 Gilson Gonçalves dos Santos
Gilson Gonçalves dos Santos Lauriane Delay1†
Lauriane Delay1† Maripat Corr
Maripat Corr- 1Department of Anesthesiology, University of California, San Diego, La Jolla, CA, United States
- 2Division of Rheumatology, Allergy and Immunology, University of California, San Diego, La Jolla, CA, United States
A high-intensity potentially tissue-injuring stimulus generates a homotopic response to escape the stimulus and is associated with an affective phenotype considered to represent pain. In the face of tissue or nerve injury, the afferent encoding systems display robust changes in the input–output function, leading to an ongoing sensation reported as painful and sensitization of the nociceptors such that an enhanced pain state is reported for a given somatic or visceral stimulus. Our understanding of the mechanisms underlying this non-linear processing of nociceptive stimuli has led to our appreciation of the role played by the functional interactions of neural and immune signaling systems in pain phenotypes. In pathological states, neural systems interact with the immune system through the actions of a variety of soluble mediators, including cytokines. Cytokines are recognized as important mediators of inflammatory and neuropathic pain, supporting system sensitization and the development of a persistent pathologic pain. Cytokines can induce a facilitation of nociceptive processing at all levels of the neuraxis including supraspinal centers where nociceptive input evokes an affective component of the pain state. We review here several key proinflammatory and anti-inflammatory cytokines/chemokines and explore their underlying actions at four levels of neuronal organization: (1) peripheral nociceptor termini; (2) dorsal root ganglia; (3) spinal cord; and (4) supraspinal areas. Thus, current thinking suggests that cytokines by this action throughout the neuraxis play key roles in the induction of pain and the maintenance of the facilitated states of pain behavior generated by tissue injury/inflammation and nerve injury.
Introduction
High-intensity mechanical or thermal stimuli will selectively increase the activity of populations of primary afferents, referred to as nociceptors, with the frequency of discharge reflecting the intensity of the stimulus. This input drives activation of second-order neurons, many of which project to the brain. The consequence of this input is to drive a pain state, which at its simplest results in a protective response (e.g., withdrawal of the affected limb) mediated by spinal and supraspinal organization (e.g., nociception) and then at higher-order levels of processing drives a state of negative affect (e.g., pain/suffering) (1). Of note, it is appreciated that in the face of persistent afferent input, as after tissue or nerve injury, there is an increased activation of the afferent and the second-order spinofugal neuron, which drives an enhanced pain response. Such “hyperalgesic” states variously reflect increased responsiveness of the primary afferent and/or the second-order projection neurons, leading to the enhanced pain report. The biology of systems underlying this change in input–output functionality of the spinal dorsal horn has been the subject of considerable interest. One underlying element of this facilitated processing reflects the role played by cytokine signaling in system function.
Classically, three considerations have characterized the actions of cytokines.
(1) They are peptides released from immunocompetent cells, notably T cells and monocyte family members (2).
(2) This increased production and release is driven by pathological conditions such as tissue injury and infection, which initiate activation of these inflammatory cells.
(3) Their perceived role is largely to engage immune signaling and pathologic targets, serving as entities involved in autocrine, paracrine, and endocrine signaling.
Advances in our understanding of cytokine biology have considerably expanded this original profile. It is now clear that aside from immune cells (resident and recruited macrophages, lymphocytes, and mast cells present throughout the neuraxis), cytokines are released from peripheral afferents (Schwann cells and peripheral termini of sensory fibers), as well as from cells within the dorsal root ganglia (DRG) and spinal cord (3). As will be reviewed, this release can indeed be initiated by injury or inflammation, and also by neuronal activity otherwise driven by these injury conditions (4). Cytokine signaling is now known to exert direct effects upon neural signaling through eponymous receptors located on neurons, microglia, and astrocytes, in the spinal parenchyma and in the DRG and brain. Further, while neuronal activation might be the result of receptor-mediated and direct cell-to-cell contact-dependent mechanisms (e.g., gap junction contacts in DRG neurons and satellite cells) (5), soluble extracellular molecules serve to create broader gradients of paracrine- and autocrine-like regulatory networks. These cytokines thus comprise a communication network between immune and neuronal cells. In the context of high-frequency afferent traffic generated by tissue injury, a wave of inflammatory cytokines acts on the terminals of sensory nerve fibers (nociceptors), triggering activation of corresponding pain pathways while neuronal activation leads to a reciprocal activation of a variety of cytokine-generating cells (3). Of note, prolonged inflammation alters nociceptive processing in such a fashion as to yield a persistent pain phenotype even after the inflammation and wounding has resolved, creating a “neuropathic”-like phenotype (6).
A further intriguing complexity is that several of these cytokines, as will be reviewed below, act through signaling to suppress excitatory signaling (e.g., they have an anti-inflammatory phenotype). Finally, current work raises the likelihood that signaling secondary to sustained cytokine and chemokine release and the recruitment of migratory effector cells into the DRG and spinal cord can initiate a feedback loop that results in neuronal injury and subsequently chronic pain. Thus, the balance between repair and proinflammatory factors may determine the rate of progression and outcome of a neurodegenerative process.
Cytokines thus play important roles at the systems level in regulating the functionality of neuraxial systems regulating neurodevelopment, neuroinflammation, and synaptic transmission. Here, we seek to provide an overview focused on a curated list of cytokines identified in the context of neuronal modulation and damage, to play a role in changes in pain processing after tissue and nerve injury, and discuss roles that cytokines play at the interface of the neuronal and immune system interfaces divided across four levels of neuronal organization: (1) peripheral termini; (2) DRG; (3) spinal cords; and (4) supraspinal areas.
Cytokine Families
Cytokines, from the combination of two Greek words cyto (cell) and kinos (movement), are defined as a family of low-molecular-weight bioactive proteins or glycoproteins secreted by immune cells and non-neuronal cells (e.g., epithelial cells, fibroblasts, and Schwann cells). Interferon was the first cytokine discovered more than 60 years ago (7). In the absence of a unified classification, cytokines are classified by numeric order of discovery, by kinetic or functional role in inflammatory/immune responses, by primary cell of origin, or by structural homologies shared with related molecules (8). According to structural homologies, cytokines can by classified into groups: tumor necrosis factors (TNFs), interleukins (ILs), interferons (IFNs), colony-stimulating factors, transforming growth factors (TGFs), and chemoattractant cytokines, also called chemokines.
Chemokines are small proteins that direct the movement of circulating leukocytes and immune cells. They constitute a family of more than 50 structurally homologous proteins classified in four families according to the location of N-terminal cysteine residues (i.e., CXC, CC, CX3C, or XC). Chemokines affect cells by activating surface receptors that are seven-transmembrane domain G-protein-coupled receptors (GPCRs) and have been implicated in a wide range of inflammatory diseases, such as multiple sclerosis and atherosclerosis (9). These ligands and their respective receptors participate in neuronal and microglial crosstalk (10, 11). The temporal expression of chemokines and their receptors may directly or indirectly contribute to the development of acute pain and the maintenance of chronic pain states.
Historically, cytokines were simply classified according to the functional T-helper (Th) cell group (Th1 or Th2) that produced them. However, recent studies show that cytokines and chemokine display anti-inflammatory and proinflammatory properties producing inhibitory and stimulatory effects in the immune system. As shown in Table 1, properties of a cytokine are dependent on the microenvironment, and most have dual effects according to their context (38, 112, 113). For example, IL-1β is considered a proinflammatory cytokine and can increase neuronal sensitization (17, 18), but it can also regulate inhibitory neurotransmission (15, 16). IL-10 is typically considered to be an immunosuppressive cytokine, which attenuates proinflammatory cytokine release and can reduce antigen presentation. However, IL-10 can also support the activation and proliferation of B cells (39), which can sustain autoimmune attacks. One of the most complex cytokines is TGF-β, which under certain conditions is involved in the differentiation of regulatory T cells (Treg) or in conjunction with IL-6 can drive the differentiation of proinflammatory T cells that produce IL-17 (Th17) (38). Hence, cytokines are characterized by (1) pleiotropy (i.e., a specific cytokine can affect several types of cells), (2) redundancy (i.e., overlapping functions), and (3) cascading signal activation (i.e., one cytokine stimulates the production of additional cytokines) (113, 114).
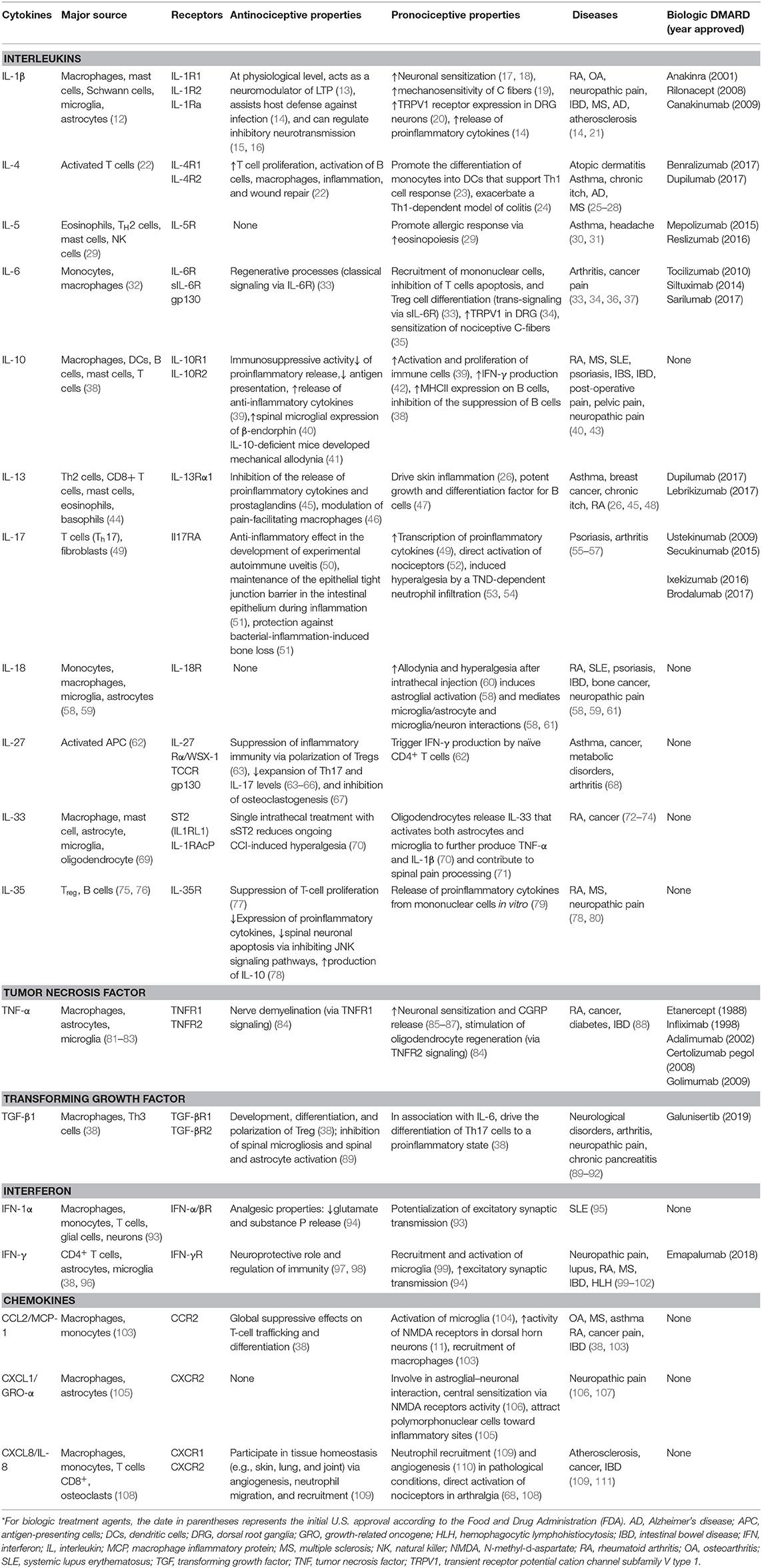
Table 1. Dual effects of cytokines involved in chronic pain*.
Physiologically, cytokines are involved in multiple biological functions such as cell differentiation, survival, growth, and metabolism (115). Although broad characterizations of cytokine behavior were aligned with adaptive immune functions, cytokine responses of the innate immune system are important to prevent damage during and following autoimmune attack, inflammation, and infections. In pathological conditions, the imbalance of cytokines participates in the development of the disease and progression leading to damage (114). In the context of the nervous system, some cytokines are considered to function as pain mediators as well as messengers of the immune system. This level of pleiotropy underscores the elegant role these molecules play in communication between the immune system and the nervous system.
Signaling Pathways
Although the receptors for individual cytokines display specificity for their respective ligands, the subsequent signaling pathways often converge, resulting in nuclear translocation of transcription factors and a secondary transcription of additional downstream mediators. Common signaling pathways activated following cytokine receptor ligation and activation include (1) nuclear factor-κB (NF-κB), (2) the mitogen-activated protein kinases (MAPKs), (3) the Janus kinase (JAK) and signal transducer and activator of transcription (STAT), and (4) the Smad family signaling pathways (114, 116).
NF-κB Signaling
The most widely studied signaling cascade associated with cytokine signaling is the NF-κB (NF kappa light chain enhancer of activated B cells) family (117). These are a family of highly conserved transcription factors including NF-κB2 p52/p100, NF-κB1 p50/p105, c-Rel, RelA/p65, and RelB, which form functional dimers. Receptors that can activate this cascade include IL-1R and the TNF receptors. In the cytoplasm, NF-κB family members are bound to IκB. In the classical or canonical pathways, proinflammatory cytokine receptors activate an IκB kinase complex (IKKβ, IKKα, and NEMO), which phosphorylates IκB proteins, leading to IκB degradation and the release and translocation of the NF-κB/Rel complexes to the nucleus. In the nucleus, these transcription factors can induce gene transcription alone or in combination with other transcription factors including AP-1 and STATs (118).
Some of the target genes include other proinflammatory cytokines, like IL-6 and IL-8. In some instances, there is an alternative pathway through signaling of cytokine receptors like the lymphotoxin-β receptor (LTβR and TNFRSF3) (119). These activate Nck-interacting kinase [NIK; MAPK kinase kinase (MAPKKK) 14], which in turn activates IKKα complexes that phosphorylate NF-κB2 p100. Phosphorylation of NF-κB2 p100 then leads to its ubiquitination and proteasomal processing to NF-κB2 p52/RelB complexes that translocate to the nucleus and induce target gene expression (117).
MAPK Signaling
The MAPKs are generally divided into the p38 stress-activated protein kinase (SAPK)/Jun amino-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK) pathways (120). These kinase pathways are activated by a variety of environmental stresses, growth factors, GPCR agonists, and inflammatory cytokines. In the MAPK cascades, there are tiered activation steps. The membrane proximal MAPKKK kinases (MAPKKKKs) or GTPases activate MAPKKK, which mediate phosphorylation and activation of MAPK kinases (MAPKKs), which in turn phosphorylate and activate MAPK. p38 MAPK is activated by MKK3/MKK6 and is involved in the regulation of HSP27, MAPKAPK-2 (MK2), MAPKAPK-3 (MK3), and several transcription factors including ATF-2, Stat1, the Max/Myc complex, MEF-2, Elk-1, and indirectly CREB via activation of MSK1 (121).
Stress signals are delivered to the JNK family cascade by small GTPases of the Rho family (Rac, Rho, and cdc42). As with the other MAPKs, the membrane proximal kinase is a MAPKKK, typically MEKK1–MEKK4, or a member of the mixed lineage kinases (MLKs) that phosphorylate and activate MKK4 (SEK) or MKK7 and that phosphorylate the SAPK/JNK kinases, which then translocate to the nucleus where they can regulate the activity of multiple other transcription factors (122).
The ERK signaling cascade is activated by receptors involved in growth and differentiation including receptor tyrosine kinases (RTKs), integrins, and ion channels. The receptors signal through cascades that include small GTP-binding proteins (Ras and Rap1), which in turn activate a MAPKKK (Raf), a MAPKK (MEK1/MEK2), and then Erk MAPK (123). Erk dimers can regulate targets in the cytosol and also translocate to the nucleus where they phosphorylate a variety of transcription factors regulating gene expression related to growth, migration, and differentiation. As an example of signaling complexity for cytokines, TNF acts through two receptors, TNFR1 and TNFR2, which drive MAP kinase activation and enhance inflammatory responses by secondary IL-1, IL-6, and IL-8 release following the transcription of their target genes (124).
Activation of neuronal TNF receptors drives MAPK activation, which enhances inflammatory response by increasing IL-1, IL-6, and IL-8 release. IL-1 for instance is involved with cyclooxygenase (COX) upregulation within the DRG, inducing neuronal sensitization. Moreover, sensitization of ion channels in neuronal cells is involved with pain processing (125). IL-6 has been shown to induce JAK and protein kinase C (PKC) activation, which enhances the ion channel transient receptor potential (TRP) cation channel subfamily V member (TRPV1) sensitivity. In fact, JAK and PKC inhibitors decrease TRPV1 sensitization (126, 127). However, not only does this sensitization apply for the primary afferent, but it also seems that cytokines can induce neuronal sensitization in other anatomical levels such as cells in the DRG, dorsal horn of the spinal cord, and supraspinal areas (128). In fact, peripheral inflammation increases the expression of IL-1β and COX in the DRG, cascades known to be involved with neuronal network sensitization (18). It is thus noteworthy that TNF and IL-1β induced sensitization of cells in the dorsal horn and increased pain hypersensitivity (hyperalgesia) by enhancing α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)- or N-methyl-D-aspartate (NMDA)-induced currents. Further, IL-1β and IL-6 suppressed typical inhibitory gamma-aminobutyric acid (GABA)- and glycine-induced currents (17). Accordingly, TNF and IL-1β enhanced NMDA receptor phosphorylation in the trigeminal nucleus in mesencephalic (trigeminal nucleus) and also increased NMDA current in the hippocampus (129).
Smad Signaling
The Smad family of transcription factors is largely downstream of the TGF-β and bone morphogenetic protein (BMP) superfamilies (130). In general, signaling is initiated with ligand-induced activation of serine/threonine receptor kinases and phosphorylation of the cytoplasmic signaling molecules Smad2/Smad3 for the TGF-β/activin pathway, and Smad1/Smad5/Smad9 for the BMP pathway. Activated Smads regulate diverse biological effects by partnering with other transcription factors, resulting in transcription of specific cell state-associated target genes (131). This family has inherent regulatory negative feedback loops with inhibitory Smads (I-Smads) 6 and 7, which are also induced by both TGF-β and BMP signaling. The TGF-β and BMP pathways are cross-regulated by MAPK signaling. Moreover, in certain contexts, TGF-β signaling can also affect the Erk, SAPK/JNK, and p38 MAPK pathways independent of Smad activation (116).
JAK/STAT Signaling
Over 50 cytokines and growth factors use the JAK/STAT pathway for signaling. After receptor ligation, the JAK proteins are phosphorylated, and activated JAKs then phosphorylate STAT monomers, leading to dimerization, nuclear translocation, and DNA binding. Although there are four JAKs (JAK1, JAK2, JAK3, and TYK2) and seven STATs (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6) in mammals, the number of potential combinations alone does not fully explain the pleiotropy in signaling (22, 34, 132). For example, IL-6R and IL-10R signaling both result in STAT3 activation through JAK activation; however, one cytokine is mostly proinflammatory, and the other is considered anti-inflammatory. There may be differences in STAT use for some cytokines, whereas other cytokines like IL-27 strongly activate more than one STAT protein (STAT1 and STAT3). To add to the complexity, multiple STATs may bind to the same target site due to shared specificities. In addition STAT proteins can be phosphorylated on serine residues to influence their DNA binding pattern, and STAT signaling influences epigenetic changes. Some STAT proteins have extra nuclear functions, for instance, at the mitochondrion level (133, 134).
Cytokine-Induced Peripheral Transcription
While it is well-documented that nuclear translocation of transcription factors is initiated by cytokine signaling, it has recently been demonstrated that cytokines also can induce nociceptive plasticity by local protein synthesis in the peripheral processes of sensory afferents (135). It is well-established that IL-6 expression is increased in arthritis and peripheral nerve injury. Likewise, nerve growth factor (NGF) levels are elevated in inflammatory and neuropathic pain states (36). IL-6- and NGF-induced mechanical hyperalgesia is reversed by rapamycin (135). Moreover, the IL-6R signals through the ERK pathway and the NGF receptor signals primarily through the AKT/mTOR pathway leading to phosphorylation of the CAP-binding protein eIF4E. Finally, phosphorylation of eIF4E enhances rapid changes in the translational control of gene expression in sensory neurons, and this effect is linked to mechanical allodynia (136). Although cytokines can induce nociceptive plasticity by local protein synthesis in the peripheral processes of sensory afferents, it remains unclear how these pathways directly intersect with the activities of ion channels or GPCR. In Figure 1, we schematically illustrate these pathways and actions in the cell body of a nociceptor (Figure 1A) and the peripheral terminus (Figure 1B).
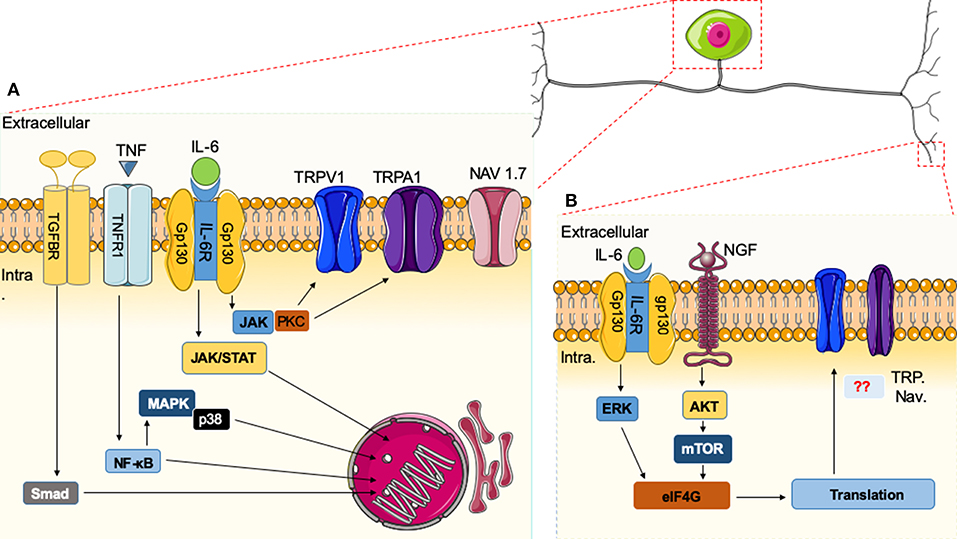
Figure 1. Immune system and nociceptor activation. (A) Cytokines released in the vicinity of the cell body of nociceptors can induce specific receptor activation and signaling cascades. Upon activation of nuclear factor (NF)-κB, mitogen-activated protein kinase (MAPK)/Janus kinase (JAK), and Smad transcription factors present in the cytoplasm are phosphorylated and translocated to the nucleus, leading to the expression of target genes, resulting in biological responses. Alternatively, downstream modulators like protein kinase C (PKC) can sensitize neurons through effects on ion channels (e.g., TRPV1 or Na+ channels). (B) In the peripheral terminus, there are additional cytokines that signal through the extracellular signal-regulated kinase (ERK) pathway and the AKT/mTOR pathways leading to phosphorylation of eIF4E, which can regulate local protein synthesis in the peripheral processes of sensory afferents. It remains unknown how these pathways directly intersect with the activities of ion channels.
Peripheral Nociceptor Terminal
Irritants or immunogens such as carrageenan administered in the hindpaw induce a transient hyperalgesia that is prevented by non-steroidal anti-inflammatory drugs (NSAIDs). However, inflammatory mediators such as prostaglandins into the same site induced prolonged hyperalgesia, described as latent or “primed” state, that is not prevented by NSAIDs (85, 137). In the same manner, during chronic pathophysiological conditions such as arthritis, cytokines induce neuronal sensitization and priming of the primary afferent (85), of the DRG (18), and as reviewed below at supraspinal levels (128). Indeed, in an arthritic murine model [K/BxN passive serum transfer (138)], the mouse shows an early phase characterized by inflammation, increased TNF production, and pain and a late phase characterized by the inflammation's resolution, decreased TNF, but persistent pain (41). These data suggest that cytokines play a role in priming peripheral nociceptors.
Peripheral termini of nociceptors form arborization-like structures in the skin, muscles, bone, joints, and viscera (139). These locations, in proximity to many different cells (e.g., keratinocytes and immune cells), facilitate the immune modulation of nociceptor function. Among the many different inflammatory diseases, arthritic diseases like rheumatoid arthritis (RA) have been studied for the pathophysiology of localized peripheral inflammation and pain. Notably, arthritic diseases have been described to involve an imbalance of cytokines (114, 140). In RA, studies highlight an early alteration of cytokine and chemokine levels months to years before the onset of joint swelling, particularly in patients with arthralgia (i.e., joint pain without observable clinical signs of disease such as joint redness or swelling) (48, 141, 142). Serum cytokine profiles also differ in RA patients with specific autoantibodies. Anti-citrullinated peptide autoantibodies (ACPAs) are present in a major subset of RA patients (around 60–70%) (143), are serological markers for the diagnosis of RA (144), and are prognostic factors for more aggressive joint diseases (145). In the sera of RA patients with ACPAs, there was an increase in the IL-5, IFN-1α, TNF, and IL-13 levels. In contrast, there were specific increases in eotaxin and RANTES (i.e., regulated on activation normal T cell expressed and secreted) levels in the sera of RA patients who did not have any detectable ACPAs (48). In animal models of arthritis, including antigen-induced arthritis (AIA), collagen antibody-induced arthritis (CAIA), and collagen-induced arthritis (CIA), IL-6 has been implicated as a key factor in peripheral pain mechanisms (36). Direct IL-6 or IL-6/sIL-6R administration into knee joints in rodents induced a long-lasting sensitization of nociceptive C-fibers, contributing to mechanical hypersensitivity (35).
In models of inflammatory pain, such as carrageenan administration in the hindpaw, infiltration of macrophages and the local release of TNF play a key role in the development and sensitization of peripheral afferents (85). Activation of neuronal TNF receptors increases the production of IL-1β, IL-6, and CCL2 [formerly known as monocyte chemoattractant protein-1 (MCP-1)]. Both IL-1β and IL-6 have been shown to have activity in acute inflammatory and chronic pain (146–148). Indeed IL-1β enhanced pain transduction and conduction via modulation of ion channels such as TRP ankyrin 1 (TRPA1), TRPV1, and Nav1.7. CCL2 also contributes to macrophage recruitment (104, 149). Thus, these factors serve to perpetuate a feedback loop between neuronal sensitization and cytokine production during tissue injury or inflammation.
Other studies have suggested that additional cytokines play key roles in the induction of inflammatory pain in models of arthritis, including IL-8, IL-17, and IL-27 (55, 58, 68). The chemokine IL-8 and its receptor CXCR2 are involved in sensitization of afferent nociceptors in ACPA-induced arthralgia. Interestingly, recent studies demonstrated that a single injection of human IgG ACPA or monoclonal murinized IgG ACPA antibodies isolated from RA patients is capable of initiating pain without paw swelling when injected into rodents (68). This effect of ACPAs is associated with osteoclast activation, at least in vitro, which via the release of the chemokine CXCL1 (an analog to human IL-8) mediates their pronociceptive effects (68, 108). IL-8 has also been implicated in conditions such as chronic low back pain (LBP). Krock et al. showed a specific upregulation of this chemokine in the cerebrospinal fluid of LBP patients with degenerating disks and a reduction of disk degeneration and chronic back pain in a mouse model (148). In a neuropathic orofacial pain condition, the burning mouth syndrome, patients present an elevated level of plasma IL-8, and this signature directly correlates with pain and depressive symptomatology (150). Hence, IL-8 has been implicated in the periphery for several different pain phenotypes.
As described above, IL-6 has pleiotropic roles associated with pain and inflammation. Another cytokine that also shares the gp130 common receptor chain and signals through the JAK/STAT pathway is IL-27. IL-27 is a heterodimer formed by the Epstein–Barr virus-induced gene 3 (EBI3) and IL-27 p28 subunits, which binds to a receptor composed of the gp130 common receptor chain and IL-27Rα (i.e., WSX-1 or TCCR) (62). Sasaguri et al. showed that IL-27 signaling constitutively contributes to control of thermal (heat and cold) and mechanical sensitivity (151). In an arthritis model, IL-27 attenuates disease development and histological disease severity (i.e., cell infiltration in the joint, synovial hyperplasia, and joint erosion) by reducing the expansion of Th17 cells and IL-17 levels (63–66), which can reduce nociception (Table 1). In osteoimmunology, IL-27 plays a critical role in limiting bone erosion by inhibiting osteoclastogenesis (67). Osteoclast activity is directly involved in pain development and reduced with the use of osteoclast inhibitors such as bisphosphonates or denosumab (152). The suggested mechanisms of this action include mechanical stabilization in bone pain from trauma and also changes in pH and acidosis in bone pain from cancer. Nociception could be promoted by acidosis in which H+ protons can directly activate specific ionic or receptors sensitive to protons such as TRPV1 and the acid-sensing ion channel (ASIC) family (153, 154).
Dorsal Root Ganglia
It has become apparent that the excitability of the afferent input circuitry reflects the functional complexity of the DRG system. DRG neurons are supplied by a fenestrated vasculature that lies outside the blood–brain barrier (BBB), slowing the passage of molecules that are normally excluded from the neuraxis (155). This exposure of the afferent cell bodies to circulating products, including cytokines, may partly explain why circulating neurotoxic agents (e.g., chemotherapeutics) preferentially accumulate and injure cells within the DRG, inducing a sensory rather than a motor neuropathy (156). DRG neurons are additionally supported by satellite glial cells (SGCs), which envelop them and display gap junction linkages between these two cell types (157). During inflammation, SGCs display enhanced activation, increased TNF production, and neuronal excitability (73). An increase of gap junctions has been observed in pain-generating conditions, and this correlates with enhanced neuronal excitability (157). Importantly, peripheral inflammation or nerve injury causes DRG neuronal sensitization, leading to a spreading activation of SGCs through gap junctions and to the expression and release of IL-1β from SGCs.
Current work highlights an important role of macrophages in response to inflammation and cytokine signaling. A major source of endogenous cytokine production is from these resident and migratory DRG macrophages. In arthritic conditions (e.g., osteoarthritis and RA), macrophages infiltrate into the DRG and acquire a phenotype resembling that of TNF-stimulated macrophages, suggesting a role of these cells in the maintenance of arthritic pain (158). In vitro, macrophages stimulated with TNF promote release of calcitonin gene-related peptide (CGRP) by nociceptors, which is consistent with their pronociceptive effect (159). In vivo, TNF is involved with sensitization of nociceptive fibers and elicits a rapid increase of CGRP release from the peripheral termini of nociceptors (86, 87). Other than macrophages, during inflammation, SGCs become activated, increasing TNF production, and enhancing neuronal excitability (73). In neuropathic pain, a key role of TNF in the DRG has been demonstrated by lentivirus-mediated silencing of TNF in DRG, which attenuated the pain phenotype and reduced neuronal cell death in mice with an L5 transection (160).
DRG cells produce additional cytokines such as IL-1β and IL-6 and chemokines such as CCL2 or CXCL1 within the DRGs that are involved in pain signaling (3, 161). Interestingly in osteoarthritis, a high CCL2 production is associated with elevated numbers of macrophages in the DRG and a high level of CGRP in DRG neurons (162, 163). Mice lacking the CCR2 receptor (global knockout) fail to develop mechanical allodynia in nerve injury models (164). Accordingly, we suggest that during inflammation, a neuro-crosstalk can occur in the DRG where inflammation triggers cytokine and chemokine release from local/infiltrating macrophages and neurons, contributing to development and maintenance of facilitated states of excitability in the local DRG circuit. Such enhanced excitability would contribute to an enhanced afferent input into dorsal horn second-order neurons.
Several cytokines, IL-6 for example, can excite DRG neurons directly by rapid effects that do not require gene transcription but are likely to involve phosphorylation of different ion channels, such as the TRP family. IL-6 is a pleiotropic cytokine with a pivotal role in the pathophysiology of arthritis and pain sensitization through increasing neuronal calcium mobilization, action potential generation, and ion channel sensitization. IL-6 acting through IL-6R and gp130 drives JAK and PKC activation, which enhances TRPV1, inducing excitability of nociceptive TRPV1+ DRG neurons (34, 124). Correspondingly JAK and PKC inhibitors decrease TRPV1 sensitization (34).
Gap junctions in the DRG can provide direct communications between neuronal cell bodies and SGCs. An increase in gap junctions has been observed in pain condition and seems to enhance neuronal excitability and thus elicit pain (157). Importantly, peripheral inflammation or nerve injury causes sensitization of neurons, innervating peripheral tissues, and spreading of activation of SGCs through gap junctions, which leads to the expression and release of IL-1β from SGCs. IL-1β has been shown to increase TRPV1 expression in DRG neurons. Moreover, IL-1RI antagonism reduces thermal hyperalgesia antigen-induced arthritis (20). IL-1β has been shown to act in a p38 MAPK-dependent manner, to increase the excitability of nociceptors. Indeed, IL-1β relieves resting slow inactivation of tetrodotoxin-resistant voltage-gated sodium channels and also enhances persistent TTX-resistant current near the threshold (165). These IL-1β actions on nociceptors have facilitatory effects in neurotransmission, which at least in part explains the hyperalgesic effect seen with the direct application of IL-1β or the endogenous production and release of IL-1β within the DRG.
Beyond the effect of IL-1β on ion channel sensitization, it has been shown that intraplantar IL-1β can induce persistent hyperalgesia, which is dependent on GPCR kinase 2 (GRK2) and IL-10 downregulation. GRK2 plays a regulatory role in the inflammatory response as studied in arthritis models. Reduction of GRK2 in peripheral macrophages markedly prolonged hyperalgesia and pain behavior in response to an intraplantar injection of IL-1β or the inflammatory agent carrageenan (166). The reduction of GRK2 in macrophages is associated with the transition from acute to persistent hyperalgesia due to the lack of IL-10 production. Moreover, local anti-IL-10 treatment in the paw did not influence IL-1β-induced hyperalgesia, indicating that IL-10 signaling in the spinal cord or DRG is required for spontaneous resolution of hyperalgesia. Corroborating these data, our group recently showed that mice deficient in IL-10 rapidly developed mechanical allodynia that did not recover, suggesting that this cytokine also plays a key role in the acute and chronic phases of pain-like behavior (41).
These data suggest that beyond changes in the peripheral terminus nociceptor, the DRG plays an important role in the development of pain states. Thus, cytokines produced by local cells or released into the DRG are involved with the facilitation of nociceptive stimulus, inducing subsequent dorsal horn spinal activity.
Spinal Cord
The peripheral nociceptor forms an excitatory synapse with second-order neurons in the dorsal horn of the spinal cord to initiate transmission in the central nervous system (CNS). This synapse can be modulated by local interneurons, microglia, and astrocytes. These last cells constitute a component serving to alter the input–output function of the dorsal horn. Inflammatory cytokines maintain enhanced pain signaling through modulating the central terminals of nociceptors and/or spinal cord neurons. In naïve animals, intrathecal delivery of cytokines can induce a direct pronociceptive effect, leading to mechanical, and/or thermal hyperalgesia, which has been observed after intrathecal injection of IL-1β (15, 16, 167, 168), IL-18 (60), TNF (15, 16, 167), IL-6 (15), CXCL1 (107), CX3CL1/fractalkine (169), or CCL2/MCP-1 (170) among others. Conversely, a pronociceptive phenotype is reduced in mice lacking such cytokine or chemokine signaling or after the administration of anti-cytokine antibodies or cytokine receptor antagonists. In pathologic conditions, chronic intrathecal administration of IL-1 receptor antagonist (IL-1ra) prevents pain induced by nerve injury in mice, and a single intrathecal injection of IL-1ra induced a long-lasting attenuation of mechanical hypersensitivity in the same model (171). Intrathecally, coadministration of IFN-β and anti-TNF antibodies permanently reversed mechanical allodynia in males in the murine K/BxN serum transfer model of RA (41). Moreover, intrathecal delivery of cytokines such as TGF-β or IL-13 can also induce an antinociceptive effect in neuropathic pain condition (46, 89, 158). TGF-β can inhibit excitatory synaptic transmission in the spinal cord (172) and neuropathic pain along with glial activation and neuroinflammation in the spinal cord (89, 172).
Considering that cytokines play a role in the development and maintenance of pain at the spinal cord level, we raise the question of where these neuraxial cytokines are produced and where they act. Microglia serve as spinal-resident macrophage-like cells and display a rapid response to increased afferent traffic and to pathological changes in the CNS. Spinal microglial activation has been demonstrated in several pain conditions [for review, see (173), including mononeuropathies after peripheral nerve injury (174), polyneuropathies after chemotherapy (175, 176), or in diabetic models (177), chronic inflammatory pain (178), and cancer pain (179), although some data are conflicting (180)]. Microglia participate notably in the regulation of neuroinflammation that contributes to the pathogenesis of pain (173). Signals that activate microglia converge on intracellular signaling cascades frequently involving the phosphorylation of p38 MAPK, triggering production and release of TNF, IL-1β, and IL-18; increased expression of COX; and subsequent synthesis of prostaglandin E2 (PGE2) (18, 181, 182). These neuromodulators then lead to enhanced dorsal horn excitability, which ultimately serves to enhance receptive fields, increase output of the pain-relevant sensory message to supraspinal areas, and enhance pain behavior.
Astrocytes have a key role in neurotransmitter recycling, regulation of blood flow, energy metabolism, synaptogenesis, and synaptic transmission (183–187). Astrocytes signal physically through gap junctions (e.g., connexin Cx43), facilitating intercellular transmission (188, 189). Moreover, it has been demonstrated that upregulation of Cx43 triggers release of chemokines, ATP, and glutamate, which ultimately induces nociceptor sensitization (185, 186). Astrocytes have been shown to play both beneficial and detrimental roles, depending on the nature of the injury or disease, that differ in their functions (190). Thus, spinal cord astrocytes can generate IFN-α, which have an antinociceptive effect mediated through the mu opioid receptor (94). In addition, recent data from our group have shown that the post-inflammatory allodynia from an arthritis model may be robustly regulated by downstream effectors activated through IFN-β and interferon-inducible factors, including IL-10 and IL-1ra (41). In this model, the allodynia only subsided when anti-TNF therapy was combined with supplemental IFN-β, indicating that chronic pain treatment might require modulation of multiple pathways (41).
Astrocytes can also modulate pain through IL-33 production. IL-33 is a member of the IL-1 superfamily but is active after transcription and is deactivated by caspase cleavage. It binds to the ST2 (Il1rl1) receptor, encoded by the IL1RL1 gene, and the coreceptor IL-1 receptor accessory protein (IL-1RAcP). After receptor engagement, the MyD88 signal cascade is activated similarly as after IL-1R and IL-18R activation. This cytokine has a pronociceptive effect with intrathecal injection (191, 192). In addition, inhibiting the IL-33/ST2 pathway reduced nociceptive behavior in murine models of pain, including cancer and chemically induced inflammation (72, 192, 193).
The effects of the intrathecal injection of cytokines cited above directly support the key roles played by spinal cord cytokines in pain states. As these cytokines are not acting independently, it is not surprising that these agents display important interactions in different pain states reflecting their facilitatory and suppressive interactions. As an example, intrathecal administration of recombinant IL-27 induced antinociception dependent on IL-10 during the maintenance phase of peripheral neuropathy (194). Also, intrathecal delivery of IL-35 in an experimental murine model of autoimmune encephalomyelitis (EAE) reduced pain behaviors (i.e., facial allodynia and grimacing), which was noted to occur through an upregulation of an inflammatory cytokine, IL-10 (195). IL-35 has been very recently highlighted as a candidate target for diabetic neuropathic pain (DNP) treatment (78). In fact, in a streptozotocin-induced DNP rat model, other than a protective effect against inflammatory response, IL-35 injected intrathecally reduced allodynia via inhibition of JNK signaling (78).
Second-order dorsal horn neurons express cytokine receptors such as TNFR1, TNFR2, IL-1R, and IL-6R. Cytokine released at the spinal cord level by resident cells (e.g., microglia/astrocytes) can induce sensitization of the secondary neuron, leading to supraspinal areas of activation where pain is processed and perceived as an uncomfortable sensation (139). In fact, IL-6, TNF, and IL-β enhance spontaneous post-synaptic current (sEPSCs) in the spinal cord by both increasing excitatory synaptic neurotransmission and suppressing inhibitory synaptic transmissions (160). Taken together, these studies show that therapeutically targeting peripheral inflammation will not necessarily affect persistent pain. However, modulating multiple pathways at the spinal level might be an effective way to prevent the development of chronic pain and to alleviate ongoing pain.
Supraspinal Areas
Changes in higher-order functions such as anxiety or depression are critical components of pain phenotypes, especially in the context of a chronic pain state (196, 197). Studies in animal models (99, 198, 199) and in vivo positron emission tomography (PET) associated with magnetic resonance imaging (MRI) in humans (200, 201) are consistent with the fact that chronic pain states lead to an alteration of glial function in the brain. Current thinking emphasizes the critical role that cytokines can play in regulating depressive states, through their effects upon key aminergic pharmacological systems regulating depressive states such as increased monoamine transporter activity (202), reduced cofactor availability (203), and reduced expression of glutamate transporters and increased glutamate release from astrocytes (204).
Cytokines play key roles in supraspinal modulation of pain transduction. Consistent with this assertion are the findings that intracerebroventricular (ICV) injection of TNF, IL-1β, and IL-6 induced hyperalgesia (205–207) and that blocking these cytokines in the brain reduced the behaviorally defined expression of a pain state (208). Of note, mechanistically, it is not clear whether this pain state reflects the role of an enhanced supraspinal response to the ascending pain stimulus or an activation of descending facilitatory/loss of descending inhibitory system (209).
Microglial activation (Iba1 marker) after nerve injury is observed in specific brain regions involved in pain and affect. These regions include not only the thalamus and somatosensory cortex but also limbic regions considered to be affiliated with the affective component of the pain response (210). Importantly, as in the spinal cord, glial cells are thought to be a major source of cytokines and chemokines in the brain (204), and the activation of these cells is considered to play a key role in anxiety and depression, comorbidities associated with chronic pain states (211, 212). Other works support the involvement of microglial activation in prefrontal cortex, amygdala, and hippocampal neurons associated with an overproduction of TNF in neuropathic pain and chronic-pain-associated depression (211, 213). Moreover, upregulation of IL-10 and IL-1β is found in the contralateral–ventrolateral orbital cortex (VLO) of rats with spared nerve injury (SNI), and IL-1β expression and glutamatergic neurotransmission are enhanced in the prefrontal cortex (PFC) of mice with neuropathic pain (214).
Cytokine activation in the brain on the affective-motivation component of pain processing is an exciting component of the role played by cytokines in pain processing. Considerable work has demonstrated that circulating inflammatory markers (e.g., IL-1β, TNF, IL-6, and C-reactive protein) are important covariates for depression and anxiety in humans (215, 216).
Cytokines as Therapeutic Targets
Although not the focus of this review, it would seem remiss not to briefly comment on some of the advances seen with the advent of approved biologic therapies. As described previously, cytokines and chemokines have a key role in disease-associated pain and therapies exerting an action on cytokine release or activity, which is really effective [Table 1; (182, 183)]. Accumulating data suggest that in a variety of pain states, there is a strong covariance between circulating proinflammatory cytokine messages and the pain states in fibromyalgia (217) or painful (vs. non-painful) neuropathies (218, 219). Of note, TNF antagonism improved depressive symptoms in patients with high baseline inflammatory biomarkers (220). While these endpoints do not directly impact upon pain signaling, they provide an important covariate between chronic pain and comorbidities that can enhance the chronic pain states (221).
The bulk of clinical data describe the relief of pain in the treatment of inflammatory states like RA using agents that block the activity of key cytokines like IL-6 and TNF. Conventional synthetic disease-modifying antirheumatic drugs (csDMARDs) such as methotrexate or sulfasalazine can attenuate cytokine release (222–226), but the biologic disease-modifying antirheumatic drugs (bDMARDs), which have a direct effect on the function and levels of circulating cytokines, have been shown to be more effective alone or in combination with csDMARDs [Table 1; (124, 227–229)]. The success of these agents is reflected in the development of agents with similar targets. Notable groups of agents include anti-TNF (including infliximab, adalimumab, certolizumab pegol, etanercept, or golimumab) and anti-IL-6 or IL-6 receptor antagonists (i.e., tocilizumab, sarilumab, clazakizumab, and olokizumab) (115). However, remission of inflammation by clinical parameters has not universally been associated with complete relief of pain, and residual pain with neuropathic features can persist (178). More recently, agents that intercede with signaling, notably the JAK inhibitors, may add to the armamentarium of agents that can reduce pain as well as inflammation (230, 231). The development of therapeutic agents that target individual cytokines and their signaling pathways is a promising and exciting area that has been extensively reviewed by others (232, 233). These therapies hold significant promise for the future, and further investigations into their level of anatomic activity will hopefully yield insights into individualized therapeutic plans.
Conclusion
The primary emphasis of this review has been on reviewing the role of cytokines at the levels of the peripheral terminus, the DRG, the spinal dorsal horn, and supraspinal circuits (Figure 2). Although pain arises from different conditions, some mechanistic components are conserved across pain states:
(i) Peripheral nerve fibers (nociceptors) are directly exposed to circulating products like cytokines and detect environmental stimuli (thermal, mechanical, or chemical nature) and stimulate excitation of second-order neurons at the spinal cord.
(ii) In the DRG, the somata of nociceptors are surrounded by SGCs and macrophages. As noted, the DRG lies outside of the conventional BBB restriction, leading it to be directly exposed to these circulating proteins and other danger signals. These peripheral stimuli drive cytokine secretion from SGCs and macrophages, contributing to inflammatory signaling cascades and persistent pain.
(iii) The dorsal horn in the spinal cord receives information from nociceptors.
(iv) The incoming information is processed by complex circuits involving excitatory and inhibitory interneurons and transmitted to projection neurons to several supraspinal areas in the CNS. Spinal cord astrocytes and microglia are described as key cells in the mechanism of pain processing in several pain models (234).
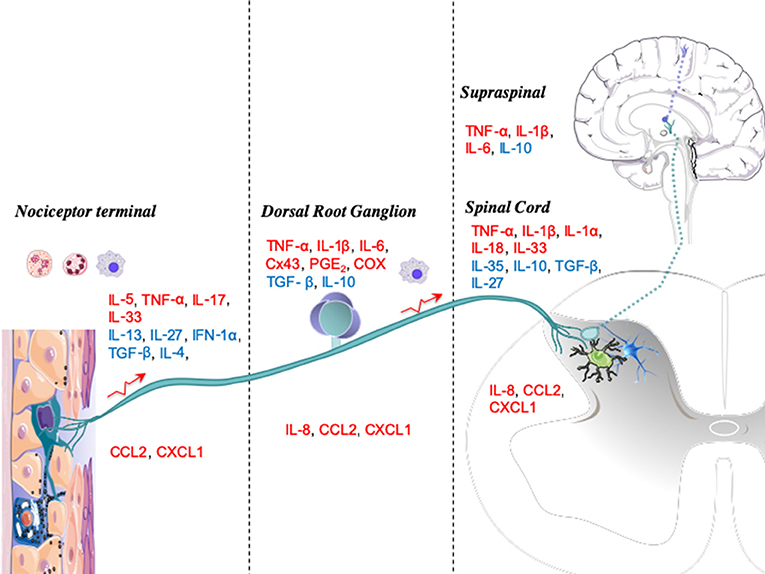
Figure 2. Anatomic levels of cytokine interactions with nociceptive processing. Upon injury or infection, mediators such as immune cytokines are released locally by resident or blood-derived immune cells. The peripheral terminals of nociceptors, dorsal root ganglia (DRGs), and spinal cords have several receptors for these mediators, which activate signaling cascades that modulate nociceptive activity. Cytokines are displayed as pronociceptive (red) or antinociceptive (blue).
In Figure 2, we graphically summarize key cytokines that play a role in these four major neuraxial components (i.e., the peripheral terminal, DRG, spinal cord, and brain). At each anatomic level, we note the relevance of the several local systems (neuronal, glial, and inflammatory cells) that contribute both as a source of cytokines and as a target for these molecules functioning in an autocrine-/paracrine-like fashion.
This review serves to emphasize the multiple levels at which cytokines may be released and act to alter the nociceptive phenotype and reflect the role of a local paracrine or autocrine function. It is clear, however, that this position does not exclude the likelihood that the circulating cytokine profile observed in a variety of inflammatory and injury states might contribute to the abnormal pain and depression though a circulating delivery. Of note, the presence of these circulating proinflammatory products and the accessibility of these products to neuraxial components such as the peripheral terminal and the DRG point to potential interactions. The ability of glia such as astrocytes and perivascular macrophages to sample circulating products, along with the evident role played by glia in CNS function, points to the likelihood that circulating products can modify function throughout the neuraxis. We therefore conclude that targeting inflammatory cytokine and chemokine signaling may provide additional strategies in the therapeutic intervention of chronic pain. However, we note that despite recent promising advances, any single agent is unlikely to be uniformly effective, and future studies in this area are warranted.
Author Contributions
All authors contributed to the drafting and revision of the manuscript.
Funding
This work was funded in part by grants from NIH/NINDS 1 R01 NS099338 (TY) and 1 R01 NS102432-01.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Catani M, Dell'Acqua F, Thiebaut de Schotten M. A revised limbic system model for memory, emotion and behaviour. Neurosci Biobehav Rev. (2013) 37:1724–37. doi: 10.1016/j.neubiorev.2013.07.001
2. Rittner HL, Machelska H, Stein C. Immune system, pain and analgesia. In: Masland RH, editor. The Senses: A Comprehensive Reference. New York, NY: Elsevier (2008). p. 407–27.
3. Miller RJ, Jung H, Bhangoo SK, White FA. Cytokine and chemokine regulation of sensory neuron function. Handb Exp Pharmacol. (2009) 194:417–49. doi: 10.1007/978-3-540-79090-7_12
4. Baral P, Udit S, Chiu IM. Pain and immunity: implications for host defence. Nat Rev Immunol. (2019) 19:433–47. doi: 10.1038/s41577-019-0147-2
5. Lapato AS, Tiwari-Woodruff SK. Connexins and pannexins: at the junction of neuro-glial homeostasis & disease. J Neurosci Res. (2018) 96:31–44. doi: 10.1002/jnr.24088
6. Woller SA, Eddinger KA, Corr M, Yaksh TL. An overview of pathways encoding nociception. Clin Exp Rheumatol. (2017) 35(Suppl. 1):40–6.
7. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc London Ser B Biol Sci. (1957) 147:258–67. doi: 10.1098/rspb.1957.0048
9. Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. (2006) 354:610–21. doi: 10.1056/NEJMra052723
10. Abbadie C, Bhangoo S, De Koninck Y, Malcangio M, Melik-Parsadaniantz S, White FA. Chemokines and pain mechanisms. Brain Res Rev. (2009) 60:125–34. doi: 10.1016/j.brainresrev.2008.12.002
11. Gao Y-J, Ji R-R. Chemokines, neuronal-glial interactions, and central processing of neuropathic pain. Pharmacol Ther. (2010) 126:56–68. doi: 10.1016/j.pharmthera.2010.01.002
12. Ren K, Dubner R. Interactions between the immune and nervous systems in pain. Nat Med. (2010) 16:1267–76. doi: 10.1038/nm.2234
13. Hewett SJ, Jackman NA, Claycomb RJ. Interleukin-1β in central nervous system injury and repair. Eur J Neurodegener Dis. (2012) 1:195–211.
14. Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev. (2018) 281:8–27. doi: 10.1111/imr.12621
15. Kawasaki Y, Zhang L, Cheng J-K, Ji R-R. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci. (2008) 28:5189–94. doi: 10.1523/JNEUROSCI.3338-07.2008
16. Gruber-Schoffnegger D, Drdla-Schutting R, Hönigsperger C, Wunderbaldinger G, Gassner M, Sandkühler J. Induction of thermal hyperalgesia and synaptic long-term potentiation in the spinal cord lamina I by TNF-α and IL-1β is mediated by glial cells. J Neurosci. (2013) 33:6540–51. doi: 10.1523/JNEUROSCI.5087-12.2013
17. Liu T, Jiang CY, Fujita T, Luo SW, Kumamoto E. Enhancement by interleukin-1β of AMPA and NMDA receptor-mediated currents in adult rat spinal superficial dorsal horn neurons. Mol Pain. (2013) 9:16. doi: 10.1186/1744-8069-9-16
18. Araldi D, Ferrari LF, Lotufo CM, Vieira AS, Athié MCP, Figueiredo JG, et al. Peripheral inflammatory hyperalgesia depends on the COX increase in the dorsal root ganglion. Proc Natl Acad Sci. (2013) 110:3603–8. doi: 10.1073/pnas.1220668110
19. Cook AD, Christensen AD, Tewari D, McMahon SB, Hamilton JA. Immune cytokines and their receptors in inflammatory pain. Trends Immunol. (2018) 39:240–55. doi: 10.1016/j.it.2017.12.003
20. Ebbinghaus M, Uhlig B, Richter F, Von Banchet GS, Gajda M, Bräuer R, et al. The role of interleukin-1β in arthritic pain: main involvement in thermal, but not mechanical, hyperalgesia in rat antigen-induced arthritis. Arthritis Rheum. (2012) 64:3897–907. doi: 10.1002/art.34675
21. Ren K, Torres R. Role of interleukin-1beta during pain and inflammation. Brain Res Rev. (2009) 60:57–64. doi: 10.1016/j.brainresrev.2008.12.020
22. Busch-Dienstfertig M, González-Rodríguez S. IL-4, JAK-STAT signaling, and pain. JAK-STAT. (2014) 2:e27638. doi: 10.4161/jkst.27638
23. Major J, Fletcher JE, Hamilton TA. IL-4 pretreatment selectively enhances cytokine and chemokine production in lipopolysaccharide-stimulated mouse peritoneal macrophages. J Immunol. (2002) 168:2456–63. doi: 10.4049/jimmunol.168.5.2456
24. Fort MM, Lesley R, Davidson NJ, Menon S, Brombacher F, Leach MW, et al. IL-4 exacerbates disease in a Th1 cell transfer model of colitis. J Immunol. (2001) 166:2793–800. doi: 10.4049/jimmunol.166.4.2793
25. Gadani SP, Cronk JC, Norris GT, Kipnis J. IL-4 in the brain: a cytokine to remember. J Immunol. (2012) 189:4213–9. doi: 10.4049/jimmunol.1202246
26. Oetjen LK, Mack MR, Feng J, Whelan TM, Niu H, Guo CJ, et al. Sensory neurons co-opt classical immune signaling pathways to mediate chronic itch. Cell. (2017) 171:217–28.e13. doi: 10.1016/j.cell.2017.08.006
27. Kim J, Kim BE, Leung DYM. Pathophysiology of atopic dermatitis: clinical implications. Allergy asthma Proc. (2019) 40:84–92. doi: 10.2500/aap.2019.40.4202
28. Steinke JW, Borish L. Th2 cytokines and asthma. Interleukin-4: its role in the pathogenesis of asthma, and targeting it for asthma treatment with interleukin-4 receptor antagonists. Respir Res. (2001) 2:66–70.
29. Bagnasco D, Ferrando M, Varricchi G, Puggioni F, Passalacqua G, Canonica GW. Anti-Interleukin 5 (IL-5) and IL-5Ra biological drugs: efficacy, safety, and future perspectives in severe eosinophilic asthma. Front Med. (2017) 4:135. doi: 10.3389/fmed.2017.00135
30. Munno I, Centonze V, Marinaro M, Bassi A, Lacedra G, Causarano V, et al. Cytokines and migraine: increase of IL-5 and IL-4 plasma levels. Headache. (1998) 38:465–7. doi: 10.1046/j.1526-4610.1998.3806465.x
31. Talbot S, Foster SL, Woolf CJ. Neuroimmunity: physiology and pathology. Annu Rev Immunol. (2016) 34:421–47. doi: 10.1146/annurev-immunol-041015-055340
32. Gabay C. Interleukin-6 and chronic inflammation. Arthritis Res Ther. (2006) 8 (Suppl. 2):S3. doi: 10.1186/ar1917
33. Schett G. Physiological effects of modulating the interleukin-6 axis. Rheumatology. (2018) 57:ii43–50. doi: 10.1093/rheumatology/kex513
34. Fang D, Kong L-Y, Cai J, Li S, Liu X-D, Han J-S, et al. Interleukin-6-mediated functional upregulation of TRPV1 receptors in dorsal root ganglion neurons through the activation of JAK/PI3K signaling pathway: roles in the development of bone cancer pain in a rat model. Pain. (2015) 156:1124–44. doi: 10.1097/j.pain.0000000000000158
35. Brenn D, Richter F, Schaible H-G. Sensitization of unmyelinated sensory fibers of the joint nerve to mechanical stimuli by interleukin-6 in the rat: an inflammatory mechanism of joint pain. Arthritis Rheum. (2007) 56:351–9. doi: 10.1002/art.22282
36. Krock E, Jurczak A, Svensson CI. Pain pathogenesis in rheumatoid arthritis-what have we learned from animal models? Pain. (2018) 159(Suppl. 1):S98–109. doi: 10.1097/j.pain.0000000000001333
37. Choy EHS, Calabrese LH. Neuroendocrine and neurophysiological effects of interleukin 6 in rheumatoid arthritis. Rheumatology. (2018) 57:1885–95. doi: 10.1093/rheumatology/kex391
38. Shachar I, Karin N. The dual roles of inflammatory cytokines and chemokines in the regulation of autoimmune diseases and their clinical implications. J Leukoc Biol. (2013) 93:51–61. doi: 10.1189/jlb.0612293
39. Iyer SS, Cheng G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit Rev Immunol. (2012) 32:23–63. doi: 10.1615/CritRevImmunol.v32.i1.30
40. Wu H-Y, Mao X-F, Tang X-Q, Ali U, Apryani E, Liu H, et al. Spinal interleukin-10 produces antinociception in neuropathy through microglial β-endorphin expression, separated from antineuroinflammation. Brain Behav Immun. (2018) 73:504–19. doi: 10.1016/j.bbi.2018.06.015
41. Woller SA, Ocheltree C, Wong SY, Bui A, Fujita Y, Gonçalves dos Santos G, et al. Neuraxial TNF and IFN-beta co-modulate persistent allodynia in arthritic mice. Brain Behav Immun. (2019) 76:151–8. doi: 10.1016/j.bbi.2018.11.014
42. Lauw FN, Pajkrt D, Hack CE, Kurimoto M, van Deventer SJH, van der Poll T. Proinflammatory effects of IL-10 during human endotoxemia. J Immunol. (2000) 165:2783–9. doi: 10.4049/jimmunol.165.5.2783
43. Tian G, Li J-L, Wang D-G, Zhou D. Targeting IL-10 in auto-immune diseases. Cell Biochem Biophys. (2014) 70:37–49. doi: 10.1007/s12013-014-9903-x
44. Fasoulakis Z, Kolios G, Papamanolis V, Kontomanolis EN. Interleukins associated with breast cancer. Cureus. (2018) 10:e3549. doi: 10.7759/cureus.3549
45. McCann B, Miaskowski C, Koetters T, Baggott C, West C, Levine JD, et al. Associations between pro- and anti-inflammatory cytokine genes and breast pain in women prior to breast cancer surgery. J Pain. (2012) 13:425–37. doi: 10.1016/j.jpain.2011.02.358
46. Kiguchi N, Sakaguchi H, Kadowaki Y, Saika F, Fukazawa Y, Matsuzaki S, et al. Peripheral administration of interleukin-13 reverses inflammatory macrophage and tactile allodynia in mice with partial sciatic nerve ligation. J Pharmacol Sci. (2017) 133:53–56. doi: 10.1016/j.jphs.2016.11.005
48. Chalan P, Bijzet J, van den Berg A, Kluiver J, Kroesen B-J, Boots AMH, et al. Analysis of serum immune markers in seropositive and seronegative rheumatoid arthritis and in high-risk seropositive arthralgia patients. Sci Rep. (2016) 6:26021. doi: 10.1038/srep26021
49. Moynes DM, Vanner SJ, Lomax AE. Participation of interleukin 17A in neuroimmune interactions. Brain Behav Immun. (2014) 41:1–9. doi: 10.1016/j.bbi.2014.03.004
50. Ke Y, Liu K, Huang G-Q, Cui Y, Kaplan HJ, Shao H, et al. Anti-inflammatory role of IL-17 in experimental autoimmune uveitis. J Immunol. (2009) 182:3183–90. doi: 10.4049/jimmunol.0802487
51. McGeachy MJ, Cua DJ, Gaffen SL. The IL-17 family of cytokines in health and disease. Immunity. (2019) 50:892–906. doi: 10.1016/j.immuni.2019.03.021
52. Richter F, Natura G, Ebbinghaus M, von Banchet GS, Hensellek S, König C, et al. Interleukin-17 sensitizes joint nociceptors to mechanical stimuli and contributes to arthritic pain through neuronal interleukin-17 receptors in rodents. Arthritis Rheum. (2012) 64:4125–34. doi: 10.1002/art.37695
53. Pinto LG, Cunha TM, Vieira SM, Lemos HP, Verri WA, Cunha FQ, et al. IL-17 mediates articular hypernociception in antigen-induced arthritis in mice. Pain. (2010) 148:247–56. doi: 10.1016/j.pain.2009.11.006
54. McNamee KE, Alzabin S, Hughes JP, Anand P, Feldmann M, Williams RO, et al. IL-17 induces hyperalgesia via TNF-dependent neutrophil infiltration. Pain. (2011) 152:1838–45. doi: 10.1016/j.pain.2011.03.035
55. Mens LJJ, Sande MGH, Menegatti S, Chen S, Blijdorp ICJ, Jong HM, et al. Brief report: interleukin-17 blockade with secukinumab in peripheral spondyloarthritis impacts synovial immunopathology without compromising systemic immune responses. Arthritis Rheumatol. (2018) 70:1994–2002. doi: 10.1002/art.40581
56. Chiricozzi A, Krueger JG. IL-17 targeted therapies for psoriasis. Expert Opin Investig Drugs. (2013) 22:993–1005. doi: 10.1517/13543784.2013.806483
57. McInnes IB, Mease PJ, Ritchlin CT, Rahman P, Gottlieb AB, Kirkham B, et al. Secukinumab sustains improvement in signs and symptoms of psoriatic arthritis: 2 year results from the phase 3 FUTURE 2 study. Rheumatology. (2017) 56:1993–2003. doi: 10.1093/rheumatology/kex301
58. Miyoshi K, Obata K, Kondo T, Okamura H, Noguchi K. Interleukin-18-mediated microglia/astrocyte interaction in the spinal cord enhances neuropathic pain processing after nerve injury. J Neurosci. (2008) 28:12775–87. doi: 10.1523/JNEUROSCI.3512-08.2008
59. Dinarello CA, Novick D, Kim S, Kaplanski G. Interleukin-18 and IL-18 binding protein. Front Immunol. (2013) 4:289. doi: 10.3389/fimmu.2013.00289
60. Yang Y, Li H, Li T-T, Luo H, Gu X-Y, Lü N, et al. Delayed activation of spinal microglia contributes to the maintenance of bone cancer pain in female Wistar rats via P2X7 receptor and IL-18. J Neurosci. (2015) 35:7950–63. doi: 10.1523/JNEUROSCI.5250-14.2015
61. Liu S, Liu Y-P, Lv Y, Yao J-L, Yue D-M, Zhang M-Y, et al. IL-18 Contributes to bone cancer pain by regulating glia cells and neuron interaction. J Pain. (2018) 19:186–95. doi: 10.1016/j.jpain.2017.10.003
62. Pflanz S, Timans JC, Cheung J, Rosales R, Kanzler H, Gilbert J, et al. IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4+ T cells. Immunity. (2002) 16:779–90. doi: 10.1016/S1074-7613(02)00324-2
63. Pickens SR, Chamberlain ND, Volin MV, Mandelin AM, Agrawal H, Matsui M, et al. Local expression of interleukin-27 ameliorates collagen-induced arthritis. Arthritis Rheum. (2011) 63:2289–98. doi: 10.1002/art.30324
64. Niedbala W, Cai B, Wei X, Patakas A, Leung BP, McInnes IB, et al. Interleukin 27 attenuates collagen-induced arthritis. Ann Rheum Dis. (2008) 67:1474–9. doi: 10.1136/ard.2007.083360
65. Jones GW, Bombardieri M, Greenhill CJ, McLeod L, Nerviani A, Rocher-Ros V, et al. Interleukin-27 inhibits ectopic lymphoid-like structure development in early inflammatory arthritis. J Exp Med. (2015) 212:1793–802. doi: 10.1084/jem.20132307
66. Jones GW, Hill DG, Cardus A, Jones SA. IL-27: a double agent in the IL-6 family. Clin Exp Immunol. (2018) 193:37–46. doi: 10.1111/cei.13116
67. Kalliolias GD, Zhao B, Triantafyllopoulou A, Park-Min K-H, Ivashkiv LB. Interleukin-27 inhibits human osteoclastogenesis by abrogating RANKL-mediated induction of nuclear factor of activated T cells c1 and suppressing proximal RANK signaling. Arthritis Rheum. (2010) 62:402–13. doi: 10.1002/art.27200
68. Wigerblad G, Bas DB, Fernades-Cerqueira C, Krishnamurthy A, Nandakumar KS, Rogoz K, et al. Autoantibodies to citrullinated proteins induce joint pain independent of inflammation via a chemokine-dependent mechanism. Ann Rheum Dis. (2016) 75:730–8. doi: 10.1136/annrheumdis-2015-208094
69. Fattori V, Hohmann MSN, Rossaneis AC, Manchope MF, Alves-Filho JC, Cunha TM, et al. Targeting IL-33/ST2 signaling: regulation of immune function and analgesia. Expert Opin Ther Targets. (2017) 21:1141–52. doi: 10.1080/14728222.2017.1398734
70. Zarpelon AC, Rodrigues FC, Lopes AH, Souza GR, Carvalho TT, Pinto LG, et al. Spinal cord oligodendrocyte-derived alarmin IL-33 mediates neuropathic pain. FASEB J. (2016) 30:54–65. doi: 10.1096/fj.14-267146
71. Liu S, Mi W-L, Li Q, Zhang M-T, Han P, Hu S, et al. Spinal IL-33/ST2 signaling contributes to neuropathic pain via neuronal CaMKII-CREB and astroglial JAK2-STAT3 cascades in mice. Anesthesiology. (2015) 123:1154–69. doi: 10.1097/ALN.0000000000000850
72. Zhao J, Zhang H, Liu S-B, Han P, Hu S, Li Q, et al. Spinal interleukin-33 and its receptor ST2 contribute to bone cancer-induced pain in mice. Neuroscience. (2013) 253:172–82. doi: 10.1016/j.neuroscience.2013.08.026
73. Ji R-R, Chamessian A, Zhang Y-Q. Pain regulation by non-neuronal cells and inflammation. Science. (2016) 354:572–7. doi: 10.1126/science.aaf8924
74. Fattori V, Amaral FA, Verri WA. Neutrophils and arthritis: role in disease and pharmacological perspectives. Pharmacol Res. (2016) 112:84–98. doi: 10.1016/j.phrs.2016.01.027
75. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. (2007) 450:566–9. doi: 10.1038/nature06306
76. Wang R-X, Yu C-R, Dambuza IM, Mahdi RM, Dolinska MB, Sergeev YV, et al. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat Med. (2014) 20:633–41. doi: 10.1038/nm.3554
77. Huang A, Cheng L, He M, Nie J, Wang J, Jiang K. Interleukin-35 on B cell and T cell induction and regulation. J Inflamm. (2017) 14:16. doi: 10.1186/s12950-017-0164-5
78. Jiang Y, Wang J, Li H, Xia L. IL-35 alleviates inflammation progression in a rat model of diabetic neuropathic pain via inhibition of JNK signaling. J Inflamm. (2019) 16:19. doi: 10.1186/s12950-019-0217-z
79. Filková M, Vernerová Z, Hulejová H, Prajzlerová K, Veigl D, Pavelka K, et al. Pro-inflammatory effects of interleukin-35 in rheumatoid arthritis. Cytokine. (2015) 73:36–43. doi: 10.1016/j.cyto.2015.01.019
80. Bello RO, Chin VK, Abd Rachman Isnadi MF, Abd Majid R, Atmadini Abdullah M, Lee TY, et al. The role, involvement and function(s) of interleukin-35 and interleukin-37 in disease pathogenesis. Int J Mol Sci. (2018) 19:E1149. doi: 10.3390/ijms19041149
81. Parameswaran N, Patial S. Tumor necrosis factor-a signaling in macrophages. Crit Rev Eukaryot Gene Expr. (2010) 20:87–103. doi: 10.1615/CritRevEukarGeneExpr.v20.i2.10
82. Chung IY, Benveniste EN. Tumor necrosis factor-alpha production by astrocytes. Induction by lipopolysaccharide, IFN-gamma, and IL-1 beta. J Immunol. (1990) 144:2999–3007.
83. Berta T, Park C-K, Xu Z-Z, Xie R-G, Liu T, Lü N, et al. Extracellular caspase-6 drives murine inflammatory pain via microglial TNF-α secretion. J Clin Invest. (2014) 124:1173–86. doi: 10.1172/JCI72230
84. Dong Y, Fischer R, Naudé PJW, Maier O, Nyakas C, Duffey M, et al. Essential protective role of tumor necrosis factor receptor 2 in neurodegeneration. Proc Natl Acad Sci USA. (2016) 113:12304–9. doi: 10.1073/pnas.1605195113
85. Parada CA, Yeh JJ, Joseph EK, Levine JD. Tumor necrosis factor receptor type-1 in sensory neurons contributes to induction of chronic enhancement of inflammatory hyperalgesia in rat. Eur J Neurosci. (2003) 17:1847–52. doi: 10.1046/j.1460-9568.2003.02626.x
86. Qin X, Wan Y, Wang X. CCL2 and CXCL1 trigger calcitonin gene-related peptide release by exciting primary neciceptive neurons. J Neurosci Res. (2005) 82:51–62. doi: 10.1002/jnr.20612
87. Kidd BL, Urban LA. Mechanisms of inflammatory pain. Br J Anaesth. (2001) 87:3–11. doi: 10.1093/bja/87.1.3
88. Bradley J. TNF-mediated inflammatory disease. J Pathol. (2008) 214:149–60. doi: 10.1002/path.2287
89. Echeverry S, Shi XQ, Haw A, Liu H, Zhang ZW, Zhang J. Transforming growth factor-β1 impairs neuropathic pain through pleiotropic effects. Mol Pain. (2009) 5:16. doi: 10.1186/1744-8069-5-16
90. Zhang X, Zheng H, Zhu HY, Hu S, Wang S, Jiang X, et al. Acute effects of transforming growth factor-β1 on neuronal excitability and involvement in the pain of rats with chronic pancreatitis. J Neurogastroenterol Motil. (2016) 22:333–43. doi: 10.5056/jnm15127
91. Kashima R, Hata A. The role of TGF-β superfamily signaling in neurological disorders. Acta Biochim Biophys Sin. (2018) 50:106–20. doi: 10.1093/abbs/gmx124
92. Cheon H, Yu S-J, Yoo DH, Chae IJ, Song GG, Sohn J. Increased expression of pro-inflammatory cytokines and metalloproteinase-1 by TGF-beta1 in synovial fibroblasts from rheumatoid arthritis and normal individuals. Clin Exp Immunol. (2002) 127:547–52. doi: 10.1046/j.1365-2249.2002.01785.x
93. Qin Z-F, Hou D-Y, Fang Y-Q, Xiao H-J, Wang J, Li K-C. Interferon-alpha enhances excitatory transmission in substantia gelatinosa neurons of rat spinal cord. Neuroimmunomodulation. (2012) 19:235–40. doi: 10.1159/000335167
94. Liu C-C, Gao Y-J, Luo H, Berta T, Xu Z-Z, Ji R-R, et al. Interferon alpha inhibits spinal cord synaptic and nociceptive transmission via neuronal-glial interactions. Sci Rep. (2016) 6:34356. doi: 10.1038/srep34356
95. Choubey D, Moudgil KD. Interferons in autoimmune and inflammatory diseases: regulation and roles. J Interferon Cytokine Res. (2011) 31:857–65. doi: 10.1089/jir.2011.0101
96. Racz I, Nadal X, Alferink J, Banos JE, Rehnelt J, Martin M, et al. Interferon- is a critical modulator of CB2 cannabinoid receptor signaling during neuropathic pain. J Neurosci. (2008) 28:12136–45. doi: 10.1523/JNEUROSCI.3402-08.2008
97. Sun L, Li Y, Jia X, Wang Q, Li Y, Hu M, et al. Neuroprotection by IFN-γ via astrocyte-secreted IL-6 in acute neuroinflammation. Oncotarget. (2017) 8:40065–78. doi: 10.18632/oncotarget.16990
98. Savarin C, Hinton DR, Valentin-Torres A, Chen Z, Trapp BD, Bergmann CC, et al. Astrocyte response to IFN-γ limits IL-6-mediated microglia activation and progressive autoimmune encephalomyelitis. J Neuroinflammation. (2015) 12:79. doi: 10.1186/s12974-015-0293-9
99. Tsuda M, Masuda T, Kitano J, Shimoyama H, Tozaki-Saitoh H, Inoue K. IFN-gamma receptor signaling mediates spinal microglia activation driving neuropathic pain. Proc Natl Acad Sci USA. (2009) 106:8032–7. doi: 10.1073/pnas.0810420106
100. Miller CHT, Maher SG, Young HA. Clinical use of interferon-gamma. Ann N Y Acad Sci. (2009) 1182:69–79. doi: 10.1111/j.1749-6632.2009.05069.x
101. Theofilopoulos AN, Koundouris S, Kono DH, Lawson BR. The role of IFN-gamma in systemic lupus erythematosus: a challenge to the Th1/Th2 paradigm in autoimmunity. Arthritis Res. (2001) 3:136–41. doi: 10.1186/ar290
102. Miyake K, Nakashima H, Akahoshi M, Inoue Y, Nagano S, Tanaka Y, et al. Genetically determined interferon-gamma production influences the histological phenotype of lupus nephritis. Rheumatology. (2002) 41:518–24. doi: 10.1093/rheumatology/41.5.518
103. Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. (2009) 29:313–26. doi: 10.1089/jir.2008.0027
104. Bäckryd E, Lind A-L, Thulin M, Larsson A, Gerdle B, Gordh T. High levels of cerebrospinal fluid chemokines point to the presence of neuroinflammation in peripheral neuropathic pain: a cross-sectional study of 2 cohorts of patients compared with healthy controls. Pain. (2017) 158:2487–95. doi: 10.1097/j.pain.0000000000001061
105. Silva RL, Lopes AH, Guimarães RM, Cunha TM. CXCL1/CXCR2 signaling in pathological pain: role in peripheral and central sensitization. Neurobiol Dis. (2017) 105:109–16. doi: 10.1016/j.nbd.2017.06.001
106. Cao D-L, Zhang Z-J, Xie R-G, Jiang B-C, Ji R-R, Gao Y-J. Chemokine CXCL1 enhances inflammatory pain and increases NMDA receptor activity and COX-2 expression in spinal cord neurons via activation of CXCR2. Exp Neurol. (2014) 261:328–36. doi: 10.1016/j.expneurol.2014.05.014
107. Zhang Z-J, Cao D-L, Zhang X, Ji R-R, Gao Y-J. Chemokine contribution to neuropathic pain: respective induction of CXCL1 and CXCR2 in spinal cord astrocytes and neurons. Pain. (2013) 154:2185–97. doi: 10.1016/j.pain.2013.07.002
108. Krishnamurthy A, Joshua V, Haj Hensvold A, Jin T, Sun M, Vivar N, et al. Identification of a novel chemokine-dependent molecular mechanism underlying rheumatoid arthritis-associated autoantibody-mediated bone loss. Ann Rheum Dis. (2016) 75:721–9. doi: 10.1136/annrheumdis-2015-208093
109. Russo RC, Garcia CC, Teixeira MM, Amaral FA. The CXCL8/IL-8 chemokine family and its receptors in inflammatory diseases. Expert Rev Clin Immunol. (2014) 10:593–619. doi: 10.1586/1744666X.2014.894886
110. Heidemann J, Ogawa H, Dwinell MB, Rafiee P, Maaser C, Gockel HR, et al. Angiogenic effects of interleukin 8 (CXCL8) in human intestinal microvascular endothelial cells are mediated by CXCR2. J Biol Chem. (2003) 278:8508–15. doi: 10.1074/jbc.M208231200
111. Simonini A, Moscucci M, Muller DWM, Bates ER, Pagani FD, Burdick MD, et al. IL-8 is an angiogenic factor in human coronary atherectomy tissue. Circulation. (2000) 101:1519–26. doi: 10.1161/01.CIR.101.13.1519
112. Üçeyler N, Sommer C. Cytokine regulation in animal models of neuropathic pain and in human diseases. Neurosci Lett. (2008) 437:194–8. doi: 10.1016/j.neulet.2008.03.050
113. de Oliveira CMB, Sakata RK, Issy AM, Gerola LR, Salomão R. Cytokines and pain. Brazilian J Anesthesiol. (2011) 61:255–65. doi: 10.1016/S0034-7094(11)70029-0
114. Sikandar S, Sommer C. Neurotrophins, Cytokines, and Pain. In: Wood JN, editor. The Oxford Handbook of the Neurobiology of Pain. Oxford University Press. doi: 10.1093/oxfordhb/9780190860509.013.25
115. Verri WA, Cunha TM, Parada CA, Poole S, Cunha FQ, Ferreira SH. Hypernociceptive role of cytokines and chemokines: Targets for analgesic drug development? Pharmacol Ther. (2006) 112:116–38. doi: 10.1016/j.pharmthera.2006.04.001
116. Heldin CH, Moustakas A. Signaling receptors for TGF-β family members. Cold Spring Harb Perspect Biol. (2016) 8:a022053 doi: 10.1101/cshperspect.a022053
117. Liu T, Zhang L, Joo D, Sun S-C. NF-κB signaling in inflammation. Signal Transduct Target Ther. (2017) 2:17023. doi: 10.1038/sigtrans.2017.23
118. Hayden MS, Ghosh S. Shared principles in NF-κB signaling. Cell. (2008) 132:344–62. doi: 10.1016/j.cell.2008.01.020
119. Sun S-C. The noncanonical NF-κB pathway. Immunol Rev. (2012) 246:125–40. doi: 10.1111/j.1600-065X.2011.01088.x
120. Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. (2011) 75:50–83. doi: 10.1128/MMBR.00031-10
121. Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J. (2010) 429:403–17. doi: 10.1042/BJ20100323
122. Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. (2001) 81:807–69. doi: 10.1152/physrev.2001.81.2.807
123. Caunt CJ, Finch AR, Sedgley KR, McArdle CA. Seven-transmembrane receptor signalling and ERK compartmentalization. Trends Endocrinol Metab. (2006) 17:276–83. doi: 10.1016/j.tem.2006.07.008
124. Kim GW, Lee NR, Pi RH, Lim YS, Lee YM, Lee JM, et al. IL-6 inhibitors for treatment of rheumatoid arthritis: Past, present, and future. Arch Pharm Res. (2015) 38:575–84. doi: 10.1007/s12272-015-0569-8
125. Moriyama T, Higashi T, Togashi K, Iida T, Segi E, Sugimoto Y, et al. Sensitization of TRPV1 by EP 1 and IP reveals peripheral nociceptive mechanism of prostaglandins. Mol Pain. (2005) 1:3. doi: 10.1186/1744-8069-1-3
126. Schnizler K, Shutov LP, Van Kanegan MJ, Merrill MA, Nichols B, McKnight GS, et al. Protein kinase A anchoring via AKAP150 is essential for TRPV1 modulation by forskolin and prostaglandin E2 in mouse sensory neurons. J Neurosci. (2008) 28:4904–17. doi: 10.1523/JNEUROSCI.0233-08.2008
127. Vellani V, Mapplebeck S, Moriondo A, Davis JB, McNaughton PA. Protein kinase C activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamide. J Physiol. (2001) 534:813–25. doi: 10.1111/j.1469-7793.2001.00813.x
128. Ta TT, Dikmen HO, Schilling S, Chausse B, Lewen A, Hollnagel JO, et al. Priming of microglia with IFN-γ slows neuronal gamma oscillations in situ. Proc Natl Acad Sci USA. (2019) 116:4637–42. doi: 10.1073/pnas.1813562116
129. Oka T, Aou S, Hori T. Intracerebroventricular injection of interleukin-1β enhances nociceptive neuronal responses of the trigeminal nucleus caudalis in rats. Brain Res. (1994) 656:236–44. doi: 10.1016/0006-8993(94)91466-4
130. Kitisin K, Saha T, Blake T, Golestaneh N, Deng M, Kim C, et al. Tgf-Beta signaling in development. Sci STKE. (2007) 2007:cm1. doi: 10.1126/stke.3992007cm1
131. Schmierer B, Hill CS. TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. (2007) 8:970–82. doi: 10.1038/nrm2297
132. Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O'Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov. (2017) 17:78. doi: 10.1038/nrd.2017.201
133. Levy DE, Darnell JE. STATs: transcriptional control and biological impact. Nat Rev Mol Cell Biol. (2002) 3:651–62. doi: 10.1038/nrm909
134. Meier JA, Larner AC. Toward a new STATe: the role of STATs in mitochondrial function. Semin Immunol. (2014) 26:20–8. doi: 10.1016/j.smim.2013.12.005
135. Kays J, Zhang YH, Khorodova A, Strichartz G, Nicol GD. Peripheral synthesis of an atypical protein kinase C mediates the enhancement of excitability and the development of mechanical hyperalgesia produced by nerve growth factor. Neuroscience. (2018) 371:420–32. doi: 10.1016/j.neuroscience.2017.12.030
136. Melemedjian OK, Asiedu MN, Tillu DV, Peebles KA, Yan J, Ertz N, et al. IL-6- and NGF-induced rapid control of protein synthesis and nociceptive plasticity via convergent signaling to the eIF4F complex. J Neurosci. (2010) 30:15113–23. doi: 10.1523/JNEUROSCI.3947-10.2010
137. Aley KO, Messing RO, Mochly-Rosen D, Levine JD. Chronic hypersensitivity for inflammatory nociceptor sensitization mediated by the ε isozyme of protein kinase C. J Neurosci. (2000) 20:4680–5. doi: 10.1523/JNEUROSCI.20-12-04680.2000
138. Korganow AS, Ji H, Mangialaio S, Duchatelle V, Pelanda R, Martin T, et al. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity. (1999) 10:451–61. doi: 10.1016/S1074-7613(00)80045-X
139. Julius D. Molecular mechanisms of nociception. Nature. (2001) 413:203–10. doi: 10.1038/35093019
140. Gladman. Les traumatismes et l'arthrite inflammatoire (2008). Toronto, ON: Gouvernement de L'Ontario, Ministère du Travaul.
141. Deane KD, O'Donnell CI, Hueber W, Majka DS, Lazar AA, Derber LA, et al. The number of elevated cytokines and chemokines in preclinical seropositive rheumatoid arthritis predicts time to diagnosis in an age-dependent manner. Arthritis Rheum. Ontario: Gouvernement de L'Ontario, Ministère du Travaul (2010) 62:3161–72. doi: 10.1002/art.27638
142. Kokkonen H, Söderström I, Rocklöv J, Hallmans G, Lejon K, Rantapää Dahlqvist S. Up-regulation of cytokines and chemokines predates the onset of rheumatoid arthritis. Arthritis Rheum. (2010) 62:383–91. doi: 10.1002/art.27186
143. van Zanten A, Arends S, Roozendaal C, Limburg PC, Maas F, Trouw LA, et al. Presence of anticitrullinated protein antibodies in a large population-based cohort from the Netherlands. Ann Rheum Dis. (2017) 76:1184–90. doi: 10.1136/annrheumdis-2016-209991
144. Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. (2010) 62:2569–81. doi: 10.1002/art.27584
145. Sokolove J, Pisetsky D. Bone loss, pain and inflammation: three faces of ACPA in RA pathogenesis. Ann Rheum Dis. (2016) 75:637–9. doi: 10.1136/annrheumdis-2015-208308
146. Zhang C, Ward J, Dauch JR, Tanzi RE, Cheng HT. Cytokine-mediated inflammation mediates painful neuropathy from metabolic syndrome. PLoS ONE. (2018) 13:e0192333. doi: 10.1371/journal.pone.0192333
147. Ramer MS, Thompson SWN, McMahon SB. Causes and consequences of sympathetic basket formation in dorsal root ganglia. Pain. (1999) 82(Suppl. 6):S111–20. doi: 10.1016/S0304-3959(99)00144-X
148. Krock E, Millecamps M, Anderson KM, Srivastava A, Reihsen TE, Hari P, et al. Interleukin-8 as a therapeutic target for chronic low back pain: upregulation in human cerebrospinal fluid and pre-clinical validation with chronic reparixin in the SPARC-null mouse model. EBioMedicine. (2019) 43:487–500. doi: 10.1016/j.ebiom.2019.04.032
149. Fuzzati-Armentero MT, Cerri S, Blandini F. Peripheral-central neuroimmune crosstalk in Parkinson's disease: what do patients and animal models tell Us? Front Neurol. (2019) 10:232. doi: 10.3389/fneur.2019.00232
150. Barry A, O'Halloran KD, McKenna JP, McCreary C, Downer EJ. Plasma IL-8 signature correlates with pain and depressive symptomatology in patients with burning mouth syndrome: Results from a pilot study. J Oral Pathol Med. (2018) 47:158–65. doi: 10.1111/jop.12666
151. Sasaguri T, Taguchi T, Murata Y, Kobayashi K, Iizasa S, Iizasa E, et al. Interleukin-27 controls basal pain threshold in physiological and pathological conditions. Sci Rep. (2018) 8:11022. doi: 10.1038/s41598-018-29398-3
152. Porta-Sales J, Garzón-Rodríguez C, Llorens-Torromé S, Brunelli C, Pigni A, Caraceni A. Evidence on the analgesic role of bisphosphonates and denosumab in the treatment of pain due to bone metastases: a systematic review within the European Association for Palliative Care guidelines project. Palliat Med. (2017) 31:5–25. doi: 10.1177/0269216316639793
153. Sun WH, Chen CC. Roles of proton-sensing receptors in the transition from acute to chronic pain. J Dent Res. (2016) 95:135–42. doi: 10.1177/0022034515618382
154. Mantyh PW. Mechanisms that drive bone pain across the lifespan. Br J Clin Pharmacol. (2019) 85:1103–13. doi: 10.1111/bcp.13801
155. Jimenez-Andrade JM, Herrera MB, Ghilardi JR, Vardanyan M, Melemedjian OK, Mantyh PW. Vascularization of the dorsal root ganglia and peripheral nerve of the mouse: Implications for chemical-induced peripheral sensory neuropathies. Mol Pain. (2008) 4:10. doi: 10.1186/1744-8069-4-10
156. Flatters SJL, Bennett GJ. Studies of peripheral sensory nerves in paclitaxel-induced painful peripheral neuropathy: evidence for mitochondrial dysfunction. Pain. (2006) 122:245–57. doi: 10.1016/j.pain.2006.01.037
157. Ravindra Deshmukh V. Gap junctions in the dorsal root ganglia. In: Aranda Abreu GE, editor. Neurons - Dendrites and Axons. London: Intechopen (2018). p. 1–22. doi: 10.5772/intechopen.82128
158. Raoof R, Willemen HLDM, Eijkelkamp N. Divergent roles of immune cells and their mediators in pain. Rheumatology. (2018) 57:429–40. doi: 10.1093/rheumatology/kex308
159. Schaible H-G, Von Banchet GS, Boettger MK, Bräuer R, Gajda M, Richter F, et al. The role of proinflammatory cytokines in the generation and maintenance of joint pain. Ann N Y Acad Sci. (2010) 1193:60–9. doi: 10.1111/j.1749-6632.2009.05301.x
160. Ogawa N, Kawai H, Terashima T, Kojima H, Oka K, Chan L, et al. Gene therapy for neuropathic pain by silencing of TNF-α expression with lentiviral vectors targeting the dorsal root ganglion in mice. PLoS ONE. (2014) 9:e92073. doi: 10.1371/journal.pone.0092073
161. McMahon SB, Cafferty WBJ, Marchand F. Immune and glial cell factors as pain mediators and modulators. Exp Neurol. (2005) 192:444–62. doi: 10.1016/j.expneurol.2004.11.001
162. Miller RE, Tran PB, Das R, Ghoreishi-Haack N, Ren D, Miller RJ, et al. CCR2 chemokine receptor signaling mediates pain in experimental osteoarthritis. Proc Natl Acad Sci USA. (2012) 109:20602–7. doi: 10.1073/pnas.1209294110
163. Zhang H, Li Y, de Carvalho-Barbosa M, Kavelaars A, Heijnen CJ, Albrecht PJ, et al. Dorsal root ganglion infiltration by macrophages contributes to paclitaxel chemotherapy-induced peripheral neuropathy. J Pain. (2016) 17:775–86. doi: 10.1016/j.jpain.2016.02.011
164. Abbadie C, Lindia JA, Cumiskey AM, Peterson LB, Mudgett JS, Bayne EK, et al. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci USA. (2003) 100:7947–52. doi: 10.1073/pnas.1331358100
165. Zhang P, Bi RY, Gan YH. Glial interleukin-1β upregulates neuronal sodium channel 1.7 in trigeminal ganglion contributing to temporomandibular joint inflammatory hypernociception in rats. J Neuroinflammation. (2018) 15:117. doi: 10.1186/s12974-018-1154-0
166. Willemen HLDM, Eijkelkamp N, Garza Carbajal A, Wang H, Mack M, Zijlstra J, et al. Monocytes/macrophages control resolution of transient inflammatory pain. J Pain. (2014) 15:496–506. doi: 10.1016/j.jpain.2014.01.491
167. Reeve AJ, Patel S, Fox A, Walker K, Urban L. Intrathecally administered endotoxin or cytokines produce allodynia, hyperalgesia and changes in spinal cord neuronal responses to nociceptive stimuli in the rat. Eur J Pain. (2000) 4:247–57. doi: 10.1053/eujp.2000.0177
168. Sung C-S, Wen Z-H, Chang W-K, Ho S-T, Tsai S-K, Chang Y-C, et al. Intrathecal interleukin-1β administration induces thermal hyperalgesia by activating inducible nitric oxide synthase expression in the rat spinal cord. Brain Res. (2004) 1015:145–53. doi: 10.1016/j.brainres.2004.04.068
169. Milligan E, Zapata V, Schoeniger D, Chacur M, Green P, Poole S, et al. An initial investigation of spinal mechanisms underlying pain enhancement induced by fractalkine, a neuronally released chemokine. Eur J Neurosci. (2005) 22:2775–82. doi: 10.1111/j.1460-9568.2005.04470.x
170. Tanaka T, Minami M, Nakagawa T, Satoh M. Enhanced production of monocyte chemoattractant protein-1 in the dorsal root ganglia in a rat model of neuropathic pain: possible involvement in the development of neuropathic pain. Neurosci Res. (2004) 48:463–9. doi: 10.1016/j.neures.2004.01.004
171. Gabay E, Wolf G, Shavit Y, Yirmiya R, Tal M. Chronic blockade of interleukin-1 (IL-1) prevents and attenuates neuropathic pain behavior and spontaneous ectopic neuronal activity following nerve injury. Eur J Pain. (2011) 15:242–8. doi: 10.1016/j.ejpain.2010.07.012
172. Chen G, Park C-K, Xie R-G, Ji R-R. Intrathecal bone marrow stromal cells inhibit neuropathic pain via TGF-β secretion. J Clin Invest. (2015) 125:3226–40. doi: 10.1172/JCI80883
173. Chen G, Zhang Y-Q, Qadri YJ, Serhan CN, Ji R-R. Microglia in pain: detrimental and protective roles in pathogenesis and resolution of pain. Neuron. (2018) 100:1292–311. doi: 10.1016/j.neuron.2018.11.009
174. Li Z, Wei H, Piirainen S, Chen Z, Kalso E, Pertovaara A, et al. Spinal versus brain microglial and macrophage activation traits determine the differential neuroinflammatory responses and analgesic effect of minocycline in chronic neuropathic pain. Brain Behav Immun. (2016) 58:107–17. doi: 10.1016/j.bbi.2016.05.021
175. Peters CM, Jimenez-Andrade JM, Jonas BM, Sevcik MA, Koewler NJ, Ghilardi JR, et al. Intravenous paclitaxel administration in the rat induces a peripheral sensory neuropathy characterized by macrophage infiltration and injury to sensory neurons and their supporting cells. Exp Neurol. (2007) 203:42–54. doi: 10.1016/j.expneurol.2006.07.022
176. Park HJ, Stokes JA, Pirie E, Skahen J, Shtaerman Y, Yaksh TL. Persistent hyperalgesia in the cisplatin-treated mouse as defined by threshold measures, the conditioned place preference paradigm, and changes in dorsal root ganglia activated transcription factor 3: the effects of gabapentin, ketorolac, and etanercept. Anesth Analg. (2013) 116:224–31. doi: 10.1213/ANE.0b013e31826e1007
177. Wang D, Couture R, Hong Y. Activated microglia in the spinal cord underlies diabetic neuropathic pain. Eur J Pharmacol. (2014) 728:59–66. doi: 10.1016/j.ejphar.2014.01.057
178. Christianson CA, Corr M, Firestein GS, Mobargha A, Yaksh TL, Svensson CI. Characterization of the acute and persistent pain state present in K/BxN serum transfer arthritis. Pain. (2010) 151:394–403. doi: 10.1016/j.pain.2010.07.030
179. Liu M, Yao M, Wang H, Xu L, Zheng Y, Huang B, et al. P2Y12 receptor-mediated activation of spinal microglia and p38MAPK pathway contribute to cancer-induced bone pain. J Pain Res. (2017) 10:417–26. doi: 10.2147/JPR.S124326
180. Imoto A, Mitsunaga S, Inagaki M, Aoyagi K, Sasaki H, Ikeda M, et al. Neural invasion induces cachexia via astrocytic activation of neural route in pancreatic cancer. Int J Cancer. (2012) 131:2795–807. doi: 10.1002/ijc.27594
181. Fitzsimmons BL, Zattoni M, Svensson CI, Steinauer J, Hua X-Y, Yaksh TL. Role of spinal p38α and β MAPK in inflammatory hyperalgesia and spinal COX-2 expression. Neuroreport. (2010) 21:313–7. doi: 10.1097/WNR.0b013e32833774bf
182. Rider P, Carmi Y, Cohen I. Biologics for targeting inflammatory cytokines, clinical uses, and limitations. Int J Cell Biol. (2016) 2016:9259646. doi: 10.1155/2016/9259646
183. Old EA, Clark AK, Malcangio M. The role of glia in the spinal cord in neuropathic and inflammatory pain. In: Schaible HG, editor. Handbook of Experimental Pharmacology. Vol. 227. Berlin; Heidelberg: Springer (2015). p. 145–70. doi: 10.1007/978-3-662-46450-2_8
184. Yoon H, Walters G, Paulsen AR, Scarisbrick IA. Astrocyte heterogeneity across the brain and spinal cord occurs developmentally, in adulthood and in response to demyelination. PLoS ONE. (2017) 12:e0180697. doi: 10.1371/journal.pone.0180697
185. Choi SR, Roh DH, Yoon SY, Choi HS, Kang SY, Han HJ, et al. Astrocyte sigma-1 receptors modulate connexin 43 expression leading to the induction of below-level mechanical allodynia in spinal cord injured mice. Neuropharmacology. (2016) 111:34–46. doi: 10.1016/j.neuropharm.2016.08.027
186. Chen G, Park CK, Xie RG, Berta T, Nedergaard M, Ji RR. Connexin-43 induces chemokine release from spinal cord astrocytes to maintain late-phase neuropathic pain in mice. Brain. (2014) 137(Pt 8):2193–209. doi: 10.1093/brain/awu140
187. Delpech JC, Herron S, Botros MB, Ikezu T. Neuroimmune crosstalk through extracellular vesicles in health and disease. Trends Neurosci. (2019) 42:361–72. doi: 10.1016/j.tins.2019.02.007
188. Ribeiro-Rodrigues TM, Martins-Marques T, Morel S, Kwak BR, Girão H. Role of connexin 43 in different forms of intercellular communication – gap junctions, extracellular vesicles and tunnelling nanotubes. J Cell Sci. (2017) 130:3619–30. doi: 10.1242/jcs.200667
189. Kotini M, Barriga EH, Leslie J, Gentzel M, Rauschenberger V, Schambon A, et al. Gap junction protein Connexin-43 is a direct transcriptional regulator of N-cadherin in vivo. Nat Commun. (2018) 9:3846. doi: 10.1038/s41467-018-06368-x
190. Zamanian JL, Foo LC, Nouri N, Zhou L, Barres BA, Xu L, et al. Genomic analysis of reactive astrogliosis. J Neurosci. (2012) 32:6391–410. doi: 10.1523/JNEUROSCI.6221-11.2012
191. Han P, Zhao J, Liu S-B, Yang C-J, Wang Y-Q, Wu G-C, et al. Interleukin-33 mediates formalin-induced inflammatory pain in mice. Neuroscience. (2013) 241:59–66. doi: 10.1016/j.neuroscience.2013.03.019
192. Han P, Liu S, Zhang M, Zhao J, Wang Y, Wu G, et al. Inhibition of spinal interlukin-33/ST2 signaling and downstream ERK and JNK pathways in electroacupuncture analgesia in formalin mice. PLoS ONE. (2015) 10:e0129576. doi: 10.1371/journal.pone.0129576
193. Zarpelon AC, Cunha TM, Alves-Filho JC, Pinto LG, Ferreira SH, McInnes IB, et al. IL-33/ST2 signalling contributes to carrageenin-induced innate inflammation and inflammatory pain: role of cytokines, endothelin-1 and prostaglandin E2. Br J Pharmacol. (2013) 169:90–101. doi: 10.1111/bph.12110
194. Fonseca MM, Santa-Cecilia F, Kusuda R, Ferreira D, Bezerra F, Ferreira M, et al. (153) the interleukin 27 (IL-27) protects mice from neuropathic pain development through up-regulation of anti-inflammatory cytokine IL-10. J Pain. (2017) 18:S14–15. doi: 10.1016/j.jpain.2017.02.059
195. Duffy SS, Keating BA, Perera CJ, Lees JG, Tonkin RS, Makker PGS, et al. Regulatory T cells and their derived cytokine, interleukin-35, reduce pain in experimental autoimmune encephalomyelitis. J Neurosci(2019) 39:2326–46. doi: 10.1523/JNEUROSCI.1815-18.2019
196. Kroenke K, Bair MJ, Damush TM, Wu J, Hoke S, Sutherland J, et al. Optimized antidepressant therapy and pain self-management in primary care patients with depression and musculoskeletal pain. JAMA. (2009) 301:2099. doi: 10.1001/jama.2009.723
197. Page GG, Opp MR, Kozachik SL. Reduced sleep, stress responsivity, and female sex contribute to persistent inflammation-induced mechanical hypersensitivity in rats. Brain Behav Immun. (2014) 40:244–51. doi: 10.1016/j.bbi.2014.02.013
198. Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, et al. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. (2003) 424:778–83. doi: 10.1038/nature01786
199. Ji R-R, Berta T, Nedergaard M. Glia and pain: Is chronic pain a gliopathy? Pain. (2013) 154:S10–28. doi: 10.1016/j.pain.2013.06.022
200. Loggia ML, Chonde DB, Akeju O, Arabasz G, Catana C, Edwards RR, et al. Evidence for brain glial activation in chronic pain patients. Brain. (2015) 138:604–15. doi: 10.1093/brain/awu377
201. Albrecht DS, Forsberg A, Sandstrom A, Bergan C, Kadetoff D, Protsenko E, et al. Brain glial activation in fibromyalgia - a multi-site positron emission tomography investigation. Brain Behav Immun. (2019) 75:72. doi: 10.1016/j.bbi.2018.09.018
202. Zhu C Bin, Blakely RD, Hewlett WA. The proinflammatory cytokines interleukin-1beta and tumor necrosis factor-alpha activate serotonin transporters. Neuropsychopharmacology. (2006) 31:2121–31. doi: 10.1038/sj.npp.1301029
203. Haroon E, Raison CL, Miller AH. Psychoneuroimmunology meets neuropsychopharmacology: translational implications of the impact of inflammation on behavior. Neuropsychopharmacology. (2012) 37:137–62. doi: 10.1038/npp.2011.205
204. Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. (2009) 65:732–41. doi: 10.1016/j.biopsych.2008.11.029
205. Oka T, Wakugawa Y, Hosoi M, Oka K, Hori T. Intracerebroventricular injection of tumor necrosis factor-αInduces thermal hyperalgesia in rats. Neuroimmunomodulation. (1996) 3:135–40. doi: 10.1159/000097238
206. Hori T, Oka T, Hosoi M, Aou S. Pain modulatory actions of cytokines and prostaglandin E2 in the brain. Ann N Y Acad Sci. (1998) 840:269–81. doi: 10.1111/j.1749-6632.1998.tb09567.x
207. Coelho A-M, Fioramonti J, Bueno L. Brain interleukin-1β and tumor necrosis factor-α are involved in lipopolysaccharide-induced delayed rectal allodynia in awake rats. Brain Res Bull. (2000) 52:223–8. doi: 10.1016/S0361-9230(00)00269-0
208. Wei F, Guo W, Zou S, Ren K, Dubner R. Supraspinal glial-neuronal interactions contribute to descending pain facilitation. J Neurosci. (2008) 28:10482–95. doi: 10.1523/JNEUROSCI.3593-08.2008
209. Zeidan F, Salomons T, Farris SR, Emerson NM, Adler-Neal A, Jung Y, et al. Neural mechanisms supporting the relationship between dispositional mindfulness and pain. Pain. (2018) 159:2477–85. doi: 10.1097/j.pain.0000000000001344
210. Taylor AMW, Mehrabani S, Liu S, Taylor AJ, Cahill CM. Topography of microglial activation in sensory- and affect-related brain regions in chronic pain. J Neurosci Res. (2017) 95:1330–5. doi: 10.1002/jnr.23883
211. Barcelon EE, Cho W-H, Jun SB, Lee SJ. Brain microglial activation in chronic pain-associated affective disorder. Front Neurosci. (2019) 13:213. doi: 10.3389/fnins.2019.00213
212. Yirmiya R, Rimmerman N, Reshef R. Depression as a microglial disease. Trends Neurosci. (2015) 38:637–58. doi: 10.1016/j.tins.2015.08.001
213. Liu Y, Zhou L-J, Wang J, Li D, Ren W-J, Peng J, et al. TNF-α differentially regulates synaptic plasticity in the hippocampus and spinal cord by microglia-dependent mechanisms after peripheral nerve injury. J Neurosci. (2017) 37:871–81. doi: 10.1523/JNEUROSCI.2235-16.2016
214. Shao Q, Li Y, Wang Q, Zhao J. IL-10 and IL-1β Mediate neuropathic-pain like behavior in the ventrolateral orbital cortex. Neurochem Res. (2015) 40:733–9. doi: 10.1007/s11064-015-1521-5
215. Roman M, Irwin MR. Novel neuroimmunologic therapeutics in depression: a clinical perspective on what we know so far. Brain Behav Immun. (2019) 83:7–21. doi: 10.1016/j.bbi.2019.09.016
216. Lee SM, Te S, Breen EC, Olmstead R, Irwin MR, Cho JH. Circulating versus lipopolysaccharide-induced inflammatory markers as correlates of subthreshold depressive symptoms in older adults. World J Biol Psychiatry. (2019) 1–8. doi: 10.1080/15622975.2019.1671608. [Epub ahead of print].
217. Bäckryd E, Tanum L, Lind AL, Larsson A, Gordh T. Evidence of both systemic inflammation and neuroinflammation in fibromyalgia patients, as assessed by a multiplex protein panel applied to the cerebrospinal fluid and to plasma. J Pain Res. (2017) 10:515–25 doi: 10.2147/JPR.S128508
218. Üçeyler N, Rogausch JP, Toyka KV, Sommer C. Differential expression of cytokines in painful and painless neuropathies. Neurology. (2007) 69:42–9. doi: 10.1212/01.wnl.0000265062.92340.a5
219. Langjahr M, Schubert A-L, Sommer C, Üçeyler N. Increased pro-inflammatory cytokine gene expression in peripheral blood mononuclear cells of patients with polyneuropathies. J Neurol. (2018) 265:618–27. doi: 10.1007/s00415-018-8748-4
220. Raison CL, Rutherford RE, Woolwine BJ, Shuo C, Schettler P, Drake DF, et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. Arch Gen Psychiatry. (2013) 70:31–41. doi: 10.1001/2013.jamapsychiatry.4
221. Medina-Rodriguez EM, Lowell JA, Worthen RJ, Syed SA, Beurel E. Involvement of innate and adaptive immune systems alterations in the pathophysiology and treatment of depression. Front Neurosci. (2018) 12:547. doi: 10.3389/fnins.2018.00547
222. Brown PM, Pratt AG, Isaacs JD. Mechanism of action of methotrexate in rheumatoid arthritis and the search for biomarkers. Nat Rev Rheumatol. (2016) 12:731–42. doi: 10.1038/nrrheum.2016.175
223. Davila L, Ranganathan P. Pharmacogenetics: implications for therapy in rheumatic diseases. Nat Rev Rheumatol. (2011) 7:537–50. doi: 10.1038/nrrheum.2011.117
224. Smedegård G, Björk J. Sulphasalazine: mechanism of action in rheumatoid arthritis. Br J Rheumatol. (1995) 34(Suppl. 2):7–15. doi: 10.1093/rheumatology/XXXIV.suppl_2.7
225. Tian H, Cronstein BN. Understanding the mechanisms of action of methotrexate: implications for the treatment of rheumatoid arthritis. Bull NYU Hosp Jt Dis. (2007) 65:168–73.
226. Kaltsonoudis E, Papagoras C, Drosos AA. Current and future role of methotrexate in the therapeutic armamentarium for rheumatoid arthritis. Int J Clin Rheumtol. (2012) 7:179–89. doi: 10.2217/ijr.12.10
227. Radner H, Aletaha D. Anti-TNF in rheumatoid arthritis: an overview. Wiener Medizinische Wochenschrift. (2015) 165:3–9. doi: 10.1007/s10354-015-0344-y
228. Gabay C, Hasler P, Kyburz D, So A, Villiger P, von Kempis J, et al. Biological agents in monotherapy for the treatment of rheumatoid arthritis. Swiss Med Wkly. (2014) 144:w13950. doi: 10.4414/smw.2014.13950
229. Calabrò A, Caterino AL, Elefante E, Valentini V, Vitale A, Talarico R, et al. One year in review 2016: novelties in the treatment of rheumatoid arthritis. Clin Exp Rheumatol. (2016) 34:357–72.
230. Fleischmann R, Kremer J, Cush J, Schulze-Koops H, Connell CA, Bradley JD, et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med. (2012) 367:495–507. doi: 10.1056/NEJMoa1109071
231. Lee EB, Fleischmann R, Hall S, Wilkinson B, Bradley JD, Gruben D, et al. Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med. (2014) 370:2377–86. doi: 10.1056/NEJMoa1310476
232. Smolen JS, Landewé R, Bijlsma J, Burmester G, Chatzidionysiou K, Dougados M, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. (2017) 76:935–38. doi: 10.1136/annrheumdis-2016-210715
233. Senolt L. Emerging therapies in rheumatoid arthritis: focus on monoclonal antibodies. F1000Research. (2019) 8:1549. doi: 10.12688/f1000research.18688.1
Keywords: cytokine, chemokine, pain, neuroimmune crosstalk, neuraxis
Citation: Gonçalves dos Santos G, Delay L, Yaksh TL and Corr M (2020) Neuraxial Cytokines in Pain States. Front. Immunol. 10:3061. doi: 10.3389/fimmu.2019.03061
Received: 14 October 2019; Accepted: 16 December 2019;
Published: 28 January 2020.
Edited by:
Waldiceu A. Verri, State University of Londrina, BrazilReviewed by:
R. R. Ji, Duke University, United StatesDjane Braz Duarte, University of Brasilia, Brazil
Copyright © 2020 Gonçalves dos Santos, Delay, Yaksh and Corr. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maripat Corr, bXBjb3JyQHVjc2QuZWR1
†These authors have contributed equally to this work