 Elisa Lahtela
Elisa Lahtela Matti Kankainen
Matti Kankainen Juha Sinisalo
Juha Sinisalo Olof Selroos
Olof Selroos Marja-Liisa Lokki
Marja-Liisa Lokki- 1Transplantation Laboratory, Department of Pathology, University of Helsinki, Helsinki, Finland
- 2Institute for Molecular Medicine Finland, University of Helsinki, Helsinki, Finland
- 3Medical and Clinical Genetics, Helsinki University Hospital, University of Hesinki, Helsinki, Finland
- 4Heart and Lung Center, University of Helsinki and Helsinki University Hospital, Helsinki, Finland
- 5University of Helsinki, Helsinki, Finland
Many sarcoidosis-associating immunological genes have been shown to be shared between other immune-mediated diseases. In Finnish sarcoidosis patients, good prognosis subjects more commonly have HLA-DRB1*03:01 and/or HLA-DRB1*04:01-DPB1*04:01 haplotype, but no marker for persistent disease have been found. The objective was to further pinpoint genetic differences between prognosis subgroups in relation to the HLA markers. Whole-exome sequencing was conducted for 72 patients selected based on disease activity (resolved disease, n = 36; persistent disease, n = 36). Both groups were further divided by the HLA markers (one/both markers, n = 18; neither of the markers, n = 18). The Finnish exome data from the Genome Aggregation Database was used as a control population in the WES sample. Statistical analyses included single-variant analysis for common variants and gene level analysis for rare variants. We attempted to replicate associated variants in 181 Finnish sarcoidosis patients and 150 controls. An association was found in chromosome 1p36.21 (AADACL3 and C1orf158), which has recently been associated with sarcoidosis in another WES study. In our study, variations in these genes were associated with resolved disease (AADACL3, p = 0.0001 and p = 0.0003; C1orf158, p = 7.03E-05). Another interesting chromosomal region also peaked, Leucocyte Receptor Complex in 19q13.42, but the association diminished in the replication sample. In conclusion, this WES study supports the previously found association in the region 1p36.21. Furthermore, a novel to sarcoidosis region was found, but additional studies are warranted to verify this association.
Introduction
Sarcoidosis is granulomatous disease characterized by the presence of non-caseating granulomas in affected organs. Sarcoidosis can affect any organ in the body and the disease course can be self-limited or chronic (1). These different outcomes have led to the classification of sarcoidosis based on prognosis: resolved disease, i.e., duration of disease <2 years and persistent disease with a longer duration (≥2 years) (2).
Exact etiology of sarcoidosis is still unclear. The heterogeneity of sarcoidosis in clinical course and organ involvement has led to the hypothesis that sarcoidosis might not be one disease, but consists of several disease entities, each with distinct genetic associations (3). This may explain why no single sarcoidosis-associated variant has been found, but a wide range of genes with relatively small effects. With a current knowledge it seems, that the genetic susceptibility to sarcoidosis is mainly affected by the genetic variation in the genes and pathways related to granuloma formation and immune response. The MHC region in chromosome 6p21.3 contains multiple genes essential for the immune system. The association for sarcoidosis in the MHC have been found in all classes through I to III (4, 5). In our previous studies in Finnish patients, an association between class II HLA-DRB1*03:01 and the haplotype consisting of HLA-DRB1*04:01-DPB1*04:01 and good prognosis compared to poor prognosis was found (44.9 vs. 22.7%; p = 0.001; OR = 2.78; 95% CI = 1.45–5.24) (6, 7).
Besides the MHC, other susceptible sarcoidosis risk/protective chromosome regions and genes have been found throughout the genome. Beside classical candidate-gene approaches, genome-wide association analyses (GWAS) have become method of choice nowadays. However, as sarcoidosis is a rare disease, its prevalence in Finland being 28 per 100,000 (8), collections of large case-control materials for GWAS are demanding. A possible new method for finding causality in genetics behind sarcoidosis is whole-exome sequencing (WES). The exome sequences encompass only about 2% of the human genome but harbors about 85% of all described disease-causing variants (9), making smaller sample sizes sufficient for identification of novel genes.
Aim of this study was to further characterize genetic differences between Finnish sarcoidosis prognosis utilizing whole-exome sequencing method in the subset of 72 patients and to replicate the findings in a bigger data set of 188 Finnish patients. In Finnish patients, subjects with good prognosis are more likely to have above mentioned class II HLA markers, but no genetic markers have been found for persistent disease. The objective was to further pinpoint genetic variety of sarcoidosis prognosis in relation to the HLA markers.
Materials and Methods
Study Subjects and Selection Criteria
Study subjects and their characteristics have been previously described (7). In summary, a total of 188 Finnish patients with verified pulmonary sarcoidosis had been followed-up for 5–15 years (Supplementary Table 1). After follow-up the patients were clinically divided into those with disease resolved within 2 years (n = 90) and to those with persisting activity after 2 years (n = 98). Disease activity was evaluated using the generally accepted WASOG (World Association of Sarcoidosis and Other Granulomatous diseases) criteria (10). The clinical examinations included a chest radiograph, a lung function test (spirometry, diffusion capacity), electrocardiography (ECG), liver enzymes, serum calcium, creatinine, serum lysozyme, and serum ACE.
For the WES study, a subset of 72 patients were chosen (Figure 1). Patients were selected based on disease activity (resolved disease, n = 36; persistent disease, n = 36). These subsets were further divided by the HLA markers previously known to influence disease prognosis in Finnish patients (HLA-DRB1*03:01 and HLA-DRB1*04:01-DPB1*04:01) yielding in 18 patients with one or both markers (HLA+) and 18 patients with neither of the markers (HLA–). Same selection was used in both resolved and persistent patient groups. The Genome Aggregation Database (gnomAD) (11) was used as control population in the WES study. GnomAD database consists of exome data from 11 150 Finnish subjects, with no known diseases.
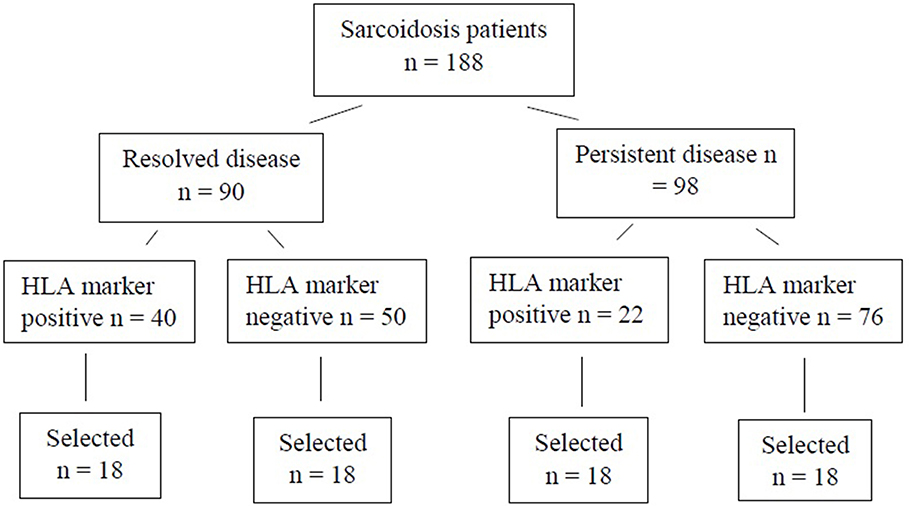
Figure 1. Finnish sarcoidosis patients. Selection criteria based on sarcoidosis prognosis and the presence or absence of the HLA markers (HLA-DRB1*03:01 and/or HLA-DRB1*04:01-DPB1*04:01 haplotype). From each of the HLA positive/negative groups, 18 samples were randomly selected for this study.
For the replication of the SNPs found in the WES study, the rest of the original above mentioned 188 Finnish sarcoidosis patients were genotyped. A control population of 150 healthy subjects representing the Finnish population (12) was included for the replication phase.
The study protocol was approved by the Ethics Committee of the Department of Internal Medicine, Hospital District of Helsinki and Uusimaa, Helsinki, Finland. All subjects provided written informed consent for their participation in genetic association studies.
Whole Exome Sequencing
The DNA extraction have been previously explained (7). The WES was performed in Institute for Molecular Medicine Finland (FIMM). 150 ng of gDNA was fragmented with Covaris E220 evolution instrument (Covaris, Woburn, MA, USA). Sample libraries and MedExome Enrichment were processed according to SeqCapEZ HyperCap Workflow User's Guide (Roche Nimblegen, Madison, WI, USA). Enrichment was performed in 4 samples Multiplexed DNA Sample Library Pools using 1 μg of each library. The amplified library was purified with 1.8x Agencourt AMPure XP beads and eluted to 200 μl EB-buffer. Library was quantified for sequencing using 2100 Bioanalyzer High sensitivity kit. Sequencing was performed with Illumina HiSeq2500 system in HiSeq High Output mode using v4 kits (Illumina, San Diego, CA, USA). Read length for the paired-end run was 2 × 101 bp. 91% of all samples had the target coverage of 20x.
Variant Calling
Sequencing data were pre-processed using Trimmomatic (leading:3, trailing:3, sliding window:4:15, illuminaclip:2:30:10, minlen:36). Paired-end reads passing the filtering were mapped to the reference genome (GRCh38) using the BWA-MEM algorithm. Reads were sorted by coordinate and duplicates were marked with the Picard tools (http://broadinstitute.github.io/picard/). Base qualities were recalibrated and local indel realignment performed around indels at the BAM-level using Genome Analysis Toolkit (GATK) followed by merging of data by sample and remarking of duplicates with the Picard tools. Joint genotyping was performed on the whole cohort of 72 samples using GATK HaplotypeCaller and variants were recalibrated using GATK VariantRecalibrator. GATK tools were applied as recommended by GATK guidelines and using GATK resource files that had been converted from GRCh37 to GRCh38 using CrossMap and chain files from EnsEMBL. Quality control analysis of sequencing data was performed using the FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) with default settings. Additionally, frameshift indels were excluded due to recognized difficulties calling from WES data (13).
Annotation and filtering of variants was performed using the Annovar tool against the RefGene database. Filtering quality control was as follows: coverage >10, variant quality value >40, frequency <1%, and assumed to impair protein function. The mean coverage of exomic regions for each variant was 43X with individual variant reads varying between 17 and 153X. Reads were manually checked using Integrative Genomics Viewer (IGV). Variants with unknown frequency in Finnish population in the gnomAD database were excluded. Version information and references of tools used in variant calling are given in Supplementary Table 2.
Testing for Genetic Association
We analyzed associations for single variants and variant groups within genes. The associations were evaluated between all the dichotomous groups (persistent vs. resolved disease, HLA+ persistent vs. resolved disease, and HLA- persistent vs. resolved disease). For single-variant analysis, the common variants [minor allele frequency (MAF) > 0.01] were included. The hypergeometric distribution method in EPACTS software pipeline (https://genome.sph.umich.edu/wiki/EPACTS) was used. The gene-based tests were assessed to find whether variants in each gene are jointly associated with the phenotype. Gene-based association improves the power to detect rare variants (MAF < 0.05) in limited sample size. Two methods were used to test the associations: hypergeometric distribution and SKAT-O (14) methods implemented in the EPACTS software. SKAT-O test was used, because it assumes that each variant can have either positive or negative effect to the phenotype. The p were corrected for multiple comparisons (number of studied variants) by using False Discovery Rate (FDR) method and Bonferroni correction. A value of p < 0.05 was considered statistically significant. Age, gender, and extrapulmonary manifestations were used as covariates in all statistical tests. To assess the functionality of the found variations in protein level, we used Sift (15) and PolyPhen (16) databases, which both predict possible impact of an amino acid substitution on the structure and function of a human protein using straightforward physical and comparative considerations. To further investigate the possibly functional effects of the significant SNPs, we used the Genotype-Tissue Expression (GTEx) Portal (17) to study the expression quantitative trait loci (eQTL).
For the replication, the SNPs that reached the significant association level in single-variant and gene-based analysis were included (Supplementary Table 3). The SNP genotyping was performed with the Agena Bioscience (Sequenom) MassARRAY System (Agena Biosciences, San Diego, California) at the Institute for Molecular Medicine Finland (FIMM), Helsinki, Finland, with standard protocols. Genotypes were called using Sequenom's MassARRAY Typer software. The allele frequencies between different groups were compared by a case-control association analysis (Chi-square χ2 test, PLINK software) (18). We applied the following quality control filters: minimum call rate per sample of 90%, SNP minor allele frequency (MAF) > 0.01 and Hardy Weinberg equilibrium (HWE) > 0.001. Total success rate for accepted SNP arrays was 95% in the replication samples.
Results
Single-Variant Analysis
Figure 2 shows the Manhattan plot from the single-variant association tests between resolved and persistent patients showing the highest associating peak in the chromosome 19, although not reaching the exome-significance level. Supplementary Figure 1 shows the quantile-quantile (Q-Q) plot for the single-variant tests, which demonstrates the similarity between observed and expected significance values. Table 1 shows the single variants that yielded the strongest associations in analyses between resolved and persistent, and between HLA+ and HLA– prognosis groups. In analysis between resolved and persistent patients altogether seven variants associated with the prognosis. Interestingly, five of these variants were located in two chromosomal regions: three in chromosome 1 (1p36.21) and two in chromosome 19 (19q13.42). In HLA+ group, the associations in these two chromosomal locations remained. In HLA- patients the chromosomal locations 1p36.21 and 19q13.42 did not associate with the prognosis. None of these associations remained significant after correcting for multiple testing with FDR or Bonferroni correction.
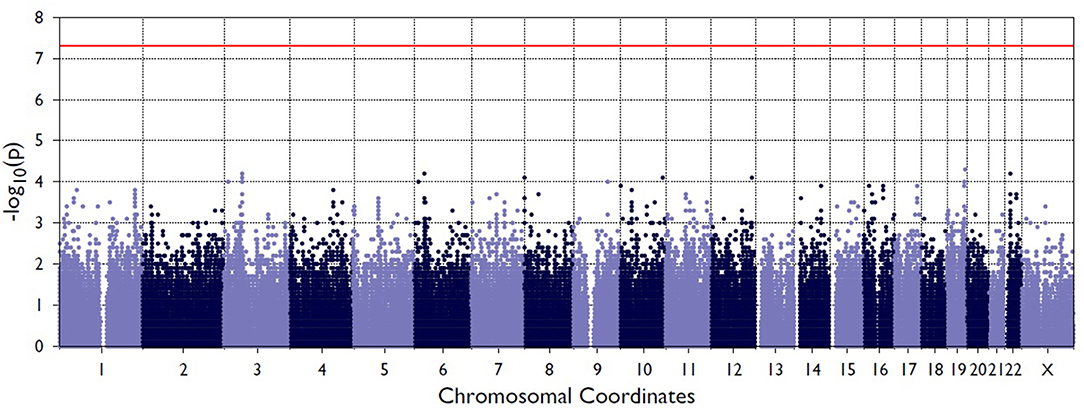
Figure 2. Manhattan plot from the single-variant association tests between persistent and resolved patients. The X-axis shows chromosome position the Y-axis shows the negative log of p-values so that higher values represent stronger significance levels. The horizontal red line shows the threshold value for significance.
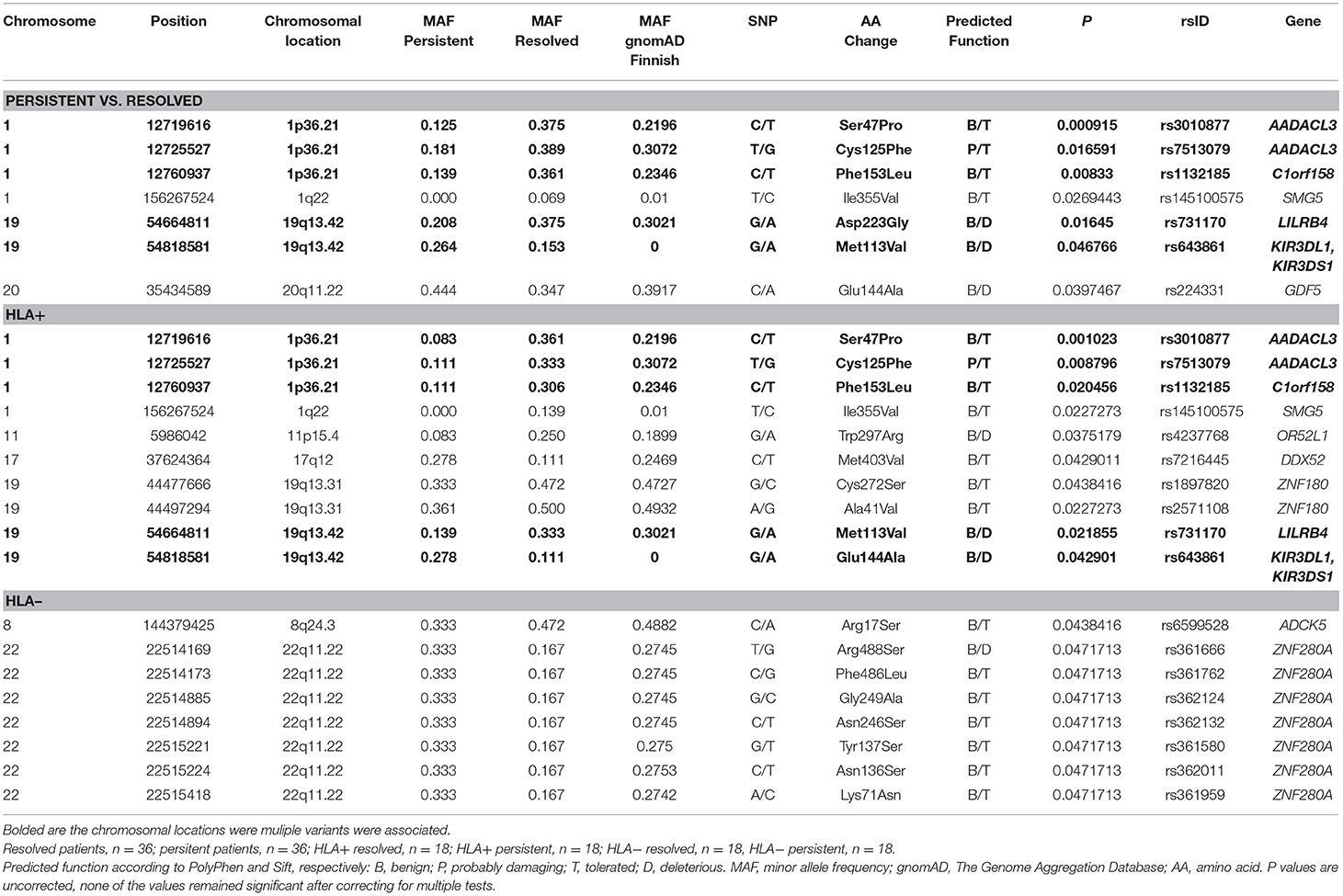
Table 1. Associated variations in single-variant analysis in different Finnish sarcoidosis subgroups based on disease prognosis (persistent, resolved) and HLA markers (HLA+ = HLA–DRB1*03:01 and/or HLA-DRB1*04:01-DPB1*04:01; HLA– = without either of the markers).
Gene-Based Analysis
In gene-based tests, all variants found in the same gene are grouped to assess the association with the phenotype. Also, in gene-based analysis the locations 1p36.21 and 19q13.42 showed associations while not remained significant after correcting with FDR or Bonferroni correction (Table 2). In location 19q13.42, variants in gene LAIR1 were also significantly associated with the prognosis, in addition to previously seen LILRB4 and KIR3DL1/KIRSDS1. Manhattan plot from the gene-based analysis between HLA+ persistent and resolved patients is seen in Supplementary Figure 2. No association in locations 1p36.21 and 19q13.42 was seen in HLA- patients. Another chromosomal location was also seen: in 2p22.3, two genes were shown to associate with disease prognosis; RASGRP3 with persistent disease and LTBP1 in resolved disease. However, these genes did not show significant association in any of the other subgroup analysis.
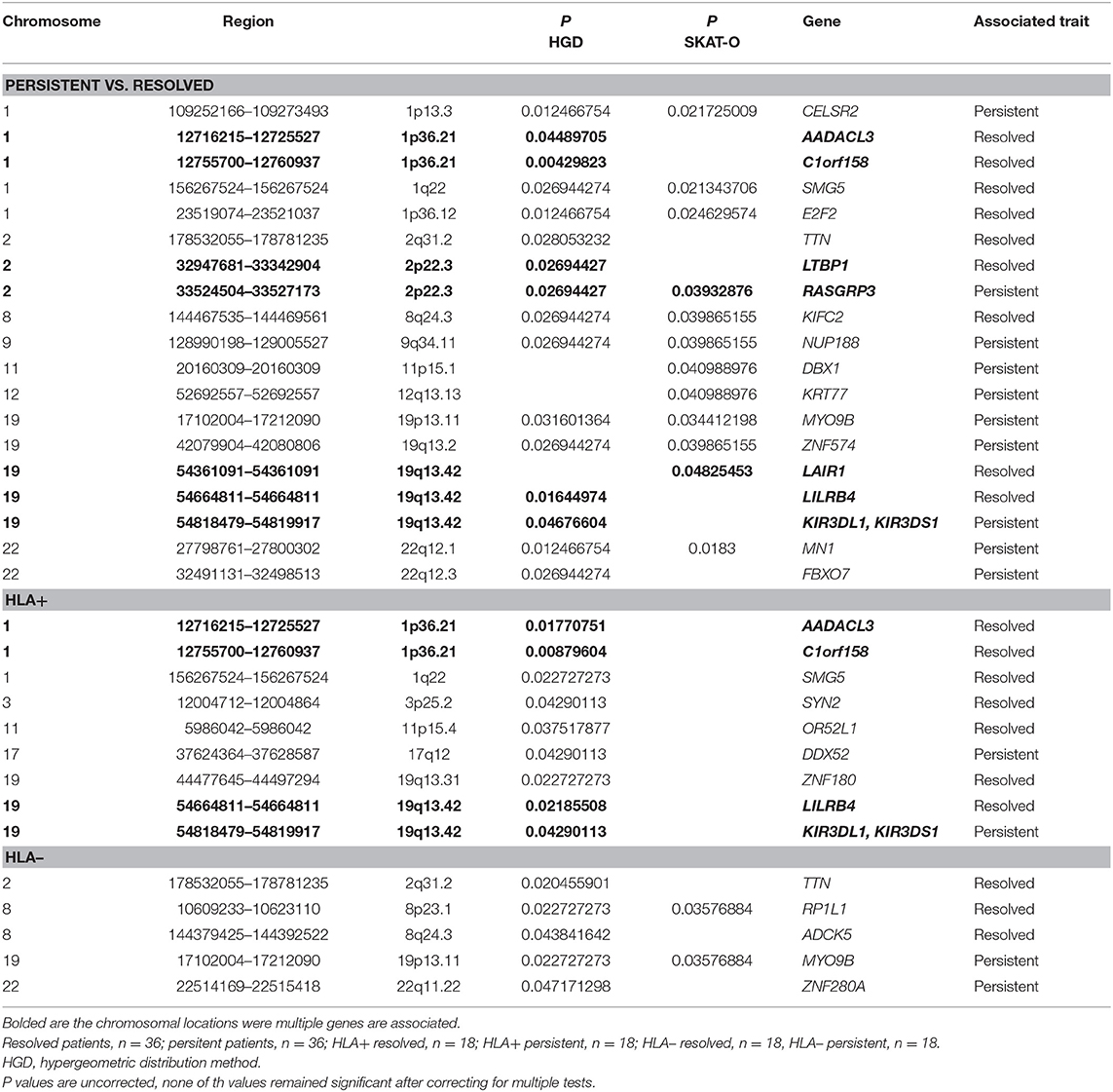
Table 2. Associated genes in gene-based analysis (hypergeometric distribution and SKAT-O methods) in different Finnish sarcoidosis subgroups based on disease prognosis (persistent, resolved) and HLA markers (HLA+ = HLA-DRB1*03:01 and/or HLA-DRB1*04:01-DPB1*04:01; HLA– = without either of the markers).
Replication Analysis
For the replication analysis, the rest of the Finnish sample set of 188 samples and 150 Finnish control material were included. After quality control, total of 150 controls and 181 cases (72 from the WES study and 109 from the replication study) were included in the analysis. In the replication analysis between persistent and resolved sarcoidosis patients, the SNPs in the chromosomal location 1p36.21 were statistically significant as in the WES study (Table 3). The association in these SNPs was also found in the analysis between HLA + persistent and resolved patients, and in contrast to the WES study, these SNPs were also significantly associated in the HLA-disease subgroup. When combining the data from the WES and replication studies, the associations became even stronger in all subgroup analysis. In chromosomal location 19q13.42, KIR3DL1/KIR3DS1 SNPs did not replicate and LILRB4 SNPs associated significantly in the HLA + resolved patients in the combined data set (Table 3). None of the associations remained statistically significant after correction with SNP count (data not shown). However, the MAFs in the control population corresponded to the ones in the gnomAD Finnish control population except for the KIR3DL1/KIR3DS1 SNP rs643861, which lacked allele frequency data in the gnomAD database (Table 3). However, in the NCBI dbSNP database (https://www.ncbi.nlm.nih.gov/snp/) MAF from TopMed database is A = 0.185, which resembles MAF found in our study. TOPMed uses hg38 as a reference genome, in gnomAD the hg19 is used as a reference genome which might explain the differences.
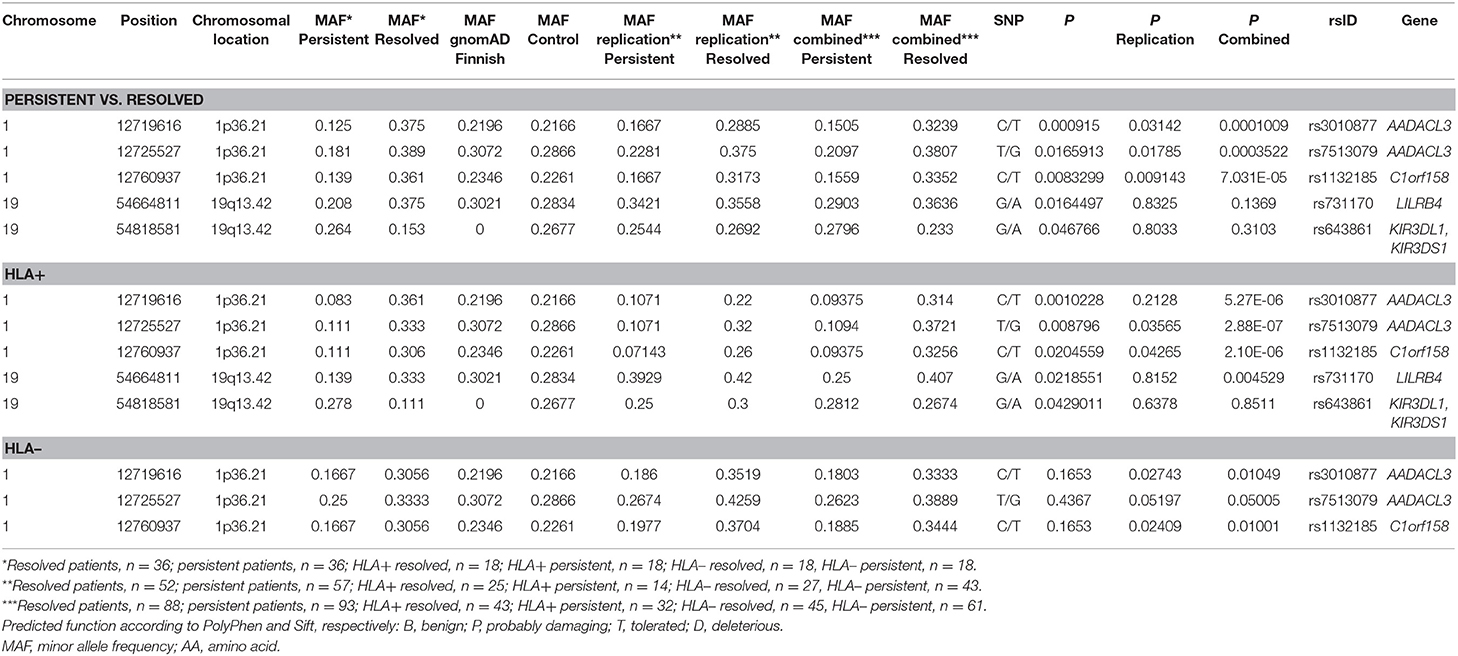
Table 3. Replication of SNPs found in the preliminary Whole-exome sequencing analysis in different Finnish sarcoidosis subgroups based on disease prognosis (persistent, resolved) and HLA markers (HLA+ = HLA–DRB1*03:01 and/or HLA–DRB1*04:01-DPB1*04:01; HLA– = without either of the markers) and Finnish control population.
The WES study associations were found in single-variation and gene levels in the chromosomal locations 1p36.21 and 19q13.42. In the replication sample, the associations in the location 1p36.21 were found only in the SNPs associating in the single-variation analysis (Supplementary Table 3). In chromosomal location 19q13.42, none of the other SNPs in the KIR3DL1/KIR3DS1 associated with the disease (data not shown). LAIR1 and KIR3DL3 SNPs did not meet the Sequenom study design criteria and could not be replicated (Supplementary Table 3). In chromosomal location 2p22.3, two genes were shown to associate with the disease prognosis in the WES study; RASGRP3 with persistent disease and LTBP1 in resolved disease. In the replication study, only one SNP in the RASGRP3 and LTBP1 passed the quality control (Supplementary Table 3) and none of these SNPs associated with the trait (data not shown).
Discussion
The heterogeneity of sarcoidosis in clinical course and development of organ involvement suggests that manifestation of the disease phenotype is a result of multiple genetic variations. There is a special interest in finding genetic differences between sarcoidosis patients with different prognosis, because at the moment prediction of the disease course at the time of diagnosis is difficult. There are also significant ethnic differences in disease development and prognosis in different populations (19). These differences represent a compelling need for ethnic-selective biomarkers to assess disease progression in diverse populations.
Here we present results from WES study in Finnish sarcoidosis patients. The strategy was to sequence groups of sarcoidosis patients with known differences in prognosis to identify variants that could alter functional properties of proteins. In Finnish sarcoidosis patients, certain HLA markers (HLA-DRB1*03:01, HLA-DRB1*04:01-DQB1*04:01) have been shown to be more common in patients with sarcoidosis disease resolving within 2 years, i.e., good prognosis disease. The aim of this study was to further pinpoint genetic variety in different sarcoidosis prognosis groups in relation to these HLA markers.
The most strongly associated and replicated chromosomal location was found in 1p36.21, containing two genes (AADACL3 and C1orf158). This region has also been found to associate with familial sarcoidosis in a recent WES study in German patients (20). However, in French WES study of familial sarcoidosis, no association with this region was found (21). In our study, variations in these genes were associated with disease resolution independent of the HLA markers DRB1*03:01 and DRB1*04:01. The found variations in AADACL3 are predicted to be benign, but the variation in C1orf158 is thought to alter the function of the protein. However, the functionality of these variants in sarcoidosis predisposition is unclear at the moment. The variants rs3010877 in AADACL3 and rs1132185 in C1orf158 are known to act as eQTLs in not sun exposed skin, but no clear in sarcoidosis pathogenesis is evident. Due to LD, it is also possible that association derives from elsewhere in the chromosome. However, no functionally plausible genes reside in near proximity of these genes, while the further region in 1p36 has been associated with resolution of chest findings in sarcoidosis patients (22). Therefore, further studies are needed to evaluate the possible causality of these genes and sarcoidosis susceptibility.
In the WES study, another interesting chromosomal region was seen in 19q13.42 (LILRB4, KIR3DL1/KIR3DS1, and LAIR1). In the replication part of the study, only association with LILRB4 remained statistically significant when WES and replication data were combined, the other associations were weakened or could not been replicated. It should be noted, that the size of the replication material is still relatively small, and possible true causality cannot be ruled out.
The 19q13.42 region, called the Leukocyte Receptor Complex (LRC), comprises a large set of genes encoding immunoglobulin superfamily receptors (23). The leukocyte immunoglobulin-like receptors (LILR) can interact with the HLA class I (24) and are expressed by a range of immunologically active cells, including natural killer (NK) and antigen-presenting cells (APCs) (25). LILRs have both activating and inhibitory effects (26). Previously no associations have been found between LILRB4 and sarcoidosis, but variations in this gene have shown to increase risk of another immune-mediated disease, systemic lupus erythematosus (SLE) (27). In our study, variations in this gene associated with good prognosis patients having the good prognosis HLA Class II markers. LILRB4 belongs to inhibitory receptors expressed on dendritic cells and has been shown to interact with the HLA-G. (28). LILRB4-HLA-G interaction has been demonstrated to limit the activation of dendritic cells (29), which are important APCs in sarcoidosis inflammation. In sarcoidosis, the association with HLA-II is thought to be more important, but it should be noted that limited information exists in relation to HLA-G in sarcoidosis. In one study, the HLA-G variation was seen in sarcoidosis patients (30). Also, there is a strong LD in the MHC region, and it is possible that patients with the HLA class II markers have haplotypes containing the HLA class I as well. The combination of these MHC variations, with the variants in the LILRB4, could alter the immune reaction in sarcoidosis and lead to self-resolving disease.
In addition to LILRB4, in the WES study, association was found with variants in LAIR1 and KIR3DL1/KIR3DS1. Leukocyte-associated immunoglobulin-like receptor-1 (LAIR1) has been shown to inhibit T and NK cell activation (31), but there are no known HLA ligands for this receptor. In our study, association with variants in the LAIR1 was seen when persistent and resolved patients were compared. No association was evident with the HLA markers, which is in concordance with the notion that there are no known HLA ligands for this protein. No association have been found between this gene and sarcoidosis, but also this immune-modulator gene has been shown to have effect in SLE (32). However, this association was seen only in gene-based analysis and with single method. Due to technical issues, the variation could not be replicated, so the effect should be considered cautiously. The other association seen only in the WES study encode KIRs, receptors expressed mainly on the surface of natural killer (NK) cells. Most diverse of the KIR genes is locus KIR3DL1/KIR3DS1 which associated with persistent sarcoidosis in our study. The gene encodes both inhibitory (3DL1) and activating (3DS1) receptors (33), so either or both of these functions could be affected, shifting the course of the disease toward chronic. In a recent WES study of three families sharing pediatric sarcoidosis, the KIR3DL1/KIR3DS1 in gene level was found to be shared in these families (34). To our knowledge, no case-control study has found association in KIR genes and sarcoidosis.
There are some limitations in this study. Sarcoidosis being a relatively rare disease, the collection of large data sets is demanding in a small country as Finland. Therefore, due to relatively small sample size, the power might have not been sufficient to detect all associations, however, the gene-based analysis in the WES study and the replication sample increases the power. Because of the patient selection based on the HLA markers, the analysis between all persistent and resolved patients has some statistical limitations. However, in the replication study, the whole Finnish sarcoidosis sample is involved. The other confounding factors: gender, age and extrapulmonary manifestations, were conditioned in all analyses.
In summary, we used WES to further characterize genetic differences between persistent and resolved sarcoidosis patients and to test the hypothesis whether different variations are associated in relation to the class II markers (HLA-DRB1*03:01/HLA-DRB1*04:01-DPB1*04:01). An association with resolved disease raised in the chromosomal region 1p36.21, a region which has recently been associated with sarcoidosis in another WES study. Another interesting chromosomal region peaked, 19q13.42, but in the replication study the association weakened. However, due to relatively small sample size, the true causality of these variants should be evaluated in a larger data set.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Department of Internal Medicine, in the Hospital District of Helsinki and Uusimaa, Helsinki, Finland (Approval Dnro 362/E5/05). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
EL, MK, JS, OS, and M-LL: study design and data analysis and manuscript writing. The manuscript has been read and approved by all named authors.
Funding
This study was supported by the Nummela Foundation for Medical Research.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank clinician Anne Pietinalho for aiding in collection of the original clinical sarcoidosis material of 188 patients. We would also like to thank laboratory technician Lauri Snellman in aiding in the all sample preparations.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.02964/full#supplementary-material
References
1. Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med. (2007) 357:2153–65. doi: 10.1056/NEJMra071714
2. Valeyre D, Prasse A, Nunes H, Uzunhan Y, Brillet PY, Muller-Quernheim J. Sarcoidosis. Lancet. (2014) 383:1155–67. doi: 10.1016/S0140-6736(13)60680-7
3. Spagnolo P, du Bois RM. Genetics of sarcoidosis. Clin Dermatol. (2007) 25:242–9. doi: 10.1016/j.clindermatol.2007.03.001
4. Grunewald J, Eklund A, Olerup O. Human leukocyte antigen class I alleles and the disease course in sarcoidosis patients. Am J Respir Crit Care Med. (2004) 169:696–702. doi: 10.1164/rccm.200303-459OC
5. Sato H, Woodhead FA, Ahmad T, Grutters JC, Spagnolo P, van den Bosch JM, et al. Sarcoidosis HLA class II genotyping distinguishes differences of clinical phenotype across ethnic groups. Hum Mol Genet. (2010) 19:4100–11. doi: 10.1093/hmg/ddq325
6. Wennerstrom A, Pietinalho A, Lasota J, Salli K, Surakka I, Seppanen M, et al. Major histocompatibility complex class II and BTNL2 associations in sarcoidosis. Eur Respir J. (2013) 42:550–3. doi: 10.1183/09031936.00035213
7. Wennerstrom A, Pietinalho A, Vauhkonen H, Lahtela L, Palikhe A, Hedman J, et al. HLA-DRB1 allele frequencies and C4 copy number variation in Finnish sarcoidosis patients and associations with disease prognosis. Hum Immunol. (2012) 73:93–100. doi: 10.1016/j.humimm.2011.10.016
8. Pietinalho A, Ohmichi M, Hiraga Y, Lofroos AB, Selroos O. The mode of presentation of sarcoidosis in Finland and Hokkaido, Japan. A comparative analysis of 571 Finnish and 686 Japanese patients. Sarcoidosis Vasc Diffuse Lung Dis. (1996) 13:159–66.
9. Clamp M, Fry B, Kamal M, Xie X, Cuff J, Lin MF, et al. Distinguishing protein-coding and noncoding genes in the human genome. Proc Natl Acad Sci USA. (2007) 104:19428–33. doi: 10.1073/pnas.0709013104
10. Hunninghake GW, Costabel U, Ando M, Baughman R, Cordier JF, du Bois R, et al. ATS/ERS/WASOG statement on sarcoidosis. American Thoracic Society/European Respiratory Society/World Association of Sarcoidosis and other granulomatous disorders. Sarcoidosis Vasc Diffuse Lung Dis. (1999) 16:149–73.
11. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. (2016) 536:285–91.
12. Seppanen M, Suvilehto J, Lokki ML, Notkola IL, Jarvinen A, Jarva H, et al. Immunoglobulins and complement factor C4 in adult rhinosinusitis. Clin Exp Immunol. (2006) 145:219–27. doi: 10.1111/j.1365-2249.2006.03134.x
13. Fang H, Wu Y, Narzisi G, O'Rawe JA, Barron LT, Rosenbaum J, et al. Reducing INDEL calling errors in whole genome and exome sequencing data. Genome Med. (2014) 6:89. doi: 10.1186/PREACCEPT-1179619571327140
14. Lee S, Emond MJ, Bamshad MJ, Barnes KC, Rieder MJ, Nickerson DA, et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet. (2012) 91:224–37. doi: 10.1016/j.ajhg.2012.06.007
15. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. (2009) 4:1073–81. doi: 10.1038/nprot.2009.86
16. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. (2010) 7:248–9. doi: 10.1038/nmeth0410-248
18. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a toolset for whole-genome association and population-based linkage analysis. Am J Human Genet. (2007) 81:559–75. doi: 10.1086/519795
19. Rybicki BA, Major M, Popovich J Jr, Maliarik MJ, Iannuzzi MC. Racial differences in sarcoidosis incidence: a 5-year study in a health maintenance organization. Am J Epidemiol. (1997) 145:234–41. doi: 10.1093/oxfordjournals.aje.a009096
20. Kishore A, Petersen BS, Nutsua M, Muller-Quernheim J, Franke A, Fischer A, et al. Whole-exome sequencing identifies rare genetic variations in German families with pulmonary sarcoidosis. Hum Genet. (2018) 137:705–16. doi: 10.1007/s00439-018-1915-y
21. Alain C, Clarice XL, Weichhart T, Buisson A, Besnard V, Antoine Rollat-Farnier P, et al. Exome sequencing and pathogenicity-network analysis of 5 French families implicate TOR signalling and autophagy in familial sarcoidosis. Eur Respir J. (2019) 54:1900430. doi: 10.1183/13993003.00430-2019
22. Rybicki BA, Sinha R, Iyengar S, Gray-McGuire C, Elston RC, Iannuzzi MC, et al. Genetic linkage analysis of sarcoidosis phenotypes: the sarcoidosis genetic analysis (SAGA) study. Genes Immun. (2007) 8:379–86. doi: 10.1038/sj.gene.6364396
23. Beziat V, Hilton HG, Norman PJ, Traherne JA. Deciphering the killer-cell immunoglobulin-like receptor system at super-resolution for natural killer and T-cell biology. Immunology. (2017) 150:248–64. doi: 10.1111/imm.12684
24. Cosman D, Fanger N, Borges L, Kubin M, Chin W, Peterson L, et al. A novel immunoglobulin superfamily receptor for cellular and viral MHC class I molecules. Immunity. (1997) 7:273–82. doi: 10.1016/S1074-7613(00)80529-4
25. Cella M, Dohring C, Samaridis J, Dessing M, Brockhaus M, Lanzavecchia A, et al. A novel inhibitory receptor (ILT3) expressed on monocytes, macrophages, and dendritic cells involved in antigen processing. J Exp Med. (1997) 185:1743–51. doi: 10.1084/jem.185.10.1743
26. Fanger NA, Borges L, Cosman D. The leukocyte immunoglobulin-like receptors (LIRs): a new family of immune regulators. J Leukoc Biol. (1999) 66:231–6. doi: 10.1002/jlb.66.2.231
27. Jensen MA, Patterson KC, Kumar AA, Kumabe M, Franek BS, Niewold TB. Functional genetic polymorphisms in ILT3 are associated with decreased surface expression on dendritic cells and increased serum cytokines in lupus patients. Ann Rheum Dis. (2013) 72:596–601. doi: 10.1136/annrheumdis-2012-202024
28. LeMaoult J, Zafaranloo K, Le Danff C, Carosella ED. HLA-G up-regulates ILT2, ILT3, ILT4, and KIR2DL4 in antigen presenting cells, NK cells, and T cells. FASEB J. (2005) 19:662–4. doi: 10.1096/fj.04-1617fje
29. Ketroussi F, Giuliani M, Bahri R, Azzarone B, Charpentier B, Durrbach A. Lymphocyte cell-cycle inhibition by HLA-G is mediated by phosphatase SHP-2 and acts on the mTOR pathway. PLoS ONE. (2011) 6:e22776. doi: 10.1371/journal.pone.0022776
30. Hviid TV, Milman N, Hylenius S, Jakobsen K, Jensen MS, Larsen LG. HLA-G polymorphisms and HLA-G expression in sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. (2006) 23:30–7.
31. Jansen CA, Cruijsen CW, de Ruiter T, Nanlohy N, Willems N, Janssens-Korpela PL, et al. Regulated expression of the inhibitory receptor LAIR-1 on human peripheral T cells during T cell activation and differentiation. Eur J Immunol. (2007) 37:914–24. doi: 10.1002/eji.200636678
32. Colombo BM, Canevali P, Magnani O, Rossi E, Puppo F, Zocchi MR, et al. Defective expression and function of the leukocyte associated Ig-like receptor 1 in B lymphocytes from systemic lupus erythematosus patients. PLoS ONE. (2012) 7:e31903. doi: 10.1371/journal.pone.0031903
33. O'Connor GM, Guinan KJ, Cunningham RT, Middleton D, Parham P, Gardiner CM. Functional polymorphism of the KIR3DL1/S1 receptor on human NK cells. J Immunol. (2007) 178:235–41. doi: 10.4049/jimmunol.178.1.235
Keywords: sarcoidosis, prognosis, MHC, whole exome sequencing, 1p36.21, leucocyte receptor complex
Citation: Lahtela E, Kankainen M, Sinisalo J, Selroos O and Lokki M-L (2019) Exome Sequencing Identifies Susceptibility Loci for Sarcoidosis Prognosis. Front. Immunol. 10:2964. doi: 10.3389/fimmu.2019.02964
Received: 05 August 2019; Accepted: 03 December 2019;
Published: 24 December 2019.
Edited by:
Alexandre Belot, Université Claude Bernard Lyon 1, FranceReviewed by:
Alain Calender, Hospices Civils de Lyon, FranceBritt-Sabina Löscher, University Hospital Schleswig-Holstein, Germany
Copyright © 2019 Lahtela, Kankainen, Sinisalo, Selroos and Lokki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elisa Lahtela, TGF1cmEubGFodGVsYUBoZWxzaW5raS5maQ==