
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 24 January 2020
Sec. T Cell Biology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.03103
This article is part of the Research Topic Mycobacteria-Host Interactions: Genetics, Immunity, Pathology View all 15 articles
 Lu Huang1
Lu Huang1 Kaixiong Ye2,3
Kaixiong Ye2,3 Michael C. McGee4
Michael C. McGee4 Natalie F. Nidetz4
Natalie F. Nidetz4 Jessica P. Elmore1
Jessica P. Elmore1 Candice B. Limper1
Candice B. Limper1 Teresa L. Southard5
Teresa L. Southard5 David G. Russell1
David G. Russell1 Avery August1*
Avery August1* Weishan Huang1,4*
Weishan Huang1,4*Interleukin-2 (IL-2) inducible T-cell kinase (ITK) is a non-receptor tyrosine kinase highly expressed in T-cell lineages and regulates multiple aspects of T-cell development and function, mainly through its function downstream of the T-cell receptor. Itk deficiency can lead to CD4 lymphopenia and Epstein-Bar virus (EBV)-associated lymphoproliferation and recurrent pulmonary infections in humans. However, the role of the ITK signaling pathway in pulmonary responses in active tuberculosis due to Mtb infection is not known. We show here that human lungs with active tuberculosis exhibit altered T-cell receptor/ITK signaling and that Itk deficiency impaired early protection against Mtb in mice, accompanied by defective development of IL-17A-producing γδ T cells in the lungs. These findings have important implications of human genetics associated with susceptibility to Mtb due to altered immune responses and molecular signals modulating host immunity that controls Mtb activity. Enhancing ITK signaling pathways may be an alternative strategy to target Mtb infection, especially in cases with highly virulent strains in which IL-17A plays an essential protective role.
Mycobacterium tuberculosis (Mtb) is the causative agent of what remains one of the most insidious and invasive human infections, responsible for millions of cases of active lung diseases and deaths per year in the world [WHO Global Tuberculosis Report, (1) and O'Garra et al. (2)]. The immune responses by macrophages, neutrophils, and T-cell populations contribute to protective immunity controlling Mtb expansion in the host and transmission to others (2, 3). Genetic and environmental factors of the host associated with primary and acquired immunodeficiency can lead to an increased risk of developing active tuberculosis that presents severe pulmonary illness in the clinic (2, 4). Our knowledge of the molecular pathways of innate and adaptive immune effector functions remains incomplete, and a better understanding of potential host factors underlying the pulmonary complications could lead to the development of more innovative therapeutic strategies.
Interleukin-2 (IL-2)-inducible T-cell kinase (ITK) is a non-receptor tyrosine kinase highly expressed in T cells. ITK functions downstream of the T-cell receptor (TCR) and regulates multiple aspects of T-cell development and function (5). ITK deficiency in humans is associated with primary immunodeficiency, progressive natural killer T (NKT) and CD4+ T-cell lymphopenia, elevated susceptibility to Epstein-Bar virus (EBV), and EBV-driven lymphoproliferative diseases, in which frequent pulmonary involvement has emerged as a clinical hallmark (6–13). Recurrent progressive pulmonary infection, airway obstruction, and respiratory failure in ITK-deficient patients pose significant threats that can eventually result in deaths of the patients at their early ages (12). Human T cells from patients with ITK deficiency exhibit impaired responses to TCR activation, with reduced generation of Th17 cells and production of the associated cytokines IL-17A, IL-22, and granulocyte-macrophage colony-stimulating factor (14). A murine model of Itk deficiency reveals a similar NKT and T-cell lymphopenia as observed in human patients with ITK deficiency. Mice lacking Itk have a marked reduction in NKT cells (15–18). Despite relatively normal number (trending the lower range) of CD8+ αβ T cells, Itk−/− mice exhibited CD4+ αβ T-cell lymphopenia, with reduced proportion of naive and increased memory αβ T cells (19–31). In the absence of Itk, mouse CD4+ T cells are impaired in Th2 (producing IL-4/5/13) (32–38), Th9 (producing IL-9) (39), Th17 (producing IL-17) (35, 40–42), and Tr1 (producing IL-10) cell responses (43), while they are enhanced in Th1 (producing IFN-γ) cell response (32, 34, 38, 44, 45). Analysis of Itk−/− mice also reveals altered γδ T-cell development (46–49); however, the presence and function of γδ T cells has not been evaluated in ITK-deficient humans.
Epidemiological analysis of single-nucleotide polymorphism has also revealed a connection between greater ITK promoter activity and higher risk of asthmatic incidence in humans, which might be associated with the function of ITK in promoting T-cell activation (50). In murine models of allergic asthma, the expression of ITK is critical for the activation and development of Th2 and Th17 cells and the associated airway and tracheal inflammation (40, 51). Interestingly, a genome-wide association study of susceptibility to Mycobacterium avium subspecies paratuberculosis in Holstein cattle identified chromosomal regions that included the ITK gene (52). However, the role of ITK signaling pathway in pulmonary responses in active tuberculosis due to Mtb infection is unknown.
Here, we show that the TCR/ITK signaling pathway is enriched in human lungs with active tuberculosis and that Itk deficiency impaired early protection against Mtb in mice, accompanied by defective development of IL-17A-producing γδ T cells in the lungs. Furthermore, ITK appears to regulate the dynamics of lung myeloid cells, which may further contribute to immune control of Mtb at the early stage of infection.
All mice were on the C57BL/6 background. Both female and male mice at the age of 6–12 weeks were used. All experiments were approved by the Office of Research Protections Institutional Animal Care and Use Committee at Cornell University.
Microarray data from lung normal tissue and caseous granulomas from active tuberculosis (TB) patient was generated as previously described (53, 54). Microarray data is available in the Gene Expression Omnibus under accession number GSE20050. In brief, tissues were fixed, and areas of interest were dissected using laser capture microdissection on the Leica AS LMD system (Leica, Buffalo Grove, IL). Total RNA was isolated and used on the GeneChip Human X3P Array (Affymetrix, Santa Clare, CA) following the manufacturer's instruction. Data analysis was performed in R (version 3.5.3) and Bioconductor (version 3.8). Probe intensities were log2 transformed and median centered. Differentially expressed genes were identified with limma (version 3.38.3) (55). In the case of multiple probes mapped to a gene, the probe with the maximum fold change was selected to represent the gene. Gene set enrichment analyses (GSEA) to determine over- and underrepresented gene sets were performed using the Kyoto Encyclopedia of Genes and Genomes pathway database as reference in gage (version 2.32.1) (56). All pathways that exhibited an up- or downregulated trend in caseum samples compared to normal tissues are summarized in the Supplementary Material (Supplementary Table 1). Enrichment score and core genes that drive the score are determined using the GSEA platform developed by the Broad Institute (57). Visualization of TCR signaling pathway with differential gene expression was performed in pathview (version 1.22.3) (Supplementary Figure 1) (58).
Mice were inoculated intranasally with ~1,000 CFUs of Erdman Mtb constitutively expressing mCherry (mCherry-Mtb) (59) in 25 μl of phosphate-buffered saline (PBS) containing 0.05% Tween-80. Mice were euthanized after 2 and 4 weeks of infection. The left lung lobe and the accessory lobe of the right lung were removed and homogenized in PBS containing 0.05% Tween-80. Bacterial loads were determined by plating serial dilutions of the homogenates on 7H10 agar plates.
Lung samples were fixed in 4% paraformaldehyde overnight, followed by hematoxylin and eosin (H&E) staining. Histological images were analyzed using DP2-BSW software (Olympus, Waltham, MA) to quantify the percentage of affected tissue area and score the severity of pathology (60).
Mice were euthanized at the indicated time points, and lungs were aseptically removed. To obtain a single-cell suspension, lungs were minced and digested in 5% fetal bovine serum/PBS solution containing 250 U/ml collagenase IV (Worthington, Lakewood, NJ) and 20 U/ml DNase (Roche, Indianapolis, IN) for 30 min at 37°C. Lung digestions were then passed through a 70-μm cell strainer, and red blood cells were lysed with ammonium-chloride-potassium buffer.
Fluorescent antibodies are listed in the format of “Fluorophore-target (clone)”: eFluor 450-CD4 (GK1.5), Phycoerythrin (PE)-Foxp3 (FJK-16s), PE-eFluor 610-NK1.1 (PK136), allophycocyanin (APC)-IL-17A (eBio17B7), APC-CD11c (N418), PerCP-eFluor 710-CD49b (DX5), PE-Cy7-NK1.1 (PK136), PE-Cy7-IFN-γ (XMG1.2), and APC-eFluor 780-MHCII (M5/114.15.2) were from eBioscience (San Diego, CA). CD16/32 (93; i.e., Fc block), Brilliant Violet 421-CD64 (X54-5/7.1), Alexa Fluor 488-TCRγδ (GL3), and APC-Cy7-TCRβ (H57-597) were from BioLegend (San Diego, CA). FITC-Ly6G (1A8), PE-Siglec-F (E50-2440), PE-TNF-α (MP6-XT22), PE-CF594 CD8α (53-6.7), Alexa Fluor 700-Ki67 (B56), PerCP-Cy5.5-CD8α (53-6.7), and PerCP-Cy5.5-CD11b (M1/70) were from BD Biosciences (San Jose, CA).
To activate bulk T cells, cells were stimulated with phorbol 12-myristate 13-acetate (PMA) (50 ng/ml, Sigma) and Ionomycin (0.5 μM, Sigma); to activate Mtb-specific CD4+ T cells, cells were stimulated with ESAT-64−17 peptide (MHCII-restricted presentation; synthesized by GenScript, purity > 95%; 5 μg/ml); to activate Mtb-specific CD8+ T cells, cells were stimulated with TB10.44−11 peptide (MHCI-restricted presentation; synthesized by GenScript, purity > 95%; 5 μg/ml). All stimulations were done in full RPMI-1640 media in the presence of Brefeldin A (5 μg/ml, Sigma) and Monensin (2 μM, Sigma), at 37°C for 5 h.
Surface protein staining was done with antibodies for surface markers in PBS, in the presence of Fc Block (BioLegend) and fixable viability dye (Tonbo Biosciences). To determine cytokine production, cells were stimulated as indicated, followed by surface staining, then were fixed with 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA), and permeabilized and stained with antibodies in PBS containing 0.3% saponin (Sigma). To stain for nuclear transcription factors Foxp3 and Ki67, following surface staining, cells were fixed, permeabilized, and stained using Foxp3 staining buffer set (eBioscience). All flow cytometry data were acquired on LSRII (BD Biosciences) and analyzed in FlowJo (Tree Star, Ashland, OR).
Two-tailed Student's t-test and two-way analysis of variance (ANOVA) between groups were performed using Prism (GraphPad, San Diego, CA), with p < 0.05 considered statistically significant. “NS” indicates differences that are not significant.
The progression of human active TB disease and transmission involves the development of the caseous granuloma, in which both Mtb and the immune response are active (53). We have previously isolated human granulomata from patients with active TB (Caseum) and analyzed the transcriptomic profile in comparison to uninvolved lung tissue (normal) (53). Using pathway enrichment analyses, we found that genes of the TCR signaling were significantly enriched in the caseum tissue that was subjected to active TB, compared to uninvolved lung tissue (Figure 1). Among the enrichment score-driving critical genes of the TCR signaling, the levels of transcripts for ITK and its signaling components (5) such as LCK, GRB2, SLP76, NCK1, FYN, and PLCG are significantly upregulated in caseated granulomas compared to uninvolved lung tissue (Figure 1B). Among the genes that are significantly enriched in active TB, ITK locates in the hub of the TCR signaling pathway (Supplementary Figure 1). These data imply a role for ITK signaling in host immune activity during active TB.
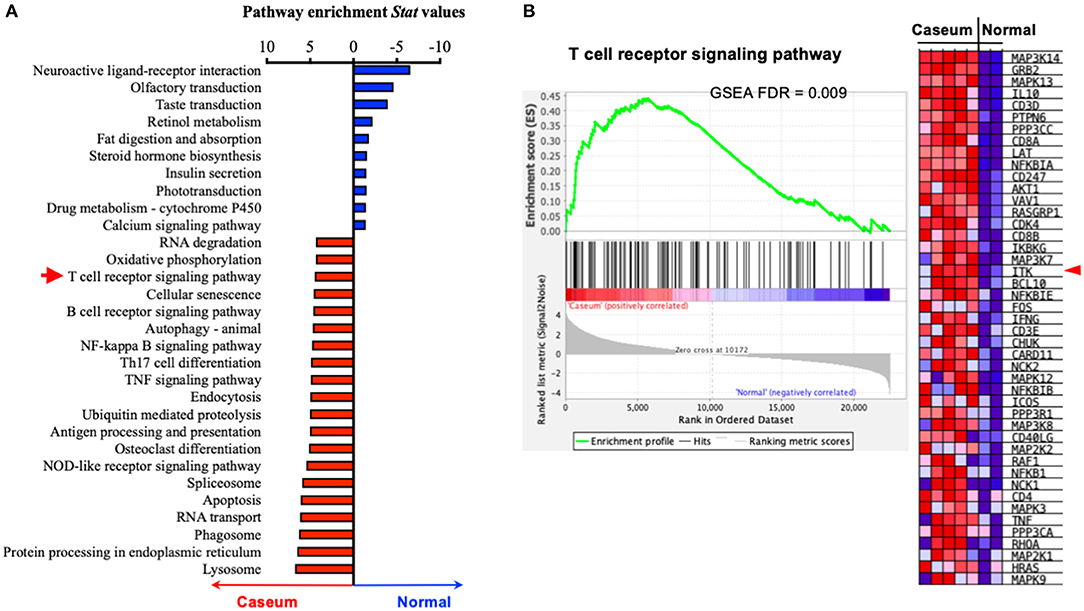
Figure 1. ITK expression is altered in human lungs with active TB. (A) Pathway enrichment analyses of human lung tissues with active TB (“Caseum,” N = 5), compared to normal lung tissues (“Normal,” N = 2). Pathway enrichment statistical values were for pathways enriched in “Caseum” group were shown as positive values (upregulated in “Caseum” compared to “Normal”), while those enriched in normal lungs were shown as negative values (upregulated in “Normal” compared to “Caseum”). Twenty most enriched pathways in the “Caseum” group (out of 145 pathways with FDR < 0.05), and 10 most enriched pathways in “Normal” group (out of 11 pathways with FDR < 0.05) are shown. Red arrow indicates T-cell receptor signaling pathway. (B) Gene set enrichment score, profile of T-cell signaling receptor pathway and heatmap of the levels of expression of the score-driving genes in T-cell receptor signaling pathway. Red arrow indicates ITK. FDR, false discover rate.
Given the observation that the TCR signaling pathway was upregulated in the face of active TB in human lungs and that ITK is a critical score-driving gene for the pathway enrichment (Figure 1), we sought to determine the role of ITK in host responses to Mtb infection. In murine models of Itk deficiency, despite no difference in animal mortality, we found that Mtb bacterial burden was significantly higher in the lungs in the absence of ITK 4 weeks post-Mtb infection (Figure 2A). Moreover, pulmonary pathology was elevated in the absence of ITK, with significantly larger areas in the airway affected at higher pathological scores (Figure 2B). Notably, compared to wild-type (WT) mice, the relative kinetics of bacterial growth in the lungs of Itk−/− and Rag−/− mice are similar (61, 62). These data suggest that the TCR/ITK signaling pathway regulates immune responses that contribute to limiting Mtb growth and controlling pulmonary inflammation 4 weeks postinfection.
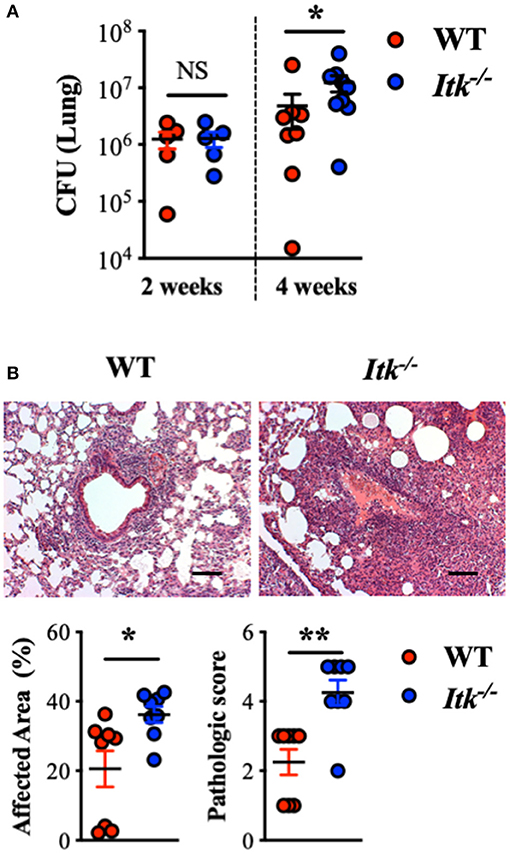
Figure 2. Itk deficiency results in impaired bacterial clearance and increased pulmonary pathology post-Mtb infection in mice. WT and Itk−/− mice were infected with 103 CFU of Mtb (intranasal delivery) and analyzed 2 and 4 weeks postinfection. (A) CFU in WT and Itk−/− mice 2 and 4 weeks postinfection. (B) Representative histology, summary of affected area, and histological scoring of WT and Itk−/− lungs of mice 4 weeks post-Mtb infection. *p < 0.05, **p < 0.01 by Student's t-test. Scale bar = 100 μm. N = 5–10. Data were presented as mean ± SEM and represent results of three independent experiments.
The progression of human active TB disease and transmission involves the development of both innate and adaptive immunity. The relative abundance of lung phagocyte populations is extremely dynamic at the early stage of Mtb infection (63). Importantly, the various phagocytes in the lung provide different environments for Mtb and reveal distinct permissiveness for the growth of Mtb (64, 65). We thus analyzed populations of innate immune cells in both WT and Itk-deficient mice infected with Mtb that constitutively express fluorescent protein mCherry, which allows mapping of the cellular location of intracellular bacteria. During early-stage infection, we observed increased total numbers of neutrophils and alveolar macrophages in the lung at 2 weeks postinfection in both WT and Itk−/− mice (Figure 3A). Further analysis of the pulmonary immune cell populations revealed that, in the absence of ITK, the proportion of Mtb-infected alveolar and interstitial macrophages was significantly higher early after infection (2 weeks); in addition, the proportion of Mtb-infected neutrophils was significantly higher 4 weeks after infection (Figure 3). Effective removal of Mtb-infected apoptotic neutrophils by macrophages, or efferocytosis, is considered beneficial for host defense (66). The failed clearance of infected neutrophils in Itk-deficient mice at 4 weeks suggests that Itk may be involved in regulating efferocytosis of lung macrophages. Both alveolar macrophages and neutrophils have been demonstrated as permissive cell types in Mtb infection by providing a hospitable environment for optimal bacterial growth (64, 65). The increased proportion of Mtb-infected neutrophils in the absence of ITK is associated with the increased bacterial burden at 4 weeks, suggesting that bacterial burden may be attributed to this difference. Therefore, these data suggest that ITK regulates the dynamic of lung phagocytes and contributes to host protection against Mtb infection.
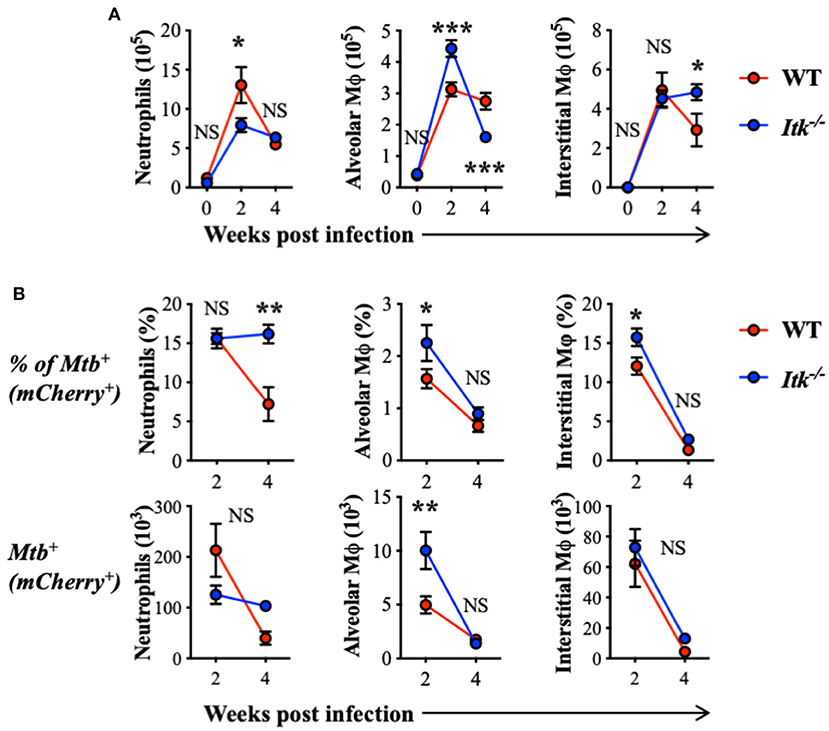
Figure 3. ITK regulates innate myeloid immune response in mice during Mtb infection. WT and Itk−/− mice were infected with 103 CFU of mCherry-Mtb, and lungs were analyzed at the indicated time points. After gating on viable singlet cells, neutrophils (Ly6G+CD11b+), alveolar macrophages (Ly6G−CD11c+Siglec-F+), and interstitial macrophages (Ly6G−Siglec-F− CD11b+MHCII+CD64+) were analyzed. (A) Number of total neutrophils, alveolar macrophages, and interstitial macrophages isolated from the lungs of infected mice. (B) Percentage and number of mCherry-Mtb positive neutrophils, alveolar macrophages, and interstitial macrophages in the lung of the infected mice. NS, not significant; *p < 0.05, **p < 0.01, ***p < 0.001 by two-way ANOVA with post hoc test. N = 5. Data were presented as mean ± SEM and represent results of three independent experiments.
ITK is highly expressed in T-cell lineages including γδ and αβ T cells. To further determine whether the absence of ITK affects T-cell and other related lymphocyte responses during Mtb infection, we also analyzed the abundance of γδ T cells, NK cells, NKT cells, and αβ T cells, including total αβ T cells, CD4+ conventional αβ T cells, CD8+ αβ T cells, and CD4+ Foxp3+ regulatory T (Treg) cells. Comparable numbers of γδ T cells, NKT cells, and CD8+ αβ T cells were observed in WT and Itk-deficient mice infected with Mtb, while NK cells and CD4+ αβ T cells were significantly increased, and Foxp3+ Treg cells were significantly reduced in the absence of ITK (Figure 4). These results seemed surprising, as a reduced number of Treg cells accompanied by increased numbers of CD4+ αβ T cells may suggest a more active immune response. The hosts' ability to limit bacterial growth and control pulmonary inflammation was, however, impaired in the absence of ITK. A possible explanation could be that the effector activity of the lymphocytes observed in the infected airway against Mtb may differ. These effector immune responses could involve innate lymphocyte activities, as well as Mtb antigen-specific adaptive lymphocyte functions.
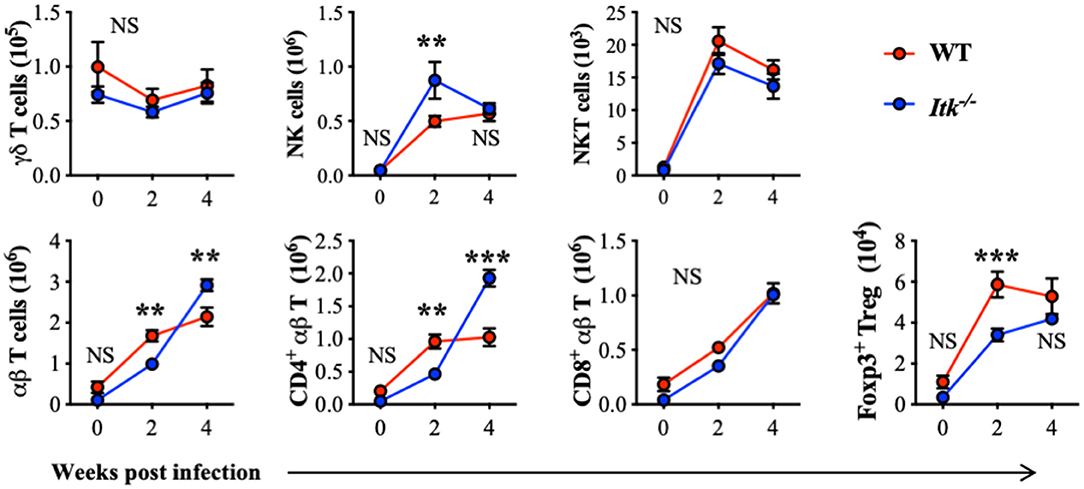
Figure 4. Itk deficiency alters lymphocyte profile in the lung during Mtb infection. WT and Itk−/− mice were infected as in Figure 3, and lungs were analyzed at the indicated time points. After gating on viable singlet cells, lymphocytes including γδT cells (TCRγδ+TCRβ−), NK cells (TCRγδ−TCRβ−NK1.1+), NKT cells (TCRγδ−TCRβ+NK1.1+), and αβT cells (TCRγδ−TCRβ+), and among αβT cells, CD4+ conventional T cells, CD8+ cells, and CD4+ Foxp3+ Treg cells were analyzed. N = 5–9. Numbers of the indicated cell types were presented as mean ± SEM and represent results of three independent experiments. NS, not significant; **p < 0.01, ***p < 0.001 by two-way ANOVA with post hoc test.
It has been reported that IL-17A is protective during primary infection of virulent Mtb (67), and IL-17A is predominantly produced by γδ T cells in the lungs early after Mtb infection (68). Indeed, while we detected significant IL-17A production by γδ T cells in the lungs of WT mice infected with Mtb (red foreground in Figures 5A–C), very limited IL-17A was produced by the conventional CD4+ αβ Th17 cells (gray background in Figure 5A, as well as in Figure 5D). Interestingly, in the absence of ITK, there was a marked reduction in IL-17A-producing γδ T cells in the lung (Figures 5A–C). In contrast to the significant levels of IL-17A production in γδ T cells, IL-17A production by CD4+ and CD8+ αβ T cells during Mtb infection was minimal (Figure 5B vs. Figure 5D). Furthermore, in the absence of ITK, unlike γδ T-cell-derived IL-17A that was significantly impaired, IL-17A production by CD4+ and CD8+ αβ T cells did not exhibit overt differences, although the proportion of those cells making IL-17A was quite low (Figure 5D).
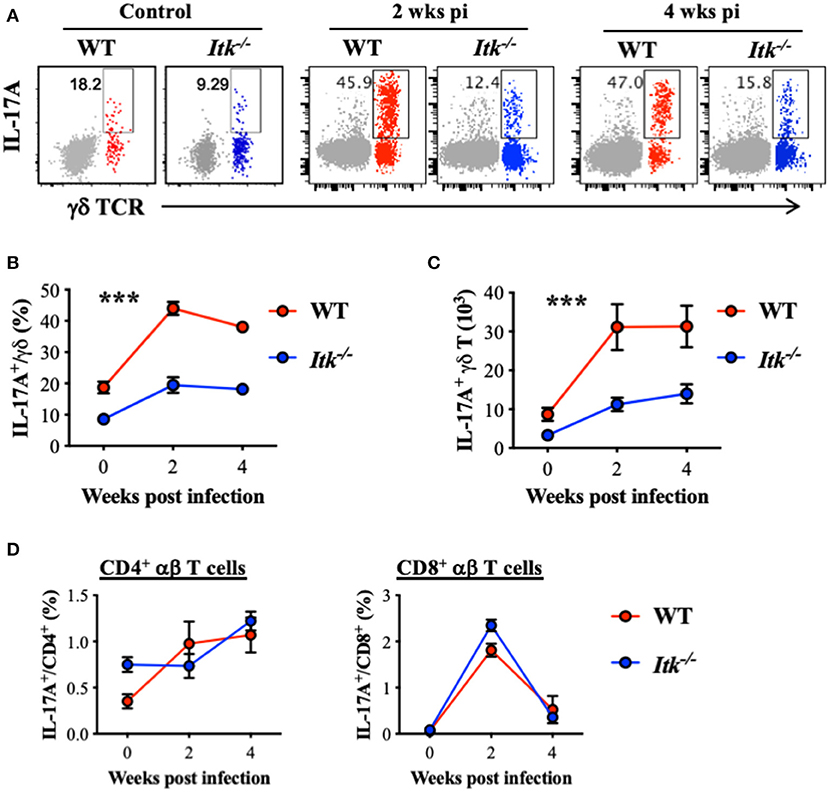
Figure 5. Itk deficiency impairs lung γδ T-cell-derived IL-17A production during Mtb infection. WT and Itk−/− mice were infected as in Figure 3, and lungs were analyzed at the indicated time points. Cells isolated from the lungs were stimulated with PMA and ionomycin, in the presence of BFA and monensin, followed by intracellular cytokine staining. (A) Representative FACS plots for IL-17A production by γδ T cells. Production of CD4+ αβ T cells is shown as gray background. (B) Percentages and (C) number of IL-17A-producing γδ T cells. (D) Percentages of IL-17A-producing CD4+ and CD8+ αβ T cells. ***p < 0.001 by two-way ANOVA with post hoc test. N = 5–9. Numbers of the indicated cell types were presented as mean ± SEM and represent results of three independent experiments.
Our data above supports a strong role of ITK in promoting the IL-17A-producing effector γδ T cells during Mtb infection. It is possible that αβ T-cell effector functions were also altered in the absence of ITK and further contributed to the impaired bacterial clearance and enhanced lung pathology as observed in Figure 2. To determine whether ITK regulates αβ T-cell effector function, we stimulated cells isolated from the lungs of the infected mice 4 weeks postinfection. We found that bulk T-cell activation by PMA and ionomycin suggested that ITK is not required for CD4+ and CD8+ αβ T cells to produce effector cytokines tumor necrosis factor alpha (TNF-α) and interferon-gamma (IFN-γ) during Mtb infection (Figure 6A). Moreover, to our surprise, stimulation of Mtb antigen-specific CD4+ αβ T cells with ESAT-64−17 (MHCII-restricted epitope), or CD8+ αβ T cells with TB10.44−11 (MHCI-restricted epitope) revealed no difference in production of TNF-α and IFN-γ by these cells in the absence of ITK (Figure 6B). Along with the data above, our results suggest that the major protective role of ITK during Mtb infection might be executed through ITK-mediated IL-17A production by γδ T cells.
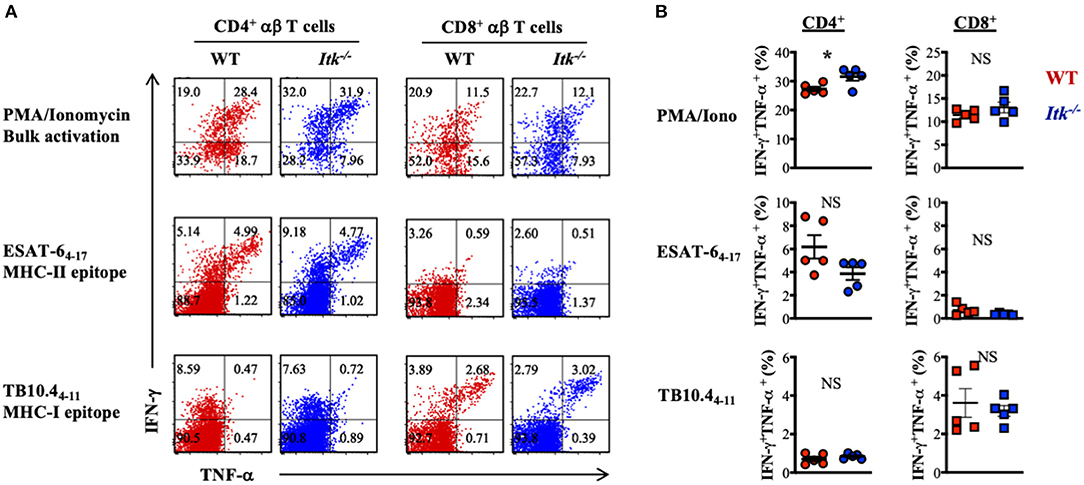
Figure 6. Itk deficiency has minimal impact on lung antigen-specific αβ effector T cells during Mtb infection. WT and Itk−/− mice were infected as in Figure 3 and analyzed 4 weeks postinfection. Cells isolated from the lungs were stimulated with PMA and ionomycin (for bulk T-cell activation), MHCII-restricted Mtb epitope ESAT-64−17 (for antigen-specific CD4+ T-cell activation), or MHCI- restricted Mtb epitope TB10.44−11 (for antigen-specific CD8+ T-cell activation), in the presence of BFA and monensin, followed by intracellular cytokine staining. (A) Representative FACS plots for IFN-γ and TNF-α production by CD4+ and CD8+ αβ T cells under the indicated stimulating conditions. (B) Summary of percentages of IFN-γ and TNF-α-producing CD4+ and CD8+ αβ T cells under the indicated stimulating conditions. N = 5. Data were presented as mean ± SEM and represent results of three independent experiments. NS, not significant; *p < 0.05 by Student's t-test.
Itk deficiency led to significantly impaired effector γδ T cells (Figure 5) but not αβ T cells (Figure 6). These might be the results of differential requirement of ITK signaling in T-cell expansion. Using proliferative maker Ki67 to detect T-cell proliferation, we found that Mtb-driven early proliferation of γδ T cells was severely impaired in ITK deficiency (Figure 7, top panel), but not the CD4+ and CD8+ αβ T-cell subsets (Figure 7, middle and bottom panels). These data, in part, explain the selective defect of effector γδ T-cell development in Itk-deficient mice during Mtb infection.
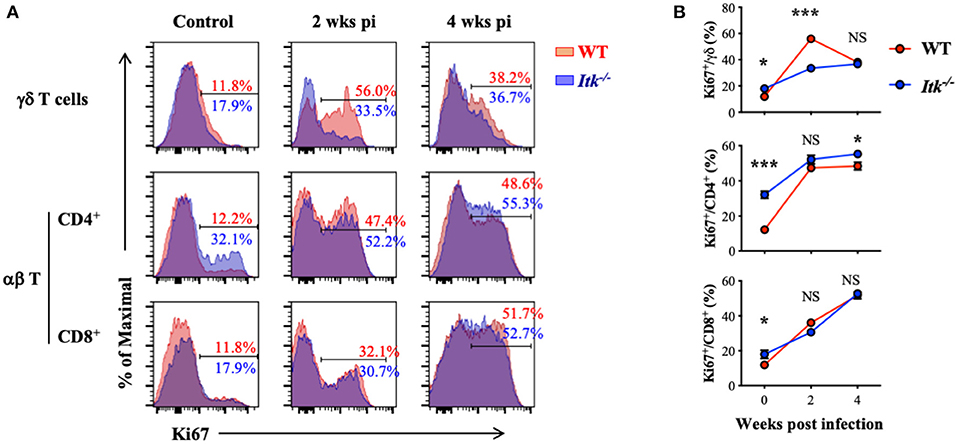
Figure 7. Itk deficiency impairs lung γδ but not αβ T-cell proliferation during Mtb infection. WT and Itk−/− mice were infected as in Figure 3 and analyzed at the indicated time points. Cells isolated from the lungs were analyzed via nuclear staining. (A) Representative FACS plots for Ki67 expression by γδ T cells and CD4+ and CD8+ αβ T cells at the indicated time points postinfection. (B) Summary of percentages of KI67+ cells among γδ T cells and CD4+ and CD8+ αβ T cells. N = 5–9. Data were presented as mean ± SEM and represent results of three independent experiments. NS, not significant; *p < 0.05, ***p < 0.001 by two-way ANOVA with post hoc test.
The immune cells that control Mtb include macrophages, neutrophils, and T-cell populations (2, 3). The TCR pathway is elevated in active Mtb infection in human lung, and ITK is a major component of this pathway; however, its role in the T-cell response to Mtb is not known. Here, we demonstrate a protective role of ITK in Mtb infections in murine models, with a particular role in the ability of lung γδ T cells to produce IL-17A, which is associated with Mtb residence in lung neutrophils. These findings have important implications for understanding the T-cell immune response to Mtb and the role of ITK in this process.
In humans, Mtb infects via inhalation of a low dose of aerosolized bacteria; therefore, low-dose aerosol inoculation would better mimic the nature of Mtb infection in humans. However, in the absence of the ability to perform aerosolized infections, the standard experimental protocol of Mtb infection in mice is 1,000 CFU via intranasal inoculation (69–72). Monocytes play an essential role in initiating T-cell responses in the lung against Mtb infection (73), and using this standard protocol of Mtb infection in mice, we previously observed monocytosis (64), similar to what has been reported in other studies using aerosolized low-dose infection protocols (74, 75). Using intranasal inoculation with 1,000 CFU of Mtb in mice, we observed that the numbers of host myeloid cells infiltrating to the lung during Mtb infection are significantly altered in the absence of ITK. Alveolar macrophages are considered as the preferred replicating niche for Mtb and promote the early stage of infection (64, 76). In Itk-deficient mice, there are more alveolar macrophages harboring Mtb, suggesting a more permissive cellular environment in the lung. Moreover, although fewer neutrophils are present in the lung in the absence of ITK, there are more Mtb resident in neutrophils at 4 weeks postinfection. The redistribution of Mtb in different phagocytes in the absence of ITK appears to correlate with the increased bacterial burden in the lung 4 weeks postinfection. Other than T cells, ITK has been indicated to regulate functions of innate immune cells such as mast cells (77, 78). However, potential intrinsic functions of ITK in lung macrophages and neutrophils during Mtb infection would merit further studies.
Activation of γδ T cells for IL-17A production is severely impaired in Itk-deficient mice and very likely responsible for the resultant neutrophil recruitment early after infection(s), as well as the increased neutrophil resident Mtb population later in infection. Indeed, we have shown that ITK can regulate γδ T cell development and function in mouse (48), and while there was no apparent difference in peripheral blood γδ T-cell numbers in humans carrying ITK mutations (12), the normal range for γδ T cells varies quite widely by anatomical location, as well as by geography and ethnicity (79). Despite relatively normal antigen-specific αβ T-cell responses, our results identify ITK signaling as an essential player for the IL-17A production by γδ T cells, the predominant source of IL-17A in Mtb-infected lungs (68). ITK exhibited a γδ T cell-specific function, as compared to αβ T cells, in driving T-cell proliferation during early immune responses to Mtb infections. Our work suggests that ITK deficiency in humans may lead to γδ T-cell deficiency in expansion and production of IL-17A, in the face of Mtb infection.
We and others have previously reported a role for ITK signaling in regulating Foxp3+ regulatory T (Treg) cell development and function (41, 80). In the absence of ITK, the proportion of Treg cells among CD4+ T cells in the lymphoid organs of naive mice is increased, and the differentiation of inducible Treg cells from CD4+ naive T cells in vitro is also enhanced (41, 80). The role of ITK in Treg cell responses in the lung during Mtb infection was, however, unclear. In mouse model of Mtb infection, we observed higher Mtb burdens, higher lung pathological scores and reduced numbers of Treg cells found in the lungs in Itk-deficient mice, compared to those in the infected WT mice (Figure 4). Tissue damage may be explained by excessive growth of the pathogen and/or immunopathology. Indeed, we observed impaired γδ T cell expansion and γδ T cell-derived IL-17A production in Mtb-infected Itk-deficient mice but no overt differences in other immune effectors. The decrease in Treg numbers in the lung during Mtb infection in the absence of ITK may explain higher levels of immunopathology potentially due to impaired immunomodulatory function. Future research using murine models with conditional deletion of Itk specifically in Foxp3+ Treg cells would allow more in-depth investigation of the role of ITK in Treg cells during Mtb infection.
Taken together, these findings suggest a potential role for ITK in active TB in humans, in addition to its known connection with primary immunodeficiency, susceptibility to EBV, lymphoproliferative diseases, and asthma (12, 50, 51). These findings also have important implications for human genetics associated with susceptibility to Mtb due to altered immune responses and molecular signals modulating host immunity that controls the progression of active tuberculosis.
Our findings support a role of ITK signaling in promoting protective immune responses against Mtb, in particular, γδ T cell expansion and production of IL-17A, which could contribute to the modulation of tuberculosis, especially in infections with highly virulent bacterial strains in which IL-17A has been shown to play an essential protective role (67). However, our work also sounds a note of caution for the potential use of compounds such as Ibrutinib that inhibit the related kinase BTK as well as ITK (81). Given the potential for inhibition of ITK, patients being treated with Ibrutinib may need to be monitored for infection or potential reactivation of latent Mtb.
The datasets generated for this study can be found in the Gene Expression Omnibus under accession number GSE20050.
The animal study was reviewed and approved by the Institutional Animal Care and Use Committees at Cornell University.
LH, MM, NN, JE, CL, and WH performed the experiments. LH, TS, AA, and WH analyzed and interpreted data. LH, AA, and WH wrote the manuscript. KY performed bioinformatic analyses. DR contributed reagents and intellectual input. AA and WH conceived research and designed experiments.
This work was supported in part by grants from the National Institutes of Health (AI120701 and AI138570 to AA, AI129422 to AA and WH, GM130555 Sub-6610 to WH, and AI134183 to DR), and a Pilot Grant (to WH) from LSU-Tulane COBRE Center for Experimental Infectious Disease Research (funded by NIH P30GM110760). JE was supported by a training program in Biomedical and Bioengineering at Cornell University (funded by NIH T32EB023860). MM is a recipient of the Pathobiological Sciences Graduate Program Fellowship in the School of Veterinary Medicine at the Louisiana State University.
AA receives research support from 3M Corporation.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Amie Redko for the animal care and Ling Zhang for the technical assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.03103/full#supplementary-material
Supplementary Figure 1. Gene enrichment profile of the TCR/ITK signaling in human lungs with active TB. Genes in the TCR signaling pathways were rendered with their positions and connections in the pathway, with colored codes indicating the log2-fold change comparing levels of gene expression in caseous to those in normal tissues. Red indicates upregulation in caseous samples while blue indicates downregulation. Note that ITK is one of the central components of the TCR signaling pathway.
Supplementary Table 1. Summary of pathways that exhibited an up- or down-regulated trend in caseum samples compared to normal tissues.
CFU, colony-forming unit; GSEA, gene set enrichment analysis; ITK, IL-2-inducible T-cell kinase; Mtb, Mycobacterium tuberculosis; NK, natural killer; TCR, T-cell receptor; Treg cells, Foxp3-expressing regulatory T cells.
2. O'Garra A, Redford PS, McNab FW, Bloom CI, Wilkinson RJ, Berry MP. The immune response in tuberculosis. Annu Rev Immunol. (2013) 31:475–527. doi: 10.1146/annurev-immunol-032712-095939
3. de Martino M, Lodi L, Galli L, Chiappini E. Immune response to Mycobacterium tuberculosis: a narrative review. Front Pediatr. (2019) 7:350. doi: 10.3389/fped.2019.00350
4. Boisson-Dupuis S, Bustamante J, El-Baghdadi J, Camcioglu Y, Parvaneh N, El Azbaoui S, et al. Inherited and acquired immunodeficiencies underlying tuberculosis in childhood. Immunol Rev. (2015) 264:103–20. doi: 10.1111/imr.12272
5. Andreotti AH, Schwartzberg PL, Joseph RE, Berg LJ. T-cell signaling regulated by the Tec family kinase, Itk. Cold Spring Harb Perspect Biol. (2010) 2:a002287. doi: 10.1101/cshperspect.a002287
6. Tangye SG, Palendira U, Edwards ES. Human immunity against EBV-lessons from the clinic. J Exp Med. (2017) 214:269–83. doi: 10.1084/jem.20161846
7. Mansouri D, Mahdaviani SA, Khalilzadeh S, Mohajerani SA, Hasanzad M, Sadr S, et al. IL-2-inducible T-cell kinase deficiency with pulmonary manifestations due to disseminated Epstein-Barr virus infection. Int Arch Allergy Immunol. (2012) 158:418–22. doi: 10.1159/000333472
8. Linka RM, Risse SL, Bienemann K, Werner M, Linka Y, Krux F, et al. Loss-of-function mutations within the IL-2 inducible kinase ITK in patients with EBV-associated lymphoproliferative diseases. Leukemia. (2012) 26:963–71. doi: 10.1038/leu.2011.371
9. Huck K, Feyen O, Niehues T, Ruschendorf F, Hubner N, Laws HJ, et al. Girls homozygous for an IL-2-inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV-associated lymphoproliferation. J Clin Invest. (2009) 119:1350–8. doi: 10.1172/JCI37901
10. Bienemann K, Borkhardt A, Klapper W, Oschlies I. High incidence of Epstein-Barr virus (EBV)-positive Hodgkin lymphoma and Hodgkin lymphoma-like B-cell lymphoproliferations with EBV latency profile 2 in children with interleukin-2-inducible T-cell kinase deficiency. Histopathology. (2015) 67:607–16. doi: 10.1111/his.12677
11. Veillette A, Perez-Quintero LA, Latour S. X-linked lymphoproliferative syndromes and related autosomal recessive disorders. Curr Opin Allergy Clin Immunol. (2013) 13:614–22. doi: 10.1097/ACI.0000000000000008
12. Ghosh S, Bienemann K, Boztug K, Borkhardt A. Interleukin-2-inducible T-cell kinase (ITK) deficiency - clinical and molecular aspects. J Clin Immunol. (2014) 34:892–9. doi: 10.1007/s10875-014-0110-8
13. Cohen JI. Primary immunodeficiencies associated with EBV disease. Curr Top Microbiol Immunol. (2015) 390:241–65. doi: 10.1007/978-3-319-22822-8_10
14. Eken A, Cansever M, Somekh I, Mizoguchi Y, Zietara N, Okus FZ, et al. Genetic deficiency and biochemical inhibition of ITK affect human Th17, Treg, and innate lymphoid cells. J Clin Immunol. (2019) 39:391–400. doi: 10.1007/s10875-019-00632-5
15. Qi Q, Huang W, Bai Y, Balmus G, Weiss RS, August A. A unique role for ITK in survival of invariant NKT cells associated with the p53-dependent pathway in mice. J Immunol. (2012) 188:3611–9. doi: 10.4049/jimmunol.1102475
16. Qi Q, Xia M, Bai Y, Yu S, Cantorna M, August A. Interleukin-2-inducible T cell kinase (Itk) network edge dependence for the maturation of iNKT cell. J Biol Chem. (2011) 286:138–46. doi: 10.1074/jbc.M110.148205
17. Felices M, Berg LJ. The Tec kinases Itk and Rlk regulate NKT cell maturation, cytokine production, and survival. J Immunol. (2008) 180:3007–18. doi: 10.4049/jimmunol.180.5.3007
18. Gadue P, Stein PL. NK T cell precursors exhibit differential cytokine regulation and require Itk for efficient maturation. J Immunol. (2002) 169:2397–406. doi: 10.4049/jimmunol.169.5.2397
19. Liao XC, Littman DR. Altered T cell receptor signaling and disrupted T cell development in mice lacking Itk. Immunity. (1995) 3:757–69. doi: 10.1016/1074-7613(95)90065-9
20. Bachmann MF, Littman DR, Liao XC. Antiviral immune responses in Itk-deficient mice. J Virol. (1997) 71:7253–7. doi: 10.1128/JVI.71.10.7253-7257.1997
21. Liao XC, Littman DR, Weiss A. Itk and Fyn make independent contributions to T cell activation. J Exp Med. (1997) 186:2069–73. doi: 10.1084/jem.186.12.2069
22. Schaeffer EM, Debnath J, Yap G, McVicar D, Liao XC, Littman DR, et al. Requirement for Tec kinases Rlk and Itk in T cell receptor signaling and immunity. Science. (1999) 284:638–41. doi: 10.1126/science.284.5414.638
23. Liu KQ, Bunnell SC, Gurniak CB, Berg LJ. T cell receptor-initiated calcium release is uncoupled from capacitative calcium entry in Itk-deficient T cells. J Exp Med. (1998) 187:1721–7. doi: 10.1084/jem.187.10.1721
24. Atherly LO, Lucas JA, Felices M, Yin CC, Reiner SL, Berg LJ. The Tec family tyrosine kinases Itk and Rlk regulate the development of conventional CD8+ T cells. Immunity. (2006) 25:79–91. doi: 10.1016/j.immuni.2006.05.012
25. Broussard C, Fleischacker C, Horai R, Chetana M, Venegas AM, Sharp LL, et al. Altered development of CD8+ T cell lineages in mice deficient for the Tec kinases Itk and Rlk. Immunity. (2006) 25:93–104. doi: 10.1016/j.immuni.2006.05.011
26. Prince AL, Yin CC, Enos ME, Felices M, Berg LJ. The Tec kinases Itk and Rlk regulate conventional versus innate T-cell development. Immunol Rev. (2009) 228:115–31. doi: 10.1111/j.1600-065X.2008.00746.x
27. Prince AL, Watkin LB, Yin CC, Selin LK, Kang J, Schwartzberg PL, et al. Innate PLZF+CD4+ alphabeta T cells develop and expand in the absence of Itk. J Immunol. (2014) 193:673–87. doi: 10.4049/jimmunol.1302058
28. Prince AL, Kraus Z, Carty SA, Ng C, Yin CC, Jordan MS, et al. Development of innate CD4+ and CD8+ T cells in Itk-deficient mice is regulated by distinct pathways. J Immunol. (2014) 193:688–99. doi: 10.4049/jimmunol.1302059
29. Hu J, Sahu N, Walsh E, August A. Memory phenotype CD8+ T cells with innate function selectively develop in the absence of active Itk. Eur J Immunol. (2007) 37:2892–9. doi: 10.1002/eji.200737311
30. Hu J, August A. Naive and innate memory phenotype CD4+ T cells have different requirements for active Itk for their development. J Immunol. (2008) 180:6544–52. doi: 10.4049/jimmunol.180.10.6544
31. Huang W, Huang F, Kannan AK, Hu J, August A. ITK tunes IL-4-induced development of innate memory CD8+ T cells in a gammadelta T and invariant NKT cell-independent manner. J Leukoc Biol. (2014) 96:55–63. doi: 10.1189/jlb.1AB0913-484RR
32. Miller A, Wilcox H, Lai Z, Berg L. Signaling through Itk promotes T helper 2 differentiation via negative regulation of T-bet. Immunity. (2004) 21:67–80. doi: 10.1016/j.immuni.2004.06.009
33. Mueller C, August A. Attenuation of immunological symptoms of allergic asthma in mice lacking the tyrosine kinase ITK. J Immunol. (2003) 170:5056–63. doi: 10.4049/jimmunol.170.10.5056
34. Kannan A, Sahu N, Mohanan S, Mohinta S, August A. Itk modulates allergic airway inflammation by suppressing IFNγ in naïve CD4+ T-cells. J Allergy Clin Immunol. (2013) 132:811–20.e1–5. doi: 10.1016/j.jaci.2013.04.033
35. Kannan A, Lee Y, Qi Q, Huang W, Jeong AR, Ohnigian S, et al. Allele-sensitive mutant, Itkas, reveals that Itk kinase activity is required for Th1, Th2, Th17, and iNKT-cell cytokine production. Eur J Immunol. (2015) 45:2276–85. doi: 10.1002/eji.201445087
36. Sahu N, Venegas AM, Jankovic D, Mitzner W, Gomez-Rodriguez J, Cannons JL, et al. Selective expression rather than specific function of Txk and Itk regulate Th1 and Th2 responses. J Immunol. (2008) 181:6125–31. doi: 10.4049/jimmunol.181.9.6125
37. Sahu N, Mueller C, Fischer A, August A. Differential sensitivity to Itk kinase signals for T helper 2 cytokine production and chemokine-mediated migration. J Immunol. (2008) 180:3833–8. doi: 10.4049/jimmunol.180.6.3833
38. Schaeffer EM, Yap GS, Lewis CM, Czar MJ, McVicar DW, Cheever AW, et al. Mutation of Tec family kinases alters T helper cell differentiation. Nat Immunol. (2001) 2:1183–8. doi: 10.1038/ni734
39. Gomez-Rodriguez J, Meylan F, Handon R, Hayes ET, Anderson SM, Kirby MR, et al. Itk is required for Th9 differentiation via TCR-mediated induction of IL-2 and IRF4. Nat Commun. (2016) 7:10857. doi: 10.1038/ncomms10857
40. Gomez-Rodriguez J, Sahu N, Handon R, Davidson TS, Anderson SM, Kirby MR, et al. Differential expression of interleukin-17A and−17F is coupled to T cell receptor signaling via inducible T cell kinase. Immunity. (2009) 31:587–97. doi: 10.1016/j.immuni.2009.07.009
41. Gomez-Rodriguez J, Wohlfert EA, Handon R, Meylan F, Wu JZ, Anderson SM, et al. Itk-mediated integration of T cell receptor and cytokine signaling regulates the balance between Th17 and regulatory T cells. J Exp Med. (2014) 211:529–43. doi: 10.1084/jem.20131459
42. Kannan AK, Kim DG, August A, Bynoe MS. Itk signals promote neuroinflammation by regulating CD4+ T-cell activation and trafficking. J Neurosci. (2015) 35:221–33. doi: 10.1523/JNEUROSCI.1957-14.2015
43. Huang W, Solouki S, Koylass N, Zheng SG, August A. ITK signalling via the Ras/IRF4 pathway regulates the development and function of Tr1 cells. Nat Commun. (2017) 8:15871. doi: 10.1038/ncomms15871
44. Kannan AK, Mohinta S, Huang W, Huang L, Koylass N, Appleton JA, et al. T-Bet independent development of IFNgamma secreting natural T helper 1 cell population in the absence of Itk. Sci Rep. (2017) 7:45935. doi: 10.1038/srep45935
45. Czar MJ, Debnath J, Schaeffer EM, Lewis CM, Schwartzberg PL. Biochemical and genetic analyses of the Tec kinases Itk and Rlk/Txk. Biochem Soc Trans. (2001) 29:863–7. doi: 10.1042/bst0290863
46. Yin CC, Cho OH, Sylvia KE, Narayan K, Prince AL, Evans JW, et al. The Tec kinase ITK regulates thymic expansion, emigration, and maturation of gammadelta NKT cells. J Immunol. (2013) 190:2659–69. doi: 10.4049/jimmunol.1202531
47. Felices M, Yin C, Kosaka Y, Kang J, Berg L. Tec kinase Itk in gammadelta T cells is pivotal for controlling IgE production in vivo. Proc Natl Acad Sci USA. (2009) 106:8308–13. doi: 10.1073/pnas.0808459106
48. Qi Q, Xia M, Hu J, Hicks E, Iyer A, Xiong N, et al. Enhanced development of CD4+ gammadelta T cells in the absence of Itk results in elevated IgE production. Blood. (2009) 114:564–71. doi: 10.1182/blood-2008-12-196345
49. Xia M, Qi Q, Jin Y, Wiest DL, August A, Xiong N. Differential roles of IL-2-inducible T cell kinase-mediated TCR signals in tissue-specific localization and maintenance of skin intraepithelial T cells. J Immunol. (2010) 184:6807–14. doi: 10.4049/jimmunol.1000453
50. Lee SH, Chang HS, Jang AS, Park SW, Park JS, Uh ST, et al. The association of a single-nucleotide polymorphism of the IL-2 inducible T-cell Kinase gene with asthma. Ann Hum Genet. (2011) 75:359–69. doi: 10.1111/j.1469-1809.2010.00637.x
51. Ferrara TJ, Mueller C, Sahu N, Ben-Jebria A, August A. Reduced airway hyperresponsiveness and tracheal responses during allergic asthma in mice lacking tyrosine kinase inducible T-cell kinase. J Allergy Clin Immunol. (2006) 117:780–6. doi: 10.1016/j.jaci.2005.12.1330
52. Alpay F, Zare Y, Kamalludin MH, Huang X, Shi X, Shook GE, et al. Genome-wide association study of susceptibility to infection by Mycobacterium avium subspecies paratuberculosis in Holstein cattle. PLoS ONE. (2014) 9:e111704. doi: 10.1371/journal.pone.0111704
53. Kim MJ, Wainwright HC, Locketz M, Bekker LG, Walther GB, Dittrich C, et al. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol Med. (2010) 2:258–74. doi: 10.1002/emmm.201000079
54. Seimon TA, Kim MJ, Blumenthal A, Koo J, Ehrt S, Wainwright H, et al. Induction of ER stress in macrophages of tuberculosis granulomas. PLoS ONE. (2010) 5:e12772. doi: 10.1371/journal.pone.0012772
55. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. (2015) 43:e47. doi: 10.1093/nar/gkv007
56. Luo W, Friedman MS, Shedden K, Hankenson KD, Woolf PJ. GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics. (2009) 10:161. doi: 10.1186/1471-2105-10-161
57. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. (2005) 102:15545–50. doi: 10.1073/pnas.0506580102
58. Luo W, Brouwer C. Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics. (2013) 29:1830–1. doi: 10.1093/bioinformatics/btt285
59. Carroll P, Schreuder LJ, Muwanguzi-Karugaba J, Wiles S, Robertson BD, Ripoll J, et al. Sensitive detection of gene expression in mycobacteria under replicating and non-replicating conditions using optimized far-red reporters. PLoS ONE. (2010) 5:e9823. doi: 10.1371/journal.pone.0009823
60. Reece JJ, Siracusa MC, Southard TL, Brayton CF, Urban JF Jr, Scott AL. Hookworm-induced persistent changes to the immunological environment of the lung. Infect Immun. (2008) 76:3511–24. doi: 10.1128/IAI.00192-08
61. Chackerian AA, Alt JM, Perera TV, Dascher CC, Behar SM. Dissemination of Mycobacterium tuberculosis is influenced by host factors and precedes the initiation of T-cell immunity. Infect Immun. (2002) 70:4501–9. doi: 10.1128/IAI.70.8.4501-4509.2002
62. Feng CG, Kaviratne M, Rothfuchs AG, Cheever A, Hieny S, Young HA, et al. NK cell-derived IFN-gamma differentially regulates innate resistance and neutrophil response in T cell-deficient hosts infected with Mycobacterium tuberculosis. J Immunol. (2006) 177:7086–93. doi: 10.4049/jimmunol.177.10.7086
63. Srivastava S, Ernst JD, Desvignes L. Beyond macrophages: the diversity of mononuclear cells in tuberculosis. Immunol Rev. (2014) 262:179–92. doi: 10.1111/imr.12217
64. Huang L, Nazarova EV, Tan S, Liu Y, Russell DG. Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J Exp Med. (2018) 215:1135–52. doi: 10.1084/jem.20172020
65. Mishra BB, Lovewell RR, Olive AJ, Zhang G, Wang W, Eugenin E, et al. Nitric oxide prevents a pathogen-permissive granulocytic inflammation during tuberculosis. Nat Microbiol. (2017) 2:17072. doi: 10.1038/nmicrobiol.2017.72
66. Dallenga T, Repnik U, Corleis B, Eich J, Reimer R, Griffiths GW, et al. M. tuberculosis-induced necrosis of infected neutrophils promotes bacterial growth following phagocytosis by macrophages. Cell Host Microbe. (2017) 22:519–30.e3. doi: 10.1016/j.chom.2017.09.003
67. Gopal R, Monin L, Slight S, Uche U, Blanchard E, Fallert Junecko BA, et al. Unexpected role for IL-17 in protective immunity against hypervirulent Mycobacterium tuberculosis HN878 infection. PLoS Pathog. (2014) 10:e1004099. doi: 10.1371/journal.ppat.1004099
68. Lockhart E, Green AM, Flynn JL. IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J Immunol. (2006) 177:4662–9. doi: 10.4049/jimmunol.177.7.4662
69. Horvath CN, Shaler CR, Jeyanathan M, Zganiacz A, Xing Z. Mechanisms of delayed anti-tuberculosis protection in the lung of parenteral BCG-vaccinated hosts: a critical role of airway luminal T cells. Mucosal Immunol. (2012) 5:420–31. doi: 10.1038/mi.2012.19
70. Segueni N, Tritto E, Bourigault ML, Rose S, Erard F, Le Bert M, et al. Controlled Mycobacterium tuberculosis infection in mice under treatment with anti-IL-17A or IL-17F antibodies, in contrast to TNFalpha neutralization. Sci Rep. (2016) 6:36923. doi: 10.1038/srep36923
71. Segueni N, Benmerzoug S, Rose S, Gauthier A, Bourigault ML, Reverchon F, et al. Innate myeloid cell TNFR1 mediates first line defence against primary Mycobacterium tuberculosis infection. Sci Rep. (2016) 6:22454. doi: 10.1038/srep22454
72. Benmerzoug S, Bounab B, Rose S, Gosset D, Biet F, Cochard T, et al. Sterile lung inflammation induced by silica exacerbates Mycobacterium tuberculosis infection via STING-dependent type 2 immunity. Cell Rep. (2019) 27:2649–64.e5. doi: 10.1016/j.celrep.2019.04.110
73. Samstein M, Schreiber HA, Leiner IM, Susac B, Glickman MS, Pamer EG. Essential yet limited role for CCR2(+) inflammatory monocytes during Mycobacterium tuberculosis-specific T cell priming. Elife. (2013) 2:e01086. doi: 10.7554/eLife.01086.013
74. Antonelli LR, Gigliotti Rothfuchs A, Goncalves R, Roffe E, Cheever AW, Bafica A, et al. Intranasal Poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J Clin Invest. (2010) 120:1674–82. doi: 10.1172/JCI40817
75. La Manna MP, Orlando V, Dieli F, Di Carlo P, Cascio A, Cuzzi G, et al. Quantitative and qualitative profiles of circulating monocytes may help identifying tuberculosis infection and disease stages. PLoS ONE. (2017) 12:e0171358. doi: 10.1371/journal.pone.0171358
76. Cohen SB, Gern BH, Delahaye JL, Adams KN, Plumlee CR, Winkler JK, et al. Alveolar macrophages provide an early Mycobacterium tuberculosis niche and initiate dissemination. Cell Host Microbe. (2018) 24:439–46.e4. doi: 10.1016/j.chom.2018.08.001
77. Huang W, Morales JL, Gazivoda VP, August A. Nonreceptor tyrosine kinases ITK and BTK negatively regulate mast cell proinflammatory responses to lipopolysaccharide. J Allergy Clin Immunol. (2016) 137:1197–205. doi: 10.1016/j.jaci.2015.08.056
78. Iyer AS, Morales JL, Huang W, Ojo F, Ning G, Wills E, et al. Absence of Tec family kinases interleukin-2 inducible T cell kinase (Itk) and Bruton's tyrosine kinase (Btk) severely impairs Fc epsilonRI-dependent mast cell responses. J Biol Chem. (2011) 286:9503–13. doi: 10.1074/jbc.M110.165613
79. Esin S, Shigematsu M, Nagai S, Eklund A, Wigzell H, Grunewald J. Different percentages of peripheral blood gamma delta + T cells in healthy individuals from different areas of the world. Scand J Immunol. (1996) 43:593–6. doi: 10.1046/j.1365-3083.1996.d01-79.x
80. Huang W, Jeong AR, Kannan AK, Huang L, August A. IL-2-inducible T cell kinase tunes T regulatory cell development and is required for suppressive function. J Immunol. (2014) 193:2267–72. doi: 10.4049/jimmunol.1400968
Keywords: active tuberculosis, transcriptomic analysis, non-receptor tyrosine kinase, IL-17A, IFN-γ, γδ T cells
Citation: Huang L, Ye K, McGee MC, Nidetz NF, Elmore JP, Limper CB, Southard TL, Russell DG, August A and Huang W (2020) Interleukin-2-Inducible T-Cell Kinase Deficiency Impairs Early Pulmonary Protection Against Mycobacterium tuberculosis Infection. Front. Immunol. 10:3103. doi: 10.3389/fimmu.2019.03103
Received: 30 October 2019; Accepted: 19 December 2019;
Published: 24 January 2020.
Edited by:
Igor Kramnik, Boston University, United StatesReviewed by:
Chinnaswamy Jagannath, University of Texas Health Science Center at Houston, United StatesCopyright © 2020 Huang, Ye, McGee, Nidetz, Elmore, Limper, Southard, Russell, August and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Avery August, YXZlcnlhdWd1c3RAY29ybmVsbC5lZHU=; Weishan Huang, aHVhbmcxQGxzdS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.