 Kaycie S. Lawson
Kaycie S. Lawson Anil Prasad
Anil Prasad Jerome E. Groopman
Jerome E. Groopman- Division of Experimental Medicine, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, MA, United States
Methamphetamine (Meth) abuse is a worldwide public health problem and contributes to HIV-1 pathobiology and poor adherence to anti-retroviral therapies. Specifically, Meth is posited to alter molecular mechanisms to provide a more conducive environment for HIV-1 replication and spread. Enhanced expression of inflammatory cytokines, such as Interleukin-1β (IL-1β), has been shown to be important for HIV-1 pathobiology. In addition, microRNAs (miRNAs) play integral roles in fine-tuning the innate immune response. Notably, the effects of Meth abuse on miRNA expression are largely unknown. We studied the effects of Meth on IL-1β and miR-146a, a well-characterized member of the innate immune signaling network. We found that Meth induces miR-146a and triggers an IL-1β auto-regulatory loop to modulate innate immune signaling in CD4+ T-cells. We also found that Meth enhances HIV-1 replication via IL-1 signaling. Our results indicate that Meth activates an IL-1β feedback loop to alter innate immune pathways and favor HIV-1 replication. These observations offer a framework for designing targeted therapies in HIV-infected, Meth using hosts.
Introduction
Methamphetamine (Meth) is an illicit drug abused worldwide, posing major public health challenges (1, 2). Meth use increases the spread and replication of Human Immunodeficiency Virus (HIV-1) by fostering risky sexual behaviors, and by facilitating viral infection and rapid progression to AIDS (3–7). Furthermore, Meth abuse is associated with poor adherence to anti-retroviral therapies (8, 9). A clearer understanding of the mechanisms that mediate the molecular effects of Meth on immune defenses should enhance the design of therapies targeted to patients who abuse the drug.
Distinct from HIV-1 infection, Meth has also been shown to alter the expression of inflammatory cytokines in several murine tissues, in the serum of self-administering rats, and in the plasma of human subjects in early recovery from addiction (10–12). Specifically, Meth increased Interleukin-1β (IL-1β) mRNA and protein expression in dendritic cells, and in the rat hypothalamus (13, 14). Chronic increased expression of IL-1β can result in deleterious over-stimulation of the innate immune response, and tissue damage (15). Fine-tuning the innate immune response is crucial for effective immunity. Type I Interferons (IFNs) have been shown to participate in a counter-regulatory antagonistic relationship with IL-1β to maintain the balance necessary for innate immunity (15). Interestingly, Meth suppressed expression of IFNα, a type I IFN, in macrophages (4). Notably, increased IL-1β expression and down regulation of IFN stimulated genes, including TRAF6, are associated with enhanced HIV-1 infection and replication (4, 16, 17). MicroRNAs (miRNAs) participate in transcriptional and translational regulation, cellular homeostasis and feedback regulation, and can serve as indicators of disease states (18–21). For example, miRNA signatures have been established as markers for different cancers, and can act either as oncogenes or tumor suppressors (22). miRNAs contain a highly conserved sequence at their 5′ end, known as a seed region, which binds a target sequence to cause translational repression and mRNA decay (23). Targeted transcripts are highly sensitive to changes in miRNA expression (23–25). In particular, miR-146a has garnered attention recently for its roles in immune regulation (19, 26–28). miR-146a can target TNF Receptor-Associated Factor 6 (TRAF6) and Interleukin-1 Receptor-Associated Kinase 1 (IRAK1) to suppress innate immune responses via negative feedback regulation (19). TRAF6 and IRAK1 serve as fundamental molecules for effective signal transduction resulting from innate immune stimuli (29). Several studies indicate a role for IL-1β in the induction of miR-146a (30, 31). Furthermore, miR-146a plays an important role in T-cell homeostasis, and overexpression of miR-146a in mice results in an autoimmune-like T-cell profile in the periphery (32). These functions of miR-146a merit its exploration it as a target of Meth in HIV-1 pathobiology.
To date, very little is known about the effects of Meth on miRNA expression outside of the central nervous system (CNS). We found that in CD4+ T-cells, miR-146a is up-regulated by Meth exposure in an IL-1β dependent manner. Upon Meth treatment, there was altered expression of miR-146a targets, specifically TRAF6. Furthermore, we observed that Meth activated an IL-1β positive auto-regulatory loop, which resulted in enhanced HIV-1 replication. To our knowledge, this is the first report that Meth induces an IL-1β feedback loop linked to increased HIV-1 replication and dysregulation of key innate immune pathways.
Materials and Methods
Cell Culture, HIV-1
CD4+ T-cells were isolated from healthy human donor buffy coats by ficoll-paque density gradient followed by negative selection using a CD4+ isolation kit (Stem Cell Technologies, Vancouver Canada). Isolated CD4+ cells were cultured in RPMI medium containing 10% fetal bovine serum (FBS), and 1% penicillin-streptomycin, referred to hereafter as complete RPMI. For stimulation, complete RPMI was supplemented with phytohemagglutinin (PHA-L) and interleukin-2 (IL-2). Following 3 days of stimulation, the media was changed to complete RPMI supplemented with only IL-2 to proliferate CD4+ T-cells.
HIV-1 BaL was obtained from the NIH AIDS Research and Reference Reagent Program, National Institute of Allergy and Infectious Diseases, NIH. The HIV-1 strain BaL was used for infection of CD4+ T-cells consistent with guidelines from the NIH AIDS Reagent Program, and previous studies (33, 34).
Reagents, Treatment of Cells
For methamphetamine (Meth) treatment, cells were plated at an initial density of 2 × 106cells/mL. Meth was administered once per day at a final concentration of 100 μM, consistent with observed concentrations of Meth in samples from Meth abusers (35). For cultures with HIV-1, cells were pretreated with Meth for 24 h, followed by infection with HIV-1. Cells treated with IL-1RA were given 200 or 400 ng/mL IL-1RA as described (Shenandoah Biotechnology Inc., Warwick, PA), followed by treatment with 100 μM Meth 2 h later if applicable. Cells treated with both HIV-1 and IL-1RA were pretreated for 24 h with IL-1RA before becoming infected with HIV-1. Cells treated with IFNα2 were administered 100U/mL IFNα2 (PBL Assay Science, Piscataway, NJ) for 2 h prior to treatment with 100 μM Meth. Cells treated with both HIV-1 and IFNα2 were pretreated for 24 h with IFNα2 before becoming infected with HIV-1.
Western Blotting (WB)
Primary IRAK1 and TRAF6 antibodies were obtained from Cell Signaling Technology (Danvers, MA). GAPDH was used as a loading control (Santa Cruz Biotechnology, Dallas, TX). Western Blotting was performed as described previously (36). Briefly, cells were collected and lysed, and protein was separated using NuPAGE pre-cast gels (Life Technologies Corp., Carlsbad, CA). Gels were transferred via semi-dry electrotransfer to 0.45 μm nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA), and probed with designated primary antibodies. Blots were then probed with LI-COR IRDye secondary antibodies and imaged using LI-COR Odyssey CLX according to the manufacturer's instructions (LI-COR, Lincoln, Nebraska). Analysis and relative quantification of gel bands was carried out using ImageJ software (NIH, Bethesda, MD) (37).
Quantitative RT-PCR (Real Time-Polymerase Chain Reaction)
RNA was isolated from CD4+ T-cells using TRIzol™ reagent according to the manufacturer's instructions (Thermo Fisher Scientific, Waltham, MA). DNase treatment was performed using TURBO DNA-free kit (Ambion RNA, Carlsbad, CA). One microgram of RNA was used to prepare cDNA using iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). For miR-146a cDNA, iScript Select cDNA synthesis kit was used for specific amplification using a stem-loop primer method (Bio-Rad, Hercules, CA) (38). RT-qPCR was performed in triplicate for each sample with SYBR green based PowerUp™ SYBR™ Green Master Mix (Thermo Fisher Scientific, Waltham, MA) using 100 ng cDNA. All data were normalized to internal Tata-Box Binding Protein (TBP) control gene, and fold change was calculated using the 2−ΔΔCt method.
Primer Sequences:
miR-146a RT-SL (NR_029701.1): GTCGTATCCAGTGCAGGGTCCGAGGTA
TTCGCACTGGATACGACAACCCA
IL-1β F (NM_000576.2): AGCTGATGGCCCTAAACAGATG
IL-1β R (NM_000576.2): TTGTCCATGGCCACAACAAC
IRAK1 F (NM_001569.3): AGAAAAGTTGGGAGCATGGC
IRAK1 R (NM_001569.3): TTTTGGACACGCAAGAGGAC
TRAF6 F (NM_145803.2): ACGGAGCGCATAAAACAAGC
TRAF6 R (NM_145803.2): TCAGCCCAGCAATTCAGTTG.
ELISA
IL-1β ELISA assay was performed according to the manufacturer's protocol using cell culture supernatants (Chondrex, Inc., Redmond WA). Culture supernatants from cells incubated with HIV-1 alone or HIV-1 and Meth/IFNα/IL-1RA were harvested on days 0, 1, 3, and 7. P24 ELISA was performed using the Zeptometrix ELISA kit according to the manufacturer's protocol (Zeptometrix Corporation, Buffalo NY). Supernatants were stored at −80°C.
Transfection
Transfection of primary CD4+ T-cells was carried out using “Nucleofector Kit for T-cells” according to the manufacturer's protocol (Lonza Group, Basel Switzerland). miR-146a-5p mimic and mimic “Negative control #1” (Dharmacon, Lafayette CO) were used for transfection at a concentration of 20 nM. miR-146a-5p stem-loop miRNA inhibitor and stem-loop miRNA inhibitor “Negative control #1” (Dharmacon, Lafayette CO) were used for transfection at a concentration of 40 nM. Transfected cells were harvested at 48 h post transfection.
Flow Cytometry
Cells untreated or treated with Nigericin, Meth, or IFNα+Meth were subjected to the FAM-FLICA Caspase 1 Assay Kit according to the manufacturer's instructions (Immunochemistry Technologies, Bloomington, MN). Nigericin was used as a positive control at a final concentration of 10 μM (Immunochemistry Technologies, Bloomington, MN). Cells were analyzed using a CytoFlex flow cytometer (Beckman Coulter, Indianapolis, IN).
Statistics
All experiments were conducted at least in triplicate, using at least three different donors, and the results between experimental groups were analyzed by ANOVA. A p < 0.05 was considered to be minimally significant.
Study Approval
Healthy human donor buffy coats were obtained from the Blood Transfusion Service, Massachusetts General Hospital, Boston, MA, in compliance with the Beth Israel Deaconess Medical Center Committee on Clinical Investigations (CCI) protocol #2008-P-000418/5. Buffy coats were provided at this institution for research purposes without identifiers; therefore, no informed consent was needed. This study was approved by Beth Israel Deaconess Medical Center's CCI, Institutional Review Board, and Privacy Board appointed to review research involving human subjects. The experimental procedures were carried out in strict accordance with approved guidelines.
Results
Meth Enhances IL-1β Expression and Caspase-1 Activation in CD4+ T-Cells
Meth has been shown to alter inflammatory cytokine expression in several murine and human models, both in the periphery and the CNS (10–12, 39). In particular, Meth has been linked to enhanced IL-1β expression in dendritic cells and in the rat hypothalamus (13, 14). Thus, we first sought to study the effects of Meth treatment on IL-1β expression in CD4+ T-cells.
Healthy donor CD4+ T-cells were treated daily with 100 μM Meth, and culture supernatants were harvested on days 1 and 3. We observed significantly increased release of IL-1β on days 1 and 3 of Meth treatment (Figure 1A). These results suggested that IL-1β may be a key cytokine released during Meth exposure.
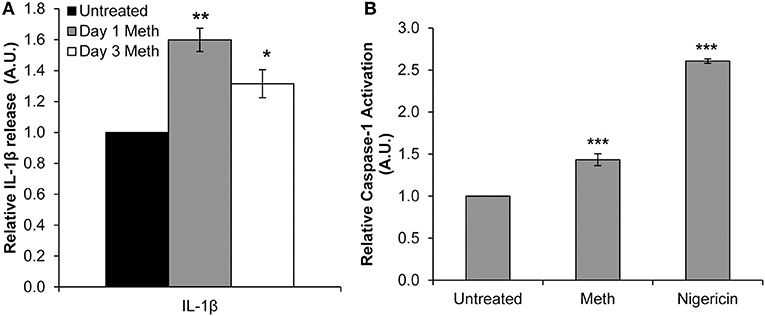
Figure 1. Meth enhances IL-1β expression and Caspase-1 activation in CD4+ T-cells. CD4+ T-cells were treated daily with or without Meth. (A) Expression of IL-1β was determined from cell culture supernatants by ELISA analysis. Relative expression was calculated by normalizing Meth treated samples to untreated control cells. Data represent the mean ± SD of 3 independent experiments, and p-values were calculated relative to untreated controls (*p < 0.05, **p < 0.01). (B) CD4+ T-cells were untreated, treated with Meth, or treated with Nigericin. Caspase-1 Activation was measured using fluorescent labeling with FAM-FLICA, and analyzed by Flow Cytometry. Data represent the mean ± SD of 3 independent experiments, and p-values were calculated relative to untreated controls (***p < 0.001).
Two steps are required for IL-1β to become its mature, released form. First, the IL-1β gene is translated to a precursor protein, known as pro-IL-1β (40). Pro-IL-1β undergoes post-translational processing by the NLRP3 Inflammasome and Caspase-1 to yield its mature form (40, 41). Interestingly, Mahajan et al. found that Meth increased expression of IL-1β in dendritic cells, and in microglial cells Meth has been shown to induce activation of the NLRP3 Inflammasome (13, 42). To assess induction of IL-1β processing in Meth treated CD4+ T-cells, we analyzed Caspase-1 activation relative to untreated cells 24 h after Meth treatment. Nigericin, a potent microbial toxin known to induce activation of Caspase-1 and the NLRP3 Inflammasome, was used as a positive control. We found that Meth treatment significantly increased the activation of Caspase-1 relative to untreated controls, concordant with increased IL-1β expression (Figure 1B).
Meth Increases miR-146a Expression and Down-Regulates TRAF6
IL-1β signaling can participate in a positive auto-regulatory loop, resulting in increased transcription of its gene (43). In addition, it has been reported that IL-1β can induce NFκB-dependent miR-146a expression to interfere with innate immune functions (31). Non-coding RNAs play important roles in regulating cellular activities and stress responses. Furthermore, Meth is known to induce activation and nuclear translocation of NFκB (44). Thus, we used RT-qPCR to identify Meth-related changes in miR-146a and IL-1β mRNA in primary CD4+ T lymphocytes.
Healthy donor CD4+ T-cells were treated daily with 100 μM Meth, and miR-146a expression was assessed. We observed that Meth significantly up-regulated miR-146a on day 3 of treatment (Figure 2A). Likewise, we assessed IL-1β mRNA levels in untreated and Meth treated cells. Unlike extracellular IL-1β, which increased after 1 day of Meth treatment, IL-1β mRNA showed increased expression only on day 3 (Figure 2A). Notably, IL-1β release and mRNA expression are controlled by distinct mechanisms (45). In addition, CD4+ T-cells constitutively express pro-IL-1β in their cytoplasm (46). As such, our results indicate that Meth first enhances release of mature IL-1β, followed by increased IL-1β gene transcription and miR-146a expression.
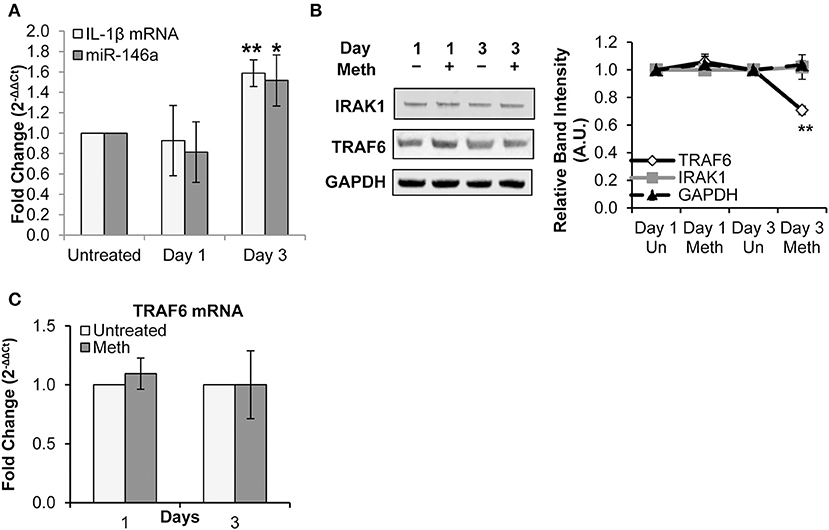
Figure 2. Meth increases miR-146a expression and downregulates of TRAF6. CD4+ T-cells were treated daily with or without Meth. (A) Expression of miR-146a and IL-1β mRNA was determined by RT-qPCR. Fold change was calculated by normalizing the Meth treated cells to untreated cells. Data represent the mean ± SD of 3 independent experiments, and p-values were calculated relative to untreated controls (*p ≤ 0.05, **p ≤ 0.01). (B) Cells treated or untreated with Meth were lysed and protein extracts were analyzed for expression of TRAF6 and IRAK1 by Western Blotting. GAPDH was used as a loading control. Relative band intensity was calculated using ImageJ software. (C) Expression of TRAF6 mRNA was determined by RT-qPCR. Fold change was calculated by normalizing the Meth treated cells to untreated cells. Data represent the mean ± SD of 3 independent experiments.
We next explored the effects of Meth on TRAF6 and IRAK1, known targets of miR-146a linked to IL-1β and innate immune signaling, and implicated in HIV-1 pathobiology (31, 47). Specifically, IRAK1 plays a key role in mediating IL-1β-induced NFκB activation, and decreased expression of TRAF6 has been implicated in increased HIV-1 replication (17, 47, 48).
By Western Blot analysis, we observed decreased TRAF6 protein expression on day 3 of Meth exposure, but no significant change in IRAK1 protein expression (Figure 2B). Through RT-qPCR, we found no significant change in TRAF6 mRNA expression (Figure 2C).
Collectively, our results— specifically decreased levels of TRAF6 protein and unchanged levels of TRAF6 mRNA— are consistent with fine tuning by miRNA, whereby translational repression precedes mRNA decay (49). Notably, decreased TRAF6 expression observed on day 3 correlated with increased miR-146a levels. These findings suggest a role for miR-146a in regulation of innate immune signaling pathways through repression of TRAF6 during Meth treatment.
We hypothesized that IL-1β signaling is responsible for the increased IL-1β mRNA levels, as well as increased miR-146a expression observed on day 3 of Meth treatment; we further hypothesized that IL-1β-dependent induction of miR-146a would target key innate immune pathways via TRAF6 inhibition.
Overexpression and Inhibition of miR-146a Confirms Immune Signaling Molecule Targets in CD4+ T-Cells
TRAF6 and IRAK1 are known direct targets of miR-146a in several cell types, including murine macrophages and liver cells, primary human monocytes, and THP-1 monocytic cells (19, 27, 50, 51). We employed a miR-146a mimic to confirm that TRAF6 and IRAK1 are targets of the miRNA in primary CD4+ T-cells. The miR-146a mimic was transfected into CD4+ T-cells, resulting in an expected significant increase in miR-146a expression (Figure 3A). We observed no change in IL-1β mRNA expression (Figure 3B). RT-qPCR analysis showed that TRAF6 and IRAK1 mRNA levels were significantly decreased, indicating that miR-146a targeted these molecules and initiated mRNA degradation pathways (Figure 3B).
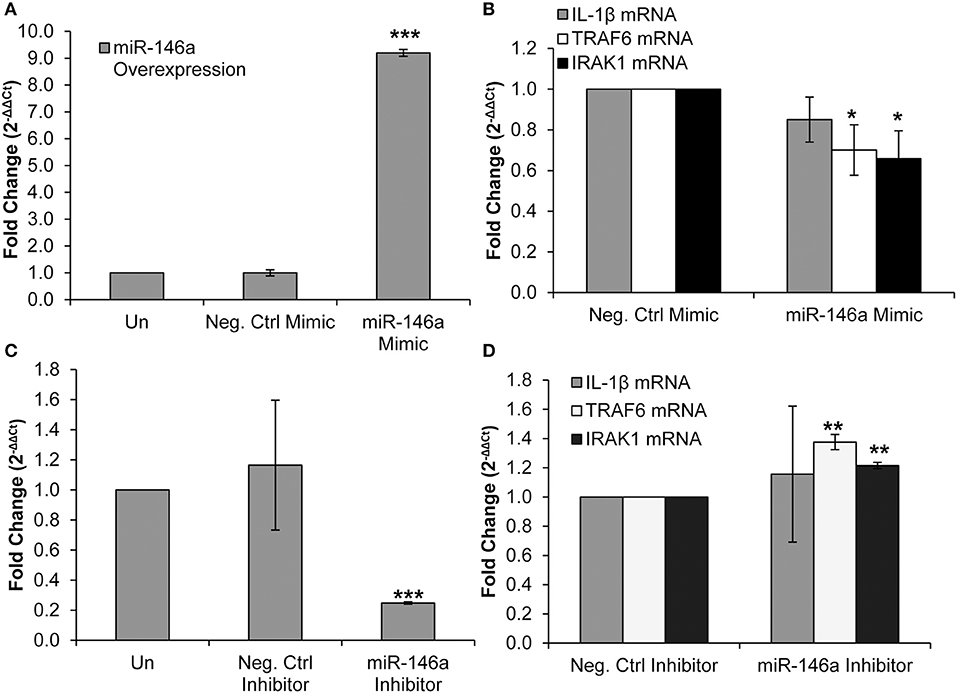
Figure 3. Overexpression and inhibition of miR-146a confirms immune signaling molecule targets in CD4+ T-cells. CD4+ T-cells were un-transfected, transfected with a negative control mimic or miR-146a mimic. (A) Successful miR-146a overexpression was confirmed by RT-qPCR. Fold change was calculated by normalizing the transfected cells to non-transfected cells. Data represent the mean ± SD of 3 independent experiments, and p-values were calculated relative to non-transfected controls (***p < 0.001). (B) Expression of IL-1β, TRAF6, and IRAK1 mRNA levels during miR-146a overexpression was determined by RT-qPCR. Fold change was calculated by normalizing miR-146a mimic-transfected cells to negative control-transfected cells. Data represent the mean ± SD of 3 independent experiments, and p-values were calculated relative to negative controls (*p < 0.05). (C) CD4+ T-cells were un-transfected, transfected with a negative control inhibitor or a miR-146a inhibitor. Successful miR-146a inhibition was confirmed by using RT-qPCR. Fold change was calculated by normalizing the transfected cells to non-transfected cells. Data represent the mean ± SD of 3 independent experiments, and p-values were calculated relative to non-transfected controls (***p < 0.001). (D) Expression of IL-1β, TRAF6, and IRAK1 mRNA levels during miR-146a inhibition was determined by RT-qPCR. Fold change was calculated by normalizing miR-146a inhibitor-transfected cells to negative control-transfected cells. Data represent the mean ± SD of 3 independent experiments, and p-values were calculated relative to negative controls (**p < 0.01).
We then used a miR-146a inhibitor and analyzed changes in TRAF6, IRAK1, and IL-1β mRNA expression. Transfection of the inhibitor significantly reduced miR-146a expression (Figure 3C). IL-1β mRNA expression was unchanged, but TRAF6 and IRAK1 mRNA levels were significantly increased (Figure 3D). Taken together, these data confirm IRAK1 and TRAF6 as targets of miR-146a in CD4+ T-cells.
Meth Increases miR-146a and IL-1β mRNA Expression via IL-1 Signaling
IL-1β has been shown to up-regulate miR-146a expression in THP-1 monocytes by activating its NFκB-dependent transcription (31). Meth treatment of CD4+ T-cells increased extracellular IL-1β levels followed by enhanced miR-146a and IL-1β mRNA expression, and decreased TRAF6 protein expression. These results suggested that Meth may modulate the innate immune response via IL-1β signaling to enhance miR-146a and IL-1β mRNA and decrease TRAF6. To address this hypothesis, we blocked IL-1 signaling by employing an IL-1 Receptor Antagonist (IL-1RA).
IL-1RA abrogated both Meth induced miR-146a overexpression and increased IL-1β mRNA levels (Figure 4A). Furthermore, TRAF6 protein expression levels, which decreased in the presence of Meth alone, were increased in Meth+IL-1RA treated samples relative to controls (Figure 4B). By ELISA analysis, we observed unchanged extracellular concentrations of IL-1β in samples treated with Meth+IL-1RA, whereas Meth alone resulted in significantly increased extracellular IL-1β levels (Figure 4C). These results support the hypothesis that IL-1 signaling mediates Meth induced miR-146a to target TRAF6. Furthermore, abrogated levels of IL-1β mRNA in cells treated with IL-1RA support the role of IL-1β signaling in a positive auto-regulatory loop.
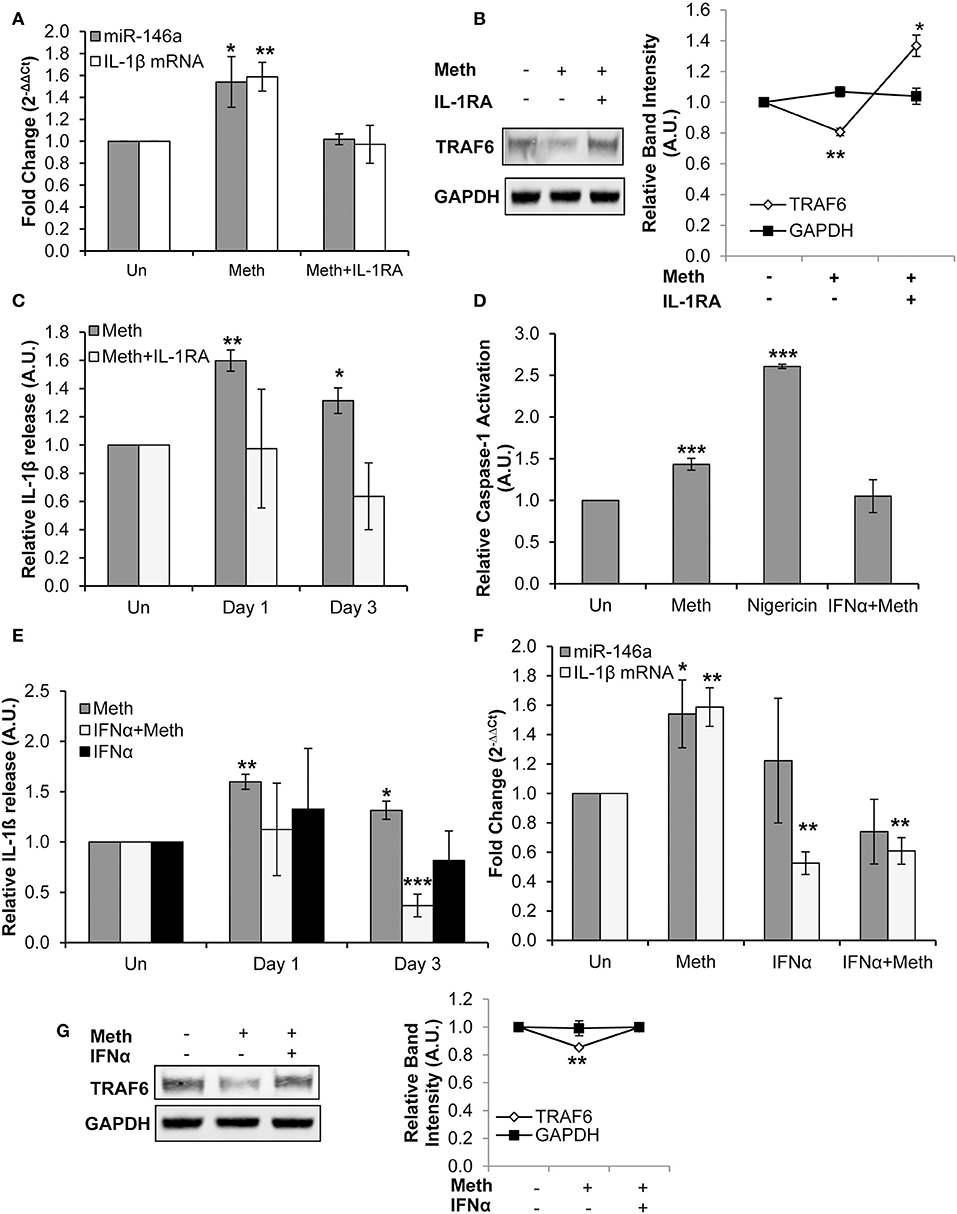
Figure 4. Meth increases miR-146a and IL-1β mRNA expression via IL-1 signaling. (A) CD4+ T-cells treated with or without Meth for 3 days and IL-1RA were analyzed for miR-146a and IL-1β mRNA expression by RT-qPCR. Fold change was calculated by normalizing Meth treated and Meth+IL-1RA treated cells to untreated controls. Data represent the mean ± SD of 3 independent experiments, and p-values were calculated relative to untreated controls (*p < 0.05, **p < 0.01). (B) Protein extracts from cells treated for 3 days with or without Meth and IL-1RA were analyzed for TRAF6 by Western Blotting. GAPDH was used as a loading control. Relative band intensity was calculated using ImageJ software, and p-values were calculated relative to untreated controls (*p < 0.05, **p < 0.01). (C) Culture supernatants were harvested after 3 days of treatment and analyzed for IL-1β by ELISA. Relative expression was calculated by normalizing Meth and Meth+IL-1RA treated samples to untreated controls. Data represent the mean ± SD of 3 independent experiments, and p-values were calculated relativeto untreated controls (*p ≤ 0.05, **p ≤ 0.01). (D) CD4+ T-cells were untreated, treated with Meth, treated with Nigericin, or treated with IFNα and Meth for 24 h. Caspase-1 Activation was measured using fluorescent labeling with FAM-FLICA, and analyzed by Flow Cytometry. Data represent the mean ± SD of 3 independent experiments, and p values were calculated relative to untreated controls (***p < 0.001). (E) CD4+ T-cells were untreated, treated with Meth, or treated with IFNα and Meth, daily for 3 days. Culture supernatants were analyzed for IL-1β expression by ELISA. Relative expression was calculated by normalizing Meth treated samples to untreated controls. Data represent the mean ± SD of 3 independent experiments, and p-values were calculated relative to untreated controls (*p < 0.05, **p < 0.01, ***p < 0.001). (F) Cells were untreated, treated with Meth, or treated with IFNα and Meth, daily for 3 days. miR-146a and IL-1β mRNA expression were determined by RT-qPCR. Fold change was calculated by normalizing Meth treated and Meth+IFNα treated cells to untreated controls. Data represent the mean ± SD of 3 independent experiments, and p-values were calculated relative to untreated controls (*p < 0.05, **p < 0.01). (G) Cells were untreated, treated with Meth, or treated with IFNα and Meth, daily for 3 days. Protein extracts were analyzed for TRAF6 by Western Blot. GAPDH was used as a loading control. Relative band intensity was calculated using ImageJ software, and p-values were calculated relative to untreated controls (**p < 0.01).
IFNα, a member of the Type I IFN family, has been shown to negatively regulate IL-1β expression, resulting in a dynamic antagonistic relationship between these cytokines (15). This occurs because Type I IFNs can inhibit Caspase-1 and Inflammasome activation (52). We observed that Meth enhanced Caspase-1 activation in CD4+ T-cells, and thus explored the effects of exogenous IFNα on the activation of Caspase-1 in CD4+ T-cells.
CD4+ T-cells were untreated, treated with Nigericin, treated with Meth, or treated with Meth and IFNα concomitantly (Meth+IFNα) for 24 h. The cells treated with Meth alone showed increased Caspase-1 activation; IFNα abrogated this effect, consistent with the antagonistic relationship between IFNα and Caspase-1 (Figure 4D). Furthermore, by ELISA analysis, we examined IL-1β release under each condition. In the presence of IFNα, there was no change in extracellular IL-1β levels on day 1, but there was significantly decreased IL-1β release on day 3 (Figure 4E). These results show that IFNα inhibits release of IL-1β in Meth treated CD4+ T-cells by inhibiting Caspase-1 activation.
After establishing the inhibitory effects of exogenously added IFNα on Caspase-1 activation and IL-1β release, we evaluated its effects on Meth mediated IL-1β mRNA and miR-146a overexpression. While Meth alone significantly increased miR-146a and IL-1β mRNA expression, when cells were treated with IFNα+Meth, miR-146a expression was unchanged and IL-1β mRNA levels were significantly decreased (Figure 4F). Further, we analyzed the expression of TRAF6 protein. Consistent with our earlier results, we observed decreased expression of TRAF6 in Meth treated cells, but IFNα abrogated this decrease (Figure 4G).
Exogenous IFNα counteracted Meth induced Caspase-1 activation and overexpression of IL-1β, in agreement with previous reports that IFNα antagonizes IL-1β (15, 53). The abrogation of Meth mediated miR-146a overexpression by IFNα further supports the role of IL-1β in Meth mediated miR-146a overexpression.
Meth Enhances HIV-1 Replication via IL-1 Signaling in CD4+ T-Cells
Since Meth was found to induce IL-1β and miR-146a expression, and these play important roles in immune regulation, we explored their effects on HIV-1 infection of CD4+ T-cells.
We first confirmed that Meth increases HIV-1 replication in CD4+ T-cells, as shown in previous studies (Figure 5A) (5, 54). Next, we assessed the effects of Meth on release of IL-1β under various conditions. Cells were pretreated for 24 h with Meth before exposure to HIV-1; Meth was then administered daily. At 1 day post infection, we observed significantly increased release of IL-1β in cells either exposed to HIV-1 or HIV+Meth (Figure 5B). However, at 2 days post infection, we observed decreased release of IL-1β.
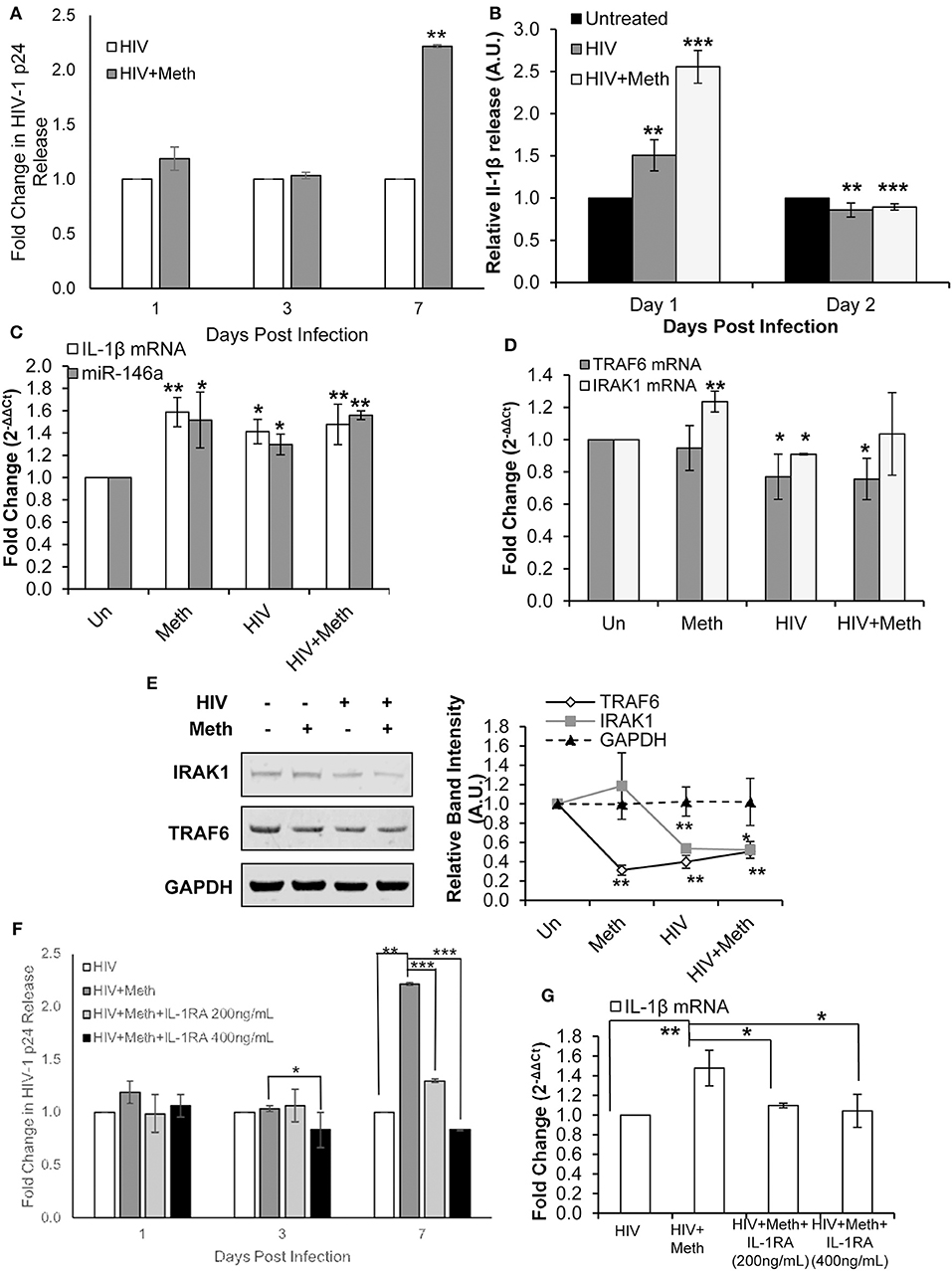
Figure 5. Methamphetamine enhances HIV-1 replication via IL-1 signaling in CD4+ T-cells. CD4+ T-cells were uninfected, infected with HIV-1, treated with Meth alone, or treated with Meth and infected with HIV-1 concomitantly. Cells were harvested at 2 days post infection and supernatants were harvested at the time points indicated on graphs. (A) Culture supernatants were analyzed for HIV-1 replication by p24 ELISA, and p-values were calculated relative to untreated controls (**p < 0.01). (B) IL-1β ELISA was used to determine the concentration of IL-1β from culture supernatants harvested on days 1 and 2 P.I. Relative expression was calculated by normalizing HIV and HIV+Meth samples to untreated controls. Data represent the mean ± SD of 3 independent experiments, and p-values were calculated relative to untreated controls (**p < 0.01, ***p < 0.001). (C) miR-146a and IL-1β mRNA expression was determined by RT-qPCR. Fold change was calculated by normalizing Meth, HIV, or HIV+Meth samples to untreated controls. Data represent the mean ± SD of 3 independent experiments, and p-values were calculated relative to untreated controls (*p < 0.05, **p < 0.01). (D) RT-qPCR was performed to assess changes in TRAF6 and IRAK1 mRNA expression on day 2 P.I. Fold change was calculated by normalizing Meth, HIV or HIV+Meth samples to untreated control cells. Data represent the mean ± SD of 3 independent experiments, and p-values were calculated relative to untreated controls (*p < 0.05, **p < 0.01). (E) Cells were harvested and lysed on day 2 P.I. Protein extracts were analyzed for IRAK1 and TRAF6 by Western Blot. GAPDH was used as a loading control. Relative band intensity was calculated using ImageJ software, and p-values were calculated relative to untreated controls (*p < 0.05, **p < 0.01). (F) CD4+ T-cells were infected with HIV-1 alone, infected with HIV-1 and treated with Meth daily, or infected with HIV-1 and treated with Meth and IL-1RA (200 or 400 ng/mL) daily. Culture supernatants were analyzed for HIV-1 replication by p24 ELISA, and p-values were calculated relative to HIV+ for HIV+Meth samples, or relative to HIV+Meth for HIV+Meth+Il-1RA samples (*p < 0.05, **p < 0.01, ***p < 0.001). (G) Cells were harvested at 2 days P.I. IL-1β mRNA expression was analyzed by RT-qPCR. Fold change was calculated by normalizing HIV+Meth or HIV+IL-1RA to HIV+ samples. Data represent the mean ± SD of 3 independent experiments, and p values were calculated relative to HIV+ for HIV+Meth samples, and relative to HIV+Meth for HIV+Meth+IL-1RA samples (*p < 0.05, **p < 0.01).
We then analyzed the expression of IL-1β mRNA and miR-146a in Meth, HIV+, and HIV+Meth treated cells. We observed significantly increased IL-1β mRNA levels as well as increased miR-146a expression across all treatments 2 days post infection (Figure 5C).
Next, we assessed the expression of TRAF6 and IRAK1. Interestingly, TRAF6 mRNA levels were unchanged in Meth treated cells, but showed significantly decreased expression in HIV+ and HIV+Meth samples (Figure 5D). In contrast, IRAK1 mRNA showed significantly increased expression in the presence of Meth alone, but significantly decreased expression in HIV+ samples (Figure 5D). When cells were treated with Meth and HIV-1 in combination, IRAK1 mRNA displayed baseline expression (Figure 5D). By Western Blot, we observed decreased TRAF6 protein levels in Meth, HIV+, and HIV+Meth samples (Figure 5E). IRAK1 protein levels showed no change in Meth treated vs. untreated cells, but decreased expression in HIV+ and HIV+Meth treated cells 2 days post infection (Figure 5E).
These data demonstrate that both HIV-1 and Meth increased IL-1β and miR-146a expression. We also observed that HIV-1 inhibited TRAF6 and IRAK1 expression. Unlike Meth treatment, HIV-1 inhibited TRAF6 at the RNA level. Moreover, IRAK1 was inhibited only in HIV-1 infected samples, consistent with previous reports (55). These findings suggest that HIV-1 inhibits expression of TRAF6 and IRAK1 independent of miR-146a expression.
Interestingly, HIV+ and HIV+Meth samples displayed increased extracellular IL-1β levels on day 1 post infection, while IL-1β mRNA levels were significantly increased at day 2 post infection. These data suggest an important role for a Meth mediated IL-1β auto-regulatory feedback loop, which may augment the inflammatory state triggered during HIV-1 infection.
To assess the involvement of Meth induced IL-1β in enhanced HIV-1 replication, we blocked IL-1 signaling using exogenous IL-1RA. Cells were pretreated with IL-1RA and/or Meth, for 24 h before exposure to HIV-1; IL-1RA and Meth were then administered daily. When HIV-1 infected CD4+ T-cells were co-treated with IL-1RA and Meth, the effect of Meth on enhancing HIV-1 replication was significantly attenuated in a dose dependent manner. We observed that while 200 ng/mL IL-1RA was sufficient to reduce the effect of Meth on HIV-1 replication, virus replication in these cells was still significantly higher than in HIV+ cells (Figure 5F). However, when cells were treated with higher concentrations of IL-1RA (400 ng/mL) prior to Meth treatment, HIV-1 replication was significantly inhibited (Figure 5F). Notably, when HIV-1 infected CD4+ T-cells were treated with IL-1RA alone, there was no change in HIV-1 replication (Figure S1A).
Next, we analyzed IL-1β mRNA expression in HIV-1 infected CD4+ T-cells that were untreated, or treated with Meth, or treated with Meth and IL-1RA together. We observed that HIV+Meth+IL-1RA200ng/mL samples showed only slightly increased IL-1β mRNA expression, while IL-1β mRNA expression remained unchanged in HIV+Meth+IL-1RA400ng/mL samples relative to HIV+ controls (Figure 5G). These results are consistent with the pattern we observed during HIV-1 replication, wherein Meth augments IL-1β mRNA and HIV-1 replication, but these effects are slightly reduced in the presence of 200 ng/mL IL-1RA, and completely abrogated in the presence of 400 ng/mL IL-1RA.
Taken together, these results demonstrate that IL-1 signaling is important for Meth mediated effects on HIV-1 replication. Specifically, our results showed that increased IL-1β expression during Meth treatment plays a role in enhanced HIV-1 replication. Further, we observed relatively unchanged expression of miR-146a, TRAF6, and IRAK1 among HIV+, HIV+Meth, HIV+Meth+IL1RA200ng/mL, and HIV+Meth+IL-1RA400ng/mL samples (Figures S1B,C), suggesting that an IL-1β positive feedback loop is central to enhanced HIV-1 replication in the presence of Meth.
IFNα Treatment Inhibits HIV-1 Replication and Disrupts IL-1β and miR-146a Expression
IFNα has been shown to inhibit both HIV-1 replication and IL-1β expression (15, 56). Thus, we next sought to probe the expression of IL-1β and miR-146a expression in IFNα treated HIV-1 infected CD4+ T-cells.
Healthy donor CD4+ T-cells were infected with HIV-1 alone (HIV+), infected with HIV-1 and treated with Meth (HIV+Meth), or infected with HIV-1 and treated with IFNα (HIV+IFNα). These experimental conditions allowed us to assess changes in IL-1β expression in baseline, enhanced, or diminished HIV-1 infection, respectively.
We first confirmed that HIV-1 replication was diminished in the presence of IFNα (Figure 6A). Next, we assessed IL-1β mRNA and miR-146a expression in CD4+ T-cells treated under each condition. We observed, relative to HIV+ cells, significantly decreased miR-146a and IL-1β mRNA expression with IFNα treatment (Figure 6B). There was significantly increased IL-1β release in HIV+Meth treated cells, and significantly decreased IL-1β release in HIV+IFNα cells relative to HIV+ cells (Figure 6C). TRAF6 and IRAK1 mRNA expression were unchanged in HIV+ and HIV+Meth conditions, but significantly increased expression of IRAK1 mRNA was observed in cells treated with HIV+IFNα (Figure 6D). Finally, TRAF6 protein was maintained at low levels of expression among HIV+, HIV+Meth and HIV+IFNα samples (Figure 6E). However, IRAK1 protein showed increased expression in cells treated with HIV+IFNα compared to both HIV+ and HIV+Meth treated cells (Figure 6E).
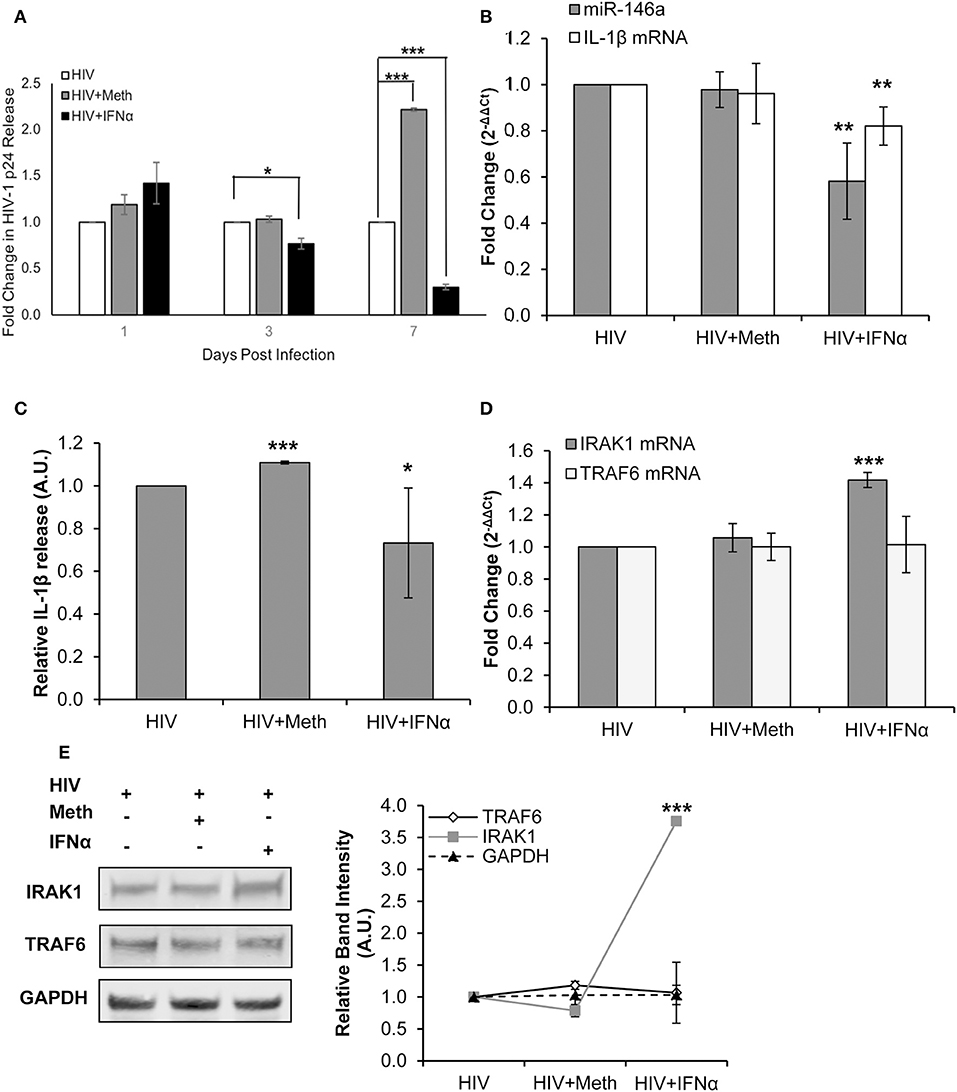
Figure 6. IFNα treatment inhibits HIV-1 replication and disrupts IL-1β and miR-146a expression. CD4+ T-cells were infected with HIV-1 alone, infected with HIV-1 and treated with Meth daily, or infected with HIV-1 and treated with IFNα daily. (A) Culture supernatants were analyzed for HIV-1 replication by p24 ELISA, and p-values were calculated relative to HIV+ controls (*p < 0.05, **p < 0.01, ***p < 0.001). (B) Cells were harvested at 2 days P.I. miR-146a and IL-1β mRNA expression was analyzed by RT-qPCR. Fold change was calculated by normalizing HIV+Meth or HIV+IFNα to HIV+ samples. Data represent the mean ± SD of 3 independent experiments, and p-values were calculated relative to HIV+ controls (**p < 0.01). (C) Culture supernatants were collected 2 days P.I. and analyzed for IL-1β concentration by ELISA. (*p < 0.05, ***p < 0.001). (D) Cells harvested at 2 days P.I. were analyzed for TRAF6 and IRAK1 mRNA expression by RT-qPCR. Fold change was calculated by normalizing HIV+Meth or HIV+IFNα to HIV+ samples. Data represent the mean ± SD of 3 independent experiments, and p-values were calculated relative to HIV+ controls (***p < 0.001). (E) Protein extracts from cells harvested at 2 days P.I. were analyzed for TRAF6 and IRAK1 expression by Western Blot analysis. GAPDH was used as a loading control. Relative band intensity was calculated using ImageJ software, and p-values were calculated relative to HIV+ controls (***p < 0.001).
Taken together, these results demonstrate that immunomodulation by IFNα inhibited HIV-1 replication concordantly with decreased IL-1β and miR-146a expression in CD4+ T-cells. Interestingly, TRAF6 protein and mRNA levels were unchanged among HIV+, HIV+Meth, and HIV+IFNα samples. Moreover, IFNα significantly increased the expression of IRAK1 at RNA and protein levels concordant with decreased miR-146a expression.
Discussion
Meth abuse is a well-established risk factor for HIV/AIDS (5, 57). The drug can enhance virus replication and clinical progression, often promoting poor adherence to anti-retroviral therapies. Meth has also been shown to modulate inflammatory cytokine expression (10–12). Specifically, Meth has been reported to enhance IL-1β expression in dendritic cells and the rat hypothalamus (13, 14). The effects of Meth on host defenses and the miRNAs that regulate these processes, are not well-defined (3–5, 9, 18, 23, 58). In addition, miRNA targets are highly sensitive to changes in their expression (23, 24). IL-1β has been shown to participate in an auto-regulatory loop, and stimulate NFκB-dependent miR-146a to disrupt key inflammatory responses (31, 43). Several direct targets of miR-146a have been implicated in HIV-1 pathobiology (47, 48, 59). Notably, miR-146a targets both TRAF6 and IRAK1, signaling molecules that facilitate innate immune responses (26, 27, 48, 60). HIV-1 can induce expression of IL-1β, which is associated with progression of HIV/AIDS, and in microglial cells HIV-1 infection resulted in overexpression of miR-146a (16, 61).
Here, we demonstrate that Meth can induce expression of IL-1β and miR-146a in CD4+ T-cells, with overexpression of the cytokine leading to increased expression of miR-146a. Meth mediated miR-146a overexpression targeted TRAF6 to modulate innate immune signaling pathways. Based on our findings, we hypothesized that IL-1β signaling results in increased IL-1β mRNA levels and increased miR-146a expression. We further hypothesized that IL-1β-dependent induction of miR-146a would target key innate immune pathways by decreasing TRAF6 expression (Figure S2). Although there are several known targets of miR-146a, only TRAF6 was significantly affected during Meth treatment. We also observed that induction of an IL-1β auto-regulatory loop contributed to Meth mediated increases in HIV-1 replication.
Meth treatment of CD4+ T-cells augmented Caspase-1 activation and enhanced IL-1β release. Subsequently, both IL-1β mRNA and miR-146a levels rose, indicating that IL-1β increased expression of these transcripts. We also explored changes in TRAF6 and IRAK1 proteins, both direct targets of miR-146a. While we found no change in IRAK1 protein expression, there was decreased TRAF6 expression, indicating selective inhibition of TRAF6 by Meth via miR-146a.
To further elucidate the role of Meth in IL-1β and miR-146a overexpression, we blocked IL-1β signaling at two steps. Cells were treated with either IL-1RA to block binding of IL-1β to its receptor, or exogenous IFNα, a type I IFN known to inhibit Caspase-1 activation and antagonize mature IL-1β release. We observed decreased IL-1β release after exogenous IFNα treatment, along with decreased Caspase-1 activation. Blocking either the IL-1 receptor with IL-1RA, or release of IL-1β with IFNα, abrogated Meth mediated effects on miR-146a and IL-1β mRNA. Our data demonstrate that Caspase-1 activation, and increased IL-1β release and signaling are critical for Meth mediated miR-146a overexpression and enhanced IL-1β mRNA expression. Notably, Caspase-1 and IL-1β are associated with pryoptosis, or inflammation mediated apoptosis; pryoptosis is linked to progression of HIV-1 infection via CD4+ T-cell depletion (62–64).
At day 1 of HIV-1 infection, we observed enhanced release of IL-1β, followed by increased IL-1β mRNA and miR-146a expression at day two. Decreased TRAF6 expression, a known miR-146a target, is associated with increased HIV-1 replication (47). We observed decreased TRAF6 expression upon infection with HIV-1 at the gene expression level. IRAK1 protein expression levels were also decreased following HIV-1 infection. These findings are consistent with previous reports that HIV-1 decreases IRAK1 and TRAF6 expression (47, 55).
In sum, we found that Meth enhanced HIV-1 replication through an IL-1β positive auto-regulatory loop. By blocking IL-1 signaling using IL-1RA, we observed dose dependent decreased HIV-1 replication after Meth treatment. We also observed that Meth enhanced IL-1β mRNA levels were abrogated in a dose dependent manner upon treatment with IL-1RA. These results support the involvement of an IL-1β auto-regulatory loop in Meth mediated enhanced HIV-1 replication. Alternatively, when HIV-1 replication was inhibited using IFNα, we found decreased IL-1β and miR-146a expression. Our results indicate that increased levels of IL-1β directly contribute to Meth mediated increased HIV-1 replication in CD4+ T-cells. Based on our results, it appears that Meth mediated increased IL-1β expression acts to prime cells to be more susceptible to infection with HIV-1.
These studies unveil unique effects of Meth on IL-1β to dysregulate innate immune signaling pathways and enhance HIV-1 infection. This novel mechanism of action of Meth points to potential therapeutic targets relevant to drug abusing hosts.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Ethics Statement
The studies involving human participants were reviewed and approved by Beth Israel Deaconess Medical Center Committee on Clinical Investigations (CCI). Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author Contributions
KL: conceptualization, investigation, and writing. AP: methodology and writing. JG: supervision, funding acquisition, and writing.
Funding
This work was supported by the National Institutes of Health [National Institute on Drug Abuse (http://www.drugabuse.gov)], 1R01DA039566 (JEG).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The following reagent was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: HIV-1Ba-L from Dr. Suzanne Gartner, Dr. Mikulas Popovic and Dr. Robert Gallo.
The authors acknowledge Dr. Shuxian Jiang for her technical role in obtaining Western Blot images.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00136/full#supplementary-material
Figure S1. No effect of IL-1RA on HIV-1 replication, TRAF6, miR-146a, or IRAK1 expression. CD4+ T-cells were infected with HIV-1 alone, or infected with HIV-1 and treated with Meth or IL-1RA at varying concentrations. (A) Culture supernatants were analyzed for HIV-1 replication by p24 ELISA. Statistical analyses were performed to assess changes relative to HIV+ controls. No significant changes were observed. (B) miR-146a expression was analyzed by RT-qPCR. Fold change was calculated by normalizing HIV+Meth, HIV+IL-1RA200 ng/mL, and HIV+IL-1RA400 ng/mL to HIV+ samples. Data represent the mean ± SD of 3 independent experiments (***p < 0.001). p-values were calculated relative to HIV+ controls. (C) Protein extracts from cells harvested at 2 days P.I. were analyzed for TRAF6 and IRAK1 expression by Western Blot analysis. GAPDH was used as a loading control. Relative band intensity was calculated using ImageJ software (***p < 0.001). p-values were calculated relative to untreated controls.
Figure S2. Hypothetical relationship between IL-1β signaling and miR-146a expression and inhibition of TRAF6. Visual representation of hypothesized pathway for Meth induced effects on miR-146a and downstream targets via IL-1β.
References
1. Gonzales R, Mooney L, Rawson RA. The methamphetamine problem in the United States. Annu Rev Public Health. (2010) 31:385–98. doi: 10.1146/annurev.publhealth.012809.103600
2. Chomchai C, Chomchai S. Global patterns of methamphetamine use. Curr Opin Psychiatr. (2015) 28:269–74. doi: 10.1097/YCO.0000000000000168
3. Ellis RJ, Childers ME, Cherner M, Lazzaretto D, Letendre S, Grant I, et al. Increased human immunodeficiency virus loads in active methamphetamine users are explained by reduced effectiveness of antiretroviral therapy. J Infect Dis. (2003) 188:1820–6. doi: 10.1086/379894
4. Liang H, Wang X, Chen H, Song L, Ye L, Wang SH, et al. Methamphetamine enhances HIV infection of macrophages. Am J Pathol. (2008) 172:1617–24. doi: 10.2353/ajpath.2008.070971
5. Potula R, Persidsky Y. Adding fuel to the fire: methamphetamine enhances HIV infection. Am J Pathol. (2008) 172:1467–70. doi: 10.2353/ajpath.2008.080130
6. Toussi SS, Joseph A, Zheng JH, Dutta M, Santambrogio L, Goldstein H. Short communication: methamphetamine treatment increases in vitro and in vivo HIV replication. AIDS Res Hum Retrovirus. (2009) 25:1117–21. doi: 10.1089/aid.2008.0282
7. Rajasingham R, Mimiaga MJ, White JM, Pinkston MM, Baden RP, Mitty JA. A systematic review of behavioral and treatment outcome studies among HIV-infected men who have sex with men who abuse crystal methamphetamine. AIDS Patient Care STDs. (2011) 26:36–52. doi: 10.1089/apc.2011.0153
8. Massanella M, Gianella S, Schrier R, Dan JM, Pérez-Santiago J, Oliveira MF, et al. Methamphetamine use in HIV-infected Individuals affects T-cell function and viral outcome during suppressive antiretroviral therapy. Sci Rep. (2015) 5:13179. doi: 10.1038/srep13179
9. Cachay ER, Moini N, Kosakovsky Pond SL, Pesano R, Lie YS, Aiem H, et al. Active methamphetamine use is associated with transmitted drug resis-tance to non-nucleoside reverse transcriptase inhibitors in individuals with HIV infection of unknown duration. Open AIDS J. (2007) 1:5–10. doi: 10.2174/1874613600701010005
10. Peerzada H, Gandhi JA, Guimaraes AJ, Nosanchuk JD, Martinez LR. Methamphetamine administration modifies leukocyte proliferation and cytokine production in murine tissues. Immunobiology. (2013) 218:1063–8. doi: 10.1016/j.imbio.2013.02.001
11. Mata MM, Napier TC, Graves SM, Mahmood F, Raeisi S, Baum LL. Methamphetamine decreases CD4 T cell frequency and alters pro-inflammatory cytokine production in a model of drug abuse. Eur J Pharmacol. (2015) 752:26–33. doi: 10.1016/j.ejphar.2015.02.002
12. Loftis JM, Choi D, Hoffman W, Huckans MS. Methamphetamine causes persistent immune dysregulation: a cross-species, translational report. Neurotox Res. (2011) 20:59–68. doi: 10.1007/s12640-010-9223-x
13. Mahajan SD, Hu Z, Reynolds JL, Aalinkeel R, Schwartz SA, Nair MP. Methamphetamine modulates gene expression patterns in monocyte derived mature dendritic cells. Mol Diagn Ther. (2006) 10:257–69. doi: 10.1007/BF03256465
14. Yamaguchi T, Kuraishi Y, Minami M, Nakai S, Hirai Y, Satoh M. Methamphetamine-induced expression of interleukin-1β mRNA in the rat hypothalamus. Neurosci Lett. (1991) 128:90–2. doi: 10.1016/0304-3940(91)90766-M
15. Mayer-Barber KD, Yan B. Clash of the cytokine titans: counter-regulation of interleukin-1 and type I interferon-mediated inflammatory responses. Cell Mol Immunol. (2017) 14:22–35. doi: 10.1038/cmi.2016.25
16. Guo H, Gao J, Taxman DJ, Ting JP, Su L. HIV-1 infection induces interleukin-1β production via TLR8 protein-dependent and NLRP3 inflammasome mechanisms in human monocytes. J Biol Chem. (2014) 289:21716–26. doi: 10.1074/jbc.M114.566620
17. Wie SH, Du P, Luong TQ, Rought SE, Beliakova-Bethell N, Lozach J, et al. HIV downregulates interferon-stimulated genes in primary macrophages. J. Interferon Cytokine Res. (2013) 33:90–5. doi: 10.1089/jir.2012.0052
18. Pillai RS. MicroRNA function: multiple mechanisms for a tiny RNA?. RNA. (2005) 11:1753–61. doi: 10.1261/rna.2248605
19. Hou J, Wang P, Lin L, Liu X, Ma F, An H, et al. MicroRNA-146a feedback inhibits RIG-I-dependent type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J Immunol. (2009) 183:2150–8. doi: 10.4049/jimmunol.0900707
20. Lin C, Li X, Zhang Y, Guo Y, Zhou J, Gao K, et al. The microRNA feedback regulation of p63 in cancer progression. Oncotarget. (2015) 6:8434–53. doi: 10.18632/oncotarget.3020
21. Siomi H, Siomi MC. Posttranscriptional regulation of MicroRNA biogenesis in animals. Mol Cell. (2010) 38:323–32. doi: 10.1016/j.molcel.2010.03.013
22. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. (2006) 6:857–66. doi: 10.1038/nrc1997
23. Ebert MS, Sharp PA. Roles for MicroRNAs in conferring robustness to biological processes. Cell. (2012) 149:515–24. doi: 10.1016/j.cell.2012.04.005
24. Rzepiela AJ, Ghosh S, Breda J, Vina-Vilaseca A, Syed AP, Gruber AJ, et al. Single-cell mRNA profiling reveals the hierarchical response of miRNA targets to miRNA induction. Mol Syst Biol. (2018) 14:e8266. doi: 10.15252/msb.20188266
25. Gantier MP, Sadler AJ, Williams BR. Fine-tuning of the innate immune response by microRNAs. Immunol Cell Biol. (2007) 85:458–62. doi: 10.1038/sj.icb.7100091
26. Labbaye C, Testa U. The emerging role of MIR-146A in the control of hematopoiesis, immune function and cancer. J Hematol Oncol. (2012) 5:13. doi: 10.1186/1756-8722-5-13
27. Wu S, He L, Li Y, Wang T, Feng L, Jiang L, et al. miR-146a facilitates replication of dengue virus by dampening interferon induction by targeting TRAF6. J Infect. (2013) 67:329–41. doi: 10.1016/j.jinf.2013.05.003
28. Li L, Chen X-P, Li Y-J. MicroRNA-146a and human disease. Scand J Immunol. (2010) 71:227–31. doi: 10.1111/j.1365-3083.2010.02383.x
29. Kawai T, Akira S. Signaling to NF-κB by Toll-like receptors. Trends Mol Med. (2007) 13:460–9. doi: 10.1016/j.molmed.2007.09.002
30. Liu L, Roberts AA, Ganz T. By IL-1 signaling, monocyte-derived cells dramatically enhance the epidermal antimicrobial response to lipopolysaccharide. J Immunol. (2003) 170:575–80. doi: 10.4049/jimmunol.170.1.575
31. Nahid MA, Satoh M, Chan EK. Interleukin 1β-responsive MicroRNA-146a is critical for the cytokine-induced tolerance and cross-tolerance to toll-like receptor ligands. J Innate Immun. (2015) 7:428–40. doi: 10.1159/000371517
32. Li Z, Zhang S, Wan Y, Cai M, Wang W, Zhu Y, et al. MicroRNA-146a overexpression impairs the positive selection during T cell development. Front Immunol. (2018) 8:2006. doi: 10.3389/fimmu.2017.02006
33. Reagent Datasheet Detail: Catalog 510 - HIV-1 Ba-L Virus - NIH AIDS Reagent Program. Available online at: https://aidsreagent.org/reagentdetail.cfm?t=viruses&id=728
34. Baxter AE, Russell RA, Duncan CJ, Moore MD, Willberg CB, Pablos JL, et al. Macrophage infection via selective capture of HIV-1-infected CD4+ T cells. Cell Host Microbe. (2014) 16:711–21. doi: 10.1016/j.chom.2014.10.010
35. Potula R, Hawkins BJ, Cenna JM, Fan S, Dykstra H, Ramirez SH, et al. Methamphetamine causes mitrochondrial oxidative damage in human T lymphocytes leading to functional impairment. J Immunol. (2010) 185:2867–76. doi: 10.4049/jimmunol.0903691
36. Prasad A, Kuzontkoski PM, Shrivastava A, Zhu W, Li DY, Groopman JE. Slit2N/Robo1 inhibit HIV-gp120-induced migration and podosome formation in immature dendritic cells by sequestering LSP1 and WASp. PLoS ONE. (2012) 7:e48854. doi: 10.1371/journal.pone.0048854
37. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. (2012) 9:671–5. doi: 10.1038/nmeth.2089
38. Kramer MF. Stem-loop RT-qPCR for miRNAs. Curr Protoc Mol Biol. (2011) 15:15.10. doi: 10.1002/0471142727.mb1510s95
39. Turowski P, Kenny BA. The blood-brain barrier and methamphetamine: open sesame? [Internet]. Front Neurosci. (2015) 9:156. doi: 10.3389/fnins.2015.00156
40. Lopez-Castejon G, Brough D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. (2011) 22:189–95. doi: 10.1016/j.cytogfr.2011.10.001
41. He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. (2016) 41:1012–21. doi: 10.1016/j.tibs.2016.09.002
42. Du L, Shen K, Bai Y, Chao J, Hu G, Zhang Y, et al. Involvement of NLRP3 inflammasome in methamphetamine-induced microglial activation through miR-143/PUMA axis. Toxicol Lett. (2019) 301:53–63. doi: 10.1016/j.toxlet.2018.10.020
43. Hiscott J, Marois J, Garoufalis J, D'Addario M, Roulston A, Kwan I, et al. Characterization of a functional NF-kappa B site in the human interleukin 1 beta promoter: evidence for a positive autoregulatory loop. Mol Cell Biol. (1993) 13:6231–40. doi: 10.1128/MCB.13.10.6231
44. Wires ES, Alvarez D, Dobrowolski C, Wang Y, Morales M, Karn J, et al. Methamphetamine activates nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and induces human immunodeficiency virus (HIV) transcription in human microglial cells. J Neurovirol. (2012)18:400–10. doi: 10.1007/s13365-012-0103-4
45. Pulugulla SH, Packard TA, Galloway NLK, Grimmett ZW, Doitsh G, Adamik J, et al. Distinct mechanisms regulate IL1B gene transcription in lymphoid CD4 T cells and monocytes. Cytokine. (2018)111:373–81. doi: 10.1016/j.cyto.2018.10.001
46. Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nat Lond. (2014) 505:509–14. doi: 10.1038/nature12940
47. Sirois M, Robitaille L, Allary R, Shah M, Woelk CH, Estaquier J, et al. TRAF6 and IRF7 control HIV replication in macrophages. PLoS ONE. (2011) 6:e28125. doi: 10.1371/journal.pone.0028125
48. Gottipati S, Rao NL, Fung-Leung WP. IRAK1: a critical signaling mediator of innate immunity. Cell Signal. (2008) 20:269–76. doi: 10.1016/j.cellsig.2007.08.009
49. Djuranovic S, Nahvi A, Green R. miRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science. (2012) 336:237–40. doi: 10.1126/science.1215691
50. Taganov KD, Boldin MP, Chang K-J, Baltimore D. NF- B-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA. (2006) 103:12481–6. doi: 10.1073/pnas.0605298103
51. Jiang W, Kong L, Ni Q, Lu Y, Ding W, Liu G, et al. miR-146a ameliorates liver ischemia/reperfusion injury by suppressing IRAK1 and TRAF6. PLoS ONE. (2014) 9:e101530. doi: 10.1371/journal.pone.0101530
52. Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Förster I, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. (2011) 34:213–23. doi: 10.1016/j.immuni.2011.02.006
53. Yu X, Du Y, Cai C, Cai B, Zhu M, Xing C, et al. Inflammasome activation negatively regulates MyD88-IRF7 type I IFN signaling and anti-malaria immunity. Nat Commun. (2018) 9:4964. doi: 10.1038/s41467-018-07384-7
54. Prasad A, Kulkarni R, Shrivastava A, Jiang S, Lawson K, Groopman JE. Methamphetamine functions as a novel CD4+ T-cell activator via the sigma-1 receptor to enhance HIV-1 infection. Sci Rep. (2019) 9:958. doi: 10.1038/s41598-018-35757-x
55. Dhamija N, Choudhary D, Ladha JS, Pillai B, Mitra D. Tat predominantly associates with host promoter elements in HIV-1-infected T-cells – regulatory basis of transcriptional repression of c-Rel. FEBS J. (2015) 282:595–610. doi: 10.1111/febs.13168
56. Soper A, Kimura I, Nagaoka S, Konno Y, Yamamoto K, Koyanagi Y, et al. Type I interferon responses by HIV-1 infection: association with disease progression and control. Front Immunol. (2018) 8:1823 doi: 10.3389/fimmu.2017.01823
57. Nair MP, Saiyed ZM. Effect of methamphetamine on expression of HIV coreceptors and CC-chemokines by dendritic cells. Life Sci. (2011) 88:987–94. doi: 10.1016/j.lfs.2010.09.019
58. Lodish HF, Zhou B, Liu G, Chen CZ. Micromanagement of the immune system by microRNAs. Nat Rev Immunol. (2008) 8:120–30. doi: 10.1038/nri2252
59. Quaranta MT, Olivetta E, Sanchez M, Spinello I, Paolillo R, Arenaccio C, et al. miR-146a controls CXCR4 expression in a pathway that involves PLZF and can be used to inhibit HIV-1 infection of CD4+ T lymphocytes. Virology. (2015) 478:27–38. doi: 10.1016/j.virol.2015.01.016
60. King CG, Kobayashi T, Cejas PJ, Kim T, Yoon K, Kim GK, et al. TRAF6 is a T cell–intrinsic negative regulator required for the maintenance of immune homeostasis. Nat Med. (2006) 12:1088–92. doi: 10.1038/nm1449
61. Rom S, Rom I, Passiatore G, Pacifici M, Radhakrishnan S, Del Valle L, et al. CCL8/MCP-2 is a target for mir-146a in HIV-1-infected human microglial cells. FASEB J. (2010) 24:2292–300. doi: 10.1096/fj.09-143503
62. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. (2009) 7:99–109. doi: 10.1038/nrmicro2070
63. Cai R, Liu L, Luo B, Wang J, Shen J, Shen Y, et al. Caspase-1 activity in CD4 T cells is downregulated following antiretroviral therapy for HIV-1 infection. AIDS Res Hum Retrovirus. (2016) 33:164–71. doi: 10.1089/aid.2016.0234
Keywords: HIV, methamphetamine, inflammation, innate immunity, miRNA
Citation: Lawson KS, Prasad A and Groopman JE (2020) Methamphetamine Enhances HIV-1 Replication in CD4+ T-Cells via a Novel IL-1β Auto-Regulatory Loop. Front. Immunol. 11:136. doi: 10.3389/fimmu.2020.00136
Received: 16 October 2019; Accepted: 20 January 2020;
Published: 07 February 2020.
Edited by:
Shokrollah Elahi, University of Alberta, CanadaReviewed by:
Gurudutt Pendyala, University of Nebraska Medical Center, United StatesLaura Fantuzzi, Istituto Superiore di Sanità (ISS), Italy
Copyright © 2020 Lawson, Prasad and Groopman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jerome E. Groopman, amdyb29wbWFAYmlkbWMuaGFydmFyZC5lZHU=