 Shuhong Jiang
Shuhong Jiang Marie Kametani
Marie Kametani Dong Feng Chen
Dong Feng Chen- 1Schepens Eye Research Institute of Massachusetts Eye and Ear, Department of Ophthalmology, Harvard Medical School, Boston, MA, United States
- 2State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou, China
Glaucoma is a globally unmet medical challenge and the most prevalent neurodegenerative disease, which permanently damages the optic nerve and retinal ganglion cells (RGCs), leading to irreversible blindness. Present therapies target solely at lowering intraocular ocular pressure (IOP), a major risk factor of the disease; however, elevated IOP is neither necessary nor sufficient to cause glaucoma. Glaucomatous RGC and nerve fiber loss also occur in individuals with normal IOP. Recent studies have provided evidence indicating a link between elevated IOP and T cell-mediated autoimmune responses, particularly that are specific to heat shock proteins (HSPs), underlying the pathogenesis of neurodegeneration in glaucoma. Reactive glial responses and low-grade inflammation may initially represent an adaptive reaction of the retina to primary stress stimuli; whereas, sustained and excessive glial reactions lead to expanded immune responses, including adaptive immunity, that contribute to progressive neural damage in glaucoma. Emerging data suggest a similar mechanism in play in causing neurodegeneration of other forms of optic neuropathy, such as that resulted from acute ischemia and traumatic injuries. These studies may lead to the paradigm shift and offer a new basis for the development of novel mechanism-based diagnosis, therapy, and preventive interventions for glaucoma. As HSPs are induced under various conditions of neural stress and damage in the brain and spinal cord, these findings may have broader implications for our elucidating of the etiology of other neurodegenerative disorders in the central nervous system.
Introduction
Glaucoma, which leads to progressive and irreversible vision loss, presents a critical medical challenge, partly due to the poorly studied mechanisms that damage the optic nerve and lead to the death of retinal ganglioncells (RGCs). While current therapy targets solely on lowering the intraocular pressure (IOP), studies have indicated that the pathogenesis of the disease is multifactorial (1). Factors such as genetics, age, and immunity are shown to be critical contributors. In addition, glaucoma can occur in patients with normal IOP, so called normal tension glaucoma (NTG). The therapeutic approach that aims to lower the IOP is insufficient to halt, sometime limited slowing down of the progression of the disease (2). There is strong evidence supported by both experimental and clinical research that elevated IOP triggers secondary responses which are responsible for RGC degeneration in glaucoma. In this review, we examine the evidence regarding the involvement of innate and adaptive immunities, including the induction of T and B cell-mediated responses and microglial activation in the acquisition of pathogenicity in glaucoma. Furthermore, the involvement of heat-shock proteins (HSPs), such as HSP27 and HSP60, both as neuroprotective and degenerative roles in the disease progression is discussed.
The Eye as an Immune Privileged Site and the Case of Glaucoma
The eye is recognized as an immune privileged site. Immune regulation in the eye is characterized by its active local immunosuppression, which is achieved, in part, through the blood-aqueous and blood-retina barriers, the unique feature of the pigment epithelial cells, and the local production of immunosuppressive cytokines and neuropeptides (3, 4). Immune cells that enter the eye in response to infection or injury usually are induced to undergo apoptosis via the activation of Fas-FasL signaling without causing inflammation or tissue damage (5). Antigenic material being introduced into the eye elicits immune deviation or suppression of T cell-mediated immunity that leads to peripheral immune tolerance to the antigens, a mechanism termed Anterior Chamber Associated Immune Deviation (6). Such immune privilege is thought to protect the retina, which has limited ability for regeneration and self-repair, from the damaging effects of an uncontrolled immune responses (7).
The eye's immune privileged status, however, is affected by diseased conditions, such as in the case of glaucoma, when the blood-retina barrier is compromised and cytokine productions are often altered (8). Activation of both innate and adaptive immune responses is evident in glaucoma. While systemic immune responses to the retina is strictly controlled, residential glial cells, including microglia, astrocytes, and Müller cells, play the roles of immune surveillance in the retina. They are found to become activated in the early stage of glaucoma (9). Recent evidence also reveals the critical involvement of systemic adaptive immune responses in the pathogenesis of glaucoma. Complex patterns of retinal proteins and autoantibodies against retinal specific antigens have been detected in the sera of patients with glaucoma (10, 11). Such patterns of serum protein and antibody profiles may be used for early diagnosis and detection of glaucoma and/or assessment of the progression of the disease (11–13). Further supporting a compromised blood-retinal barrier in glaucoma, autoantibodies were found to have access to the retina, and infiltration of inflammatory leukocytes and macrophages were noted preceding clinical symptoms of glaucoma (14, 15). Increased expression of matrix metalloproteinase in the astrocytes of the optic nerve head (ONH) is thought to associate with the loss of the retinal immune privilege, thus antibody penetration into the eye (16, 17). To date, elucidating the exact involvement of innate and adaptive immunity in pathogenesis of glaucoma remains the key to understanding of the disease etiology (18).
Innate Immune Responses in Glaucoma
The innate immunity is body's first line of defense against foreign organisms, and it offers a quick response that does not confer long-lasting protection against the same pathogen. In glaucoma, it is elevated IOP, not necessarily a foreign antigen, that triggers an innate immune response, which usually involves resident immune cells, such as microglia, as well as the infiltration of macrophages/monocytes (9). Neuroinflammatory responses generated by microglia are thought to play a leading role in glaucomatous pathophysiology. Studies show that glial activations occur at the early stage of the disease in glaucoma patients and animal models (19–21). Increased microglial activity, cell density, and their expression of complement C1q were noted in the retina and optic nerve in experimental model of glaucoma before RGC and axonal loss is observed (22, 23) and thought to be detrimental to retinal neurons. Support for this notion was the elevation of TNFα in the aqueous humor of glaucoma patients and rodent models of glaucoma, correlating with the worsening of RGC loss (24, 25). Suppression of microglial activation with minocycline or neutralization of TNFα protected RGCs from elevated IOP-induced cell death in rodents (26, 27).
Astrocytes and Müller glia, together called astroglia, also respond to the elevated IOP by developing reactive gliosis, which is characterized by the upregulation of glial fibrillary acidic protein and releasing of chemokines and cytokines, including TNFα. This response is also believed to be a pathological element that contributes to neural damage in glaucoma. More recently, studies revealed that reactive gliosis and TNFα production could be neuroprotective to RGCs. The timing of TNFα expression appears to be crucial as that the early induction of TNFα correlates with RGC survival, while longer-term expression causes RGC degeneration (28). In addition, reactive glial cells produce a variety of neuroprotective molecules, such as insulin-like growth factor-1 (IGF1), to protect RGCs from neural damage (29). Overall, the available evidence strongly suggests that reactive glia are involved in the pathology of glaucoma. To date, the key inflammatory signals that lead to glaucomatous neurodegeneration remain unknown.
Involvement of T Cells in Glaucoma
Activation of the innate immune system initiates and directs the adaptive immune responses, which involve T and B lymphocytes and are also featured in the glaucomatous pathogenesis. In contrast to the innate immune system, the adaptive immune system requires up to 7 days to be activated. Recent report on the involvement of heat shock protein (HSP)-specific T cells offers the key evidence support a role for autoimmunity in glaucoma (30). However, analysis of T cells in patients and experimental models of glaucoma remains limited; there are only few studies focusing on the presence of specific T lymphocyte subsets in the sera of glaucoma patients. In part, this is because pathogenic antigens in the retina are long thought to induce T regulatory (Treg) cells through a mechanism of ACAID, therefore maintaining the ocular immune privilege. Recent studies of the peripheral blood of glaucoma patients revealed that the immunity activated in glaucoma may not be counterbalanced by an efficient immune suppression (30, 31). Patients with glaucoma exhibited a trend of decreased frequency of Treg and their CD4+ T cells presented a greater stimulation response characterized by increased proliferation and proinflammatory cytokine secretion. Elevated frequencies of CD3+CD8+ lymphocytes in both patients with NTG and primary open-angled glaucoma were noted, and CD8+HLA-DR+ lymphocytes were particularly prevalent in NTG. This was accompanied by the increased expre ssion of the soluble interleukin-2 (IL-2) receptor, a marker of T cell activation (32, 33).
The pathogenic role of T cells in glaucomatous neurodegeneration is supported by the evidence that adoptive transfer of T cells from glaucomatous mice results in a progressive loss of RGCs and their axons in recipient mice with a normal IOP (34). A link between elevated IOP and induction of anti-HSP autoimmunity has also been suggested (14, 35). HSP27 and HSP60 immunization in rats induced a pattern of RGC and axon degeneration similar to that was seen in patients with glaucoma (36). A transient infiltration of T cells in the retina was noted 2 weeks after the immunization. In vitro study further demonstrated T cell activation following HSP-immunization, which initiated the production of inflammatory cytokine and FasL, resulting in RGC apoptosis. Recently, using mice deficient in CD4+ αβ T cells, it was reported that CD4+ T cells play a crucial role in propagating RGC degeneration, particularly during the prolonged period of progressive neural damage, in glaucoma (30). Uncovering the association between T cell-mediated autoimmunity and progressive neuron loss in glaucoma may allow the development of novel therapeutic interventions that eventually offer a cure for the disease.
HSPs as Pathogenic Autoantigens in T Cell-Mediated Glaucomatous Neurodegeneration
Adaptive immune responses are elicited by specific antigen stimulation, and HSPs have been identified as pathogenic autoantigens which evoke T cell responses in glaucoma (30). The stress response is a highly conserved mechanism of cellular responses to a wide variety of physiological challenges (37). The response is characterized by the induction of specific cellular proteins with protective functions, such as HSPs. Intracellular HSPs function as molecular chaperones to prevent protein aggregation and facilitate refolding of dysfunctional proteins, which is critical to the survival of all organisms (38). They are some most abundant intracellular proteins that protect cells from destruction and facilitate neural repair through astrocyte and microglial recruitment in the CNS. However, evidence reveals that their release into the extracellular environment is an indication of loss of cellular integrity, thus acts as “danger signals” and elicit both the innate and adaptive immune responses (39). Extracellular HSPs may activate microglia to secrete pro-inflammatory cytokines, such as IL-1β, IL-6, and TNF-α and trigger innate immune responses through toll like receptor 2 (TLR2) and TLR4 (40–42). They can also be processed and presented by antigen presenting cells to stimulate T cell responses. Continued and prolonged IOP elevation leads to HSP upregulation, autoantibody formation, and immune responses in glaucomatous eyes. Overexpression of HSP27 in neurons exacerbated RGC loss following IOP elevation without affecting RGC numbers under a normal IOP (30). These data suggest that elevated IOP not only upregulates HSPs, but also triggers their extracellular release and evoke immune responses.
Tezel et al. have demonstrated increased immunostaining of HSPs in the glaucomatous eyes (43). In a laser-induced rat and non-human primate models of glaucoma, elevated IOP induced expression of HSP27 and HSP70 (44–47). This elevation of HSPs following increased IOP is suggested to be neuroprotective, due in part to the study investigating the effects of geranylgeranylacetone, an HSP70 inducer developed as an anti-ulcer drug. Geranylgeranylacetone decreased elevated IOP-induced neuronal damage by reducing RGC apoptosis and axon loss (44). Subsequent studies by Wax et al. reported that immunization of rats with HSP27 or HSP60 induced significant RGC loss that was in conformity with the RGC- and nerve bundle-specific lesions observed in patients with NTG (36). Recent studies further demonstrate HSP-specific T cell responses in patients with glaucoma (30). Remarkably, mice raised in the absence of commensal microflora (germ-free mice) did not harbor HSP-specific T cells nor did they develop glaucomatous neurodegeneration after IOP elevation (30). These results are in line with the clinical observation that IOP is neither necessary nor sufficient for glaucomatous neuronal damage. It is the subsequent stress-induced events involving retinal inflammation and T cell-mediated responses that are keys to the pathogenesis of glaucoma. Taken together, family of HSPs are critical modulators of both the homeostatic and cytoprotective as well as pathogenic immune response and neurodegenerative arms of the retina and are thus integral to our understanding of neurodegeneration in glaucoma. Identification of key pathogenic autoantigens associated with glaucomatous T cell responses may also provide a foundation for future exploration of tolerance-based clinical intervention for preventing or treating the disease.
Concluding Remarks
Glaucoma is a globally unmet medical challenge due to its prevalence and debilitating consequences. The lack of cure for such a major disease reflects poor understanding of the disease's mechanisms. Numerous clinical and experimental data are now pointing to an unexpected interaction among elevated IOP, HSP-specific T cell responses and glaucomatous neurodegeneration and suggesting the crucial involvement of adaptive immunity in neurodegeneration associated with glaucoma. Although the primary response may be favorable in protecting the eye, the proceeding events that lead to long-lasting activation of glial cells and adaptive immune responses can be destructive. They disrupt the homeostasis of the retina and result in the dysfunction of the immune privilege status of the eye (Figure 1). The mechanisms of immune regulation in glaucoma demonstrate certain patterns which are similar to those seen in autoimmune diseases. The antigens and complex antibodies involved in the activation of immunity response are found in the sera of the patients. Although it remains unclear if the antibody production contributes to a cause or consequence of glaucoma, detection of these antibodies may serve as early diagnostic markers for the disease that may allow for proper and effective treatment prior to the late stage of the disease where progression has already occurred. Through careful examination of factors, including the activation of the glial cells, upregulation of HSPs, and the presence of different lymphocytes, development of new therapeutic treatment methods that aim to restore physiological mechanisms of self-tolerance, as well as an early detection of the disease may be possible. These studies may also have a broad impact on uncovering the pathogenesis of neurodegenerative disorders in the brain and spinal cord.
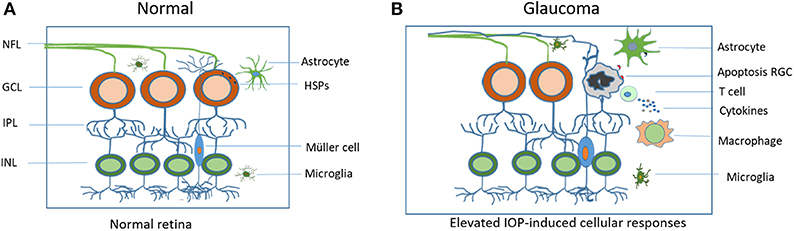
Figure 1. Schematic illustration of elevated IOP-induced cellular responses in the retina. (A) Residential glial cells maintain cellular homeostasis in the normal retina. (B) In the glaucomatous retina, elevated IOP upregulates heat shock proteins (HSPs), initiates both the innate and adaptive immune responses that include microglial activation and T cell infiltration. Sustained and excessive immune responses lead to RGC apoptosis and vision loss. IOP, intraocular pressure; RGC, retinal ganglion cell; NFL, retinal nerve fiber layer.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by NIH/NEI grants EY025913, EY025259, and P30EY003790 and Massachusetts Lions Foundation grant; National Natural Science Foundation of China 81760177 and Natural Science Foundation of Inner Mongolia A.R. 2011BS1107.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Doucette LP, Rasnitsyn A, Seifi M, Walter MA. The interactions of genes, age, and environment in glaucoma pathogenesis. Surv Ophthalmol. (2015) 60:310–26. doi: 10.1016/j.survophthal.2015.01.004
2. Adams CM, Stacy R, Rangaswamy N, Bigelow C, Grosskreutz CL, Prasanna G. Glaucoma-next generation therapeutics: impossible to possible. Pharm Res. (2019) 36:25. doi: 10.1007/s11095-018-2557-4
3. Stein-Streilein J. Immune regulation and the eye. Trends Immunol. (2008) 29:548–54. doi: 10.1016/j.it.2008.08.002
4. Streilein JW, Wilbanks GA, Cousins SW. Immunoregulatory mechanisms of the eye. J Neuroimmunol. (1992) 39:185–200. doi: 10.1016/0165-5728(92)90253-H
5. Griffith TS, Brunner T, Fletcher SM, Green DR, Ferguson TA. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science. (1995) 270:1189–92. doi: 10.1126/science.270.5239.1189
6. Wilbanks G, Streilein J. Characterization of suppressor cells in anterior chamber-associated immune deviation (ACAID) induced by soluble antigen. Evidence of two functionally and phenotypically distinct T-suppressor cell populations. Immunology. (1990) 71:383.
7. Moalem G, Leibowitz–Amit R, Yoles E, Mor F, Cohen IR, Schwartz M. Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med. (1999) 5:49. doi: 10.1038/4734
8. Perez VL, Caspi RR. Immune mechanisms in inflammatory and degenerative eye disease. Trends Immunol. (2015) 36:354–63. doi: 10.1016/j.it.2015.04.003
9. Wei X, Cho KS, Thee EF, Jager MJ, Chen DF. Neuroinflammation and microglia in glaucoma: time for a paradigm shift. J Neurosci Res. (2019) 97:70–6. doi: 10.1002/jnr.24256
10. Tezel G, Seigel GM, Wax MB. Autoantibodies to small heat shock proteins in glaucoma. Invest Ophthalmol Vis Sci. (1998) 39:2277–87.
11. Beutgen VM, Perumal N, Pfeiffer N, Grus FH. Autoantibody biomarker discovery in primary open angle glaucoma using serological proteome analysis. Front Immunol. (2019) 10:381. doi: 10.3389/fimmu.2019.00381
12. Flammer J, Mozaffarieh M. What is the present pathogenetic concept of glaucomatous optic neuropathy? Surv Ophthalmol. (2007) 52:S162–73. doi: 10.1016/j.survophthal.2007.08.012
13. Thornton I, Luo C, Yang X, Soltau J, Tezel G. Immunoproteomic analysis of glaucomatous patient serum and aqueous humor antibodies: differential immunoreactivity against glaucomatous versus non-glaucomatous retinal proteins. Invest Ophthalmol Vis Sci. (2010) 51:2673. Available online at: https://iovs.arvojournals.org/article.aspx?articleid=2371330
14. Gramlich OW, Beck S, Hohenstein-Blaul NvT, Boehm N, Ziegler A, Vetter JM, et al. Enhanced insight into the autoimmune component of glaucoma: IgG autoantibody accumulation and pro-inflammatory conditions in human glaucomatous retina. PLoS ONE. (2013) 8:e57557. doi: 10.1371/journal.pone.0057557
15. Callahan MK, Ransohoff RM. Analysis of leukocyte extravasation across the blood-brain barrier: conceptual and technical aspects. Curr Allergy Asthma Rep. (2004) 4:65–73. doi: 10.1007/s11882-004-0046-9
16. Agapova OA, Ricard CS, Salvador-Silva M, Hernandez MR. Expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in human optic nerve head astrocytes. Glia. (2001) 33:205–16. doi: 10.1002/1098-1136(200103)33:3<205::AID-GLIA1019>3.0.CO;2-D
17. Yan X, Tezel G, Wax MB, Edward DP. Matrix metalloproteinases and tumor necrosis factor α in glaucomatous optic nerve head. Arch Ophthalmol. (2000) 118:666–73. doi: 10.1001/archopht.118.5.666
18. Vu TK, Jager MJ, Chen DF. The immunology of glaucoma. Asia Pac J Ophthalmol. (2012) 1:303–11. doi: 10.1097/APO.0b013e31826f57a3
19. Howell GR, MacNicoll KH, Braine CE, Soto I, Macalinao DG, Sousa GL, et al. Combinatorial targeting of early pathways profoundly inhibits neurodegeneration in a mouse model of glaucoma. Neurobiol Dis. (2014) 71:44–52. doi: 10.1016/j.nbd.2014.07.016
20. Bosco A, Romero CO, Breen KT, Chagovetz AA, Steele MR, Ambati BK, et al. Neurodegeneration severity can be predicted from early microglia alterations monitored in vivo in a mouse model of chronic glaucoma. Dis Models Mech. (2015) 8:443–55. doi: 10.1242/dmm.018788
21. Ramirez AI, de Hoz R, Salobrar-Garcia E, Salazar JJ, Rojas B, Ajoy D, et al. The role of microglia in retinal neurodegeneration: Alzheimer's disease, Parkinson, and glaucoma. Front Aging Neurosci. (2017) 9:214. doi: 10.3389/fnagi.2017.00214
22. Ebneter A, Casson RJ, Wood JP, Chidlow G. Microglial activation in the visual pathway in experimental glaucoma: spatiotemporal characterization and correlation with axonal injury. Invest Ophthalmol Vis Sci. (2010) 51:6448–60. doi: 10.1167/iovs.10-5284
23. Howell GR, Macalinao DG, Sousa GL, Walden M, Soto I, Kneeland SC, et al. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J Clin Invest. (2011) 121:1429–44. doi: 10.1172/JCI44646
24. Tezel Gln, Li LY, Patil RV, Wax MB. TNF-α and TNF-α receptor-1 in the retina of normal and glaucomatous eyes. Invest Ophthalmol Vis Sci. (2001) 42:1787–94.
25. Yuan L, Neufeld AH. Tumor necrosis factor-α: A potentially neurodestructive cytokine produced by glia in the human glaucomatous optic nerve head. Glia. (2000) 32:42–50. doi: 10.1002/1098-1136(200010)32:1<42::AID-GLIA40>3.0.CO;2-3
26. Bosco A, Steele M, Vetter M. Microglia cell activation and clustering herald the onset of glaucoma in DBA/2J retina. Invest Ophthalmol Vis Sci. (2008) 49:5492. doi: 10.1167/iovs.07-1337
27. Roh JH, Huang Y, Bero AW, Kasten T, Stewart FR, Bateman RJ, et al. Disruption of the sleep-wake cycle and diurnal fluctuation of β-amyloid in mice with Alzheimer's disease pathology. Sci Transl Med. (2012) 4:150ra22. doi: 10.1126/scitranslmed.3004291
28. Mac Nair CE, Nickells RW. Neuroinflammation in glaucoma and optic nerve damage. Prog Mol Biol Transl Sci. (2015) 134:343–63. doi: 10.1016/bs.pmbts.2015.06.010
29. Arroba AI, Campos-Caro A, Aguilar-Diosdado M, Valverde AM. IGF-1, inflammation and retinal degeneration: a close network. Front Aging Neurosci. (2018) 10:203. doi: 10.3389/fnagi.2018.00203
30. Chen H, Cho K-S, Vu TK, Shen C-H, Kaur M, Chen G, et al. Commensal microflora-induced T cell responses mediate progressive neurodegeneration in glaucoma. Nat Commun. (2018) 9:3209. doi: 10.1038/s41467-018-06428-2
31. Yang X, Zeng Q, Göktaş E, Gopal K, Al-Aswad L, Blumberg DM, et al. T-lymphocyte subset distribution and activity in patients with glaucoma. Invest Ophthalmol Vis Sci. (2019) 60:877–88. doi: 10.1167/iovs.18-26129
32. Mariotti S, Barbesino G, Caturegli P, Marino M, Manetti L, Fugazzola L, et al. Serum soluble interleukin 2 (IL-2) receptor (sIL-2R) in differentiated thyroid carcinoma. J Endocrinol Invest. (1994) 17:861–7. doi: 10.1007/BF03347792
33. Yang J, Patil RV, Yu H, Gordon M, Wax MB. T cell subsets and sIL-2R/IL-2 levels in patients with glaucoma. Am J Ophthalmol. (2001) 131:421–6. doi: 10.1016/S0002-9394(00)00862-X
34. Gramlich OW, Ding QJ, Zhu W, Cook A, Anderson MG, Kuehn MH. Adoptive transfer of immune cells from glaucomatous mice provokes retinal ganglion cell loss in recipients. Acta Neuropathol Commun. (2015) 3:56. doi: 10.1186/s40478-015-0234-y
35. Joachim SC, Grus FH, Kraft D, White-Farrar K, Barnes G, Barbeck M, et al. Complex antibody profile changes in an experimental autoimmune glaucoma animal model. Invest Ophthalmol Vis Sci. (2009) 50:4734–42. doi: 10.1167/iovs.08-3144
36. Wax MB, Tezel G, Yang J, Peng G, Patil RV, Agarwal N, et al. Induced autoimmunity to heat shock proteins elicits glaucomatous loss of retinal ganglion cell neurons via activated T-cell-derived fas-ligand. J Neurosci. (2008) 28:12085–96. doi: 10.1523/JNEUROSCI.3200-08.2008
37. Morimoto RI. Cells in stress: transcriptional activation of heat shock genes. Science. (1993) 259:1409. doi: 10.1126/science.8451637
38. Fink AL. Chaperone-mediated protein folding. Physiol Rev. (1999) 79:425–49. doi: 10.1152/physrev.1999.79.2.425
39. Van Eden W, Jansen MA, Ludwig I, van Kooten P, Van Der Zee R, Broere F. The enigma of heat shock proteins in immune tolerance. Front Immunol. (2017) 8:1599. doi: 10.3389/fimmu.2017.01599
40. Jin C, Cleveland JC, Ao L, Li J, Zeng Q, Fullerton DA, et al. Human myocardium releases heat shock protein 27 (HSP27) after global ischemia: the proinflammatory effect of extracellular HSP27 through toll-like receptor (TLR)-2 and TLR4. Mol Med. (2014) 20:280–9. doi: 10.2119/molmed.2014.00058
41. Rosenberger K, Dembny P, Derkow K, Engel O, Krüger C, Wolf SA, et al. Intrathecal heat shock protein 60 mediates neurodegeneration and demyelination in the CNS through a TLR4-and MyD88-dependent pathway. Mol Neurodegener. (2015) 10:5. doi: 10.1186/s13024-015-0003-1
42. Swaroop S, Sengupta N, Suryawanshi AR, Adlakha YK, Basu A. HSP60 plays a regulatory role in IL-1β-induced microglial inflammation via TLR4-p38 MAPK axis. J Neuroinflammation. (2016) 13:27. doi: 10.1186/s12974-016-0486-x
43. Tezel G, Hernandez MR, Wax MB. Immunostaining of heat shock proteins in the retina and optic nerve head of normal and glaucomatous eyes. Arch Ophthalmol. (2000) 118:511–8. doi: 10.1001/archopht.118.4.511
44. Ishii Y, Kwong JM, Caprioli J. Retinal ganglion cell protection with geranylgeranylacetone, a heat shock protein inducer, in a rat glaucoma model. Invest Ophthalmol Vis Sci. (2003) 44:1982–92. doi: 10.1167/iovs.02-0912
45. Qing G, Duan X, Jiang Y. Heat shock protein 72 protects retinal ganglion cells in rat model of acute glaucoma. Yan ke Xue Bao. (2005) 21:163–8.
46. Sakai M, Sakai H, Nakamura Y, Fukuchi T, Sawaguchi S. Immunolocalization of heat shock proteins in the retina of normal monkey eyes and monkey eyes with laser-induced glaucoma. Jpn J Ophthalmol. (2003) 47:42–52. doi: 10.1016/S0021-5155(02)00627-5
Keywords: glaucoma, optic neuropathy, heat shock proteins, T cells, glial response, neuroinflammation
Citation: Jiang S, Kametani M and Chen DF (2020) Adaptive Immunity: New Aspects of Pathogenesis Underlying Neurodegeneration in Glaucoma and Optic Neuropathy. Front. Immunol. 11:65. doi: 10.3389/fimmu.2020.00065
Received: 02 November 2019; Accepted: 10 January 2020;
Published: 13 February 2020.
Edited by:
Heping Xu, Queen's University Belfast, United KingdomReviewed by:
Thomas Langmann, Universität zu Köln, GermanyStephanie C. Joachim, Ruhr University Bochum, Germany
Copyright © 2020 Jiang, Kametani and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dong Feng Chen, RG9uZ2ZlbmdfY2hlbkBtZWVpLmhhcnZhcmQuZWR1