 Federico Nicolás Penas1,2
Federico Nicolás Penas1,2 Davide Carta3
Davide Carta3 Ágata Carolina Cevey1,2
Ágata Carolina Cevey1,2 María Jimena Rada1,2
María Jimena Rada1,2 Azul Victoria Pieralisi1,2
Azul Victoria Pieralisi1,2 María Grazia Ferlin3María Elena Sales4
María Grazia Ferlin3María Elena Sales4 Gerardo A. Mirkin1,5
Gerardo A. Mirkin1,5 Nora Beatriz Goren1,2*
Nora Beatriz Goren1,2*- 1Departamento de Microbiología, Parasitología e Inmunología, Facultad de Medicina, Universidad de Buenos Aires, Buenos Aires, Argentina
- 2Instituto de Investigaciones Biomédicas en Retrovirus y Sida (INBIRS), CONICET-Universidad de Buenos Aires, Buenos Aires, Argentina
- 3Department of Pharmaceutical and Pharmacological Sciences, University of Padova, Padova, Italy
- 4Centro de Estudios Farmacológicos y Botánicos (CEFyBO), CONICET-Universidad de Buenos Aires, Buenos Aires, Argentina
- 5Instituto de Investigaciones en Microbiología y Parasitología Médica (IMPaM), CONICET-Universidad de Buenos Aires, Buenos Aires, Argentina
Chagas disease is caused by Trypanosoma cruzi infection and represents an important public health concern in Latin America. Macrophages are one of the main infiltrating leukocytes in response to infection. Parasite persistence could trigger a sustained activation of these cells, contributing to the damage observed in this pathology, particularly in the heart. HP24, a pyridinecarboxylic acid derivative, is a new PPARγ ligand that exerts anti-inflammatory and pro-angiogenic effects. The aim of this work was to deepen the study of the mechanisms involved in the pro-angiogenic and anti-inflammatory effects of HP24 in T. cruzi-infected macrophages, which have not yet been elucidated. We show for the first time that HP24 increases expression of VEGF-A and eNOS through PI3K/AKT/mTOR and PPARγ pathways and that HP24 inhibits iNOS expression and NO release, a pro-inflammatory mediator, through PPARγ-dependent mechanisms. Furthermore, this study shows that HP24 modulates H2O2 production in a PPARγ-dependent manner. It is also demonstrated that this new PPARγ ligand inhibits the NF-κB pathway. HP24 inhibits IKK phosphorylation and IκB-α degradation, as well as p65 translocation to the nucleus in a PPARγ-dependent manner. In Chagas disease, both the sustained increment in pro-inflammatory mediators and microvascular abnormalities are crucial aspects for the generation of cardiac damage. Elucidating the mechanism of action of new PPARγ ligands is highly attractive, given the fact that it can be used as an adjuvant therapy, particularly in the case of Chagas disease in which inflammation and tissue remodeling play an important role in the pathophysiology of this disease.
Introduction
Trypanosoma cruzi (T. cruzi) is a protozoan parasite that causes Chagas disease, the main cause of infectious dilated cardiomyopathy all over the world (1). This vector-borne disease affects millions of people in South America and, in recent years, it has been regarded as a risk factor for transfusion and vertical transmission in countries without vector-borne disease transmission control. During the acute stage, inflammation is involved in protection but parasite persistence leads to chronic inflammation. This impairs adequate repair leading to cumulative damage that ultimately may cause death due to cardiac insufficiency (2). Likely, chronic Chagas cardiomyopathy (CCC) is the most important clinical manifestation of Chagas disease. Clinical manifestations are characterized by conduction system disturbances, atrial and ventricular arrhythmias, congestive heart failure, systemic and pulmonary thromboembolism, and microvascular dysfunction (3). Although the mechanisms underlying progression to CCC have not been fully understood, it is generally accepted that inflammation persistence plays a predominant role (4).
Depending on their levels, oxidative species (mainly H2O2) can promote cell redox signaling or cytotoxicity (5). T. cruzi infection, together with pro-inflammatory cytokines, H2O2 and NO production by cardiac, endothelial, and immune cells, leads to an increase in nitroxidative stress that may account for host cell and tissue damage (6–8).
In response to the infection, monocytes can differentiate into macrophages and are one of the main infiltrating leukocytes to reach the myocardium earlier (9). These cells play important roles in the infection outcome and are essential for the orchestration of immunity and cardiac homeostasis. Due to their functional and phenotypic versatility, manipulating specific macrophage subsets can be crucial in collaborating with vital cardiovascular functions, such as tissue repair and defense against the infection (10). PPARγ are key nuclear receptors and therapeutic targets for the treatment of metabolic diseases through the regulation of insulin resistance, diabetes, and dyslipidemia (11). Moreover, in recent decades, it has been shown that PPARγ and its ligands can repress inflammatory genes in activated macrophages (12–14) and T. cruzi-infected cardiomyocytes (15, 16), including the inducible NO synthase (iNOS), cyclooxygenase 2 (COX2), IL-6, and TNF-α, via the nuclear factor kB (NF-κB) pathway. Although a few drugs that target PPARγ, such as troglitazone, rosiglitazone, and pioglitazone, have been approved for different pathologies, severe adverse effects have led to discover diverse and novel compounds that target PPARγ (17). In this regard, Brun et al. synthesized a new PPARγ agonist with potent anti-inflammatory properties and without cytotoxic activity, tested in macrophages stimulated with LPS, and in a murine model of dextran-induced colitis (18). This compound is a 2,4-substituted 3-hydroxy-4-pyridinecarboxylic acid derivative (HP24), an aza-analog of salicylic acid and structurally close to other potent anti-inflammatory pyridine compounds, such as aminopyridinylmethanols and aminopyridinamines. In a recent work, we demonstrated that HP24 interacts with PPARγ in T. cruzi-infected macrophages, using a molecular docking approach. Also, we showed that treatment with a HP24 increased the expression of pro-angiogenic molecules, like endothelial NO synthase (eNOS) and VEGF-A, inhibited pro-inflammatory mediators, and reduced fibrosis in the heart of T. cruzi-infected mice (19). Furthermore, it has been reported that PPARγ agonists improve the vascular function, which is partially dependent on eNOS activation through the AKT pathway (20–22). In the context of T. cruzi infection, PI3Kγ signaling in myeloid cells restricts heart parasitism and avoids heart damage and death of mice (23). Moreover, along the infection, PI3K/AKT signaling activation is able to prevent infected cells from dying by repressing the apoptotic machinery (24). The aim of this work is to deepen into the mechanisms involved in the pro-angiogenic and anti-inflammatory effects of HP24 in T. cruzi-infected macrophages, as they have not yet been elucidated. Our results reveal that HP24 increases pro-angiogenic mediators (eNOS and VEGF-A) through PI3K/AKT/mTOR and PPARγ signaling. Lastly, we provide, for the first time, evidence that HP24 inhibits reactive nitrogen and oxygen species production and the NF-κB pathway by PPARγ-dependent mechanisms in peritoneal macrophages from T. cruzi-infected mice.
Materials and Methods
Ethics Statement
To carry out this work, BALB/c mouse were bred and maintained in the animal facility at the Department of Microbiology, Parasitology and Immunology, School of Medicine, University of Buenos Aires. All the procedures were approved by the Institutional Committee for the Care and Use of Laboratory Animals (CICUAL, School of Medicine, University of Buenos Aires, CD N° 2271/2014), in line with guidelines provided by the Argentinean National Administration of Drugs, Food and Medical Devices (ANMAT), the Argentinean National Service of Sanity and Agrifoods Quality (SENASA), and also based on the US NIH Guide for the Care and Use of Laboratory Animals.
Mice and Infection
All mice were provided with a 12-h day/night cycle and water and fed ad libitum with a standard diet. Seven male mice per group were infected intraperitoneally with 1 × 105 bloodstream trypomastigotes of a lethal RA (pantropic/reticulotropic) subpopulation of T. cruzi (25). Euthanasia was carried out by CO2 inhalation at 9 days post-infection (dpi). Each experiment was performed at least three times.
Synthesis of 1-Methyl-3-Hydroxy-4-Pyridinecarboxylic Acid Derivative 24 (HP24)
1-Methyl-3-hydroxy-4-pyridinecarboxylic acid derivative was resynthesized following the pathway reported previously by Brun et al. (18), with some modifications in the reaction conditions for the final steps of the synthesis and the purification step that led to the desired compound HP24 in the zwitterions form instead of the chloride compound described above. 3-Hydroxy-isonicotinic acid (1 g, 7.18 mmol) was suspended in 5 ml of DMF in a 25-ml round-bottomed flask. The resulting suspension was stirred at room temperature, and 10% NaOH (7.5 ml) was added dropwise until complete dissolution of the solid (pH 9–10). Methyl iodide (2.06 g, 14.46 mmol, d = 2.28 g/ml, 0.9 ml) was added under stirring and the solution was then refluxed, monitoring the reaction progress by thin-layer chromatography (n-butanol:H2O:AcOH, 1:1:1). Once the starting material disappeared, the solvent was removed under reduced pressure, obtaining a deep orange colored solid, which was dissolved in boiling water (50 ml). The solution was acidified with 37% HCl (3.5 ml), and 10% H2O2 (1 ml) was added. Then, the iodine was exhaustively extracted with CHCl3 (5 × 15 ml) in a separating funnel. The organic phase was concentrated under pressure to dryness, obtaining an orange crude raw powdery solid (1.662 g), which was purified by reversed-phase chromatography in a Biotage Isolera Spektra Flash Chromatography apparatus equipped with prepacked C18 cartridges. The fractions containing the product were pooled and concentrated to dryness by means of a rotary evaporator, yielding a white powdery product (0.956 g, 6.21 mmol).
3-Hydroxy-1-Methylpyridin-1-Ium-4-Carboxylate (HP24)
Yield: 86.4%; mp: 236°C (decomposition); Rf: 0.13 (n-butanol:H2O:AcOH, 1:1:1); IR (KBr): ν (cm−1) = 3.432 (OH), 3.079 (= C–H), 2.850 (CH3), 1.654 (COO–), 1.480 (C = C), 1.381 (C = N), 1.300 (C–N) cm−1; 1H NMR (300 MHz, [D6] DMSO) δ = 8.43 (s, 1H, H-2), 8.01 (d, J = 6.00 Hz, 1H, H-6), 7.98 (d, J = 6.03 Hz, 1H, H-5), 4.19 ppm (s,3H, N-CH3); 13CNMR (75 MHz, [D6] DMSO) δ = 47.98 (N-CH3), 126.85 (C-5), 129.73 (C-4), 130.72 (C-6), 137.54 (C-2), 164.99 (C-3), 166.95 ppm (COO–); HRMS (ESI-MS, 140 eV): m/z [M+H+] calculated for C7H7NO3+, 154.0504; found, 154.0545; RT-HPLC, C18: tR = 5.40 min, 97.61 A%; elemental analyses: calculated for C7H7NO3, C 54.9%, H 4.61%, N 9.15%; found: C 54.47%, H 4.39%, N 8.98%.
Isolation of Peritoneal Macrophages
Macrophages were obtained by washing the peritoneal cavity of uninfected or T. cruzi-infected mice (9 dpi) with 8 ml of RPMI-1640 culture medium (Invitrogen Life Technologies, Grand Island, NY, USA), supplemented with 10% of heat-inactivated fetal bovine serum (FBS) (Internegocios S.A., Argentina) and antibiotics (50 μg/ml of PenStrep®). Cells were allowed to adhere to the plastic surface of 6-well culture plates (Greiner Bio One International AG) for 3 h at 37°C in a 5% CO2 atmosphere (26). For PPARγ silencing experiments using PPARγ siRNA and Oligofectamine®, cells were cultured up to 30% confluence with RPMI-1640 medium, without FBS and PenStrep®.
In vitro Treatment With HP24 and LY294002
Peritoneal macrophages were isolated from uninfected or T. cruzi-infected mice (9 dpi) as indicated above. Cells were treated in vitro with HP24 (100 μM in PBS) or the specific PI3K inhibitor LY294002 (30 μM in DMSO) (Sigma-Aldrich Co., St. Louis, USA). For in vitro experiments, treatments were performed 30 min prior to infection. After the different treatments, cell viability was examined by a Trypan blue dye exclusion test.
PPARγ Knock-Down With Small-Interfering RNA (siRNA)
Macrophages were cultured up to 30–50% of confluence in RPMI-1640 medium without FBS and antibiotics for 24 h. Thereafter, cells were transfected with 20 μM of either of two different Stealth siRNA (7863 or 7864, Invitrogen Life Technologies, Grand Island, NY, USA) that target PPARγ mRNA, following the manufacturer's instructions. Transfections were performed with Oligofectamine, as specified by the manufacturer. Assays for gene activity were performed at 72 h post-transfection. The impact of PPARγ-siRNA interference on PPARγ mRNA was evaluated by RT-qPCR. PPARγ Stealth siRNA sequence: PPARGMSS-7863 forward: 5′-CCAGGAGAUCUACAAGGACUUGUAU-3′, reverse: 5′-AUACAAGUCCUUGUAGAUCUCCUGG-3′; PARGMSS-7864 forward: 5′-UCAAGGGUGCCAGUUUCGAUCCGUA-3′, reverse: 5′-UACGGAUCGAAACUGGCACCCUUGA-3′.
RNA Purification
Total RNA was obtained from macrophages using Quickzol reagent (Kalium Technologies, Argentina), treated with RQ1 RNase-Free DNase (Promega Co., USA). Total RNA was reverse-transcribed using M-MLV Reverse Transcriptase (Promega Co., USA), according to the manufacturer's instructions.
Quantitative Reverse Transcription Polymerase Chain Reaction (RT-qPCR)
mRNA expression was determined using 5 × HOT FIREPOL EVAGREEN qPCR (Solis BioDyne, Estonia) in a StepOnePlus Real-Time PCR System. Parameters were as follows: 52°C for 2 min, 95°C for 15 min, and 40 cycles at 95°C for 15 s, 54°C (for PPARγ) or 60°C (for 18S) for 30 s and 72°C for 1 min. Normalization was carried out using 18S rRNA. Quantification was performed using the comparative threshold cycle (Ct) method, as all the primer pairs (target gene/reference gene) were amplified using comparable efficiencies (relative quantity, 2−ΔΔCt) (27, 28). Primer sequences: 18S forward: 5′-AACACGGGAAACCTCACCC-3′, reverse: 5′-CCACCAACTAAGAACGGCCA-3′; PPARγ forward: 5′-ATCTACACGATGCTGGC-3′, reverse: 5′-GGATGTCCTCGATGGG-3′.
Protein Extraction and Western Blot Analysis
Total and cytosolic protein extracts were prepared as described previously by our group (19, 29). Protein concentration was determined by the Bradford method using a commercial protein assay (Bio-Rad, USA) and bovine serum albumin (BSA, Sigma-Aldrich Co, USA) as a standard (30). Fifty micrograms of protein extracts separated in 8–12% SDS-PAGE gels was blotted onto a Hybond-P membrane (GE Health-care, Spain) and incubated with the following specific antibodies: anti-PPARγ (Santa Cruz Biotechnology, CA, USA; Cat#sc-7273), anti-phospho-AKT (Ser 473) (Biolegend Inc., USA; Cat#649001), anti-AKT (Biolegend Inc., USA; Cat#680302), anti-phospho-p70S6K (Ser 411) (Santa Cruz Biotechnology, CA, USA; Cat#sc-8416), anti-p70S6K (Santa Cruz Biotechnology, CA, USA; Cat#sc-8418), anti-VEGF-A (Santa Cruz Biotechnology, CA, USA; Cat#sc-1836), anti-eNOS (Santa Cruz Biotechnology, CA, USA; Cat#sc-654), anti-iNOS (Santa Cruz Biotechnology, CA, USA; Cat#sc-650), anti-phospho-IKK (Ser 180) (Santa Cruz Biotechnology, CA, USA; Cat#sc-23470), anti-IKK (Santa Cruz Biotechnology, CA, USA; Cat#sc-7606), anti-IκB-α (Santa Cruz Biotechnology, CA, USA; Cat#sc-371), and anti-α-actin (Santa Cruz Biotechnology, CA, USA; Cat#sc-1615). All specific antibodies were diluted 1:500 in PBS. Blots were revealed by enhanced chemoluminescence in a BioSpectrum® Imaging System (UVP, Analytik Jena Company, USA). Band intensity was analyzed using the NIH Image J software (ImageJ).
Immunocytochemistry and Confocal Laser Microscopy Imaging
Macrophages were grown on round glass coverslips and fixed with methanol and blocked with 3% BSA in PBS. The expression of iNOS was determined by immunofluorescence, as described previously (31). The expression of p65 was determined by confocal microscopy. For these purposes, rabbit polyclonal IgG anti-iNOS (Santa Cruz Biotechnology, CA, USA; Cat#sc-650), rabbit polyclonal IgG anti-p65 (Santa Cruz Biotechnology, CA, USA; Cat#sc-109), and a rabbit polyclonal IgG directed to T. cruzi developed in our laboratory were used as primary antibodies at a 1:50 dilution, and goat anti-rabbit IgG Alexa Fluor 488 nm (for iNOS) (Jackson ImmunoResearch Labs; Cat# 111-545-003) or goat anti-rabbit IgG Alexa Fluor 647 nm (for p65 and T. cruzi) (Jackson ImmunoResearch Labs; Cat# 111-605-003) was used at a 1:500 dilution as secondary antibodies. The coverslips mounted with DAPI-Fluoromount-G (SouthernBiotech) were examined under a confocal microscope (ZEISS LSM800) using a Plan Apochromat 63× 1.42 numerical aperture (NA) oil immersion objective or under an Eclipse Ti-S fluorescence microscope (Nikon) using a Plan Apochromat 100 × 1.42 NA oil immersion objective. To quantify parasitism, the percentage of infected cells and the number of parasites per cell were determined by analyzing the presence of intracellular amastigotes and trypomastigotes in at least 30 random microscopic fields. Mean fluorescence intensity (MFI) were quantified using the Fiji version of the open source Image J software (NIH, USA) (32).
NO Measurement
To determine the amount of NO released into the culture medium, nitrate was reduced to nitrite and measured spectrophotometrically using the Griess reaction (33, 34). The amount of NO in culture supernatants was calculated by interpolation of the samples absorbance at 540 nm using a standard curve of NaNO2.
Detection of H2O2 Generation
Macrophages were plated onto 96-well polystyrene plates, up to 30% confluence, and silencing of PPARγ was performed. After 72 h, macrophages were pre-treated with HP24 and infected with T. cruzi. Cellular H2O2 production was measured using a DCFDA assay kit (Abcam) according to the manufacturer's protocol. DCFDA (2′,7′-dichlorofluorescein diacetate), a cell-permeable fluorogenic dye, is deacetylated by cellular esterases to a non-fluorescent compound, and later oxidized by ROS to highly fluorescent 2′,7′-dichlorofluorescein (DCF), which measures peroxyl, hydroxyl, and other ROS activities within the cell. DCF can be detected by fluorescence spectroscopy with maximum excitation and emission spectra of 495 and 529 nm, respectively. After 24 h post-infection, cells were washed twice with PBS, incubated with 25 μM DCFDA in RPMI medium at 37°C for 45 min, and evaluated by flow cytometry using a FACSCanto (BD Biosciences). The percentage of positive cells and MFI were analyzed with FlowJo X software (TreeStar).
Statistical Analysis
Data are expressed as the mean of three independent experiments ± SEM (n = 3) (six mice/group) for each experimental group. One-way ANOVA was used to analyze the statistical significance of the differences observed between infected, treated, and untreated groups. The Tukey post-hoc test was performed to compare every mean with every other mean. Differences were considered statistically significant when P < 0.05. All analyses were performed using the Prism 7.0 Software (GraphPad Prism).
Results
Neither HP24 nor PPARγ Modifies Parasitism in T. cruzi-Infected Macrophages
Recently, we have shown that treatment with HP24 affects neither parasitemia nor the number of amastigote nests in the heart of infected mice (19). To test whether signaling of HP24 through PPARγ correlates with changes in cell parasitism, we performed silencing assays to knock down PPARγ. First, the silencing effectiveness of PPARγ siRNA 7863 and 7864 was verified in peritoneal macrophages infected in vitro with T. cruzi (parasite:cell ratio 5:1) (Figure 1A), as well as in peritoneal macrophages from T. cruzi-infected mice (9 dpi) (Figure 1B). RT-qPCR assays revealed that, in both models, the maximum level of silencing was obtained with siRNA 7863 (70–60% of silencing). To confirm these results, we performed Western blot assays and determined that PPARγ siRNA 7863 was effective in silencing the expression of this receptor, and thus this was the siRNA used for further studies (Figures 1A,B).
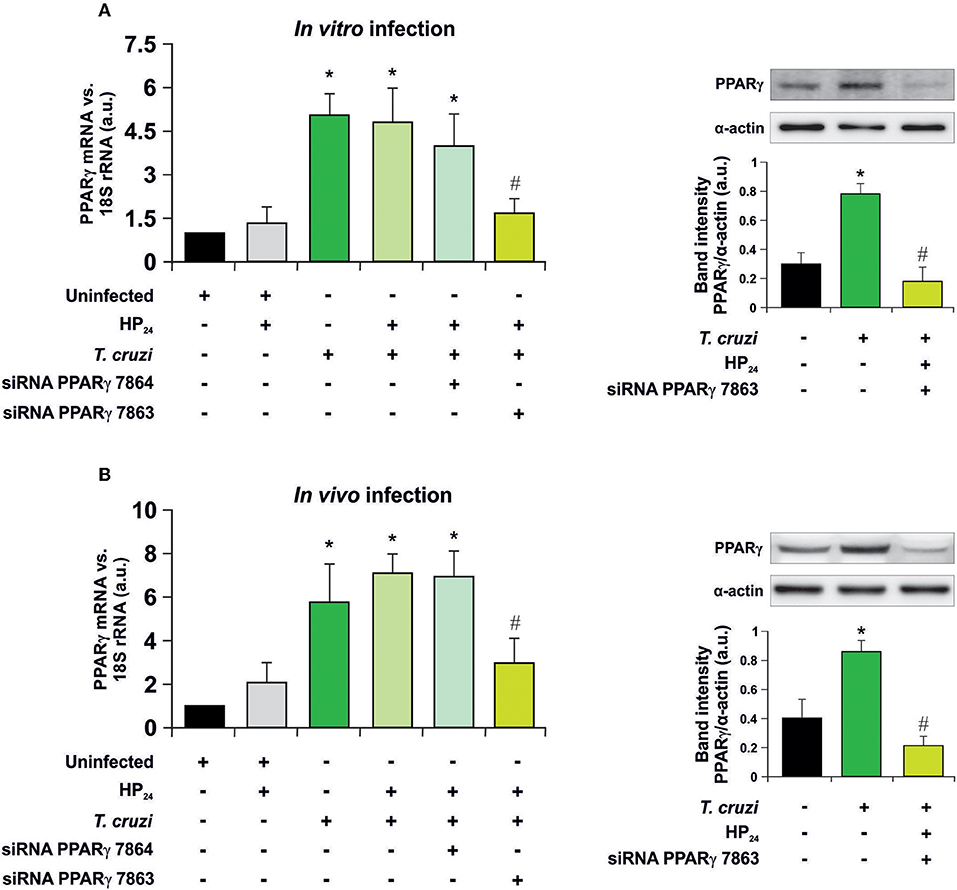
Figure 1. PPARγ silencing. (A) Uninfected peritoneal macrophages were obtained and, alternatively, transfected with two different PPARγ-siRNA during 72 h. Transfected or non-transfected cells were treated with HP24 (100 μM) since 30 min before infection. These cells were infected for 48 h with T. cruzi (parasite:cell ratio 5:1). (B) Peritoneal macrophages from T. cruzi-infected mice (9 dpi) were obtained. These cells were alternatively transfected with two different PPARγ-siRNA for 72 h. Transfected or non-transfected cells were treated with HP24 (100 μM) for 48 h. PPARγ expression was analyzed by RT-qPCR (six mice/group). Expression of PPARγ was determined by Western blot, and protein levels were normalized against α-actin. For both models, RNA and proteins were isolated 48 h after HP24 treatment. Results represent the mean ± SEM of three independent experiments *P < 0.05 vs. uninfected cells, #P < 0.05 vs. T. cruzi-infected cells.
Next, the involvement of HP24 and PPARγ in the possible changes in parasite load was evaluated. To this aim, isolated macrophages were infected in vitro with T. cruzi and the percentage of infected cells and the number of parasites per cell were evaluated by confocal microscopy. We observed that neither HP24 nor PPARγ silencing affects the parasitism of macrophages (Figure 2).
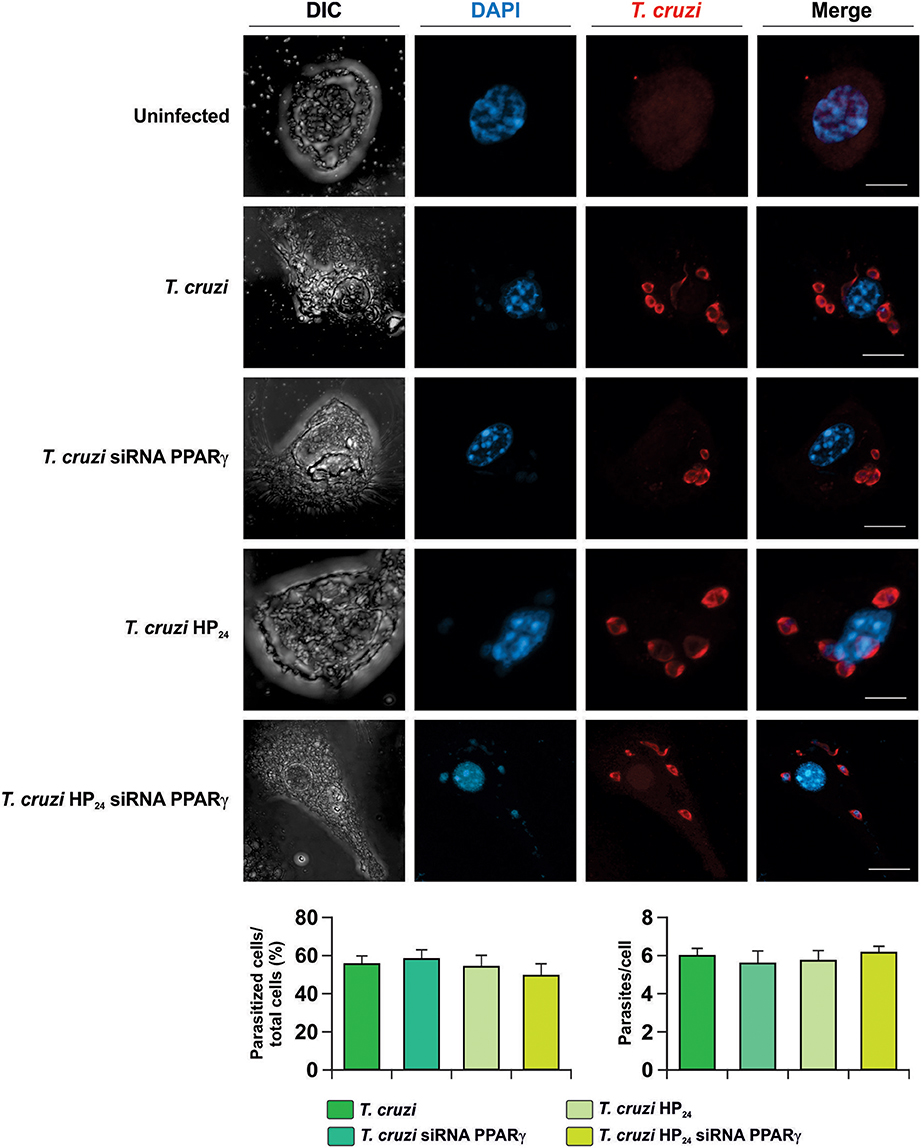
Figure 2. Neither HP24 nor PPARγ modifies parasitism in T. cruzi-infected macrophages. Peritoneal macrophages were obtained and transfected with PPARγ-siRNA during 72 h. Transfected or non-transfected cells were treated with HP24 (100 μM). Then, these cells were infected with T. cruzi for 48 h (parasite:cell ratio 5:1). T. cruzi was detected by confocal microscopy with a rabbit polyclonal anti-T. cruzi antibody and a secondary goat anti-rabbit Alexa 647-labeled antibody. Cell nuclei were stained with DAPI. Representative microphotographs are shown. The percentage of infected cells and the number of parasites per cell were determined by analyzing the presence of intracellular amastigotes and trypomastigotes in at least 30 random microscopic fields. Scale bar: 5 μm. Results represent the mean ± SEM of three independent experiments.
HP24 Regulates VEGF-A and eNOS Expression by PI3K/AKT/mTOR and PPARγ Signaling in T. cruzi-Infected Macrophages
Recently, we described that a new PPARγ ligand, HP24, increases the expression of VEGF-A and eNOS in T. cruzi-infected macrophages. These effects were prevented in the presence of T0070907, a specific PPARγ antagonist (19), suggesting ligand-dependent activity. It has been widely reported that the PI3K/AKT/mTOR pathway plays a prominent role in regulating angiogenesis (35). Therefore, the involvement of the PI3K/AKT/mTOR and PPARγ pathways in the regulation of VEGF-A and eNOS expression by HP24 was analyzed in T. cruzi-infected macrophages. Cells were infected with T. cruzi and either treated with LY294002 (PI3K inhibitor) or transfected with PPARγ-siRNA, or both. Infection of macrophages augmented expression of VEGF-A but not eNOS (Figures 3A,B). To test whether the PPARγ and PI3K/AKT/mTOR pathways are involved in the increased expression of VEGF-A in infected cells, PPARγ was silenced or treated with LY294002. In the absence of the receptor, no changes in the expression of VEGF-A were observed. Moreover, inhibition of the PI3K signaling impeded the increase of VEGF-A. This suggests that the PI3K/AKT/mTOR pathway is associated to the elevation of VEGF-A induced by T. cruzi infection. Then, the effect of HP24 on VEGF-A and eNOS expression was evaluated. The PPARγ ligand significantly increased the expression of both pro-angiogenic mediators in macrophages. The participation of PPARγ was demonstrated, since HP24 could not increase pro-angiogenic mediators in infected and transfected cells. Moreover, upon PI3K inhibition with LY294002 of T. cruzi-infected cells, HP24 could not increase both pro-angiogenic mediators. Lastly, when PPARγ-siRNA transfected cells were pre-incubated with LY294002 and treated with HP24, similar results were observed for eNOS and VEGF-A. These results suggest independent, though not necessarily additive, pro-angiogenic effects of HP24 involving the PI3K/AKT/mTOR and PPARγ pathways (Figure 3).
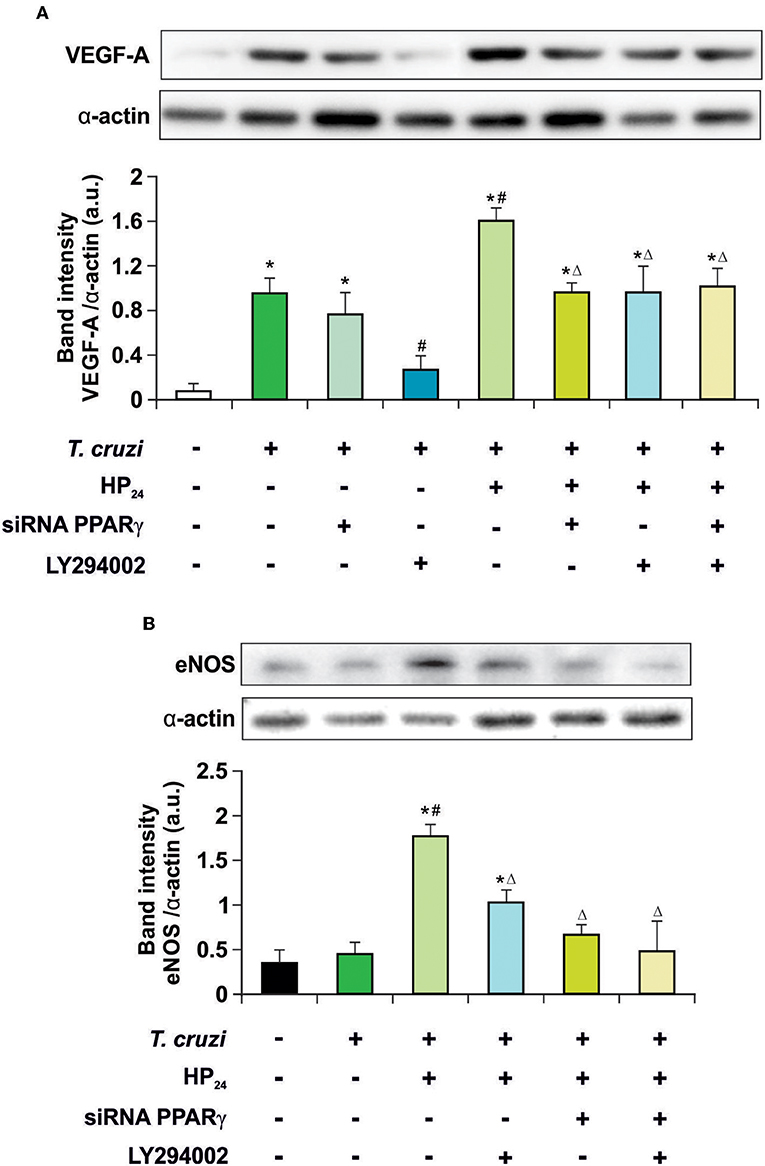
Figure 3. HP24 regulates eNOS and VEGF-A expression by PPARγ and AKT signaling pathway. Peritoneal macrophages from T. cruzi-infected mice (9 dpi) were obtained. Cells were transfected with PPARγ-siRNA during 72 h. Transfected or non-transfected cells were treated with HP24 (100 μM) or HP24 (100 μM) plus LY294002 (30 μM) for 48 h. (A,B) Expression of eNOS and VEGF-A was determined by Western blot, and protein levels were normalized against α-actin. Results represent the mean ± SEM of three independent experiments (six mice/group). *P < 0.05 vs. uninfected cells, #P < 0.05 vs. T. cruzi-infected cells. ΔP < 0.05 vs. T. cruzi-infected HP24-treated cells.
Then, we kept delving into the participation of PPARγ in the effect of HP24 on the PI3K/AKT/mTOR pathway. For this purpose, we analyzed AKT and the ribosomal protein S6 kinase (p70S6K) phosphorylation, since they are considered to be the hallmark of the PI3K/AKT/mTOR pathway activation. Macrophages were treated with HP24 (100 μM) for 45 min before T. cruzi infection (parasite:cell ratio 5:1). Increased phosphorylation of AKT but not p70S6K was observed in macrophages after 30 min of infection (Figure 4). When infected macrophages were treated with HP24, AKT phosphorylation increased further and p70S6K phosphorylation showed significant phosphorylation in comparison with infected untreated cells (Figure 4). PPARγ silencing prevented the effect of HP24 on the phosphorylation of AKT and p70S6K. As expected, phosphorylation of AKT and p70S6K was inhibited in cells treated with LY294002 (data not shown). These results indicate that PPARγ is required for the effects of HP24 to take place on the PI3K/AKT/mTOR pathway (Figure 4).
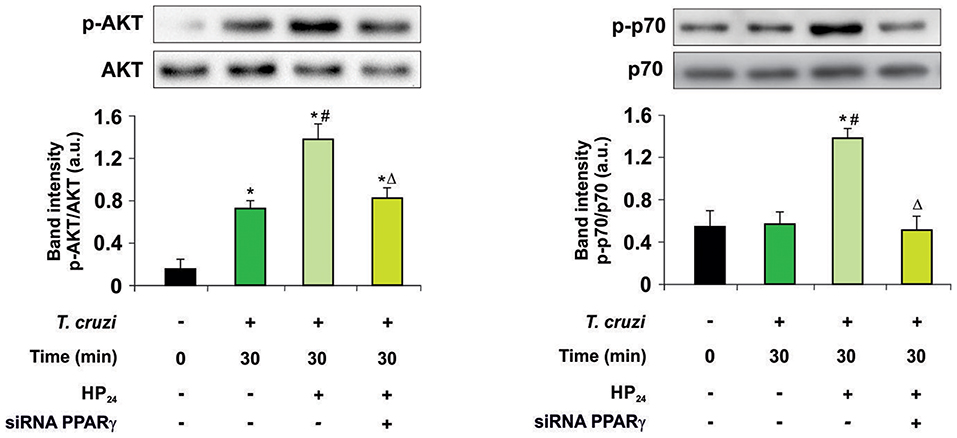
Figure 4. PPARγ is required for the HP24 effects on PI3K/AKT/mTOR signaling pathway. Peritoneal macrophages were obtained and transfected with PPARγ-siRNA during 72 h. Transfected or non-transfected cells were treated with HP24 (100 μM) since 30 min before infection. Then, these cells were infected with T. cruzi for 30 min (parasite:cell ratio 5:1). Western blot analyses were carried out in cytosolic extracts, and p-AKT (Ser 473)/total AKT and p-p70 (Ser 411)/total p70 expression was analyzed. Results represent the mean ± SEM of three independent experiments. *P < 0.05 vs. uninfected cells, #P < 0.05 vs. T. cruzi-infected cells. ΔP < 0.05 vs. T. cruzi-infected and HP24-treated cells.
HP24 Inhibits Reactive Nitrogen and Oxygen Species in T. cruzi-Infected Macrophages Through PPARγ
To determine whether HP24 modulates iNOS expression and activity through PPARγ, immunocytochemistry (ICQ) analysis was performed as well as Western blot assays and detection of NO release by the Griess reaction on T. cruzi-infected macrophages. iNOS expression was significantly increased in infected macrophages in comparison with uninfected ones. Moreover, HP24 treatment inhibited iNOS expression, while it was restored in PPARγ siRNA-transfected cells (Figure 5A). Western blot analysis confirmed these findings, since iNOS expression increased upon infection, decreased after treatment with PPARγ agonist and this decline was reversed in silenced cells (Figure 5B). Lastly, the activity of iNOS, as evidenced by the release of NO, paralleled the expression of iNOS observed by ICQ and Western blot (Figure 5C).
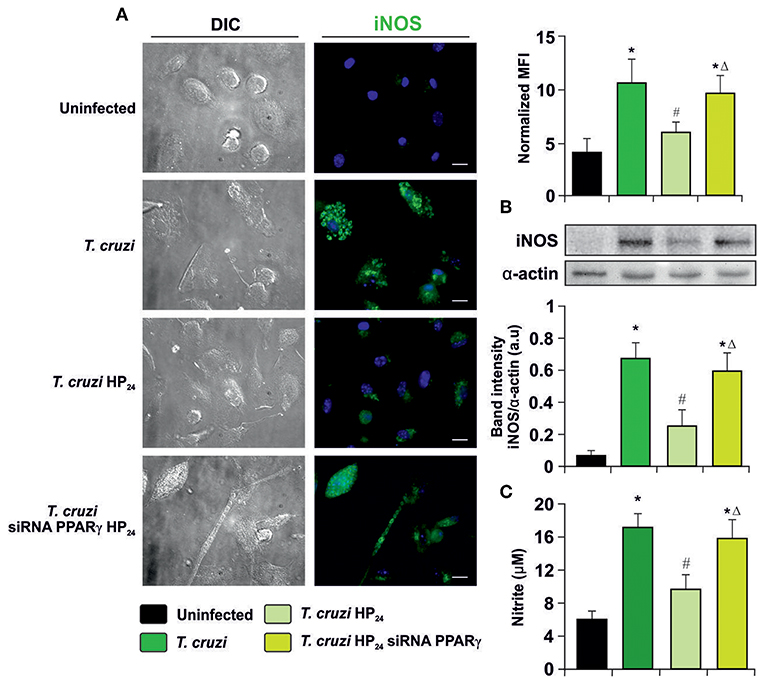
Figure 5. Participation of PPARγ in iNOS inhibition by HP24. (A) Peritoneal macrophages from T. cruzi-infected mice (9 dpi) were obtained. Cells were transfected with PPARγ-siRNA during 72 h. Transfected or non-transfected cells were treated with HP24 (100 μM) for 48 h. iNOS expression was detected by immunofluorescence with a rabbit polyclonal anti-iNOS antibody and with a secondary goat anti-rabbit Alexa 488-labeled antibody. Cell nuclei were stained with DAPI. Mean fluorescence intensity (MFI) represents iNOS expression. Representative microphotographs are shown. Scale bar: 10 μm. (B) Western blot analysis was carried out and iNOS expression was determined. Protein levels were normalized against α-actin. (C) NO release to culture supernatants was analyzed by the Griess method. Results are expressed as the mean of three independent experiments (six mice/group). *P < 0.05 vs. uninfected cells, #P < 0.05 vs. T. cruzi-infected cells. ΔP < 0.05 vs. T. cruzi-infected HP24-treated cells.
In light of these results, we aimed to determine whether HP24 affects H2O2 production (reactive oxygen species). Macrophages were infected in vitro with T. cruzi for 24 h. A high content of H2O2 was observed, as measured by flow cytometry, using the redox-sensitive fluorescent probe, 2′,7′-dichlorofluorescein-diacetate (DCFDA). HP24 treatment significantly inhibited H2O2 production. Moreover, when PPARγ was silenced, HP24 could not exert this effect in T. cruzi-infected cells. These results suggest that HP24 modulates H2O2 production through PPARγ (Figure 6).
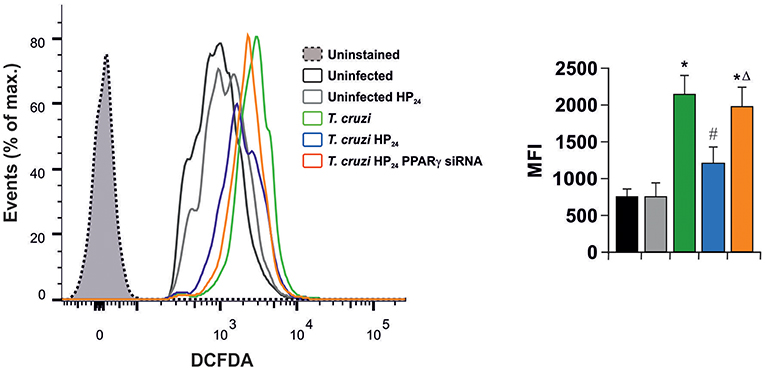
Figure 6. HP24 inhibits H2O2 production through PPARγ in T. cruzi-infected macrophages. Peritoneal macrophages were obtained and transfected with PPARγ-siRNA for 72 h. Transfected or non-transfected cells were treated with HP24 (100 μM) since 30 min before infection. Then, these cells were infected with T. cruzi for 24 h (parasite:cell ratio 5:1). Sample histogram shows H2O2 generation measured by flow cytometry using 2′,7′dichlorofluorescein-diacetate (DCFDA). Ten thousand events were acquired for each group. Data are presented as the mean MFI ± SEM of three independent experiments. *P < 0.05 vs. uninfected cells, #P < 0.05 vs. T. cruzi-infected cells. ΔP < 0.05 vs. T. cruzi-infected HP24-treated cells.
HP24 Inhibits the NF-κB Pathway by PPARγ-Dependent Mechanisms
Previously, we demonstrated that 15dPGJ2, a natural ligand of PPARγ, is a potent modulator of the inflammatory process through PPARγ-dependent and -independent pathways in T. cruzi-infected cardiac cells (15, 16). In this work, we analyzed whether the new PPARγ ligand HP24 exerts its anti-inflammatory effects through the NF-κB pathway in a PPARγ-dependent manner. For that aim, two different approaches were designed: Firstly, we evaluated the effects of HP24 on IKK and IκB-α, two cytosolic components of NF-κB pathway, by Western blot. HP24 treatment inhibits IKK phosphorylation and IκB-α degradation in in vitro infected T. cruzi macrophages. Moreover, in PPARγ-silenced cells, HP24 was unable to exert its inhibitory effect on both components (Figure 7).
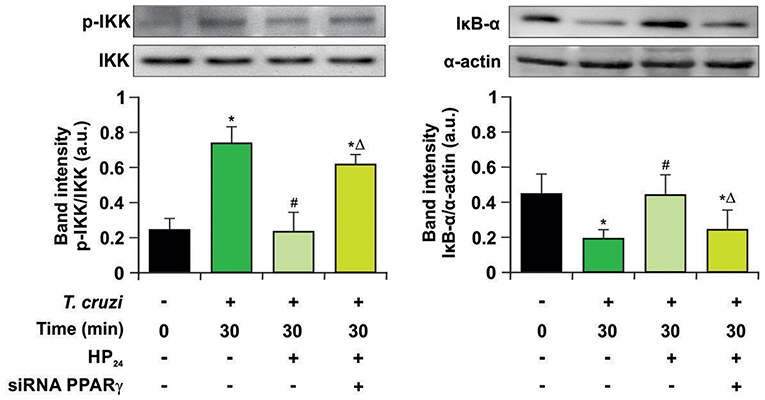
Figure 7. HP24 inhibits IKK and IκB-α phosphorylation by PPARγ-dependent mechanisms. Peritoneal macrophages were obtained and transfected with PPARγ-siRNA for 72 h. Transfected or non-transfected cells were treated with HP24 (100 μM) since 30 min before infection. Then, these cells were infected for 30 min with T. cruzi (parasite:cell ratio 5:1). Cytosolic expression of p-IKK (Ser 180) and IκB-α was determined by Western blot, and protein levels were normalized against IKK total and α-actin. Results are expressed as mean of three independent experiments *P < 0.05 vs. uninfected cells, #P < 0.05 vs. T. cruzi-infected cells. ΔP < 0.05 vs. T. cruzi-infected HP24-treated cells.
Secondly, we measured the translocation of p65 subunit of NF-κB to the nucleus by confocal microscopy in PPARγ-silenced cells. Figure 8 shows that p65 translocates to the nucleus at 30 min after infection, and HP24 treatment inhibits this translocation. However, under silencing conditions, HP24 was unable to inhibit NF-κB activation, confirming that HP24 regulates NF-κB pathway in a PPARγ-dependent manner (Figure 8).
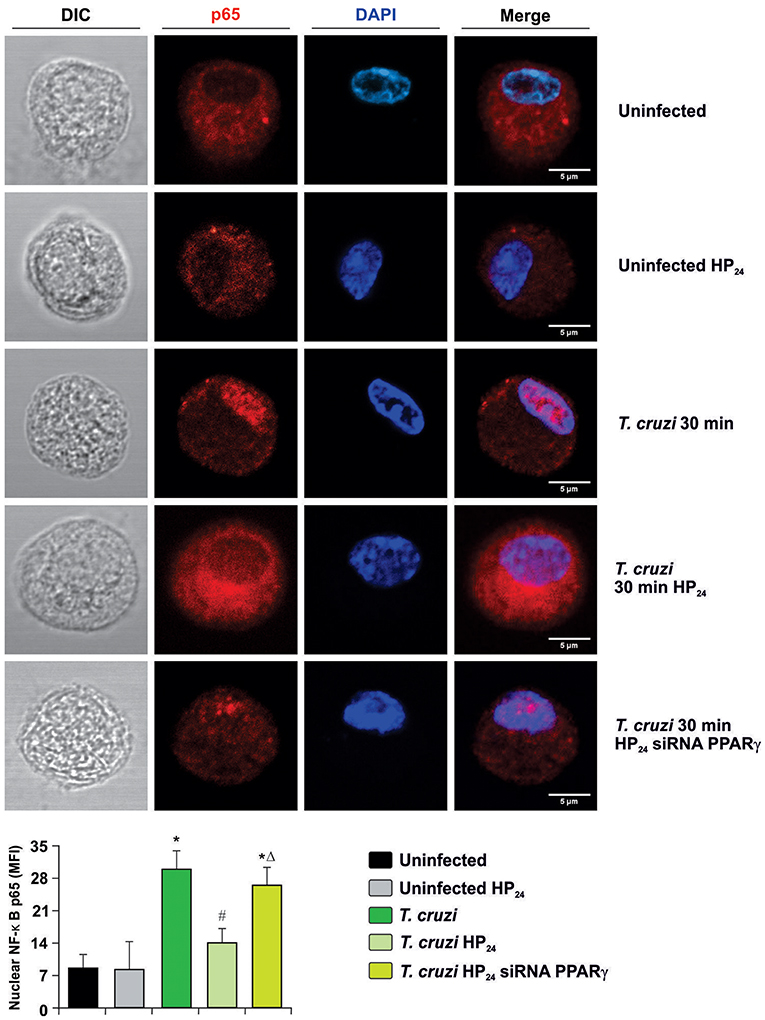
Figure 8. HP24 inhibits the NF-κB pathway by PPARγ-dependent mechanisms. Peritoneal macrophages were obtained and transfected with PPARγ-siRNA for 72 h. Transfected or non-transfected cells were treated with HP24 (100 μM) since 30 min before infection. Then, these cells were infected for 30 min with T. cruzi (parasite:cell ratio 5:1). p65 expression was detected by confocal microscopy with a rabbit polyclonal anti-p65 antibody and a secondary goat anti-rabbit Alexa 647-labeled antibody. Cell nuclei were stained with DAPI. Representative microphotographs are shown. MFI represents nuclear p65. Scale bar: 5 μm. Results are expressed as the mean MFI of three independent experiments. *P < 0.05 vs. uninfected cells, #P < 0.05 vs. T. cruzi-infected cells. ΔP < 0.05 vs. T. cruzi-infected HP24-treated cells.
Discussion
This work describes for the first time the signaling pathways of HP24, a new synthetic PPARγ ligand, in T. cruzi-infected macrophages. This compound is derived from 3-hydroxy-4-pyridinecarboxylic acid (18). Although we recently published that HP24 has important pro-angiogenic and anti-inflammatory properties (19), the signaling cascades involved in these effects have not been studied yet.
In this work, we demonstrate that treatment with HP24 does not modify parasite load, in comparison with infected untreated cells. This is in agreement with previous results from our group using HP24 using an in vivo model of acute infection (19). Also, we showed similar results using a PPARα ligand, in an in vivo model of chronic infection (29). Besides, Rodrigues et al. show reduction of skeletal muscle parasitism (36).
Macrophages infected with T. cruzi and treated with HP24 display activation of the PI3K/AKT/mTOR and PPARγ signaling pathways, leading to induction of VEGF-A and eNOS expression. Consistent with this, it has been described that pioglitazone treatment up-regulates VEGF-A and eNOS expression, significantly ameliorates endothelial dysfunction, and enhances blood flow recovery after tissue ischemia in diabetic mice, via AKT phosphorylation (37). Moreover, it has been shown that rosiglitazone restores endothelial dysfunction in a rat model of metabolic syndrome, through PPARγ- and PPARδ-dependent phosphorylation of eNOS and AKT (38). In this study, we show that HP24 can regulate the PI3K/AKT/mTOR signaling pathway at 30 min in a PPARγ-dependent manner in T. cruzi-infected macrophages. Moreover, our results show that in PPARγ-siRNA transfected cells, HP24 is unable to increase phosphorylation levels of AKT and p70S6K, strongly suggesting a link between PI3K/AKT/mTOR signaling and the role of PPARγ as a transcription regulator.
Recognition of the pathogens by resident and recruited macrophages activates several signaling pathways, including PI3K signaling. This pathway is involved in different cellular processes, such as cytoskeletal rearrangement, membrane trafficking, and endosome fusion through the phosphorylation of lipids and proteins (39, 40). Regarding T. cruzi, several authors have demonstrated that infection activates PI3K signaling in human and mouse macrophages leading to increased infection and anti-apoptotic pathways that allow for intracellular parasite multiplication and survival (24, 41, 42). Recently, it has been demonstrated that canonical PI3Kγ signaling in myeloid cells is essential to restrict T. cruzi heart parasitism and ultimately to avoid myocarditis, heart damage, and death in mice (23). Furthermore, in a human in vitro model of osteoarthritis characterized by increased advanced glycation end (AGE) products accumulation and reduced autophagy, it was demonstrated that pioglitazone, a PPARγ ligand, increased AKT/mTOR phosphorylation in a dose-dependent manner, leading to better cell viability and inducing increased chondrocyte autophagy (43). However, several works on cancer and neurodegenerative diseases have shown that PPARγ activation inhibits the PI3K/AKT/mTOR signaling pathway (44–48). Likewise, it has been described that the PI3K/AKT/mTOR-p70S6K activation pathway is critical in restricting pro-inflammatory and promoting anti-inflammatory responses in TLR-stimulated macrophages (49).
We have previously shown that different PPAR ligands are potent inhibitors of inflammatory mediators like NO, IL-1β, IL-6, and TNFα, through PPAR-dependent and -independent pathways in T. cruzi-infected macrophages and cardiomyocytes (15, 16, 26, 29). In the present study, we determined the anti-inflammatory efficacy of HP24. We observed that this new compound inhibits the production of H2O2 and the release of NO by reducing iNOS expression in a PPARγ-dependent manner in T. cruzi-infected macrophages. It is well-documented that the activation of the respiratory burst of macrophages in response to infection with T. cruzi inflicts oxidative damage to host tissues. ROS and nitric oxide (NO) combine to form peroxynitrite, participating in the destruction of parasites phagocytosed by activated macrophages (50). Nevertheless, if the parasites are not completely eliminated, this phagocytic cell response may persist in the chronic stage of the infection and contribute to oxidative damage that impairs heart function (51). It has been reported that PPARγ ligands induce anti-inflammatory effects, mainly mediated by antioxidant properties. PPARγ activation by β3-adrenergic receptor inhibits the activation of the NADPH oxidase and leads to the expression of catalase in macrophages and myometrial cells in an in vitro model of preterm labor (52). Furthermore, the role of PPARγ in the protection of muscle fibers against oxidative stress caused by excessive acute exercise in Sprague–Dawley rats has been demonstrated (53). Moreover, Liu Z. and coworkers established that PPARγ alleviates oxidative stress and adipose inflammation by binding to Mark4 promoter region in Kunming male mice (54). Over the past decade, several authors have demonstrated that during the early acute phase, ROS production could favor T. cruzi infection in macrophages (8, 55, 56). Based on these results, it could be suggested that HP24 affects the course of infection, since it inhibits the production of H2O2 and NO. However, in this work, we demonstrate that HP24 treatment does not affect the parasite load. These results are in agreement with a recent work where we showed that HP24 treatment affects neither parasitemia nor the number of amastigote nests in the heart of acutely infected mice (19).
It has been widely demonstrated that NF-κB is involved in the pro-inflammatory response in different models of T. cruzi infection (15, 29, 31, 57, 58). This transcriptional factor regulates the expression of several pro-inflammatory cytokines and chemokines such as TNFα, IL-1β, IL-6, and pro-inflammatory gene expression such as iNOS, COX2, and metalloproteases, among others (59). Several authors have shown that different PPARγ ligands exert anti-inflammatory effects through the inhibition of NF-κB-dependent inflammatory genes (60–63). In the same line of evidence, we demonstrated the anti-inflammatory properties of several PPAR ligands in different models of T. cruzi infection (15, 16, 29). Herein, we evidenced that HP24 inhibits NF-κB activation in T. cruzi-infected macrophages. Moreover, we showed that knockdown of PPARγ by specific siRNA impedes the HP24-mediated inhibition of NF-κB in macrophages infected with T. cruzi. These findings indicate the specificity of the HP24 effects and the inhibition of NF-κB in a PPARγ-dependent manner.
In Chagas disease, both the sustained increase in pro-inflammatory mediators as a result of parasite persistence and microvascular abnormalities are crucial aspects in the generation of cardiac damage. In this context, elucidating the mechanism of action of new PPARγ ligands as an alternative to thiazolidinediones is highly attractive since they may serve as possible adjuvants in anti-inflammatory therapy in combination with anti-parasitic treatments used currently, to avoid or delay irreversible tissue damage in the host.
Overall, this work demonstrates for the first time that this new PPARγ ligand, HP24, exerts anti-inflammatory and pro-angiogenic effects through PI3K/AKT/mTOR and PPARγ signaling (Figure 9).
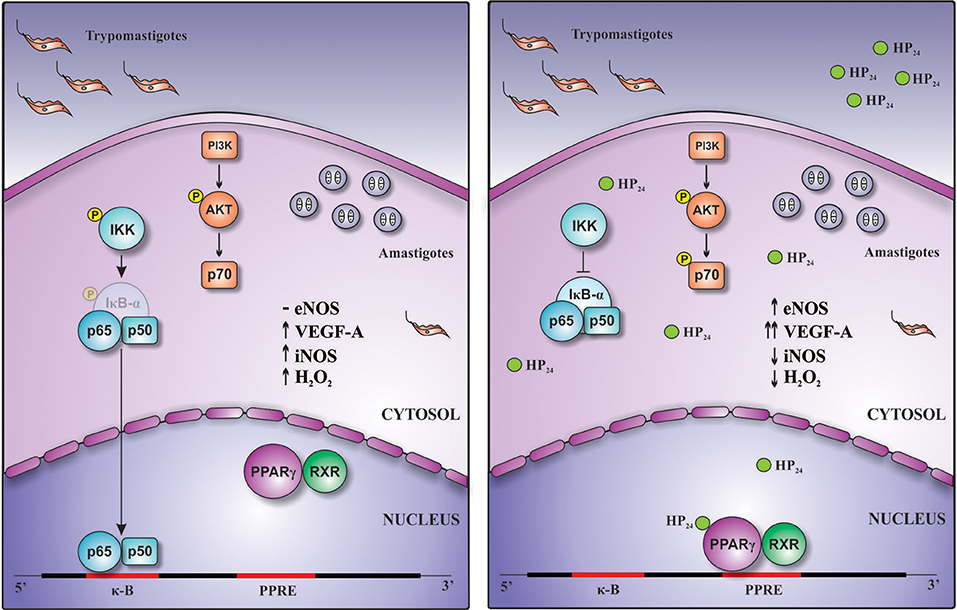
Figure 9. Schematic representation of the pro-angiogenic and anti-inflammatory actions of HP24 in T. cruzi-infected macrophages. Treatment of T. cruzi-infected macrophages with HP24 exerts pro-angiogenic effects, increasing the expression of eNOS and of VEGF-A. This effect depends on both PPARγ and PI3K/AKT/mTOR pathways. Besides, HP24 avoids activation of NF-κB, thereby preventing iNOS expression, NO release, and H2O2 production. This anti-inflammatory effect occurs in a PPARγ-dependent manner.
Data Availability Statement
All datasets generated for this study are included in the article.
Ethics Statement
The animal study was reviewed and approved by the Institutional Committee for the Care and Use of Laboratory Animals (CICUAL, School of Medicine, University of Buenos Aires, CD N° 2271/2014).
Author Contributions
NG and FP designed the experiment. FP, NG, ÁC, and GM contributed to the writing of the manuscript. FP, ÁC, MR, and AP did experiments. FP, NG, and GM analyzed the data. MF and DC provided the 3-hydroxy-4-pyridinecarboxylic acid derivative (HP24). NG, MS, GM, and FP contributed to the final approval of the version to be published.
Funding
This work was supported by the Universidad de Buenos Aires (Grant number: 20020170100562BA) and Agencia Nacional de Promoción Científica y Tecnológica (Grant number: PICT 2016-0629).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Mr. Eduardo Alejandro Giménez and Mr. Ricardo Chung for their technical assistance. We are grateful to Carla Pascuale, Ph.D., Pehuén Pereyra Gerber, Ph.D., and Fernando Erra Díaz, M.D., for their excellent technical assistance performing confocal microscopy analysis. The authors would also like to thank Mr. Sergio Mazzini for his assistance in English grammar and spelling corrections.
References
1. WHO - World Health Organization. Chagas Disease (American trypanosomiasis). WHO (2019). Available online at: https://www.who.int/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis) (accessed April 17, 2019).
2. López-Muñoz RA, Molina-Berríos A, Campos-Estrada C, Abarca-Sanhueza P, Urrutia-Llancaqueo L, Peña-Espinoza M, et al. Inflammatory and pro-resolving lipids in trypanosomatid infections: a key to understanding parasite control. Front Microbiol. (2018) 9:1961. doi: 10.3389/fmicb.2018.01961
3. Rassi A, Rassi A, Marin-Neto JA. Chagas disease. Lancet. (2010) 375:1388–402. doi: 10.1016/S0140-6736(10)60061-X
4. Trachtenberg BH, Hare JM. Inflammatory cardiomyopathic syndromes. Circ Res. (2017) 121:803–18. doi: 10.1161/CIRCRESAHA.117.310221
5. Sies H. Role of metabolic H2O2 generation: redox signaling and oxidative stress. J Biol Chem. (2014) 289:8735–41. doi: 10.1074/jbc.R113.544635
6. Estrada D, Specker G, Martínez A, Dias PP, Hissa B, Andrade LO, et al. Cardiomyocyte diffusible redox mediators control Trypanosoma cruzi infection: role of parasite mitochondrial iron superoxide dismutase. Biochem J. (2018) 475:1235–51. doi: 10.1042/BCJ20170698
7. Dias PP, Capila RF, Do Couto NF, Estrada D, Gadelha FR, Radi R, et al. Cardiomyocyte oxidants production may signal to T. cruzi intracellular development. PLoS Negl Trop Dis. (2017) 11:1–23. doi: 10.1371/journal.pntd.0005852
8. Paiva CN, Feijó DF, Dutra FF, Carneiro VC, Freitas GB, Alves LS, et al. Oxidative stress fuels Trypanosoma cruzi infection in mice. J Clin Invest. (2012) 122:2531–42. doi: 10.1172/JCI58525
9. Melo RCN. Acute heart inflammation: ultrastructural and functional aspects of macrophages elicited by Trypanosoma cruzi infection. J Cell Mol Med. (2009) 13:279–94. doi: 10.1111/j.1582-4934.2008.00388.x
10. Sanmarco LM, Eberhardt N, Ponce NE, Cano RC, Bonacci G, Aoki MP. New insights into the immunobiology of mononuclear phagocytic cells and their relevance to the pathogenesis of cardiovascular diseases. Front Immunol. (2018) 8:1921. doi: 10.3389/fimmu.2017.01921
11. Silva AR, Gonçalves-de-Albuquerque CF, Pérez AR, de Frias Carvalho V. Immune-endocrine interactions related to a high risk of infections in chronic metabolic diseases: the role of PPAR gamma. Eur J Pharmacol. (2019) 854:272–81. doi: 10.1016/j.ejphar.2019.04.008
12. Seed B, Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. (1998) 391:82. doi: 10.1038/34184
13. Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. (1998) 391:79–82. doi: 10.1038/34178
14. Tontonoz P, Nagy L, Alvarez JG., Thomazy VA, Evans RM. PPARγ promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. (1998) 93:241–52. doi: 10.1016/S0092-8674(00)81575-5
15. Penas F, Mirkin GA, Hovsepian E, Cevey Á, Caccuri R, Sales ME, et al. PPARγ ligand treatment inhibits cardiac inflammatory mediators induced by infection with different lethality strains of Trypanosoma cruzi. Biochim Biophys Acta Mol Basis Dis. (2013) 1832:239–48. doi: 10.1016/j.bbadis.2012.08.007
16. Hovsepian E, Mirkin GA, Penas F, Manzano A, Bartrons R, Goren NB. Modulation of inflammatory response and parasitism by 15-Deoxy-Δ(12,14) prostaglandin J(2) in Trypanosoma cruzi-infected cardiomyocytes. Int J Parasitol. (2011) 41:553–62. doi: 10.1016/j.ijpara.2010.12.002
17. Garcia-Vallvé S, Guasch L, Tomas-Hernández S, Del Bas JM, Ollendorff V, Arola L, et al. Peroxisome proliferator-activated receptor γ (PPARγ) and ligand choreography: newcomers take the stage. J Med Chem. (2015) 58:5381–94. doi: 10.1021/jm501155f
18. Brun P, Dean A, Di Marco V, Surajit P, Castagliuolo I, Carta D, et al. Peroxisome proliferator-activated receptor-γ mediates the anti-inflammatory effect of 3-hydroxy-4-pyridinecarboxylic acid derivatives: synthesis and biological evaluation. Eur J Med Chem. (2013) 62:486–97. doi: 10.1016/j.ejmech.2013.01.024
19. Penas FN, Carta D, Dmytrenko G, Mirkin GA, Modenutti CP, Cevey ÁC, et al. Treatment with a new peroxisome proliferator-activated receptor gamma agonist, pyridinecarboxylic acid derivative, increases angiogenesis and reduces inflammatory mediators in the heart of Trypanosoma cruzi-infected mice. Front Immunol. (2017) 8:1738. doi: 10.3389/fimmu.2017.01738
20. Jimenez R, Sanchez M, Zarzuelo MJ, Romero M, Quintela AM, Lopez-Sepulveda R, et al. Endothelium-dependent vasodilator effects of peroxisome proliferator-activated receptor agonists via the phosphatidyl-inositol-3 kinase-akt pathway. J Pharmacol Exp Ther. (2010) 332:554–61. doi: 10.1124/jpet.109.159806
21. Cho DH, Choi YJ, Jo SA, Jo I. Nitric oxide production and regulation of endothelial nitric-oxide synthase phosphorylation by prolonged treatment with troglitazone: evidence for involvement of peroxisome proliferator-activated receptor (PPAR) γ-dependent and PPARγ-independent signaling. J Biol Chem. (2004) 279:2499–506. doi: 10.1074/jbc.M309451200
22. Ryan MJ, Didion SP, Mathur S, Faraci FM, Sigmund CD. PPARγ agonist rosiglitazone improves vascular function and lowers blood pressure in hypertensive transgenic mice. Hypertension. (2004) 43:661–6. doi: 10.1161/01.HYP.0000116303.71408.c2
23. Silva MC, Davoli-Ferreira M, Medina TS, Sesti-Costa R, Silva GK, Lopes CD, et al. Canonical PI3Kγ signaling in myeloid cells restricts Trypanosoma cruzi infection and dampens chagasic myocarditis. Nat Commun. (2018) 9:1513. doi: 10.1038/s41467-018-03986-3
24. Chuenkova MV, PereiraPerrin M. Trypanosoma cruzi targets Akt in host cells as an intracellular antiapoptotic strategy. Sci Signal. (2009) 2:ra74. doi: 10.1126/scisignal.2000374
25. Mirkin GA, Jones M, Sanz OP, Rey R, Sica RE, González Cappa SM. Experimental Chagas' disease: electrophysiology and cell composition of the neuromyopathic inflammatory lesions in mice infected with a myotropic and a pantropic strain of Trypanosoma cruzi. Clin Immunol Immunopathol. (1994) 73:69–79. doi: 10.1006/clin.1994.1171
26. Penas F, Mirkin GA, Vera M, Cevey ÁC, González CD, Gómez MI, et al. Treatment in vitro with PPARα and PPARγ ligands drives M1-to-M2 polarization of macrophages from T. cruzi-infected mice. Biochim Biophys Acta. (2015) 1852:893–904. doi: 10.1016/j.bbadis.2014.12.019
27. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. (2008) 3:1101–8. doi: 10.1038/nprot.2008.73
28. Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. (2009) 55:611–22. doi: 10.1373/clinchem.2008.112797
29. Cevey ÁC, Mirkin GA, Donato M, Rada MJ, Penas FN, Gelpi RJ, et al. Treatment with fenofibrate plus a low dose of benznidazole attenuates cardiac dysfunction in experimental chagas disease. Int J Parasitol Drugs Drug Resist. (2017) 7:378–87. doi: 10.1016/j.ijpddr.2017.10.003
30. Kruger NJ. The Bradford method for protein quantitation. Methods Mol Biol. (1994) 32:9–15. doi: 10.1385/0-89603-268-X:9
31. Cevey ÁC, Mirkin GA, Penas FN, Goren NB. Low-dose benznidazole treatment results in parasite clearance and attenuates heart inflammatory reaction in an experimental model of infection with a highly virulent Trypanosoma cruzi strain. Int J Parasitol Drugs Drug Resist. (2016) 6:12–22. doi: 10.1016/j.ijpddr.2015.12.001
32. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. (2012) 9:676–82. doi: 10.1038/nmeth.2019
33. Bryan NS, Grisham MB. Methods to detect nitric oxide and its metabolites in biological samples. Free Radic Biol Med. (2007) 43:645–57. doi: 10.1016/j.freeradbiomed.2007.04.026
34. Díaz-Guerra MJ, Velasco M, Martín-Sanz P, Boscá L. Evidence for common mechanisms in the transcriptional control of type II nitric oxide synthase in isolated hepatocytes. Requirement of NF-kappaB activation after stimulation with bacterial cell wall products and phorbol esters. J Biol Chem. (1996) 271:30114–20. doi: 10.1074/jbc.271.47.30114
35. Davies EM, Gurung R, Le KQ, Mitchell CA. Effective angiogenesis requires regulation of phosphoinositide signaling. Adv Biol Regul. (2019) 71:69–78. doi: 10.1016/j.jbior.2018.11.008
36. Rodrigues WF, Miguel CB, Chica JEL, Napimoga MH. 15d-PGJ(2) modulates acute immune responses to Trypanosoma cruzi infection. Memórias do Inst Oswaldo Cruz. (2010) 105:137–43. doi: 10.1590/S0074-02762010000200005
37. Huang PH, Sata M, Nishimatsu H, Sumi M, Hirata Y, Nagai R. Pioglitazone ameliorates endothelial dysfunction and restores ischemia-induced angiogenesis in diabetic mice. Biomed Pharmacother. (2008) 62:46–52. doi: 10.1016/j.biopha.2007.06.014
38. Zhao Z, Luo Z, Wang P, Sun J, Yu H, Cao T, et al. Rosiglitazone restores endothelial dysfunction in a rat model of metabolic syndrome through PPAR - and PPAR δ-dependent phosphorylation of Akt and eNOS. PPAR Res. (2011) 2011:291656. doi: 10.1155/2011/291656
39. Hazeki K, Nigorikawa K, Hazeki O. Role of phosphoinositide 3-kinase in innate immunity. Biol Pharm Bull. (2007) 30:1617–23. doi: 10.1248/bpb.30.1617
40. Cantley LC. The phosphoinositide 3-kinase pathway. Science. (2002) 296:1655–7. doi: 10.1126/science.296.5573.1655
41. Wilkowsky SE, Barbieri MA, Stahl P, Isola ELD. Trypanosoma cruzi: Phosphatidylinositol 3-kinase and protein kinase B activation is associated with parasite invasion. Exp Cell Res. (2001) 264:211–8. doi: 10.1006/excr.2000.5123
42. Todorov AG, Einicker-Lamas M, De Castro SL, Oliveira MM, Guilherme A. Activation of host cell phosphatidylinositol 3-kinases by Trypanosoma cruzi infection. J Biol Chem. (2000) 275:32182–6. doi: 10.1074/jbc.M909440199
43. Wang ZJ, Zhang H-B, Chen C, Huang H, Liang JX. Effect of PPARG on AGEs-induced AKT/MTOR signaling-associated human chondrocytes autophagy. Cell Biol Int. (2018) 42:841–8. doi: 10.1002/cbin.10951
44. Lv S, Wang W, Wang H, Zhu Y, Lei C. PPARγ activation serves as therapeutic strategy against bladder cancer via inhibiting PI3K-Akt signaling pathway. BMC Cancer. (2019) 19:204. doi: 10.1186/s12885-019-5426-6
45. Wang J, Pang T, Hafko R, Benicky J, Sanchez-Lemus E, Saavedra JM. Telmisartan ameliorates glutamate-induced neurotoxicity: roles of AT 1 receptor blockade and PPARγ activation. Neuropharmacology. (2014) 79:249–61. doi: 10.1016/j.neuropharm.2013.11.022
46. Kitagishi Y, Matsuda S. Diets involved in PPAR and PI3K/AKT/PTEN pathway may contribute to neuroprotection in a traumatic brain injury. Alzheimer's Res Ther. (2013) 5:42. doi: 10.1186/alzrt208
47. Aljada A, O'Connor L, Fu YY, Mousa SA. PPARγ ligands, rosiglitazone and pioglitazone, inhibit bFGF- and VEGF-mediated angiogenesis. Angiogenesis. (2008) 11:361–7. doi: 10.1007/s10456-008-9118-0
48. Goetze S, Eilers F, Bungenstock A, Kintscher U, Stawowy P, Blaschke F, et al. PPAR activators inhibit endothelial cell migration by targeting Akt. Biochem Biophys Res Commun. (2002) 293:1431–7. doi: 10.1016/S0006-291X(02)00385-6
49. López-Peláez M, Soria-Castro I, Boscá L, Fernández M, Alemany S. Cot/tpl2 activity is required for TLR-induced activation of the Akt p70 S6k pathway in macrophages: implications for NO synthase 2 expression. Eur J Immunol. (2011) 41:1733–41. doi: 10.1002/eji.201041101
50. Paiva CN, Medei E, Bozza MT. ROS and Trypanosoma cruzi: fuel to infection, poison to the heart. PLOS Pathog. (2018) 14:e1006928. doi: 10.1371/journal.ppat.1006928
51. Wen JJ, Gupta S, Guan Z, Dhiman M, Condon D, Lui C, Garg NJ. Phenyl-α-tert-butyl-nitrone and benzonidazole treatment controlled the mitochondrial oxidative stress and evolution of cardiomyopathy in chronic chagasic rats. J Am Coll Cardiol. (2010) 55:2499–508. doi: 10.1016/j.jacc.2010.02.030
52. Hadi T, Douhard R, Dias AMM, Wendremaire M, Pezzè M, Bardou M, et al. Beta3 adrenergic receptor stimulation in human macrophages inhibits NADPHoxidase activity and induces catalase expression via PPARγ activation. Biochim Biophys Acta Mol Cell Res. (2017) 1864:1769–84. doi: 10.1016/j.bbamcr.2017.07.003
53. Kim JS, Lee YH, Chang YU, Yi HK. PPARγ regulates inflammatory reaction by inhibiting the MAPK/NF-κB pathway in C2C12 skeletal muscle cells. J Physiol Biochem. (2017) 73:49–57. doi: 10.1007/s13105-016-0523-3
54. Liu Z, Gan L, Chen Y, Luo D, Zhang Z, Cao W, et al. Mark4 promotes oxidative stress and inflammation via binding to PPARγ and activating NF-κB pathway in mice adipocytes. Sci Rep. (2016) 6:21382. doi: 10.1038/srep21382
55. Goes GR, Rocha PS, Diniz ARS, Aguiar PHN, Machado CR, Vieira LQ. Trypanosoma cruzi needs a signal provided by reactive oxygen species to infect macrophages. PLoS Negl Trop Dis. (2016) 10:e0004555. doi: 10.1371/journal.pntd.0004555
56. Paiva CN, Bozza MT. Are reactive oxygen species always detrimental to pathogens? Antioxid Redox Signal. (2014) 20:1000–37. doi: 10.1089/ars.2013.5447
57. Suman S, Rachakonda G, Mandape SN, Sakhare SS, Villalta F, Pratap S, et al. Phospho-proteomic analysis of primary human colon epithelial cells during the early Trypanosoma cruzi infection phase. PLoS Negl Trop Dis. (2018) 12:e0006792. doi: 10.1371/journal.pntd.0006792
58. Wan X, Wen JJ, Koo SJ, Liang LY, Garg NJ. SIRT1-PGC1α-NFκB pathway of oxidative and inflammatory stress during Trypanosoma cruzi infection: benefits of SIRT1-targeted therapy in improving heart function in chagas disease. PLoS Pathog. (2016) 12:1–24. doi: 10.1371/journal.ppat.1005954
59. Pahl HL. Activators and target genes of Rel/NF-κB transcription factors. Oncogene. (1999) 18:6853–66. doi: 10.1038/sj.onc.1203239
60. Hong O-Y, Youn HJ, Jang H-Y, Jung SH, Noh E-M, Chae HS, et al. Troglitazone inhibits matrix metalloproteinase-9 expression and invasion of breast cancer cell through a peroxisome proliferator-activated receptor γ-dependent mechanism. J Breast Cancer. (2018) 21:28. doi: 10.4048/jbc.2018.21.1.28
61. Chen C, Peng S, Chen F, Liu L, Li Z, Zeng G, et al. Protective effects of pioglitazone on vascular endothelial cell dysfunction induced by high glucose via inhibition of IKKα/β-NFκB signaling mediated by PPARγ in vitro. Can J Physiol Pharmacol. (2017) 95:1480–7. doi: 10.1139/cjpp-2016-0574
62. Straus DS, Pascual G, Li M, Welch JS, Ricote M, Hsiang CH, et al. 15-deoxy-delta 12,14-prostaglandin J2 inhibits multiple steps in the NF-kappa B signaling pathway. Proc Natl Acad Sci USA. (2000) 97:4844–9. doi: 10.1073/pnas.97.9.4844
Keywords: new PPARγ ligand, PI3K/AKT/mTOR, NF-κB pathway, Trypanosoma cruzi, macrophages
Citation: Penas FN, Carta D, Cevey ÁC, Rada MJ, Pieralisi AV, Ferlin MG, Sales ME, Mirkin GA and Goren NB (2020) Pyridinecarboxylic Acid Derivative Stimulates Pro-Angiogenic Mediators by PI3K/AKT/mTOR and Inhibits Reactive Nitrogen and Oxygen Species and NF-κB Activation Through a PPARγ-Dependent Pathway in T. cruzi-Infected Macrophages. Front. Immunol. 10:2955. doi: 10.3389/fimmu.2019.02955
Received: 08 August 2019; Accepted: 02 December 2019;
Published: 09 January 2020.
Edited by:
Hugo Caire Castro-Faria-Neto, Oswaldo Cruz Foundation (Fiocruz), BrazilReviewed by:
Alexandre Morrot, Federal University of Rio de Janeiro, BrazilZsuzsa Szondy, University of Debrecen, Hungary
Copyright © 2020 Penas, Carta, Cevey, Rada, Pieralisi, Ferlin, Sales, Mirkin and Goren. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nora Beatriz Goren, bmdvcmVuQGZtZWQudWJhLmFy