
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 20 December 2019
Sec. Cancer Immunity and Immunotherapy
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02990
 Teresa Lozano1†
Teresa Lozano1† Silvia Chocarro1†Celia Martin1
Silvia Chocarro1†Celia Martin1 Aritz Lasarte-Cia1
Aritz Lasarte-Cia1 Cynthia del Valle1
Cynthia del Valle1 Marta Gorraiz1Patricia Sarrión1Marina Ruiz de Galarreta2
Marta Gorraiz1Patricia Sarrión1Marina Ruiz de Galarreta2 Amaia Lujambio2
Amaia Lujambio2 Sandra Hervás-Stubbs1
Sandra Hervás-Stubbs1 Pablo Sarobe1
Pablo Sarobe1 Noelia Casares1*†
Noelia Casares1*† Juan J. Lasarte1*†
Juan J. Lasarte1*†Adoptive immunotherapy with ex vivo-expanded tumor-infiltrating lymphocytes (TILs) has achieved objective clinical responses in a significant number of patients with cancer. The failure of many patients to develop long-term tumor control may be, in part, due to exhaustion of transferred T cells in the presence of a hostile tumor microenvironment. In several tumor types, growth and survival of carcinoma cells appear to be sustained by a network of receptors/ligands of the ErbB family. We speculated that if transferred T cells could benefit from EGFR ligands produced by the tumor, they might proliferate better and exert their anti-tumor activities more efficiently. We found that CD8+ T cells transduced with a retrovirus to express EGFR responded to EGFR ligands activating the EGFR signaling pathway. These EGFR-expressing effector T cells proliferated better and produced more IFN-γ and TNF-α in the presence of EGFR ligands produced by tumor cells in vitro. EGFR-expressing CD8 T cells from OT-1 mice were more efficient killing B16-OVA cells than control OT-1 CD8 T cells. Importantly, EGFR-expressing OT-1 T cells injected into B16-OVA tumor bearing mice were recruited into the tumor, expressed lower levels of the exhaustion markers PD1, TIGIT, and LAG3, and were more efficient in delaying tumor growth. Our results suggest that genetic modification of CD8+ T cells to express EGFR might be considered in immunotherapeutic strategies based on adoptive transfer of anti-tumor T cells against cancers expressing EGFR ligands.
Adoptive cell therapy (ACT) with ex vivo-expanded tumor-infiltrating lymphocytes (TILs) has achieved objective clinical responses in a significant fraction of patients with cancer (1). The success of ACT is largely related to their capacity to infiltrate the tumor, persist in the host, and exert their anti-tumoral activity after their transfer. The failure of many patients to develop long-term tumor control may be, in part, due to exhaustion and apoptosis of the transferred T cells. The in vivo persistence of transferred T cells can vary from hours or days to weeks (2). T cell persistence can depend largely on the manner in which T cells were expanded in vitro, the conditions of cell administration, and importantly, on the tumor microenvironment.
The tumor microenvironment is a hostile medium with an abundance of inhibitory factors for lymphocytes. However, tumor microenvironment may contain factors that can favor cell proliferation. This fact prompted several investigations aimed at assembling T cells with cytokine receptors such as CSF-1 (3) or inverted cytokine receptors IL-4Ralpha ectodomain/IL-2 beta(c) endodomain (4), IL4/IL7 (5), or IL4/IL21 (6) among others, to respond positively to ligands expressed into the tumor. In this work, we focussed on the epidermal growth factor receptor (EGFR, also known as ErbB1 or HER-1) and their ligands. Indeed, their overexpression confer a proliferative advantage to tumor cells and have been correlated with progression to invasion and metastasis in a wide variety of cancers, including esophageal (7), gastric (8), hepatocellular (9, 10), head and neck (11) lung (12), or colorectal carcinomas (13, 14). Thus, in addition to overexpression of the EGF receptor, its ligands epidermal growth factor (EGF), transforming growth factor-α TGF-α, amphiregulin (AR), betacellulin (BTC), heparin-binding EGF-like growth factor (HB-EGF), and epiregulin (EREG) have been reported to be upregulated in tumor tissues (9–15). For these reasons, EGFR and its downstream signaling molecules are considered as targets for therapeutic interventions in cancer (16).
The ErbB ligands are synthesized as membrane-anchored precursors (17) that are released as active factors from the cell surface by transmembrane proteases of the “disintegrin and metalloproteinases” (ADAM) type (18, 19). The soluble growth factors may bind their cognate receptors in an autocrine or paracrine manner, although membrane anchored ErbB ligands can also signal to adjacent cells in a juxtacrine fashion (17). These auto/justa/paracrine loops lead to signaling via the tyrosine kinase domain and may provide an advantage for tumor development and metastatic progression.
EGFR is well-established to be ubiquitously expressed but, in general, is thought to be absent in the hematopoietic cell linage with the exception of sporadic expression described in monocytes (20) or plasma cells (21). Recent reports have also shown that Foxp3+ regulatory T (Treg) cells express EGFR under inflammatory conditions. The EGFR ligand amphiregulin (AR) markedly enhances Treg cell function in vitro and in vivo (22–24). These findings reveal EGFR as a component in the regulation of local immune responses as well as in tissue protection after damage (22, 25). Curiously, conventional effector T cells do not express EGFR (22) and thus, they do not benefit from the presence of EGFR ligands in the tumor microenvironment. A recent report has shown that, under certain circumstances Th2 cells can express EGFR allowing them to produce inflammatory cytokines that may protect the host from infections (26). We speculated that if adoptively transferred T cells could respond to these ligands they might be able to persist and proliferate better within the tumor microenvironment. We modified genetically anti-tumor CD8+ T cells to express EGFR and studied the effect of EGFR ligands on their function in vitro and in vivo.
EL-4, CT26, B16F10, B16-OVA, E.G7.OVA, 4T1, A20, MC38 (ATCC, American Type Culture Collection), Hepa129 (provided by Dr. M Gonzalez-Carmona, Bonn, Germany), 5TGM1 (kindly provided by Dr. Oyajobi, TX), PM-299L (provided by Dr. Lujambio, NY) cell lines were cultured in complete medium (RPMI 1640 or DMEM containing 10% FCS, antibiotics, 2 mM glutamine and 50 μM 2-ME. The Platinum Ecotropic cell line (Plat-E, ATCC) was cultured in DMEM supplemented with 10% FCS and the selection antibiotics puromycin (100 ug/ml) and blasticidin (10 ug/ml).
Female C57BL/6 mice were purchased from Harlan Laboratories. OT-1 transgenic mice (C57BL/6-Tg [Tcra/Tcrb] 1100 Mjb/J) with a TCR recognizing H2-Kb-restricted OVA (257-264 SIINFEKL peptide) and CD45.1 transgenic C57BL/6 mice (B6.SJL-PtprcaPep3b/BoyJ mice) were obtained from Jackson Laboratory (Bar Harbor, ME). OT-1 mice were crossed with CD45.1 mice at our animal facility to obtain homozygous OT-1 × CD45.1 mice.
Total RNA was isolated from different tumor cell lines (5 × 106 cells) and from murine tumor biopsies (homogenized in 1 mL of Ultraspec solution, Biotex, Houston, TX) using MagMax 96 total kit (Ambion). After DNAse treatment (Invitrogen) and retrotranscription (Invitrogen), the expression of target genes was measured by a real-time PCR reaction using specific primers (Table 1) and the iQ SYBR Green Supermix (Bio-Rad, Hercules, CA). β-Actin was used to normalize gene expression. mRNA values were represented by the formula: 2ΔCt, where ΔCt. indicates the difference in the threshold cycle between β-Actin and target genes.

Table 1. Primers used for iQ-PCR.
EGF present into the tumor microenvironment was measured in tumor cell extracts obtained from mice bearing B16-OVA, PM299L, or Hepa129 derived tumors using the DuoSet Mouse EGF ELISA kit (R&D, MN, USA) according to manufacturer's instructions.
The retrovirus expression plasmid KMV IRES-GFP was kindly provided by Dr. Rao (LIAI; La Jolla, California). Plasmids expressing EGFR were prepared by Genscript (NJ, USA) and used to subclone them into the KMV IRES EGFR-GFP plasmid. The Plat-E cell line as used for retrovirus production. Packaging cells were transfected with 5 μg of retroviral plasmid (KMV-IRES-GFP or KMV-IRES-EGFR-GFP) along with 2.5 μg pCL-Eco plasmid DNA using lipofectamine (ThermoFisher Scientific, MA, USA. Retroviral supernatants were collected at 48 and 72 h. CD8+ T cells were isolated from the spleen of mice by magnetic selection (Miltenyi) and activated with dynabeads CD3/CD28 at a 1:2 bead/cell ratio. Cells were cultured at a density of 106 cell /ml in 12-well plates in RMPI culture medium supplemented with 10% FBS and antibiotics. One day later T cells were collected and resuspended in retroviral supernatant with 50 IU/mL rhIL-2 and 10 μg/mL protamine sulfate (Sigma), and spun at 2,000 g at 32°C for 90 min in 12 well plates. Infection was repeated at day 2. Lymphocytes were cultured with 100 IU rhIL-2 and subsequently split until day 5, when cells were used for functional analysis.
Five days after retroviral (RV) transfection, transduced CD8+ T cells were harvested and GFP+ cells were sorted using a FACS Aria Sorter device (BD Biosciences). After 24 h, the expression of EGFR on the surface was measured by flow cytometry using EGF-APC conjugate (Life technologies). T cells were incubated with a 1:100 dilution of the ligand for 30 min at 37°C. After one wash with PBS, cells were analyzed by flow cytometry (FACS Calibur; BD Biosciences). For signaling studies, CD8+ T cells were lysed in RIPA buffer (Sigma) and homogenates were prepared and subjected to Western blot analysis as described previously (27) using anti-p-ERK1/2 (Thr202, Tyr204) and anti GAPDH as loading control (Cell Signaling; Beverly, MA).
Functional activity of transduced GFP CD8+ T cells (controls) or EGFR-GFP CD8+ T cells was measured by in vitro experiments. First, genetically modified OT-1 CD8+ T cells were stimulated with SIINFEKL peptide at a suboptimal (0.01 pg/ml) or optimal (10 μg/ml) concentration in the presence or absence of recombinant EGF (100 nM) for 24 h. The number of IFN-γ or TNF-α producing cells was analyzed by flow cytometry. Briefly, cells were incubated with Zombie NIR Fixable dye (Biolegend) and subsequently stained with fluorochrome-conjugated monoclonal antibodies (mAbs) against CD8 (53-6.7), CD4 (RM4-5) in the presence of purified anti-CD16/32 mAb. Cells were then fixed and permeabilized (eBiosciences) and then stained with anti-IFN-γ (XMG1.2), and anti-TNF-α (MP6-XT22) (BD Biosciences) mAbs. Samples were acquired on a FACSCanto-II cytometer (BD Biosciences). Data were analyzed using FlowJo software (TreeStar).
Also, genetically modified OT-1 T cells were cocultured with irradiated B16-OVA cells for 48 h, and proliferation rate and IFN-γ production were measured by 3H-timidine incorporation (0.5 μCi per well) and ELISA, respectively, as previously described (28).
Cytotoxic activity of modified OT1 cells was measured by a Real-time cytotoxicity assay (xCELLigence). In this assay, adhesion of cells to the gold microelectrodes impedes the flow of electric current between electrodes. The impedance value is plotted as a unit-less parameter called “Cell Index,” that increases as cells proliferate until cells approach 100% confluence. After the addition of B16.OVA cells to the wells, an initial phase of cell adhesion and spreading (0–6 h) is recorded before reaching a plateau phase (around 1 arbitrary CI). At this point, effector T cells are added and changes in cell index are recorded. The curve represents the mean Cell Index value from 3 wells ± SD. B16-OVA or B16F10 target cells were seeded in culture medium at a density of 20,000 cells per well (E-Plates 96 (Roche, Grenzach-Wyhlen, Germany). Cell attachment was monitored until the plateau phase was reached. Then, OT1 cells were added at different Effector:Tumor (E:T) cell ratios. Upon addition of effector cells, impedance measurements were monitored in real-time every 15 min during 24 h. An RTCA SP (Roche) instrument and the RTCA software Version 1.1 (Roche) were used to measure and analyze the data. All experiments were performed in duplicate.
Measurement of SIINFEKL specific IFN-γ producing cells after ACT. To evaluate the behavior of the modified CD8+ T cells in vivo, CD8 T cells obtained from C56BL/6 mice expressing the CD45.1 allele were modified genetically to express GFP or EGFR-GFP and injected into CD45.2 mice bearing B16-OVA tumors. Seven days after T cell infusion, T-cells producing IFN-γ were determined by ELISPOT (BD-Biosciences) following manufacturer's instructions. Briefly, splenocytes (7 × 105/well) were stimulated with or without 1 μg/ml SIINFEKL peptide. After one day of culture the number of spot-forming cells was enumerated with an automated ELISPOT reader (CTL, Aalen, Germany).
C57BL/6 mice (6–12 weeks of age) were injected with 5 × 105 B16-OVA melanoma cells or with 5 × 105 PM-299L hepatoma cells by s.c route. Seven days later, mice were sublethally irradiated (total body irradiation) with 500 cGy and received 2–5*106 CD8+ T cells from OT-I retrovirally transduced to express GFP or EGFR-GFP. Modified CD8+ T cells were purified by flow sorting of GFP+ cells before ACT experiments. The perpendicular diameters of the tumors were subsequently measured with a caliper. Mice were sacrificed when a tumor diameter reached a value >2 cm. The estimation of tumor volume was done using the modified ellipsoid formula 1/2(Length × Width2). For characterization experiments, B16-OVA tumor bearing mice (expressing CD45.2 allele) were treated with genetically modified CD8+ T cell (2*106) from OTI × CD45.1 mice and 7 days later, mice were sacrificed to analyze by flow cytometry the presence of transferred cells into the tumors.
Excised tumors were digested with 400 U/mL collagenase D and 50 μg/mL DNase-I (Roche) for 20 min at 37°C. After washing with PBS, red cells were lysed by ACK buffer (Sigma). Spleens were mashed in PBS. For functional analyses, cells were stimulated with PMA (50 ng/ml)/Ionomycin (1 μg/ml) and GolgiStop and GolgiPlug (BD Biosciences). After 5 h, cells were incubated with Zombie NIR Fixable dye (Biolegend). Subsequently, they were stained with fluorochrome-conjugated mAbs against CD45.1 (A20), CD8 (XMG1.4), PD-1 (29F.1A12), LAG3 (C9B7W), and TIGIT (1G9) in the presence of purified anti-CD16/32 mAb. For intracellular staining, cells were fixed and permeabilized with the BD Fixation/Perm buffer (BD Biosciences) and then stained with anti-IFN-γ (XMG1.2) and with anti-KI67 (16A8) mAbs. Samples were acquired on a FACSCanto-II cytometer (BD Biosciences). Data were analyzed using FlowJo software (TreeStar).
Statistical analyses were performed using parametric (Student's t test and one-way ANOVA, and two-tailed paired T-test), and non-parametric (Mann-Whitney U and Kruskal-Wallis) tests. Kaplan-Meier survival curves were evaluated for statistical significance with the Log-rank Mantel-Cox test. For all tests a p value <0.05 was considered statistically significant. Descriptive data for continuous variables are reported as means ±SEM. GraphPad software was used for statistical analysis.
We examined the expression of EGFR and EGFR ligands using Real-time PCR in murine tumor cell lines and confirmed the broad expression of EGFR in tumors from different origin. Of note, we found a high expression of EGFR in Hepa 129, 4T1, EG7-OVA, and MC38, as compared to EL4, CT26, B16, A20, or 5TGM1 (Figure 1A). Regarding the EGFR ligands, we found that EGF was the predominant EGFR ligand in lymphoma, hepatocarcinoma, colon carcinoma, melanoma, breast cancer, myeloma and reticulum cell sarcoma cell lines (Figure 1A). For the remaining EGFR ligands, there was some heterogeneity of expression, both in cell lines and tumor biopsies obtained from mice (Figures 1A,B). The levels of EGF protein present into the tumor microenvironment were also measured by ELISA using tumor cell extracts obtained from mice bearing B16-OVA, MC38, PM299L, or Hepa129 cell line derived tumors. Interestingly, MC38, PM299L, and Hepa129 derived tumor extracts presented significantly higher EGF levels than B16-OVA melanoma extracts (Figure 1C).
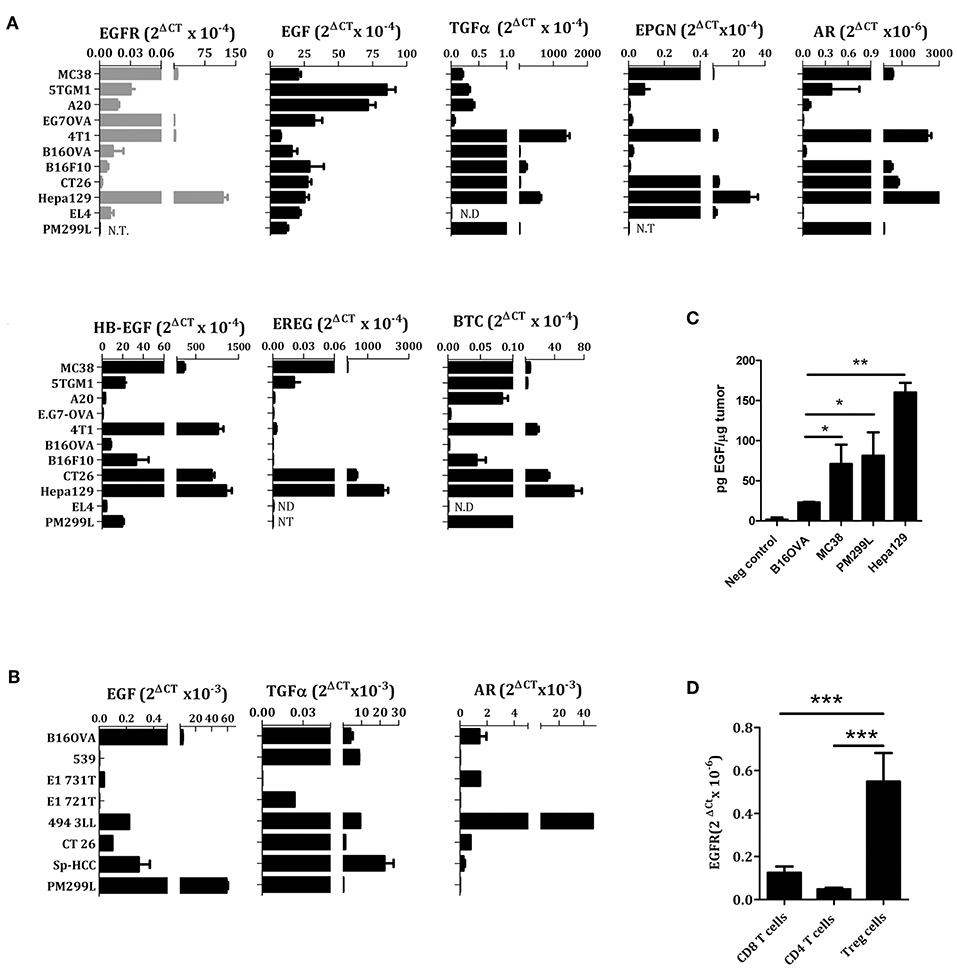
Figure 1. EGFR ligands and EGFR expression in different cell lines (A), tumor biopsies (B,C), and lymphocytes (D) analyzed by RT PCR. EL4, lymphoma; Hepa129 and PM299L, hepatocellular carcinoma; CT26, colon carcinoma; B16F10 and B16-OVA, melanoma; 4T1, breast cancer; EG7OVA, lymphoma; A20, reticulum cell sarcoma; 5TGM1, myeloma; MC38, colon carcinoma. (C) Amount of EGF in tumor cell extracts measured by ELISA, (D) EGFR expression in resting T cells. *p < 0.05; **p < 0.01; ***p < 0.005.
It has been described that conventional human CD4 or CD8+ T lymphocytes do not express EGFR. However, Treg cells can express EGFR and benefit from EGFR ligands such as amphiregulin (22, 24). We have confirmed these results of EGFR expression using conventional murine CD4+ and CD8+ T cells as well as CD4+CD25+ Treg cells (Figure 1D).
In order to modify genetically CD8+ T cells to respond to EGFR ligands, we generated a retrovirus for the simultaneous expression of EGFR and GFP using an IRES-containing bicistronic vector (RV-EGFR-GFP). As a control, we used a vector expressing only the GFP protein (RV-GFP). Infection of purified CD8+ T cells with RV-GFP vector was slightly more efficient (around 80% of transfection) than that achieved with RV-EGFR-GFP (around 65%). This difference is probably related to the size of EGFR gene. Indeed, although the cloning capacity of retroviral vectors is up to 10 kbps, the size of the transgene significantly influences its expression and viral titer (29). Also, the level of GFP expression was significantly lower in RV-EGFR-GFP than in RV-GFP as a result of the size of EGFR before the IRES sequence preceding GFP. Irrespective of these differences, in both cases (RV-EGFR-GFP and RV-GFP) the percentage of infected cells, measured by flow cytometry, was high (Figure 2A). Flow cytometry analysis using EGF labeled with APC showed that cells transduced with RV-EGFR-GFP expressed higher levels of EGFR than cells transduced with RV-GFP (Figure 2B).
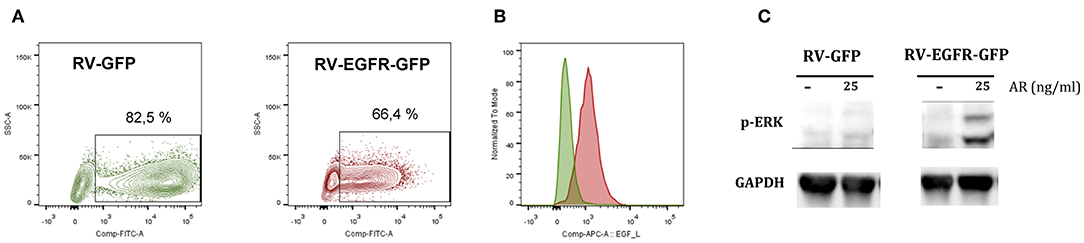
Figure 2. Genetically modified CD8 T cells express EGFR. (A) Percentage of CD8+ GFP+ cells after RV-GFP or RV_EGFR-GFP infection measured by flow cytometry. (B) EGFR expression on CD8 T cells transduced with RV-GFP and RV-EGFR-GFP cells, measured by flow cytometry using EGF-APC ligand. (C) Western blot analysis of phospho-ERK expression in CD8+ T cells transduced with RV-GFP and RV-EGFR-GFP. GAPDH was used as a loading control. Cells were left untreated or treated with 25 ng/ml Amphiregulin for 5 min. Results are representative of at least two experiments.
EGFR triggering activates the ras/raf/MEK/MAPK pathway, comprising the activation of extracellular signal-regulated kinase (ERK) and JUN N-terminal kinase [reviewed in (30)]. In order to verify the adequate signaling of the transduced EGFR, we performed a western blot assay in cell extracts from CD8+ T cells transfected with RV-GFP control virus and RV-EGFR-GFP to measure MAPK/ERK pathway. A band of 42-44 kDa, corresponding to the putative molecular weight of p-ERK, was found in CD8+ T cells transduced with RV-EGFR-GFP but not with RV-GFP in response to the EGFR ligand amphiregulin (AR) (Figure 2C), suggesting the functionality of EGFR in lymphocytes sensing exogenous EGFR ligands. GAPDH protein was measured by specific WB and used to normalize the results.
T Cell Receptor (TCR) activation promotes a number of signaling cascades that ultimately determine cell fate through regulating cytokine production, cell survival, proliferation, and differentiation. There is a potential degree of crosstalk between TCR and EGFR induced signaling cascades. We speculated that EGFR-induced signaling might allow CD8+ T cells to respond better to simultaneous TCR stimulation. Therefore, CD8+ T cells from OT-1 mice were retroviraly modified to express GFP (CD8-GFP) or EGFR and GFP (CD8-EGFR-GFP) and stimulated with SIINFEKL peptide at two different concentrations (suboptimal: 0.1 pg/ml or optimal: 0.1 ng/ml) in the presence or absence of 100 nM EGF. The number of IFN-γ and TNF-α producing cells was analyzed by flow cytometry. The addition of EGF in the absence of SIINFEKL stimulation induced only a slight increase in the number of IFN-γ or TNF-α producing cells when CD8 T cells were modified to express EGFR (Figure 3A). When SIINFEKL peptide was added at a high concentration (0.1 ng/ml), no clear improvements were observed by the addition of EGF. However, when SIINFEKL concentration was suboptimal (0.1 pg/ml), a very significant improvement in the number of IFN-γ or TNF-α producing cells was observed in CD8 T cells transduced with EGFR. This increase was also evident when the number of double positive IFN-γ and TNF-α producing cells was analyzed (pie charts in Figure 3A). We also analyzed the effect of different doses of EGF on the activity of T cells exposed to high (0.1 ng/ml) or low (0.1 pg/ml) doses of SIINFEKL peptide. As it is shown in Figure 3B, EGFR transduced OT-1 T cells did not respond efficiently to low doses (0.1 pg/ml) of SIINFEKL. However, addition of as low as 14 nM of EGF allowed the production of TNF-α in response to this peptide concentration. On the other hand, when SIINFEKL was added at high concentration (0.1 ng/ml) almost 80% of the EGFR transduced OT-1 T cells produced TNF-α and no beneficial effect was observed by the addition of EGF to the co-cultures. These results may suggest that EGFR signaling may synergize with TCR signaling when the latter is suboptimal.
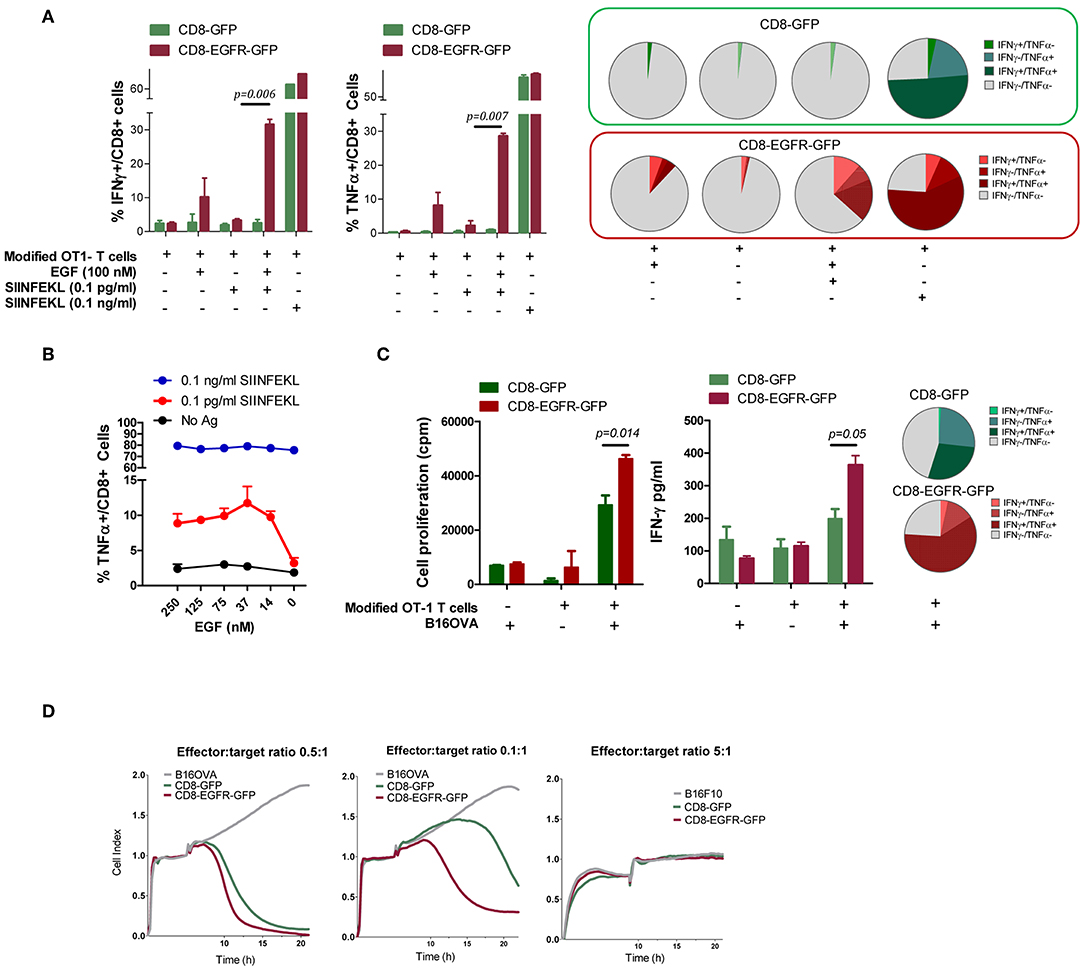
Figure 3. Effect of EGFR expression of T cell function. (A) Number of genetically modified OT-1 T cells producing IFN-γ and TNF-α after stimulation with SIINFEKL peptide in the presence/absence of EGF. Pie charts representing the percentages of T cells producing IFN-γ, TNF-α, or both cytokines. (B) Effect of different doses of EGF on CD8-EGFR-GFP T cell activation in response to high or low doses of SIINFEKL peptide. (C) T Cell proliferation and IFN-γ production of modified T cells in the presence or absence of irradiated B16-OVA cells, with a pie chart representing the percentages of T cells producing IFN-γ, TNF-α or both cytokines. (D) Capacity of modified CD8 T cells to recognize and lyse B16-OVA or B16F10 tumor cells, using the xCELLigence impedance-based system. Different T cell to tumor cell ratios were tested. Results are representative of at least two experiments.
Modified OT-1 T cells were also stimulated by co-culture with irradiated B16-OVA tumor cells and IFN-γ production and T cell proliferation was measured after 48 or 72 h, respectively. Interestingly, CD8-EGFR-GFP proliferate significantly better in response to B16-OVA cells and produce higher levels of IFN-γ than CD8-GFP transduced cells (Figure 3C). These differences were also observed in the number of double positive IFN-γ and TNF-α producing cells measured by flow cytometry, suggesting that EGFR signaling is favoring the activation of a polyfunctional immune response.
To evaluate a potential benefit that EGFR ligands produced by tumor cells might provide to modified T cells for tumor recognition, we examined the capacity of EGFR-transduced OT-1 CD8+ T cells to recognize and lyse tumor cells expressing ovalbumin. Using the xCELLigence impedance-based system we found that both CD8-GFP and CD8-EGFR-GFP T cells were able to recognize and kill B16-OVA cells in a similar way when the effector to target ratio was relatively high (0.5 effector T cells per 1 tumor cell) (Figure 3D, first graph). However, when the E:T ratio was five times lower (0.1 effector T cells per 1 tumor cell), CD8-EGFR-GFP T cells were clearly more efficient killing B16-OVA cells with respect to CD8-GFP cells (Figure 3D, second graph). This antitumor response was antigen-specific since neither CD8-EGFR-GFP or CD8-GFP were able to kill B16F10 tumor cells even at a high E:T ratio (5:1) (Figure 3D, third graph).
We wondered if EGFR-engineered CD8+ lymphocytes would exhibit an enhanced therapeutic effect in vivo in tumor-bearing mice. Thus, C57BL/6 mice were injected with B16-OVA cells and at day 7, when tumors reached 5 mm in diameter, 5 × 106 OT1 T cells modified with RV-EGFR-GFP or RV-GFP were injected i.v. into the mice. Although there was a trend to a higher reduction of tumor size immediately after the adoptive transfer of CD8-EGFR-GFP cells, compared to that found with CD8-GFP cells, this therapeutic effect was not statistically significant and it was lost soon after T cell transfer (Figures 4A,B). This lack of efficacy might be associated to the loss of OVA antigen expression in B16-OVA cells due to immune pressure, as it has been described previously (31). In a parallel experiment, we evaluated the level of expression of OVA antigen in tumors 10 days after adoptive transfer of 5 × 106 OT1 T cells. As it is shown in Figure 4C, OVA expression measured by RT-PCR was dramatically reduced (around 4 logs) in mice treated with OT-I T cells with respect to mice treated with saline (Figure 4C). These data suggest that immune pressure on the target B16.OVA tumor cells in vivo promoted the appearance of tumor cells characterized by loss target OVA antigen expression as it has been described previously (32–34).
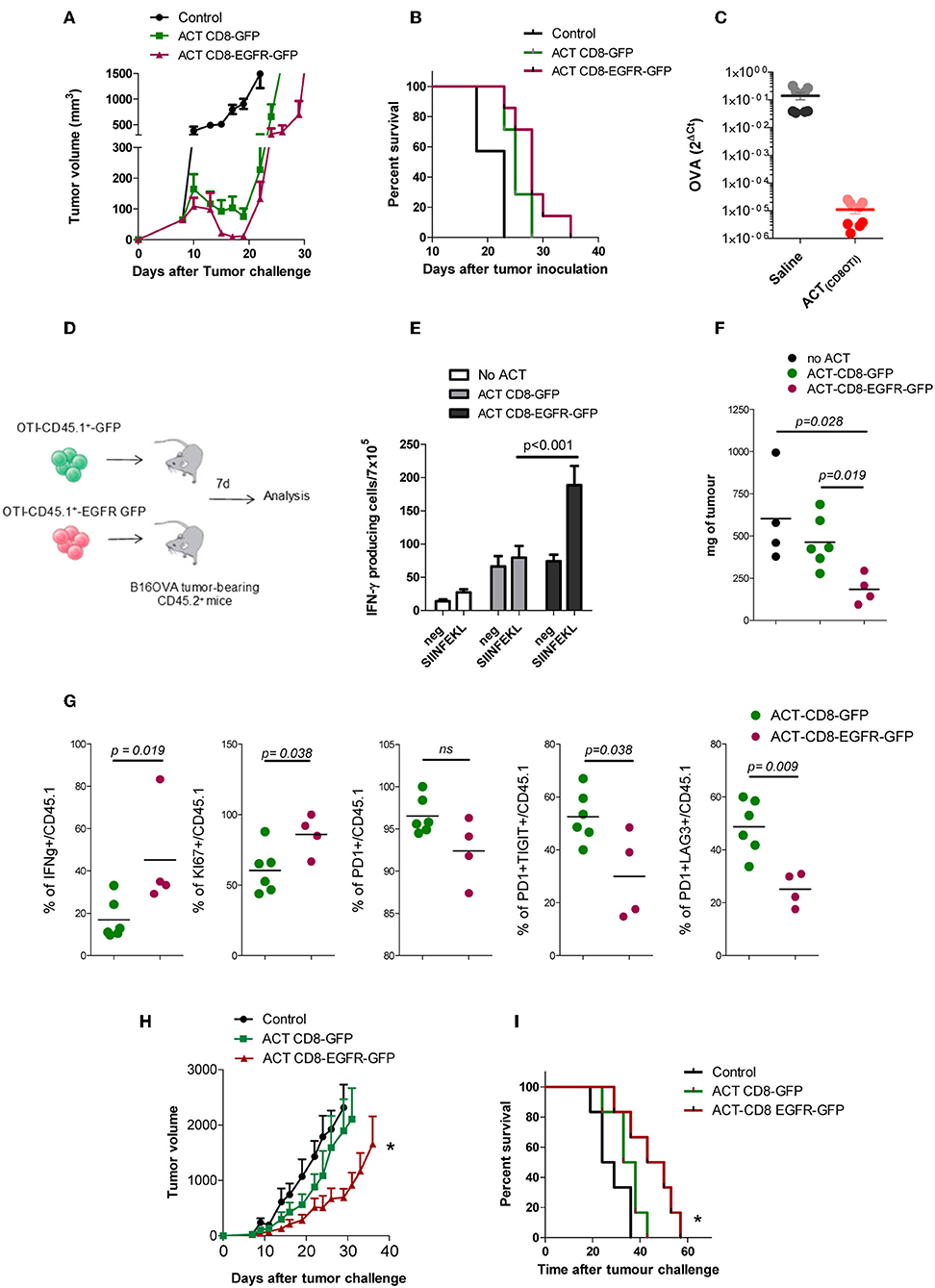
Figure 4. In vivo effect of EGFR expressing CD8+ T cells in vivo. (A) B16-OVA tumor growth after adoptive transfer of OT-1 modified T cells. (B) Kaplan–Meier plots of survival of mice bearing B16-OVA tumors. (C) Levels of OVA expression on tumor tissue isolated from B16.OVA bearing tumor mice 10 days after ACT with OT1-T cell or with saline (two mice per group, 4 experimental replicates per sample) (D) Schematic of experimental design. (E) ELISPOT analysis of IFN-γ producing cells in response to SIINFEKL peptide in splenocytes, 7 days after adoptive transfer of genetically modified CD8 OT1 T cells. (F) Tumor weight (in mg) 7 days after adoptive T cell therapy. (G) Functional and phenotypic analysis of tumor infiltrating CD45.1 T cells 7 days after adoptive T cell transfer. (H) PM299L tumor growth and (I) Kaplan Meyer survival plots after adoptive transfer of OT-1 modified T cells. Results are representative of at least two experiments. *p < 0.05.
To evaluate the behavior of the modified CD8+ T cells in vivo, we repeated the experiment of adoptive transfer but using CD8 T cells obtained from C56BL/6 mice expressing the CD45.1 allele. Thus, CD45.1 T cells were modified genetically to express GFP or EGFR-GFP and injected into CD45.2 mice bearing B16-OVA tumors (Figure 4D). Seven days after T cell infusion we analyzed by ELISPOT the capacity of T cells to produce IFN-γ in response to 1 μg/ml SIINFEKL stimulation. It was found that splenocytes from mice transferred with CD8-EGFR-GFP cells had a higher number of SIINFEKL-specific IFN-γ producing cells (Figure 4E). The size of the tumor at day 7 after T cell transfer was also significantly lower in mice transferred with CD8-EGFR-GFP cells (Figure 4F). The CD45.1 marker allowed us to analyse by flow cytometry the behavior of transferred T cells infiltrating the tumor. Although no significant changes in the number of CD8+ T cells infiltrating the tumor were observed (data not shown), we found a significantly higher percentage of CD45.1+ IFN-γ producing cells into the tumor of mice treated with CD8-EGFR-GFP as compared to that found in mice treated with control CD8-GFP T cells. The % of proliferating cells (KI67+ cells) was significantly higher in tumor infiltrating CD8-EGFR-GFP T cells and they appeared to be less dysfunctional. Indeed, a significantly lower percentage of PD1+TIGIT+ or PD1+LAG3+ CD8+ T cells was observed in tumors from mice injected with CD8-EGFR-GFP cells (Figure 4G) with respect to that found in tumors from mice transferred with CD8-GFP T cells.
We then studied if EGFR-engineered CD8+ lymphocytes would have a better therapeutic effect on mice bearing PM299L tumors, a tumor cell line derived from fresh hepatic tumors induced in C57BL/6 mice as previously described (35). PM299L tumors express SIINFEKL and grow very efficiently when they are injected s.c. in C57BL/6 mice. In this model, we found a significant delay in tumor growth (Figures 4H,I) and a statistically longer overall survival in mice adoptively transferred with CD8-EGFR-GFP T cells as compared to untreated mice or to mice receiving CD8-GFP T cells.
The efficacy of adoptive T cell therapy is highly compromised by the tumor microenvironment. It has been shown in preclinical models that, just 24 h after tumor-specific CD8+ T cell transfer, these cells become hypofunctional and express inhibitory receptor proteins that converge in cell cycle inhibition (36). Persistence and survival of the genetically modified T cells is considered a key factor contributing to the efficacy of ACT (37). We speculated that if adoptive transferred T cells could benefit from cytokines presented in the tumor microenvironment they could exert better anti-tumor responses. Among the potential alternatives we focussed on EGFR ligands.
EGFR utilizes a network of downstream signaling events leading to cell growth, proliferation, survival, angiogenesis, migration, and metastasis of many cancers. Its signaling can provide substantial advantage in tumor cell survival. We first analyzed the expression of EGFR and their ligands in a panel of tumor cell lines as well as tumor biopsies obtained from mice previously inoculated with the tumor cell lines. These experiments demonstrated that EGFR and its ligands are broadly expressed in murine tumors as occurred in humans (38, 39).
EGFR is ubiquitously expressed in normal tissues albeit at a much lower level than on tumors. But, in general, is thought to be absent in the hematopoietic cell linage, with the exception of some monocytes upon activation (20). Recently, several reports have shown that Treg cells express EGFR and enhance their immunosuppressive activity after stimulation with the EGFR ligand AR (22–24). In contrast, effectors T cells poorly express EGFR [(22), and confirmed in the present study], so that they do not benefit from the pro-proliferative and pro-survival signals provided by these ligands in the tumor microenvironment. In this scenario, the purpose of the current study was to modify genetically anti-tumor CD8+ cells to express EGFR to take advantage of the proliferative effect that EGFR ligands produced in the tumor microenvironment.
We have found that retrovirus RV-EGFR-GFP can efficiently modify CD8+ T cells to express EGFR and activate the MAPK/ERK pathway in response to EGFR ligands. It was also shown that T cells modified with RV-EGFR-GFP were able to respond to EGF improving their proliferation rate as well as the IFN-γ and TNF-α production capacity in response to a suboptimal TCR stimulation as compared to cells modified with the control RV-GFP. This improvement was not so clear when cells were treated with saturating concentration of antigen, probably because the induction of an optimal activation may make difficult to see any potential improvement induced by EGF. However, the TCR stimulation induced by tumors in vivo is by far, lower than the in vitro stimulation with saturating concentrations of SIINFEKL peptide used in our in vitro assays. Indeed, it has been reported that T cell stimulation in the tumor microenvironment is, in general, suboptimal (40). Similarly, CD8-EGFR GFP cells were able to kill more efficiently OVA expressing tumors than CD8-GFP cells when the E:T ratio was low. Thus, we believe that under the patho-physiological conditions present in many types of cancers (low E:T ratio, low antigen concentration, impaired antigen presentation and high immunosuppressive microenvironment), the presence of EGFR ligands in tumors might improve the effector functions of EGFR modified T cells.
ErbB ligands and receptors activate a complex signaling network including the ras/raf/MEK/ MAPK and the PI3K pathways and the activation of various transcription factors such as c-fos, c-Jun, c-myc, STAT, NF-kB, zinc finger transcription factor and Ets family members (41). Our in vitro experiments using antigen specific CD8+ T cells suggest that expression of EGFR on T cells might give a functional advantage in a tumor microenvironment containing EGFR ligands. EGFR-expressing effector T cells proliferate better and produce more IFN-γ and TNF-α in the presence of EGFR ligands produced by tumor cells in vitro and exerted stronger antitumor response delaying tumor growth in vivo. However, our results in vivo are not optimal, and the tumors still escape the action of the transferred T cells.
EGFR modified T cells could benefit from EGF ligands if their concentration into the tumors falls into the range of concentrations leading to augmented T cell activity. Although several works have estimated the concentration of EGFR ligands in human body fluids (42, 43) there are not clear data about their concentration in tumor tissues. We have quantified by ELISA the EGF levels in different tumors types isolated from mice previously inoculated with tumor cell lines. EGF levels ranges between 20 pg/μg tumor (in B16.OVA) to 160 pg/μg tumor (in Hepa129). A rough estimation using these data might suggest the presence of a tumor concentration of EGF 1 log below the concentration used in our in vitro assays. However, it is difficult to extrapolate these results to the in vivo setting since EGFR might respond not only to EGF but also to TGF-α, AR, EPGN, BTC, HB-EGF and EREG, which are also increased in tumor tissues (9–15). A recent study by Pinilla et al. has estimated a range of EGFR ligand concentrations in tumors as low as ~0.2 and ~0.6 ng/ml (~34 and 100 pM) (44). They have also shown that the levels of phosphorylation and ubiquitylation of EGFR in tumors in vivo closely resemble the levels observed in the same tumor cells treated with 20–200 pM EGF in vitro. The authors conclude that a small pool of active EGFRs is sufficient to activate efficiently EGFR signaling. Considering these results, we might guess that the retrovirus-transduced CD8 T cells to express EGFR could benefit from EGFR ligand concentrations found in the tumor microenvironment.
Tumors can escape from T cell attack in a variety of ways. They can promote T cell exhaustion, the expansion of suppressive cells, or become resistant to CTL killing by downregulating MHC-I expression or by losing the target antigen. In agreement with previous reports (31–33) we observed that in vivo ACT rapidly promoted the emergence of tumor cells negative for OVA antigen suggesting that potent immunotherapies can actively promote tumor evolution. Loss of tumor antigen expression in our models might have an impact in the anti-tumor activity of EGFR expressing T cells. But, in addition to this phenomenon, there are other barriers in the tumor microenvironment that need to be overcome for a better antitumor efficacy of adoptive cell therapy.
The potential effect of EGFR and its ligands on the immune system is still poorly understood. Indeed, recent reports suggest that EGFR expression by leukocytes in the functioning of the immune system might be underestimated (26, 45). Cancer patients treated with EGFR antagonists suffer not only from a wide range of side effects caused by loss of EGFR function in epithelial cells but also become more susceptible to infections (46) probably due to effects on the immune system (47). Our results suggest that the genetic modification of CD8+ T cells to express EGFR might endow them with a proliferative advantage to fight against tumors expressing EGFR ligands (Figure 5). EGFR expressing T cells could also compete for EGFR ligands with tumor cells and Treg cells as has been described for soluble EGFR (48), and thus might also have a positive effect on tumor control. Although further experiments are needed to elucidate the potential crosstalk between different signaling cascades and the final outcome of the modified T cells into the tumor microenvironment, our results suggest a potential benefit of EGFR expression on T cells that should be considered in immunotherapeutic strategies based on adoptive transfer with anti-tumor T cells.
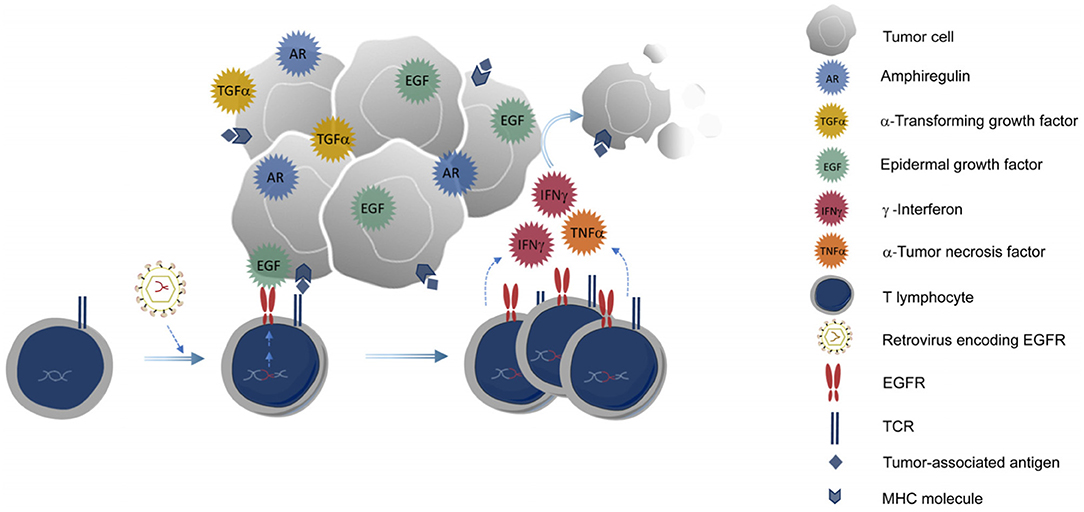
Figure 5. Genetic modification of CD8+ T cells to express EGFR. T cells expressing EGFR could benefit from EGFR ligands produced by the tumor, proliferate better, and exert their anti-tumor activities more efficiently into the tumor microenvironment.
All datasets generated for this study are included in the article/supplementary material.
Animals were housed in the accredited animal facility of the Center for Applied Medical Research, CIMA, and animal experimentations were performed after approval by and according to the guidelines of the Animal Experimentation Ethical Committee of the University of Navarra and The Local Authority for the use of laboratory animals (protocol R-131-16GN).
TL, SC, and NC performed most of the experiments. JL and NC conceived the study and participated in the design and interpretation of results. CV, MG, MR, PSarr, PSaro, AL-C, CM, AL, SH-S, and JL participated in the experiments. JL wrote the first draft and NC, TL, SC, SH-S, and PSaro participated in the manuscript writing. All authors read and approved the final manuscript.
This study was supported by grants from Ministerio de Educación y Ciencia (SAF2016-78568-R, SAF2019-08971-R to JL), Fundación Ramón Areces, Gobierno de Navarra Industria (0011-1411-2019-000079; Proyecto DESCARTHeS), and Fundación Bancaria La Caixa-Hepacare Project.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer CA declared a shared affiliation, with no collaboration, with several of the authors JL, TL, SC, CV, MG, CM, AL-C, SH-S, PSaro, PSarr, and NC to the handling editor at the time of the review.
We thank Elena Ciordia and Eneko Elizalde for excellent animal care and Drs. Uxue. Latasa, Carmen Berasain, and Matías Ávila for helpful discussion. We also thank the help from the Murchante contra el cáncer initiative.
1. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. (2015) 348:62–8. doi: 10.1126/science.aaa4967
2. Yee C. Adoptive T cell therapy: addressing challenges in cancer immunotherapy. J Transl Med. (2005) 3:17. doi: 10.1186/1479-5876-3-17
3. Lo AS, Taylor JR, Farzaneh F, Kemeny DM, Dibb NJ, Maher J. Harnessing the tumour-derived cytokine, CSF-1, to co-stimulate T-cell growth and activation. Mol Immunol. (2008) 45:1276–87. doi: 10.1016/j.molimm.2007.09.010
4. Wilkie S, Burbridge SE, Chiapero-Stanke L, Pereira AC, Cleary S, Van Der Stegen SJ, et al. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J Biol Chem. (2010) 285:25538–44. doi: 10.1074/jbc.M110.127951
5. Leen AM, Sukumaran S, Watanabe N, Mohammed S, Keirnan J, Yanagisawa R, et al. Reversal of tumor immune inhibition using a chimeric cytokine receptor. Mol Ther. (2014) 22:1211–20. doi: 10.1038/mt.2014.47
6. Wang Y, Jiang H, Luo H, Sun Y, Shi B, Sun R, et al. An IL-4/21 inverted cytokine receptor improving CAR-T cell potency in immunosuppressive solid-tumor microenvironment. Front Immunol. (2019) 10:1691. doi: 10.3389/fimmu.2019.01691
7. Hollstein MC, Smits AM, Galiana C, Yamasaki H, Bos JL, Mandard A, et al. Amplification of epidermal growth factor receptor gene but no evidence of ras mutations in primary human esophageal cancers. Cancer Res. (1988) 48:5119–23.
8. Yasui W, Sumiyoshi H, Hata J, Kameda T, Ochiai A, Ito H, et al. Expression of epidermal growth factor receptor in human gastric and colonic carcinomas. Cancer Res. (1988) 48:137–41.
9. Castillo J, Erroba E, Perugorria MJ, Santamaria M, Lee DC, Prieto J, et al. Amphiregulin contributes to the transformed phenotype of human hepatocellular carcinoma cells. Cancer Res. (2006) 66:6129–38. doi: 10.1158/0008-5472.CAN-06-0404
10. Moon WS, Park HS, Yu KH, Park MY, Kim KR, Jang KY, et al. Expression of betacellulin and epidermal growth factor receptor in hepatocellular carcinoma: implications for angiogenesis. Hum Pathol. (2006) 37:1324–32. doi: 10.1016/j.humpath.2006.04.022
11. Gao J, Ulekleiv CH, Halstensen TS. Epidermal growth factor. (EGF) receptor-ligand based molecular staging predicts prognosis in head and neck squamous cell carcinoma partly due to deregulated EGF- induced amphiregulin expression. J Exp Clin Cancer Res. (2016) 35:151. doi: 10.1186/s13046-016-0422-z
12. Rusch V, Klimstra D, Venkatraman E, Pisters PW, Langenfeld J, Dmitrovsky E. Overexpression of the epidermal growth factor receptor and its ligand transforming growth factor alpha is frequent in resectable non-small cell lung cancer but does not predict tumor progression. Clin Cancer Res. (1997) 3:515–22.
13. Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol. (2007) 25:3230–7. doi: 10.1200/JCO.2006.10.5437
14. Seligmann JF, Elliott F, Richman SD, Jacobs B, Hemmings G, Brown S, et al. Combined epiregulin and amphiregulin expression levels as a predictive biomarker for panitumumab therapy benefit or lack of benefit in patients with RAS wild-type advanced colorectal cancer. JAMA Oncol. (2016) 2:633–42. doi: 10.1001/jamaoncol.2015.6065
15. Sagmeister S, Drucker C, Losert A, Grusch M, Daryabeigi A, Parzefall W, et al. HB-EGF is a paracrine growth stimulator for early tumor prestages in inflammation-associated hepatocarcinogenesis. J Hepatol. (2008) 49:955–64. doi: 10.1016/j.jhep.2008.06.031
16. Kwapiszewski R, Pawlak SD, Adamkiewicz K. Anti-EGFR agents: current status, forecasts and future directions. Target Oncol. (2016) 11:739–52. doi: 10.1007/s11523-016-0456-3
17. Schneider MR, Wolf E. The epidermal growth factor receptor ligands at a glance. J Cell Physiol. (2009) 218:460–6. doi: 10.1002/jcp.21635
18. Blobel CP. ADAMs: key components in EGFR signalling and development. Nat Rev Mol Cell Biol. (2005) 6:32–43. doi: 10.1038/nrm1548
19. Blobel CP, Carpenter G, Freeman M. The role of protease activity in ErbB biology. Exp Cell Res. (2009) 315:671–82. doi: 10.1016/j.yexcr.2008.10.011
20. Chan G, Nogalski MT, Yurochko AD. Activation of EGFR on monocytes is required for human cytomegalovirus entry and mediates cellular motility. Proc Natl Acad Sci USA. (2009) 106:22369–74. doi: 10.1073/pnas.0908787106
21. Mahtouk K, Hose D, Reme T, De Vos J, Jourdan M, Moreaux J, et al. Expression of EGF-family receptors and amphiregulin in multiple myeloma. Amphiregulin is a growth factor for myeloma cells. Oncogene. (2005) 24:3512–24. doi: 10.1038/sj.onc.1208536
22. Zaiss DM, Van Loosdregt J, Gorlani A, Bekker CP, Grone A, Sibilia M, et al. Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity. (2013) 38:275–84. doi: 10.1016/j.immuni.2012.09.023
23. Dai K, Huang L, Chen J, Yang L, Gong Z. Amphiregulin promotes the immunosuppressive activity of intrahepatic CD4(+) regulatory T cells to impair CD8(+) T-cell immunity against hepatitis B virus infection. Immunology. (2015) 144:506–17. doi: 10.1111/imm.12400
24. Wang S, Zhang Y, Wang Y, Ye P, Li J, Li H, et al. Amphiregulin confers regulatory t cell suppressive function and tumor invasion via the EGFR/GSK-3β/Foxp3 axis. J Biol Chem. (2016) 291:21085–95. doi: 10.1074/jbc.M116.717892
25. Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DM, Artis D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc Natl Acad Sci USA. (2015) 112:10762–7. doi: 10.1073/pnas.1509070112
26. Minutti CM, Drube S, Blair N, Schwartz C, Mccrae JC, Mckenzie AN, et al. Epidermal growth factor receptor expression licenses type-2 helper T cells to function in a T cell receptor-independent fashion. Immunity. (2017) 47:e716. doi: 10.1016/j.immuni.2017.09.013
27. Berasain C, Nicou A, Garcia-Irigoyen O, Latasa MU, Urtasun R, Elizalde M, et al. Epidermal growth factor receptor signaling in hepatocellular carcinoma: inflammatory activation and a new intracellular regulatory mechanism. Dig Dis. (2012) 30:524–31. doi: 10.1159/000341705
28. Casares N, Rudilla F, Arribillaga L, Llopiz D, Riezu-Boj JI, Lozano T, et al. A peptide inhibitor of FOXP3 impairs regulatory T cell activity and improves vaccine efficacy in mice. J Immunol. (2010) 185:5150–9. doi: 10.4049/jimmunol.1001114
29. Maetzig T, Galla M, Baum C, Schambach A. Gammaretroviral vectors: biology, technology and application. Viruses. (2011) 3:677–713. doi: 10.3390/v3060677
30. Berasain C, Avila MA. The EGFR signalling system in the liver: from hepatoprotection to hepatocarcinogenesis. J Gastroenterol. (2014) 49:9–23. doi: 10.1007/s00535-013-0907-x
31. Kaluza KM, Kottke T, Diaz RM, Rommelfanger D, Thompson J, Vile R. Adoptive transfer of cytotoxic T lymphocytes targeting two different antigens limits antigen loss and tumor escape. Hum Gene Ther. (2012) 23:1054–64. doi: 10.1089/hum.2012.030
32. Sanchez-Perez L, Kottke T, Diaz RM, Ahmed A, Thompson J, Chong H, et al. Potent selection of antigen loss variants of B16 melanoma following inflammatory killing of melanocytes in vivo. Cancer Res. (2005) 65:2009–17. doi: 10.1158/0008-5472.CAN-04-3216
33. Bai XF, Liu JQ, Joshi PS, Wang L, Yin L, Labanowska J, et al. Different lineages of P1A-expressing cancer cells use divergent modes of immune evasion for T-cell adoptive therapy. Cancer Res. (2006) 66:8241–9. doi: 10.1158/0008-5472.CAN-06-0279
34. Goldberger O, Volovitz I, Machlenkin A, Vadai E, Tzehoval E, Eisenbach L. Exuberated numbers of tumor-specific T cells result in tumor escape. Cancer Res. (2008) 68:3450–7. doi: 10.1158/0008-5472.CAN-07-5006
35. Ruiz De Galarreta M, Bresnahan E, Molina-Sanchez P, Lindblad KE, Maier B, Sia D, et al. β-catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov. (2019) 9:1124–41. doi: 10.1158/2159-8290.CD-19-0074
36. Waugh KA, Leach SM, Moore BL, Bruno TC, Buhrman JD, Slansky JE. Molecular profile of tumor-specific CD8+ T cell hypofunction in a transplantable murine cancer model. J Immunol. (2016) 197:1477–88. doi: 10.4049/jimmunol.1600589
37. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. (2011) 17:4550–7. doi: 10.1158/1078-0432.CCR-11-0116
38. Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol. (1995) 19:183–232. doi: 10.1016/1040-8428(94)00144-I
39. Normanno N, Bianco C, De Luca A, Maiello MR, Salomon DS. Target-based agents against ErbB receptors and their ligands: a novel approach to cancer treatment. Endocr Relat Cancer. (2003) 10:1–21. doi: 10.1677/erc.0.0100001
40. Anderson KG, Stromnes IM, Greenberg PD. Obstacles posed by the tumor microenvironment to T cell activity: a case for synergistic therapies. Cancer Cell. (2017) 31:311–25. doi: 10.1016/j.ccell.2017.02.008
41. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. (2001) 2:127–37. doi: 10.1038/35052073
42. Connolly JM, Rose DP. Epidermal growth factor-like proteins in breast fluid and human milk. Life Sci. (1988) 42:1751–6. doi: 10.1016/0024-3205(88)90041-0
43. Rich T, Zhao F, Cruciani RA, Cella D, Manola J, Fisch MJ. Association of fatigue and depression with circulating levels of proinflammatory cytokines and epidermal growth factor receptor ligands: a correlative study of a placebo-controlled fatigue trial. Cancer Manag Res. (2017) 9:1–10. doi: 10.2147/CMAR.S115835
44. Pinilla-Macua I, Grassart A, Duvvuri U, Watkins SC, Sorkin A. EGF receptor signaling, phosphorylation, ubiquitylation and endocytosis in tumors in vivo. Elife. (2017) 6:e31993. doi: 10.7554/eLife.31993.020
45. Zeboudj L, Maitre M, Guyonnet L, Laurans L, Joffre J, Lemarie J, et al. Selective EGF-receptor inhibition in CD4(+) T cells induces anergy and limits atherosclerosis. J Am Coll Cardiol. (2018) 71:160–72. doi: 10.1016/j.jacc.2017.10.084
46. Yamashita M, Chattopadhyay S, Fensterl V, Saikia P, Wetzel JL, Sen GC. Epidermal growth factor receptor is essential for Toll-like receptor 3 signaling. Sci Signal. (2012) 5:ra50. doi: 10.1126/scisignal.2002581
47. Ott PA, Adams S. Small-molecule protein kinase inhibitors and their effects on the immune system: implications for cancer treatment. Immunotherapy. (2011) 3:213–27. doi: 10.2217/imt.10.99
Keywords: epidermal growth factor receptor EGFR, EGFR ligands, adoptive cell therapy, genetic modification, CD8+ T cells, tumor microenvironment, hepatocellular carcinoma
Citation: Lozano T, Chocarro S, Martin C, Lasarte-Cia A, del Valle C, Gorraiz M, Sarrión P, Ruiz de Galarreta M, Lujambio A, Hervás-Stubbs S, Sarobe P, Casares N and Lasarte JJ (2019) Genetic Modification of CD8+ T Cells to Express EGFR: Potential Application for Adoptive T Cell Therapies. Front. Immunol. 10:2990. doi: 10.3389/fimmu.2019.02990
Received: 28 August 2019; Accepted: 05 December 2019;
Published: 20 December 2019.
Edited by:
Giovanna Schiavoni, Istituto Superiore di Sanità (ISS), ItalyReviewed by:
Chantale Bernatchez, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2019 Lozano, Chocarro, Martin, Lasarte-Cia, del Valle, Gorraiz, Sarrión, Ruiz de Galarreta, Lujambio, Hervás-Stubbs, Sarobe, Casares and Lasarte. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Noelia Casares, bmNhc2FyZXNAdW5hdi5lcw==; Juan J. Lasarte, ampsYXNhcnRlQHVuYXYuZXM=
†These authors have contributed equally to this work and share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.