 Lokesh Sharma
Lokesh Sharma Jingjing Feng1,2
Jingjing Feng1,2 Charles S. Dela Cruz
Charles S. Dela Cruz
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 11 February 2020
Sec. Microbial Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.00091
This article is part of the Research TopicOutsmarting the Host: How Bacterial Pathogens Modulate Immune Responses in The LungView all 19 articles
Bacterial lung infections are major healthcare challenges killing millions of people worldwide and resulting in a huge economic burden. Both basic and clinical research have elucidated host mechanisms that contribute to the bacterial clearance where an indispensable role of immune cells has been established. However, the role of respiratory epithelial cells in bacterial clearance has garnered limited attention due to their weak inflammatory or phagocytic ability compared to immune cells such as macrophages and neutrophils. These studies often underappreciate the fact that epithelial cells are the most abundant cells in the lung, not only serving as building blocks but also providing immune protection throughout the lung. Epithelial cells function either independently to eradicate the pathogen or communicate with immune cells to orchestrate pathogen clearance. The epithelial cells have multiple mechanisms that include mucus production, antimicrobial peptide production, muco-ciliary clearance, and phagocytosis, all of which contribute to their direct antibacterial function. Secretion of cytokines to recruit immune cells and potentiate their antimicrobial activities is a pathway by which the epithelium contributes to bacterial clearance. Successful pathogens outsmart epithelial resistance and find a way to replicate in sufficient numbers to establish infections in the airway or lung epithelial surfaces. In this mini-review, we discuss evidences that establish important roles for epithelial host defense against invading respiratory bacterial pathogens and demonstrate how pathogens outsmart these epithelial immune mechanisms to successfully establish infection. Finally, we discuss briefly how to boost epithelial immunity to improve outcomes in bacterial lung infections.
The lung surface area of more than 50 m2 consists of epithelial monolayers which cover the upper and lower airways as well as lung alveoli (1). The large epithelial surfaces are continuously exposed to particles, pollutants, and microbes. It is not surprising that the lung epithelium has a well-developed immunity to fend off these microbes. Different subsets of lung epithelium are equipped with their unique antimicrobial mechanisms (2–4). While nasal epithelium is known to secrete large amounts of antimicrobial peptides (AMPs) and type II pneumocytes secrete surfactant proteins, both of these are host protective mechanisms against bacterial infections. Ciliated airway epithelium mechanically removes invading pathogens with the help of mucus secreted by the secretory epithelium. If the epithelium fails to limit the invading pathogens itself, the epithelium signals to seek the help of resident immune cells such as alveolar macrophages or to recruit immune cells to help eradicate the invading pathogen, which often comes at a cost of tissue injury, including to the epithelium (5, 6). Lung epithelium also contributes to the removal of dead inflammatory cells to ensure restoration of lost lung function post infection. However, pathogens have developed multiple mechanisms to avoid various epithelial resistance mechanisms. Respiratory pathogens discussed here include clinically relevant pathogens such as Pseudomonas aeruginosa (infects patients with chronic lung diseases such as cystic fibrosis and COPD) (7), Streptococcus pneumoniae (affects healthy people and causes post-viral bacterial infections) (8), Staphylococcus aureus (major cause of hospital-acquired pneumonia) (9), and Klebsiella pneumoniae (opportunistic pathogen with high antimicrobial resistant) (10).
Antimicrobial functions of the lung epithelium can be divided into two main categories: direct killing and indirect control of lung infections.
Killing of the invading pathogen is the most effective strategy to remove the invading pathogen, however the primary aim of the epithelium is to survive the infection (Figure 1). To avoid unnecessary inflammatory damage, which may be unavoidable consequence of the pathogen killing, the host epithelium tolerates commensal microbes or colonizing pathogens, which are present at higher density especially in the upper respiratory tract (11). However, an appropriate level of active epithelial immunity is required to maintain the colonization status of these pathogens. Any impairment in the epithelial immunity can lead to infection from colonizing pathogens, as seen during influenza infection, where nasally colonized Streptococcus invades and establishes a lower respiratory tract infection (12). Multiple mechanisms have been attributed to influenza enhanced susceptibility including a damaged epithelial barrier, increased micronutrition due to cell death and decreased surfactant proteins (13). Other colonizing pathogens such as Staphylococcus aureus show similar infecting mechanisms post influenza, where colonizing pathogens are often associated with the disease. The detailed mechanisms of colonization are discussed in other reviews (14). The epithelium has multiple tools to limit the growth of pathogens or to remove them completely from the host.
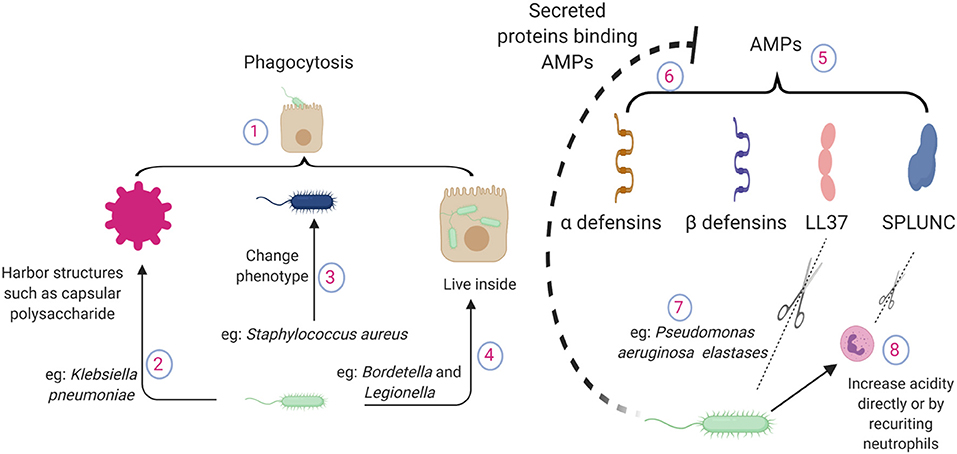
Figure 1. Pathogen mechanisms to circumvent direct killing of bacteria by lung epithelial cells. (1) Epithelial cells possess phagocytic ability. (2) Bacteria harbor structures to evade epithelial phagocytosis, such as capsular polysaccharides of Klebsiella pneumoniae. (3) Altered phenotypes in pathogens cause chronic infections e.g., small colony variant of Staphylococcus aureus. (4) Pathogens can live inside the lung epithelium e.g., Bordetella or Legionella. (5) Epithelium releases antimicrobial peptides during lung infections, which bind to the pathogen surface and neutralize the pathogen. (6) During inflammation, a large number of proteins are accumulated in the lung. AMPs interact with other proteins and lose their antimicrobial potency. (7) P. aeruginosa elastase breaks down human LL37 in a dose dependent manner. (8) Pathogen recruited neutrophils contribute to the acidity. SPLUNC loses its function in an acidic environment; also, neutrophil elastases can break down SPLUNC. AMPs, antimicrobial proteins and peptides; SPLUNC, short palate lung and nasal epithelium clone 1.
Phagocytosis is the main mechanism of bacterial killing used by professional phagocytes such as macrophages and neutrophils. However, epithelial cells also possess significant phagocytic ability, which is often referred to as “internalization” against a wide range of bacterial pathogens (15, 16). There are major differences in the capacity and mechanisms of phagocytosis among the professional phagocytes and epithelial cells. Professional phagocytes like macrophages actively phagocytose the pathogens by recognizing them using its pattern recognition receptors, where antibody mediated opsonization plays a major role in the process (17). On the other hand, epithelial phagocytosis is relatively low and often initiated by the pathogen. Pathogens use two distinct mechanisms to enter the epithelial cells: “zipper” and “trigger” mechanisms (18). In the “zipper” mechanism, pathogen attachment induces rearrangement of actin cytoskeleton and the resultant internalization. In the “trigger” method, the pathogen secretes soluble factors that cause actin fiber rearrangement to form a phagocytic cup and subsequent internalization of the pathogen. However, the epithelium expresses various pattern recognition receptors that actively recognize the invading pathogen and allow them to respond to the pathogen by releasing cytokines and chemokines. A wide range of TLRs and nod like receptors have been detected on lung epithelium on both the surfaces as well as within the cell (19–21). Although epithelial cells have lower phagocytic activity per cell compared to the professional phagocytes, given the number of epithelial cells in the lung compartment and the surface area covered by these cells, their phagocytic activity provides significant contribution to pathogen clearance.
Phagocytosis allows digestion of the bacterial components within the cell and thus limits inflammatory signaling arising from extracellular bacterial killing. Antibiotic induced bacterial killing has been shown to be highly inflammatory (22). Phagocytic ability of the epithelium also helps maintain the homeostatic lung environment, which is disrupted in the aftermath of an infectious injury by an abundance of dead cells and cellular debris from various cells, including the epithelium. Epithelial phagocytosis of apoptotic cells is dependent on small GTPase Rac-1, which allows the epithelium to secrete anti-inflammatory cytokines upon phagocytosis of cell debris to inhibit inflammatory recruitment and restore homeostasis (23).
Epithelial-mediated phagocytosis is dependent on various host and pathogen factors. Pathogens often harbor structures on their surface to evade epithelial phagocytosis as seen in Klebsiella, where the amount of capsular polysaccharide is inversely related to epithelial internalization. As expected, bacterial strains lacking capsular polysaccharides are effectively cleared by the lung epithelium (24). Pseudomonas forms multicellular complexes known as biofilms, which are enormous in size, to prevent phagocytosis (25, 26). Avoiding epithelial phagocytosis may even provide Pseudomonas an advantage by dysregulating the inflammatory response and causing systemic sepsis that is detrimental to the host but supports bacterial growth (27). Some pathogens have developed ways to reside within the epithelium, such as Bordetella which uses host tubulin activity to enter the epithelial cells. This entry provides protection not only against antibiotic treatment but also against host antimicrobial peptides (28). Pathogens also avoid fusion with acidic lysosomes, which are filled with enzymes that can lyse the bacterial cell wall by manipulating cellular trafficking. Similarly, highly pathogenic strains of Legionella reside and replicate in the lung epithelium in addition to replicating within macrophages, which are normal targets of Legionella (29). Pathogens also alter their phenotype to adjust themselves within the epithelium to cause chronic infections. A small colony variant of Staphylococcus aureus often persists intracellularly including in the epithelium due to its ability to exert minimal inflammatory response compared to wild type strains (30). These examples demonstrate a wide array of pathogen mechanisms to outsmart epithelial phagocytosis, which contributes to the disease pathogenesis.
The lung epithelium is a primary source of many AMPs released during lung infection. AMPs are charged peptides that can bind to the pathogen surface and kill them. These AMPs are separate from complement pathways, which primarily “tag” the pathogen to be identified by professional phagocytes, although some evidences of direct killing of pathogens by complements has been reported (31).
Defensins are a group of AMPs, of which β-defensins are almost exclusively secreted by the lung epithelium (32). Inflammatory cytokines produced during infection can lead to the release of AMPs (33, 34). The role of AMPs has been well established whereby they kill a wide range of pathogens including E. coli, Pseudomonas and Haemophilus influenzae in a salt dependent manner (34, 35). These antibacterial effects are confirmed in a mouse model of H. influenzae, where β-defensin deficiency impaired bacterial clearance (36). Over-expression of β-defensin 2 in a porcine model has been shown to increase the clearance of Actinobacillus pleuropneumoniae (37).
Cathelicidins, another important epithelial AMPs, are cleaved into their mature form LL37. Unlike β-defensins, only a single cathelicidin peptide has been found in the lungs of various species, including humans. The positively charged LL37 is secreted by lung epithelial cells (38) and has bactericidal activity against a variety of lung pathogens such as P. aeruginosa and S. aureus (39, 40). Besides direct antibacterial activity, LL37 also promotes bacterial phagocytosis in macrophages (41), indicating AMPs serve as important communicators between the epithelial and immune cells.
More recently, short palate lung and nasal epithelium clone 1 (SPLUNC) has been identified as an important AMP expressed in the upper airways and has a binding affinity for lipopolysaccharides (LPS) (42). SPLUNC has been shown to protect mice from infection with respiratory pathogens such as P. aeruginosa and Mycoplasma (43, 44). Furthermore, this study demonstrated that there might be a positive feedback mechanism between various AMPs since deficiency of SPLUNC1 led to decreased expression in LL37.
Successful pathogens often develop strategies to overcome AMP-mediated host defense. These strategies include cleavage of AMPs by secreted proteases such as aureolysin and V8 protease from S. aureus (45). P. aeruginosa elastase can effectively break down LL37 (46) and SPLUNC (47). Given the wide substrate range, it is very likely that various AMPs are susceptible to host neutrophil elastase mediated cleavage and subsequent loss of antibacterial activity, as neutrophil elastase levels are significantly increased during bacterial infections (48).
During inflammation, a large number of host and pathogen proteins/products are accumulated in the lung airspace. Given the charged nature of AMPs, they easily interact with other charged molecules and lose their antimicrobial potency (49). Pathogens also modulate the lung environment to decrease the toxicity of AMPs. For example, the antimicrobial properties of SPLUNC are lost in the airways in cystic fibrosis due to its acidic environment. Pathogens can directly create an acidic environment by changing their metabolic activity or by recruiting metabolically active immune cells such as neutrophils (50).
Pathogens also shed components that actively bind to AMPs and prevent their binding to the bacterial surface to avoid AMP mediated toxicity. LPS, a part of the bacterial outer membrane in gram negative pathogens, are shed during infection. β-defensin binds to LPS and blocks its signaling. At the same time, LPS effectively block the antimicrobial activity of AMPs by direct binding (51). The capsule polysaccharide from K. pneumoniae has been shown to act as a bacterial decoy for AMPs secreted by the host to abrogate antimicrobial capacity (52).
Despite having well-developed mechanisms to prevent bacterial invasion, the epithelium often fails to completely eradicate the pathogens. In these situations, the epithelium often collaborates with other cells to ensure proper removal of invading pathogens. In cases where removing the pathogen completely seems impossible, the epithelium ensures that the infection remains localized and does not spread systemically. To perform these indirect functions, the epithelium communicates with other cells either by direct contact or by releasing soluble factors like cytokines and chemokines. Mechanisms of indirect pathogens control include the following: (see Figure 2).
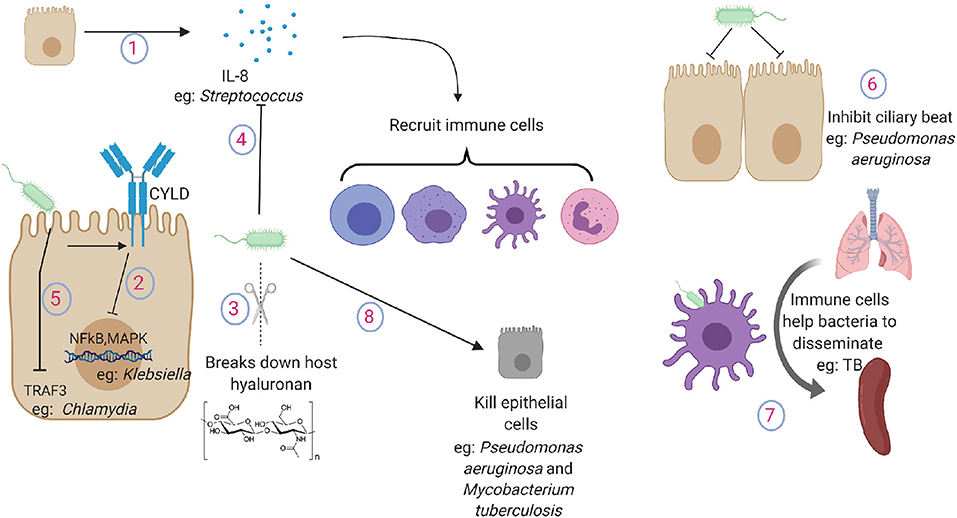
Figure 2. Pathogen tactics to avoid indirect killing during lung infections. (1) Lung epithelium secrete cytokines and chemokines to recruit immune cells in the lung. (2) Klebsiella inhibits the activation of NFkB and MAPK pathway by promoting the expression of ubiquitinating enzyme CYLD in lung epithelial cells. (3) Gram positive pathogens break down host hyaluronan. (4) Streptococcus cleaves and inactivates IL-8. (5) Chlamydia targets TRAF3 in the epithelial cells. (6) Pseudomonas aeruginosa inhibits ciliary beat function. (7) Macrophages and dendritic cells carry TB from the lung to secondary lymphoid organs. (8) Pathogens induce damage to lung cells or to the extracellular matrix. IL-8, interleukin-8; NFkB, nuclear factor kappa B; MAPK, mitogen activated protein kinase; TRAF3, TNF receptor-associated factor 3.
If the lung epithelium fails to control the growth of invading pathogens, it recruits immune cells to participate in the bacterial removal process. To sense pathogen, pattern recognition receptors are expressed on both apical and basolateral surfaces of the epithelium (20). Upon recognition of the pathogens, these cells secrete cytokines and chemokines to increase the expression of complementary adhesion molecules on the vascular endothelium and immune cells. The epithelium itself also expresses adhesion molecules such as ICAM-1 and VCAM1 (53) to facilitate the recruitment of immune cells in the lung. Epithelial-macrophage interactions can further amplify the secretion of cytokines to mount a robust inflammatory response; for example, epithelial release of CXCL8 can be enhanced by incubation with macrophages or macrophage supernatants (54). Upon recruitment, immune cells eliminate the pathogens by phagocytosis or extracellular killing by neutrophils and macrophage extracellular traps (55–57). Immune cells are highly effective in pathogen clearance but may cause lung injury by proteolytic enzymes contained within the immune cells. Upon pathogen clearance, the epithelium also contributes to clearance of the infiltrated immune cells. Conventionally, macrophages have been credited to clear the dead cells present in the lung post inflammatory response; however, recent studies have also implicated important roles of the lung epithelium in clearing the dead cells and cellular debris from the lung and preventing persistent inflammation (58). Blocking epithelial phagocytosis by inducible mutation of small GTPase Rac1 leads to persistent lung inflammation (23).
As inflammation is essential for effective clearance of invading bacterial pathogens, the pathogens can evade this inflammation by either suppressing the inflammatory response or by causing excessive inflammatory response. K. pneumoniae lung infections often induce very limited early inflammatory response, and it successfully disseminates across organs before inducing a robust inflammatory response (59). Klebsiella inhibits the activation of NFkB and MAPK pathway by promoting the expression of ubiquitinating enzyme CYLD in lung epithelial cells (60). Streptococcus secretes hyaluronidases that break down host hyaluronan into small components that inhibit not only inflammatory host hyaluronan fragments but also other TLR ligands such as LPS (61). Epithelial cells can recognize the pathogens and secrete IL-8, which is a potent chemokine in the lungs (62). Streptococcus species that cause aggressive disseminating disease have developed mechanisms by which they can cleave and inactivate IL-8 to blunt the inflammatory response through cell envelope proteinase (63). Chlamydia pneumoniae manipulates epithelial immunity by targeting TRAF3 in the epithelial cells and thus impairing TRAF3 mediated immunity (64). In contrast, some pathogens can use the presence of inflammatory cytokines as growth factors and survive excessive inflammation (65).
The lung epithelium is a continuous barrier between the environment and circulation. This barrier ensures that pathogens in the lung do not achieve access to the circulation. Pathogens often strive to reach the blood circulation, likely due to the high amount of nutrients present in the blood. It is well documented that systemic spread of lung infection results in higher mortality compared to localized lung infection (66, 67).
The tight junctions between the lung epithelium are maintained by various junction proteins such as occludin family members and ZO-1 (68). Various enhancements in the lung epithelium increases the effectiveness of the barrier. These include muco-ciliary clearance in the upper airways (69) and surfactant proteins secreted by type II pneumocytes, which can promote bacterial killing including Klebsiella (70). Together these physical and biochemical structures ensure that lung infections do not extend outside the lung compartment.
Ciliary beat function serves as one of the first physical defenses to prevent the pathogen from reaching the lung and subsequently causing systemic infection. It has been shown that virulence factors from P. aeruginosa can significantly inhibit ciliary beat, possibly allowing the bacteria to stick to the lung epithelium (71). Despite maintaining the uniform monolayer with sealed tight junctions, the epithelium provides regulated passage to immune cells such as those recruited during infections. This passage is provided by expression of complementary adhesion molecules on both immune and epithelial cells (72). Pathogens often take advantage of this host mechanism to spread by hiding within the immune cells. Antigen presenting cells such as macrophages and dendritic cells are well known to sense the pathogen and leave the lung compartment to present the antigen to lymphoid organs. However, recent evidence suggests that neutrophils also leave the lung and return to the bone marrow through the circulation once the inflammation is resolved (73). Macrophages and dendritic cells are believed to carry Mycobacteria tuberculosis from the lung to secondary lymphoid organs such as spleen (74). Other mechanisms of bacterial dissemination across the lung epithelium such as “ejectosomes” observed in amoeba have not yet been described in mammals (75). Pathogens also use TLR pathways to downregulate expression of tight junction protein claudins 7 and 10, which are essential to maintain intercellular junctions (76). Inhibition of claudins allows the pathogen to escape the lung by increasing epithelial permeability and causing systemic disease.
Various pathogens also induce damage to the lung structure by either directly killing epithelial cells or by digesting the extracellular matrix. These effects can be directly caused by pathogens or by pathogen induced host responses (77). P. aeruginosa has been shown to inject Exos, a potent toxin, using its type III secretion system to induce localized epithelial cell death to facilitate bacterial dissemination (78). Pore forming toxins secreted by a wide range of pathogens have also been shown to cause necroptotic cell death in the lung epithelium (79). Damage to the lung epithelium by other exposures such as hyperoxia or exposure to environmental particles increases the bacterial dissemination for P. aeruginosa (80). M. tuberculosis can directly cause epithelial death by secreting virulence factors such as ESAT6, leading to its dissemination (81).
Antibiotics remain the current first line therapy for bacterial infections, but their efficacy is being challenged with current rising of antimicrobial resistance (82). Given the urgency, it is essential to develop alternatives to treat lung infections. As the epithelium provides important host defense, it is logical to boost their function to improve outcomes during infections. Boosting the epithelial host defense can provide an important immunomodulatory strategy, especially in patients who have either a limited number of immune cells (genetic or acquired immunodeficiency, chemotherapy), a defect in immunity derived particularly from defects in the lung epithelium (such as CF), a recipient of lung transplant (where immunosuppression is required to prevent lung rejection) or those with extremes of the age. Boosting epithelial immunity can not only prevent the collateral damage that can be caused by recruited immune cells but also circumvent the need to remove the immune cells from tissue once the invading bacteria is cleared.
A large number of mouse studies have demonstrated the usefulness of promoting epithelial resistance to help the host clear the invading pathogens. These strategies involve stimulation with TLR ligands. These include single reagents such as flagellin (for TLR5) (83), poly I:C (84), aminoalkyl glucosaminide phosphate (synthetic TLR4 ligand) (85), CpG ODN (TLR 9 ligand) (86) or a combination of two or more ligands such as Pam2CSK4 + ODNM62 (87). Possible mechanisms of this trained immunity include early recruitment of immune cells (88) and promoting epithelial resistance by epigenetic changes (89) or promoting reactive oxygen species production (90).
Although these strategies have been highly successful in the mouse models, their use in susceptible human populations still needs to be confirmed. Major challenges in their success remain to be their tolerability in subjects and their provision of sufficient protection in the lungs against these infections.
In summary, the epithelium provides an array of host defense mechanisms to prevent and control bacterial lung infections. However, pathogens often circumvent these mechanisms to establish successful infections. Our aim should be to improve the epithelial host defense to boost host immunity and improve survival in lung infections.
LS drafted the manuscript. JF prepared the figures. CB Provided critical comments. CD revised the manuscript and provided critical comments.
This project was supported by funding from the American Thoracic Society through the Unrestricted Critical Care award (to LS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Hasleton PS. The internal surface area of the adult human lung. J Anat. (1972) 112(Pt 3):391–400.
2. Knowles MR, Boucher RC. Mucus clearance as a primary innate defense mechanism for mammalian airways. J Clin Invest. (2002) 109:571–7. doi: 10.1172/JCI0215217
3. Mason RJ. Biology of alveolar type II cells. Respirology. (2006) 11:S12–5. doi: 10.1111/j.1440-1843.2006.00800.x
4. Leiva-Juárez MM, Kolls JK, Evans SE. Lung epithelial cells: therapeutically inducible effectors of antimicrobial defense. Mucosal Immunol. (2018) 11:21–34. doi: 10.1038/mi.2017.71
5. Moldoveanu B, Otmishi P, Jani P, Walker J, Sarmiento X, Guardiola J, Saad M, Yu J. Inflammatory mechanisms in the lung. J Inflamm Res. (2009) 2:1−11.
6. Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med. (2011) 17:293. doi: 10.2119/molmed.2010.00138
7. Gellatly SL, Hancock RE. Pseudomonas aeruginosa: new insights into pathogenesis and host defenses. Pathog Dis. (2013) 67:159–73. doi: 10.1111/2049-632X.12033
8. van der Poll T, Opal SM. Pathogenesis, treatment, and prevention of pneumococcal pneumonia. Lancet. (2009) 374:1543–56. doi: 10.1016/S0140-6736(09)61114-4
9. Jones RN. Microbial etiologies of hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia. Clin Infect Dis. (2010) 51(Suppl_1):S81–7. doi: 10.1086/653053
10. Munoz-Price LS, Poirel L, Bonomo RA, Schwaber MJ, Daikos GL, Cormican M, et al. Clinical epidemiology of the global expansion of Klebsiella pneumoniae carbapenemases. Lancet Infect Dis. (2013) 13:785–96. doi: 10.1016/S1473-3099(13)70190-7
11. Kirst ME, Baker D, Li E, Abu-Hasan M, Wang GP. Upper versus lower airway microbiome and metagenome in children with cystic fibrosis and their correlation with lung inflammation. PLoS ONE. (2019) 14:e0222323. doi: 10.1371/journal.pone.0222323
12. Small CL, Shaler CR, McCormick S, Jeyanathan M, Damjanovic D, Brown EG, et al. Influenza infection leads to increased susceptibility to subsequent bacterial superinfection by impairing NK cell responses in the lung. J Immunol. (2010) 184:2048–56. doi: 10.4049/jimmunol.0902772
13. McCullers JA. The co-pathogenesis of influenza viruses with bacteria in the lung. Nat Rev Microbiol. (2014) 12:252. doi: 10.1038/nrmicro3231
14. Siegel SJ, Weiser JN. Mechanisms of bacterial colonization of the respiratory tract. Annu Rev Microbiol. (2015) 69:425–44. doi: 10.1146/annurev-micro-091014-104209
15. Plotkowski MC, de Bentzmann S, Pereira SHM, Zahm J, Bajolet-Laudinat O, Roger P, et al. Pseudomonas aeruginosa internalization by human epithelial respiratory cells depends on cell differentiation, polarity, and junctional complex integrity. Am J Respir Cell Mol Biol. (1999) 20:880–90. doi: 10.1165/ajrcmb.20.5.3408
16. Fumagalli O, Tall BD, Schipper C, Oelschlaeger TA. N-glycosylated proteins are involved in efficient internalization of Klebsiella pneumoniae by cultured human epithelial cells. Infect Immun. (1997) 65:4445–51. doi: 10.1128/IAI.65.11.4445-4451.1997
17. Mosser DM, Zhang X. Measuring opsonic phagocytosis via fcγ receptors and complement receptors on macrophages. Curr Protoc Immunol. (2011) 95:14.27. doi: 10.1002/0471142735.im1427s95
18. Günther J, Seyfert H. The first line of defence: insights into mechanisms and relevance of phagocytosis in epithelial cells. Semin Immunopathol. (2018) 40:555–65. doi: 10.1007/s00281-018-0701-1
19. Fukata M, Vamadevan AS, Abreu MT. Toll-like receptors (TLRs) and nod-like receptors (NLRs) in inflammatory disorders. Semin Immunol. (2009) 21:242–53. doi: 10.1016/j.smim.2009.06.005
20. Ioannidis I, Ye F, McNally B, Willette M, Flano E. Toll-like receptor expression and induction of type I and type III interferons in primary airway epithelial cells. J Virol. (2013) 87:3261–70. doi: 10.1128/JVI.01956-12
21. Ritter M, Mennerich D, Weith A, Seither P. Characterization of toll-like receptors in primary lung epithelial cells: strong impact of the TLR3 ligand poly (I: C) on the regulation of toll-like receptors, adaptor proteins and inflammatory response. J Inflamm. (2005) 2:16. doi: 10.1186/1476-9255-2-16
22. Silverstein R, Wood JG, Xue Q, Norimatsu M, Horn DL, Morrison DC. Differential host inflammatory responses to viable versus antibiotic-killed bacteria in experimental microbial sepsis. Infect Immun. (2000) 68:2301–8. doi: 10.1128/IAI.68.4.2301-2308.2000
23. Juncadella IJ, Kadl A, Sharma AK, Shim YM, Hochreiter-Hufford A, Borish L, et al. Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nature. (2013) 493:547. doi: 10.1038/nature11714
24. Cortes G, Alvarez D, Saus C, Alberti S. Role of lung epithelial cells in defense against Klebsiella pneumoniae pneumonia. Infect Immun. (2002) 70:1075–80. doi: 10.1128/IAI.70.3.1075-1080.2002
25. Høiby N, Ciofu O, Bjarnsholt T. Pseudomonas aeruginosa biofilms in cystic fibrosis. Future Microbiol. (2010) 5:1663–74. doi: 10.2217/fmb.10.125
26. Murray TS, Egan M, Kazmierczak BI. Pseudomonas aeruginosa chronic colonization in cystic fibrosis patients. Curr Opin Pediatr. (2007) 19:83–8. doi: 10.1097/MOP.0b013e3280123a5d
27. Grassme H, Jendrossek V, Riehle A, Von Kürthy G, Berger J, Schwarz H, et al. Host defense against Pseudomonas aeruginosa requires ceramide-rich membrane rafts. Nat Med. (2003) 9:322. doi: 10.1038/nm823
28. Lamberti Y, Gorgojo J, Massillo C, Rodriguez ME. Bordetella pertussis entry into respiratory epithelial cells and intracellular survival. Pathog Dis. (2013) 69:194–204. doi: 10.1111/2049-632X.12072
29. Maruta K, Miyamoto H, Hamada T, Ogawa M, Taniguchi H, Yoshida S. Entry and intracellular growth of Legionella dumoffii in alveolar epithelial cells. Am J Respir Crit Care Med. (1998) 157:1967–74. doi: 10.1164/ajrccm.157.6.9710108
30. Ou JJ, Drilling AJ, Cooksley C, Bassiouni A, Kidd SP, Psaltis AJ, et al. Reduced innate immune response to a Staphylococcus aureus small colony variant compared to its wild-type parent strain. Front Cell Infect Microbiol. (2016) 6: 187. doi: 10.3389/fcimb.2016.00187
31. Nordahl EA, Rydengard V, Nyberg P, Nitsche DP, Morgelin M, Malmsten M, et al. Activation of the complement system generates antibacterial peptides. Proc Natl Acad Sci USA. (2004) 101:16879–84. doi: 10.1073/pnas.0406678101
32. Fellermann K, Stange EF. Defensins–innate immunity at the epithelial frontier. Eur J Gastroenterol Hepatol. (2001) 13:771–6. doi: 10.1097/00042737-200107000-00003
33. Scharf S, Zahlten J, Szymanski K, Hippenstiel S, Suttorp N, N'Guessan PD. Streptococcus pneumoniae induces human β-defensin-2 and-3 in human lung epithelium. Exp Lung Res. (2012) 38:100–10. doi: 10.3109/01902148.2011.652802
34. Singh PK, Jia HP, Wiles K, Hesselberth J, Liu L, Conway BA, et al. Production of beta-defensins by human airway epithelia. Proc Natl Acad Sci USA. (1998) 95:14961–6. doi: 10.1073/pnas.95.25.14961
35. Morrison G, Kilanowski F, Davidson D, Dorin J. Characterization of the mouse beta defensin 1, Defb1, mutant mouse model. Infect Immun. (2002) 70:3053–60. doi: 10.1128/IAI.70.6.3053-3060.2002
36. Moser C, Weiner DJ, Lysenko E, Bals R, Weiser JN, Wilson JM. Beta-defensin 1 contributes to pulmonary innate immunity in mice. Infect Immun. (2002) 70:3068–72. doi: 10.1128/IAI.70.6.3068-3072.2002
37. Yang X, Cheng YT, Tan MF, Zhang HW, Liu WQ, Zou G, et al. Overexpression of porcine beta-defensin 2 enhances resistance to actinobacillus pleuropneumoniae infection in pigs. Infect Immun. (2015) 83:2836–43. doi: 10.1128/IAI.03101-14
38. Rivas-Santiago B, Hernandez-Pando R, Carranza C, Juarez E, Contreras JL, Aguilar-Leon D, et al. Expression of cathelicidin LL-37 during mycobacterium tuberculosis infection in human alveolar macrophages, monocytes, neutrophils, and epithelial cells. Infect Immun. (2008) 76:935–41. doi: 10.1128/IAI.01218-07
39. Bals R, Wang X, Zasloff M, Wilson JM. The peptide antibiotic LL-37/hCAP-18 is expressed in epithelia of the human lung where it has broad antimicrobial activity at the airway surface. Proc Natl Acad Sci USA. (1998) 95:9541–6. doi: 10.1073/pnas.95.16.9541
40. Dürr UH, Sudheendra U, Ramamoorthy A. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim Biophys Acta. (2006) 1758:1408–25. doi: 10.1016/j.bbamem.2006.03.030
41. Wan M, van der Does Anne M, Tang X, Lindbom L, Agerberth B, et al. Antimicrobial peptide LL-37 promotes bacterial phagocytosis by human macrophages. J Leukoc Biol. (2014) 95:971–81. doi: 10.1189/jlb.0513304
42. Walton WG, Ahmad S, Little MS, Kim CS, Tyrrell J, Lin Q, et al. Structural features essential to the antimicrobial functions of human SPLUNC1. Biochemistry. (2016) 55:2979–91. doi: 10.1021/acs.biochem.6b00271
43. Gally F, Di YP, Smith SK, Minor MN, Liu Y, Bratton DL, et al. SPLUNC1 promotes lung innate defense against mycoplasma pneumoniae infection in mice. American J Pathol. (2011) 178:2159–67. doi: 10.1016/j.ajpath.2011.01.026
44. Lukinskiene L, Liu Y, Reynolds SD, Steele C, Stripp BR, Leikauf GD, et al. Antimicrobial activity of PLUNC protects against Pseudomonas aeruginosa infection. J Immunol. (2011) 187:382–90. doi: 10.4049/jimmunol.1001769
45. Sieprawska-Lupa M, Mydel P, Krawczyk K, Wojcik K, Puklo M, Lupa B, et al. Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob Agents Chemother. (2004) 48:4673–9. doi: 10.1128/AAC.48.12.4673-4679.2004
46. Schmidtchen A, Frick I, Andersson E, Tapper H, Björck L. Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol Microbiol. (2002) 46:157–68. doi: 10.1046/j.1365-2958.2002.03146.x
47. Jiang D, Wenzel SE, Wu Q, Bowler RP, Schnell C, Chu HW. Human neutrophil elastase degrades SPLUNC1 and impairs airway epithelial defense against bacteria. PLoS ONE. (2013) 8:e64689. doi: 10.1371/journal.pone.0064689
48. Domon H, Nagai K, Maekawa T, Oda M, Yonezawa D, Takeda W, et al. Neutrophil elastase subverts the immune response by cleaving toll-like receptors and cytokines in pneumococcal pneumonia. Front Immunol. (2018) 9:732. doi: 10.3389/fimmu.2018.00732
49. Burris D, Sharma L, Mantell L. Effects of bacterial endotoxins on the toxicity of extracellular histones to mammalian cells. FASEB J. (2015) 29(1_Suppl):718.3.
50. Ahmad S, Gilmore RC, Alexis NE, Tarran R. SPLUNC1 loses its antimicrobial activity in acidic cystic fibrosis airway secretions. Am J Respir Crit Care Med. (2019) 200:633–6. doi: 10.1164/rccm.201812-2303LE
51. Liu H, Yu H, Gu Y, Xin A, Zhang Y, Diao H, et al. Human beta-defensin DEFB126 is capable of inhibiting LPS-mediated inflammation. Appl Microbiol Biotechnol. (2013) 97:3395–408. doi: 10.1007/s00253-012-4588-9
52. Llobet E, Tomas JM, Bengoechea JA. Capsule polysaccharide is a bacterial decoy for antimicrobial peptides. Microbiology. (2008) 154:3877–86. doi: 10.1099/mic.0.2008/022301-0
53. Atsuta J, Sterbinsky SA, Plitt J, Schwiebert LM, Bochner BS, Schleimer RP. Phenotyping and cytokine regulation of the BEAS-2B human bronchial epithelial cell: demonstration of inducible expression of the adhesion molecules VCAM-1 and ICAM-1. Am J Respir Cell Mol Biol. (1997) 17:571–82. doi: 10.1165/ajrcmb.17.5.2685
54. Marriott HM, Gascoyne KA, Gowda R, Geary I, Nicklin MJ, Iannelli F, et al. Interleukin-1beta regulates CXCL8 release and influences disease outcome in response to Streptococcus pneumoniae, defining intercellular cooperation between pulmonary epithelial cells and macrophages. Infect Immun. (2012) 80:1140–9. doi: 10.1128/IAI.05697-11
55. Byrne AJ, Mathie SA, Gregory LG, Lloyd CM. Pulmonary macrophages: key players in the innate defence of the airways. Thorax. (2015) 70:1189–96. doi: 10.1136/thoraxjnl-2015-207020
56. Chow OA, von Köckritz-Blickwede M, Bright AT, Hensler ME, Zinkernagel AS, Cogen AL, et al. Statins enhance formation of phagocyte extracellular traps. Cell Host Microbe. (2010) 8:445–54. doi: 10.1016/j.chom.2010.10.005
57. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
58. Penberthy KK, Juncadella IJ, Ravichandran KS. Apoptosis and engulfment by bronchial epithelial cells. Implications for allergic airway inflammation. Ann Am Thorac Soc. (2014) 11(Suppl. 5):S259–62. doi: 10.1513/AnnalsATS.201405-200AW
59. Sharma L, Amick AK, Vasudevan S, Lee SW, Marion CR, Liu W, et al. Regulation and role of chitotriosidase during lung infection with Klebsiella pneumoniae. J Immunol. (2018) 201:615–26. doi: 10.4049/jimmunol.1701782
60. Regueiro V, Moranta D, Frank CG, Larrarte E, Margareto J, March C, et al. Klebsiella pneumoniae subverts the activation of inflammatory responses in a NOD1-dependent manner. Cell Microbiol. (2011) 13:135–53. doi: 10.1111/j.1462-5822.2010.01526.x
61. Kolar SL, Kyme P, Tseng CW, Soliman A, Kaplan A, Liang J, et al. Group B Streptococcus evades host immunity by degrading hyaluronan. Cell Host Microbe. (2015) 18:694–704. doi: 10.1016/j.chom.2015.11.001
62. Eckmann L, Kagnoff MF, Fierer J. Epithelial cells secrete the chemokine interleukin-8 in response to bacterial entry. Infect Immun. (1993) 61:4569–74. doi: 10.1128/IAI.61.11.4569-4574.1993
63. Edwards RJ, Taylor GW, Ferguson M, Murray S, Rendell N, Wrigley A, et al. Specific C-terminal cleavage and inactivation of interleukin-8 by invasive disease isolates of Streptococcus pyogenes. J Infect Dis. (2005) 192:783–90. doi: 10.1086/432485
64. Wolf K, Fields KA. Chlamydia pneumoniae impairs the innate immune response in infected epithelial cells by targeting TRAF3. J Immunol. (2013) 190:1695–701. doi: 10.4049/jimmunol.1202443
65. Meduri GU, Kanangat S, Stefan J, Tolley E, Schaberg D. Cytokines IL-1 β, IL-6, and TNF-α enhance in vitro growth of bacteria. Am J Respir Crit Care Med. (1999) 160:961–7. doi: 10.1164/ajrccm.160.3.9807080
66. Borer A, Saidel-Odes L, Riesenberg K, Eskira S, Peled N, Nativ R, et al. Attributable mortality rate for carbapenem-resistant Klebsiella pneumoniae bacteremia. Infect Control Hosp Epidemiol. (2009) 30:972–6. doi: 10.1086/605922
67. Tsay R, Siu L, Fung C, Chang F. Characteristics of bacteremia between community-acquired and nosocomial Klebsiella pneumoniae infection: risk factor for mortality and the impact of capsular serotypes as a herald for community-acquired infection. Arch Intern Med. (2002) 162:1021–7. doi: 10.1001/archinte.162.9.1021
68. Brune K, Frank J, Schwingshackl A, Finigan J, Sidhaye VK. Pulmonary epithelial barrier function: some new players and mechanisms. Am J Physiol Lung Cell Mol Physiol. (2015) 308:L731–745. doi: 10.1152/ajplung.00309.2014
69. Munkholm M, Mortensen J. Mucociliary clearance: pathophysiological aspects. Clin Physiol Funct Imaging. (2014) 34:171–7. doi: 10.1111/cpf.12085
70. Ofek I, Mesika A, Kalina M, Keisari Y, Podschun R, Sahly H, et al. Surfactant protein D enhances phagocytosis and killing of unencapsulated phase variants of Klebsiella pneumoniae. Infect Immun. (2001) 69:24–33. doi: 10.1128/IAI.69.1.24-33.2001
71. Wilson R, Pitt T, Taylor G, Watson D, MacDermot J, Sykes D, et al. Pyocyanin and 1-hydroxyphenazine produced by Pseudomonas aeruginosa inhibit the beating of human respiratory cilia in vitro. J Clin Invest. (1987) 79:221–9. doi: 10.1172/JCI112787
72. Rosseau S, Selhorst J, Wiechmann K, Leissner K, Maus U, Mayer K, et al. Monocyte migration through the alveolar epithelial barrier: adhesion molecule mechanisms and impact of chemokines. J Immunol. (2000) 164:427–35. doi: 10.4049/jimmunol.164.1.427
73. Wang J, Hossain M, Thanabalasuriar A, Gunzer M, Meininger C, Kubes P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science. (2017) 358:111–16. doi: 10.1126/science.aam9690
74. Krishnan N, Robertson BD, Thwaites G. The mechanisms and consequences of the extra-pulmonary dissemination of mycobacterium tuberculosis. Tuberculosis. (2010) 90:361–6. doi: 10.1016/j.tube.2010.08.005
75. Hagedorn M, Rohde KH, Russell DG, Soldati T. Infection by tubercular mycobacteria is spread by nonlytic ejection from their amoeba hosts. Science. (2009) 323:1729–33. doi: 10.1126/science.1169381
76. Clarke TB, Francella N, Huegel A, Weiser JN. Invasive bacterial pathogens exploit TLR-mediated downregulation of tight junction components to facilitate translocation across the epithelium. Cell Host Microbe. (2011) 9:404–14. doi: 10.1016/j.chom.2011.04.012
77. Rosendahl A, Bergmann S, Hammerschmidt S, Goldmann O, Medina E. Lung dendritic cells facilitate extrapulmonary bacterial dissemination during pneumococcal pneumonia. Front Cell Infect Microbiol. (2013) 3:21. doi: 10.3389/fcimb.2013.00021
78. Rangel SM, Diaz MH, Knoten CA, Zhang A, Hauser AR. The role of ExoS in dissemination of Pseudomonas aeruginosa during pneumonia. PLoS Pathog. (2015) 11:e1004945. doi: 10.1371/journal.ppat.1004945
79. González-Juarbe N, Bradley KM, Shenoy AT, Gilley RP, Reyes LF, Hinojosa CA, et al. Pore-forming toxin-mediated ion dysregulation leads to death receptor-independent necroptosis of lung epithelial cells during bacterial pneumonia. Cell Death Differ. (2017) 24:917. doi: 10.1038/cdd.2017.49
80. Kikuchi Y, Tateda K, Fuse ET, Matsumoto T, Gotoh N, Fukushima J, et al. Hyperoxia exaggerates bacterial dissemination and lethality in Pseudomonas aeruginosa pneumonia. Pulm Pharmacol Ther. (2009) 22:333–9. doi: 10.1016/j.pupt.2008.12.021
81. Kinhikar AG, Verma I, Chandra D, Singh KK, Weldingh K, Andersen P, et al. Potential role for ESAT6 in dissemination of M. tuberculosis via human lung epithelial cells. Mol Microbiol. (2010) 75:92–106. doi: 10.1111/j.1365-2958.2009.06959.x
82. Ten Threats to Global Health in 2019,. (2019). Available online at: https://www.who.int/emergencies/ten-threats-to-global-health-in-2019 (accessed September 26, 2019).
83. Porte R, Fougeron D, Munoz-Wolf N, Tabareau J, Georgel AF, Wallet F, et al. A toll-like receptor 5 agonist improves the efficacy of antibiotics in treatment of primary and influenza virus-associated pneumococcal mouse infections. Antimicrob Agents Chemother. (2015) 59:6064–72. doi: 10.1128/AAC.01210-15
84. Davis CG, Chang K, Osborne D, Walton AH, Ghosh S, Dunne WM, et al. TLR3 agonist improves survival to secondary pneumonia in a double injury model. J Surg Res. (2013) 182:270–6. doi: 10.1016/j.jss.2012.09.039
85. Lembo A, Pelletier M, Iyer R, Timko M, Dudda JC, West TE, et al. Administration of a synthetic TLR4 agonist protects mice from pneumonic tularemia. J Immunol. (2008) 180:7574–81. doi: 10.4049/jimmunol.180.11.7574
86. Wu CC, Sabet M, Hayashi T, Tawatao R, Fierer J, Carson DA, et al. In vivo efficacy of a phosphodiester TLR-9 aptamer and its beneficial effect in a pulmonary anthrax infection model. Cell Immunol. (2008) 251:78–85. doi: 10.1016/j.cellimm.2008.04.001
87. Duggan JM, You D, Cleaver JO, Larson DT, Garza RJ, Guzman Pruneda FA, et al. Synergistic interactions of TLR2/6 and TLR9 induce a high level of resistance to lung infection in mice. J Immunol. (2011) 186:5916–26. doi: 10.4049/jimmunol.1002122
88. Jean D, Rezaiguia-Delclaux S, Delacourt C, Leclercq R, Lafuma C, Brun-Buisson C, et al. Protective effect of endotoxin instillation on subsequent bacteria-induced acute lung injury in rats. Am J Respir Crit Care Med. (1998) 158:1702–8. doi: 10.1164/ajrccm.158.6.9709122
89. Bigot J, Guillot L, Guitard J, Ruffin M, Corvol H, Chignard M, et al. Respiratory epithelial cells can remember infection: a proof-of-concept study. J Infect Dis. (2019) jiz569. doi: 10.1093/infdis/jiz569
Keywords: bacterial dissemination, host pathogen interaction, epithelial immunity, pathogen evolution, lung epithelium
Citation: Sharma L, Feng J, Britto CJ and Dela Cruz CS (2020) Mechanisms of Epithelial Immunity Evasion by Respiratory Bacterial Pathogens. Front. Immunol. 11:91. doi: 10.3389/fimmu.2020.00091
Received: 11 November 2019; Accepted: 14 January 2020;
Published: 11 February 2020.
Edited by:
Markus M. Heimesaat, Charité Medical University of Berlin, GermanyReviewed by:
David Dockrell, University of Edinburgh, United KingdomCopyright © 2020 Sharma, Feng, Britto and Dela Cruz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Charles S. Dela Cruz, Y2hhcmxlcy5kZWxhY3J1ekB5YWxlLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.