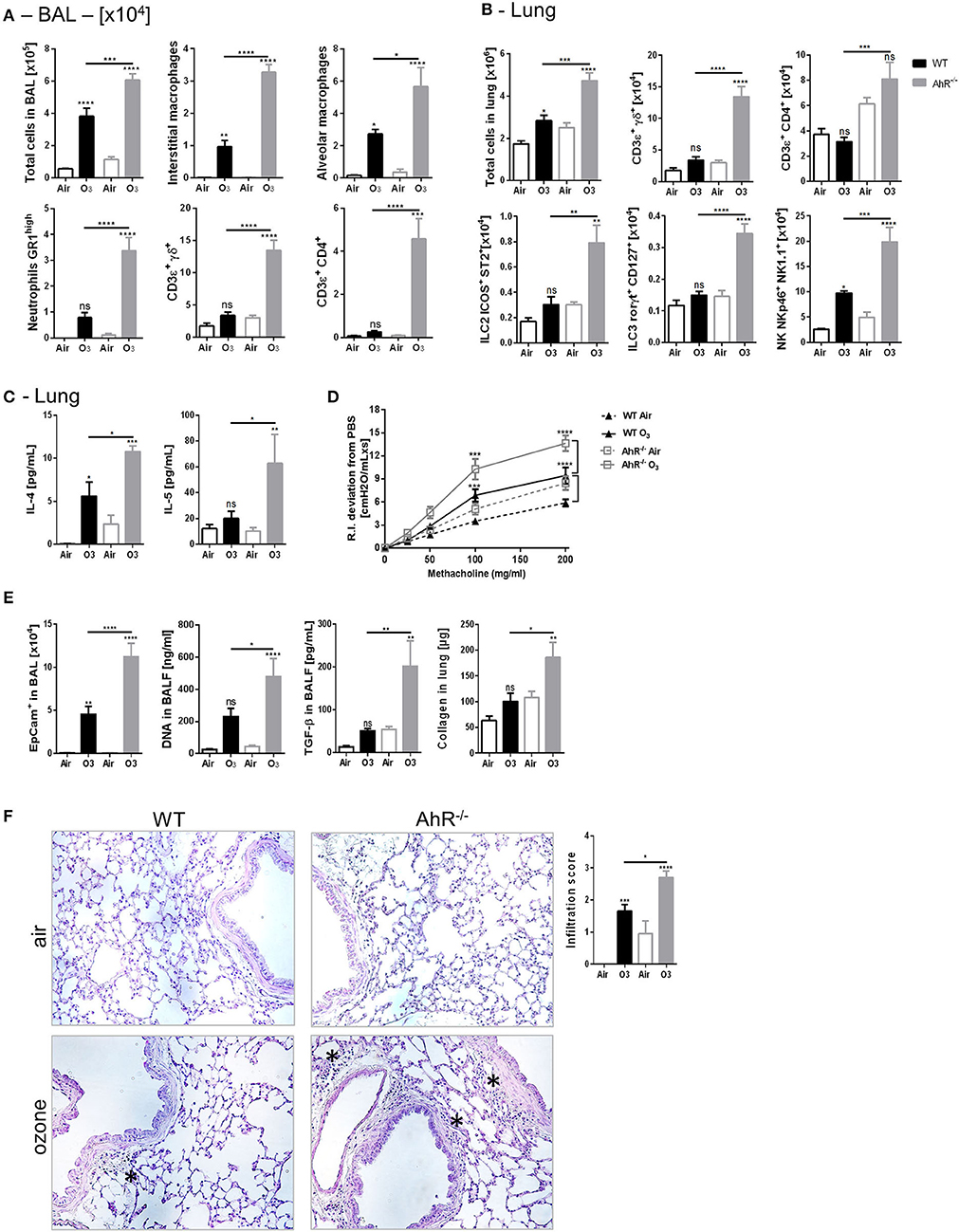
Figure 2. Inflammation, AHR, and remodeling parameters in AhR deficient mice. BAL cells recruitment (total cells, macrophages, neutrophils, and T cells) (A). Lung cells recruitment (total cells, neutrophils, macrophages, ILC2, ILC3, αβ, and γδ T cells) (B), TH2 cytokines (IL-4, IL-5) (C), AHR (D), and remodeling parameters (epithelial cells, DNA, and TGF-β in BAL; collagen in lung) (E); lung histology representative pictures (400× magnification, asterisk show cell infiltration) and cell infiltration score (F) after ozone exposure in WT and AhR deficient mice. The data are representative for one of two independent experiments with n = 5–6 mice per group. Values are expressed as mean ± SEM.
Considering the known effect of ozone on airway hyperresponsiveness, we investigated whether chronic ozone exposure exacerbates airway hyperresponsiveness. Compared to air-exposed mice, ozone-exposed WT mice exhibit a significantly increased lung resistance (RI) in response to methacholine (Figure 2D), which is significantly increased in AhR−/− mice (Figure 2D). Since ozone disrupts the epithelial barrier (41, 47–49), we analyzed the desquamation of epithelial cells by microscopic assessment of the lung integrity. DNA release was used as a marker of cell death in the alveolar space, and TGF-β (50) and collagen deposition were used as indicators of repair with fibrosis in the lung. An increased number of epithelial cells, DNA, and TGF-β levels in BALF were observed (Figure 2E). Collagen was also augmented in the lung homogenate of WT mice, which was further increased in AhR−/− mice (Figure 2E). The microscopic investigations indicated a chronic inflammatory cell infiltration, which was enhanced in the lung of AhR−/− mice in comparison to WT mice (Figure 2F). Mucus-producing cells were increased after ozone exposure, but not different in absence of AhR (data not shown).
Taken together, these data show an increased cell recruitment, airway hyperreactivity, epithelial cell injury, inflammation, and fibrosis in AhR-deficient mice, suggesting that AhR regulates lung inflammation and epithelial damage after ozone exposure.
AhR Expressed by T Cells Is Critical to Control Ozone Induced Lung Inflammation
The effect of AhR on T cell differentiation and plasticity of TH17 and Treg cells has been reported (23). To understand whether AhR solely expressed by T cells is sufficient to regulate ozone induced lung inflammation, AhR-deficient mice in T lymphocytes (AhRCD4cre) were generated by crossing AhRflox/flox with CD4cre mice (WTCD4cre), and T cell-specific AhR depletion was checked in these mice (Supplemental Figure 4).
Increases of ILC2, ILC3, and CD4+ T cells were observed in the lung (Figure 3B) to same extent as in both AhRCD4cre and the AhR−/− mice, in comparison to the AhRflox/flox control mice and WT mice, after ozone exposure, but not for BAL (Figure 3A). In contrast, interstitial macrophages and neutrophils were lower in the BAL in AhR CD4cre mice in comparison to AhR−/− mice, as well as ILC2 and γδ T cells in the lung (Figures 3A,B). Moreover with regards to TH2 cytokine level, IL-4 from AhRCD4cre mice was similar to WT mice, but not for IL-5 (Figure 3C).
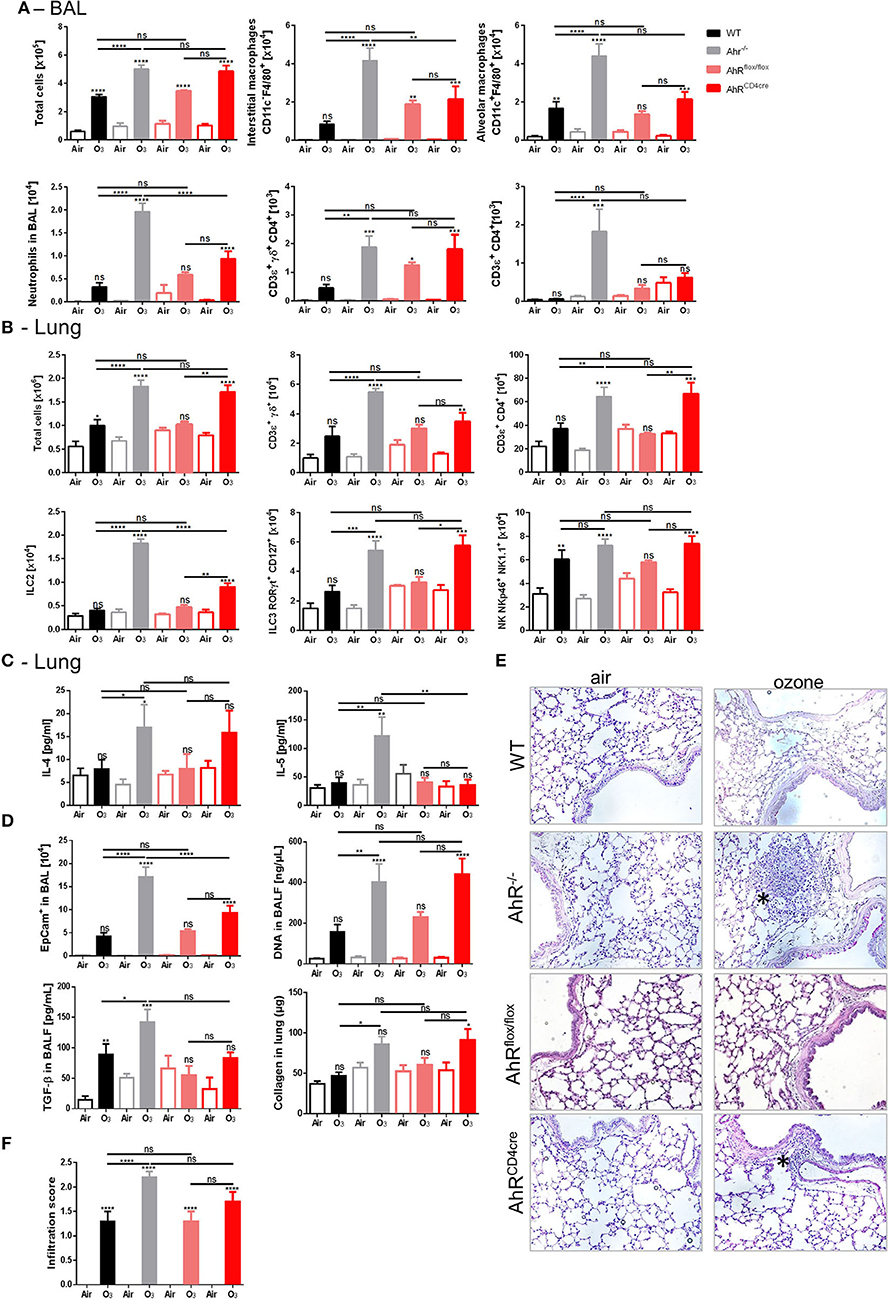
Figure 3. Specific AhR depletion in T cell affects inflammatory parameters. BAL cells recruitment (total cells, macrophages, neutrophils, and T cells) (A). Lung cells recruitment (total cells, neutrophils, macrophages, ILC2, ILC3, αβ, and γδ T cells) (B), TH2 cytokines (IL-4 and IL-5) (C) and remodeling parameters (epithelial cells, DNA, and TGF-β in BAL; collagen in lung) (D); lung histology representative pictures (400× magnification, asterisks show cell infiltration) (E), and cell infiltration score (F) after ozone exposure in WT and AhR deficient mice. The data are representative for one of two independent experiments with n = 5–6 mice per group. Values are expressed as mean ± SEM.
In addition, the increased epithelial barrier damage observed in the AhRCD4cre mice was similar to AhR−/− mice, as shown by increased DNA release in the BAL and the collagen deposition in the lung (Figure 3D). The epithelial cell desquamation level in the BAL was reduced in the AhRCD4cre mice compared to the AhR−/− mice (Figure 3D). Microscopic observation revealed that inflammatory cell infiltration was comparable in AhR−/− and AhRCD4cre mice (Figures 3E,F). To exclude any dysfunction of AhR by introducing loxP sites in the control AhRflox/flox mice, AhRflox/flox mice were compared with WT mice (Figures 3A–F) and no relevant differences were observed between them, suggesting no impact from genetic modification.
Therefore, the data obtained from AhRCD4cre mice suggest that AhR deficiency in T cells recapitulates partially the data from AhR−/− mice (notably in lung cells recruitment), and hence, AhR expressed in T cells is partially necessary to control ozone-induced inflammation in lung.
AhR Activation During Chronic Ozone Exposure Suppresses the Recruitment of IL-17A and IL-22 Producing Cells
AhR has a broad-spectrum of biological activity including on the production of IL-17A and IL-22 (22, 51–53). IL-17A is involved in the inflammatory process after acute (28, 29) and chronic ozone exposure (31). IL-22, in view of its protective role in the epithelial barrier, might be involved in ozone injury. Therefore, we assessed the role of AhR on IL-17A and IL-22 production during chronic ozone exposure.
First, the capacity of cells deficient for AhR to produce IL-17A and IL-22 was assessed. With ex vivo stimulation of isolated lung cells from WT or AhR−/− mice, enhanced IL-22 production was observed in WT mice, which is abrogated in AhR−/− mice, while the production IL-17A was increased (Figure 4A). These findings suggest that IL-22 and IL-17A may be differentially regulated by AhR. Moreover, increased levels of IL-17A and IL-22 were detected in the lung homogenate of the AhR−/− exposed mice in comparison to ozone exposed WT mice (Figure 4B). After 6 weeks of ozone exposure, IL-17A and IL-22 production was higher in the lung when AhR gene was missing, suggesting an impact of AhR on the recruitment of IL-22-producing cells. This increase was lost for IL-17A in AhRflox/flox and AhRCD4cre mice, but conserved in AhRCD4cre mice for IL-22 (Figure 4B).
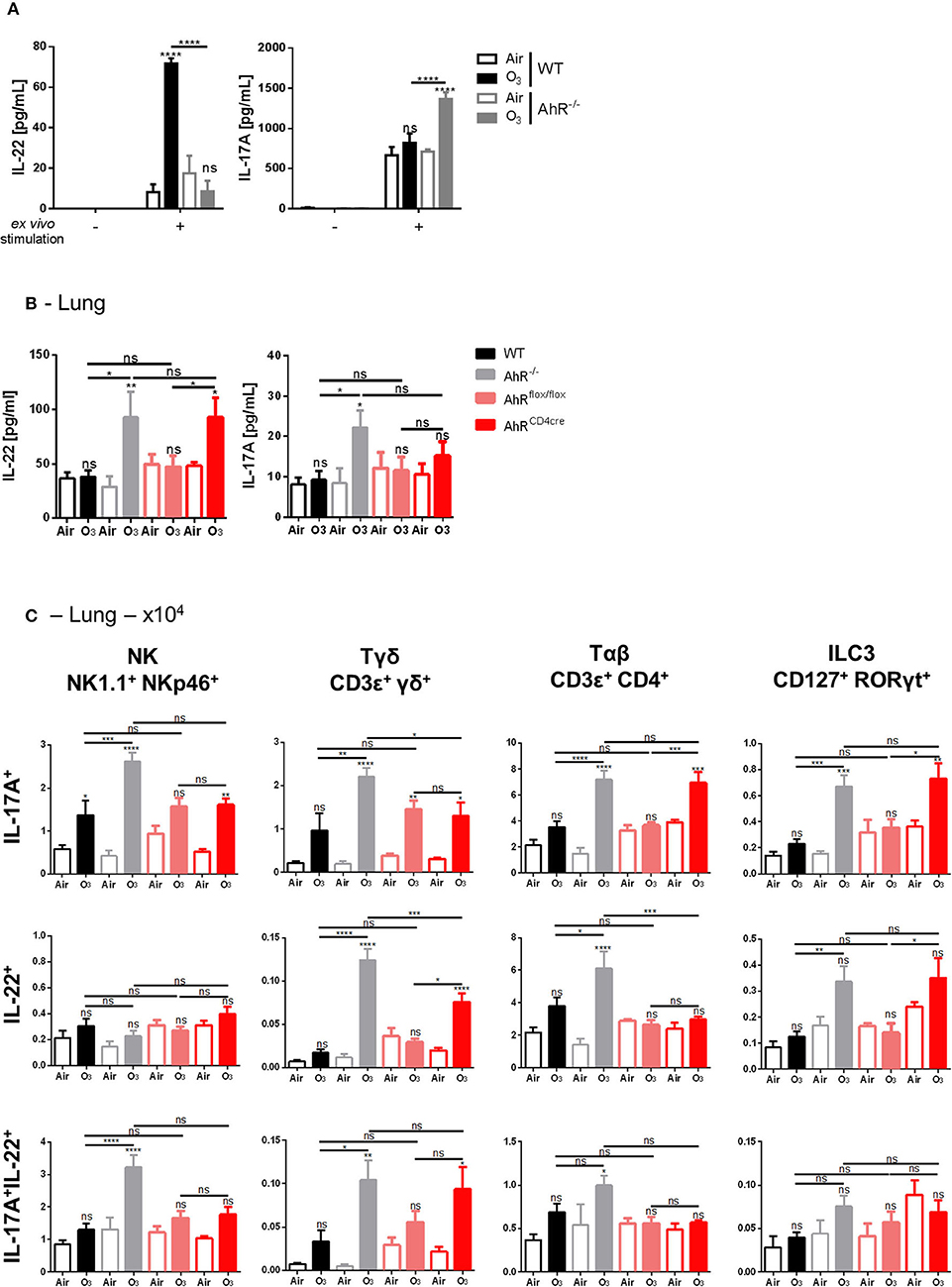
Figure 4. Influence of AhR on IL-17A and IL-22 production. IL-22 and IL-17A release after ex vivo stimulation of lung cells (A). Levels of IL-17A and IL-22 in WT and AhR deficient mice (B). Absolute number (C) of NK (NK1.1+NKp46+), ILC3 (CD45+Lin−CD127+Rorγt+), αβ T cells (CD45+CD3 ε+CD4+), γδ T cells (CD3ε+panγδ+), IL-17A+, IL-22+, and IL-17A+IL-22+ producing cells. The data are representative for one of independent experiments with n = 5–6 mice per group. Values are expressed as mean ± SEM. Statistical significance was defined at a p-value **** < 0.0001, *** < 0.001, ** < 0.01, and * < 0.05.
To understand the mechanism involved in IL-22 and IL-17 increase, we identified the cell types expressing IL-17A and IL-22 in mononuclear cells isolated from the lungs of ozone-exposed mice. NK, ILC3, αβ, and γδ T cells expressed IL-17A or IL-22 or both IL-17A/IL-22 upon ozone exposure (Figure 4C). IL-17A expression was increased in NK cells, ILC3, αβ, and γδ T cells in AhR−/− mice, in comparison to WT mice, and an increased IL-22 expression was observed in T cells and ILC3 (Figure 4C). In addition, a lower expression of IL-22 in T cells and a slight decrease of IL-17A expression in NK and γδ T cells in AhRCD4cre mice were observed in comparison to AhR−/− mice (Figure 4C). For double positive cells for IL-17A and IL-22, we observed an increase of NK, γδ, and αβ T cells in the AhR−/− mice in comparison to WT mice. A similar phenotype to the one observed in the AhR−/− mice was found for AhRCD4cre mice for the γδ T cells. Therefore, the presence of AhR in CD4+ cells is necessary for the recruitment of the IL-17A+ αβ T cells and the IL-22+ and IL-17A+ ILC3 and partially for the IL-22+ and of IL-17A+/IL-22+ γδ T cells (Figure 4C).
Thus, the data suggest that AhR downregulates indirectly IL-22 production via IL-22-producing cells recruitment, like NK, ILC3, and T cells, after chronic ozone exposure. For IL-17A, AhR acts directly on cell production and indirectly on cell recruitment as for IL-22.
Role of IL-17A and IL-22 Axis Upon Ozone Exposure and AhR Dependence
The possible involvement of IL-17A and IL-22 in dampening the lung inflammation in AhR−/− mice was then investigated. IL-22−/− (Supplemental Figures 6A–F), IL-22Rα−/−, and IL-22 × IL-17Rα−/− mice (Figure 5, Supplemental Figure 5) were exposed to ozone, and different lung inflammation parameters were evaluated. Reduced cell infiltration in all gene deficient mice used was observed after chronic exposure (Figures 5A,B, Supplemental Figures 6A,B), with diminished macrophages and neutrophils recruitment in the BAL (Figure 5A), and NK and ILC2 cells in the lung, but not significantly (Figure 5B). Moreover, TH2 cytokines, including IL-4 and IL-5, were reduced after chronic ozone exposure in IL-22Rα−/− and in IL-22 × IL-17Rα−/− mice in comparison to WT mice (Figure 5C). Airway hyperresponsiveness was reduced in the IL-22Rα−/− and IL-22−/− mice in comparison to WT mice (Figure 5D, Supplemental Figure 6D). Epithelial cells desquamation in the BAL, DNA, and TGF-β levels in BALF, as well as collagen in the lung were reduced in IL-22Rα−/− and IL-22 × IL-17Rα−/− mice (Figure 5E). Histological analysis of the lung tissue revealed reduced cell infiltration in parenchyma, in the IL-22−/−, IL-22Rα−/−, and IL-22 × IL-17Rα−/− mice after chronic ozone exposure, in comparison to WT mice (Figures 5F,G). Anti-IL-22 alone is sufficient to recapitulate the phenotype, the anti-IL-17A does not increase the effect observed with the anti-IL-22 (Supplemental Figure 7A). A decrease in interstitial macrophages and γδ T cells recruitment was observed in the BAL (Supplemental Figure 7A), as well as decreased T and NK cells in the lung (Supplemental Figure 7B). Lung remodeling parameters, such as epithelial cells desquamation, DNA release, and collagen production, were reduced in comparison to WT mice (Supplemental Figures 7C,D). Moreover, using the same neutralizing antibodies in AhR-deficient mice, we showed that the increased inflammatory parameters were dependent on IL-22/IL-17A signaling. In the BAL, IL-22 antibody neutralization was sufficient to reduce interstitial macrophages, neutrophils, and T cells, in comparison to what was observed in the AhR deficient mice (Figure 6A). Similar reductions of the T cells, ILC2, ILC3, and NK were observed in the lung (Figure 6B). Moreover, desquamation, DNA release, collagen production, and inflammatory score were reduced in AhR-deficient mice that were treated by anti-IL22 and anti-IL-17A antibodies weekly (Figures 6C–E).
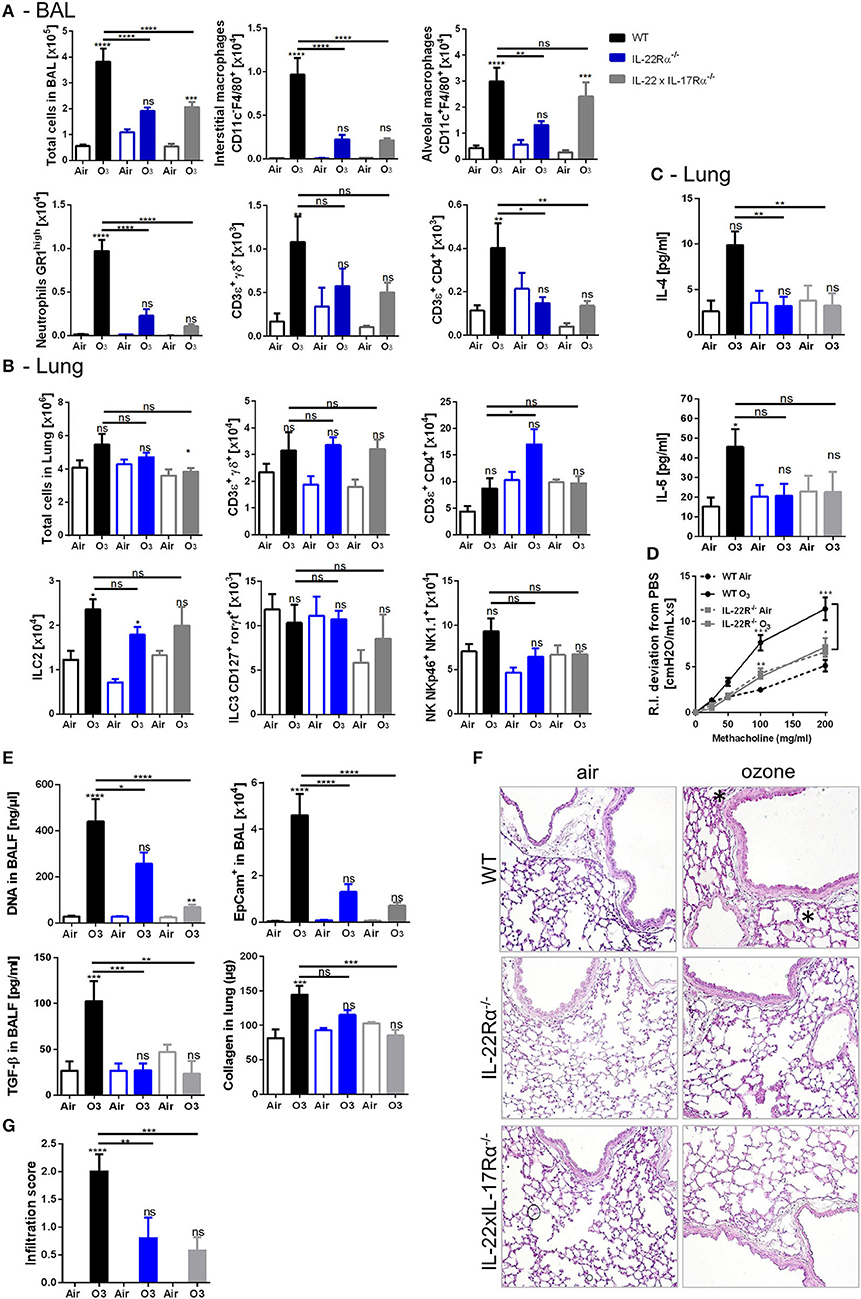
Figure 5. Inflammatory parameters in IL-22R and IL-22/17Rα deficient mice. BAL cells recruitment (total cells, macrophages, neutrophils, and T cells) (A). Lung cell recruitment (total cells, neutrophils, macrophages, ILC2, ILC3, αβ, and γδ T cells) (B), TH2 cytokines (IL-4, IL-5) (C), AHR (D), and remodeling parameters (epithelial cells, DNA and TGF-β in BAL; collagen in lung) (E); lung histology (400× magnification, asterisk show cell infiltration) (F) and cell infiltration score (G) after ozone exposure in WT and AhR-deficient mice. The data are representative of one from two independent experiments with n = 5–6 mice per group. Values are expressed as mean ± SEM.
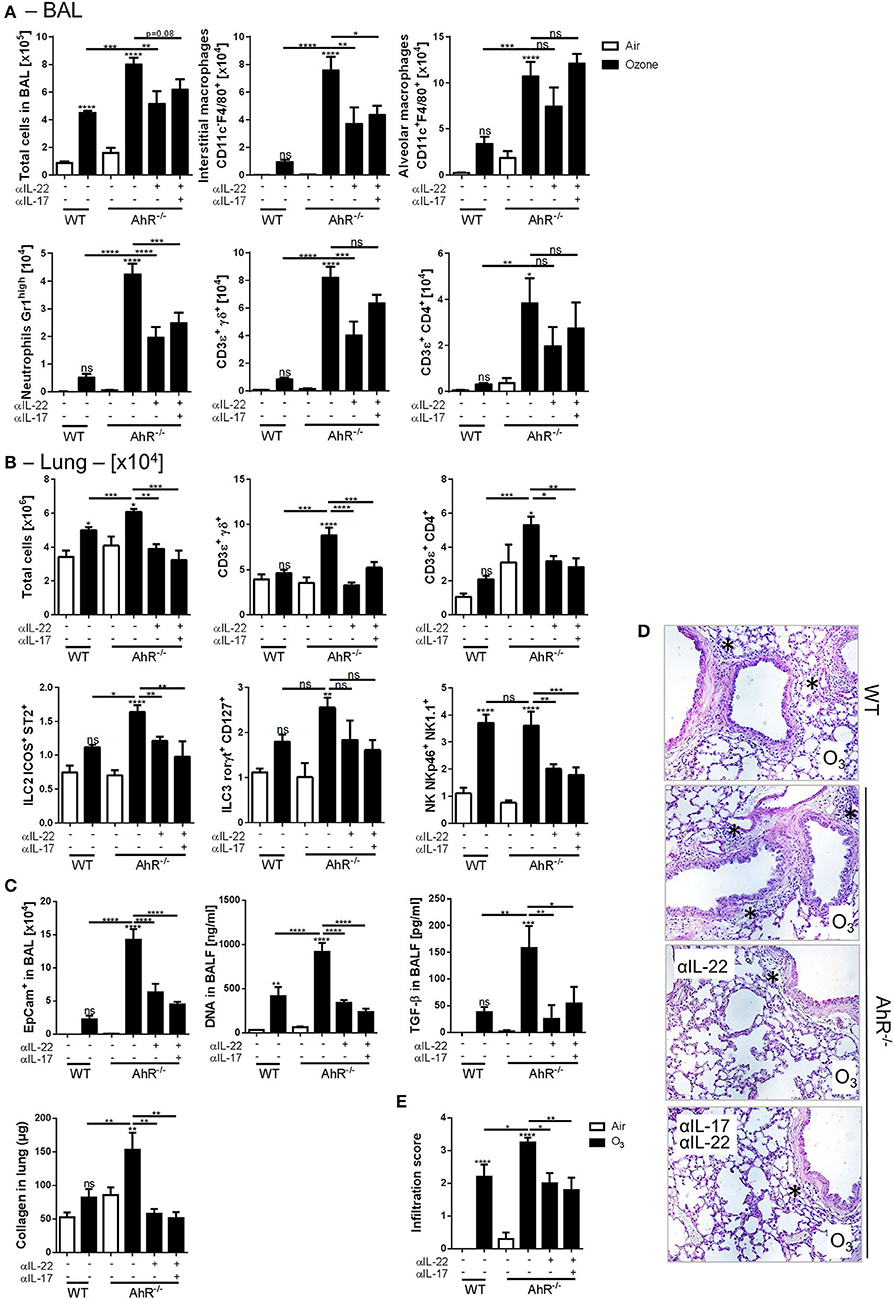
Figure 6. IL-22 and IL-17A neutralization in AhR deficient mice. BAL cells recruitment (total cells, macrophages, neutrophils, and T cells) (A). Lung cell recruitment (total cells, neutrophils, macrophages, ILC2, ILC3, αβ, and γδ T cells) (B), and remodeling parameters (epithelial cells, DNA, and collagen in lung) (C); lung histology (400× magnification; asterisks show cell infiltration) (D) and cell infiltration score (E) after ozone exposure in WT and AhR-deficient mice. The data are representative of one from two independent experiments with n = 5–6 mice per group. Values are expressed as mean ± SEM.
Therefore, the data demonstrated that the absence of IL-22 reduced inflammation, suggesting a pro-inflammatory effect of this cytokine in this model. Since IL-22 expression is enhanced in absence of AhR, we hypothesized that AhR modulate inflammation including the recruitment of IL-22-producing cells, which play a major role in ozone induced lung injury.
Discussion
Environmental air pollution plays an important role in chronic respiratory diseases (54). Ozone is a major air pollutant contributing to the development of allergic and non-allergic asthma and chronic lung diseases, such as COPD and emphysema.
Here, we report that tryptophan produced upon ozone exposure leads to AhR activation and IL-22 and IL-17A production in the lung, probably through the production of tryptophan metabolites. AhR plays a protective role on lung inflammation, airway hyperresponsiveness, and tissue remodeling after chronic ozone exposure (1.5 ppm, twice a week for 2 h). The protection via AhR had previously been described in several models of lung inflammation including the COPD model induced by cigarette smoke (55, 56) and idiopathic pneumonia syndrome (1) or in the DSS-induced colitis model (57). AhR acts on several pathways, including cell cycle, cytokine production (51, 58), metabolism (20), and the maintenance of ILC3 (19). The type of immune response induced by AhR activation depends on the nature of the ligand. Several studies showed differential cytokine production due to AhR activation after PBMC stimulation with TCDD or PGE2 (22) supporting this hypothesis.
After ozone exposure, we identified two molecules released in the lung: tryptophan and LXA4, which potentially activate AhR. AhR plays a key role of tryptophan metabolism. Since the 1980s, several studies reported a role of tryptophan metabolites in the lung, including in cancer (59) and chronic inflammatory lung diseases affecting the parenchyma, such as sarcoidosis and idiopathic fibrosis (1, 60). The indole/tryptamine pathway could induce the production of several indole from tryptophan that may activate AhR. For example, in the intestine, IAld is known to induce the activation of AhR and the production of IL-22 (53). In such conditions, IAld is produced by microbiota, in particular the Lactobacilli. Lung/gut microbiota in the context of ozone exposure had been recently studied (61); moreover, ozone is commonly used in sterilization protocols, and it has an impact on plants (62), suggesting an impact of ozone on microbiota. Stress of the lung microbiome (63) induced by inflammatory cells and epithelial cells desquamation after ozone exposure may lead to an increased release of tryptophan in the lung that can be converted in various metabolites, but we cannot exclude an initial synthesis in gut. Moreover, we identified another AhR ligand, the lipoxin A4. The amount of lipoxin A4 was five times higher than the tryptophan itself in the lung tissue after chronic ozone exposure. This class of ligand has been described for the first time in 1999 (64). Lipoxin 44 could be produced in different cell types, as neutrophils, eosinophils, alveolar macrophages, and epithelial cells (65). Lipoxin A4 effects are mediated through its binding to FPR2/ALX membrane receptor or to AhR. Lipoxins are known to participate to inflammation resolution. In experimental asthma, lipoxins regulate NK or ILC2 activation (66), and in experimental psoriasis model induced by imiquimod, they reduce IL-17A and IL-22 production via HMGB1 modulation (67).
Here, we show that, AhR activation induced the recruitment of several immune cells, including macrophages, γδ T cells, αβ T cells, NK, ILC2, and ILC3 (Graphical Abstract). Upon chronic ozone exposure in AhR-deficient mice, we observed a type 2 response, as demonstrated by the production of TH2 cytokines (IL-4 and IL-5). This response is associated with an increased IL-6 production. Airway hyperresponsiveness after chronic ozone exposure, a key feature of ozone exposure (28), is significantly enhanced in absence of AhR, as expected in a TH2 response. In the intestine, AhR maintenance is involved in the recruitment (19) of ILC3; however, the link between AhR and the ILC3 have not been investigated in the lung yet. Compared to the ILC2, ILC3 cells are very few in the lungs. But after ozone exposure, the quantity of ILC2 and ILC3 are very similar. Thus, we hypothesize that ILC3 had migrated from intestine to other tissues, particularly in the lung tissue after chronic ozone exposure. The deletion of AhR, in the CD4+ cells, controls the recruitment of the ILC3 and CD4+ T cells and is partly involved in the recruitment of the ILC2. Various direct or indirect mediators can drive this effect on different cell populations. Moreover, AhR may act on iNKT function (68), on the generation of Treg (69) or TH17 cells (23), and in the balance between TH1 and TH2 (70) as AhR activation suppresses the TH2 differentiation.
Furthermore, AhR regulates IL-17A and IL-22 production (Graphical Abstract). Here, in AhR-deficient mice, IL-17A, and IL-22 levels were increased as well as the amount of NK, ILC3, and γδ T cells expressing IL-17A and IL-22. By using AhRCD4cre mice we observed that the AhR-specific deficiency in T cells influenced ILC3 numbers and increased their capability to produce IL-17A and IL-22. IL-17A is known to be a pro-inflammatory cytokine that can collaborate with IL-22 to induce inflammation (71, 72). We hypothesized that the protective effect of AhR is related to its capacity to repress the recruitment of the cells secreting IL-17A and IL-22 likely mediated the production of an endogenous ligand induced by ozone. This repression could be driven by the LXA4. Indeed, AhR deficient mice present a reduced LXA4 expression (73). As shown by Zhang et al. (31), IL-17A is pro-inflammatory in the context of chronic exposure to ozone, confirmed in our lab with utilization of IL-17Rα−/− mice (data not shown). In the IL-22Rα−/− mice, we observed a reduction of cell infiltration and TH2 cytokines production. Moreover, airway hyperresponsiveness and tissue remodeling parameters (epithelial cells, DNA release, and collagen deposition) were reduced. Thus, our data confirm the pro-inflammatory role of both IL-17A and IL-22 in this model, but our data suggest that the phenotype observed in AhR−/− mice is only dependent of IL-22.
In conclusion, ozone exposure induces tryptophan metabolites and LXA4 that activates AhR. This activation likely control cell recruitment, TH2 and TH17/22 response, airway hyperresponsiveness and tissue remodeling. Altogether, our data show that AhR plays a protective role upon chronic exposure to ozone by modulating IL-22-producing cells recruitment leading to the control inflammation (Graphical Abstract).
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Ethics Statement
The animal study was reviewed and approved by The French Institutional Committee under agreement CLE CCO 2015-1088.
Author Contributions
CM, FB, LF, IM, CC, and MS conducted the experiments. CM, BR, and DT designed the experiments. CM, BR, DT, CC, JS, VQ, HS, AC-M, and LD wrote the paper or had a critical regard on manuscript.
Funding
This work was supported by le Centre National de la Recherche Scientifique (CNRS), by French Institute of Health and Medical Research (INSERM), European Regional Development Fund (FEDER n°2016-00110366), and la Région Centre (Respir_Ozone, n°2014-00091905).
Conflict of Interest
DT and LF are employees at ArtImmune.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Marc Le Bert for advice and technical support. Thanks to EUCOM for Il22Ra1tm1a(EUCOM)Wtsi mice, Pr Liu for Dppa3tm1(cre)Peli mice and the TAAM for ROSA26Fki mice. We thank Claire Mackowiak, Quentin Marquant, Delphine Sedda, Mégane Nascimento, and Ana Espino for technical support. We thank Nathalie Froux, Caroline Bertrand, Tamara Durand, and ML Dessain for the excellent technical support of the staff of the animal unit including. The graphical abstract was created and adapted with Servier Medical Art by Servier and is licensed under a Creative Commons Attribution 3.0 Unported License.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00144/full#supplementary-material
Abbreviations
AhR, Aryl hydrocarbon receptor; AhRR, AhR repressor; AREG, Amphiregulin; ARNT, Aryl hydrocarbon nuclear translocator; BAL, Broncho alveolar lavage; IDO, Indoleamine 2,3-Dioxygenase; ILC, Innate lymphoid cells; iNKT, invariant NK T cells; MPO, Myeloperoxidase; NK, Natural killer; PGE2, Prostaglandin E2; PBMC, Peripheral blood mononuclear cells; WT, Wild type (C57BL/6).
References
1. Lee SM, Park HY, Suh YS, Yoon EH, Kim J, Jang WH, et al. Inhibition of acute lethal pulmonary inflammation by the IDO-AhR pathway. Proc Natl Acad Sci USA. (2017) 114:E5881–90. doi: 10.1073/pnas.1615280114
2. Katsouyanni K, Samet JM, Anderson HR, Atkinson R, Le Tertre A, Medina S, et al. Air pollution and health: a European and North American approach (APHENA). Res Rep Health Eff Inst. (2009) 5–90.
3. Stafoggia M, Forastiere F, Faustini A, Biggeri A, Bisanti L, Cadum E, et al. Susceptibility factors to ozone-related mortality: a population-based case-crossover analysis. Am J Respir Crit Care Med. (2010) 182:376–84. doi: 10.1164/rccm.200908-1269OC
4. Ji M, Cohan DS, Bell ML. Meta-analysis of the Association between Short-Term Exposure to Ambient Ozone and Respiratory Hospital Admissions. Environ Res Lett. (2011) 6:024006. doi: 10.1088/1748-9326/6/2/024006
5. Miller RL, Peden DB. Environmental effects on immune responses in patients with atopy and asthma. J Allergy Clin Immunol. (2014) 134:1001–8. doi: 10.1016/j.jaci.2014.07.064
6. Mathews JA, Krishnamoorthy N, Kasahara DI, Cho Y, Wurmbrand AP, Ribeiro L, et al. IL-33 drives augmented responses to ozone in obese mice. Environ Health Perspect. (2017) 125:246–53. doi: 10.1289/EHP272
7. Peng RD, Samoli E, Pham L, Dominici F, Touloumi G, Ramsay T, et al. Acute effects of ambient ozone on mortality in Europe and North America: results from the APHENA study. Air Qual Atmos Health. (2013) 6:445–53. doi: 10.1007/s11869-012-0180-9
8. Al-Hegelan M, Tighe RM, Castillo C, Hollingsworth JW. Ambient ozone and pulmonary innate immunity. Immunol Res. (2011) 49:173–91. doi: 10.1007/s12026-010-8180-z
9. Wiegman CH, Michaeloudes C, Haji G, Narang P, Clarke CJ, Russell KE, et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. (2015) 136:769–80. doi: 10.1016/j.jaci.2015.01.046
10. Kasahara DI, Kim HY, Williams AS, Verbout NG, Tran J, Si H, et al. Pulmonary inflammation induced by subacute ozone is augmented in adiponectin-deficient mice: role of IL-17A. J Immunol. (2012) 188:4558–67. doi: 10.4049/jimmunol.1102363
11. Ledford DK, Burks AW, Ballow M, Cox L, Wood RA, Casale TB. Statement regarding “The public health benefits of air pollution control”. J Allergy Clin Immunol. (2012) 130:24. doi: 10.1016/j.jaci.2012.02.055
12. Michaudel C, Couturier-Maillard A, Chenuet P, Maillet I, Mura C, Couillin I, et al. Inflammasome, IL-1 and inflammation in ozone-induced lung injury. Am J Clin Exp Immunol. (2016) 5:33–40.
13. Michaudel C, Fauconnier L, Jule Y, Ryffel B. Functional and morphological differences of the lung upon acute and chronic ozone exposure in mice. Sci Rep. (2018) 8:10611. doi: 10.1038/s41598-018-28261-9
14. Michaudel C, Mackowiak C, Maillet I, Fauconnier L, Akdis CA, Sokolowska M, et al. Ozone exposure induces respiratory barrier biphasic injury and inflammation controlled by IL-33. J Allergy Clin Immunol. (2018) 142:942–58. doi: 10.1016/j.jaci.2017.11.044
15. Stockinger B, Di Meglio P, Gialitakis M, Duarte JH. The aryl hydrocarbon receptor: multitasking in the immune system. Annu Rev Immunol. (2014) 32:403–32. doi: 10.1146/annurev-immunol-032713-120245
16. Lamas B, Richard ML, Leducq V, Pham HP, Michel ML, Da Costa G, et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med. (2016) 22:598–605. doi: 10.1038/nm.4102
17. Gutierrez-Vazquez C, Quintana FJ. Regulation of the immune response by the aryl hydrocarbon receptor. Immunity. (2018) 48:19–33. doi: 10.1016/j.immuni.2017.12.012
18. Kimura A, Naka T, Nakahama T, Chinen I, Masuda K, Nohara K, et al. Aryl hydrocarbon receptor in combination with Stat1 regulates LPS-induced inflammatory responses. J Exp Med. (2009) 206:2027–35. doi: 10.1084/jem.20090560
19. Schiering C, Wincent E, Metidji A, Iseppon A, Li Y, Potocnik AJ, et al. Feedback control of AHR signalling regulates intestinal immunity. Nature. (2017) 542:242–5. doi: 10.1038/nature21080
20. Kalthoff S, Ehmer U, Freiberg N, Manns MP, Strassburg CP. Interaction between oxidative stress sensor Nrf2 and xenobiotic-activated aryl hydrocarbon receptor in the regulation of the human phase II detoxifying UDP-glucuronosyltransferase 1A10. J Biol Chem. (2010) 285:5993–6002. doi: 10.1074/jbc.M109.075770
21. Zelante T, Iannitti RG, Fallarino F, Gargaro M, De Luca A, Moretti S, et al. Tryptophan Feeding of the IDO1-AhR Axis in Host-Microbial Symbiosis. Front Immunol. (2014) 5:640. doi: 10.3389/fimmu.2014.00640
22. Ramirez JM, Brembilla NC, Sorg O, Chicheportiche R, Matthes T, Dayer JM, et al. Activation of the aryl hydrocarbon receptor reveals distinct requirements for IL-22 and IL-17 production by human T helper cells. Eur J Immunol. (2010) 40:2450–9. doi: 10.1002/eji.201040461
23. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. (2008) 453:65–71. doi: 10.1038/nature06880
24. Neavin DR, Liu D, Ray B, Weinshilboum RM. The role of the Aryl Hydrocarbon Receptor (AHR) in immune and inflammatory diseases. Int J Mol Sci. (2018) 19:E3851. doi: 10.3390/ijms19123851
25. Quesniaux V, Ryffel B, Di Padova F. Th 17 Cells: Role in Inflammation and Autoimmune Disease. (2013). Available online at: https://books.google.fr/books?id=9TzX882x0X4C&pg=PA48&dq=Quesniaux+V,+Ryffel+B,+Di+Padova++Th17+cell&hl=fr&sa=X&ved=0ahUKEwj0i6rxsNvnAhVCxIUKHaL3As4Q6AEIMzAB#v=onepage&q=Quesniaux%20V%2C%20Ryffel%20B%2C%20Di%20Padova%20%20Th17%20cells&f=false
26. Gurczynski SJ, Moore BB. IL-17 in the lung: the good, the bad, and the ugly. Am J Physiol Lung Cell Mol Physiol. (2018) 314:L6–16. doi: 10.1152/ajplung.00344.2017
27. Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov. (2012) 11:763–76. doi: 10.1038/nrd3794
28. Pichavant M, Goya S, Meyer EH, Johnston RA, Kim HY, Matangkasombut P, et al. Ozone exposure in a mouse model induces airway hyperreactivity that requires the presence of natural killer T cells and IL-17. J Exp Med. (2008) 205:385–93. doi: 10.1084/jem.20071507
29. Mathews JA, Williams AS, Brand JD, Wurmbrand AP, Chen L, Ninin FM, et al. gammadelta T cells are required for pulmonary IL-17A expression after ozone exposure in mice: role of TNFalpha. PLoS ONE. (2014) 9:e97707. doi: 10.1371/journal.pone.0097707
30. Pinart M, Zhang M, Li F, Hussain F, Zhu J, Wiegman C, et al. IL-17A modulates oxidant stress-induced airway hyperresponsiveness but not emphysema. PLoS ONE. (2013) 8:e58452. doi: 10.1371/journal.pone.0058452
31. Zhang M, Fei X, Zhang GQ, Zhang PY, Li F, Bao WP, et al. Role of neutralizing anti-murine interleukin-17A monoclonal antibody on chronic ozone-induced airway inflammation in mice. Biomed Pharmacother. (2016) 83:247–56. doi: 10.1016/j.biopha.2016.06.041
32. Mathews JA, Kasahara DI, Ribeiro L, Wurmbrand AP, Ninin FM, Shore SA. gammadelta T cells are required for M2 macrophage polarization and resolution of ozone-induced pulmonary inflammation in mice. PLoS ONE. (2015) 10:e0131236. doi: 10.1371/journal.pone.0131236
33. Ribot JC, deBarros A, Pang DJ, Neves JF, Peperzak V, Roberts SJ, et al. CD27 is a thymic determinant of the balance between interferon-gamma- and interleukin 17-producing gammadelta T cell subsets. Nat Immunol. (2009) 10:427–36. doi: 10.1038/ni.1717
34. Omenetti S, Pizarro TT. The Treg/Th17 Axis: a dynamic balance regulated by the gut microbiome. Front Immunol. (2015) 6:639. doi: 10.3389/fimmu.2015.00639
35. Andoh A, Zhang Z, Inatomi O, Fujino S, Deguchi Y, Araki Y, et al. Interleukin-22, a member of the IL-10 subfamily, induces inflammatory responses in colonic subepithelial myofibroblasts. Gastroenterology. (2005) 129:969–84. doi: 10.1053/j.gastro.2005.06.071
36. Boniface K, Bernard FX, Garcia M, Gurney AL, Lecron JC, Morel F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol. (2005) 174:3695–702. doi: 10.4049/jimmunol.174.6.3695
37. Fernandez-Salguero P, Pineau T, Hilbert DM, McPhail T, Lee SS, Kimura S, et al. Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science. (1995) 268:722–6. doi: 10.1126/science.7732381
38. Kreymborg K, Etzensperger R, Dumoutier L, Haak S, Rebollo A, Buch T, et al. IL-22 is expressed by Th17 cells in an IL-23-dependent fashion, but not required for the development of autoimmune encephalomyelitis. J Immunol. (2007) 179:8098–104. doi: 10.4049/jimmunol.179.12.8098
39. Liu H, Wang W, Chew SK, Lee SC, Li J, Vassiliou GS, et al. Stella-Cre mice are highly efficient Cre deleters. Genesis. (2011) 49:689–95. doi: 10.1002/dvg.20741
40. Couch C, Franxman T, Greenhawt M. The economic effect and outcome of delaying oral food challenges. Ann Allergy Asthma Immunol. (2016) 116:420–4. doi: 10.1016/j.anai.2016.02.016
41. Reddy AS, Reddy N, Dinapadu S, Reddy M, Pasari S. Role of ozone therapy in minimal intervention dentistry and endodontics - a review. J Int Oral Health. (2013) 5:102–8.
42. Demyanets S, Konya V, Kastl SP, Kaun C, Rauscher S, Niessner A, et al. Interleukin-33 induces expression of adhesion molecules and inflammatory activation in human endothelial cells and in human atherosclerotic plaques. Arterioscler Thromb Vasc Biol. (2011) 31:2080–9. doi: 10.1161/ATVBAHA.111.231431
43. Madouri F, Guillou N, Fauconnier L, Marchiol T, Rouxel N, Chenuet P, et al. Caspase-1 activation by NLRP3 inflammasome dampens IL-33-dependent house dust mite-induced allergic lung inflammation. J Mol Cell Biol. (2015) 7:351–65. doi: 10.1093/jmcb/mjv012
44. Chandrasekharan JA, Sharma-Walia N. Lipoxins: nature's way to resolve inflammation. J Inflamm Res. (2015) 8:181–92. doi: 10.2147/JIR.S90380
45. Marginean A, Sharma-Walia N. Lipoxins exert antiangiogenic and anti-inflammatory effects on Kaposi's sarcoma cells. Transl Res. (2015) 166:111–33. doi: 10.1016/j.trsl.2015.02.009
46. Romano M, Cianci E, Simiele F, Recchiuti A. Lipoxins and aspirin-triggered lipoxins in resolution of inflammation. Eur J Pharmacol. (2015) 760:49–63. doi: 10.1016/j.ejphar.2015.03.083
47. Barr BC, Hyde DM, Plopper CG, Dungworth DL. A comparison of terminal airway remodeling in chronic daily versus episodic ozone exposure. Toxicol Appl Pharmacol. (1990) 106:384–407. doi: 10.1016/0041-008X(90)90335-R
48. Hyde DM, Miller LA, McDonald RJ, Stovall MY, Wong V, Pinkerton KE, et al. Neutrophils enhance clearance of necrotic epithelial cells in ozone-induced lung injury in rhesus monkeys. Am J Physiol. (1999) 277(Pt 1):L1190–8. doi: 10.1152/ajplung.1999.277.6.L1190
49. Song H, Tan W, Zhang X. Ozone induces inflammation in bronchial epithelial cells. J Asthma. (2011) 48:79–83. doi: 10.3109/02770903.2010.529224
50. Makinde T, Murphy RF, Agrawal DK. The regulatory role of TGF-beta in airway remodeling in asthma. Immunol Cell Biol. (2007) 85:348–56. doi: 10.1038/sj.icb.7100044
51. Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. (2008) 453:106–9. doi: 10.1038/nature06881
52. Monteleone I, Rizzo A, Sarra M, Sica G, Sileri P, Biancone L, et al. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology. (2011) 141:237–48, 248.e231. doi: 10.1053/j.gastro.2011.04.007
53. Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity. (2013) 39:372–85. doi: 10.1016/j.immuni.2013.08.003
54. Kurt OK, Zhang J, Pinkerton KE. Pulmonary health effects of air pollution. Curr Opin Pulm Med. (2016) 22:138–43. doi: 10.1097/MCP.0000000000000248
55. Rogers S, de Souza AR, Zago M, Iu M, Guerrina N, Gomez A, et al. Aryl hydrocarbon receptor (AhR)-dependent regulation of pulmonary miRNA by chronic cigarette smoke exposure. Sci Rep. (2017) 7:40539. doi: 10.1038/srep40539
56. Zago M, Sheridan JA, Traboulsi H, Hecht E, Zhang Y, Guerrina N, et al. Low levels of the AhR in chronic obstructive pulmonary disease (COPD)-derived lung cells increases COX-2 protein by altering mRNA stability. PLoS ONE. (2017) 12:e0180881. doi: 10.1371/journal.pone.0180881
57. Islam J, Sato S, Watanabe K, Watanabe T, Ardiansyah Hirahara K, et al. Dietary tryptophan alleviates dextran sodium sulfate-induced colitis through aryl hydrocarbon receptor in mice. J Nutr Biochem. (2017) 42:43–50. doi: 10.1016/j.jnutbio.2016.12.019
58. Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y, Kishimoto T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc Natl Acad Sci USA. (2008) 105:9721–6. doi: 10.1073/pnas.0804231105
59. Yasui H, Takai K, Yoshida R, Hayaishi O. Interferon enhances tryptophan metabolism by inducing pulmonary indoleamine 2,3-dioxygenase: its possible occurrence in cancer patients. Proc Natl Acad Sci USA. (1986) 83:6622–6. doi: 10.1073/pnas.83.17.6622
60. Meyer KC, Arend RA, Kalayoglu MV, Rosenthal NS, Byrne GI, Brown RR. Tryptophan metabolism in chronic inflammatory lung disease. J Lab Clin Med. (1995) 126:530–40.
61. Kasahara DI, Wilkinson JE, Cho Y, Cardoso AP, Huttenhower C, Shore SA. The interleukin-33 receptor contributes to pulmonary responses to ozone in male mice: role of the microbiome. Respir Res. (2019) 20:197. doi: 10.1186/s12931-019-1168-x
62. Ueda Y, Frindte K, Knief C, Ashrafuzzaman M, Frei M. Effects of elevated tropospheric ozone concentration on the bacterial community in the phyllosphere and rhizoplane of rice. PLoS ONE. (2016) 11:e0163178. doi: 10.1371/journal.pone.0163178
63. Huffnagle GB, Dickson RP, Lukacs NW. The respiratory tract microbiome and lung inflammation: a two-way street. Mucosal Immunol. (2017) 10:299–306. doi: 10.1038/mi.2016.108
64. Schaldach CM, Riby J, Bjeldanes LF. Lipoxin A4: a new class of ligand for the Ah receptor. Biochemistry. (1999) 38:7594–600. doi: 10.1021/bi982861e
65. Higgins G, Ringholz F, Buchanan P, McNally P, Urbach V. Physiological impact of abnormal lipoxin A(4) production on cystic fibrosis airway epithelium and therapeutic potential. Biomed Res Int. (2015) 2015:781087. doi: 10.1155/2015/781087
66. Barnig C, Cernadas M, Dutile S, Liu X, Perrella MA, Kazani S, et al. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci Transl Med. (2013) 5:174ra126. doi: 10.1126/scitranslmed.3004812
67. Liu X, Wang X, Duan X, Poorun D, Xu J, Zhang S, et al. Lipoxin A4 and its analog suppress inflammation by modulating HMGB1 translocation and expression in psoriasis. Sci Rep. (2017) 7:7100. doi: 10.1038/s41598-017-07485-1
68. Moreira-Teixeira L, Resende M, Coffre M, Devergne O, Herbeuval JP, Hermine O, et al. Proinflammatory environment dictates the IL-17-producing capacity of human invariant NKT cells. J Immunol. (2011) 186:5758–65. doi: 10.4049/jimmunol.1003043
69. Funatake CJ, Marshall NB, Steppan LB, Mourich DV, Kerkvliet NI. Cutting edge: activation of the aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin generates a population of CD4+ CD25+ cells with characteristics of regulatory T cells. J Immunol. (2005) 175:4184–8. doi: 10.4049/jimmunol.175.7.4184
70. Negishi T, Kato Y, Ooneda O, Mimura J, Takada T, Mochizuki H, et al. Effects of aryl hydrocarbon receptor signaling on the modulation of TH1/TH2 balance. J Immunol. (2005) 175:7348–56. doi: 10.4049/jimmunol.175.11.7348
71. Sonnenberg GF, Nair MG, Kirn TJ, Zaph C, Fouser LA, Artis D. Pathological versus protective functions of IL-22 in airway inflammation are regulated by IL-17A. J Exp Med. (2010) 207:1293–305. doi: 10.1084/jem.20092054
72. Cho KA, Kim JY, Woo SY, Park HJ, Lee KH, Pae CU. Interleukin-17 and interleukin-22 induced proinflammatory cytokine production in keratinocytes via inhibitor of nuclear factor kappaB kinase-alpha expression. Ann Dermatol. (2012) 24:398–405. doi: 10.5021/ad.2012.24.4.398
Keywords: ozone, AhR, ILC3, T cells, inflammation, IL-17, IL-22
Citation: Michaudel C, Bataille F, Maillet I, Fauconnier L, Colas C, Sokol H, Straube M, Couturier-Maillard A, Dumoutier L, van Snick J, Quesniaux VF, Togbe D and Ryffel B (2020) Ozone-Induced Aryl Hydrocarbon Receptor Activation Controls Lung Inflammation via Interleukin-22 Modulation. Front. Immunol. 11:144. doi: 10.3389/fimmu.2020.00144
Received: 27 October 2019; Accepted: 20 January 2020;
Published: 25 February 2020.
Edited by:
Takayuki Yoshimoto, Tokyo Medical University, JapanReviewed by:
Feng Li, Shanghai Jiaotong University, ChinaDavid Kasahara, Harvard University, United States
Copyright © 2020 Michaudel, Bataille, Maillet, Fauconnier, Colas, Sokol, Straube, Couturier-Maillard, Dumoutier, van Snick, Quesniaux, Togbe and Ryffel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bernhard Ryffel, YmVybmhhcmQucnlmZmVsQGNucnMtb3JsZWFucy5mcg==