 Rita G. Domingues
Rita G. Domingues Matthew R. Hepworth
Matthew R. Hepworth
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 06 February 2020
Sec. NK and Innate Lymphoid Cell Biology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.00116
This article is part of the Research Topic Circuits of Resident Immunity Regulating Tissue Adaptation and Organ Homeostasis View all 12 articles
Recent years have seen a revolution in our understanding of how cells of the immune system are modulated and regulated not only via complex interactions with other immune cells, but also through a range of potent inputs derived from diverse and varied biological systems. Within complex tissue environments, such as the gastrointestinal tract and lung, these systems act to orchestrate and temporally align immune responses, regulate cellular function, and ensure tissue homeostasis and protective immunity. Group 3 Innate Lymphoid Cells (ILC3s) are key sentinels of barrier tissue homeostasis and critical regulators of host-commensal mutualism—and respond rapidly to damage, inflammation and infection to restore tissue health. Recent findings place ILC3s as strategic integrators of environmental signals. As a consequence, ILC3s are ideally positioned to detect perturbations in cues derived from the environment—such as the diet and microbiota—as well as signals produced by the host nervous, endocrine and circadian systems. Together these cues act in concert to induce ILC3 effector function, and form critical sensory circuits that continually function to reinforce tissue homeostasis. In this review we will take a holistic, organismal view of ILC3 biology and explore the tissue sensory circuits that regulate ILC3 function and align ILC3 responses with changes within the intestinal environment.
Innate lymphoid Cells (ILCs) are a family of innate immune effectors that localize mainly to mucosal surfaces and which play critical roles in regulating tissue immunity and homeostasis. The ILC family can be divided into three main subsets—group 1 ILC (ILC1), ILC2, and ILC3 based on their expression of master transcription factors and associated effector cytokine profiles [Reviewed extensively elsewhere (1–8)]. In this review we will focus on group 3 ILC (ILC3), a group of ILC that act constitutively to maintain intestinal health through regulation of the intestinal barrier and commensal microbiota, and through protective immune responses against extracellular microbial pathogens.
ILC3s are characterized by the expression of the retinoid-related orphan receptor γt (RORγt) (1, 5, 6) and they can be further sub-divided into at least two sub-groups in adults (9). These subsets are developmentally, transcriptionally and functionally heterogeneous and include lymphoid tissue inducer cells (LTi)-like ILC3s; characterized by surface expression of CCR6, c-kit (CD117), Neuropilin-1, and variable expression of CD4, in addition to natural cytotoxicity receptor expressing (NCR)+ ILC3s—which lack LTi-associated markers but express a range of NCR (e.g., NKp46 in mice) while further co-expressing the transcription factor T-bet (10, 11). The characteristics and differences between ILC3 subsets have been discussed in detail elsewhere (9) and as such, for the sake of clarity, we will largely refer to ILC3 cumulatively in this review without distinguishing the specific subset.
As discussed in detail below, ILC3s are at the center of multiple tissue regulatory circuits in which a variety of inputs (in the form of environmental and host-derived cues) are sensed and interpreted by ILC3 and give rise to functional outputs that culminate in the downstream modulation of tissue physiology to maintain health and homeostasis. While the inputs of these sensory circuits vary, and will be discussed in detail below, a major common ILC3-associated output is the secretion of effector cytokines including IL-22, IL-17A, IL-17F, and GM-CSF and lymphotoxin (LT) (1, 4, 7, 8) (Figure 1). These soluble mediators in turn act upon both neighboring tissue-resident immune cells and non-hematopoietic cells—such as epithelia and stroma. In this review, we will comprehensively discuss the major tissue circuits through which ILC3 function is regulated, and through which ILC3 propagate these signals to regulate and orchestrate the wider immune response and to promote optimal tissue function, mediate protective immune responses and maintain health.
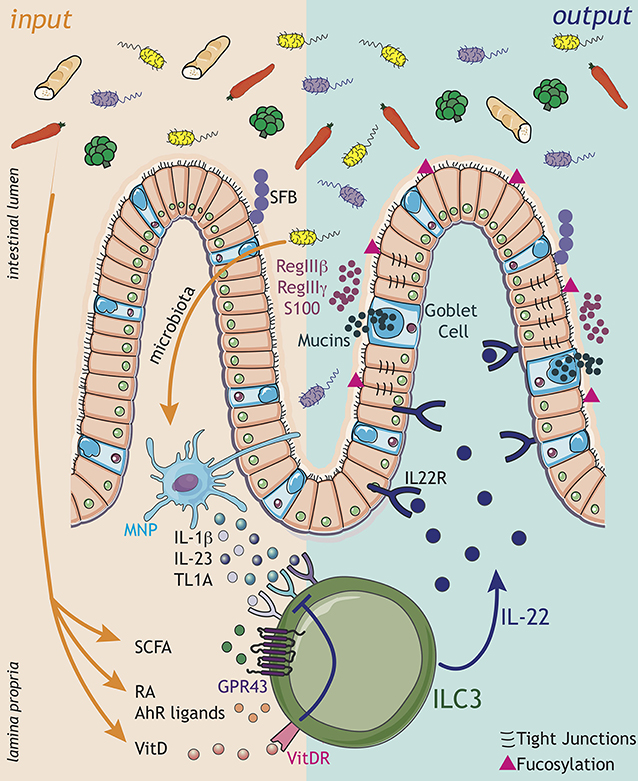
Figure 1. ILC3 engage in complex sensory circuits in order to integrate microbial and dietary cues and enforce mucosal homeostasis. Inputs (orange arrows): ILC3s act as innate immune sentinels of the gastrointestinal tract, and respond rapidly to changes in the tissue environment. Environmental signals, comprising microbial and dietary cues, are sensed either via myeloid cell intermediaries [e.g., dendritic cells (DC), macrophages, also known as mononuclear phagocytes (MNP)], which release cytokine cues (IL-1β, IL-23, TL1A) to modulate ILC3 function, or through direct sensing of metabolites and dietary ligands. Microbial metabolites, such as short chain fatty acids (SCFA), signal directly to modulate ILC3 function though the receptor GPR43. Additionally, ILC3 integrate dietary cues in the form of the vitamin A metabolite retinoic acid (RA) and AhR ligands, which together promote ILC3 development and effector cytokine responses. In contrast, vitamin D acts as a negative regulator of ILC3 activation by suppressing the ability of ILC3 to sense myeloid cues—such as IL-23. Within the complex tissue microenvironment ILC3 are likely exposed to multiple signals in parallel, which must be appropriately integrated to maintain intestinal homeostasis. Outputs (dark blue arrows): Signals translated by ILC3 are propagated in the form of ILC3-derived outputs—most notably cytokine signals, which are received by other immune and non-immune cells within the local environment. In particular, ILC3-derived IL-22 acts on epithelial cells to enforce intestinal barrier integrity and induce the production of antimicrobial peptides (AMPs) such as RegIIIβ, RegIIIγ, and S100 family proteins, secretion of mucins by goblet cells, modulation of tight junctions and epithelial cell fucosylation. IL-22-dependent pathways further regulate the growth of specific commensal bacteria species that are intimately associated with the host, such as segmented filamentous bacteria (SFB). Together, the balance of signals perceived by ILC3 determine the strength of the effector response, regulate the balance of the commensal microbiota and ensure their spatial segregation from the underlying intestinal tissue. In the context of disease, dysregulation of these signals may dramatically alter ILC3 responses and result in a loss of barrier function and translocation of the bacteria from the lumen, thus precipating or exacerbating inflammatory disease.
Mammals have evolved multiple complimentary immunological mechanisms to promote the anatomical containment of commensal bacteria. These mechanisms enforce tolerance, suppress inflammation and maximize mutualism with the microbiota, and ILC3s have key roles in this process (12–15). ILC3s are enriched within gastrointestinal (GI) tract where they are ideally positioned to promote barrier repair and to prevent bacterial translocation (15). ILC3 produce a range of soluble mediators that enable them to continually reinforce the barrier and maintain the containment and physical segregation of commensal microorganisms. Chief amongst these mediators is the cytokine interleukin (IL)-22, which binds to the heterodimeric receptor IL22RA1-IL10RB (IL-22R) expressed by cells of the non-haematopoietic lineage, most notably intestinal epithelial cells (Figure 1: outputs). IL-22 signaling induces the production of antibacterial peptides such as RegIIIβ and RegIIIγ and S100 family members, which in turn regulate the commensal microbiota and limit access to the epithelial and mucosal niche (16, 17). IL-22 also promotes the physical exclusion of commensal bacteria through induction of mucins and goblet cell hyperplasia, and by regulating the expression of tight-junction components (15, 17, 18). Moreover, ILC3s induce fucosylation of intestinal epithelial cells through an IL-22 and LTα driven process, which in turn favors colonization by mutualistic bacterial species at the expense of potential pathogens (Figure 1: outputs) (19–21). In addition, IL-22 produced by ILC3s acts to regulate epithelial turnover and intestinal crypt stem cell maintenance, and has been ascribed both pro- and anti-tumorogenic functions, most recently being shown to promote DNA damage response (DDR) mechanisms in order to prevent tumor formation (22–25). IL-22 also modulates nutrient uptake via the intestinal epithelia, in particular lipid uptake (26). In line with this central role for ILC3 and IL-22 in maintaining intestinal barrier function and tissue homeostasis, loss of IL-22 production by ILC3s in mice results in dysbiosis, barrier disruption and an increased susceptibility to experimental induced colitis (27, 28). Moreover, depletion of intestinal ILC3 leads to peripheral dissemination of intestinal bacteria and systemic inflammation that can be rescued by providing exogenous IL-22 (15). Thus, a central function, and key output, of ILC3-mediated effector responses is the orchestration of host-microbiota interactions (Figure 1: outputs).
Intestinal homeostasis and host-commensal interactions are also modulated by the type 3 cytokines IL-17A and IL-17F, both of which are also produced by ILC3 (1, 4, 7, 8). Similar to IL-22, IL-17A/F promote tissue integrity by enhancing the synthesis of tight junctions and antimicrobial peptides, including β-defensins, REG proteins, S100 proteins, lipocalins and lactoferrins (29). Additionally IL-17A/F act in part to attract myeloid cells to the tissue site, through the induction of chemokines and growth factor expression by epithelial cells (30, 31). While ILC3 have been reported to be a potent source of IL-17A/F in early life, expression of these cytokines appears to be somewhat limited at steady state in adult tissues (28, 32). In contrast, during infection and inflammation ILC3 produce IL-17 in response to myeloid-derived cues including IL-23 and IL-1β (33, 34), and ILC3-derived IL-17 has been attributed critical roles in immunity to fungal and bacterial pathogens (34–37). In particular, IL-17 production by ILC3s has been implicated in immunity against fungal pathogens, specifically in response to Candida albicans (34). Interestingly, HIV patients commonly manifest oropharyngeal candidiasis, and loss of IL-17 production by ILC3s was observed in tonsils and buccal mucosa during SIV infection in macaques (38, 39).
While homeostatic IL-17 production has been attributed protective functions in intestinal health and host-commensal microbe interactions, elevated IL-17A/F production has also been associated with the pathogenesis of inflammatory bowel disease (IBD). Indeed, ILC3-derived IL-17A and IL-17F are increased during intestinal inflammation in both mice and humans (40, 41). Together, IL-17A/F production by intestinal ILC3—in addition to Th17 and γδ T cell populations—has highly contextual roles in intestinal health, immunity and inflammation.
Conversely, the microbiota itself is also increasingly appreciated to act reciprocally to modulate ILC3 function (Figure 1: inputs). Indeed, early studies suggested microbial colonization of the neonatal intestine regulates the composition and size of the ILC3 pool within the intestinal tract. Pups born to germ free mothers were reported to have reduced frequencies of ILC3s—indicating a role of microbial signals in promoting tissue seeding by ILC3 subsets (28). However, in contrast to these findings IL-22 producing ILC3 numbers were found to be suppressed in a microbiota dependent manner through epithelial expression of IL-25 (32). Despite these discrepancies, the dialogue between the microbiota and ILC3s within the intestine has emerged as a critical circuit of intestinal immunity and tissue homeostasis.
Recent studies have begun to shed light on the microbial-derived metabolites that mediate this immune regulatory on ILC3. For example, ILC3s have the capacity to sense and respond to short chain fatty acids (SCFA)—including butyrate, acetate and propionate—critical regulators of immune responses which are metabolized from dietary fiber by commensal microbes (Figure 1: inputs) (42, 43). Levels of butyrate differ along the intestinal tract, in line with differing densities of commensal microbes, and were previously correlated with reduced ILC3 cell number and cytokine production in distal regions of the small intestine (44). SCFA can signal via multiple G-coupled protein receptors, as well as via histone deacetylase enzymes (HDAC) (43), and despite these advances the mechanisms through which SCFA regulate ILC3 are still being delineated. The SCFA receptor GPR109a was implicated in the microbiota-associated regulation of ILC3 cytokine production via the modulation of dendritic cell (DC)-derived IL-23 in the colon, although these studies largely utilized a GPR109a agonist—leaving the precise contribution of endogenous SCFA unclear (45). Interestingly, a recent study highlighted ILC3-intrinsic expression of the SCFA receptor Gpr43 (Ffar2) in the modulation of intestinal ILC3 responses (Figure 1: inputs) (46). Triggering of GPR43 with the SCFAs propionate and acetate (but not butyrate) selectively promoted colonic ILC3 proliferation and expansion and production of IL-22, subsequently protecting mice from chemically induced colitis and from enteric bacterial infection (46).
Cues derived from mutualistic microbiota establish a critical dialogue between the host and it's environment and regulate the intestinal immune system—including ILC3. In addition to the microbiota, the intestine is also continually exposed to metabolites and phytochemicals derived from the diet (Figure 1: inputs). As highlighted above, the availability and liberation of many dietary metabolites is also determined in part by mutualistic, commensal microbes within the intestine—while conversely the diet itself can modulate microbial composition and thus, determine the nature of host-commensal interactions. For example, the feeding of high fat diet (HFD) to pregnant mice was found to modify the expansion of ILC3 in the intestines of progeny through the modification of the mothers microbiota (47).
Similarly, Aryl hydrocarbon receptor (AhR) ligands are normally liberated from cruciferous vegetables in the diet—such as broccoli and cabbage, but they can also be microbially derived (48). AhR is a dietary-sensing nuclear receptor that is expressed by ILC3s and has critical roles in the development, transcription and function of these cells (Figure 1: inputs). Indeed, ILC3 are highly AhR dependent, and present severe functional impairments in the absence of cell-intrinsic AhR expression (14, 49–51). As a result of ILC3 defects, AhR-deficient mice fail to form tissue-associated lymphoid structures, such as cryptopatches (CP) and are unable to control infections with the extracellular pathogen Citrobacter rodentium (49, 52). Intriguingly, the development and seeding of intestinal ILC3 in neonates was demonstrated to be dependent upon the mothers microbiota and the transfer of antibody-bound AhR ligands through the mothers milk (48), suggesting maternal transfer of dietary ligands to neonates may play critical roles in the development of the immune system, microbial colonization and protection from infections in early life.
Indeed, maternal transfer of dietary ligands is increasingly appreciated to be a determinant of neonatal immunity and ILC3 development. In utero exposure to the Vitamin A metabolite retinoic acid (RA) impacts directly on secondary lymphoid organ development with long-term immunological consequences (53). Mice genetically modified to have hematopoietic cell-intrinsic deficiency in RA lacked PP or exhibited impairment in LN formation and maturation as a result of defective ILC3 differentiation (Figure 1: inputs). Moreover, it was shown that RA directly regulates the master transcription factor of ILC3, RORγt, and in the absence of maternal retinoids ILC3 failed to develop correctly (53). In addition to maternally derived RA signals, deprivation of vitamin A in adulthood also results in the collapse of the intestinal ILC3 populations and, as a consequence, results in susceptibility to Citrobacter rodentium infection (54, 55). In addition to direct effects of RA on ILC3 development, RA produced by DCs was also found to regulate the homing properties of ILC3s by imprinting expression of the intestinal homing markers CCR9 and α4β7 (56).
The importance of dietary vitamins in ILC3 effector circuits is further supported by evidence that vitamin D also plays a role in intestinal ILC3 homeostasis (Figure 1: inputs). ILC3 numbers in the small intestine of mice deficient for the vitamin D receptor (VDR—KO mice) were shown to be increased, as was IL-22 expression, resulting in enhanced resistance to infection with Citrobacter rodentium (57). Consistently, human ILC3s stimulated with IL-23 and IL-1β upregulate the VDR, and VDR signaling subsequently acts to downregulate the IL-23 signaling pathway—suggesting vitamin D acts as a negative regulator and suppressive feedback loop to control ILC3 activation (Figure 1: inputs) (58). Vitamin D availability has also been implicated in the pathogenesis of IBD, as patients are reported to have lower plasma levels of vitamin D than healthy subjects, and exhibit an upregulation of the IL-23 signaling pathway which could potentially explain exacerbated ILC3 responses that are associated with intestinal inflammation in IBD (58). In contrast to these studies, mice lacking Cyp27B1—an enzyme required for the conversion of vitamin D to it's chemically active form—exhibit reduced colonic ILC3 numbers and IL-22 production suggesting a more nuanced role for vitamin D in the regulation of ILC3 function (59). Together these findings highlight the importance of dietary cues in regulating ILC3 function and intestinal homeostasis. An increased understanding of the complex dialogue between diet, microbiota and host is likely to reveal novel immune regulatory circuits and clarify how environmental cues act as risk factors, and contribute to the onset of metabolic and inflammatory disorders.
While ILC3 are potently regulated by the microbiota and diet within the intestinal environment, it remains unclear the extent to which they are able to directly sense these cues, beyond the pathways detailed above. Indeed, the majority of evidence suggests third party sensory cells of the myeloid lineage are required to directly sense, translate and communicate environmental information to ILC3. Classically, tissue-resident mononuclear phagocytes (MNPs) act as key intermediaries and signal to ILC3 via the release of cytokine mediators during both homeostatic and protective immune responses (60, 61). Indeed, intestinal myeloid populations are well-equipped to directly sense microbial metabolites, pathogen associated molecular patterns (PAMPs) and danger signals and to transfer this information to ILC3 (Figure 1: inputs). In particular, CX3CR1+ intestinal MNPs cluster with ILC3 in distinct, organized lymphoid structures, such as CPs (62, 63). Microbiota sensing by CX3CR1+ MNPs was shown to result in local production of IL-1β and IL-23, which are key activating cytokines of ILC3 and which potently induce IL-22 secretion (Figure 1: inputs) (63). Depletion of CX3CR1+ MNPs resulted in impaired IL-22 production by ILC3 and failure to control Citrobacter rodentium infection (62, 64, 65). In addition to the provision of the activating signals IL-23 and IL-1β, CX3CR1+ MNP-derived TL1A further acts to augment IL-22 production from ILC3 (Figure 1: inputs) (62).
While microbial sensing by intestinal MNP and conserved crosstalk with ILC3 appear to be a major sensory circuit of intestinal immunity, emerging evidence suggests diverse sensory mechanisms across multiple biological systems provide additional inputs to regulate ILC3 function. In particular, the central and enteric nervous systems are rapidly being appreciated as critical sensory and immunoregulatory systems.
It has been suggested that the immune and nervous systems are evolutionary linked, since they share functional similarities (66, 67). Both nervous and immune systems rely on similar processes to for cellular communication; such as cell-cell contact and synapse formation, release of soluble mediators and sensing of circulating metabolites. Recent evidence suggests immune and neuronal cells are positioned in close proximity, and form conserved interactions that have been termed “neuro-immune cell units” (NICUs) (67). NICUS can form through interactions with both the central and peripheral nervous system and are increasingly being described in peripheral tissues such as the gastrointestinal tract and lung.
Neuroimmune interactions are evident very early in life—and during the embryonic period the development of the enteric nervous system (ENS) and SLO organogenesis share many parallels. Notably, the neurotrophic factor receptor RET is essential for the development of Peyer's patches (PP) and also the ENS (68, 69). Moreover, RET expression by CD11c+ cells present in the anlagen initiates a cascade of immune cell recruitment, in particular of fetal ILC3s, through sensing of neurotrophic factors that drive the formation of primordial lymphoid clusters (68, 69). Moreover, increasing evidence suggests ILC3 can directly sense these neuronal derived inputs and respond during both development as well as in the adult intestine (Figure 2: inputs). As mentioned previously, fetal and adult ILC3 development and function relies on RA signaling (53). Intriguingly, neurons have been suggested to be a physiological source of RA (70), surprisingly suggesting RA may be derived not only from the diet but also from the host nervous system.
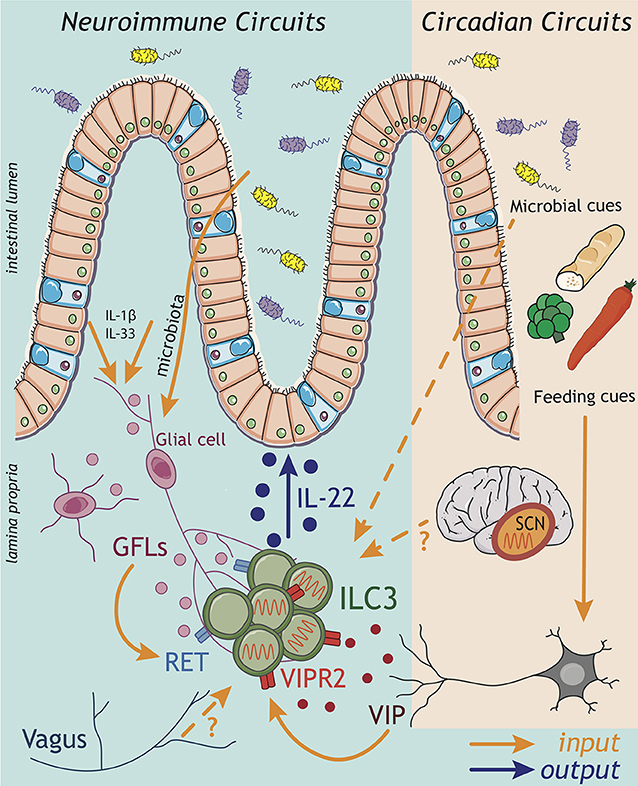
Figure 2. ILC3 neuroimmune and circadian circuits. Emerging findings implicate inputs from the nervous system in the regulation of ILC3 circuits within the gastrointestinal tract. Both the central (CNS) and enteric (ENS) nervous systems have the capacity to sense perturbations within the intestinal environment and relay this information via the release of neuropeptides to influence the ILC3 response. Strikingly, enteric glial cells are able to directly sense microbial patterns and alarmins released within the tissue, and respond by producing glial-derived neurotrophic factors (GDNF family of ligands; GFL) that directly activate the production of IL-22 by ILC3 through the tyrosine kinase RET. Indeed, recent evidence suggests a broader spectrum of neuropeptides may act to regulate ILC3 function including vasointestinal peptide (VIP) produced by enteric neurons in response to feeding cues. Signals transmitted by the nervous system also play critical roles in aligning ILC3 effector function with periods of activity and high risks of environmental exposure and pathogen encounter over the course of a 24 h day. In this regard, circadian rhythms entrained by light—and sensed via the suprachiasmatic nucleus (SCN) of the brain—trigger a cascade of molecular transcriptional-translational feedback loops of clock genes, which orchestrate rhythms in the ILC3 response. While the “central clock” within the CNS appears to be a central entrainer of ILC3 oscillatory function in the gut, the mechanisms through which the CNS transmits this information to regulate ILC3 function peripherally remain unknown. Nonetheless, inputs from the CNS have previously been shown to be relayed to ILC3 via the vagus nerve. Together cues from both the CNS and ENS have the potential to entrain intestinal ILC3 function, while circadian rhythms in ILC3 may be imprinted through a combination of central clock-mediated light entrainment, feeding-associated neuronal feedback and environmental cues from the microbiota.
The ENS is increasingly appreciated to regulate tissue-resident immune functions (71), include those of ILC3. One pioneer study demonstrated that a glial-ILC3-epithelial axis is required to regulate enteric defense against bacterial infection (72). Like myeloid cells, intestinal glial cells also have the capacity to sense microbial cues and alarmins in a Myd88-dependent manner; thus, implicating the enteric nervous system as a key player in environmental sensing circuits. In response to these cues glial cells secrete neurotrophic factors, which directly act on adult ILC3 cytokine production via cell-intrinsic RET expression (Figure 2: inputs). Ablation of Ret in ILC3s led to a reduction in IL-22, consequently impairing epithelial function and host defense to enteric bacterial infection (72). In addition to ENS cues, CNS–derived signals propagated by the vagus nerve—via release of acetylcholine—have also been implicated in the regulation of ILC3 responses to bacterial infections in the peritoneal cavity (73). Vagal disruption was shown to lead to dysregulated ILC3 cell numbers in the peritoneal lavage (73). Mechanistically, acetylcholine acted to promote the release of pro-resolving lipid mediators—generated via ILC3-intrinsic expression of the PCTR biosynthetic pathway—which subsequently promoted protective immunity during E. coli-driven sepsis (73). Together, these studies illustrate the importance of neuronal inputs in regulating ILC3 outputs during infection (Figure 2).
Recent studies suggest that the number of neuropeptides with immunoregulatory activity may be broader than previously appreciated. Vasoactive intestinal peptide (VIP) release by enteric neurons was also shown to regulate ILC3-derived IL-22 production through signaling via VIPR2, triggered in part by feeding and dietary cues (Figure 2: inputs) (74, 75) (discussed in detail below). However, despite the strategic location of ILC3s within the CPs, which are enveloped by glial cell nervous fiber bundles and neuronal projections, the full extent of neuroimmune interactions that regulate ILC3 function are still to be determined. Indeed, recent years have seen an explosion in our understanding of neuroimmune signals that regulate other immune cells, including other members of the ILC family—most notably ILC2s (76–82). These studies have opened up new avenues of research and expanded our understanding of crosstalk between diverse biological systems, thus provoking the need for further studies to fully elucidate neuroimmune sensory circuits in the regulation of ILC3 responses, intestinal immunity and host-microbiota interactions.
In addition to local environmental cues, mammals are also constantly exposed to a range of external stimuli and pressures such as fluctuations in temperature, oxygen levels and the daily light cycle. As a result many organisms have evolved circadian rhythms to align core biological processes with time of day, which are imprinted by an internal biological clock. Specifically, circadian rhythms are driven by cell-autonomous transcriptional feedback loops (“clocks”), which enable organisms to anticipate and adapt to temporal changes in their environment (e.g., changing seasons, jet lag, shift work) and regulate metabolically demanding biological processes including body temperature, locomotor activity, endocrine responses, and feeding behavior—while on the cellular level circadian clocks regulate cellular metabolism and cell cycle (83, 84). In line with this, it is increasingly appreciated that circadian rhythms also regulate immune cell responses (85), and immune cells exhibit circadian oscillations in leukocyte trafficking, priming, effector function and host-pathogen interactions (85).
In mammals, circadian rhythms are controlled by the central circadian pacemaker or master clock—located in the suprachiasmatic nucleus (SCN) of the brain (86). The SCN acts to interpret and propagate light cues received via the optical nerve and subsequently, cell autonomous circadian rhythms are imprinted by systemic signals that act to align oscillations in a tripartite system of transcriptional-translational feedback-loops (85, 87, 88). The induction of the loop starts with the transcriptional activators CLOCK and BMAL1 promoting the expression of the repressors Period (Per) and Cryptochrome (Cry), which in time translocate back into the nucleus and inhibit their own expression (85, 87, 88). The second loop is composed by nuclear receptors RAR-related orphan receptors (RORs) (α, β, γ) and REV-ERBs (α, β), which exert opposing effects on the clock through transcription factor binding to the promoter of Arntl (encoding BMAL1) (85, 87, 88). Finally, the third loop consists of transcriptional activator albumin D-box binding protein (DBP) and the repressor nuclear factor for interleukin 3 (NFIL3), which act synergistically to regulate the expression of D-box genes including that of Per (85, 87, 88). Upon establishment of the transcriptional loops, the SCN keeps peripheral clocks in synchrony via neuronal sympathetic/parasympathetic transmission and through the hypothalamus pituitary adrenal (HPA) axis, including the release of catecholamines (epinephrine and norepinephrine) and glucocorticoids (84). Remarkably, similar circadian molecular mechanisms are found in the periphery. However, while the SCN network allows for the generation of sustained oscillations and time-of-day alignments, perturbations from environmental inputs such as temperature changes, the microbiota and feeding cues can also impact on peripheral, cell-intrinsic clocks (84).
Many constitutive innate immune processes, including the maintenance of intestinal barrier function via steady state IL-22 release from ILC3, come with significant metabolic costs for the host. Thus, circadian rhythms are thought to have evolved to align these processes with anticipated challenges and times of highest risk—most notably during waking activity and feeding where exposure to microbes, dietary antigens and potential pathogens is highest. Intriguingly, several components of the transcriptional circadian clock machinery including NFIL3 and RORγ/α are also key transcriptional regulators of ILC3 development and function, suggesting the possibility that these cells may also be regulated in a circadian manner (89–92). Moreover, ILC3 and IL-22 are critical regulators of the intestinal microbiota, with oscillations also reported amongst levels of commensal microbes in the intestinal tract (93, 94).
In line with this, several recent studies have demonstrated circadian control and oscillatory ILC3 responses, which are regulated by the master clock gene Arntl (Bmal1) in a cell-autonomous fashion (95, 96). Deletion of Arntl in ILC3s resulted in an altered epigenetic landscape, dysregulated cell numbers and IL-22 expression, and subsequently contributed to alterations in steady-state oscillations in the microbiome itself (95–97). Moreover, disrupted ILC3 responses resulted in altered epithelial responses and disrupted lipid uptake within the intestine (74, 95, 96). Of note, while deletion of Arntl led to a broad impairment of total ILC3 numbers (95), deletion of the related clock gene Nr1d1 (also known as Rev-erbα) resulted in altered ILC3 subset development—with mice exhibiting a marked reduction NCR+ ILC3s, while LTi-like ILC3 were unperturbed (97). Moreover, lack of Nr1d1 increased expression of Il17 in ILC3s, a mechanism previously reported in in Th17 cells (97, 98). Interestingly, ILC3s isolated from the inflamed intestine of patients with IBD presented with alterations in expression of several circadian-related genes, including Nr1d1, suggesting circadian clock disruptions—such as those seen in shift workers—may act to disrupt normal immune function and have relevance in the onset and/or pathogenesis of chronic inflammatory diseases (96). Of note however, the role of Nr1d1 as a transcriptional regulator of both Nfil3 and Rorc suggests clock-associated genes may have additional roles that are independent of circadian regulation (97).
Circadian rhythms may be imprinted by a range of systemic and environmental cues. Within the intestinal tract feeding cues were shown to contribute to the entrainment of oscillatory function in ILC3 (Figure 2: inputs). Time-specific feeding altered daily circadian rhythms in clock related genes (95) and IL-22 expression oscillated across the day between active and resting phases (74, 75). Interestingly, signals from the ENS appear to be critical in sensing feeding cues to entrain circadian rhythms in ILC3 (Figure 2) (74, 75). Feeding was found to induce VIP release from enteric neurons, consequently triggering VIPR2 signaling in ILC3s and enhancing IL-22 production and the barrier function of the epithelium. In contrast during fasting this neuropeptide cue waned, resulting in decreased IL-22 production by ILC3s and thus, imprinting diurnal rhythms onto intestinal ILC3s (74). In contrast, another study (pre-print currently under review) reported that VIP release from enteric neurons upon feeding rather decreases production of IL-22 by ILC3s, allowing for the outgrowth of the epithelial-associated commensal microbe SFB (75). Despite these discrepancies, both studies clearly implicate the sensing of feeding cues by the enteric nervous system as a key entrainer of circadian rhythmicity in ILC3. One possible explanation for the apparent differences in these findings is that complex interplay with the host microbiota may further augment ENS cues or act directly on ILC3 to provide complimentary or competing inputs, which then combine with cues from the central clock to tune anticipatory rhythms. In line with this, the microbiota was also shown to have an impact on circadian gene expression in ILC3s—adding another layer of complexity in the crosstalk between ILC3s and the commensal microbiota (95, 96).
While these studies all implicate peripheral cues in the entrainment of anticipatory ILC3 responses, light signals derived from the central clock (in the brain) are also known to be central in aligning many biological processes and in imprinting circadian rhythms. Indeed, signals from the central clock were shown to be a key regulator of ILC3 rhythmicity (Figure 2: inputs) (95). Utilizing mice in which the central clock was surgically ablated, or mice genetically deficient for Arntl only in the SCN, ILC3s developed disrupted cytokine oscillations and an altered phenotype—including the downregulation of intestinal homing markers which could partially explain time of day differences in ILC3 numbers within the gastrointestinal tract (95). The mechanisms through which the central clock in the SCN mechanistically aligns biological processes with light cues vary, but can include the release of hormonal cues—most notably glucocorticoids (84). While it remains to be determined whether this mechanism acts on ILC3 in the context of the central circadian clock, glucocorticoids have been shown to suppress ILC3s IL-22 production in vitro (99). Together these findings suggest that long-range and local circadian cues may directly regulate ILC3 numbers and function during homeostasis or following infection, mediating ILC3 interactions with the microbiota and regulation of intestinal barrier function.
Unlike cells of the adaptive immune system, ILC3 are one of the first immune cells to colonize the intestine during the embryonic period and are critical for the formation of SLOs (100). In this regard one of the most fundamental circuits through which ILC3 contribute to barrier immunity is through the orchestration of organized interactions between the innate and adaptive immune system. In contrast to the sensory circuits described above, where inputs derived from third party cells stimulate outputs in ILC3, during both embryogenesis and adult life ILC3 provide the input and stimulatory cues to stromal cells to initiate a cascade of events that lead to the formation of secondary and tertiary lymphoid tissues.
The formation of LN and PP is initiated via specialized stromal cells, known as lymphoid tissue organizer cells (LTo) that start to express chemokines such as CXCL13, CCL19, CCL21, as well as the adhesion molecules VCAM-1, ICAM-1, and MadCAM-1 (101, 102). The expression of these factors creates a gradient to recruit bona fide fetal lymphoid tissue inducer cells (LTis; fetal members of the ILC family, referred to here as fetal ILC3), which cluster with the LTo forming the primitive anlagen of the SLO (103). Fetal ILC3s at this stage express CXCR5, CCR7, and α4β7; homing markers that are important for fetal ILC3 recruitment and which were shown to mediate migration toward LTo-derived chemokines and adhesion molecules, respectively. In fact, full maturation of LTo and development of lymphoid tissue is dependent on recruitment of fetal ILC3s and provision of lymphotoxin (LT) (103, 104). Conversely, LTo also provide critical survival signals for fetal LTi/ILC3 with IL-7 expression shown to be necessary for ILC3 maintenance, while IL-7R blockade in adults also resulted in a rapid loss of normal migration of B and T cells to the LN (105). This stromal IL-7 circuit is likely also active at other sites such as in the fetal liver and bone marrow, where stroma derived IL-7 signaling could trigger the expression of NFIL3 (91). In addition, the same stromal-ILC3 circuit acts to restore normal lymph node architecture following infection-induced disruption of lymphoid microanatomy (106). Therefore, the crosstalk between ILC3s and lymph node-associated stroma is reactivated in in adulthood and crucial to enable adaptive immune responses during secondary infections (106). Thus, a key sensory circuit and stimulatory loop formed between ILC3 and stromal cells is critical for the formation of lymphoid tissues, and to facilitate the action of the broader innate and adaptive immune system.
Postnatally, a large number of organized lymphoid structures designated as tertiary lymphoid structures start to form under the influence of environmental stimuli. These immune cell clusters include cryptopatches (CP), which are confined to bottom of the crypts within the intestinal lamina propria. CP formation is driven through similar molecular mechanisms to SLO, including via interactions between ILC3-associated LTα1β2 with the LTβR expressed by stromal cells and IL-7 signaling (107, 108). CPs can further give rise to isolated lymphoid follicles (ILFs) in a CCR6 and LTα1β2-dependent manner (109, 110), resulting in up-regulation of secretory antibody (Immunoglobulin A; IgA) synthesis in response to changes in the composition of microbiota (111, 112).
A unique feature of ILF development in comparison to LN, PP, and CPs is the requirement for microbial exposure. Intestinal bacteria are sensed by myeloid cells which increase the interactions between ILC3s and LTos, also via a LTα1β2 dependent axis, leading to increased expression of adhesion molecules by the stroma and recruitment of B cells to these structures (113, 114). ILFs are largely absent in a microbiota free environment, and are restored upon recolonization with commensal microbes (115). Similarly inflammation and intestinal barrier disruption results in increased numbers of ILFs in the colon, and intriguingly mice deficient in the transcription factor RORγt develop more ILFs than their wild type counterparts in the context of intestinal inflammation, suggesting a potential regulatory role for type 3 immune responses, such as ILC3, in this setting (116).
Interactions between ILC3 and stroma also provide important cues to localize ILC3 to defined tissue microenvironments, and to facilitate interactions with adaptive immunity (discussed in detail below). Within the intestine-draining mesenteric lymph node multiple distinct stromal populations have been identified with differential capacities to attract immune populations and orchestrate immune cell crosstalk (117). One such population expresses the enzyme Ch25h, which acts to generate the cholesterol metabolite 7,α25-OH—a key migratory ligand for multiple immune cells including ILC3 (117–120). This stromal cue is sensed by ILC3 via the receptor EBI2 (Gpr183), and facilitates localization of not only ILC3 but also T follicular helper cells, DCs and B cells to the follicular border of lymph nodes (118, 120–125). Similarly, within the intestinal tissue stromal generation and breakdown of cholesterol ligand cues create a migratory gradient required to recruit ILC3 to CP in a Gpr183-dependent manner (119). Together these studies indicate that a stromal ILC3 circuit is a key regulator not only of lymphoid organogenesis but also of ILC3 localization and function, which together facilitate the interactions between ILC3 and adaptive immune cells and foster modulatory crosstalk.
ILC3s are also emerging as key orchestrators and regulators of adaptive immune responses [Reviewed in detail in (126)]. This regulation is mediated by ILC3 either through indirect modulation of bystander cells that subsequently modulate the adaptive immune response or directly via both soluble mediators and cell contact-dependent interactions with adaptive lymphocytes.
As discussed above, ILC3s contribute to the formation of lymphoid structures and were found to be strategically positioned in clusters within lymph nodes where they have potential to interact with both T and B cells both directly and indirectly (127). Many of the same mechanisms employed by ILC3 to induce lymphoid organogenesis during early life are similarly employed in adult tissues to regulate the adaptive immune system. For example, ILC3s can support the production of IgA by B cells in the PP, in part through both soluble LTα3 and surface bound LTα1β2 interactions with DCs (128, 129). Similarly, in the spleen, production of LTα1β2, GM-CSF, and BAFF/APRIL production by ILC3s also acts to support B cell responses (Figure 3: outputs) (130).
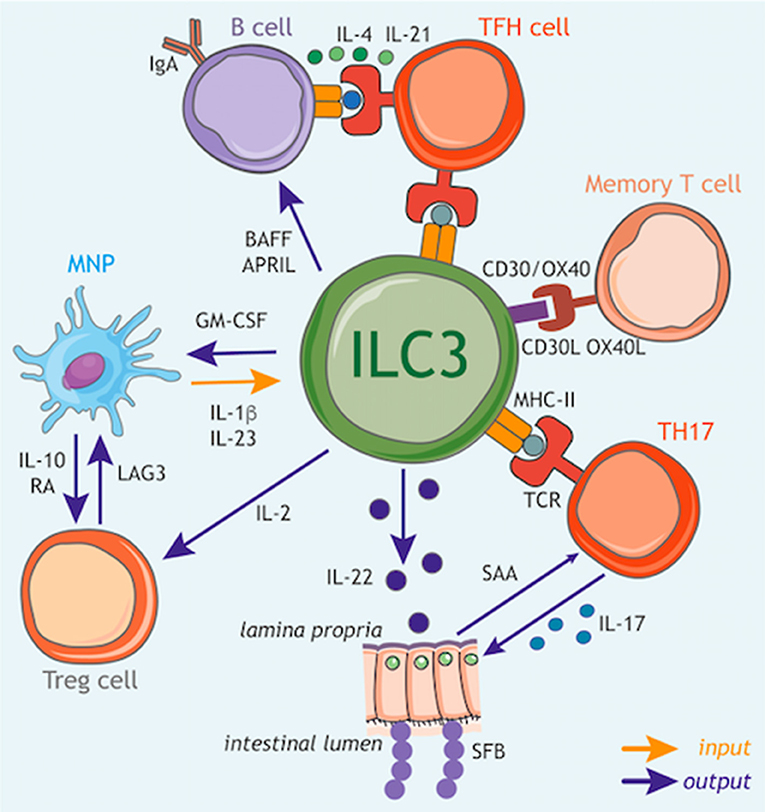
Figure 3. ILC3 circuits orchestrate adaptive immune responses. In addition to their function as tissue-resident cytokine producing cells, ILC3s have the capacity to participate in multiple cellular circuits through direct cell–cell modulation of T cell responses, as well as the release of soluble mediators that augment adaptive immune function and development. ILC3s can control the magnitude and quality of the CD4+ T cell response via antigen presentation in the context of MHC class II (MHCII). At steady state ILC3s lack co-stimulatory molecule expression and appear to limit CD4+ T cell responses, however this interaction may be altered in inflammatory scenarios via upregulation of co-stimulatory molecules such as CD40, CD80, and CD86, which favor the promotion of T cell response. Furthermore, ILC3s act to modulate the survival of recirculating memory CD4+ T cells via interactions via OX40L and CD30L, although it is unknown whether this process also requires MHCII-dependent antigen presentation. In addition, ILC3 regulation of T follicular helper (TFH) cell responses has consequences for the priming of germinal center B cells and the induction of T-dependent IgA responses toward colon-dwelling commensal microbes. ILC3s can also modulate adaptive immune cells through the production of regulatory cytokines and growth factors. In line with this, ILC3 directly support B cell responses in the spleen through provision of critical growth factors such as BAFF/APRIL. Similarly, ILC3 also modulate the magnitude of the T cell response within the intestinal tract through the production of soluble mediators. For example, ILC3-derived IL-22 induces epithelial serum amyloid A (SAA) protein, which subsequently promotes local Th17 responses and acts to limit colonization with segmented filamentous bacteria (SFB) via the induction of antimicrobial peptides. In addition, ILC3 facilitate the establishment of a regulatory and tolerogenic environment in the gut by promoting regulatory T cell (Treg) responses. ILC3 crosstalk with tissue-resident myeloid cell populations establishes a feedback circuit whereby ILC3-derived GM-CSF promotes IL-10 and RA production by myeloid cells to promote Treg conversion. Conversely, Treg, myeloid cells and ILC3 may feedback on each other through a variety of soluble and cell-cell interactions suggesting a dynamic and malleable communication loop to ensure tolerance and tissue homeostasis. Finally, ILC3 subsets are a potent source of IL-2 in the small intestine that provides survival signals for Treg. Together these tissue-resident immune circuits place ILC3 at the center of a number of pathways through which they regulate adaptive immune responses to promote tissue health and homeostatic interactions with the microbiota.
In line with these findings, ILC3s have the capacity to crosstalk both directly and indirectly with the adaptive immune system through the production of multiple soluble factors. Following exposure to the commensal microbiota IL-22 produced by ILC3s acts to support homeostatic tissue Th17 responses through the induction of serum amyloid protein A (SAA) from epithelial cells (Figure 3: outputs) (131). Interestingly, ILC3 derived IL-22 can also prevent the activation of T cells in an AhR-dependent manner to limit immune activation or tissue damage (132). Conversely, T cells may also regulate the magnitude of ILC3-derived IL-22 production (26, 133), suggesting complex crosstalk between T cells and ILC3 in determining the level of IL-22 produced in the tissue.
As highlighted previously, sensing of the microbiota by myeloid cells is a critical regulator of ILC3 responses, and has consequences for adaptive immunity. IL-1β induction of GM-CSF production by ILC3s feeds back on tissue-resident MNP to trigger IL-10 and RA production by intestinal macrophages and DCs—resulting in the induction and maintenance of tissue regulatory T cells (Treg) and reinforcing immune tolerance (Figure 3: inputs/outputs) (134). Similarly, IL-1β produced by intestinal MNP further induces ILC3 to produce IL-2, a critical growth signal that helps to support peripherally induced Tregs in the small intestine and to maintain intestinal tolerance (Figure 3: inputs/outputs) (135). Conversely Treg interactions with MNP may limit IL-23 production to prevent ILC3-driven inflammation via a LAG3-dependent mechanism (Figure 3: outputs) (136), implicating a bidirectional axis involving ILC3, MNP, and Treg in determining the immune tone of the intestinal tract.
ILC3s are increasingly appreciated to also act as a direct orchestrator of tissue immune responses through their ability to act as antigen-presenting cells. ILC3 are also endowed with a broad array of accessory co-activating and co–inhibitory molecules that enable further modulation and tuning of adaptive immune cell function. Thus, when coupled with their strategic localization within lymphoid structures, ILC3 have the potential to potently regulate adaptive immune responses. At steady state, ILC3s in the mLNs and large intestine constitutively express MHC class II (MHCII) molecules at levels comparable with other professional antigen-presenting cells and can acquire, process and present antigens (Figure 3: inputs/outputs) (137). However, under homeostatic circumstances these interactions do not induce T cell proliferation, due in part to the absence of classical co-stimulatory molecules such as CD40, CD80, and CD86 on the cell surface (137). In contrast MHCII+ ILC3s were found to suppress effector CD4+ T cell responses toward the microbiota in the intestine (137–139). In line with a suppressive function for ILC3-associated antigen presentation, deletion of ILC3-intrinsic MHCII also disrupts crosstalk between ILC3 and adaptive immune cells at the interfollicular border of the mLN resulting in a spontaneous T follicular helper response that subsequently drives increased IgA responses against mucosal-dwelling commensals, and results in an altered intestinal metabolome (Figure 3: outputs) (120). While these findings suggest a suppressive and regulatory role for antigen-presenting ILC3 in the context of health, additional reports suggest that in contrast during immunization or infection tissue-specific inflammatory cues act to alter the nature—and consequences—of ILC3 antigen presentation. Indeed, activation of ILC3 by IL-1β resulted in antigen-presentation dependent promotion of T cell responses as a result of upregulated expression of classical co-stimulatory molecules (CD80/CD86) on ILC3 (140). In addition to antigen-presentation to CD4+ T cell subsets, ILC3 also express CD1d—conferring the ability to present lipid antigens to invariant (i) NKT cell populations, and promote their functionality (141).
Indeed, ILC3 have the capacity to modulate a broad variety of specialized adaptive immune responses through cell-cell interactions via additional non-classical co-stimulatory and co-inhibitory molecules. Seminal early studies in the field demonstrated a critical role for ILC3-associated CD30L and OX40L in the modulation of T cell memory through cognate interactions with CD30/OX40 (Figure 3: inputs/outputs) (126, 142, 143). Recent studies have expanded upon these observations to demonstrate a role for tissue-resident MNP-derived TL1A in regulating the expression of OX40L on ILC3, which was subsequently demonstrated to enable ILC3 to promote inflammatory effector T cell responses in the context of colitis (144). In addition ILC3 have been reported to express co-inhibitory and immune checkpoint molecules (e.g., PD1, PDL1) suggesting further immunoregulatory functions for these cells—although further investigation is required to determine the functional relevance of this receptor repertoire (145, 146). As investigation into this aspect of ILC3 function increases, the nature and breadth of interactions with both innate and adaptive immunity are likely to expand and present new intervention possibilities for the modulation of tissue immune responses.
The maintenance of mucosal homeostasis is mediated through a complex interplay between the host and its environment, between immune and non-immune cells and by the balance of pathogenic and commensal microbes. Here we have highlighted the contributions of sensory circuits within the intestinal tract, which culminate in the activation and regulation of ILC3s. ILC3 display connectivity with an increasing number of physiological systems, many of which are likely to act simultaneously within the tissue in the context of health and disease—and ultimately to regulate the same range of ILC3-derived outputs. Thus, despite recent advances, one future challenge will be to understand how ILC3 integrate multiple concurrent signals from varying biological systems within a given tissue niche, and to determine how these cues are translated into cell fate decisions to determine the magnitude or quality of an ILC3 response. Many signaling pathways downstream of both cytokine and neuropeptide receptors converge upon core regulators of cell function—such as the mammalian target of rapamycin (mTOR) (147). Moreover, the appropriate licensing and modulation of anabolic cell metabolism pathways in order to generate new cellular biomass, effector proteins and facilitate proliferation is a central checkpoint of cellular function, critical to regulate immune cell function and controlled in part through mTOR activation (148). In line with this, a recent report demonstrated the induction of an mTOR complex 1-dependent programme of glycolytic metabolism as a central rate-limiting step in the production of ILC3-derived cytokines and proliferation (149). Engagement of glycolysis was also associated with the expression of the oxygen-sensing transcription factor HIF1α, suggesting other tissue-specific environmental factors may augment ILC3 responses via licensing of glycolysis and anabolic metabolism.
Ultimately, an increased knowledge of the network of inputs and outputs—and importantly the mechanisms through which these multiple sensory circuits are integrated and interpreted—will allow for new approaches to target this mucosal immune sentinel in the context of health and disease. Indeed, while ILC3 mediate many protective processes at homeostasis, dysregulated ILC3 responses have been implicated in a wide range of chronic inflammatory and metabolic diseases and have increasingly been suggested to play roles in cancer development and progression. Most notably, disruption of ILC3 responses is associated with the pathogenesis of inflammatory bowel disease (IBD) (1, 27, 40, 41, 62, 138, 150). Interestingly, lifestyles associated with disruption of sleep cycles and circadian rhythms (e.g., shift work, jet lag) have been suggested as potential triggers for IBD flares (151). Thus, while there have been recent major achievements in the understanding of how ILC3 sense signals from the CNS and ENS and perceive circadian cues (72, 74, 75, 95–97), the physiological impact of these systems on ILC3 function in the context of IBD could prove important in beginning to decode the multitude of factors that lead to disease onset and progression.
In conclusion, ILC3 are strategically positioned within mucosal sites where they act as a hub of multiple distinct, yet complementary, sensory circuits. Together, these circuits act to continually survey the intestinal tract for perturbations in microbial, dietary and external environmental cues and enable the rapid communication and translation of this information, resulting in protective effector responses that continually reinforce normal tissue function and health. Strategies aimed at exploiting these cues and sensory circuits to promote or restore homeostatic ILC3 function, while simultaneously suppressing the dysregulated signaling associated with maladapted immune function, may lead to novel therapeutic intervention strategies in a number of human diseases.
MH and RD conceived of and contributed to the writing of the manuscript. RD constructed the figures.
The Hepworth Laboratory was supported by a Royal Society and Wellcome Trust Sir Henry Dale Fellowship (Award 105644/Z/14/Z) and a Lister Institute of Preventative Medicine Prize.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank members of the Hepworth laboratory for the discussions and critical reading of the manuscript.
1. Klose CSN, Artis D. Innate lymphoid cells as regulators of immunity, inflammation, and tissue homeostasis. Nat Immunol. (2016) 17:765–74. doi: 10.1038/ni.3489
2. Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol. (2011) 12:21–7. doi: 10.1038/ni.1962
3. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells—a proposal for uniform nomenclature. Nat Rev Immunol. (2013) 13:145–9. doi: 10.1038/nri3365
4. Spits H, Cupedo T. Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annu Rev Immunol. (2012) 30:647–75. doi: 10.1146/annurev-immunol-020711-075053
5. Artis D, Spits H. The biology of innate lymphoid cells. Nature. (2015) 517:293–301. doi: 10.1038/nature14189
6. McKenzie ANJ, Spits H, Eberl G. Innate lymphoid cells in inflammation and immunity. Immunity. (2014) 41:366–74. doi: 10.1016/j.immuni.2014.09.006
7. Eberl G, Colonna M, Di Santo JP, McKenzie ANJ. Innate lymphoid cells: a new paradigm in immunology. Science. (2015) 348:aaa6566. doi: 10.1126/science.aaa6566
8. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell. (2018) 174:1054–66. doi: 10.1016/j.cell.2018.07.017
9. Melo-Gonzalez F, Hepworth MR. Functional and phenotypic heterogeneity of group 3 innate lymphoid cells. Immunology. (2017) 150:265–75. doi: 10.1111/imm.12697
10. Klose CSN, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, D'Hargues Y, et al. A T-bet gradient controls the fate and function of CCR6-RORγt + innate lymphoid cells. Nature. (2013) 494:261–5. doi: 10.1038/nature11813
11. Vonarbourg C, Mortha A, Bui VL, Hernandez PP, Kiss EA, Hoyler T, et al. Regulated expression of nuclear receptor RORγt confers distinct functional fates to NK cell receptor-expressing RORγt+ innate lymphocytes. Immunity. (2010) 33:736–51. doi: 10.1016/j.immuni.2010.10.017
12. Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JKM, et al. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature. (2009) 457:722–5. doi: 10.1038/nature07537
13. Tumanov AV, Koroleva EP, Guo X, Wang Y, Kruglov A, Nedospasov S, et al. Lymphotoxin controls the IL-22 protection pathway in gut innate lymphoid cells during mucosal pathogen challenge. Cell Host Microbe. (2011) 10:44–53. doi: 10.1016/j.chom.2011.06.002
14. Qiu J, Heller JJ, Guo X, Chen ZME, Fish K, Fu YX, et al. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity. (2012) 36:92–104. doi: 10.1016/j.immuni.2011.11.011
15. Sonnenberg GF, Monticelli LA, Alenghat T, Fung TC, Hutnick NA, Kunisawa J, et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science. (2012) 336:1321–5. doi: 10.1126/science.1222551
16. Guo X, Qiu J, Tu T, Yang X, Deng L, Anders RA, et al. Induction of innate lymphoid cell-derived interleukin-22 by the transcription factor STAT3 mediates protection against intestinal infection. Immunity. (2014) 40:25–39. doi: 10.1016/j.immuni.2013.10.021
17. Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol. (2011) 12:383–90. doi: 10.1038/ni.2025
18. Turner J-E, Morrison PJ, Wilhelm C, Wilson M, Ahlfors H, Renauld J-C, et al. IL-9–mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J Exp Med. (2013) 210:2951–65. doi: 10.1084/jem.20130071
19. Goto Y, Obata T, Kunisawa J, Sato S, Ivanov II, Lamichhane A, et al. Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science. (2014) 345:1254009. doi: 10.1126/science.1254009
20. Pickard JM, Maurice CF, Kinnebrew MA, Abt MC, Schenten D, Golovkina TV, et al. Rapid fucosylation of intestinal epithelium sustains host–commensal symbiosis in sickness. Nature. (2014) 514:638–41. doi: 10.1038/nature13823
21. Pham TAN, Clare S, Goulding D, Arasteh JM, Stares MD, Browne HP, et al. Epithelial IL-22RA1-mediated fucosylation promotes intestinal colonization resistance to an opportunistic pathogen. Cell Host Microbe. (2014) 16:504–16. doi: 10.1016/j.chom.2014.08.017
22. Gronke K, Hernández PP, Zimmermann J, Klose CSN, Kofoed-Branzk M, Guendel F, et al. Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature. (2019) 566:249–53. doi: 10.1038/s41586-019-0899-7
23. Aparicio-Domingo P, Romera-Hernandez M, Karrich JJ, Cornelissen F, Papazian N, Lindenbergh-Kortleve DJ, et al. Type 3 innate lymphoid cells maintain intestinal epithelial stem cells after tissue damage. J Exp Med. (2015) 212:1783–91. doi: 10.1084/jem.20150318
24. Lindemans CA, Calafiore M, Mertelsmann AM, O'Connor MH, Dudakov JA, Jenq RR, et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature. (2015) 528:560–4. doi: 10.1038/nature16460
25. Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature. (2012) 491:259–63. doi: 10.1038/nature11535
26. Mao K, Baptista AP, Tamoutounour S, Zhuang L, Bouladoux N, Martins AJ, et al. Innate and adaptive lymphocytes sequentially shape the gut microbiota and lipid metabolism. Nature. (2018) 554:255–259. doi: 10.1038/nature25437
27. Takayama T, Kamada N, Chinen H, Okamoto S, Kitazume MT, Chang J, et al. Imbalance of NKp44+NKp46- and NKp44 -NKp46+ natural killer cells in the intestinal mucosa of patients with Crohn's disease. Gastroenterology. (2010) 139:882–92.e3. doi: 10.1053/j.gastro.2010.05.040
28. Satoh-Takayama N, Vosshenrich CAJ, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity. (2008) 29:958–70. doi: 10.1016/j.immuni.2008.11.001
29. Kinugasa T, Sakaguchi T, Gu X, Reinecker HC. Claudins regulate the intestinal barrier in response to immune mediators. Gastroenterology. (2000) 118:1001–11. doi: 10.1016/S0016-5085(00)70351-9
30. Onishi RM, Gaffen SL. Interleukin-17 and its target genes: mechanisms of interleukin-17 function in disease. Immunology. (2010) 129:311–21. doi: 10.1111/j.1365-2567.2009.03240.x
31. Gaffen SL, Jain R, Garg AV, Cua DJ. IL-23-IL-17 immune axis: discovery, mechanistic understanding, and clinical testing. Nat Rev Immunol. (2014) 14:585–600. doi: 10.1038/nri3707
32. Sawa S, Lochner M, Satoh-Takayama N, Dulauroy S, Bérard M, Kleinschek M, et al. RORγt+innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat Immunol. (2011) 12:320–8. doi: 10.1038/ni.2002
33. Coccia M, Harrison OJ, Schiering C, Asquith MJ, Becher B, Powrie F, et al. IL-1β mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4 + Th17 cells. J Exp Med. (2012) 209:1595–609. doi: 10.1084/jem.20111453
34. Gladiator A, Wangler N, Trautwein-Weidner K, LeibundGut-Landmann S. Cutting Edge: IL-17–secreting innate lymphoid cells are essential for host defense against fungal infection. J Immunol. (2013) 190:521–5. doi: 10.4049/jimmunol.1202924
35. Xiong H, Keith JW, Samilo DW, Carter RA, Leiner IM, Pamer EG. Innate lymphocyte/Ly6Chi monocyte crosstalk promotes klebsiella pneumoniae clearance. Cell. (2016) 165:679–89. doi: 10.1016/j.cell.2016.03.017
36. Ardain A, Domingo-Gonzalez R, Das S, Kazer SW, Howard NC, Singh A, et al. Group 3 innate lymphoid cells mediate early protective immunity against tuberculosis. Nature. (2019) 570:528–32. doi: 10.1038/s41586-019-1276-2
37. Nakagawa S, Matsumoto M, Katayama Y, Oguma R, Wakabayashi S, Nygaard T, et al. Staphylococcus aureus virulent PSMα peptides induce keratinocyte alarmin release to orchestrate IL-17-dependent skin inflammation. Cell Host Microbe. (2017) 22:667–77.e5. doi: 10.1016/j.chom.2017.10.008
38. Xu H, Wang X, Liu DX, Moroney-Rasmussen T, Lackner AA, Veazey RS. IL-17-producing innate lymphoid cells are restricted to mucosal tissues and are depleted in SIV-infected macaques. Mucosal Immunol. (2012) 5:658–69. doi: 10.1038/mi.2012.39
39. Li H, Reeves RK. Functional perturbation of classical natural killer and innate lymphoid cells in the oral mucosa during SIV infection. Front Immunol. (2012) 3:417. doi: 10.3389/fimmu.2012.00417
40. Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology: commentary. Nature. (2010) 464:1371–5. doi: 10.1038/nature08949
41. Geremia A, Arancibia-Cárcamo CV, Fleming MPP, Rust N, Singh B, Mortensen NJ, et al. IL-23–responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med. (2011) 208:1127–33. doi: 10.1084/jem.20101712
42. Rooks MG, Garrett WS. Gut microbiota, metabolites and host immunity. Nat Rev Immunol. (2016) 16:341–52. doi: 10.1038/nri.2016.42
43. Honda K, Littman DR. The microbiota in adaptive immune homeostasis and disease. Nature. (2016) 535:75–84. doi: 10.1038/nature18848
44. Kim SH, Cho BH, Kiyono H, Jang YS. Microbiota-derived butyrate suppresses group 3 innate lymphoid cells in terminal ileal Peyer's patches. Sci Rep. (2017) 7:1–12. doi: 10.1038/s41598-017-02729-6
45. Bhatt B, Zeng P, Zhu H, Sivaprakasam S, Li S, Xiao H, et al. Gpr109a limits microbiota-induced IL-23 production to constrain ILC3-mediated colonic inflammation. J Immunol. (2018) 200:2905–14. doi: 10.4049/jimmunol.1701625
46. Eunyoung C, Sydney L, Fonseca-pereira D, Bae S, Michaud M, Hoveyda HR, et al. Metabolite-sensing receptor Ffar2 regulates colonic group 3 innate lymphoid cells and gut article metabolite-sensing receptor Ffar2 regulates colonic group 3 innate lymphoid cells and gut immunity. Immunity. (2019) 51:871–84.e6. doi: 10.1016/j.immuni.2019.09.014
47. Babu ST, Niu X, Raetz M, Savani RC, Hooper LV, Mirpuri J. Maternal high-fat diet results in microbiota-dependent expansion of ILC3s in mice offspring. JCI Insight. (2018) 3:1–12. doi: 10.1172/jci.insight.99223
48. Gomez de Aguero M, Ganal-Vonarburg SC, Fuhrer T, Rupp S, Uchimura Y, Li H, et al. The maternal microbiota drives early postnatal innate immune development. Science. (2016) 351:1296–302. doi: 10.1126/science.aad2571
49. Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science. (2011) 334:1561–5. doi: 10.1126/science.1214914
50. Kiss EA, Diefenbach A. Role of the aryl hydrocarbon receptor in controlling maintenance and functional programs of RORγt+ innate lymphoid cells and intraepithelial lymphocytes. Front Immunol. (2012) 3:124. doi: 10.3389/fimmu.2012.00124
51. Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol. (2011) 13:144–51. doi: 10.1038/ni.2187
52. Li S, Bostick JW, Ye J, Qiu J, Zhang B, Urban JF, et al. Aryl hydrocarbon receptor signaling cell intrinsically inhibits intestinal group 2 innate lymphoid cell function. Immunity. (2018) 49:915–28.e5. doi: 10.1016/j.immuni.2018.09.015
53. van de Pavert SA, Ferreira M, Domingues RG, Ribeiro H, Molenaar R, Moreira-Santos L, et al. Maternal retinoids control type 3 innate lymphoid cells and set the offspring immunity. Nature. (2014) 508:123–7. doi: 10.1038/nature13158
54. Spencer SP, Wilhelm C, Yang Q, Hall JA, Bouladoux N, Boyd A, et al. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science. (2014) 343:432–7. doi: 10.1126/science.1247606
55. Goverse G, Labao-Almeida C, Ferreira M, Molenaar R, Wahlen S, Konijn T, et al. Vitamin A controls the presence of RORγ + innate lymphoid cells and lymphoid tissue in the small intestine. J Immunol. (2016) 196:5148–55. doi: 10.4049/jimmunol.1501106
56. Kim MH, Taparowsky EJ, Kim CH. Retinoic acid differentially regulates the migration of innate lymphoid cell subsets to the gut. Immunity. (2015) 43:107–19. doi: 10.1016/j.immuni.2015.06.009
57. Chen J, Waddell A, Lin YD, Cantorna MT. Dysbiosis caused by vitamin D receptor deficiency confers colonization resistance to Citrobacter rodentium through modulation of innate lymphoid cells. Mucosal Immunol. (2015) 8:618–26. doi: 10.1038/mi.2014.94
58. Konya V, Czarnewski P, Forkel M, Rao A, Kokkinou E, Villablanca EJ, et al. Vitamin D downregulates the IL-23 receptor pathway in human mucosal group 3 innate lymphoid cells. J Allergy Clin Immunol. (2018) 141:279–92. doi: 10.1016/j.jaci.2017.01.045
59. Lin YD, Arora J, Diehl K, Bora SA, Cantorna MT. Vitamin D is required for ILC3 derived IL-22 and protection from Citrobacter rodentium infection. Front Immunol. (2019) 10:1. doi: 10.3389/fimmu.2019.00001
60. Takatori H, Kanno Y, Watford WT, Tato CM, Weiss G, Ivanov II, et al. Lymphoid tissue inducer–like cells are an innate source of IL-17 and IL-22. J Exp Med. (2009) 206:35–41. doi: 10.1084/jem.20072713
61. Kinnebrew MA, Buffie CG, Diehl GE, Zenewicz LA, Leiner I, Hohl TM, et al. Interleukin 23 production by intestinal CD103 +CD11b + dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity. (2012) 36:276–87. doi: 10.1016/j.immuni.2011.12.011
62. Longman RS, Diehl GE, Victorio DA, Huh JR, Galan C, Miraldi ER, et al. CX3CR1+ mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J Exp Med. (2014) 211:1571–83. doi: 10.1084/jem.20140678
63. Savage AK, Liang H-E, Locksley RM. The development of steady-state activation hubs between adult LTi ILC3s and primed macrophages in small intestine. J Immunol. (2017) 199:1912–22. doi: 10.4049/jimmunol.1700155
64. Manta C, Heupel E, Radulovic K, Rossini V, Garbi N, Riedel CU, et al. CX3CR1+ macrophages support IL-22 production by innate lymphoid cells during infection with Citrobacter rodentium. Mucosal Immunol. (2013) 6:177–88. doi: 10.1016/S0016-5085(12)62628-6
65. Satoh-Takayama N, Serafini N, Verrier T, Rekiki A, Renauld JC, Frankel G, et al. The chemokine receptor CXCR6 controls the functional topography of interleukin-22 producing intestinal innate lymphoid cells. Immunity. (2014) 41:776–88. doi: 10.1016/j.immuni.2014.10.007
66. Jonathan Kipnis. Multifaceted interactions between adaptive immunity and the central nervous system. Neuroimmunol Rev. (2016) 353:766–71. doi: 10.1126/science.aag2638
67. Veiga-Fernandes H, Pachnis V. Neuroimmune regulation during intestinal development and homeostasis. Nat Immunol. (2017) 18:116–22. doi: 10.1038/ni.3634
68. Veiga-Fernandes H, Coles MC, Foster KE, Patel A, Williams A, Natarajan D, et al. Tyrosine kinase receptor RET is a key regulator of Peyer's Patch organogenesis. Nature. (2007) 446:547–51. doi: 10.1038/nature05597
69. Patel A, Harker N, Moreira-Santos L, Ferreira M, Alden K, Timmis J, et al. Differential RET signaling pathways drive development of the enteric lymphoid and nervous systems. Sci Signal. (2012) 5:ra55. doi: 10.1126/scisignal.2002734
70. Van De Pavert SA, Olivier BJ, Goverse G, Vondenhoff MF, Greuter M, Beke P, et al. Chemokine cxcl13 is essential for lymph node initiation and is induced by retinoic acid and neuronal stimulation. Nat Immunol. (2009) 10:1193–9. doi: 10.1038/ni.1789
71. Obata Y, Pachnis V. The effect of microbiota and the immune system on the development and organization of the enteric nervous system. Gastroenterology. (2016) 151:836–44. doi: 10.1053/j.gastro.2016.07.044
72. Ibiza S, García-Cassani B, Ribeiro H, Carvalho T, Almeida L, Marques R, et al. Glial-cell-derived neuroregulators control type 3 innate lymphoid cells and gut defence. Nature. (2016) 535:440–3. doi: 10.1038/nature18644
73. Dalli J, Colas RA, Arnardottir H, Serhan CN. Vagal regulation of group 3 innate lymphoid cells and the immunoresolvent PCTR1 controls infection resolution. Immunity. (2017) 46:92–105. doi: 10.1016/j.immuni.2016.12.009
74. Seillet C, Luong K, Tellier J, Jacquelot N, Shen RDS, Hickey P, et al. Vasoactive intestinal peptide confers anticipatory mucosal immunity by regulating ILC3 activity. Nat Immunol. (2019) 21:168–77. doi: 10.1038/s41590-019-0567-y
75. Talbot J, Hahn P, Kroehling L, Nguyen H, Li D, Littman DR. VIP-producing enteric neurons interact with innate lymphoid cells to regulate feeding-dependent intestinal epithelial barrier functions. bioRxiv. [Preprint]. (2019). doi: 10.1101/721464
76. Wallrapp A, Riesenfeld SJ, Burkett PR, Abdulnour REE, Nyman J, Dionne D, et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature. (2017) 549:351–6. doi: 10.1038/nature24029
77. Klose CSN, Mahlakõiv T, Moeller JB, Rankin LC, Flamar AL, Kabata H, et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature. (2017) 549:282–6. doi: 10.1038/nature23676
78. Cardoso V, Chesné J, Ribeiro H, García-Cassani B, Carvalho T, Bouchery T, et al. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature. (2017) 549:277–81. doi: 10.1038/nature23469
79. Nagashima H, Mahlakõiv T, Shih H-Y, Davis FP, Meylan F, Huang Y, et al. Neuropeptide CGRP limits group 2 innate lymphoid cell responses and constrains type 2 inflammation. Immunity. (2019) 51:682–95.e6. doi: 10.1016/j.immuni.2019.06.009
80. Wallrapp A, Burkett PR, Riesenfeld SJ, Kim S-J, Christian E, Abdulnour R-EE, et al. Calcitonin gene-related peptide negatively regulates alarmin-driven type 2 innate lymphoid cell responses. Immunity. (2019) 51:709–23. doi: 10.1016/j.immuni.2019.09.005
81. Moriyama S, Brestoff JR, Flamar AL, Moeller JB, Klose CSN, Rankin LC, et al. β2-adrenergic receptor-mediated negative regulation of group 2 innate lymphoid cell responses. Science. (2018) 359:1056–61. doi: 10.1126/science.aan4829
82. Nussbaum JC, Van Dyken SJ, Von Moltke J, Cheng LE, Mohapatra A, Molofsky AB, et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature. (2013) 502:245–8. doi: 10.1038/nature12526
83. Feng D, Lazar MA. Clocks, Metabolism, and the epigenome. Mol Cell. (2012) 47:158–67. doi: 10.1016/j.molcel.2012.06.026
84. Mohawk JA, Green CB, Takahashi JS. Central and peripheral circadian clocks in mammals. Annu Rev Neurosci. (2012) 35:445–62. doi: 10.1146/annurev-neuro-060909-153128
85. Scheiermann C, Gibbs J, Ince L, Loudon A. Clocking in to immunity. Nat Rev Immunol. (2018) 18:423–37. doi: 10.1038/s41577-018-0008-4
86. Welsh DK, Takahashi JS, Kay SA. Suprachiasmatic nucleus: cell autonomy and network properties. Annu Rev Physiol. (2010) 72:551–77. doi: 10.1146/annurev-physiol-021909-135919
87. Curtis AM, Bellet MM, Sassone-Corsi P, O'Neill LAJ. Circadian clock proteins and immunity. Immunity. (2014) 40:178–86. doi: 10.1016/j.immuni.2014.02.002
88. Takahashi JS. Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet. (2016) 18:164–79. doi: 10.1038/nrg.2016.150
89. Seillet C, Rankin LC, Groom JR, Mielke LA, Tellier J, Chopin M, et al. Nfil3 is required for the development of all innate lymphoid cell subsets. J Exp Med. (2014) 211:1733–40. doi: 10.1084/jem.20140145
90. Geiger TL, Abt MC, Gasteiger G, Firth MA, O'Connor MH, Geary CD, et al. Nfil3 is crucial for development of innate lymphoid cells and host protection against intestinal pathogens. J Exp Med. (2014) 211:1723–31. doi: 10.1084/jem.20140212
91. Xu W, Domingues RG, Fonseca-Pereira D, Ferreira M, Ribeiro H, Lopez-Lastra S, et al. NFIL3 orchestrates the emergence of common helper innate lymphoid cell precursors. Cell Rep. (2015) 10:2043–54. doi: 10.1016/j.celrep.2015.02.057
92. Yu X, Wang Y, Deng M, Li Y, Ruhn KA, Zhang CC, et al. The basic leucine zipper transcription factor NFIL3 directs the development of a common innate lymphoid cell precursor. Elife. (2014) 3:e04406. doi: 10.7554/eLife.04406
93. Thaiss CA, Zeevi D, Levy M, Zilberman-Schapira G, Suez J, Tengeler AC, et al. Transkingdom control of microbiota diurnal oscillations promotes metabolic homeostasis. Cell. (2014) 159:514–29. doi: 10.1016/j.cell.2014.09.048
94. Thaiss CA, Levy M, Korem T, Dohnalová L, Shapiro H, Jaitin DA, et al. Microbiota diurnal rhythmicity programs host transcriptome oscillations. Cell. (2016) 167:1495–510.e12. doi: 10.1016/j.cell.2016.11.003
95. Godinho-Silva C, Domingues RG, Rendas M, Raposo B, Ribeiro H, da Silva JA, et al. Light-entrained and brain-tuned circadian circuits regulate ILC3s and gut homeostasis. Nature. (2019) 574:254–8. doi: 10.1101/723932
96. Teng F, Goc J, Zhou L, Chu C, Shah MA, Eberl G, et al. A circadian clock is essential for homeostasis of group 3 innate lymphoid cells in the gut. Sci Immunol. (2019) 4:eaax1215. doi: 10.1126/sciimmunol.aax1215
97. Wang Q, Robinette ML, Billon C, Collins PL, Bando JK, Fachi JL, et al. Circadian rhythm–dependent and circadian rhythm–independent impacts of the molecular clock on type 3 innate lymphoid cells. Sci Immunol. (2019) 4:eaay7501. doi: 10.1126/sciimmunol.aay7501
98. Yu X, Rollins D, Ruhn KA, Stubblefield JJ, Green CB, Kashiwada M, et al. TH17 cell differentiation is regulated by the circadian clock. Science. (2013) 342:727–30. doi: 10.1126/science.1243884
99. Seshadri S, Pope RL, Zenewicz LA. Glucocorticoids inhibit group 3 innate lymphocyte IL-22 production. J Immunol. (2018) 201:1267–74. doi: 10.4049/jimmunol.1800484
100. Eberl G, Littman DR. The role of the nuclear hormone receptor RORγt in the development of lymph nodes and Peyer's patches. Immunol Rev. (2003) 195:81–90. doi: 10.1034/j.1600-065X.2003.00074.x
101. Cupedo T, Vondenhoff MFR, Heeregrave EJ, de Weerd AE, Jansen W, Jackson DG, et al. Presumptive lymph node organizers are differentially represented in developing mesenteric and peripheral nodes. J Immunol. (2004) 173:2968–75. doi: 10.4049/jimmunol.173.5.2968
102. Onder L, Danuser R, Scandella E, Firner S, Chai Q, Hehlgans T, et al. Endothelial cell-specific lymphotoxin-β receptor signaling is critical for lymph node and high endothelial venule formation. J Exp Med. (2013) 210:465–73. doi: 10.1084/jem.20121462
103. Adachi S, Yoshida H, Honda K, Maki K, Saijo K, Ikuta K, et al. Essential role of IL-7 receptor α in the formation of Peyer's patch anlage. Int Immunol. (1998) 10:1–6. doi: 10.1093/intimm/10.1.1
104. Bénézech C, White A, Mader E, Serre K, Parnell S, Pfeffer K, et al. Ontogeny of stromal organizer cells during lymph node development. J Immunol. (2010) 184:4521–30. doi: 10.4049/jimmunol.0903113
105. Yang J, Cornelissen F, Papazian N, Reijmers RM, Llorian M, Cupedo T, et al. IL-7-dependent maintenance of ILC3s is required for normal entry of lymphocytes into lymph nodes. J Exp Med. (2018) 215:1069–77. doi: 10.1084/jem.20170518
106. Scandella E, Bolinger B, Lattmann E, Miller S, Favre S, Littman DR, et al. Restoration of lymphoid organ integrity through the interaction of lymphoid tissue-inducer cells with stroma of the T cell zone. Nat Immunol. (2008) 9:667–75. doi: 10.1038/ni.1605
107. Taylor RT, Lugering A, Newell KA, Williams IR. Intestinal cryptopatch formation in mice requires lymphotoxin and the lymphotoxin receptor. J Immunol. (2004) 173:7183–9. doi: 10.4049/jimmunol.173.12.7183
108. Lügering A, Ross M, Sieker M, Heidemann J, Williams IR, Domschke W, et al. CCR6 identifies lymphoid tissue inducer cells within cryptopatches. Clin Exp Immunol. (2010) 160:440–9. doi: 10.1111/j.1365-2249.2010.04103.x
109. Lorenz RG, Chaplin DD, McDonald KG, McDonough JS, Newberry RD. Isolated lymphoid follicle formation is inducible and dependent upon lymphotoxin-sufficient B lymphocytes, lymphotoxin receptor, and TNF receptor I function. J Immunol. (2003) 170:5475–82. doi: 10.4049/jimmunol.170.11.5475
110. McDonald KG, McDonough JS, Wang C, Kucharzik T, Williams IR, Newberry RD. CC chemokine receptor 6 expression by B lymphocytes is essential for the development of isolated lymphoid follicles. Am J Pathol. (2007) 170:1229–40. doi: 10.2353/ajpath.2007.060817
111. Macpherson AJ, Köller Y, McCoy KD. The bilateral responsiveness between intestinal microbes and IgA. Trends Immunol. (2015) 36:460–70. doi: 10.1016/j.it.2015.06.006
112. Kubinak JL, Round JL. Do antibodies select a healthy microbiota? Nat Rev Immunol. (2016) 16:767–74. doi: 10.1038/nri.2016.114
113. Tsuji M, Suzuki K, Kitamura H, Maruya M, Kinoshita K, Ivanov II, et al. Requirement for lymphoid tissue-inducer cells in isolated follicle formation and T cell-independent immunoglobulin a generation in the gut. Immunity. (2008) 29:261–71. doi: 10.1016/j.immuni.2008.05.014
114. Bouskra D, Brézillon C, Bérard M, Werts C, Varona R, Boneca IG, et al. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature. (2008) 456:507–10. doi: 10.1038/nature07450
115. Pabst O, Herbrand H, Friedrichsen M, Velaga S, Dorsch M, Berhardt G, et al. Adaptation of solitary intestinal lymphoid tissue in response to microbiota and chemokine receptor CCR7 signaling. J Immunol. (2006) 177:6824–32. doi: 10.4049/jimmunol.177.10.6824
116. Lochner M, Ohnmacht C, Presley L, Bruhns P, Si-Tahar M, Sawa S, et al. Microbiota-induced tertiary lymphoid tissues aggravate inflammatory disease in the absence of RORγt and LTi cells. J Exp Med. (2011) 208:125–34. doi: 10.1084/jem.20100052
117. Rodda LB, Lu E, Bennett ML, Sokol CL, Wang X, Luther SA, et al. Single-cell RNA sequencing of lymph node stromal cells reveals niche-associated heterogeneity. Immunity. (2018) 48:1014–28.e6. doi: 10.1016/j.immuni.2018.04.006
118. Chu C, Moriyama S, Li Z, Zhou L, Flamar AL, Klose CSN, et al. Anti-microbial functions of group 3 innate lymphoid cells in gut-associated lymphoid tissues are regulated by G-protein-coupled receptor 183. Cell Rep. (2018) 23:3750–8. doi: 10.1016/j.celrep.2018.05.099
119. Emgård J, Kammoun H, García-Cassani B, Chesné J, Parigi SM, Jacob JM, et al. Oxysterol sensing through the receptor GPR183 promotes the lymphoid-tissue-inducing function of innate lymphoid cells and colonic inflammation. Immunity. (2018) 48:120–132.e8. doi: 10.1016/j.immuni.2017.11.020
120. Melo-Gonzalez F, Kammoun H, Evren E, Dutton EE, Papadopoulou M, Bradford BM, et al. Antigen-presenting ILC3 regulate T cell-dependent IgA responses to colonic mucosal bacteria. J Exp Med. (2019) 216:728–42. doi: 10.1084/jem.20180871
121. Yi T, Cyster JG. EBI2-mediated bridging channel positioning supports splenic dendritic cell homeostasis and particulate antigen capture. Elife. (2013) 2:e00757. doi: 10.7554/eLife.00757
122. Yi T, Wang X, Kelly LM, An J, Xu Y, Sailer AW, et al. Oxysterol gradient generation by lymphoid stromal cells guides activated B cell movement during humoral responses. Immunity. (2012) 37:535–48. doi: 10.1016/j.immuni.2012.06.015
123. Kelly LM, Pereira JP, Yi T, Xu Y, Cyster JG. EBI2 guides serial movements of activated B cells and ligand activity is detectable in lymphoid and non-lymphoid tissues. J Immunol. (2011) 187:3026–32. doi: 10.4049/jimmunol.1101262
124. Lu E, Dang EV, McDonald JG, Cyster JG. Distinct oxysterol requirements for positioning naïve and activated dendritic cells in the spleen. Sci Immunol. (2017) 2:eaal5237. doi: 10.1126/sciimmunol.aal5237
125. Li J, Lu E, Yi T, Cyster JG. EBI2 augments Tfh cell fate by promoting interaction with IL-2-quenching dendritic cells. Nature. (2016) 533:110–4. doi: 10.1038/nature17947
126. Sonnenberg GF, Hepworth MR. Functional interactions between innate lymphoid cells and adaptive immunity. Nat Rev Immunol. (2019) 19:599–613. doi: 10.1038/s41577-019-0194-8
127. Mackley EC, Houston S, Marriott CL, Halford EE, Lucas B, Cerovic V, et al. CCR7-dependent trafficking of RORγ+ ILCs creates a unique microenvironment within mucosal draining lymph nodes. Nat Commun. (2015) 6:5862. doi: 10.1038/ncomms6862
128. Kruglov AA, Grivennikov SI, Kuprash DV, Winsauer C, Prepens S, Seleznik GM, et al. Non-redundant function of soluble LTα3 produced by innate lymphoid cells in intestinal homeostasis. Science. (2013) 342:1243–6. doi: 10.1126/science.1243364
129. Reboldi A, Arnon TI, Rodda LB, Atakilit A, Sheppard D, Cyster JG. Mucosal immunology: IgA production requires B cell interaction with subepithelial dendritic cells in Peyer's patches. Science. (2016) 352:aaf4822. doi: 10.1126/science.aaf4822
130. Magri G, Miyajima M, Bascones S, Mortha A, Puga I, Cassis L, et al. Innate lymphoid cells integrate stromal and immunological signals to enhance antibody production by splenic marginal zone B cells. Nat Immunol. (2014) 15:354–64. doi: 10.1038/ni.2830
131. Sano T, Huang W, Hall JA, Yang Y, Chen A, Gavzy SJ, et al. An IL-23R/IL-22 circuit regulates epithelial serum amyloid a to promote local effector Th17 responses. Cell. (2015) 163:381–93. doi: 10.1016/j.cell.2015.08.061
132. Qiu J, Guo X, Chen Z, He L, Sonnenberg GF, Artis D, et al. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity. (2013) 39:386–99. doi: 10.1016/j.immuni.2013.08.002
133. Korn LL, Thomas HL, Hubbeling HG, Spencer SP, Sinha R, Simkins HM, et al. Conventional CD4+ T cells regulate IL-22-producing intestinal innate lymphoid cells. Mucosal Immunol. (2014) 7:1045–57. doi: 10.1038/mi.2013.121
134. Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y, et al. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science. (2014) 343:1249288. doi: 10.1126/science.1249288
135. Zhou L, Chu C, Teng F, Bessman NJ, Goc J, Santosa EK, et al. Innate lymphoid cells support regulatory T cells in the intestine through interleukin-2. Nature. (2019) 568:405–9. doi: 10.1038/s41586-019-1082-x
136. Bauché D, Joyce-Shaikh B, Jain R, Grein J, Ku KS, Blumenschein WM, et al. LAG3 + regulatory T cells restrain interleukin-23-producing CX3CR1 + gut-resident macrophages during group 3 innate lymphoid cell-driven colitis. Immunity. (2018) 49:342–52.e5. doi: 10.1016/j.immuni.2018.07.007
137. Hepworth MR, Monticelli LA, Fung TC, Ziegler CGK, Grunberg S, Sinha R, et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature. (2013) 498:113–7. doi: 10.1038/nature12240
138. Hepworth MR, Fung TC, Masur SH, Kelsen JR, McConnell FM, Dubrot J, et al. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4 + T cells. Science. (2015) 348:1031–5. doi: 10.1126/science.aaa4812
139. Goto Y, Panea C, Nakato G, Cebula A, Lee C, Diez MG, et al. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity. (2014) 40:594–607. doi: 10.1016/j.immuni.2014.03.005
140. von Burg N, Chappaz S, Baerenwaldt A, Horvath E, Bose Dasgupta S, Ashok D, et al. Activated group 3 innate lymphoid cells promote T-cell-mediated immune responses. Proc Natl Acad Sci USA. (2014) 111:12835–40. doi: 10.1073/pnas.1406908111
141. Saez de Guinoa J, Jimeno R, Farhadi N, Jervis PJ, Cox LR, Besra GS, et al. CD1d-mediated activation of group 3 innate lymphoid cells drives IL-22 production. EMBO Rep. (2017) 18:39–47. doi: 10.15252/embr.201642412
142. Withers DR, Jaensson E, Gaspal F, McConnell FM, Eksteen B, Anderson G, et al. The survival of memory CD4 + T cells within the gut lamina propria requires OX40 and CD30 signals. J Immunol. (2009) 183:5079–84. doi: 10.4049/jimmunol.0901514
143. Withers DR, Gaspal FM, Bekiaris V, McConnell FM, Kim M, Anderson G, et al. OX40 and CD30 signals in CD4 + T-cell effector and memory function: a distinct role for lymphoid tissue inducer cells in maintaining CD4 + T-cell memory but not effector function. Immunol Rev. (2011) 244:134–48. doi: 10.1111/j.1600-065X.2011.01057.x
144. Castellanos JG, Woo V, Viladomiu M, Putzel G, Lima S, Diehl GE, et al. Microbiota-induced TNF-like ligand 1A drives group 3 innate lymphoid cell-mediated barrier protection and intestinal T cell activation during colitis. Immunity. (2018) 49:1077–89.e5. doi: 10.1016/j.immuni.2018.10.014
145. Vacca P, Pesce S, Greppi M, Fulcheri E, Munari E, Olive D, et al. PD-1 is expressed by and regulates human group 3 innate lymphoid cells in human decidua. Mucosal Immunol. (2019) 12:624–31. doi: 10.1038/s41385-019-0141-9
146. Mariotti FR, Quatrini L, Munari E, Vacca P, Moretta L. Innate lymphoid cells: expression of PD-1 and other checkpoints in normal and pathological conditions. Front Immunol. (2019) 10:910. doi: 10.3389/fimmu.2019.00910
147. Weichhart T, Hengstschläger M, Linke M. Regulation of innate immune cell function by mTOR. Nat Rev Immunol. (2015) 15:599–614. doi: 10.1038/nri3901
148. O'Neill LAJ, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. (2016) 213:15–23. doi: 10.1083/jcb.2121OIA306
149. Di Luccia B, Gilfillan S, Cella M, Colonna M, Huang SC-C. ILC3s integrate glycolysis and mitochondrial production of reactive oxygen species to fulfill activation demands. J Exp Med. (2019) 216:2231–41. doi: 10.1084/jem.20180549
150. Eken A, Singh AK, Treuting PM, Oukka M. IL-23R+ innate lymphoid cells induce colitis via interleukin-22-dependent mechanism. Mucosal Immunol. (2014) 7:143–54. doi: 10.1038/mi.2013.33
Keywords: innate lymphoid cells, ILC, mucosal immunology, neuroimmune, circadian, immune circuits
Citation: Domingues RG and Hepworth MR (2020) Immunoregulatory Sensory Circuits in Group 3 Innate Lymphoid Cell (ILC3) Function and Tissue Homeostasis. Front. Immunol. 11:116. doi: 10.3389/fimmu.2020.00116
Received: 12 November 2019; Accepted: 16 January 2020;
Published: 06 February 2020.
Edited by:
Arthur Mortha, University of Toronto, CanadaReviewed by:
Jennifer Bando, Washington University in St. Louis, United StatesCopyright © 2020 Domingues and Hepworth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew R. Hepworth, bWF0dGhldy5oZXB3b3J0aEBtYW5jaGVzdGVyLmFjLnVr
†ORCID: Rita G. Domingues orcid.org/0000-0001-8571-1132
Matthew R. Hepworth orcid.org/0000-0002-9613-7858
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.