 Marco Quaglia1,2
Marco Quaglia1,2 Sergio Dellepiane1,2,3
Sergio Dellepiane1,2,3 Gabriele Guglielmetti1,2
Gabriele Guglielmetti1,2 Guido Merlotti1,2Giuseppe Castellano4
Guido Merlotti1,2Giuseppe Castellano4 Vincenzo Cantaluppi1,2*
Vincenzo Cantaluppi1,2*- 1Nephrology and Kidney Transplantation Unit, Department of Translational Medicine, University of Piemonte Orientale (UPO), Novara, Italy
- 2Center for Autoimmune and Allergic Diseases (CAAD), University of Piemonte Orientale (UPO), Novara, Italy
- 3Division of Hematology/Medical Oncology, Icahn School of Medicine at Mount Sinai Hospital, The Tisch Cancer Institute, New York, NY, United States
- 4Nephrology Dialysis and Transplantation Unit, Department of Medical and Surgical Sciences, University of Foggia, Foggia, Italy
Extracellular vesicles (EVs) are known immune-modulators exerting a critical role in kidney transplantation (KT). EV bioactive cargo includes graft antigens, costimulatory/inhibitory molecules, cytokines, growth factors, and functional microRNAs (miRNAs) that may modulate expression of recipient cell genes. As paracrine factors, neutrophil- and macrophage-derived EVs exert immunosuppressive and immune-stimulating effects on dendritic cells, respectively. Dendritic cell-derived EVs mediate alloantigen spreading and modulate antigen presentation to T lymphocytes. At systemic level, EVs exert pleiotropic effects on complement and coagulation. Depending on their biogenesis, they can amplify complement activation or shed complement inhibitors and prevent cell lysis. Likewise, endothelial- and platelet-derived EVs can exert procoagulant/prothrombotic effects and also promote endothelial survival and angiogenesis after ischemic injury. Kidney endothelial- and tubular-derived EVs play a key role in ischemia–reperfusion injury (IRI) and during the healing process; additionally, they can trigger rejection by inducing both alloimmune and autoimmune responses. Endothelial EVs have procoagulant/pro-inflammatory effects and can release sequestered self-antigens, generating a tissue-specific autoimmunity. Renal tubule-derived EVs shuttle pro-fibrotic mediators (TGF-β and miR-21) to interstitial fibroblasts and modulate neutrophil and T-lymphocyte influx. These processes can lead to peritubular capillary rarefaction and interstitial fibrosis–tubular atrophy. Different EVs, including those from mesenchymal stromal cells (MSCs), have been employed as a therapeutic tool in experimental models of rejection and IRI. These particles protect tubular and endothelial cells (by inhibition of apoptosis and inflammation–fibrogenesis or by inducing autophagy) and stimulate tissue regeneration (by triggering angiogenesis, cell proliferation, and migration). Finally, urinary and serum EVs represent potential biomarkers for delayed graft function (DGF) and acute rejection. In conclusion, EVs sustain an intricate crosstalk between graft tissue and innate/adaptive immune systems. EVs play a major role in allorecognition, IRI, autoimmunity, and alloimmunity and are promising as biomarkers and therapeutic tools in KT.
Introduction
Extracellular vesicles (EVs or microparticles) is a general term that refers to membrane structures released by all cell types through different biogenesis pathways; EVs are secreted after fusion of endosomes with the plasma membrane (exosomes), shed from plasma membrane (microvesicles), or released during apoptosis (apoptotic bodies). These three entities differ in size (exosomes, 30–150 nm; shedding microvesicles, 150 nm−1 μm; apoptotic bodies, 1–5 μm) and partly in content (1–4). In this review, we will employ the umbrella term “EVs” to include all the above-mentioned types of secreted membrane vesicles.
After cellular shedding, EVs are rapidly taken up by neighboring or distant target cells (paracrine and endocrine effects) through a variety of mechanisms, such as endocytosis, phagocytosis/pinocytosis, membrane fusion, and receptor-mediated endocytosis (2).
EVs are involved in a wide range of physiological and pathological processes (4–7), including acute kidney injury (AKI), chronic kidney disease (CKD), thrombotic microangiopathies, and vasculitis (2, 3). EVs play a key role in all these settings by shuttling their bioactive cargo between cells. Most of their effects are mediated by microRNAs (miRNAs), which modulate gene expression in target cells and induce epigenetic reprogramming (3). Additionally, EVs carry a wide variety of immune modulatory molecules (e.g., cytokines, costimulatory/inhibitory molecules, and growth factors). Packing of nucleic acids and other contents into EVs is coordinated by multiple signals from EVs themselves or from cellular/extracellular environment (8–10). For example, TNFα modulates miRNA content of endothelial particles (11). Of interest, most EVs do not express human leukocyte antigens (HLAs) and escape the immune system; moreover, they cross numerous biological barriers (8), including glomerular endothelium basement membrane (12). Homing and uptake of EVs are mediated by signals and receptors on target cells (13) and influenced by local factors such as pH and electric charge (14). After intake, their complex biocargo exerts multiple effects: mRNAs are translated; miRNAs activate or silence protein expression (1, 2, 8); surface receptors are transferred from one cell to another (15, 16) and bacterial, viral, or graft alloantigens can be exchanged among immune cells (17, 18). A detailed analysis of EV general properties has been covered by recent reviews (1, 6, 8) (Figure 1).
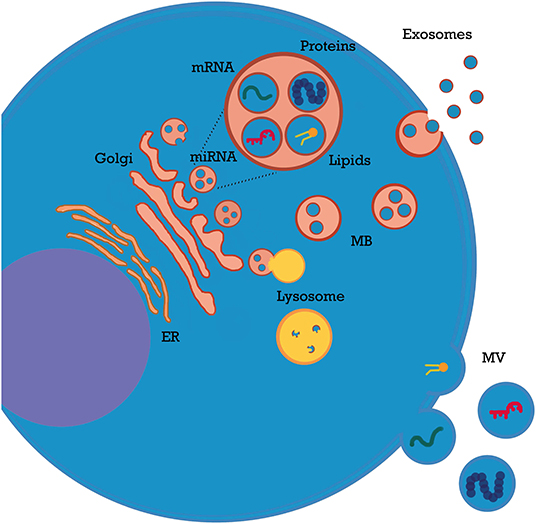
Figure 1. Extracellular vesicle (EV) biogenesis.
EVs released from innate immune cells, such as macrophages, dendritic cells (DCs), or natural killer (NK) cells, are involved in the regulation of innate immune response mainly as pro-inflammatory and paracrine mediators (4, 19). However, their immunomodulatory role is probably far more complex and includes anti-inflammatory and immunosuppressive effects.
The role of innate immunity as a trigger for acute rejection has been the focus of intense research over the last years (20, 21), and the possibility of manipulating EVs as a therapeutic tool or employing them as biomarkers is opening new paths in solid organ transplantation (22).
The aim of this review is to outline the role of EVs in innate immunity by analyzing different aspects of kidney transplantation (KT) biology. After analyzing EVs as mediators among different innate immune cell types, we will describe the role in complement and coagulation, two pivotal systems in innate immunity, and in other key settings such as allorecognition, ischemia–reperfusion injury (IRI), and the autoimmune component of antibody-mediated rejection. Finally, we will review recent evidence about the role of EVs as potential therapeutic tools and biomarkers in KT.
A general overview of immune-modulating effects of innate cell-derived EVs on different immune system cells or molecular targets is outlined in Table 1.
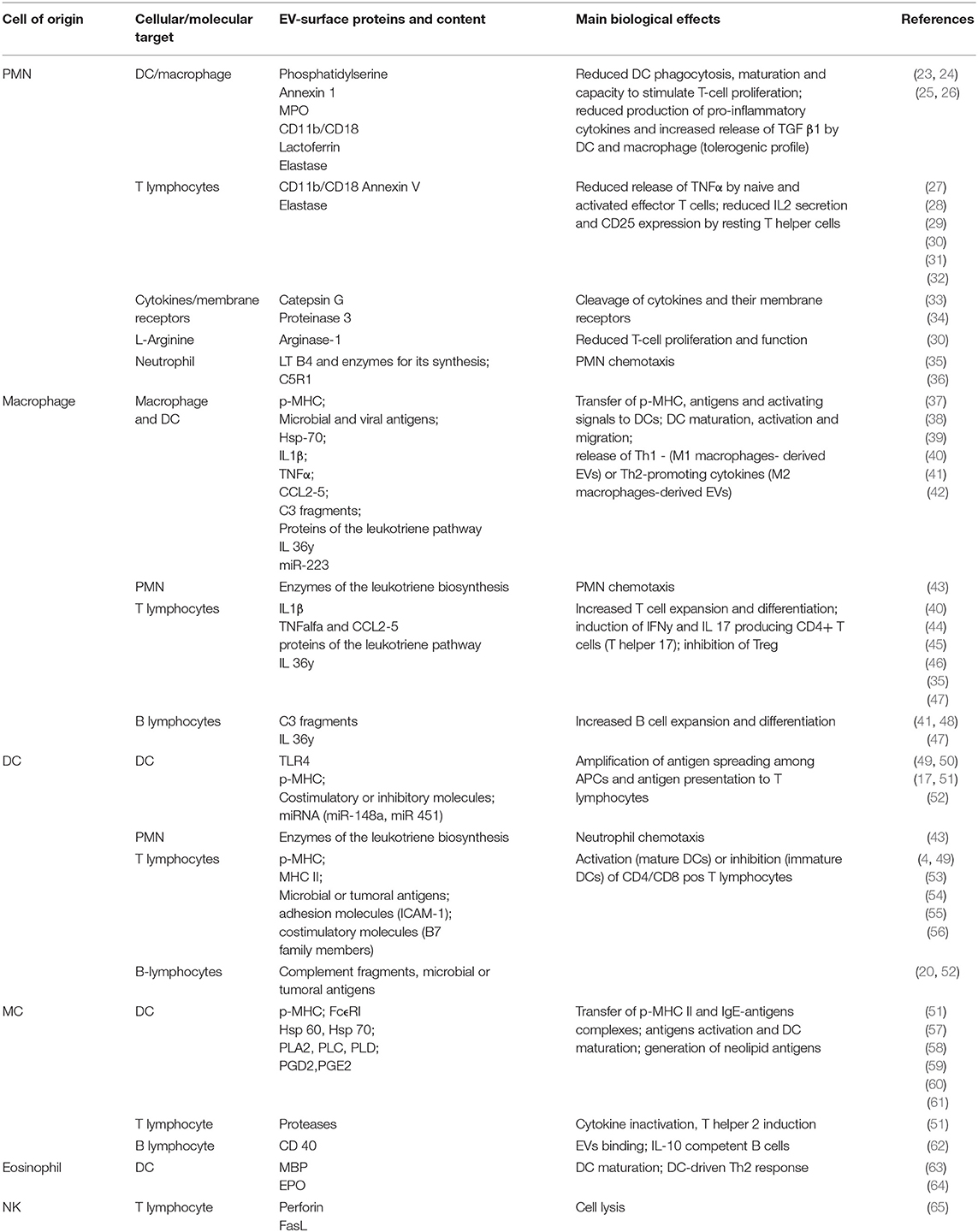
Table 1. Immune-modulating effects of innate cell-derived EVs on other immune system cells or molecular targets.
Neutrophil-Derived Extracellular Vesicles
Far from being mere final effectors of the inflammatory response, neutrophils [or polymorphonuclear cells (PMN)] exert several modulating effects on both innate and adaptive immune cells and can migrate to secondary lymphoid organs. These actions are partly mediated by EVs (23).
In general, PMN-derived EVs have anti-inflammatory and immunosuppressive effects, mainly on DCs and macrophages. EVs released from apoptotic PMNs also extend their actions on T-lymphocyte subsets, blunting their activation (24).
Neutrophil-derived EVs can inhibit lipopolysaccharide (LPS)-activated DCs and macrophages by reducing their phagocytic capacity, their maturation, and the release of pro-inflammatory cytokines (IL-8, IL-10, IL-12, and TNFα) while increasing TGF-β1 excretion. This cytokine plays a key role in suppressing immune response: it promotes anti-inflammatory DC, suppresses CD4+ and CD8+ T cells and induces T reg expansion (25, 26).
EVs released by apoptotic human PMN suppress T-cell proliferation, IL-2 production, and IL-2 receptor upregulation on activated T cells (27). The binding of these EVs to activated T cells seems to occur through Mac-1 (CD11b/CD18), an integrin also involved in immunological synapse formation (28).
The bioactive cargo of PMN-derived EVs includes numerous immune modulatory molecules: annexin V (induction of Tregs) (29), arginase-1 (depletion of arginine with inhibition of T-cell proliferation), lactoferrin (inhibition of DC migration to lymph nodes) (30), myeloperoxidase (inhibition of DCs) (31), elastase (conversion of human immature DCs into TGF-β1-secreting cells) (32), and other proteases such as cathepsin G and proteinase 3, which can inactivate pro-inflammatory cytokines (IL-2, IL-6, and TNFα) (33) and cleave their receptors from the plasma membrane (34). Additionally, PMN EVs regulate inflammatory cell trafficking; leukotriene B4 (LTB4) activates PMN chemotaxis and is particularly enriched in their EVs (35). Conversely, during sepsis, PNM shed C5a receptor 1 into their EVs and reduce their response to complement activation (36).
Macrophage-Derived Extracellular Vesicles
In general, macrophage-derived EVs exert pro-inflammatory effects, mainly directed toward DCs, macrophages, PMNs, and T lymphocytes.
Infected macrophages release EVs loaded with pathogens' proteins that can activate other antigen-presenting cells (APCs). Depending on the microenvironment, targeted macrophages activate either M1 or M2 polarization (37), whereas DCs process and present the antigens to T cells, thus promoting allorecognition and adaptive immunity. In addition to microbial or viral antigens, macrophage-derived EVs also carry peptide–major histocompatibility complex (MHC) complexes and costimulatory molecules, further enhancing alloantigen spreading among innate immune cells (38).
The cargo of macrophage-derived EVs includes several molecules with immunomodulatory functions, such as Hsp 70 (pro-inflammatory or tolerogenic effect depending on coexistent signals) (39), IL-1 β (DC migration and expansion of T/B lymphocytes) (40, 41), TNFα, and several chemokines (CCL2, CCL3, CCL4, and CCL5) (44–46). Complement C3 fragments are expressed on EV surface and interact with T cells during antigen presentation (48). Proteins involved in leukotriene synthesis were isolated in human macrophages, converting LTA4 into LTB4 and LTC4 and potentially activating DCs and CD4/CD8 T cells (43). IL-36γ was found in EVs released by infected pulmonary macrophages, with possible impact on DC maturation and T-cell activation [T helper (Th)1 or Th17 development and inhibition of Tregs] (47). Finally miR-223, a regulator of myeloid differentiation, was found in macrophage-derived EVs (42).
Zhang et al. stimulated macrophages in vitro with different protocols and performed an extensive proteomic profiling of their EVs. When the inflammasome complex was activated, EVs had a higher immunogenicity and induced NF-κB signaling in neighboring immune cells, thus amplifying inflammation (44). The inflammasome is a multimeric caspase-activating complex that can modulate a wide range of pathways in response to pathogens and activate both innate and adaptive immunity.
This is relevant to KT because IRI determines tissue damage, release of EVs, and inflammasome activation (44). These aspects will discussed in Extracellular Vesicles in Ischemia–Reperfusion Injury and in the Autoimmune Component of Rejection.
Finally, glucocorticoid therapy and long-term LPS exposure (mimicking chronic infection) can trigger macrophage release of toll-like receptor-2-containing EVs; these particles act as decoy receptors to antagonize toll-like receptor-2 signaling and blunt inflammation (66).
Dendritic Cell-Derived Extracellular Vesicles: Early Inflammatory Response and T-Lymphocyte Activation
Dendritic Cell Extracellular Vesicle and Innate Immunity
DCs highly express pattern recognition receptors and represent a pivotal link between innate and adaptive immunity (49). Toll-like receptors belong to pattern recognition receptors family and play a key role in the early inflammatory response; indeed, toll-like receptors avidly bind damage-associated molecular patterns, a wide group of molecules released by damaged tissues (e.g., during IRI) (67, 68). Toll-like receptor 4 is transferred via EVs among bone marrow DCs (BM-DCs) and activate NF-κB signaling pathway (50). Moreover, EV-mediated transfer of miRNAs among DCs contributes to enhance their mutual activation during inflammation (17, 69).
As described above (PMN paragraph), DC-derived EVs also carry enzymes of the leukotriene biosynthesis, which stimulate PMN chemotaxis (43).
Antigen Presentation to T Lymphocytes
DC-derived EVs also play a pivotal role in allorecognition (4, 49). DCs capture EVs released from graft tissue. Graft particles carry surface class I and II MHC molecules, non-HLA donor antigens, costimulatory and adhesion molecules, and pro-inflammatory cytokines such as IL-1β (52). The DC–EVs axis plays a pivotal role in all the three antigen presentation pathways described in transplant immunology, as reported in Figure 2 (53, 68, 70, 71):
• Direct antigen presentation: In this setting, donor APCs interact with recipient T cells. Of note, donor DC-derived EVs contain high density of allogeneic peptides complexed with donor MHC (p-MHC) and can interact directly with CD8+ and CD4+ T cells.
• Indirect antigen presentation: In this pathway, recipient APCs interact with recipient T cells. Graft EVs are internalized into the recipient APC and transfer their peptides to MHC class II molecules. These complexes are then exposed to APC surface for indirect presentation to T lymphocytes.
• Indirect antigen presentation by “cross-dressing” APCs (semi-direct antigen presentation): Donor-derived EVs containing p-MHC complexes are captured by recipient APC on their surface and then presented directly to T cells without any p-MHC reprocessing, a phenomenon referred to as “cross-dressing.”
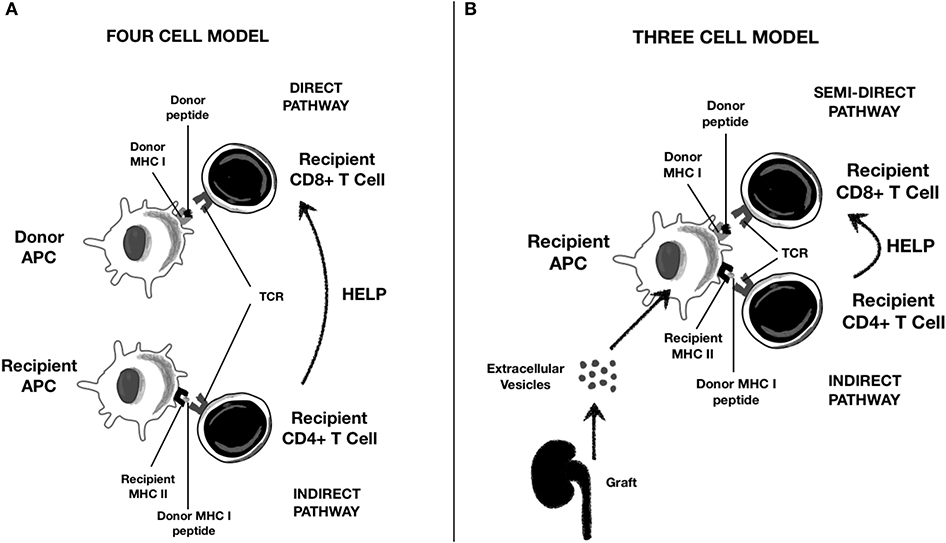
Figure 2. Role of Extracellular Vescicles (EVs) in alloantigen presentation to T lymphocytes. (A) Classical direct and indirect presentation; (B) “semi-direct” presentation trough cross-dressing of recipient APC with graft-derived EVs.
Recent evidence suggests that donor DC transplanted with the graft are antigen transporting rather than antigen presenting cells and that “cross-dressing” rather than “passenger leukocyte” is the main mechanism of alloantigen presentation from donor APC (70, 71). Although semi-direct modality rapidly initiates alloresponse and leads to acute rejection, indirect T-cell activation has been associated with chronic antibody-mediated rejection (72). “Cross-dressing” is also typical of follicular DCs, key players in germinal center reactions (54).
The effectiveness of DC-derived EVs in p-MHC presentation depends on the coexistence of other molecules in their cargo (MHC class II, CD86, and ICAM) and on parental cell maturation (20):
• Mature DC-derived EVs are characterized by higher expression of surface MHC, adhesion, and costimulatory molecules (55, 73) and present antigens to CD4+ T lymphocytes through “cross-dressing,” promoting Th1 phenotype (56, 74).
• Immature DC-derived EVs are efficiently internalized by mature APCs and transfer their antigens to the target cell MHC. Thus, the antigen is indirectly presented to CD4+ T lymphocytes, skewing them toward a Th2 phenotype. Additionally, immature DC can release immunoregulatory EVs loaded with anti-inflammatory cytokines such as TGF-β1 (4) and can target other DCs to amplify a tolerogenic response (75).
Therefore, donor EVs target recipient cells and generate a chimerism that can determine either DC activation or DC inhibition depending on their content (76, 77). For example, EV-derived CD86, a costimulatory molecule, activates T cells through direct or semi-direct pathway, whereas the indirect pathway vehicles miRNAs upregulating PD-L1 and induces CD4 T cells anergy (78). Indeed, graft-infiltrating PD-L1hi cross-dressed DCs blunted T-cell response in a mouse model of liver transplantation (77).
Finally, the relationship between DCs and adaptive immunity is bidirectional: for example, DCs are targeted by Treg-derived EVs that induce a tolerogenic phenotype trough transfer of miRNAs (miR-150-5p and miR-142-3p) (79).
Mast Cell-Derived Extracellular Vesicles
MC-derived EVs contain p-MHC complexes or endocytosis-derived antigens and can be released by both activated and resting BM-MCs. The main target of these particles is DCs and other professional APCs (51, 80, 81). Skokos et al. investigated the role of MCs in allo-antigen presentation; the authors observed that ovalbumin was more effectively recognized by T cell if taken up by MCs and then transferred to DCs rather than presented directly by DCs (57). Indeed, MCs and DCs form a highly structured immune synapsis devoted to antigen transfer through EVs (58).
Several molecules with immunomodulatory roles have been isolated in MC-derived EVs. Heat shock proteins (Hsp 60 and 70) are essential for antigen loading and EV uptake by DCs (57, 59) and are capable of inducing BM-DC maturation; FceRI–IgE complexes contribute to horizontal antigen transfer among MCs; additionally, phospholipases (PLA-A2, C, and D2), neolipid antigens, and lysophospholipids (60, 61) inhibit DC functions (e.g., phosphatidic acid) and induce Th2 response [lysophosphatidylcholine (LPC)] (60, 82). MC-derived EVs also carry proteases that inactivate cytokines and also target T-cell proteins (34, 60). Finally, CD40L-positive EVs from BM-derived MCs generate IL-10 competent B cells (62, 83).
Other Innate Immune Cell-Derived Extracellular Vesicles
There is a paucity of data about EVs generated by other innate immune cells.
Eosinophils can release EVs containing major basic protein (MBP) and eosinophil peroxidase (EPO) when stimulated with IFNγ; both promote DC maturation (63, 64, 84).
NK cells can release EVs loaded with several cytotoxic proteins (85, 86), including perforin and FasL, which can induce lysis of activated T lymphocyte and thus possibly blunt inflammation (65, 87).
The main immune-modulating effects of innate cell-derived EVs on other immune system cells or molecular targets are summarized in Table 1.
Extracellular Vesicles and the Complement System
EVs can play a dual role in this setting, either activating or inhibiting the complement cascade (88).
This function is extremely relevant to the transplant setting, as EVs play a role in complement attack on ECs in both antibody-mediated rejection (89) and IRI (90).
a) Extracellular Vesicles as Complement Activators
T cell-derived EVs can activate complement through immunoglobulin binding, whereas other types of EVs do so directly, through interactions between C1q and their membrane lipids (91, 92). For instance, both PMN- and erythrocyte-derived EVs (93, 94) can provide a platform for C1q deposition, with consequent activation of classic pathway on their surface.
Activated endothelial cells can shed EVs under inflammatory conditions; this phenomenon has been observed after complement activation and membrane attack complex formation on endothelial cell surface (95). These endothelial-derived EVs express membrane attack complex and have a strong procoagulant phenotype, which further triggers complement activation through thrombin formation. This creates a vicious circle of endothelial complement-mediated damage and endothelial shedding of complement-enhancing EVs (88).
b) Extracellular Vesicles as Complement Inhibitors
On the other hand, EV shedding could also represent a mechanism to protect cells from complement attack: indeed, EVs remove complement molecules from cell surface acting as “scavengers” and allowing complement evasion (88). Complement-induced EVs shedding has been demonstrated in PMNs, erythrocytes, and glomerular endothelial and epithelial cells (96).
Consistently, complement-coated EVs from leukocytes can be rapidly phagocytosed by PMNs. Clearance of these opsonized EVs is also facilitated by complement receptor 1, expressed on erythrocytes; as such, red blood cells bind EVs and transport them to the liver and spleen (94).
EVs also carry several complement inhibitors that allow them to transport activated complement factors without being lysed: CR1, CD55, or decay-accelerating factor (modulation of C3 and C5 convertase), CD59 (direct MAC inhibitor), and membrane cofactor protein (MCP or CD46) (2, 97). Interestingly, endothelial EVs are also rich in complement inhibitor mRNA and prevent glomerular injury in experimental models of glomerulonephritis (98).
Extracellular Vesicles and the Coagulation System
Complement and coagulation cascades are key components of innate immunity and are tightly connected to each other; their simultaneous activation has been extensively studied in transplant rejection and IRI (99–102). EVs released from endothelial cells and platelets (PLTs) are critical promoters of coagulation in renal disease (89); besides carrying inflammatory and chemotactic proteins, these vesicles release also a number of growth factors [e.g., PLT-derived growth factor (PDGF)] and promote tissue regeneration.
a) Endothelial Extracellular Vesicles
When shed after complement activation, endothelial cell EVs have procoagulant and PLT-activating effects (95, 103). They expose phosphatidylserine and binding sites for factor Va and tissue factor (TF) (104, 105); the latter triggers extrinsic pathway determining thrombin generation (106). Thrombin directly cleaves complement components C3 and C5 into C3/C5 convertase, further amplifying the cascade (107). Endothelial EVs can also transfer TF to monocytes and PLTs (108). On the other hand, these EVs preserve endothelial cell survival in physiological condition (caspase-3 removal and protein C receptor exposure) (109), and EVs derived from endothelial progenitor cells can promote angiogenesis (110).
b) Platelet-Derived Extracellular Vesicles
These play a key role in hemostasis and coagulation (111) through a variety of mechanisms summarized in Table 2 (112–119). Of note, PLT-derived EVs have a 50- to 100-fold stronger procoagulant/prothrombotic effect than have PLTs (120). On the other hand, they promote angiogenesis and endothelial cell regeneration after vascular injury (2, 121).
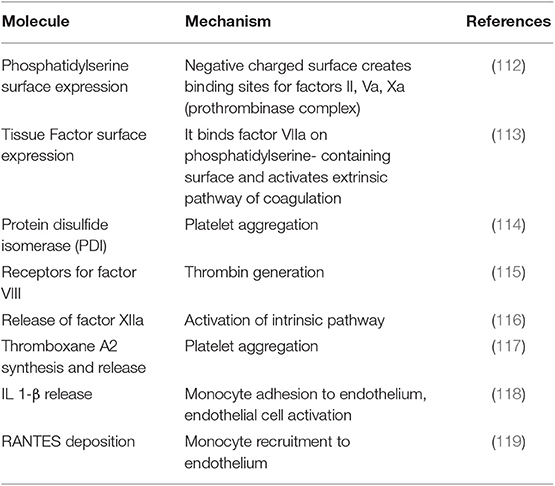
Table 2. Platelet-derived EVs procoagulant and prothrombotic effects.
Extracellular Vesicles in Ischemia–Reperfusion Injury and in the Autoimmune Component of Rejection
IRI is the main cause of delayed graft function (DGF), which determines an increased risk of acute rejection and progression to chronic allograft dysfunction (122). IRI triggers a complex, alloantigen-independent immune response characterized by crosstalk between PMNs, macrophages, and DCs (123). All these cells release EVs with pro-inflammatory and anti-inflammatory effects (see above) (19).
Two other cell types release critical EVs in this condition: endothelial cells and renal tubular epithelial cells. Both release EV when exposed to hypoxia, oxidative stress, acidic pH, or inflammation. Hypoxia determines an accumulation of hypoxia-inducible factor (HIF)-α subunit, which dimerizes with HIF-β to form HIF, a transcription factor that can activate over 70 target genes. This results in changes in surface receptors and remodeling of plasma membrane, which triggers release of EVs (124). Furthermore, HIF increases Rab22, an essential element for EV biogenesis (125).
Endothelial Cells
IRI induces a complex vascular phenotype characterized by a progressive spectrum of functional and structural alterations: vasoconstriction, vascular inflammation, microvascular rarefaction of peritubular capillaries, chronic hypoxia, interstitial fibrosis, and tubular atrophy (126, 127). Microvascular lesions appear to be a key driver of fibrosis after IRI, with a predominant effect over tubular ones (128).
Transplant procedure itself is characterized by tissue damage and some degree of ischemia, resulting in activation of different cell death programs (apoptosis, necrosis, necroptosis, pyroptosis, and autophagy-associated cell death) with release of damage-associated molecular patterns. Bacterial and viral components can also be released during transplant surgery or in infections after KT (122, 123). Both damage-associated molecular patterns and pathogen-associated molecular patterns bind a wide range of innate pattern recognition receptors expressed on several cells, including macrophages, DCs, and endothelia (129). Pattern recognition receptor activation triggers inflammatory response and EV release (126).
Caspase-3 is a pivotal regulator of cell apoptosis (128); under physiological conditions, endothelial EVs protect parental cell by removing caspase-3 (130). During IRI, caspase-3 hyper-activation can overtake EV clearance and cause cell death. In this scenario, endothelial cell generate both “classical” apoptotic bodies and smaller exosome-like vesicles; both are overloaded with caspase-3 and can propagate cell death. Additionally, these exosome-like vesicles carry activated 20S proteasome; this complex recruits adaptive immune cells and induces the production of auto-antibodies toward perlecan/LG3, angiotensin-1 receptor, and dsDNA, further aggravating vascular inflammation (46, 127, 131). Reperfusion has also been associated with the occurrence of a broad range of IgM “natural antibodies,” targeting “neo-epitopes” on ischemic tissues and activating complement (123). Thus, EVs shed by an activated or injured endothelium can trigger mechanisms of alloimmunity and autoimmunity.
The role of EVs in the autoimmune component of rejection has been the focus of recent studies. Tissue-specific self-antigens were found in circulating EVs released by apoptotic cells in the lung, heart, islet, and KT recipients while rejection is developing, whereas they were not detected in control grafts (132). For example, EVs from KT recipients with transplant glomerulopathy have an increased expression of fibronectin and type IV collagen than have EVs from stable KT recipients (133).
Innate immune response generates graft tissue damage, which can favor continuous release of sequestered self-antigens through EVs, with secondary activation of self-reactive T lymphocytes and development of a tissue-restricted form of autoimmunity (46, 72).
It must be emphasized that only in an inflammatory environment (e.g., IRI) can adaptive cells determine autoimmunity. Consistently, Sharma et al. showed that anti-cardiac myosin (CM) antibodies trigger graft rejection in syngeneic heart transplantation only when administered at time of surgery, but not 1 week after it (134).
Renal Tubular Epithelial Cells
In general, whereas EVs from injured cells can promote tubule interstitial inflammation and fibrosis, those derived from cells with regenerative properties can promote cell proliferation and tissue repair. However, this distinction is blurred, as injured renal tubular epithelial cells can also stimulate repair (as detailed below), whereas mesenchymal stromal cell (MSC)- or endothelial progenitor cell-derived EVs can have harmful effects (135).
With this caveat, we will now focus on actions mediated by EVs released by ischemic renal tubular epithelial cells, whereas EV potential to repair tissue damage will be dealt with in a specific paragraph.
Renal proximal tubular epithelial cells are especially prone to ischemic damage because they depend on mitochondrial metabolism for ATP production owing to their modest glycolytic capacity.
Under hypoxic conditions, HIF-1 mediates EV release by renal proximal tubular epithelial cells (136), which modulate severity of kidney injury by targeting neighboring cells (3).
Furthermore, renal proximal tubular epithelial cells express receptors for complement fractions C3a and C5a and toll-like receptors, making them responsive to innate immune activation (137). Damage-associated molecular patterns can be transferred into renal tubular epithelial cells trough EVs (138) and prevent tubular recovery (139, 140). Pathogen-associated molecular patterns, such as LPS, upregulate the expression of DC-SIGN and toll-like receptor 4, stimulating tubular secretion of IL-6 and TNFα (141).
In this early inflammatory phase, tubular EVs containing cytokines, growth factors, and complement fractions can recruit innate immune cells such as PMNs, M1 macrophages, and NK cells (135). EVs released by hypoxic renal tubular epithelial cells are characterized by a decreased content of miR-7641-2-3p, a downregulator of chemoattractant CXCL1, resulting in increased PMN influx (142).
Injured hypoxic tubular cells can transfer TGF-β-containing EVs across disrupted basement membrane to interstitial fibroblasts, activating them and mediating progression to CKD (143). Furthermore, TGF-β itself stimulates renal tubular epithelial cells in an autocrine way to secrete EVs enriched for miR-21, which targets recipient tubules enhancing Akt-mTOR proliferative pathway and consequently exacerbating interstitial fibrosis (144); of note, miR-21 is also released by several other types of human cells through toll-like receptor 3 activation (145).
Also, miR-155 worsens tubular damage during IRI, as it promotes tubular pyroptosis by upregulating expression of caspase-1 and downregulating FoxO3a expression together with its downstream protein “apoptosis repressor with caspase recruitment domain” (146, 147).
In addition to hypoxia, also albuminuria triggers release of CCL2-containing EVs, activating interstitial macrophages and promoting tubule interstitial inflammation (148). Proteases and glycosidase on EV surface may contribute to interstitial fibrosis by degrading extracellular matrix (149).
Furthermore, besides tubular-interstitial diffusion, EVs from renal proximal epithelial cells can also move downstream through urinary tract to target distal tubule or collecting duct (3), although with largely unknown effects.
Tubule-derived EVs can mediate anti-inflammatory and pro-angiogenic actions, for example, secreting IL-10, which polarizes macrophages toward an M2 phenotype, and galectin-1 and CD73, which promote Treg function (135, 150–152). Tubule-derived EVs can also directly interact with T lymphocytes through T-cell immunoglobulin- and mucin-containing molecules Tim-1 and Tim-4; interestingly, the same receptor Tim-1 (also called KIM-1) is expressed on renal tubular epithelial cells surface and mediates suppression of NF-κB (153).
EVs also mediate a less defined crosstalk between endothelia and renal tubular epithelial cells. On the one hand, tubule-derived EVs transport ApoA1, which inhibits ICAM-1 and P-selectin and alleviates ischemic damage and PMN retention (154); on the other hand, endothelial EVs can pass into urinary space upregulating HIFα/VEGFα signaling in renal tubular epithelial cells (155).
Extracellular Vesicles as a Therapeutic Tool in Renal Transplantation
Most studies on EVs as a therapeutic tool in renal transplantation have employed MSC-derived EVs (MSC-EVs) and have focused especially on IRI.
MSCs themselves have drawn much interest in transplantation, mainly because of their capacity to stimulate tissue repair after ischemic injury and their immunomodulatory properties (156).
Injected MSCs can inhibit tubular cell apoptosis and interstitial fibrosis while stimulating proliferation of tissue-specific progenitor cells. Although MSCs can engraft in renal tubular and endothelial cells, regenerative actions are primarily mediated by EVs (157, 158).
Additionally, MSC modulate both innate (DCs, monocytes, and NK cells) and adaptive (T and B lymphocytes) immune cells, with predominantly anti-inflammatory and immunosuppressive effects, which may play a role in preventing or counteracting rejection. Also these effects are predominantly mediated by MSC-EVs (159).
Since side effects and practical challenges of MSC therapy have been reported (160), MSC-EVs have been proposed by several studies as a safer, cell-free alternative. Nevertheless, they have shown similar or even potentially additive regenerative and immunomodulatory properties (161).
Recent evidence suggests that innate immune EVs and MCS EVs play opposite roles in immune system regulation: whereas the former can carry and spread alloantigens, stimulating allorecognition and rejection, the latter can exert immunosuppressive and tolerogenic effects. In particular, MSC-EVs inhibit DC maturation and NK function and skew T lymphocytes toward a Treg phenotype. Of note, MSC-EV proteomic analysis has identified 938 proteins, which could be relevant to MSC-EV interaction with immune cells (159, 162).
MSC-EVs and immune cell EVs are phenotypically different, as they reflect profile of surface molecules of respective parental cell; in particular, MSCs are defined by the expression of CD73, D90, and CD105 and lack CD14, CD34, and CD45 markers (163). However, MSC-EV cargo is not merely a reflection of their parental cell, as it is characterized by a peculiar enrichment in mRNA, miRNAs, and proteins involved in key processes, such as cell cycle regulation, cell differentiation, and immune regulation (157).
a) Ischemia–Reperfusion Injury
Mesenchymal Stromal Cell-Derived Extracellular Vesicles
MSCs can be isolated from different tissues, as shown in Figure 3. MSC-EVs recapitulate beneficial properties of origin cells, which are mediated by a variety of mRNAs, miRNAs, and proteins. These molecules are horizontally shuttled into recipient cells and activate signaling pathways related to the following (164):
• renal protection: inhibition of apoptosis/necrosis, inflammation, oxidative stress, fibrogenesis, and promotion of autophagy (165); and
• renal regeneration: stimulation of cell proliferation, migration, tubular dedifferentiation, and angiogenesis.
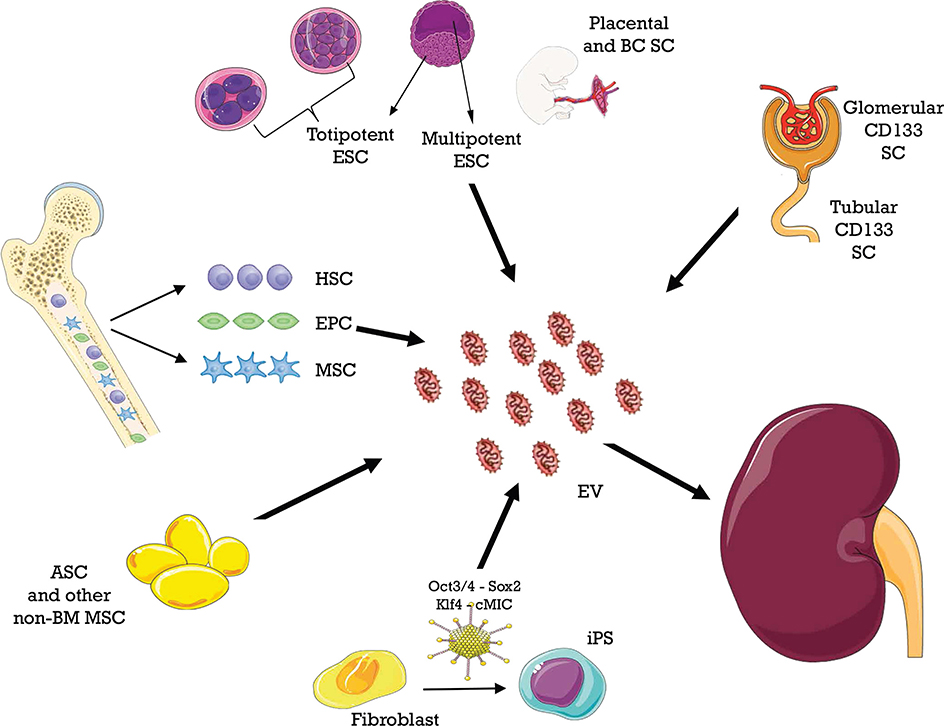
Figure 3. Different sources of extracellular vesicles (EVs) employed as therapy in ischemia–reperfusion injury.
Importantly, pretreatment with RNAase abolishes these effects, indicating that mRNAs and/or miRNAs account for them (166).
Ferguson et al. identified 23 top miRNAs, which account for over 79% of total miRNA load in MCS exosomes and seem to mediate the predominant effects, targeting 5,481 genes (167). Different miRNAs carried by MSC-EVs are extensively reviewed elsewhere (168–171).
The main miRNAs involved in renal protection from IRI, type of secreting cell, and mechanisms of action are outlined in Table 3 (172–181).
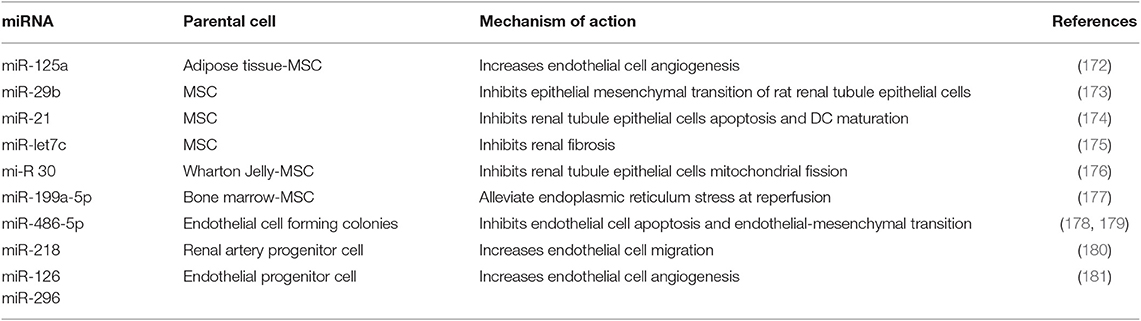
Table 3. Main miRNAs involved in renal protection from IRI.
Specific functions of these miRNAs are being defined: miR-125a can promote endothelial cell angiogenesis (172); miR-29b inhibits angiotensin II-induced epithelial-to-mesenchymal transition of rat RTECs (173) and blunts inflammation by inhibiting NF-κB; miR-21 prevents renal tubule epithelial cell apoptosis and inhibits DC maturation (174); and miR-199a-5p alleviates endoplasmic reticulum stress at very early reperfusion stages (8–16 h after reperfusion in vivo) (177).
Murine studies in which MSC-EVs were employed as a therapeutic tool for IRI are summarized in Table 4 (166, 177, 182–194). In all of them, administration of MSC-EVs improved renal function and/or decreased tubular injury through multiple mechanisms (164). Most studies were performed with BM- and umbilical cord-derived EVs; however, other MSC have been used including kidney resident populations (189, 190) and adipose tissue (191). Of interest, i.v. administered human MSC-EVs were effective in alleviating renal damage in rats that had received KT from cardiac death donor, a procedure characterized by severe IRI (194) (Table 3).

Table 4. Studies on MSC-derived EVs as therapeutic tool in AKI from IRI.
Trophic factors carried in MSC-EVs depend on the parental cell and the surrounding milieu, such as inflammation and hypoxia (136, 164, 195).
Hypoxia has a profound impact on EV properties. In general, ischemic conditioning (preconditioning, postconditioning, and remote conditioning) provides positive results in the setting of myocardial infarction, and hypoxic EVs appear to mediate these effects (124, 125, 195).
Hypoxic EVs derived from BM MSCs can exert protective effects in experimental models of AKI through several mechanisms: inhibition of renal tubule and endothelial cell apoptosis, stimulation of endothelial cell proliferation, reduction of inflammation and PMN infiltration, and inhibition of renal fibrosis (124).
Of interest, hypoxia can stimulate the secretion of EVs by adipose tissue-derived MSCs and can enhance their regenerative properties; specific anti-apoptotic, anti-oxidative, anti-inflammatory and pro-angiogenic pathways are activated by hypoxic EVs, and a distinct proteomic pattern is determined by this type of EVs in renal proximal tubule epithelial cells (196).
In the study by Collino et al. (196), four effects were specifically enhanced in hypoxic EV and could blunt progression of ischemic AKI to CKD: downregulation of fibroblast growth factor receptor 1 (FGFR-1), which mediates TGF-β1-induced epithelial-to-mesenchymal transition, and inhibition of maladaptive repair and fibrogenesis (197); angiogenesis stimulation, alleviating renal microvasculature rarefaction under hypoxia (198); translocation of Nrf-2 into the nucleus, activating antioxidant genes such as HO-1 (199); and downregulation of IL-6, blunting macrophage infiltration and polarization toward a M2 phenotype (200).
Moreover, hypoxic EVs carry respiratory complexes, supporting a non-mitochondrial aerobic metabolism when mitochondrial respiratory capacity is impaired (201); they reestablish intracellular ATP levels and reverse pre-apoptotic changes like histone H2 and H2B upregulation (202); they favor cell proliferation through JNK pathway activation (203) and downregulate calnexin, a NADPH oxidase NOX4-interacting protein, reducing reactive oxygen radical formation (204–206).
However, remote ischemic preconditioning on KT recipients has not proven to be as clinically effective as in ischemic heart disease, and further studies are needed to implement these findings into clinical tools (207, 208).
Another therapeutic approach is MSC transfection with specific miRNA. These engineered EVs proved to be more effective than those derived from naïve MSCs (209).
Other Cell Type-Derived Extracellular Vesicles
Cell types other than mesenchymal stromal cells also release reno-protective extracellular vesicles.
Under hypoxic conditions, endothelial colony-forming cells inhibit endothelial cell apoptosis and endothelial mesenchymal transition through EV containing miR-486-5p (178, 179), whereas renal artery progenitor cells increase endothelial cell migration through EV containing miR-218 (180).
Endothelial progenitor cells inhibit capillary rarefaction and progression toward chronic lesions in ischemic AKI; this effect was lost after depletion of pro-angiogenic miR-126 and miR-296 by transfection with specific antagomirs (181).
EVs from renal tubule cells also are capable of accelerating recovery of established renal ischemic damage (210).
b) Acute Rejection
Mesenchymal Stromal Cell-Derived Extracellular Vesicles
Studies using EVs from stem cells and tumors have shown immunosuppressive effects of their transcription factors and miRNAs (159).
In an MHC-mismatched rat model of kidney transplant, injection of recipient MSC-EVs on day 7 after transplant has reduced NK infiltrates and almost completely abolished intra-graft TNFα expression. However, B- and T-lymphocyte infiltrates were higher in EV-treated rats, whereas there was no difference in macrophage populations. Importantly, no difference was observed in antibody response against the donor, which occurred in both groups. These data suggest that MSC-EVs mainly affect some type of innate immunity cells (NK cells and related cytokines, such as TNFα), whereas they do not suppress adaptive immunity and rejection in a strong alloreactive model (162, 211).
Immune Cell-Derived Extracellular Vesicles
Immunosuppressive properties of EVs (75–79) could be exploited to inhibit innate component of rejection, for example, skewing DC function and maturation toward a tolerogenic profile (212–214). EVs released from Treg lymphocytes modulated DC maturation and prolonged kidney allograft survival in a rat model (215).
In a study on heart transplant rat model, DC-derived EVs were administered together with LF-15-0195, a DC maturation blocker. This approach determined a donor-specific tolerance with significantly blunted anti-donor proliferative response and chronic rejection, resulting in prolonged graft survival (2, 211).
Extracellular Vesicles as Biomarkers in Kidney Transplantation
EVs have also been investigated as possible biomarkers in KT. Plasma and urinary EVs have been studied in different transplant settings and will be discussed separately (216).
a) Acute Rejection
Plasma Extracellular Vesicles
Plasma EVs are one of the most promising biomarkers for solid organ transplantation, reducing or even obviating the need for renal biopsy (216–218).
In a recent study, Zhang et al. compared levels of mRNA transcripts carried by plasma EVs of patients with antibody-mediated rejection, T-cell mediated rejection, and no rejection and their related genes, identifying those that were significantly overexpressed in EVs from patients with antibody-mediated rejection. On this basis, they created a gene combination score elaborated from mRNA transcripts of four genes (gp130, SH2D1B, TNFα, and CCL4), which was able to predict imminent antibody-mediated rejection (219).
In a study on 231 KT patients, circulating endothelial microparticles were analyzed before and periodically after KT (up to 2 months); plasma levels increased during antibody-mediated rejection episodes and decreased after therapy, with a slower decline in patients with peritubular capillary C4d staining (220).
In another study, quantification was carried out of plasma C4d+CD144+ EVs released from endothelial cells associated with antibody-mediated rejection (11-fold increase in concentration compared with that in patients with no rejection), its severity, and response to treatment (over 70% decrease in concentration after successful anti-rejection therapy) (100).
Urinary Extracellular Vesicles
In one study (221), 11 proteins were significantly enriched in urinary EVs from patients with T cell-mediated rejection; of note, the association was lost when the whole urinary protein fraction was analyzed. This finding highlighted the impact of “background noise” from uromodulin and proteinuria, suggesting that urinary EVs are a more selective source of biomarkers. Despite this, little evidence has been produced on urinary EV RNAs so far, as most papers have focused on total, cell-derived, or cell-free urinary transcripts (222).
In a more recent study, increased expression of 17 urinary EV proteins was found in patients with T cell-mediated rejection and two proteins—tetraspanin-1 and hemopexin—were proposed as biomarkers (223).
Finally, a urine-based platform termed IKEA (“integrated kidney exosome analysis”), detecting EVs shed by T cells into urine, revealed high levels of CD3-positive EVs in patients with rejection, with an accuracy of over 90% for T cell-mediated rejection (224).
b) Delayed Graft Function and Other Settings
Plasma Extracellular Vesicles
In the already mentioned study by Qamri et al., circulating endothelial microparticles decreased within 2 months of KT, paralleling renal function recovery, only in patients with specific types of causal nephropathies such as diabetic nephropathy or glomerulonephritis secondary to autoimmune disorders (220).
Consistently, Al Massarani et al. found a progressive decrease in serum EV concentration and in their procoagulant activity after KT. This evolution was independent from the type of immunosuppression, whereas it seemed to be influenced by history of cardiovascular disease and CMV infection (225, 226). In a Brazilian cohort of 91 KT patients, PLT and endothelial EV size and concentration were significantly different depending on renal function and time from KT (227).
Taken together, these data suggest that decreased endothelial EVs after KT reflect not only antibody- or T cell-mediated rejection but also improvement of preexisting endothelial dysfunction and of cardiovascular risk factors, paralleling recovery of renal function after KT (227, 228).
Urinary Extracellular Vesicles
Urinary EVs have been proposed an enriched source of biomarkers of DGF. For example, neutrophil gelatinase-associated lipocalin expression in EVs was higher than in urinary cells and correlated with DGF (229).
Urinary CD133+ EVs appear to be decreased in KT patients with slow graft function and vascular damage, suggesting possible damage to renal stem cell compartment (230).
Likewise, a reduction in urinary aquaporin-1- and aquaporin-2-containing EVs was observed in rat model of IRI, probably reflecting impaired trafficking and expression of these proteins in renal tubule epithelial cells (231), confirming previous finding of decreased abundance of aquaporin-1 in KT recipients in the immediate postoperative days (232).
Limits, Perspectives, and Conclusions
Despite the large volume of literature, our knowledge of innate immunity EVs is still limited (233). Further studies are needed to widen our understanding in graft antigen spreading and processing by DCs (53, 70–72) and to clarify their tolerogenic potential (75–78). Little evidence has been produced on PMN or macrophage vesicles. Additionally, few studies identified the target genes of EV miRNAs.
Finally, a major limit of EV analysis is the lack of standardization and consistency (234): based on different techniques, diverse markers with almost no overlapping results have been proposed. Of note, most housekeeping genes used for cellular assay normalization (e.g., β-actin or GAPDH) are not consistently expressed in EVs. Normalization of urinary EV proteins with tetraspanins (CD9, CD63, or CD81) is not a validated approach, and mRNA analysis remains problematic (235). Finally, most urinary markers should be standardized for urinary creatinine, but not all studies have adopted this method.
Despite these barriers, EVs appear promising as both biomarkers and therapeutic agents in KT. Enhancement of MSC-EVs therapeutic potential through stimulation with biophysical or biochemical cues (i.e., LPS, hypoxia, inflammatory cytokines, growth factors, hormones such as erythropoietin, nitric oxide, and EVs from other cells such as endothelial cells) is an attractive perspective (236). Genetically engineered EVs overexpressing specific proteins or miRNAs acquire stronger therapeutic properties: for example, HIF-α-overexpressing MSCs have enhanced angiogenic activity and repaired more efficiently cardiac tissue in a mouse model compared with control EVs (237); miR-let7c-overexpressing MSCs selectively homed to damaged kidney, where they upregulated miR-let7c genes and downregulated expression of TGF-β, its receptor (TGF-β-R1), and other pro-fibrotic genes in a renal mouse model of unilateral ureteral obstruction (175).
Bioengineered EVs hold promise as targeted vehicle of drugs or miRNAs, as they naturally overcome biological barriers (209, 238). “Decoy EVs” have also been employed to antagonize inflammatory cytokines (239).
In conclusion, EVs finely tune the crosstalk among innate immune cells and graft tissue; in particular, they determine antigen spreading and “cross-dressing” in the early transplant phases, thus being a key trigger of either alloimmunity or graft tolerance. Systemically, they modulate complement and coagulation cascades during transplant related kidney injuries as antibody-mediated rejection and IRI.
Growing evidence support a potential application of EVs derived from MSCs and other cell types as therapeutic tools in different settings of renal transplantation. Finally, urinary and serum EVs are promising biomarkers of rejection and DGF, opening new paths toward a renal “liquid biopsy.”
Author Contributions
MQ and VC designed, wrote, and critically revised the Review. SD analyzed innate cells derived EVs. GG and GM analyzed EVs role in DGF and rejection. GC dealt with EVs and complement and coagulation system.
Funding
This study was funded by the AGING Project – Department of Excellence – Department of Translational Medicine, Università del Piemonte Orientale and by Ricerca sulle malattie da invecchiamento 2016 – Fondazione Cariplo.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Maas SLN, Breakefield XO, Weaver AM. Extracellular vesicles: unique intercellular delivery vehicles. Trends Cell Biol. (2017) 27:172–88. doi: 10.1016/j.tcb.2016.11.003
2. Karpman D, Ståhl AL, Arvidsson I. Extracellular vesicles in renal disease. Nat Rev Nephrol. (2017) 13:545–62. doi: 10.1038/nrneph.2017.98
3. Lv LL, Feng Y, Tang TT, Liu BC. New insight into the role of extracellular vesicles in kidney disease. J Cell Mol Med. (2019) 23:731–9. doi: 10.1111/jcmm.14101
4. Robbins PD, Dorronsoro A, Booker CN. Regulation of chronic inflammatory and immune processes by extracellular vesicles. J Clin Invest. (2016) 126:1173–80. doi: 10.1172/JCI81131
5. Caruso S, Poon IKH. Apoptotic cell-derived extracellular vesicles: more than just Debris. Front Immunol. (2018) 9:1486. doi: 10.3389/fimmu.2018.01486
6. Veerman RE, Güçlüler Akpinar G, Eldh M, Gabrielsson S. Immune cell-derived extracellular vesicles - functions and therapeutic applications. Trends Mol Med. (2019) 25:382–94. doi: 10.1016/j.molmed.2019.02.003
7. Lema DA, Burlingham WJ. Role of exosomes in tumour and transplant immune regulation. Scand J Immunol. (2019) 8:e12807. doi: 10.1111/sji.12807
8. Yáñez-Mó M, Siljander PR, Andreu Z, Zavec AB, Borràs FE, Buzas EI, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles. (2015) 4:27066. doi: 10.3402/jev.v4.27066
9. Lai RC, Tan SS, Yeo RW, Choo AB, Reiner AT, Su Y, et al. MSC secretes at least 3 EV types each with a unique permutation of membrane lipid, protein and RNA. J Extracell Vesicles. (2016) 5:29828. doi: 10.3402/jev.v5.29828
10. Laulagnier K, Motta C, Hamdi S, Roy S, Fauvelle F, Pageaux JF, et al. Mast cell- and dendritic cell-derived exosomes display a specific lipid composition and an unusual membrane organization. Biochem J. (2004) 380:161–71. doi: 10.1042/bj20031594
11. Alexy T, Rooney K, Weber M, Gray WD, Searles CD. TNF-α alters the release and transfer of microparticle-encapsulated miRNAs from endothelial cells. Physiol Genomics. (2014) 46:833–40. doi: 10.1152/physiolgenomics.00079.2014
12. Ståhl AL, Arvidsson I, Johansson KE, Chromek M, Rebetz J, Loos S, et al. A novel mechanism of bacterial toxin transfer within host blood cell-derived microvesicles. PLoS Pathog. (2015) 11:e1004619. doi: 10.1371/journal.ppat.1004619
13. Mulcahy LA, Pink RC, Carter DR. Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles. (2014) 3, 1–14. doi: 10.3402/jev.v3.24641
14. Parolini I, Federici C, Raggi C, Lugini L, Palleschi S, De Milito A, et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J Biol Chem. (2009) 284:34211–22. doi: 10.1074/jbc.M109.041152
15. Mack M, Kleinschmidt A, Brühl H, Klier C, Nelson PJ, Cihak J, et al. Transfer of the chemokine receptor CCR5 between cells by membrane-derived microparticles: a mechanism for cellular human immunodeficiency virus 1 infection. Nat Med. (2000) 6:769–75. doi: 10.1038/77498
16. Kahn R, Mossberg M, Ståhl AL, Johansson K, Lopatko Lindman I, Heijl C, et al. Microvesicle transfer of kinin B1-receptors is a novel inflammatory mechanism in vasculitis. Kidney Int. (2017) 91:96–105. doi: 10.1016/j.kint.2016.09.023
17. Montecalvo A, Shufesky WJ, Stolz DB, Sullivan MG, Wang Z, Divito SJ, et al. Exosomes as a short-range mechanism to spread alloantigen between dendritic cells during T cell allorecognition. J Immunol. (2008) 180:3081–90. doi: 10.4049/jimmunol.180.5.3081
18. Walker JD, Maier CL, Pober JS. Cytomegalovirus-infected human endothelial cells can stimulate allogeneic CD4+ memory T cells by releasing antigenic exosomes. J Immunol. (2009) 182:1548–59. doi: 10.4049/jimmunol.182.3.1548
19. Groot Kormelink T, Mol S, de Jong EC, Wauben MHM. The role of extracellular vesicles when innate meets adaptive. Semin Immunopathol. (2018) 40:439–52. doi: 10.1007/s00281-018-0681-1
20. Murphy SP, Porrett PM, Turka LA. Innate immunity in transplant tolerance and rejection. Immunol Rev. (2011) 241:39–48. doi: 10.1111/j.1600-065X.2011.01009.x
21. Mueller FB, Yang H, Lubetzky M, Verma A, Lee JR, Dadhania DM, et al. Landscape of innate immune system transcriptome and acute T cell-mediated rejection of human kidney allografts. JCI Insight. (2019) 4:e128014. doi: 10.1172/jci.insight.128014
22. Stahl PD, Raposo G. Exosomes and extracellular vesicles: the path forward. Essays Biochem. (2018) 62:119–24. doi: 10.1042/EBC20170088
23. Breedveld A, Groot Kormelink T, van Egmond M, de Jong EC. Granulocytes as modulators of dendritic cell function. J Leukoc Biol. (2017) 102:1003–16. doi: 10.1189/jlb.4MR0217-048RR
24. Eken C, Gasser O, Zenhaeusern G, Oehri I, Hess C, Schifferli JA. Polymorphonuclear neutrophil-derived ectosomes interfere with the maturation of monocyte-derived dendritic cells. J Immunol. (2008) 180:817–24. doi: 10.4049/jimmunol.180.2.817
25. Kelly A, Houston SA, Sherwood E, Casulli J, Travis MA. Regulation of innate and adaptive immunity by TGFβ. Adv Immunol. (2017) 134:137–233. doi: 10.1016/bs.ai.2017.01.001
26. Eken C, Sadallah S, Martin PJ, Treves S, Schifferli JA. Ectosomes of polymorphonuclear neutrophils activate multiple signaling pathways in macrophages. Immunobiology. (2013) 218:382–92. doi: 10.1016/j.imbio.2012.05.021
27. Shen G, Krienke S, Schiller P, Nießen A, Neu S, Eckstein V, et al. Microvesicles released by apoptotic human neutrophils suppress proliferation and IL-2/IL-2 receptor expression of resting T helper cells. Eur J Immunol. (2017) 47:900–10. doi: 10.1002/eji.201546203
28. van Spriel AB, Leusen JH, van Egmond M, Dijkman HB, Assmann KJ, Mayadas TN, et al. Mac-1 (CD11b/CD18) is essential for Fc receptor-mediated neutrophil cytotoxicity and immunologic synapse formation. Blood. (2001) 97:2478–86. doi: 10.1182/blood.V97.8.2478
29. Gavins FN, Hickey MJ. Annexin A1 and the regulation of innate and adaptive immunity. Front Immunol. (2012) 3:354. doi: 10.3389/fimmu.2012.00354
30. Mayadas TN, Cullere X, Lowell CA. The multifaceted functions of neutrophils. Annu Rev Pathol. (2014) 9:181–218. doi: 10.1146/annurev-pathol-020712-164023
31. Odobasic D, Kitching AR, Yang Y, O'Sullivan KM, Muljadi RC, Edgtton KL, et al. Neutrophil myeloperoxidase regulates T-cell-driven tissue inflammation in mice by inhibiting dendritic cell function. Blood. (2013) 121:4195–204. doi: 10.1182/blood-2012-09-456483
32. Maffia PC, Zittermann SE, Scimone ML, Tateosian N, Amiano N, Guerrieri D, et al. Neutrophil elastase converts human immature dendritic cells into transforming growth factor-beta1-secreting cells and reduces allostimulatory ability. Am J Pathol. (2007) 171:928–37. doi: 10.2353/ajpath.2007.061043
33. Bank U, Ansorge S. More than destructive: neutrophil-derived serine proteases in cytokine bioactivity control. J Leukoc Biol. (2001) 69:197–206. doi: 10.1189/jlb.69.2.197
34. Fu Z, Thorpe M, Alemayehu R, Roy A, Kervinen J, de Garavilla L, et al. Highly selective cleavage of cytokines and chemokines by the human mast cell chymase and neutrophil cathepsin G. J Immunol. (2017) 198:1474–83. doi: 10.4049/jimmunol.1601223
35. Majumdar R, Tavakoli Tameh A, Parent CA. Exosomes mediate LTB4 release during neutrophil chemotaxis. PLoS Biol. (2016) 14:e1002336. doi: 10.1371/journal.pbio.1002336
36. Unnewehr H, Rittirsch D, Sarma JV, Zetoune F, Flierl MA, Perl M, et al. Changes and regulation of the C5a receptor on neutrophils during septic shock in humans. J Immunol. (2013) 190:4215–25. doi: 10.4049/jimmunol.1200534
37. Singh PP, Smith VL, Karakousis PC, Schorey JS. Exosomes isolated from mycobacteria-infected mice or cultured macrophages can recruit and activate immune cells in vitro and in vivo. J Immunol. (2012) 189:777–85. doi: 10.4049/jimmunol.1103638
38. Cheng L, Wang Y, Huang L. Exosomes from M1-polarized macrophages potentiate the cancer vaccine by creating a pro-inflammatory microenvironment in the lymph node. Mol Ther. (2017) 25:1665–75. doi: 10.1016/j.ymthe.2017.02.007
39. Anand PK, Anand E, Bleck CK, Anes E, Griffiths G. Exosomal Hsp70 induces a pro-inflammatory response to foreign particles including mycobacteria. PLoS ONE. (2010) 5:e10136. doi: 10.1371/journal.pone.0010136
40. Ben-Sasson SZ, Wang K, Cohen J, Paul WE. IL-1β strikingly enhances antigen-driven CD4 and CD8 T-cell responses. Cold Spring Harb Symp Quant Biol. (2013) 78:117–24. doi: 10.1101/sqb.2013.78.021246
41. Rosser EC, Oleinika K, Tonon S, Doyle R, Bosma A, Carter NA, et al. Regulatory B cells are induced by gut microbiota-driven interleukin-1β and interleukin-6 production. Nat Med. (2014) 20:1334–9. doi: 10.1038/nm.3680
42. Ismail N, Wang Y, Dakhlallah D, Moldovan L, Agarwal K, Batte K, et al. Macrophage microvesicles induce macrophage differentiation and miR-223 transfer. Blood. (2013) 121:984–95. doi: 10.1182/blood-2011-08-374793
43. Esser J, Gehrmann U, D'Alexandri FL, Hidalgo-Estévez AM, Wheelock CE, Scheynius A, et al. Exosomes from human macrophages and dendritic cells contain enzymes for leukotriene biosynthesis and promote granulocyte migration. J Allergy Clin Immunol. (2010) 126:1032–40. doi: 10.1016/j.jaci.2010.06.039
44. Zhang Y, Liu F, Yuan Y, Jin C, Chang C, Zhu Y, et al. Inflammasome-derived exosomes activate NF-κB signaling in macrophages. J Proteome Res. (2017) 16:170–8. doi: 10.1021/acs.jproteome.6b00599
45. McDonald MK, Tian Y, Qureshi RA, Gormley M, Ertel A, Gao R, et al. Functional significance of macrophage-derived exosomes in inflammation and pain. Pain. (2014) 155:1527–39. doi: 10.1016/j.pain.2014.04.029
46. Dieudé M, West LJ, Muruve DA, Gunaratnam L, Mohanakumar T, Zorn E, et al. New answers to old conundrums: what antibodies, exosomes and inflammasomes bring to the conversation. Canadian National Transplant Research Program International Summit Report. Transplantation. (2018) 102:209–14. doi: 10.1097/TP.0000000000001872
47. Bassoy EY, Towne JE, Gabay C. Regulation and function of interleukin-36 cytokines. Immunol Rev. (2018) 281:169–78. doi: 10.1111/imr.12610
48. Papp K, Végh P, Prechl J, Kerekes K, Kovács J, Csikós G, et al. B lymphocytes and macrophages release cell membrane deposited C3-fragments on exosomes with T cell response-enhancing capacity. Mol Immunol. (2008) 45:2343–51. doi: 10.1016/j.molimm.2007.11.021
49. Said A, Weindl G. Regulation of dendritic cell function in inflammation. J Immunol Res. (2015) 2015:743169. doi: 10.1155/2015/743169
50. Zhang Y, Meng J, Zhang L, Ramkrishnan S, Roy S. Extracellular vesicles with exosome-like features transfer TLRs between dendritic cells. Immunohorizons. (2019) 3:186–93. doi: 10.4049/immunohorizons.1900016
51. Groot Kormelink T, Arkesteijn GJ, van de Lest CH, Geerts WJ, Goerdayal SS, Altelaar MA, et al. Mast cell degranulation is accompanied by the release of a selective subset of extracellular vesicles that contain mast cell-specific proteases. J Immunol. (2016) 197:3382–92. doi: 10.4049/jimmunol.1600614
52. Liu Q, Rojas-Canales DM, Divito SJ, Shufesky WJ, Stolz DB, Erdos G, et al. Donor dendritic cell-derived exosomes promote allograft-targeting immune response. J Clin Invest. (2016) 126:2805–20. doi: 10.1172/JCI84577
53. Gonzalez-Nolasco B, Wang M, Prunevieille A, Benichou G. Emerging role of exosomes in allorecognition and allograft rejection. Curr Opin Organ Transplant. (2018) 23:22–7. doi: 10.1097/MOT.0000000000000489
54. Denzer K, van Eijk M, Kleijmeer MJ, Jakobson E, de Groot C, Geuze HJ. Follicular dendritic cells carry MHC class II-expressing microvesicles at their surface. J Immunol. (2000) 165:1259–65. doi: 10.4049/jimmunol.165.3.1259
55. Segura E, Amigorena S, Théry C. Mature dendritic cells secrete exosomes with strong ability to induce antigen-specific effector immune responses. Blood Cells Mol Dis. (2005) 35:89–93. doi: 10.1016/j.bcmd.2005.05.003
56. Tkach M, Kowal J, Zucchetti AE, Enserink L, Jouve M, Lankar D, et al. Qualitative differences in T-cell activation by dendritic cell-derived extracellular vesicle subtypes. EMBO J. (2017) 36:3012–28. doi: 10.15252/embj.201696003
57. Skokos D, Botros HG, Demeure C, Morin J, Peronet R, Birkenmeier G, et al. Mast cell-derived exosomes induce phenotypic and functional maturation of dendritic cells and elicit specific immune responses in vivo. J Immunol. (2003) 170:3037–45. doi: 10.4049/jimmunol.170.6.3037
58. Carroll-Portillo A, Cannon JL, te Riet J, Holmes A, Kawakami Y, Kawakami T, et al. Mast cells and dendritic cells form synapses that facilitate antigen transfer for T cell activation. J Cell Biol. (2015) 210:851–64. doi: 10.1083/jcb.201412074
59. Binder RJ. Functions of heat shock proteins in pathways of the innate and adaptive immune system. J Immunol. (2014) 193:5765–71. doi: 10.4049/jimmunol.1401417
60. Subra C, Grand D, Laulagnier K, Stella A, Lambeau G, Paillasse M, et al. Exosomes account for vesicle-mediated transcellular transport of activatable phospholipases and prostaglandins. J Lipid Res. (2010) 51:2105–20. doi: 10.1194/jlr.M003657
61. Cheung KL, Jarrett R, Subramaniam S, Salimi M, Gutowska-Owsiak D, Chen YL, et al. Psoriatic T cells recognize neolipid antigens generated by mast cell phospholipase delivered by exosomes and presented by CD1a. J Exp Med. (2016) 213:2399–412. doi: 10.1084/jem.20160258
62. Mion F, D'Incà F, Danelli L, Toffoletto B, Guarnotta C, Frossi B, et al. Mast cells control the expansion and differentiation of IL-10-competent B cells. J Immunol. (2014) 193:4568–79. doi: 10.4049/jimmunol.1302593
63. Akuthota P, Carmo LA, Bonjour K, Murphy RO, Silva TP, Gamalier JP, et al. Extracellular microvesicle production by human eosinophils activated by “inflammatory” stimuli. Front Cell Dev Biol. (2016) 4:117. doi: 10.3389/fcell.2016.00117
64. Cañas JA, Sastre B, Mazzeo C, Fernández-Nieto M, Rodrigo-Muñoz JM, González-Guerra A, et al. Exosomes from eosinophils autoregulate and promote eosinophil functions. J Leukoc Biol. (2017) 101:1191–9. doi: 10.1189/jlb.3AB0516-233RR
65. Lugini L, Cecchetti S, Huber V, Luciani F, Macchia G, Spadaro F, et al. Immune surveillance properties of human NK cell-derived exosomes. J Immunol. (2012) 189:2833–42. doi: 10.4049/jimmunol.1101988
66. Hoppstädter J, Dembek A, Linnenberger R, Dahlem C, Barghash A, Fecher-Trost C, et al. Toll-like receptor 2 release by macrophages: an anti-inflammatory program induced by glucocorticoids and lipopolysaccharide. Front Immunol. (2019) 10:1634. doi: 10.3389/fimmu.2019.01634
67. Cucchiari D, Podestà MA, Ponticelli C. The critical role of innate immunity in kidney transplantation. Nephron. (2016) 132:227–37. doi: 10.1159/000444267
68. Zhuang Q, Lakkis FG. Dendritic cells and innate immunity in kidney transplantation. Kidney Int. (2015) 87:712–8. doi: 10.1038/ki.2014.430
69. Montecalvo A, Larregina AT, Shufesky WJ, Stolz DB, Sullivan ML, Karlsson JM, et al. Mechanism of transfer of functional microRNAs between mouse denditic cells via exosomes. Blood. (2012) 119:756–66. doi: 10.1182/blood-2011-02-338004
70. Marino J, Babiker-Mohamed MH, Crosby-Bertorini P, Paster JT, LeGuern C, Germana S, et al. Donor exosomes rather than passenger leukocytes initiate alloreactive T cell responses after transplantation. Sci Immunol. (2016) 1:aaf8759. doi: 10.1126/sciimmunol.aaf8759
71. Morelli AE, Bracamonte-Baran W, Burlingham WJ. Donor-derived exosomes: the trick behind the semidirect pathway of allorecognition. Curr Opin Organ Transplant. (2017) 22:46–54. doi: 10.1097/MOT.0000000000000372
72. Benichou G, Prunevieille A. Graft-derived exosomes. When small vesicles play a big role in transplant rejection. Am J Transplant. (2018) 18:1585–6. doi: 10.1111/ajt.14720
73. Buschow SI, Nolte-'t Hoen EN, van Niel G, Pols MS, ten Broeke T, Lauwen M, et al. MHC II in dendritic cells is targeted to lysosomes or T cell-induced exosomes via distinct multivesicular body pathways. Traffic. (2009) 10:1528–42. doi: 10.1111/j.1600-0854.2009.00963.x
74. Qazi KR, Gehrmann U, Domange Jordö E, Karlsson MC, Gabrielsson S. Antigen-loaded exosomes alone induce Th1-type memory through a B-cell-dependent mechanism. Blood. (2009) 113:2673–83. doi: 10.1182/blood-2008-04-153536
75. Li X, Li JJ, Yang JY, Wang DS, Zhao W, Song WJ, et al. Tolerance induction by exosomes from immature dendritic cells and rapamycin in a mouse cardiac allograft model. PLoS ONE. (2012) 7:e44045. doi: 10.1371/journal.pone.0044045
76. Podestà MA, Cucchiari D, Ponticelli C. The diverging roles of dendritic cells in kidney allotransplantation. Transplant Rev. (2015) 29:114–20. doi: 10.1016/j.trre.2015.04.001
77. Ono Y, Perez-Gutierrez A, Nakao T, Dai H, Camirand G, Yoshida O, et al. Graft-infiltrating PD-L1hi cross-dressed dendritic cells regulate antidonor T cell responses in mouse liver transplant tolerance. Hepatology. (2018) 67:1499–515. doi: 10.1002/hep.29529
78. Bracamonte-Baran W, Florentin J, Zhou Y, Jankowska-Gan E, Haynes WJ, Zhong W, et al. Modification of host dendritic cells by microchimerism-derived extracellular vesicles generates split tolerance. Proc Natl Acad Sci USA. (2017) 114:1099–104. doi: 10.1073/pnas.1618364114
79. Tung SL, Boardman DA, Sen M, Letizia M, Peng Q, Cianci N, et al. Regulatory T cell-derived extracellular vesicles modify dendritic cell function. Sci Rep. (2018) 8:6065. doi: 10.1038/s41598-018-24531-8
80. Shefler I, Salamon P, Hershko AY, Mekori YA. Mast cells as sources and targets of membrane vesicles. Curr Pharm Des. (2011) 17:3797–804. doi: 10.2174/138161211798357836
81. Jahanyar J, Koerner MM, Loebe M, Youker KA, Torre-Amione G, Noon GP. The role of mast cells after solid organ transplantation. Transplantation. (2008) 85:1365–71. doi: 10.1097/TP.0b013e31816fc0a3
82. Panther E, Idzko M, Corinti S, Ferrari D, Herouy Y, Mockenhaupt M, et al. The influence of lysophosphatidic acid on the functions of human dendritic cells. J Immunol. (2002) 169:4129–35. doi: 10.4049/jimmunol.169.8.4129
83. Mekori YA, Hershko AY, Frossi B, Mion F, Pucillo CE. Integrating innate and adaptive immune cells: mast cells as crossroads between regulatory and effector B and T cells. Eur J Pharmacol. (2016) 778:84–9. doi: 10.1016/j.ejphar.2015.03.087
84. Lotfi R, Lotze MT. Eosinophils induce DC maturation, regulating immunity. J Leukoc Biol. (2008) 83:456–60. doi: 10.1189/jlb.0607366
85. Wen C, Seeger RC, Fabbri M, Wang L, Wayne AS, Jong AY. Biological roles and potential applications of immune cell-derived extracellular vesicles. J Extracell Vesicles. (2017) 6:1400370. doi: 10.1080/20013078.2017.1400370
86. Wu CH, Li J, Li L, Sun J, Fabbri M, Wayne AS, et al. Extracellular vesicles derived from natural killer cells use multiple cytotoxic proteins and killing mechanisms to target cancer cells. J Extracell Vesicles. (2019) 8:1588538. doi: 10.1080/20013078.2019.1588538
87. López-Botet M, Vilches C, Redondo-Pachón D, Muntasell A, Pupuleku A, Yélamos J, et al. Dual role of natural killer cells on graft rejection and control of cytomegalovirus infection in renal transplantation. Front Immunol. (2017) 8:166. doi: 10.3389/fimmu.2017.00166
88. Karasu E, Eisenhardt SU, Harant J, Huber-Lang M. Extracellular vesicles: packages sent with complement. Front Immunol. (2018) 9:721. doi: 10.3389/fimmu.2018.00721
89. Meehan SM, Limsrichamrern S, Manaligod JR, Junsanto T, Josephson MA, Thistlethwaite JR, et al. Platelets and capillary injury in acute humoral rejection of renal allografts. Hum Pathol. (2003) 34:533–40. doi: 10.1016/S0046-8177(03)00189-8
90. Yu ZX, Qi S, Lasaro MA, Bouchard K, Dow C, Moore K, et al. Targeting complement pathways during cold ischemia and reperfusion prevents delayed graft function. Am J Transplant. (2016) 16:2589–97. doi: 10.1111/ajt.13797
91. Karpman D, Ståhl AL, Arvidsson I, Johansson K, Loos S, Tati R, et al. Complement interactions with blood cells, endothelial cells and microvesicles in thrombotic and inflammatory conditions. Adv Exp Med Biol. (2015) 865:19–42. doi: 10.1007/978-3-319-18603-0_2
92. Bradley AJ, Brooks DE, Norris-Jones R, Devine DV. C1q binding to liposomes is surface charge dependent and is inhibited by peptides consisting of residues 14–26 of the human C1qA chain in a sequence independent manner. Biochim Biophys Acta. (1999) 1418:19–30.
93. Biro' E, Nieuwland R, Tak PP, Pronk LM, Schaap MC, Sturk A, et al. Activated complement components and complement activator molecules on the surface of cell-derived microparticles in patients with rheumatoid arthritis and healthy individuals. Ann Rheum Dis. (2007) 66:1085–92. doi: 10.1136/ard.2006.061309
94. Gasser O, Schifferli JA. Microparticles released by human neutrophils adhere to erythrocytes in the presence of complement. Exp Cell Res. (2005) 307:381–7. doi: 10.1016/j.yexcr.2005.03.011
95. Morel O, Morel N, Jesel L, Freyssinet JM, Toti F. Microparticles: a critical component in the nexus between inflammation, immunity, and thrombosis. Semin Immunopathol. (2011) 33:469–86. doi: 10.1007/s00281-010-0239-3
96. Bakouboula B, Morel O, Faller AL, Freyssinet JM, Toti F. Significance of membrane microparticles in solid graft and cellular transplantation. Front Biosci. (2011) 16:2499–514. doi: 10.2741/3868
97. Clayton A, Harris CL, Court J, Mason MD, Morgan BP. Antigen-presenting cell exosomes are protected from complement-mediated lysis by expression of CD55 and CD59. Eur J Immunol. (2003) 33:522–31. doi: 10.1002/immu.200310028
98. Cantaluppi V, Medica D, Mannari C, Stiaccini G, Figliolini F, Dellepiane S, et al. Endothelial progenitor cell-derived extracellular vesicles protect from complement-mediated mesangial injury in experimental anti-Thy1.1 glomerulonephritis. Nephrol Dial Transplant. (2015) 30:410–22. doi: 10.1093/ndt/gfu364
99. Gorsuch WB, Chrysanthou E, Schwaeble WJ, Stahl GL. The complement system in ischemia-reperfusion injuries. Immunobiology. (2012) 217:1026–33. doi: 10.1016/j.imbio.2012.07.024
100. Tower CM, Reyes M, Nelson K, Leca N, Kieran N, Muczynski K, et al. Plasma C4d+ endothelial microvesicles increase in acute antibody-mediated rejection. Transplantation. (2017) 101:2235–43. doi: 10.1097/TP.0000000000001572
101. Kozarcanin H, Lood C, Munthe-Fog L, Sandholm K, Hamad OA, Bengtsson AA, et al. The lectin complement pathway serine proteases (MASPs) represent a possible crossroad between the coagulation and complement systems in thromboinflammation. J Thromb Haemost. (2016) 14:531–45. doi: 10.1111/jth.13208
102. Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Ischemia/reperfusion. Compr Physiol. (2016) 7:113–70. doi: 10.1002/cphy.c160006
103. Dignat-George F, Boulanger CM. The many faces of endothelial microparticles. Arterioscler Thromb Vasc Biol. (2011) 31:27–33. doi: 10.1161/ATVBAHA.110.218123
104. Hisada Y, Mackman N. Cancer cell-derived tissue factor-positive extracellular vesicles: biomarkers of thrombosis and survival. Curr Opin Hematol. (2019) 26:349–56. doi: 10.1097/MOH.0000000000000521
105. Shustova ON, Antonova OA, Golubeva NV, Khaspekova SG, Yakushkin VV, Aksuk SA, et al. Differential procoagulant activity of microparticles derived from monocytes, granulocytes, platelets and endothelial cells: impact of active tissue factor. Blood Coagul Fibrinolysis. (2017) 28:373–82. doi: 10.1097/MBC.0000000000000609
106. Gao C, Xie R, Yu C, Ma R, Dong W, Meng H, et al. Thrombotic role of blood and endothelial cells in uremia through phosphatidylserine exposure and microparticle release. PLoS ONE. (2015) 10:e0142835. doi: 10.1371/journal.pone.0142835
107. Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. (2006) 12:682–7. doi: 10.1038/nm1419
108. Abid Hussein MN, Böing AN, Biró E, Hoek FJ, Vogel GM, Meuleman DG, et al. Phospholipid composition of in vitro endothelial microparticles and their in vivo thrombogenic properties. Thromb Res. (2008) 121:865–71. doi: 10.1016/j.thromres.2007.08.005
109. Abid Hussein MN, Böing AN, Sturk A, Hau CM, Nieuwland R. Inhibition of microparticle release triggers endothelial cell apoptosis and detachment. Thromb Haemost. (2007) 98:1096–107. doi: 10.1160/TH05-04-0231
110. Mezentsev A, Merks RM, O'Riordan E, Chen J, Mendelev N, Goligorsky MS, et al. Endothelial microparticles affect angiogenesis in vitro: role of oxidative stress. Am J Physiol Heart Circ Physiol. (2005) 289:H1106–14. doi: 10.1152/ajpheart.00265.2005
111. Melki I, Tessandier N, Zufferey A, Boilard E. Platelet microvesicles in health and disease. Platelets. (2017) 28:214–21. doi: 10.1080/09537104.2016.1265924
112. Bevers EM, Williamson PL. Getting to the outer leaflet: physiology of phosphatidylserine exposure at the plasma membrane. Physiol Rev. (2016) 96:605–45. doi: 10.1152/physrev.00020.2015
113. Ståhl AL, Sartz L, Nelsson A, Békássy ZD, Karpman D. Shiga toxin and lipopolysaccharide induce platelet-leukocyte aggregates and tissue factor release, a thrombotic mechanism in hemolytic uremic syndrome. PLoS ONE. (2009) 4:e6990. doi: 10.1371/journal.pone.0006990
114. Fan GQ, Qin RR, Li YH, Song DJ, Chen TS, Zhang W, et al. Endothelial cells microparticle-associated protein disulfide isomerase promotes platelet activation in metabolic syndrome. Oncotarget. (2016) 7:83231–40. doi: 10.18632/oncotarget.13081
115. Gilbert GE, Sims PJ, Wiedmer T, Furie B, Furie BC, Shattil SJ. Platelet-derived microparticles express high affinity receptors for factor VIII. J Biol Chem. (1991) 266:17261–8.
116. Van Der Meijden PE, Van Schilfgaarde M, Van Oerle R, Renné T, ten Cate H, Spronk HM. Platelet- and erythrocyte-derived microparticles trigger thrombin generation via factor XIIa. J Thromb Haemost. (2012) 10:1355–62. doi: 10.1111/j.1538-7836.2012.04758.x
117. Barry OP, Pratico D, Lawson JA, FitzGerald GA. Transcellular activation of platelets and endothelial cells by bioactive lipids in platelet microparticles. J Clin Invest. (1997) 99:2118–27. doi: 10.1172/JCI119385
118. Brown GT, McIntyre TM. Lipopolysaccharide signaling without a nucleus: kinase cascades stimulate platelet shedding of proinflammatory IL-1β-rich microparticles. J Immunol. (2011) 186:5489–96. doi: 10.4049/jimmunol.1001623
119. Mause SF, von Hundelshausen P, Zernecke A, Koenen RR, Weber C. Platelet microparticles: a transcellular delivery system for RANTES promoting monocyte recruitment on endothelium. Arterioscler Thromb Vasc Biol. (2005) 25:1512–8. doi: 10.1161/01.ATV.0000170133.43608.37
120. Sinauridze EI, Kireev DA, Popenko NY, Pichugin AV, Panteleev MA, Krymskaya OV, et al. Platelet microparticle membranes have 50- to 100-fold higher specific procoagulant activity than activated platelets. Thromb Haemost. (2007) 97:425–34. doi: 10.1160/TH06-06-0313
121. Kim HK, Song KS, Chung JH, Lee KR, Lee SN. Platelet microparticles induce angiogenesis in vitro. Br J Haematol. (2004) 124:376–84. doi: 10.1046/j.1365-2141.2003.04773.x
122. Ponticelli C. Ischaemia-reperfusion injury: a major protagonist in kidney transplantation. Nephrol Dial Transplant. (2014) 29:1134–40. doi: 10.1093/ndt/gft488
123. Eltzschig HK, Eckle T. Ischemia and reperfusion–from mechanism to translation. Nat Med. (2011) 17:1391–401. doi: 10.1038/nm.2507
124. Duan P, Tan J, Miao Y, Zhang Q. Potential role of exosomes in the pathophysiology, diagnosis, and treatment of hypoxic diseases. Am J Transl Res. (2019) 11:1184–201.
125. Wang T, Gilkes DM, Takano N, Xiang L, Luo W, Bishop CJ, et al. Hypoxia-inducible factors and RAB22A mediate formation of microvesicles that stimulate breast cancer invasion and metastasis. Proc Natl Acad Sci USA. (2014) 111:E3234–42. doi: 10.1073/pnas.1410041111
126. Sturtzel C. Endothelial Cells. Adv Exp Med Biol. (2017) 1003:71–91. doi: 10.1007/978-3-319-57613-8_4
127. Cardinal H, Dieudé M, Hébert MJ. Endothelial dysfunction in kidney transplantation. Front Immunol. (2018) 9:1130. doi: 10.3389/fimmu.2018.01130
128. Yang B, Lan S, Dieudé M, Sabo-Vatasescu JP, Karakeussian-Rimbaud A, Turgeon J, et al. Caspase-3 is a pivotal regulator of microvascular rarefaction and renal fibrosis after ischemia-reperfusion injury. J Am Soc Nephrol. (2018) 29:1900–16. doi: 10.1681/ASN.2017050581
129. Kouwaki T, Okamoto M, Tsukamoto H, Fukushima Y, Oshiumi H. Extracellular vesicles deliver host and virus RNA and regulate innate immune response. Int J Mol Sci. (2017) 18:666. doi: 10.3390/ijms18030666
130. Cocucci E, Racchetti G, Meldolesi J. Shedding microvesicles: artefacts no more. Trends Cell Biol. (2009) 19:43–51. doi: 10.1016/j.tcb.2008.11.003
131. Dieudé M, Bell C, Turgeon J, Beillevaire D, Pomerleau L, Yang B, et al. The 20S proteasome core, active within apoptotic exosome-like vesicles, induces autoantibody production and accelerates rejection. Sci Transl Med. (2015) 7:318ra200. doi: 10.1126/scitranslmed.aac9816
132. Bansal S, Sharma M, Ranjithkumar R, Mohanakumar T. The role of exosomes in allograft immunity. Cell Immunol. (2018) 331:85–92. doi: 10.1016/j.cellimm.2018.06.003
133. Sharma M, Ravichandran R, Bansal S, Bremner RM, Smith MA, Mohanakumar T. Tissue-associated self-antigens containing exosomes: role in allograft rejection. Hum Immunol. (2018) 79:653–8. doi: 10.1016/j.humimm.2018.06.005
134. Sharma M, Liu W, Perincheri S, Gunasekaran M, Mohanakumar T. Exosomes expressing the self-antigens myosin and vimentin play an important role in syngeneic cardiac transplant rejection induced by antibodies to cardiac myosin. Am J Transplant. (2018) 18:1626–35. doi: 10.1111/ajt.14650
135. Feigerlová E, Battaglia-Hsu SF, Hauet T, Guéant JL. Extracellular vesicles as immune mediators in response to kidney injury. Am J Physiol Renal Physiol. (2018) 314:F9–21. doi: 10.1152/ajprenal.00336.2017
136. Zhang W, Zhou X, Yao Q, Liu Y, Zhang H, Dong Z. HIF-1-mediated production of exosomes during hypoxia is protective in renal tubular cells. Am J Physiol Renal Physiol. (2017) 313:F906–13. doi: 10.1152/ajprenal.00178.2017
137. Castellano G, Franzin R, Sallustio F, Stasi A, Banelli B, Romani M, et al. Complement component C5a induces aberrant epigenetic modifications in renal tubular epithelial cells accelerating senescence by Wnt4/βcatenin signaling after ischemia/reperfusion injury. Aging. (2019) 11:4382–406. doi: 10.18632/aging.102059
138. Buzas EI, György B, Nagy G, Falus A, Gay S. Emerging role of extracellular vesicles in inflammatory diseases. Nat Rev Rheumatol. (2014) 10:356–64. doi: 10.1038/nrrheum.2014.19
139. Jansen MP, Emal D, Teske GJ, Dessing MC, Florquin S, Roelofs JJ. Release of extracellular DNA influences renal ischemia reperfusion injury by platelet activation and formation of neutrophil extracellular traps. Kidney Int. (2017) 91:352–64. doi: 10.1016/j.kint.2016.08.006
140. Lou Q, Li Y, Hou B, Liu Y, Zhang Y, Hao J, et al. Heat shock transcription factor 1 affects kidney tubular cell migration by regulating the TGF β1 Smad2/3 signaling pathway. Mol Med Rep. (2019) 20:4323–30. doi: 10.3892/mmr.2019.10689
141. Feng D, Wang Y, Liu Y, Wu L, Li X, Chen Y, et al. DC-SIGN reacts with TLR-4 and regulates inflammatory cytokine expression via NF-κB activation in renal tubular epithelial cells during acute renal injury. Clin Exp Immunol. (2018) 191:107–15. doi: 10.1111/cei.13048
142. Wang X, Wilkinson R, Kildey K, Potriquet J, Mulvenna J, Lobb RJ, et al. Unique molecular profile of exosomes derived from primary human proximal tubular epithelial cells under diseased conditions. J Extracell Vesicles. (2017) 6:1314073. doi: 10.1080/20013078.2017.1314073
143. Borges FT, Melo SA, Özdemir BC, Kato N, Revuelta I, Miller CA, et al. TGF-β1-containing exosomes from injured epithelial cells activate fibroblasts to initiate tissue regenerative responses and fibrosis. J Am Soc Nephrol. (2013) 24:385–92. doi: 10.1681/ASN.2012101031
144. Zheng SB, Zheng Y, Jin LW, Zhou ZH, Li ZY. Microvesicles containing microRNA-21 secreted by proximal tubular epithelial cells are involved in renal interstitial fibrosis by activating AKT pathway. Eur Rev Med Pharmacol Sci. (2018) 22:707–14. doi: 10.26355/eurrev_201802_14297
145. Fukushima Y, Okamoto M, Ishikawa K, Kouwaki T, Tsukamoto H, Oshiumi H. Activation of TLR3 and its adaptor TICAM-1 increases miR-21 levels in extracellular vesicles released from human cells. Biochem Biophys Res Commun. (2018) 500:744–50. doi: 10.1016/j.bbrc.2018.04.146
146. Wu H, Huang T, Ying L, Han C, Li D, Xu Y, et al. MiR-155 is involved in renal ischemia-reperfusion injury via direct targeting of FoxO3a and regulating renal tubular cell pyroptosis. Cell Physiol Biochem. (2016) 40:1692–705. doi: 10.1159/000453218
147. Zhang XB, Chen X, Li DJ, Qi GN, Dai YQ, Gu J, et al. Inhibition of miR-155 ameliorates acute kidney injury by apoptosis involving the regulation on TCF4/Wnt/β-catenin pathway. Nephron. (2019) 143:135–47. doi: 10.1159/000501038
148. Lv LL, Feng Y, Wen Y, Wu WJ, Ni HF, Li ZL, et al. Exosomal CCL2 from tubular epithelial cells is critical for albumin-induced tubulointerstitial inflammation. Am Soc Nephrol. (2018) 29:919–35. doi: 10.1681/ASN.2017050523
149. Shimoda M, Khokha R. Metalloproteinases in extracellular vesicles. Biochim Biophys Acta Mol Cell Res. (2017) 1864:1989–2000. doi: 10.1016/j.bbamcr.2017.05.027
150. Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol. (2015) 11:264–76. doi: 10.1038/nrneph.2015.3
151. Docherty MH, O'Sullivan ED, Bonventre JV, Ferenbach DA. Cellular senescence in the kidney. J Am Soc Nephrol. (2019) 30:726–36. doi: 10.1681/ASN.2018121251
152. Brinchmann MF, Patel DM, Iversen MH. The role of galectins as modulators of metabolism and inflammation. Mediat Inflamm. (2018) 2018:9186940. doi: 10.1155/2018/9186940
153. Yang L, Brooks CR, Xiao S, Sabbisetti V, Yeung MY, Hsiao LL, et al. KIM-1-mediated phagocytosis reduces acute injury to the kidney. J Clin Invest. (2015) 125:1620–36. doi: 10.1172/JCI75417
154. Shi N, Wu MP. Apolipoprotein A-I attenuates renal ischemia/reperfusion injury in rats. J Biomed Sci. (2008) 15:577–83. doi: 10.1007/s11373-008-9258-7
155. Fernandez-Martínez AB, Torija AV, Carracedo J, Ramirez R, de Lucio-Cazaña FJ. Microparticles released by vascular endothelial cells increase hypoxia inducible factor expression in human proximal tubular HK-2 cells. Int J Biochem Cell Biol. (2014) 53:334–42. doi: 10.1016/j.biocel.2014.05.025
156. Cantaluppi V, Biancone L, Quercia A, Deregibus MC, Segoloni G, Camussi G. Rationale of mesenchymal stem cell therapy in kidney injury. Am J Kidney Dis. (2013) 61:300–9. doi: 10.1053/j.ajkd.2012.05.027
157. Nargesi AA, Lerman LO, Eirin A. Mesenchymal stem cell-derived extracellular vesicles for renal repair. Curr Gene Ther. (2017) 17:29–42. doi: 10.2174/1566523217666170412110724
158. Fatima F, Ekstrom K, Nazarenko I, Maugeri M, Valadi H, Hill AF, et al. Non-coding RNAs in mesenchymal stem cell-derived extracellular vesicles: deciphering regulatory roles in stem cell potency, inflammatory resolve, and tissue regeneration. Front Genet. (2017) 8:161. doi: 10.3389/fgene.2017.00161
159. Burrello J, Monticone S, Gai C, Gomez Y, Kholia S, Camussi G. Stem cell-derived extracellular vesicles and immune-modulation. Front Cell Dev Biol. (2016) 4:83. doi: 10.3389/fcell.2016.00083
160. Haarer J, Johnson CL, Soeder Y, Dahlke MH. Caveats of mesenchymal stem cell therapy in solid organ transplantation. Transpl Int. (2015) 28:1–9. doi: 10.1111/tri.12415
161. Koch M, Lemke A, Lange C. Extracellular vesicles from MSC modulate the immune response to renal allografts in a MHC disparate rat model. Stem Cells Int. (2015) 2015:486141. doi: 10.1155/2015/486141
162. Mardpour S, Hamidieh AA, Taleahmad S, Sharifzad F, Taghikhani A, Baharvand H. Interaction between mesenchymal stromal cell-derived extracellular vesicles and immune cells by distinct protein content. J Cell Physiol. (2019) 234:8249–58. doi: 10.1002/jcp.27669
163. Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. (2006) 8:315–7. doi: 10.1080/14653240600855905
164. Tsuji K, Kitamura S, Wada J. Secretomes from mesenchymal stem cells against acute kidney injury: possible heterogeneity. Stem Cells Int. (2018) 2018:8693137. doi: 10.1155/2018/8693137
165. Kaushal GP, Shah SV. Autophagy in acute kidney injury. Kid Int. (2016) 98:779–91. doi: 10.1016/j.kint.2015.11.021
166. Gatti S, Bruno S, Deregibus MC, Sordi A, Cantaluppi V, Tetta C, et al. Microvesicles derived from human adult mesenchymal stem cells protect against ischaemia-reperfusion-induced acute and chronic kidney injury. Nephrol Dial Transplant. (2011) 26:1474–83. doi: 10.1093/ndt/gfr015
167. Ferguson SW, Wang J, Lee CJ, Liu M, Neelamegham S, Canty JM, et al. The microRNA regulatory landscape of MSC-derived exosomes: a systems view. Sci Rep. (2018) 8:1419. doi: 10.1038/s41598-018-19581-x
168. Wang SY, Hong Q, Zhang CY, Yang YJ, Cai GY, Chen XM. miRNAs in stem cell-derived extracellular vesicles for acute kidney injury treatment: comprehensive review of preclinical studies. Stem Cell Res Ther. (2019) 10:281. doi: 10.1186/s13287-019-1371-1
169. Lv LL, Wu WJ, Feng Y, Li ZL, Tang TT, Liu BC. Therapeutic application of extracellular vesicles in kidney disease: promises and challenges. J Cell Mol Med. (2018) 22:728–37. doi: 10.1111/jcmm.13407
170. Kezić A, Stajic N, Thaiss F. Innate immune response in kidney ischemia/reperfusion injury: potential target for therapy. J Immunol Res. (2017) 2017:6305439. doi: 10.1155/2017/6305439
171. Malek M, Nematbakhsh M. Renal ischemia/reperfusion injury; from pathophysiology to treatment. J Renal Inj Prev. (2015) 4:20–7. doi: 10.12861/jrip.2015.06
172. Liang X, Zhang L, Wang S, Han Q, Zhao RC. Exosomes secreted by mesenchymal stem cells promote endothelial cell angiogenesis by transferring miR-125a. J Cell Sci. (2016) 129:2182–9. doi: 10.1242/jcs.170373
173. Hu H, Hu S, Xu S, Gao Y, Zeng F, Shui H. miR-29b regulates Ang II-induced EMT of rat renal tubular epithelial cells via targeting PI3K/AKT signaling pathway. Int J Mol Med. (2018) 42:453–60. doi: 10.3892/ijmm.2018.3579
174. Song N, Zhang T, Xu X, Lu Z, Yu X, Fang Y, et al. miR-21 protects against ischemia/reperfusion-induced acute kidney injury by preventing epithelial cell apoptosis and inhibiting dendritic cell maturation. Front Physiol. (2018) 9:790. doi: 10.3389/fphys.2018.00790
175. Wang B, Yao K, Huuskes BM, Shen HH, Zhuang J, Godson C, et al. Mesenchymal stem cells deliver exogenous MicroRNA-let7c via exosomes to attenuate renal fibrosis. Mol Ther. (2016) 24:1290–301. doi: 10.1038/mt.2016.90
176. Gu D, Zou X, Ju G, Zhang G, Bao E, Zhu Y. Mesenchymal stromal cells derived extracellular vesicles ameliorate acute renal ischemia reperfusion injury by inhibition of mitochondrial fission through miR-30. Stem Cells Int. (2016) 2016:2093940. doi: 10.1155/2016/2093940
177. Wang C, Zhu G, He W, Yin H, Lin F, Gou X, et al. BMSCs protect against renal ischemia-reperfusion injury by secreting exosomes loaded with miR-199a-5p that target BIP to inhibit endoplasmic reticulum stress at the very early reperfusion stages. FASEB J. (2019) 33:5440–56. doi: 10.1096/fj.201801821R
178. Burger D, Viñas JL, Akbari S, Dehak H, Knoll W, Gutsol A, et al. Human endothelial colony-forming cells protect against acute kidney injury: role of exosomes. Am J Pathol. (2015) 185:2309–23. doi: 10.1016/j.ajpath.2015.04.010
179. Viñas JL, Burger D, Zimpelmann J, Haneef R, Knoll W, Campbell P, et al. Transfer of microRNA-486–5p from human endothelial colony forming cell-derived exosomes reduces ischemic kidney injury. Kidney Int. (2016) 90:1238–50. doi: 10.1016/j.kint.2016.07.015
180. Pang P, Abbott M, Chang SL, Abdi M, Chauhan N, Mistri M, et al. Human vascular progenitor cells derived from renal arteries are endothelial-like and assist in the repair of injured renal capillary networks. Kidney Int. (2017) 91:129–43. doi: 10.1016/j.kint.2016.07.037
181. Cantaluppi V, Gatti S, Medica D, Figliolini F, Bruno S, Deregibus MC, et al. Microvesicles derived from endothelial progenitor cells protect the kidney from ischemia-reperfusion injury by microRNA-dependent reprogramming of resident renal cells. Kidney Int. (2012) 82:412–27. doi: 10.1038/ki.2012.105
182. Wang R, Lin M, Li L, Li L, Qi G, Rong R, et al. Bone marrow mesenchymal stem cell-derived exosome protects kidney against ischemia reperfusion injury in rats. Zhonghua Yi Xue Za Zhi. (2014) 94:3298–303.
183. Zhang G, Zou X, Miao S, Chen J, Du T, Zhong L, et al. The anti-oxidative role of micro-vesicles derived from human Wharton-Jelly mesenchymal stromal cells through NOX2/gp91(phox) suppression in alleviating renal ischemia-reperfusioninjury in rats. PLoS ONE. (2014) 9:e92129. doi: 10.1371/journal.pone.0092129
184. Zhang G, Zou X, Huang Y, Wang F, Miao S, Liu G, et al. Mesenchymal stromal cell-derived extracellular vesicles protect against acute kidney injurythrough anti-oxidation by enhancing Nrf2/ARE activation in rats. Kidney Blood Press Res. (2016) 41:119–28. doi: 10.1159/000443413
185. Zou X, Zhang G, Cheng Z, Yin D, Du T, Ju G, et al. Microvesicles derived from human Wharton's Jelly mesenchymal stromal cells ameliorate renalischemia-reperfusion injury in rats by suppressing CX3CL1. Stem Cell Res Ther. (2014) 5:40. doi: 10.1186/scrt428
186. Zou X, Gu D, Zhang G, Zhong L, Cheng Z, Liu G, et al. NK cell regulatory property is involved in the protective role of MSC-derived extracellular vesicles in renal ischemic reperfusion injury. Hum Gene Ther. (2016) 27:926–35. doi: 10.1089/hum.2016.057
187. Zou X, Gu D, Xing X, Cheng Z, Gong D, Zhang G, et al. Human mesenchymal stromal cell-derived extracellular vesicles alleviate renal ischemic reperfusion injury and enhance angiogenesis in rats. Am J Transl Res. (2016) 8:4289–99.
188. Ju GQ, Cheng J, Zhong L, Wu S, Zou XY, Zhang GY, et al. Microvesicles derived from human umbilical cord mesenchymal stem cells facilitate tubular epithelial cell dedifferentiation and growth via hepatocyte growth factor induction. PLoS ONE. (2015) 10:e0121534. doi: 10.1371/journal.pone.0121534
189. Choi HY, Moon SJ, Ratliff BB, Ahn SH, Jung A, Lee M, et al. Microparticles from kidney-derived mesenchymal stem cells act as carriers of proangiogenic signals and contribute to recovery from acute kidney injury. PLoS ONE. (2014) 9:e87853. doi: 10.1371/journal.pone.0087853
190. Ranghino A, Bruno S, Bussolati B, Moggio A, Dimuccio V, Tapparo M, et al. The effects of glomerular and tubular renal progenitors and derived extracellular vesicles on recovery from acute kidney injury. Stem Cell Res Ther. (2017) 8:24. doi: 10.1186/s13287-017-0478-5
191. Lin KC, Yip HK, Shao PL, Wu SC, Chen KH, Chen YT, et al. Combination of adipose-derived mesenchymal stem cells (ADMSC) and ADMSC-derived exosomes for protecting kidney from acute ischemia-reperfusion injury. Int J Cardiol. (2016) 216:173–85. doi: 10.1016/j.ijcard.2016.04.061
192. Chen W, Yan Y, Song C, Ding Y, Du T. Microvesicles derived from human Wharton's Jelly mesenchymal stem cells ameliorate ischemia-reperfusion-induced renal fibrosis by releasing from G2/M cell cycle arrest. Biochem J. (2017) 474:4207–18. doi: 10.1042/BCJ20170682
193. Zhu G, Pei L, Lin F, Yin H, Li X, He W, et al. Exosomes from human-bone-marrow-derived mesenchymal stem cells protect against renal ischemia/reperfusion injury via transferring miR-199a-3p. J Cell Physiol. (2019) 234:23736–49. doi: 10.1002/jcp.28941
194. Wu X, Yan T, Wang Z, Wu X, Cao G, Zhang C, et al. Micro-vesicles derived from human Wharton's Jelly mesenchymal stromal cells mitigate renal ischemia-reperfusion injury in rats after cardiac death renal transplantation. J Cell Biochem. (2018) 119:1879–88. doi: 10.1002/jcb.26348
195. Zhu F, Chong Lee Shin OLS, Pei G, Hu Z, Yang J, Zhu H, et al. Adipose-derived mesenchymal stem cells employed exosomes to attenuate AKI-CKD transition through tubular epithelial cell dependent Sox9 activation. Oncotarget. (2017) 8:70707–26. doi: 10.18632/oncotarget.19979
196. Collino F, Lopes JA, Corrêa S, Abdelhay E, Takiya CM, Wendt CHC, et al. Adipose-derived mesenchymal stromal cells under hypoxia: changes in extracellular vesicles secretion and improvement of renal recovery after ischemic injury. Cell Physiol Biochem. (2019) 52:1463–83. doi: 10.33594/000000102
197. Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL, et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med. (2015) 21:998–1009. doi: 10.1038/nm.3902
198. Han Y, Ren J, Bai Y, Pei X, Han Y. Exosomes from hypoxia-treated human adipose-derived mesenchymal stem cells enhance angiogenesis through VEGF/VEGF-R. Int J Biochem Cell Biol. (2019) 109:59–68. doi: 10.1016/j.biocel.2019.01.017
199. Shokeir AA, Hussein AM, Barakat N, Abdelaziz A, Elgarba M, Awadalla A. Activation of nuclear factor erythroid 2-related factor 2 (Nrf2) and Nrf-2-dependent genes by ischaemic pre-conditioning and post-conditioning: new adaptive endogenous protective responses against renal ischaemia/reperfusion injury. Acta Physiol. (2014) 210:342–53. doi: 10.1111/apha.12164
200. Su H, Lei CT, Zhang C. Interleukin-6 signaling pathway and its role in kidney disease: an update. Front Immunol. (2017) 8:405. doi: 10.3389/fimmu.2017.00405
201. Panfoli I, Ravera S, Podestà M, Cossu C, Santucci L, Bartolucci M, et al. Exosomes from human mesenchymal stem cells conduct aerobic metabolism in term and preterm newborn infants. FASEB J. (2016) 30:1416–24. doi: 10.1096/fj.15-279679
202. Visvanathan A, Ahmed K, Even-Faitelson L, Lleres D, Bazett-Jones DP, Lamond AI. Modulation of higher order chromatin conformation in mammalian cell nuclei can be mediated by polyamines and divalent cations. PLoS ONE. (2013) 8:e67689. doi: 10.1371/journal.pone.0067689
203. Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. (2009) 9:537–49. doi: 10.1038/nrc2694
204. Prior KK, Wittig I, Leisegang MS, Groenendyk J, Weissmann N, Michalak M, et al. The endoplasmic reticulum chaperone calnexin is a NADPH oxidase NOX4 interacting protein. J Biol Chem. (2016) 291:7045–59. doi: 10.1074/jbc.M115.710772
205. Zhang L, Liu H, Xu K, Ling Z, Huang Y, Hu Q, et al. Hypoxia preconditioned renal tubular epithelial cell-derived extracellular vesicles alleviate renal ischaemia-reperfusion injury mediated by the HIF-1α/Rab22 pathway and potentially affected by microRNAs. Int J Biol Sci. (2019) 15:1161–76. doi: 10.7150/ijbs.32004
206. Zhang G, Yang Y, Huang Y, Zhang L, Ling Z, Zhu Y, et al. Hypoxia-induced extracellular vesicles mediate protection of remote ischemic preconditioning for renal ischemia-reperfusion injury. Biomed Pharmacother. (2017) 90:473–8. doi: 10.1016/j.biopha.2017.03.096
207. Krogstrup NV, Oltean M, Nieuwenhuijs-Moeke GJ, Dor FJ, Møldrup U, Krag SP, et al. Remote ischemic conditioning on recipients of deceased renal transplants does not improve early graft function: a multicenter randomized, controlled clinical trial. Am J Transplant. (2017) 17:1042–9. doi: 10.1111/ajt.14075
208. Zhou CC, Ge YZ, Yao WT, Wu R, Xin H, Lu TZ, et al. Limited clinical utility of remote ischemic conditioning in renal transplantation: a meta-analysis of randomized controlled trials. PLoS ONE. (2017) 12:e0170729. doi: 10.1371/journal.pone.0170729
209. Tapparo M, Bruno S, Collino F, Togliatto G, Deregibus MC, Provero P, et al. Renal regenerative potential of extracellular vesicles derived from miRNA-engineered mesenchymal stromal cells. Int J Mol Sci. (2019) 20:E2381. doi: 10.3390/ijms20102381
210. Dominguez JH, Liu Y, Gao H, Dominguez JM, Xie D, Kelly KJ. Renal tubular cell-derived extracellular vesicles accelerate the recovery of established renal ischemia reperfusion injury. J Am Soc Nephrol. (2017) 28:3533–44. doi: 10.1681/ASN.2016121278
211. Pêche H, Renaudin K, Beriou G, Merieau E, Amigorena S, Cuturi MC. Induction of tolerance by exosomes and short-term immunosuppression in a fully MHC-mismatched rat cardiac allograft model. Am J Transplant. (2006) 6:1541–50. doi: 10.1111/j.1600-6143.2006.01344.x
212. Ochando J, Ordikhani F, Jordan S, Boros P, Thomson AW. Tolerogenic dendritic cells in organ transplantation. Transpl Int. (2019) 33:113–27. doi: 10.1111/tri.13504
213. Reis M, Mavin E, Nicholson L, Green K, Dickinson AM, Wang XN. Mesenchymal stromal cell-derived extracellular vesicles attenuate dendritic cell maturation and function. Front Immunol. (2018) 9:2538. doi: 10.3389/fimmu.2018.02538
214. Pang XL, Pang XL, Wang ZG, Liu L, Feng YH, Wang JX, et al. Immature dendritic cells derived exosomes promotes immune tolerance by regulating T cell differentiation in renal transplantation. Aging. (2019) 11:8911–24. doi: 10.18632/aging.102346
215. Yu X, Huang C, Song B, Xiao Y, Fang M, Feng J, et al. CD4+CD25+ regulatory T cells-derived exosomes prolonged kidney allograft survival in a rat model. Cell Immunol. (2013) 285:62–8. doi: 10.1016/j.cellimm.2013.06.010
216. Verhoeven JGHP, Boer K, Van Schaik RHN, Manintveld OC, Huibers MMH, Baan CC, et al. Liquid biopsies to monitor solid organ transplant function: a review of new biomarkers. Ther Drug Monit. (2018) 40:515–25. doi: 10.1097/FTD.0000000000000549
217. Svenningsen P, Sabaratnam R, Jensen BL. Urinary extracellular vesicles: origin, role as intercellular messengers and biomarkers; efficient sorting and potential treatment options. Acta Physiol. (2019) 23:e13346. doi: 10.1111/apha.13346
218. Vallabhajosyula P, Korutla L, Habertheuer A, Yu M, Rostami S, Yuan CX, et al. Tissue-specific exosome biomarkers for noninvasively monitoring immunologic rejection of transplanted tissue. J Clin Invest. (2017) 127:1375–91. doi: 10.1172/JCI87993
219. Zhang H, Huang E, Kahwaji J, Nast CC, Li P, Mirocha J, et al. Plasma exosomes from hla-sensitized kidney transplant recipients contain mRNA transcripts which predict development of antibody-mediated rejection. Transplantation. (2017) 101:2419–28. doi: 10.1097/TP.0000000000001834
220. Qamri Z, Pelletier R, Foster J, Kumar S, Momani H, Ware K, et al. Early posttransplant changes in circulating endothelial microparticles in patients with kidney transplantation. Transpl Immunol. (2014) 31:60–4. doi: 10.1016/j.trim.2014.06.006
221. Sigdel TK, Ng YW, Lee S, Nicora CD, Qian WJ, Smith RD, et al. Perturbations in the urinary exosome in transplant rejection. Front Med. (2015) 1:57. doi: 10.3389/fmed.2014.00057
222. van de Vrie M, Deegens JK, Eikmans M, van der Vlag J, Hilbrands LB. Urinary MicroRNA as biomarker in renal transplantation. Am J Transplant. (2017) 17:1160–6. doi: 10.1111/ajt.14082
223. Lim JH, Lee CH, Kim KY, Jung HY, Choi JY, Cho JH, et al. Novel urinary exosomal biomarkers of acute T cell-mediated rejection in kidney transplant recipients: a cross-sectional study. PLoS ONE. (2018) 13:e0204204. doi: 10.1371/journal.pone.0204204
224. Park J, Lin HY, Assaker JP, Jeong S, Huang CH, Kurdi T, et al. Integrated kidney exosome analysis for the detection of kidney transplant rejection. ACS Nano. (2017) 11:11041–6. doi: 10.1021/acsnano.7b05083
225. Al-Massarani G, Vacher-Coponat H, Paul P, Widemann A, Arnaud L, Loundou A, et al. Impact of immunosuppressive treatment on endothelial biomarkers after kidney transplantation. Am J Transplant. (2008) 8:2360–7. doi: 10.1111/j.1600-6143.2008.02399.x
226. Al-Massarani G, Vacher-Coponat H, Paul P, Arnaud L, Loundou A, Robert S, et al. Kidney transplantation decreases the level and procoagulant activity of circulating microparticles. Am J Transplant. (2009) 9:550–7. doi: 10.1111/j.1600-6143.2008.02532.x
227. Martins SR, Alves LV, Cardoso CN, Silva LG, Nunes FFC, das Lucas Júnior FDM, et al. Cell-derived microparticles and Von willebrand factor in brazilian renal transplant recipients. Nephrology. (2019) 24:1304–12. doi: 10.1111/nep.13657
228. Dursun I, Yel S, Unsur E. Dynamics of circulating microparticles in chronic kidney disease and transplantation: is it really reliable marker? World J Transplant. (2015) 5:267–75. doi: 10.5500/wjt.v5.i4.267
229. Alvarez S, Suazo C, Boltansky A, Ursu M, Carvajal D, Innocenti G, et al. Urinary exosomes as a source of kidney dysfunction biomarker in renal transplantation. Transplant Proc. (2013) 45:3719–23. doi: 10.1016/j.transproceed.2013.08.079
230. Dimuccio V, Ranghino A, Praticò Barbato L, Fop F, Biancone L, Camussi G, et al. Urinary CD133+ extracellular vesicles are decreased in kidney transplanted patients with slow graft function and vascular damage. PLoS ONE. (2014) 9:e104490. doi: 10.1371/journal.pone.0104490
231. Asvapromtada S, Sonoda H, Kinouchi M, Oshikawa S, Takahashi S, Hoshino Y, et al. Characterization of urinary exosomal release of aquaporin-1 and−2 after renal ischemia-reperfusion in rats. Am J Physiol Renal Physiol. (2018) 314:F584–601. doi: 10.1152/ajprenal.00184.2017
232. Sonoda H, Yokota-Ikeda N, Oshikawa S, Kanno Y, Yoshinaga K, Uchida K, et al. Decreased abundance of urinary exosomal aquaporin-1 in renal ischemia-reperfusion injury. Am J Physiol Renal Physiol. (2009) 297:F1006–16. doi: 10.1152/ajprenal.00200.2009
233. Wiklander OPB, Brennan MÁ, Lötvall J, Breakefield XO, El Andaloussi S. Advances in therapeutic applications of extracellular vesicles. Sci Transl Med. (2019) 11:eaav8521. doi: 10.1126/scitranslmed.aav8521
234. Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. (2018) 7:1535750. doi: 10.1080/20013078.2018.1461450
235. Andreu Z, Yáñez-Mó M. Tetraspanins in extracellular vesicle formation and function. Front Immunol. (2014) 5:442. doi: 10.3389/fimmu.2014.00442
236. Park KS, Bandeira E, Shelke GV, Lässer C, Lötvall J. Enhancement of therapeutic potential of mesenchymal stem cell-derived extracellular vesicles. Stem Cell Res Ther. (2019) 10:288. doi: 10.1186/s13287-019-1398-3
237. Gonzalez-King H, García NA, Ontoria-Oviedo I, Ciria M, Montero JA, Sepúlveda P. Hypoxia inducible factor-1α potentiates jagged 1-mediated angiogenesis by mesenchymal stem cell-derived exosomes. Stem Cells. (2017) 35:1747–59. doi: 10.1002/stem.2618
238. Grange C, Skovronova R, Marabese F, Bussolati B. Stem cell-derived extracellular vesicles and kidney regeneration. Cells. (2019) 8:1240. doi: 10.3390/cells8101240
Keywords: extracellular vesicles, acute rejection, ischemia-reperfusion syndrome, autoimmunity, renal transplant, biomarker, miRNA
Citation: Quaglia M, Dellepiane S, Guglielmetti G, Merlotti G, Castellano G and Cantaluppi V (2020) Extracellular Vesicles as Mediators of Cellular Crosstalk Between Immune System and Kidney Graft. Front. Immunol. 11:74. doi: 10.3389/fimmu.2020.00074
Received: 06 November 2019; Accepted: 13 January 2020;
Published: 27 February 2020.
Edited by:
Guseppe Grandaliano, University of Foggia, ItalyReviewed by:
Xiangyang Zhu, Mayo Clinic, United StatesFrancesco Paolo Schena, University of Bari Aldo Moro, Italy
Copyright © 2020 Quaglia, Dellepiane, Guglielmetti, Merlotti, Castellano and Cantaluppi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vincenzo Cantaluppi, dmluY2Vuem8uY2FudGFsdXBwaUBtZWQudW5pdXBvLml0