 Manolo Sambucci
Manolo Sambucci Francesca Gargano
Francesca Gargano Luca Battistini
Luca Battistini Giovanna Borsellino
Giovanna Borsellino
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 20 December 2019
Sec. T Cell Biology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02947
This article is part of the Research Topic Control of Regulatory T cell Stability, Plasticity and Function in Health and Disease View all 21 articles
As the Nobel laureate Luigi Pirandello wrote in his novels, identities can be evanescent. Although a quarter of a century has passed since regulatory T cells (Treg) were first described, new studies continue to reveal surprising and contradictory features of this lymphocyte subset. Treg cells are the core of the immunological workforce engaged in the restraint of autoimmune or inflammatory reactions, and their characterization has revealed substantial heterogeneity and complexity in the phenotype and gene expression profiles, proving them to be a most versatile and adaptive cell type, as exemplified by their plasticity in fine-tuning immune responses. Defects in Treg function are associated with several autoimmune diseases, including multiple sclerosis, which is caused by an inappropriate immune reaction toward brain components; conversely, the beneficial effects of immunomodulating therapies on disease progression have been shown to partly act upon the biology of these cells. Both in animals and in humans the pool of circulating Treg cells is a mixture of natural (nTregs) and peripherally-induced Treg (pTregs). Particularly in humans, circulating Treg cells can be phenotypically subdivided into different subpopulations, which so far are not well-characterized, particularly in the context of autoimmunity. Recently, Treg cells have been rediscovered as mediators of tissue healing, and have also shown to be involved in organ homeostasis. Moreover, stability of the Treg lineage has recently been addressed by several conflicting reports, and immune-suppressive abilities of these cells have been shown to be dynamically regulated, particularly in inflammatory conditions, adding further levels of complexity to the study of this cell subset. Finally, Treg cells exert their suppressive function through different mechanisms, some of which—such as their ectoenzymatic activity—are particularly relevant in CNS autoimmunity. Here, we will review the phenotypically and functionally discernible Treg cell subpopulations in health and in multiple sclerosis, touching also upon the effects on this cell type of immunomodulatory drugs used for the treatment of this disease.
The immune system is called to respond to the environment at each beat of the heart, at each breath, and at each breaching of the organism's surface. In a healthy organism, should an exogenous and potentially dangerous entity gain access, the immediate response of the immune system's defense mechanisms is activated, and clearance of the pathogen is achieved, millions of times a day (1). In addition, immune cells interact continuously with the trillions of bacteria, fungi, and viruses which inhabit, peacefully, the intestines. The interactions with the intestinal microbiota are the second process, after thymic selection, that shapes the immune system. This notion has become clearer in recent years with the knowledge that dysbiosis, an alteration of the physiological composition of the microbial community, underlies the generation of an inflammatory environment which may raise the level of basal immune cell activation thus predisposing to the initiation of inappropriate and uncontrolled immune responses, leading to chronic inflammation and disease (2).
Normally, immune reactions are kept in check by multiple mechanisms which control the magnitude of the response, so escalation to overt inflammation and tissue damage are avoided. However, additional stimuli which may activate immune T cells come also from within, since all the organism's tissues are also highly immunogenic, as exemplified by the immediate rejection of transplants between incompatible individuals. Conveniently, efficient mechanisms of selection during T cell development in the thymus are in place for purging autoreactive cells from the repertoire, and clonal deletion eliminates most of them (3, 4). Inevitably, though, some autoreactive T cells escape this negative selection and circulate in the periphery, and indeed they have been shown to be present at similar frequencies both in healthy individuals and in those afflicted by autoimmune diseases (5–7). But autoimmunity leading to pathology represents the exception rather than the rule, so clearly these autoreactive cells are kept in check in the periphery. Several immune mechanisms exist which mediate restraint of autoreactivity (8), and among these are FoxP3+ T regulatory (Treg) cells, whose features and roles in the aberrant immune response which underlies multiple sclerosis (MS) will be discussed.
Treg cells were initially described as a CD4+CD25high T cell population whose depletion is associated with the development of severe multi-organ autoimmunity, and were defined by the expression of the transcription factor forkhead box P3 (FoxP3) (9–11).
FoxP3 is capable of binding the astounding number of 2,800 genomic sites, and interacts with more than 360 factors to form a large protein complex (12–15). Four evolutionary conserved intronic cis-elements in the FoxP3 locus regulate the foxp3 gene expression: conserved non-coding sequence (CNS) 3 is indispensable for the initiation of FoxP3 transcription through the recruitment of c-Rel; CNS2 enables the stable expression of FoxP3 in actively proliferating Tregs, and CNS1 is key for the extrathymic induction of Tregs in the periphery, and contains binding sites for TGF-β (16). Last to be discovered, but actually the pioneering element, is CNS0, crucial for the establishment of the earliest epigenetic modification controlling FoxP3 expression (17). Interestingly, methylation at these crucial sites is affected by cytokine signaling and by environmental cues, thus it is possible that the inflammation which accompanies autoimmunity may have an impact on this “basic” epigenetic regulation and stability of FoxP3 (18).
Stable FoxP3 expression also relies on epigenetic modifications of the Treg-specific demethylated region (TDSR), a non-coding region in the first intron of the foxP3 gene locus (19, 20), and this has become the marker of “true” Treg cells, allowing discrimination from activated CD4+CD25+FoxP3+ cells. The presence of DNA hypomethylation at Treg signature genes contributes to the maintenance of lineage stability, and does not occur in activated cells which transiently express FoxP3 and which lack suppressive abilities (21).
Additionally, similar to most transcription factors, FoxP3's function can be modulated by post-translational modifications (such as ubiquitination, acetylation, and phosphorylation), which couple extracellular cues to adjustments of transcriptional programmes [for a review see (22, 23)].
In humans, several splicing variants of FoxP3 have been described (24). The splicing variant containing exon 2 (FoxP3-E2) is the better equipped for interaction with RORα and RORγt, two transcription factors involved in Th17 specification (25, 26). Metabolic and cytokinic factors determine alternative splicing, and we and others have shown that, in patients with MS, Treg cells express reduced levels of FoxP3-E2 and are thus deprived of an auxiliary level of regulation (27, 28).
The Treg phenotype needs to be “locked in” and stabilized, since these cells are self-reactive and their conversion into conventional effector cells would unleash a dangerous army of autoimmune effectors (29). So how do Treg cells resist acquisition of conventional T (Tconv) cell properties, in inflammatory environments? FoxP3 prevents the expression of genes encoding effector cytokines by acting as a repressor or an activator and through the physical interaction with other transcription factors (30, 31). These aspects are discussed below.
In vivo, most Treg cells develop in the thymus (tTreg) during positive selection as an alternative to conventional CD4+ T cells, but they also may be generated in the periphery (pTreg) through extra-thymic conversion of conventional T cells.
In the thymus, T cells recognizing self-antigens are deleted, except for a small fraction of cells, which is pushed down the path of Treg differentiation, largely due to the avidity of the MHC-peptide interaction that their TCR establishes with cortical thymocytes (32). Specifically, diversion in the Treg lineage occurs when the affinity of the TCR for self-peptides ligands falls between the range of positive selection of conventional T cells and negative selection of T cells carrying TCRs with high affinity. Thus, strong TCR signaling is the prerequisite for Treg development (33). In addition to TCR signaling, other factors, such as signaling through CD28 (34), the presence of IL-2 (35) and of TGF-β (36) are required for Treg differentiation. Following this first step of TCR-dependent maturation, CD25 upregulation occurs, making tTreg precursors responsive to the IL-2 signals, which are transduced by STAT5 and are necessary for the induction of the master regulator FoxP3 (37). Interestingly, in the thymus there is limited availability of IL-2, which may be further reduced following sequestration by mature activated Treg cells recirculating to the thymus and limiting further generation of Treg cells (38). Acquisition of FoxP3 expression by thymocytes coincides with the emergence of phenotypic and functional features of mature peripheral Tregs, including inability to produce IL-2 and emergence of suppressive abilities. Genome-wide analyses of FoxP3 have shown that it binds to ~700 genes and miRNAs involved in multiple networks, acting both as a repressor and an activator (14, 39); FoxP3 also forms multiprotein complexes with over 360 proteins and acts in cooperation with FoxP1, another member of the FoxP family, in the regulation of gene expression (40, 41).
However, FoxP3 expression alone is not sufficient to establish the full Treg cell phenotype, as indicated by the fact that ectopic expression of FoxP3 fails to induce a large proportion of Treg cell signature genes, and T cell activation in humans transiently induces high levels of FoxP3 but not suppressive abilities (42–44). Thus, in addition to FoxP3 expression, a second process is necessary for the full and stable development of the Treg lineage, and that is the establishment of a specific epigenetic signature (45–47). Tregs in fact exhibit a specific DNA hypomethylation pattern (48) particularly at Treg cell signature genes, such as FoxP3, CTLA4, IL2ra, and Ikfz2. Chromatin accessibility of the genes defining Treg cell identity is facilitated by activation of super-enhancers (SE) in Treg precursors, and the genomic organizer Satb1 has a pioneering effect through “preconditioning” of the chromatin landscape and binding to Treg-SE at the early stages of thymic Treg development (17). Interestingly, the SATB1 locus contains single nucleotide polymorphisms (SNPs) associated with MS (18), suggesting that alterations in the initial events that lead to the generation of Tregs may contribute to genetic susceptibility to immune dysregulation and to disease development. The presence of DNA hypomethylation at Treg signature genes contributes to the maintenance of lineage stability, and does not occur in activated cells which transiently express FoxP3 and which lack suppressive abilities (21).
In addition to Tregs generated in the thymus, peripheral conversion of Treg cells occurs in some organs, such as the colon, where pTregs emerge following encounter with commensal bacteria and their metabolites (49–53), and in the placenta, where they mitigate maternal reactivity to the fetus (54, 55). FoxP3 induction is dependent on the FoxP3 enhancer CNS1, and selective ablation of pTregs in CNS−/− mice induces spontaneous development of pronounced Th2-type inflammation in the gastrointestinal tract and lungs, with concomitant alterations in the composition of the gut microbiota (16, 56).
It has long been known that thymectomy before day 3 after birth induces severe autoimmunity, indicating that pTregs alone are insufficient for the maintenance of cell tolerance (11). Current thinking is that pTregs suppress inflammation at mucosal barriers where they are key contributors to the maintenance of tolerance to a vast array of foreign antigens derived from the microbiota and from the diet, while tTregs control immune responses to self antigens (56, 57). A healthy immune system results from the balanced interaction between the antigenic blast present at barrier surfaces and the active suppression provided by Treg cells. The distinction between pTregs and tTreg is only one of several that can segregate these cells in different subsets, in a graded scale of differentiation and function that we describe below.
In humans, all circulating CD4+FoxP3+ tTreg cells are characterized by high levels of CD25 and low CD127. However, they comprise different phenotypic subsets, which can be separated based on expression of CD45RA and CD25 in naïve (CD45RA+CD25+FoxP3+) and effector (CD45RA−CD25+FoxP3+) cells; a third subset, which expresses low levels of FoxP3, contains a mixed population of recently activated Tconv cells which do not have suppressive functions and produce cytokines at high levels (44). Naïve Tregs are likely recent thymic emigrants, and accordingly a high fraction express CD31 (58); following activation they differentiate in effector Tregs (44). Effector Tregs express high levels of suppressive molecules, such as CTLA4. A recent study investigating the proteomics and transcriptomics of these subsets has defined both a common Treg and an effector Treg signature, which is maintained even after stimulation and expansion in vitro. The main features of the Treg signature are the stability of FoxP3 expression and the inability to produce effector cytokines, the latter characteristic being achieved by the low abundance of transcription factors, such as STAT4, a major activator of IFNγ transcription (59).
The effector and naïve phenotypes are associated with differential expression of other molecules, such as CD39, CCR6, HLA-DR, GITR, LAG3 (60, 61), which are involved in the suppressive function and migratory ability of Tregs, and which may be expressed at different times during the cells' life.
ICOS expression defines two other Treg subsets, and is associated with ability to secrete IL-10 together with TGF-β. FoxP3+ cells receive a different imprinting during thymic development, depending on the presence or absence of costimulation through ICOS and on the thymic antigen presenting cell they interact with (62).
A specialized subset of Treg cells expresses TIGIT, a coinhibitory molecule, and gene profiling of TIGIT+ and TIGIT− Treg cells indicate that TIGIT+ cells have the attributes of a highly suppressive population specialized in suppression of Th1 and Th17 responses, with increased levels of expression of CTLA4, PD-1, Tim-3, and in humans also of CD39 and CCR6 (63, 64).
Thus, Treg cells can be defined on the basis of the expression of surface markers which identify distinct stages of differentiation and which denote the suppressive competence of each subset.
Conventional T cells differentiate into distinct types of effector cells when exposed to particular combinations of cytokines produced by innate immune cells following infection with specific classes of pathogens. The expression of lineage-specific transcription factors and epigenetic modifications underlies the full differentiation of distinct T cell subsets which can optimally deal with the invading pathogens (65). Several Th subsets have been identified to date, and following the initial discovery of the Th1 and Th2 subsets, Th9, Th17, Th22, and Tr1 subsets have been subsequently described. The effector function of these Th subsets mainly consists in the specialized secretion of the most appropriate set of cytokines for each category of challenge, so Th1 cells produce mainly IFNγ in their fight against intracellular bacteria, Th2 cells secrete IL-4, IL-15, and IL-13 to contrast helminths and parasites, and IL-17 produced by Th17 cells protects from fungal infections.
It soon became clear, both in the human and in the murine systems, that also Treg cells have a considerable degree of plasticity, exemplified by the co-expression of multiple transcription factors typical of other CD4 T cell lineages (66). This allows Treg cells to adapt to their location and to be equipped with the surface receptors, effector molecules, and metabolic assets matching those expressed by activated effector cells (Figure 1). Indeed, Tregs are able to position themselves in all tissues, wherever immune cells are activated and need to be regulated. Through the vast arrays of chemokine receptors and adhesion molecules they can swarm to non-lymphoid tissues just as conventional T cells do (67), showing that also Tregs have an inherent flexibility which enables adaptation to changing environments and focused immune regulation.
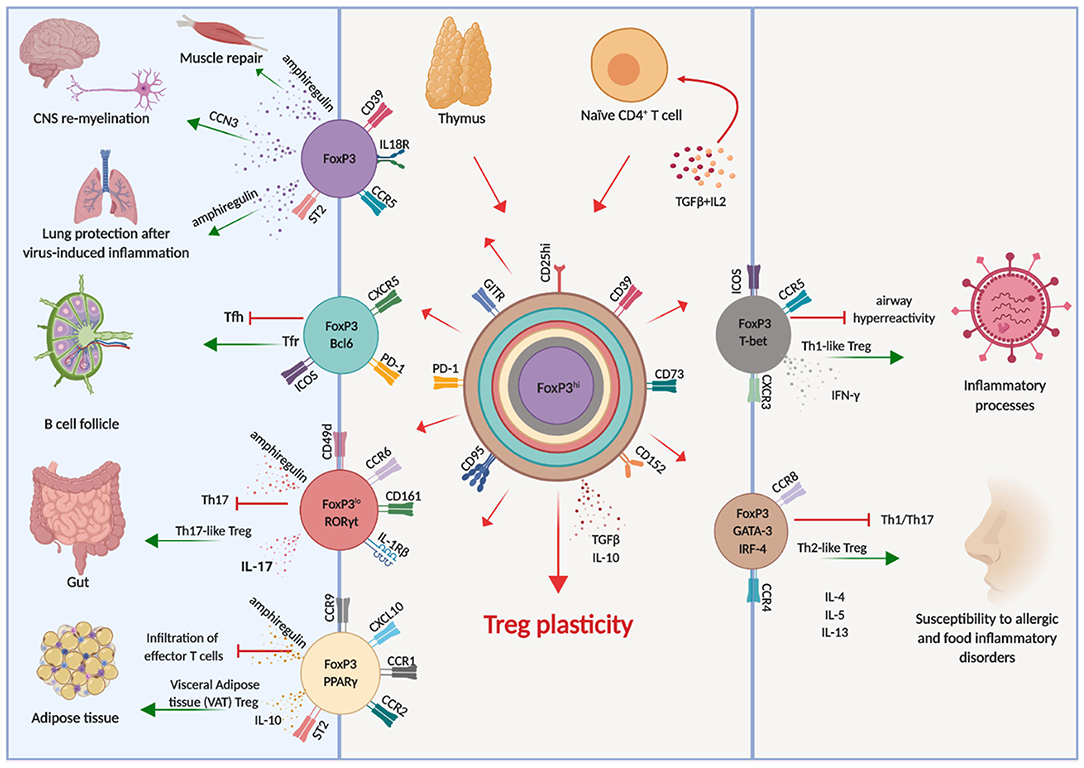
Figure 1. Treg cell plasticity. Treg cells have the potential to acquire expression of the molecular machinery which enables them to adapt to different environments, shadowing effector T cells and tissue-resident cells. This potential is schematically illustrated by the different colors, each corresponding to distinct cellular localization. Arrows indicate the known pathways of functional differentiation of Tregs and the tissues and organs where they can localize.
For instance, in response to IFNγ Treg cells express T-bet, which in turn induces expression of CXCR3 and Treg cell accumulation at sites of Th1 mediated inflammation (68), and loss of T-bet-expressing Treg cells causes severe Th1 autoimmunity (69). Relevant to immune reactions occurring in the brain, the CXCR3 chemokine receptor guides cells who express it toward CXCL9, CXCL10, and CXCL11, which are released by both neural and immune cells. Once T cells, both conventional and regulatory, have entered the brain, they are retained by CXCR3 in the perivascular zone rather than spreading in the white matter. This restricts the area of inflammation and favors the interactions between cells, enabling cell contact-dependent mechanisms of immune regulation (70).
Similarly, the microbiota in the intestine induces Treg cells expressing RORγt and STAT3 (71–73), which colocalize with RORγt-expressing Th17 cells. Moreover, high levels of expression of interferon regulatory factor-4 (IRF-4), one of the transcription factors involved in Th2 differentiation, is dependent on FoxP3 expression and is required to efficiently restrain Th2 responses (74). Treg cells can also acquire expression of Bcl6 which induces CXCR5, enabling them to accumulate in germinal centers and to regulate humoral immunity, including B cell affinity maturation and plasma cell differentiation (75).
Although Tregs respond to inflammatory cues and express the master transcriptional factors of Tconv cells, they do not produce the respective cytokines and they maintain stable expression of FoxP3 (56, 76, 77), although it is possible, in conditions of extreme inflammation, that FoxP3 expression is lost and conversion to conventional cytokine-producing cells is reached (78–80). In these cases, “ex-FoxP3” cells acquire the ability to produce IFNγ and are pathogenic in the experimental model of MS, experimental autoimmune encephalomyelitis (EAE) (78).
To—literally—gain a picture of Treg's mode of action, a good starting point is the definition of their tissue distribution and, more microscopically, of their positioning relative to the other immune cells during immune responses. tTregs circulate in the periphery and populate peripheral lymph nodes, where at the steady state they constitute 10–15% of the CD4 cell pool (81, 82). Here, Tregs aggregate in clusters with activated autoreactive effector CD4 T cells, which they can effectively “govern” given their close proximity, thus suppressing incipient autoimmunity (83). An interesting study showed that the first responders to the IL-2 produced in the initial phase of the immune responses are STAT5+ Treg cells, which are promptly activated and form a “safety net” constraining subsequent activation of conventional T cells (84). Indeed, Treg express very high levels of CD25, which enables them to capture IL-2, thereby depriving conv T cells of the primary cytokine which sustains their proliferation, and inducing apoptosis (85). This ability has earned Treg cells the epithet “IL-2 sink,” although subsequent studies challenge the notion of IL-2 deprivation as a prominent mechanism of suppression, at least in vivo (86).
Once they are positioned in close proximity of the cells they need to regulate, Tregs use different molecules to oppose Tconv activation, the most prominent of which is CTLA4, which captures its ligands CD80 and CD86 thus making them unavailable for binding to CD28 on Tconv cells (87). Moreover, CTLA4 has a higher affinity for CD80 and CD86 than does CD28, thus CTLA4+ Tregs outcompete CD28-expressing T conv cells for interaction with Dendritic Cells (DCs) (88). Also, CTLA4 induces upregulation of the tryptophan catabolizing enzyme indoleamine2,3-dioxygenase (IDO) by DC, inducing their regulatory phenotype (89). This modulation of antigen presenting cell maturation and function represents a major mechanism used by Treg cells to suppress immune responses.
Endangered or dying cells release potent proinflammatory stimuli, such as ATP, which Treg cells are able to catabolize through the ectoenzymes CD39 and CD73, which degrade ATP to AMP and adenosine (90, 91). Nucleotide catabolism achieves dampening of the immune response through both the removal of an inflammatory molecule (ATP) and the generation of highly immunosuppressive adenosine, which can bind to its receptors expressed on the surface of all immune cells (92, 93). CD39 expression identifies Tregs with an effector-memory phenotype, and these cells are more stable and present increased functionality (94). This pathway is of great interest as in addition to a general downregulation of aspecific inflammation, it also impacts on the generation of pathogenic Th17 cells. ATP is a potent activator of the inflammasome, with consequent release of the proinflammatory cytokine IL1β (95), and it also induces IL-23 release by a subset of DCs (96), two cytokines involved in Th17 differentiation. Moreover, ATP is directly involved in the generation of Th17 cells in the intestine (97), the major site of antigenic challenge and consequent cell activation. Thus, CD39+ Tregs cells at sites of inflammation may contrast the generation of pathogenic effectors. Interestingly, CD39 expression can also be induced in Th17 cells conferring immune-suppressive properties (98), and during the resolution of inflammation, Th17 cells can transdifferentiate into Tregs (99), with CD39 expression correlating with the ability to produce IL-10 (100). Thus, Th17 and Treg cells' functions intersect also in this pathway, and in this regard it is interesting to notice that while human Treg cells only express CD73 at minimal levels, if any, Th17 in inflammatory sites have high levels of CD73 on their surface (101); thus, the generation of immune-suppressive adenosine through the concerted action of CD39 and CD73 may occur when Th17 and Treg cells are in close proximity, as happens in sites of immune regulation (83).
Other mechanisms of action include the release of immune-suppressive cytokines, such as TGF-β (102), IL-10 (103), IL-35 (104), and of cytotoxic molecules, such as perforin (105), granzyme (106), and cAMP (107, 108).
Treg cells may also acquire tissue-specific abilities, not limited to immune suppression. Tregs stably express high levels of the enzyme 15-hydroxyprostaglandin dehydrogenase, particularly in the visceral adipose tissue, which enables them to catabolize PGE2 into 15-keto-PGE2, which in turn inhibits the proliferation of CD4+ Tconv cells (109). In the muscle, Tregs participate to tissue repair through the secretion of amphiregulin, which facilitates the regeneration of muscle satellite cells (110); similarly, amphiregulin production by Treg cells helps contain the damage to lung tissue following infection (111).
In the CNS, in a mouse model of lysolecithine-induced focal demyelination, Treg cells have been shown promote oligodendrocyte differentiation and remyelination through the release of CCN3, a growth-regulatory protein usually expressed in the developing brain (112). Moreover, ablation of Treg cells during acute spinal cord injury delays tissue remodeling (113).
The skin, another barrier site constantly exposed to exogenous antigens, hosts significant numbers of Treg cells (114), although curiously these are mainly represented by tTregs which colonize this tissue shortly after birth (115). Skin Treg have been shown to facilitate cutaneous wound healing and epithelial stem cell differentiation through expression of Jagged 1 (116), and to regulate fibroblast activation and fibrosis through expression of a skewed transcriptional programme dominated by GATA3 (117).
Thus, Treg cells can adopt several mechanisms for the down modulation of immune responses at both in lymphoid and non-lymphoid tissues, which generally safeguard against pathological and autoimmune reactions.
In the words of Blaser, the microbiota is at the same time self and non-self, being part of our biology but rapidly changing in response to external stimuli (118). While tolerance to self-antigens is instructed in the thymus, post-thymic education to tolerate foreign antigens occurs in the intestine, which is colonized by trillions of microbes with whom a healthy symbiotic relationship is established. The immune system shapes and preserves the ecology of the microbiota, which in turn tunes and calibrates immune cells, in a continuous homeostatic dialogue (119, 120). Changes in diet, improved sanitation, and mindless use of antibiotics have had a profound impact on the microbiota composition of contemporary populations in western countries, reducing diversity and promoting enrichment with bacterial strains capable of inducing inflammation, mainly through the generation of Th17 cells, but also through decreased induction of suppressive Tregs. These changes in microbiota composition are thought to contribute significantly to the increase in autoimmune and inflammatory diseases observed in recent times (2, 121).
Gut Tregs, which represent a large proportion of mucosal T cells interacting with the commensal microbes, are generated both through the peripheral conversion of Tconv cells in response to the microbiota (49, 51, 52, 56, 73), or following colonization and expansion of tTreg (50, 122–124), thus establishing tolerance, and actually also influencing the composition of the microbiota: Treg cell deficiency is accompanied by gut dysbiosis (125), and impaired pTreg generation leads to perturbations in the composition and metabolic function of intestinal microbiota (126).
Metabolites produced by the commensal bacteria also influence Tregs. For instance, short chain fatty acids (SCFA) including butyric and propionic acid induce the differentiation of Treg cells through H3 acetylation at the CNS3 and CNS1 regions of the FoxP3 gene locus (49, 127). Polysaccharide A (PSA) produced by the commensal Bacteroides fragilis prevents intestinal inflammatory disease and induces the generation of IL-10-producing FoxP3+ Treg cells (128–130). These expanded Treg populations express CD39, which in addition to its role in catabolizing ATP, also modulates their migratory capacity (131).
The microbiota consists also of organisms that direct pro-inflammatory immune responses and Th17 differentiation. Th17 are essential for the protection against extracellular bacteria and fungi (132–134), but their pathogenic role in inflammatory disease is also well documented. Commensal microbiota are involved in the development of intestinal Th17, with Candida albicans playing a prominent role (132) followed by segmented filamentous bacteria (SFB), which promote Th17 differentiation and brain inflammation (135, 136).
The picture that emerges is that the composition of a healthy microbiome is not accidental, and was established after millennia of coevolution. The balance between the generation of proinflammatory effector cells, which combat potentially pathogenic microbes, and immune-suppressive regulatory T cells, which oppose and downmodulate these responses, is an active process. Changes in this balance, either due to an altered composition of the microbiota or to an intrinsic defect of Tregs, may generate inflammatory responses that target organs distant from the gut, as the brain (Figure 2).
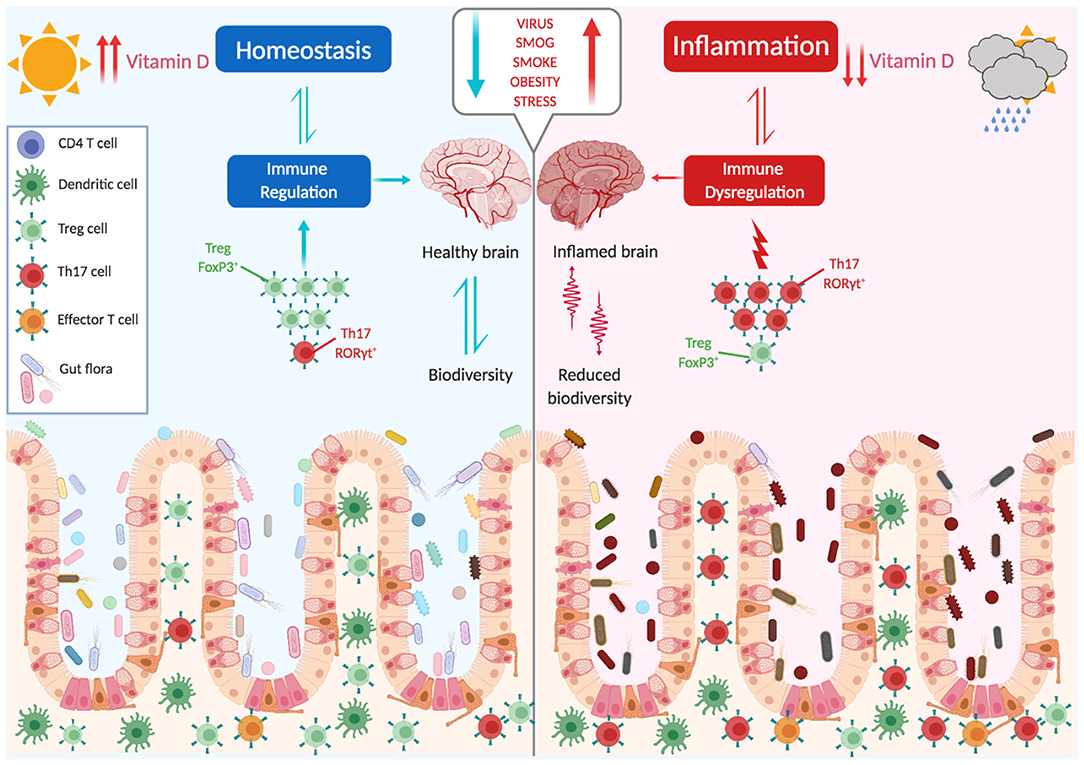
Figure 2. The microbiota shapes immune responses. The balance between proinflammatory and immune suppressive factors (dysbiosis, vit. D, stress, obesity, smoking, infections, diet) is critical for the maintenance of tolerance and avoidance of autoimmunity. Several environmental factors can influence this balance, with a main role being played by the gut microbiota.
Multiple sclerosis is an inflammatory disease of the central nervous system characterized by recurrent attacks of neurological dysfunction from which early on in the disease course patients gradually recover; in most patients, a phase characterized by progressive clinical worsening then ensues, and permanent disability is established (137–139). Pathologically, MS is characterized by the appearance of demyelinating lesions with extensive inflammatory infiltrates both in the white and in the cortical gray matter, pointing to an involvement of the immune system in disease pathogenesis. Indeed, genome-wide association studies (GWAS) have indicated that virtually all MS-associated genes are involved in immune processes (18, 140, 141), with expression of the HLA-DRB1*15:01 allele being the most dominant risk factor. This genetic association argues strongly in favor of the central role of the adaptive immune response in MS, since T cell activation sparks from the interaction of TCRs with HLA molecules.
MS arises for an unfortunate combination of factors and events, and different risk factors have been identified. The genetic component is evidenced by a high concordance rate in monozygotic twins (25–30%), but the fact that in this case the relative risk does not reach 100% points to a role of environmental factors in inducing disease. Low vitamin D levels, cigarette smoking, the presence of environmental toxins, obesity, and infection with EBV virus are the most consistent environmental factors linked to MS (142, 143). In more recent years, the influences of the microbiome in inducing or preventing MS have emerged. Alterations in the composition of the microbiome in contemporary populations is thought to be partly responsible for increases in autoimmune disorders including MS. An interesting paper by Vatanen et al. (144) showed that depending on the composition of the microbiota, LPS derived from different strains has different immunogenicity and may lead to altered immune maturation and predispose to autoimmunity. Indeed, the intestinal microbiome of MS patients shows distinct changes compared to healthy controls, particularly with a significant reduction in biodiversity (145–148). Interestingly, fecal transplants from MS patients in transgenic models of brain autoimmunity precipitate disease (149, 150), further supporting a role of microbial composition in inducing pathological immune responses.
Dysbiosis, with a shift in the microbiota composition toward a proinflammatory asset, also determines alterations in intestinal permeability, granting access of bacterial components and preceding the onset of brain autoimmunity in the experimental model (151, 152).
The CNS is kept separate from the rest of the organism and from the outside environment by several anatomical and cellular layers, in order to protect the resident cells which are particularly vulnerable to immune-mediated damage and unable to regenerate. The meninges surround the brain and contain the cerebrospinal fluid, and are also host to a functional lymphatic system which enables drainage from the CNS to the deep cervical lymph nodes (153, 154). Although it is equipped with a lymphatic system, the brain is an immunologically unique site, and it is shielded from the blood circulation by a specialized vasculature composed by endothelial cells joined together by tight junctions and surrounded by glial podocytes, thus creating a nearly impermeable boundary, the blood brain barrier (BBB) (155), which represents a limiting factor to the entry of metabolites and of immune cells into the CNS. Alterations of the permeability of the BBB clinically correspond to the formation of the typical acute gadolinium-enhancing regions visible by magnetic resonance imaging (MRI) scans. Interestingly, the BBB is vulnerable to changes in the gut microbiota (156), and coloniziation of mice with a single organism, B. fragilis, can restore permeability (157).
Activated and memory T cells in the periphery express the adhesion molecules and chemokine receptors which enable them to cross the BBB and to infiltrate the brain tissue (158, 159), and this feature is exemplified by the induction of neuroinflammation in the murine model through the transfer of myelin-specific T cells which upon injection in naïve mice rapidly accumulate in the brain, generating the typical lesions similar to those observed in MS patients. CCR6 expression has been shown to be a requirement for migration in the CNS (160), following a gradient of CCL20 produced by the choroid plexus, which allows entry in the brain ventricles. Once in the tissue, these cells can become reactivated following interaction with resident antigen-presenting cells (APCs), such as microglia or myeloid cells, and can start producing inflammatory cytokines which increase BBB permeability and initiate the cascade of infiltration by pathogenic proinflammatory cells, and thus the typical perivascular infiltrates are formed (161). CD8+ T cells also gain access to the CNS, and actually constitute the majority of the T cell infiltrate; through the release of cytotoxic molecules, such as perforin and granzyme they induce demyelination (138, 162, 163), and they have also been shown to produce IL-17 (163). “Special” CD8 T cells also participate to disease: mucosal-associated invariant T (MAIT) cells (164, 165), a subset of innate-like T cells which recognize molecules derived from bacterial metabolism and which usually reside in the gut, where they control the intestinal microbiome. These cells have been found in the infiltrating lesions of MS patients, where they produce IL-17. Also B cells have a pathogenic role in MS (166): the presence of oligoclonal bands is a hallmark of the disease, and ectopic B cell follicles are present in meninges from patients with MS (167, 168). Moreover, the spectacular results obtained in clinical trials using antibodies targeting B cells confirm the role that these cells play in the disease, including their role as APC which may reactivate autoreactive T cells crossing the BBB (169–171).
Together with these pathogenic cells, also Tregs gain access to the CNS.
The CNS is an excellent organ to study in models of inflammatory autoimmune disease, since physiologically it is free from activated immune cells, such as lymphocytes and macrophages. The induction of an autoimmune reaction directed against CNS components—typically myelin antigens—is a well-established model for human multiple sclerosis, and has provided crucial insight on the immune mechanisms underlying the disease.
The initial studies on Treg cells in MS were those performed in the murine model of brain autoimmunity, where it was discovered that their ablation aggravated disease, while adoptive transfer inhibited it (172, 173). Several studies then addressed the issue of Tregs in MS patients, which showed that Treg cell frequency as a whole, mostly determined by the gross measurement of CD4+CD25high circulating cells, was shown to be in the physiological range (174–177), or decreased (178, 179). However, investigations on the functional abilities of these cells with the identification of distinct subsets have revealed impairments at different levels which partly explain the defects in immunoregulation underlying the autoimmune attack in MS (180–182).
It has been shown that the frequency in the peripheral blood of Tregs expressing CD39 is reduced in MS patients, and that this correlates with a lower ability to catabolize ATP (90). Interestingly, CD39+ Treg cells are able to efficiently suppress IL-17 production by Th17 cells, contrary to their CD39− counterpart, thus their reduction in MS patients may participate to the ineffective control of Th17-driven inflammation in this disease (183). A recent study has shown that in MS patients experiencing clinical relapses the frequency of CD39+ Treg cells was increased (184), possibly in an attempt to counterbalance active inflammation (185). In the murine model, PSA produced by the human intestinal commensal Bacteroides fragilis, induces expansion of CD39+Treg cells with protective effects on EAE manifestations, and ablation of CD39+ abrogates these effects (186).
Treg cells from MS patients have been shown to express lower levels of FoxP3 (187), and a more detailed study later showed that in patients there is prevalent expression of the FoxP3 isoform lacking exon2, making Treg cells less efficient in inhibiting the development of proinflammatory cells (28).
Genetic polymorphisms in Treg-relevant genes may also underlie the development of MS. For example, a high risk of MS is associated with polymorphism in CD25 (188).
Also resident cells of the CNS can influence Treg cell activity. Neurons, for instance, produce TGF-β and might directly affect T cells. One study has shown that neurons participate to the conversion of encephalitogenic CD4 T cells to Treg cells expressing FoxP3 and mediating suppression through CTLA4 (189). Moreover, serotonin, a CNS neurotransmitter, favors the expansion in vitro of Treg cells from MS patients while reducing production of IL-17 by Tconv cells, suggesting that it acts as a neuroprotectant in the attempt of resolving inflammation (190).
Adding to all this, another aspect is resistance of Tconv to the suppressive action of Treg cells: effector T cells may “break free” and escape Treg suppression, mainly through accelerated production of IL-6 and STAT-3 signaling, as has been shown to happen in MS patients (191–193).
Thus, Treg impairment at different levels has been described to occur in MS patients, and likely plays an important role in determining altered immune regulation and disease susceptibility (Figure 3).
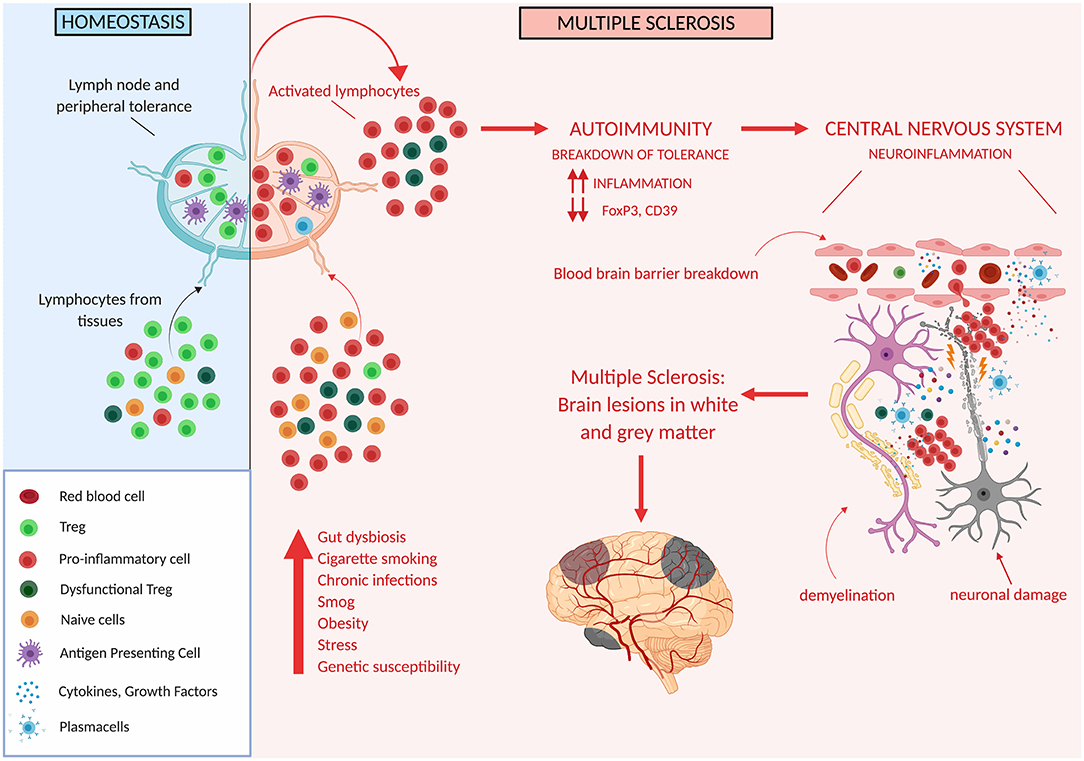
Figure 3. Tregs in multiple sclerosis. Genetic and/or environmental factors may induce hordes of proinflammatory cells and thus may overcome the ability of Treg cells to suppress inflammation. This may unleash autoreactive immune cells which cross the blood brain barrier and target different areas of the CNS ultimately determining the development of white and gray matter lesions.
Although there is no cure for MS, 15 drugs are now available for modifying disease course of the relapsing-remitting form (194, 195). Early treatment is recommended in order to achieve the goal of “no evidence of disease activity” (NEDA) (196), and currently no patient is left without therapy. All these treatments target the immune system, and most also exert an effect on Treg cells.
The first injectable drugs approved for use in MS were type I interferons and glatiramer acetate (GA), and they are used as first-line agents to slow disease progression. Type I interferons have a plethora of immunomodulatory functions, in addition to their role in viral interference (197, 198). In MS patients, treatment with this drug enhances regulatory cell function and increases FoxP3 mRNA levels, thus favoring immune suppression (199–201). GA is a synthetic polymer of aminoacids, whose complex mechanism of action are still incompletely understood, and involves both immunomodulatory and neuroprotective effects. Patients treated with GA show increased levels of FoxP3 (202) and of Treg function (203), seemingly through a direct effect on thymic output of naïve Tregs.
Natalizumab is a humanized monoclonal antibody directed against the α4 chain of α4β1 integrin (CD49d), and it interferes with lymphocyte migration in the CNS. It is highly effective in reducing the annual relapse rate (204, 205), although it may predispose to developing opportunistic infections in the CNS. Interestingly, contrary to Tconv cells Tregs express very low levels of CD49d (206) thus their migration to the CNS may not be affected by therapy with natalizumab. Indeed, given the low expression of CD49d by Treg cell, the therapeutic effects of this drug seem to be independent of a direct effect on Tregs (207).
Fingolimod was the first oral drug to be approved for use in MS. It is a S1P1 antagonist, thus it prevents T cells from exiting the lymph nodes, with the result of reduced numbers of circulating T cells (208, 209). However, it is now clear that the effects of fingolimod on immune cells go beyond the modulation of their migratory properties. In Treg cells S1P1 inhibition leads to increased thymic egress and suppressive function, through blockade of the Akt-mTOR pathway (210). Deletion of S1P1 in a mouse model induced development of systemic autoimmunity, and acute S1P1 ablation enhanced susceptibility to EAE (211). In the same study it was also shown that in patients undergoing treatment with fingolimod the phenotypic conversion from naïve Treg cells to effector T cells occurs with greater frequency. Fingolimod also affects Treg plasticity by reducing the expression of T-bet and of IFNγ and by enhancing expression of Tim-3, a marker associated with superior suppressor capacity (212). In general, these studies show that Treg function is increased following treatment with fingolimod.
Dimethyl fumarate (DMF) is an oral drug which was initially used to treat psoriasis, and was later approved for use in MS (213). The mechanism of action is still not completely understood, but seems to rely on the activation of the nuclear-related factor 2 (Nrf2) antioxidative response pathways (214). Very few studies have addressed the impact of DMF directly on Treg cells, and besides an increase of FoxP3+ cells during the first months of treatment (215), no other effect has been described.
Alemtuzumab is a humanized monoclonal antibody specific for CD52, a protein highly expressed by T and B cells and at lower levels on innate immune cells. Two trials established its safety and efficacy in MS patients (216, 217). Treatment with alemtuzumab induces long-lasting lymphopenia, and the beneficial effect on MS seems to be due on the re-equilibration of the immune system which occurs through depletion and repopulation of lymphocytes, which includes an enrichment in Treg cells (218, 219), and an increase of Treg suppressive function (220).
Cladribine (CdA) is a synthetic deoxyadenosine analog. Like deoxyadenosine, it enters lymphocytes through an efficient transport mechanism. Once in the cell, deoxyadenosine has two potential fates: irreversible deamination by adeonine deaminase (ADA), leading ultimately to the uric acid excretion pathway, or serial phosphorylation by deoxycytidine kinase (DCK) to dATP. Cladribine, on the other hand, is resistant to deamination by ADA because of its chlorinated purine ring structure and thus can only phosphorylated by DCK to its lymphocytotoxic form, 2-chlorodeoxyadenosine triphosphate, which accumulates and is incorporated into DNA. This results in DNA strand breakage and inhibition of DNA synthesis and repair, and to cell death. The selective toxicity of CdA toward both dividing and resting lymphocytes makes the drug useful as an antileukemic or immunosuppressive agent, and indeed it is used in hematologic cancers, such as B cell chronic leukemia and particularly hairy cell leukemia (221, 222). Subsequently, the drug was tested for treatment of MS. The “CLARITY” trial for relapsing remitting MS showed a significant reduction in relapse rate (223) and the drug is now approved for treatment of “rapidly evolving severe” MS (defined as at least two relapses in the previous year and an MRI scan showing new, or bigger, lesions). The beneficial effects of cladribine seem to be achieved through targeted and sustained reduction of circulating T and B lymphocytes, but it is possible that additional immunomodulatory actions participate to its effects (224). MS patients that express low levels of CD39 are likely to respond to cladribine therapy, since they lack the level of immune-regulation provided by the generation of suppressive adenosine, and thus may benefit from pharmacologically induced increases in adenosine.
From this survey of the effects of currently used immunomodulatory drugs it emerges that the most frequently measured effect is an increase in frequency and in suppressive function of Tregs, and although these cells are not the direct target of these therapies, nonetheless the improved immune regulation they provide contributes to altering the progression of the disease.
The Treg cell population is vastly heterogeneous, as most complex makings of Nature, and comprises an army of cells expressing graded levels of transcription factors and of effector molecules. FoxP3 dominates the machinery of these cells, and through the interactions with both the genes made accessible by epigenetic mechanisms and with the nuclear proteins available for binding, it regulates transcription of the quintessential Treg molecules. Treg differentiation is a plastic and actively maintained state determined by the collective activity of the whole transcriptional network, and this complexity confers elasticity and adaptability to changing environments. Treg cells swarm to areas of immune interrogation and interact with APCs in lymphoid tissues, as do naïve Tconv cells, but they can just as easily migrate in tissues together with T effector cell populations, responding to the same cues; moreover, besides adapting to the environment and downregulating the immune response, Tregs provide molecules which sustain tissue cells, actively participating to tissue protection and regeneration. One prominent site of Treg activity is the intestine, where these cells modulate immune interactions with the microbiota and help maintain tolerance. Changes in microbiota composition in present-day populations, with a prominent representation of proinflammatory microorganisms and a significant reduction of biodiversity, have been shown to underlie the steep increase of autoimmune and allergic disorders observed in western countries, raising the level of inflammation to a point where Treg cells are unable to contrast the fire. It is also possible that subtle defects in Treg function may be unmasked by excessive inflammation, and this may explain why only some individuals succumb to autoimmunity.
Many themes still need to be investigated. For instance, in recent years the role of circadian factors on immune cell function has emerged as a factor which also impacts the development of autoimmunity. Indeed, immune responses have a rhythmicity regulated by intrinsic timers (225, 226), and in animal models susceptibility to sepsis or to EAE induction are significantly and intriguingly dependent on the time of the day (227–230). Only few studies have addressed the issue of circadian rhythms on the functions of Treg cells (231–234), all suggesting that also Treg cells are under circadian control. Also the recently identified resolvins, molecules which mediate resolution of inflammation (235), are a fascinating area of investigation and have been shown to induce Treg cell polarization in humans (236) flipping the balance of immune reactions toward immune regulation and away from inflammation. Additionally, as mentioned, T cell identity is maintained not only by transcriptional programs, and post-transcriptional processes are in place to rapidly tune cell fate decisions in response to changing environments. Non-coding RNAs are RNA transcripts which do not code for protein products but instead regulate gene expression by driving post-transcriptional repression through pairing with mRNA. They include microRNA, small interfering RNA (siRNA), long non-coding RNA, and the recently discovered circular RNA (237–239). The finding that miRNA disruption selectively in Tregs leads to fatal systemic autoimmunity similar to the disease occurring in FoxP3 deficient mice indicates that non-coding mRNAs are central for maintaining Treg cell identity and function (25, 26). Indeed, studies on miRNA and on their role in Treg cell biology have identified a miRNA Treg signature which seems to be conserved across species (240, 241). miRNA are also involved in the pathogenesis of autoimmune diseases, including MS, mainly through the modulation of the Treg/Th17 balance (242–245), and are interestingly found in the extracellular vesicles released by activated lymphocytes. Moreover, they may directly participate to Treg-mediated immune suppression (246).
These areas of investigation may hold the answer to what goes wrong in CNS autoimmunity, although given the complexity of the human meta-organism and of its immune network we have many more than one question, closer to one hundred thousand.
GB and MS wrote the manuscript. FG and GG revised the literature. MS and GB conceived and edited the figures. All authors contributed to manuscript revision.
This study was partially supported by the Italian Ministry of Health (Progetto di ricerca Finalizzata RF-2018-12366111), by TEVA Italia s.r.l. (Teva 0211212014), by the Italian Foundation for Multiple Sclerosis (FISM Progetto Speciale 2018/S/5) to LB, and by the Italian Ministry of Health (Progetto di ricerca Finalizzata RF-2016-02363688) to GB. MS is partially supported by Italian Ministry of Health (GR-2016-02363725 and GR-2018-12365529).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All the figures were created with BioRender.com.
1. Ganusov VV, Auerbach J. Mathematical modeling reveals kinetics of lymphocyte recirculation in the whole organism. PLoS Comput Biol. (2014) 10:e1003586. doi: 10.1371/journal.pcbi.1003586
2. Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E. Dysbiosis and the immune system. Nat Rev Immunol. (2017) 17:7. doi: 10.1038/nri.2017.7
3. Daley SR, Hu DY, Goodnow CC. Helios marks strongly autoreactive CD4+ T cells in two major waves of thymic deletion distinguished by induction of PD-1 or NF-κB. J Exp Med. (2013) 210:269–85. doi: 10.1084/jem.20121458
4. Stritesky GL, Xing Y, Erickson JR, Kalekar LA, Wang X, Mueller DL, et al. Murine thymic selection quantified using a unique method to capture deleted T cells. Proc Natl Acad Sci USA. (2013) 110:4679–84. doi: 10.1073/pnas.1217532110
5. Cao Y, Goods BA, Raddassi K, Nepom GT, Kwok WW, Love CJ, et al. Functional inflammatory profiles distinguish myelin-reactive T cells from patients with multiple sclerosis. Sci Transl Med. (2015) 7:287ra74. doi: 10.1126/scitranslmed.aaa8038
6. Danke NA, Koelle DM, Yee C, Beheray S, Kwok WW. Autoreactive T cells in healthy individuals. J Immunol. (2004) 172:5967–72. doi: 10.4049/jimmunol.172.10.5967
7. Snir O, Rieck M, Gebe JA, Yue BB, Rawlings CA, Nepom G, et al. Identification and functional characterization of T cells reactive to citrullinated vimentin in HLA–DRB1*0401–positive humanized mice and rheumatoid arthritis patients. Arthritis Rheum. (2011) 63:2873–83. doi: 10.1002/art.30445
8. Christoffersson G, von Herrath M. Regulatory immune mechanisms beyond regulatory T cells. Trends Immunol. (2019) 40:482–91. doi: 10.1016/j.it.2019.04.005
9. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. (2003) 4:904. doi: 10.1038/ni904
10. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. (2003) 299:1057–61. doi: 10.1126/science.1079490
11. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. (1995) 155:1151–64.
12. Hori S. The Foxp3 interactome: a network perspective of Treg cells. Nat Immunol. (2012) 13:943. doi: 10.1038/ni.2424
13. Lu L, Barbi J, Pan F. The regulation of immune tolerance by FOXP3. Nat Rev Immunol. (2017) 17:75. doi: 10.1038/nri.2017.75
14. Marson A, Kretschmer K, Frampton GM, Jacobsen ES, Polansky JK, MacIsaac KD, et al. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature. (2007) 445:5478. doi: 10.1038/nature05478
15. Zheng Y, Josefowicz SZ, Kas A, Chu T-T, Gavin MA, Rudensky AY. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. (2007) 445:936–40. doi: 10.1038/nature05563
16. Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. (2010) 463:808. doi: 10.1038/nature08750
17. Kitagawa Y, Ohkura N, Kidani Y, Vandenbon A, Hirota K, Kawakami R, et al. Guidance of regulatory T cell development by Satb1-dependent super-enhancer establishment. Nat Immunol. (2016) 18:3646. doi: 10.1038/ni.3646
18. Ic I, Beecham AH, Patsopoulos NA, Xifara DK, Davis MF, Kemppinen A, et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet. (2013) 45:2770. doi: 10.1038/ng.2770
19. Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. (2007) 5:e38. doi: 10.1371/journal.pbio.0050038
20. Huehn J, Polansky JK, Hamann A. Epigenetic control of FOXP3 expression: the key to a stable regulatory T-cell lineage? Nat Rev Immunol. (2009) 9:83–9. doi: 10.1038/nri2474
21. Ohkura N, Kitagawa Y, Sakaguchi S. Development and maintenance of regulatory T cells. Immunity. (2013) 38:414–23. doi: 10.1016/j.immuni.2013.03.002
22. Deng G, Song X, Fujimoto S, Piccirillo CA, Nagai Y, Greene MI. Foxp3 post-translational modifications and Treg suppressive activity. Front. Immunol. (2019) 10:2486. doi: 10.3389/fimmu.2019.02486
23. van Loosdregt J, Coffer PJ. Post-translational modification networks regulating FOXP3 function. Trends Immunol. (2014) 35:368–78. doi: 10.1016/j.it.2014.06.005
24. Kaur G, Goodall JC, Jarvis LB, Gaston JS. Characterisation of Foxp3 splice variants in human CD4+ and CD8+ T cells—identification of Foxp3Δ7 in human regulatory T cells. Mol Immunol. (2010) 48:321–32. doi: 10.1016/j.molimm.2010.07.008
25. Du J, Huang C, Zhou B, Ziegler SF. Isoform-specific inhibition of RORα-mediated transcriptional activation by human FOXP3. J Immunol. (2008) 180:4785–92. doi: 10.4049/jimmunol.180.7.4785
26. Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, et al. TGF-β-induced Foxp3 inhibits TH17 cell differentiation by antagonizing RORγt function. Nature. (2008) 453:236. doi: 10.1038/nature06878
27. Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C, et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat Immunol. (2015) 16:3269. doi: 10.1038/ni.3269
28. Sambucci M, Gargano F, Rosa V, Bardi M, Picozza M, Placido R, et al. FoxP3 isoforms and PD-1 expression by T regulatory cells in multiple sclerosis. Sci Rep. (2018) 8:3674. doi: 10.1038/s41598-018-21861-5
29. Fu W, Ergun A, Lu T, Hill JA, Haxhinasto S, Fassett MS, et al. A multiply redundant genetic switch “locks in” the transcriptional signature of regulatory T cells. Nat Immunol. (2012) 13:972. doi: 10.1038/ni.2420
30. Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, et al. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. (2007) 445:5543. doi: 10.1038/nature05543
31. Kwon H-K, Chen H-M, Mathis D, Benoist C. Different molecular complexes that mediate transcriptional induction and repression by FoxP3. Nat Immunol. (2017) 18:1238–48. doi: 10.1038/ni.3835
32. Bevan MJ, Hogquist KA, Jameson SC. Selecting the T cell receptor repertoire. Science. (1994) 264:796–7. doi: 10.1126/science.8171333
33. Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J, et al. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J Exp Med. (2011) 208:1279–89. doi: 10.1084/jem.20110308
34. Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. (2000) 12:431–40. doi: 10.1016/S1074-7613(00)80195-8
35. Burchill MA, Yang J, Vang KB, Moon JJ, Chu HH, Lio C-WJ, et al. Linked T cell receptor and cytokine signaling govern the development of the regulatory T cell repertoire. Immunity. (2008) 28:112–21. doi: 10.1016/j.immuni.2007.11.022
36. Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol. (2008) 9:632–40. doi: 10.1038/ni.1607
37. Lio C-W, Hsieh C-S. A two-step process for thymic regulatory T cell development. Immunity. (2008) 28:100–11. doi: 10.1016/j.immuni.2007.11.021
38. Thiault N, Darrigues J, Adoue V, Gros M, Binet B, Perals C, et al. Peripheral regulatory T lymphocytes recirculating to the thymus suppress the development of their precursors. Nat Immunol. (2015) 16:628–34. doi: 10.1038/ni.3150
39. Zheng Y, Rudensky AY. Foxp3 in control of the regulatory T cell lineage. Nat Immunol. (2007) 8:457–62. doi: 10.1038/ni1455
40. Konopacki C, Pritykin Y, Rubtsov Y, Leslie CS, Rudensky AY. Transcription factor Foxp1 regulates Foxp3 chromatin binding and coordinates regulatory T cell function. Nat Immunol. (2019) 20:232–42. doi: 10.1038/s41590-018-0291-z
41. Rudra D, deRoos P, Chaudhry A, Niec RE, Arvey A, Mstein R, et al. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat Immunol. (2012) 13:1010. doi: 10.1038/ni.2402
42. Allan SE, Crome SQ, Crellin NK, Passerini L, Steiner TS, Bacchetta R, et al. Activation-induced FOXP3 in human T effector cells does not suppress proliferation or cytokine production. Int Immunol. (2007) 19:345–54. doi: 10.1093/intimm/dxm014
43. Hill JA, Feuerer M, Tash K, Haxhinasto S, Perez J, Melamed R, et al. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity. (2007) 27:786–800. doi: 10.1016/j.immuni.2007.09.010
44. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. (2009) 30:899–911. doi: 10.1016/j.immuni.2009.03.019
45. Kitagawa Y, Ohkura N, Sakaguchi S. Epigenetic control of thymic Treg-cell development. Eur J Immunol. (2015) 45:11–6. doi: 10.1002/eji.201444577
46. Morikawa H, Sakaguchi S. Genetic and epigenetic basis of Treg cell development and function: from a FoxP3-centered view to an epigenome-defined view of natural Treg cells. Immunol Rev. (2014) 259:192–205. doi: 10.1111/imr.12174
47. Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity. (2012) 37:785–99. doi: 10.1016/j.immuni.2012.09.010
48. Schmidl C, Klug M, Boeld TJ, Andreesen R, Hoffmann P, Edinger M, et al. Lineage-specific DNA methylation in T cells correlates with histone methylation and enhancer activity. Genome Res. (2009) 19:1165–74. doi: 10.1101/gr.091470.109
49. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. (2013) 504:451. doi: 10.1038/nature12726
50. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. (2011) 331:337–41. doi: 10.1126/science.1198469
51. Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio C-W, Santacruz N, et al. Peripheral education of the immune system by colonic commensal microbiota. Nature. (2011) 478:250. doi: 10.1038/nature10434
52. Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci USA. (2010) 107:12204–9. doi: 10.1073/pnas.0909122107
53. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini C, Bohlooly YM, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. (2013) 341:569–73. doi: 10.1126/science.1241165
54. Samstein RM, Josefowicz SZ, Arvey A, Treuting PM, Rudensky AY. Extrathymic generation of regulatory T cells in placental mammals mitigates maternal-fetal conflict. Cell. (2012) 150:29–38. doi: 10.1016/j.cell.2012.05.031
55. Wienke J, Brouwers L, van der Burg LM, Mokry M, Scholman RC, Nikkels PG, et al. Human regulatory T cells at the maternal-fetal interface show functional site-specific adaptation with tumor-infiltrating-like features. bioRxiv [Preprint]. (2019). doi: 10.1101/820753
56. Josefowicz SZ, Niec RE, Kim H, Treuting P, Chinen T, Zheng Y, et al. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature. (2012) 482:395. doi: 10.1038/nature10772
57. Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell. (2010) 140:845–58. doi: 10.1016/j.cell.2010.02.021
58. Valmori D, Merlo A, Souleimanian NE, Hesdorffer CS, Ayyoub M. A peripheral circulating compartment of natural naive CD4+ Tregs. J Clin Invest. (2005) 115:1953–62. doi: 10.1172/JCI23963
59. Cuadrado E, van den Biggelaar M, de Kivit S, Chen Y, Slot M, Doubal I, et al. Proteomic analyses of human regulatory T cells reveal adaptations in signaling pathways that protect cellular identity. Immunity. (2018) 48:1046–59.e6. doi: 10.1016/j.immuni.2018.04.008
60. Liang B, Workman C, Lee J, Chew C, Dale BM, Colonna L, et al. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J Immunol. (2008) 180:5916–26. doi: 10.4049/jimmunol.180.9.5916
61. McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Evach E, Collins M, et al. CD4+CD25+ immunoregulatory T cells gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. (2002) 16:311–23. doi: 10.1016/S1074-7613(02)00280-7
62. Ito T, Hanabuchi S, Wang Y-H, Park W, Arima K, Bover L, et al. Two functional subsets of FOXP3+ regulatory T cells in human thymus and periphery. Immunity. (2008) 28:870–80. doi: 10.1016/j.immuni.2008.03.018
63. Dhuban K, d'Hennezel E, Nashi E, Bar-Or A, Rieder S, Shevach EM, et al. Coexpression of TIGIT and FCRL3 identifies helios+ human memory regulatory T cells. J Immunol. (2015) 194:3687–96. doi: 10.4049/jimmunol.1401803
64. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity. (2014) 40:569–81. doi: 10.1016/j.immuni.2014.02.012
65. Schmitt N, Ueno H. Regulation of human helper T cell subset differentiation by cytokines. Curr Opin Immunol. (2015) 34:130–6. doi: 10.1016/j.coi.2015.03.007
66. Campbell DJ, Koch MA. Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat Rev Immunol. (2011) 11:119. doi: 10.1038/nri2916
67. Sather BD, Treuting P, Perdue N, Miazgowicz M, Fontenot JD, Rudensky AY, et al. Altering the distribution of Foxp3+ regulatory T cells results in tissue-specific inflammatory disease. J Exp Med. (2007) 204:1335–47. doi: 10.1084/jem.20070081
68. Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. (2009) 10:1731. doi: 10.1038/ni.1731
69. Levine AG, Mendoza A, Hemmers S, Moltedo B, Niec RE, Schizas M, et al. Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature. (2017) 546:421. doi: 10.1038/nature22360
70. Müller M, Carter SL, Hofer MJ, Manders P, Getts DR, Getts MT, et al. CXCR3 signaling reduces the severity of experimental autoimmune encephalomyelitis by controlling the parenchymal distribution of effector and regulatory T cells in the central nervous system. J Immunol. (2007) 179:2774–86. doi: 10.4049/jimmunol.179.5.2774
71. Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science. (2009) 326:986–91. doi: 10.1126/science.1172702
72. Ohnmacht C, Park J-H, Cording S, Wing JB, Atarashi K, Obata Y, et al. The microbiota regulates type 2 immunity through RORγt+ T cells. Science. (2015) 349:989–93. doi: 10.1126/science.aac4263
73. Sefik E, Geva-Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire A, et al. Individual intestinal symbionts induce a distinct population of RORγ+ regulatory T cells. Science. (2015) 349:993–7. doi: 10.1126/science.aaa9420
74. Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT, et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control TH2 responses. Nature. (2009) 458:351. doi: 10.1038/nature07674
75. Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med. (2011) 17:983. doi: 10.1038/nm.2426
76. Miyao T, Floess S, Setoguchi R, Luche H, Fehling H, Waldmann H, et al. Plasticity of Foxp3+ T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity. (2012) 36:262–75. doi: 10.1016/j.immuni.2011.12.012
77. Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, et al. Stability of the regulatory T cell lineage in vivo. Science. (2010) 329:1667–71. doi: 10.1126/science.1191996
78. Bailey-Bucktrout SL, Martinez-Llordella M, Zhou X, Anthony B, Rosenthal W, Luche H, et al. Self-antigen-driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity. (2013) 39:949–62. doi: 10.1016/j.immuni.2013.10.016
79. Chen Z, Barbi J, Bu S, Yang H-Y, Li Z, Gao Y, et al. The ubiquitin ligase stub1 negatively modulates regulatory T cell suppressive activity by promoting degradation of the transcription factor Foxp3. Immunity. (2013) 39:272–85. doi: 10.1016/j.immuni.2013.08.006
80. Gao Y, Tang J, Chen W, Li Q, Nie J, Lin F, et al. Inflammation negatively regulates FOXP3 and regulatory T-cell function via DBC1. Proc Natl Acad Sci USA. (2015) 112:E3246–54. doi: 10.1073/pnas.1421463112
81. Mempel TR, Marangoni F. Guidance factors orchestrating regulatory T cell positioning in tissues during development, homeostasis, and response. Immunol Rev. (2019) 289:129–41. doi: 10.1111/imr.12761
82. Smigiel KS, Richards E, Vastava S, Thomas KR, Dudda JC, Klonowski KD, et al. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J Exp Med. (2014) 211:121–36. doi: 10.1084/jem.20131142
83. Liu Z, Gerner MY, Panhuys N, Levine AG, Rudensky AY, Germain RN. Immune homeostasis enforced by co-localized effector and regulatory T cells. Nature. (2015) 528:225. doi: 10.1038/nature16169
84. O'Gorman WE, Dooms H, Thorne SH, Kuswanto WF, Simonds EF, Krutzik PO, et al. The initial phase of an immune response functions to activate regulatory T cells. J Immunol. (2009) 183:332–9. doi: 10.4049/jimmunol.0900691
85. Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation–mediated apoptosis of effector CD4+ T cells. Nat Immunol. (2007) 8:1536. doi: 10.1038/ni1536
86. Szymczak-Workman AL, Delgoffe GM, Green DR, Vignali DA. Cutting edge: regulatory T cells do not mediate suppression via programmed cell death pathways. J Immunol. (2011) 187:4416–20. doi: 10.4049/jimmunol.1100548
87. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. (2011) 332:600–3. doi: 10.1126/science.1202947
88. Yokosuka T, Kobayashi W, Takamatsu M, Sakata-Sogawa K, Zeng H, Hashimoto-Tane A, et al. Spatiotemporal basis of CTLA-4 costimulatory molecule-mediated negative regulation of T cell activation. Immunity. (2010) 33:326–39. doi: 10.1016/j.immuni.2010.09.006
89. Pallotta MT, Orabona C, Volpi C, Vacca C, Belladonna ML, Bianchi R, et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat Immunol. (2011) 12:870. doi: 10.1038/ni.2077
90. Borsellino G, Kleinewietfeld M, Mitri D, Sternjak A, Diamantini A, Giometto R, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. (2007) 110:1225–32. doi: 10.1182/blood-2006-12-064527
91. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. (2007) 204:1257–65. doi: 10.1084/jem.20062512
92. Burnstock G, Boeynaems J-M. Purinergic signalling and immune cells. Purinerg Signal. (2014) 10:529–64. doi: 10.1007/s11302-014-9427-2
93. Takenaka MC, Robson S, Quintana FJ. Regulation of the T cell response by CD39. Trends Immunol. (2016) 37:427–39. doi: 10.1016/j.it.2016.04.009
94. Gu J, Ni X, Pan X, Lu H, Lu Y, Zhao J, et al. Human CD39hi regulatory T cells present stronger stability and function under inflammatory conditions. Cell Mol Immunol. (2016) 14:521. doi: 10.1038/cmi.2016.30
95. Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol. (2007) 7:31–40. doi: 10.1038/nri1997
96. Schnurr M, Toy T, Shin A, Wagner M, Cebon J, Maraskovsky E. Extracellular nucleotide signaling by P2 receptors inhibits IL-12 and enhances IL-23 expression in human dendritic cells: a novel role for the cAMP pathway. Blood. (2005) 105:1582–9. doi: 10.1182/blood-2004-05-1718
97. Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, Onoue M, et al. ATP drives lamina propria TH17 cell differentiation. Nature. (2008) 455:808. doi: 10.1038/nature07240
98. Chalmin F, Mignot G, Bruchard M, Chevriaux A, Végran F, Hichami A, et al. Stat3 and Gfi-1 transcription factors control Th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity. (2012) 36:362–73. doi: 10.1016/j.immuni.2011.12.019
99. Gagliani N, Vesely M, Iseppon A, Brockmann L, Xu H, Palm NW, et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature. (2015) 523:221. doi: 10.1038/nature14452
100. Fernández D, Flores-Santibáñez F, Neira J, Osorio-Barrios F, Tejón G, Nuñez S, et al. Purinergic signaling as a regulator of Th17 cell plasticity. PLoS ONE. (2016) 11:e0157889. doi: 10.1371/journal.pone.0157889
101. Doherty GA, Bai A, Hanidziar D, Longhi MS, Lawlor GO, Putheti P, et al. CD73 is a phenotypic marker of effector memory Th17 cells in inflammatory bowel disease. Eur J Immunol. (2012) 42:3062–72. doi: 10.1002/eji.201242623
102. Nakamura K, Kitani A, Strober W. Cell contact–dependent immunosuppression by Cd4+Cd25+regulatory T cells is mediated by cell surface–bound transforming growth factor β. J Exp Med. (2001) 194:629–44. doi: 10.1084/jem.194.5.629
103. Hara M, Kingsley CI, Niimi M, Read S, Turvey SE, Bushell AR, et al. IL-10 is required for regulatory T cells to mediate tolerance to alloantigens in vivo. J Immunol. (2001) 166:3789–96. doi: 10.4049/jimmunol.166.6.3789
104. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. (2007) 450:566. doi: 10.1038/nature06306
105. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity. (2007) 27:635–46. doi: 10.1016/j.immuni.2007.08.014
106. Askenasy N. Enhanced killing activity of regulatory T cells ameliorates inflammation and autoimmunity. Autoimmun Rev. (2013) 12:972–5. doi: 10.1016/j.autrev.2013.04.005
107. Klein M, Bopp T. Cyclic AMP represents a crucial component of Treg cell-mediated immune regulation. Front Immunol. (2016) 7:315. doi: 10.3389/fimmu.2016.00315
108. Yamaguchi T, Wing JB, Sakaguchi S. Two modes of immune suppression by Foxp3+ regulatory T cells under inflammatory or non-inflammatory conditions. Semin Immunol. (2011) 23:424–30. doi: 10.1016/j.smim.2011.10.002
109. Schmidleithner L, Thabet Y, Schönfeld E, Köhne M, Sommer D, Abdullah Z, et al. Enzymatic activity of HPGD in Treg cells suppresses Tconv cells to maintain adipose tissue homeostasis and prevent metabolic dysfunction. Immunity. (2019) 50:1232–48.e14. doi: 10.1016/j.immuni.2019.03.014
110. Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al. A special population of regulatory T cells potentiates muscle repair. Cell. (2013) 155:1282–95. doi: 10.1016/j.cell.2013.10.054
111. Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, et al. A distinct function of regulatory T cells in tissue protection. Cell. (2015) 162:1078–89. doi: 10.1016/j.cell.2015.08.021
112. Dombrowski Y, O'Hagan T, Dittmer M, Penalva R, Mayoral SR, Bankhead P, et al. Regulatory T cells promote myelin regeneration in the central nervous system. Nat Neurosci. (2017) 20:674. doi: 10.1038/nn.4528
113. Raposo C, Graubardt N, Cohen M, Eitan C, London A, Berkutzki T, et al. CNS repair requires both effector and regulatory T cells with distinct temporal and spatial profiles. J Neurosci. (2014) 34:10141–55. doi: 10.1523/JNEUROSCI.0076-14.2014
114. Whibley N, Tucci A, Powrie F. Regulatory T cell adaptation in the intestine and skin. Nat Immunol. (2019) 20:386–96. doi: 10.1038/s41590-019-0351-z
115. Scharschmidt TC, Vasquez KS, Truong H-A, Gearty SV, Pauli ML, Nosbaum A, et al. A wave of regulatory T cells into neonatal skin mediates tolerance to commensal microbes. Immunity. (2015) 43:1011–21. doi: 10.1016/j.immuni.2015.10.016
116. Ali N, Zirak B, Rodriguez R, Pauli ML, Truong H-A, Lai K, et al. Regulatory T cells in skin facilitate epithelial stem cell differentiation. Cell. (2017) 169:1119–29.e11. doi: 10.1016/j.cell.2017.05.002
117. Kalekar LA, Cohen JN, Prevel N, Sandoval P, Mathur AN, Moreau JM, et al. Regulatory T cells in skin are uniquely poised to suppress profibrotic immune responses. Sci Immunol. (2019) 4:eaaw2910. doi: 10.1126/sciimmunol.aaw2910
118. Dominguez-Bello M, Godoy-Vitorino F, Knight R, Blaser MJ. Role of the microbiome in human development. Gut. (2019) 68:1108. doi: 10.1136/gutjnl-2018-317503
119. Belkaid Y, Harrison OJ. Homeostatic immunity and the microbiota. Immunity. (2017) 46:562–76. doi: 10.1016/j.immuni.2017.04.008
120. Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. (2012) 336:1268–73. doi: 10.1126/science.1223490
121. Bach J-F. The hygiene hypothesis in autoimmunity: the role of pathogens and commensals. Nat Rev Immunol. (2017) 18:105. doi: 10.1038/nri.2017.111
122. Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. (2013) 500:232. doi: 10.1038/nature12331
123. Cebula A, Seweryn M, Rempala GA, Pabla S, McIndoe RA, Denning TL, et al. Thymus-derived regulatory T cells contribute to tolerance to commensal microbiota. Nature. (2013) 497:258. doi: 10.1038/nature12079
124. Tanoue T, Atarashi K, Honda K. Development and maintenance of intestinal regulatory T cells. Nat Rev Immunol. (2016) 16:36. doi: 10.1038/nri.2016.36
125. He B, Hoang TK, Wang T, Ferris M, Taylor CM, Tian X, et al. Resetting microbiota by Lactobacillus reuteri inhibits T reg deficiency–induced autoimmunity via adenosine A2A receptors. J Exp Med. (2016) 214:20160961. doi: 10.1084/jem.20160961
126. Campbell C, Dikiy S, Bhattarai SK, Chinen T, Matheis F, Calafiore M, et al. Extrathymically generated regulatory T cells establish a niche for intestinal border-dwelling bacteria and affect physiologic metabolite balance. Immunity. (2018) 48:1245–57.e9. doi: 10.1016/j.immuni.2018.04.013
127. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. (2013) 504:446. doi: 10.1038/nature12721
128. Mazmanian SK, Liu C, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. (2005) 122:107–18. doi: 10.1016/j.cell.2005.05.007
129. Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. (2008) 453:620. doi: 10.1038/nature07008
130. Ochoa-Repáraz J, Mielcarz D, Wang Y, Begum-Haque S, Dasgupta S, Kasper D, et al. A polysaccharide from the human commensal Bacteroides fragilis protects against CNS demyelinating disease. Mucosal Immunol. (2010) 3:487. doi: 10.1038/mi.2010.29
131. Wang Y, Telesford KM, Ochoa-Repáraz J, Haque-Begum S, Christy M, Kasper EJ, et al. An intestinal commensal symbiosis factor controls neuroinflammation via TLR2-mediated CD39 signalling. Nat Commun. (2014) 5:4432. doi: 10.1038/ncomms5432
132. Bacher P, Hohnstein T, Beerbaum E, Röcker M, Blango MG, Kaufmann S, et al. Human anti-fungal Th17 immunity and pathology rely on cross-reactivity against Candida albicans. Cell. (2019) 176:1340–55.e15. doi: 10.1016/j.cell.2019.01.041
133. Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. (2009) 206:299–311. doi: 10.1084/jem.20081463
134. Hernández-Santos N, Huppler A, Peterson A, Khader S, McKenna K, Gaffen S. Th17 cells confer long-term adaptive immunity to oral mucosal Candida albicans infections. Mucosal Immunol. (2013) 6:900. doi: 10.1038/mi.2012.128
135. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. (2009) 139:485–98. doi: 10.1016/j.cell.2009.09.033
136. Lee Y, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. (2011) 108:4615–22. doi: 10.1073/pnas.1000082107
137. Baecher-Allan C, Kaskow BJ, Weiner HL. Multiple sclerosis: mechanisms and immunotherapy. Neuron. (2018) 97:742–68. doi: 10.1016/j.neuron.2018.01.021
138. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. (2015) 15:3871. doi: 10.1038/nri3871
139. Thompson AJ, Baranzini SE, Geurts J, Hemmer B, Ciccarelli O. Multiple sclerosis. Lancet. (2018) 391:1622–36. doi: 10.1016/S0140-6736(18)30481-1
140. Consortium I, Mitrovič M, Patsopoulos NA, Beecham AH, Dankowski T, Goris A, et al. Low-frequency and rare-coding variation contributes to multiple sclerosis risk. Cell. (2018) 175:1679–87.e7. doi: 10.1016/j.cell.2018.09.049
141. Moutsianas L, Jostins L, Beecham AH, Dilthey AT, Xifara DK, Ban M, et al. Class II HLA interactions modulate genetic risk for multiple sclerosis. Nat Genet. (2015) 47:1107–13. doi: 10.1038/ng.3395
142. Ascherio A, Munger KL. Epidemiology of multiple sclerosis: from risk factors to prevention–an update. Semin Neurol. (2016) 36:103–14. doi: 10.1055/s-0036-1579693
143. Olsson T, Barcellos LF, Alfredsson L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat Rev Neurol. (2016) 13:25–36. doi: 10.1038/nrneurol.2016.187
144. Vatanen T, Kostic AD, d'Hennezel E, Siljander H, Franzosa EA, Yassour M, et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell. (2016) 165:842–53. doi: 10.1016/j.cell.2016.04.007
145. Chen J, Chia N, Kalari KR, Yao JZ, Novotna M, Soldan MM, et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci Rep. (2016) 6:28484. doi: 10.1038/srep28484
146. Jangi S, Gandhi R, Cox LM, Li N, von Glehn F, Yan R, et al. Alterations of the human gut microbiome in multiple sclerosis. Nat Commun. (2016) 7:12015. doi: 10.1038/ncomms12015
147. Miyake S, Kim S, Suda W, Oshima K, Nakamura M, Matsuoka T, et al. Dysbiosis in the gut microbiota of patients with multiple sclerosis, with a striking depletion of species belonging to clostridia XIVa and IV clusters. PLoS ONE. (2015) 10:e0137429. doi: 10.1371/journal.pone.0137429
148. Tremlett H, Bauer KC, Appel-Cresswell S, Finlay BB, Waubant E. The gut microbiome in human neurological disease: a review. Ann Neurol. (2017) 81:369–82. doi: 10.1002/ana.24901
149. Berer K, Gerdes L, Cekanaviciute E, Jia X, Xiao L, Xia Z, et al. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc Natl Acad Sci USA. (2017) 114:10719–24. doi: 10.1073/pnas.1711233114
150. Cekanaviciute E, Yoo BB, Runia TF, Debelius JW, Singh S, Nelson CA, et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc Natl Acad Sci USA. (2017) 114:10713–8. doi: 10.1073/pnas.1711235114
151. Buscarinu M, Fornasiero A, Romano S, Ferraldeschi M, Mechelli R, Reniè R, et al. The contribution of gut barrier changes to multiple sclerosis pathophysiology. Front Immunol. (2019) 10:1916. doi: 10.3389/fimmu.2019.01916
152. Nouri M, Bredberg A, Weström B, Lavasani S. Intestinal barrier dysfunction develops at the onset of experimental autoimmune encephalomyelitis, and can be induced by adoptive transfer of auto-reactive T cells. PLoS ONE. (2014) 9:e106335. doi: 10.1371/journal.pone.0106335
153. Absinta M, Ha S-K, Nair G, Sati P, Luciano NJ, Palisoc M, et al. Human and nonhuman primate meninges harbor lymphatic vessels that can be visualized noninvasively by MRI. Elife. (2017) 6:e29738. doi: 10.7554/eLife.29738.018
154. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske DJ, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. (2015) 523:337. doi: 10.1038/nature14432
155. Daneman R, Prat A. The blood–brain barrier. CSH Perspect Biol. (2015) 7:a020412. doi: 10.1101/cshperspect.a020412
156. Braniste V, Al-Asmakh M, Kowal C, Anuar F, Abbaspour A, Tóth M, et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci Transl Med. (2014) 6:263ra158. doi: 10.1126/scitranslmed.3009759
157. Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. (2013) 155:1451–63. doi: 10.1016/j.cell.2013.11.024
158. Goverman J. Autoimmune T cell responses in the central nervous system. Nat Rev Immunol. (2009) 9:2550. doi: 10.1038/nri2550
159. Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol. (2012) 12:623. doi: 10.1038/nri3265
160. Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, et al. C-C chemokine receptor 6–regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol. (2009) 10:1716. doi: 10.1038/ni.1716
161. Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. (2007) 13:1173–5. doi: 10.1038/nm1651
162. Friese MA, Fugger L. Pathogenic CD8+ T cells in multiple sclerosis. Ann Neurol. (2009) 66:132–41. doi: 10.1002/ana.21744
163. Zang YC, Li S, Rivera VM, Hong J, Robinson RR, Breitbach WT, et al. Increased CD8+ cytotoxic T cell responses to myelin basic protein in multiple sclerosis. J Immunol. (2004) 172:5120–7. doi: 10.4049/jimmunol.172.8.5120
164. Abrahamsson SV, Angelini DF, Dubinsky AN, Morel E, Oh U, Jones JL, et al. Non-myeloablative autologous haematopoietic stem cell transplantation expands regulatory cells and depletes IL-17 producing mucosal-associated invariant T cells in multiple sclerosis. Brain. (2013) 136:2888–903. doi: 10.1093/brain/awt182
165. Annibali V, Ristori G, Angelini DF, Serafini B, Mechelli R, Cannoni S, et al. CD161highCD8+T cells bear pathogenetic potential in multiple sclerosis. Brain. (2011) 134:542–54. doi: 10.1093/brain/awq354
166. Wekerle H. B cells in multiple sclerosis. Autoimmunity. (2017) 50:57–60. doi: 10.1080/08916934.2017.1281914
167. Magliozzi R, Howell O, Vora A, Serafini B, Nicholas R, Puopolo M, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. (2007) 130:1089–104. doi: 10.1093/brain/awm038
168. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. (2004) 14:164–74. doi: 10.1111/j.1750-3639.2004.tb00049.x
169. Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-cell depletion with rituximab in relapsing–remitting multiple sclerosis. N Engl J Med. (2008) 358:676–88. doi: 10.1056/NEJMoa0706383
170. Jelcic I, Nimer F, Wang J, Lentsch V, Planas R, Jelcic I, et al. Memory B cells activate brain-homing, autoreactive CD4+ T cells in multiple sclerosis. Cell. (2018) 175:85–100.e23. doi: 10.1016/j.cell.2018.08.011
171. Montalban X, Hauser SL, Kappos L, Arnold DL, Bar-Or A, Comi G, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. (2017) 376:209–20. doi: 10.1056/NEJMoa1606468
172. Lowther DE, Hafler DA. Regulatory T cells in the central nervous system. Immunol Rev. (2012) 248:156–69. doi: 10.1111/j.1600-065X.2012.01130.x
173. Sakaguchi S, Wing K, Miyara M. Regulatory T cells—a brief history and perspective. Eur J Immunol. (2007) 37:S116–S123. doi: 10.1002/eji.200737593
174. Haas J, Hug A, Viehöver A, Fritzsching B, Falk CS, Filser A, et al. Reduced suppressive effect of CD4+CD25high regulatory T cells on the T cell immune response against myelin oligodendrocyte glycoprotein in patients with multiple sclerosis. Eur J Immunol. (2005) 35:3343–52. doi: 10.1002/eji.200526065
175. Kumar M, Putzki N, Limmroth V, Remus R, Lindemann M, Knop D, et al. CD4+CD25+FoxP3+ T lymphocytes fail to suppress myelin basic protein-induced proliferation in patients with multiple sclerosis. J Neuroimmunol. (2006) 180:178–84. doi: 10.1016/j.jneuroim.2006.08.003
176. Putheti P, Pettersson A, Soderstrom M, Link H, Huang Y. Circulating CD4+CD25+ T regulatory cells are not altered in multiple sclerosis and unaffected by disease-modulating drugs. J Clin Immunol. (2004) 24:155–61. doi: 10.1023/B:JOCI.0000019780.93817.82
177. Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. (2004) 199:971–9. doi: 10.1084/jem.20031579
178. Bjerg L, Brosbøl-Ravnborg A, Tørring C, Dige A, Bundgaard B, Petersen T, et al. Altered frequency of T regulatory cells is associated with disability status in relapsing–remitting multiple sclerosis patients. J Neuroimmunol. (2012) 249:76–82. doi: 10.1016/j.jneuroim.2012.04.012
179. Libera D, Mitri D, Bergami A, Centonze D, Gasperini C, Grasso M, et al. T regulatory cells are markers of disease activity in multiple sclerosis patients. PLoS ONE. (2011) 6:e21386. doi: 10.1371/journal.pone.0021386
180. Buckner J. Mechanisms of impaired regulation by CD4+CD25+FOXP3+ regulatory T cells in human autoimmune diseases. Nat Rev Immunol. (2010) 10:849. doi: 10.1038/nri2889
181. Dominguez-Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat Immunol. (2018) 19:665–73. doi: 10.1038/s41590-018-0120-4
182. O'Connor RA, Anderton SM. Foxp3+ regulatory T cells in the control of experimental CNS autoimmune disease. J Neuroimmunol. (2008) 193:1–11. doi: 10.1016/j.jneuroim.2007.11.016
183. Fletcher JM, Lonergan R, Costelloe L, Kinsella K, Moran B, O'Farrelly C, et al. CD39+Foxp3+ regulatory T cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J Immunol. (2009) 183:7602–10. doi: 10.4049/jimmunol.0901881
184. Álvarez-Sánchez N, Cruz-Chamorro I, Díaz-Sánchez M, Lardone P, Guerrero J, Carrillo-Vico A. Peripheral CD39-expressing T regulatory cells are increased and associated with relapsing-remitting multiple sclerosis in relapsing patients. Sci Rep. (2019) 9:2302. doi: 10.1038/s41598-019-38897-w
185. Peelen E, Damoiseaux J, Smolders J, Knippenberg S, Menheere P, Tervaert J, et al. Th17 expansion in MS patients is counterbalanced by an expanded CD39+ regulatory T cell population during remission but not during relapse. J Neuroimmunol. (2011) 240:97–103. doi: 10.1016/j.jneuroim.2011.09.013
186. Wang Y, Begum-Haque S, Telesford KM, Ochoa-Repáraz J, Christy M, Kasper EJ, et al. A commensal bacterial product elicits and modulates migratory capacity of CD39+ CD4 T regulatory subsets in the suppression of neuroinflammation. Gut Microbes. (2014) 5:552–61. doi: 10.4161/gmic.29797
187. Venken K, Hellings N, Thewissen M, Somers V, Hensen K, Rummens J, et al. Compromised CD4+CD25high regulatory T-cell function in patients with relapsing-remitting multiple sclerosis is correlated with a reduced frequency of FOXP3-positive cells and reduced FOXP3 expression at the single-cell level. Immunology. (2008) 123:79–89. doi: 10.1111/j.1365-2567.2007.02690.x
188. Consortium I, Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. (2007) 357:851–62. doi: 10.1056/NEJMoa073493
189. Liu Y, Teige I, Birnir B, Issazadeh-Navikas S. Neuron-mediated generation of regulatory T cells from encephalitogenic T cells suppresses EAE. Nat Med. (2006) 12:1402. doi: 10.1038/nm1402
190. Sacramento PM, Monteiro C, Dias AS, Kasahara TM, Ferreira TB, Hygino J, et al. Serotonin decreases the production of Th1/Th17 cytokines and elevates the frequency of regulatory CD4+ T-cell subsets in multiple sclerosis patients. Eur J Immunol. (2018) 48:1376–88. doi: 10.1002/eji.201847525
191. Mercadante ER, Lorenz UM. Breaking free of control: how conventional T cells overcome regulatory T cell suppression. Front Immunol. (2016) 7:193. doi: 10.3389/fimmu.2016.00193
192. Schneider A, Long S, Cerosaletti K, Ni CT, Samuels P, Kita M, et al. In active relapsing-remitting multiple sclerosis, effector T cell resistance to adaptive Tregs involves IL-6–mediated signaling. Sci Transl Med. (2013) 5:170ra15. doi: 10.1126/scitranslmed.3004970
193. Trinschek B, Luessi F, Lüssi F, Haas J, Wildemann B, Zipp F, et al. Kinetics of IL-6 production defines T effector cell responsiveness to regulatory T cells in multiple sclerosis. PLoS ONE. (2013) 8:e77634. doi: 10.1371/annotation/0e76c09e-75b2-493a-90fd-cda3187a0888
194. Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med. (2018) 378:169–80. doi: 10.1056/NEJMra1401483
195. Tintore M, Vidal-Jordana A, Sastre-Garriga J. Treatment of multiple sclerosis—success from bench to bedside. Nat Rev Neurol. (2018) 15:1. doi: 10.1038/s41582-018-0082-z
196. Ontaneda D, Tallantyre E, Kalincik T, Planchon SM, Evangelou N. Early highly effective versus escalation treatment approaches in relapsing multiple sclerosis. Lancet Neurol. (2019) 18:973–80. doi: 10.1016/S1474-4422(19)30151-6
197. González-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. (2012) 12:125. doi: 10.1038/nri3133
198. Piconese S, Pacella I, Timperi E, Barnaba V. Divergent effects of type-I interferons on regulatory T cells. Cytokine Growth F R. (2015) 26:133–41. doi: 10.1016/j.cytogfr.2014.10.012
199. Ebrahimi M, Ganji A, Zahedi S, Nourbakhsh P, Ghasami K, Mosayebi G. Characterization of regulatory T-cells in multiple sclerosis patients treated with interferon beta-1a. CNS Neurol Disord Drug Targets. (2018) 17:113–8. doi: 10.2174/1871527317666180327122435
200. Namdar A, Nikbin B, Ghabaee M, Bayati A, Izad M. Effect of IFN-ß therapy on the frequency and function of CD4+CD25+ regulatory T cells and Foxp3 gene expression in relapsing–remitting multiple sclerosis (RRMS): a preliminary study. J Neuroimmunol. (2010) 218:120–4. doi: 10.1016/j.jneuroim.2009.10.013
201. Vandenbark AA, Huan J, Agotsch M, Tocha D, Goelz S, Offner H, et al. Interferon-beta-1a treatment increases CD56bright natural killer cells and CD4+CD25+ Foxp3 expression in subjects with multiple sclerosis. J Neuroimmunol. (2009) 215:125–8. doi: 10.1016/j.jneuroim.2009.08.007
202. Hong J, Li N, Zhang X, Zheng B, Zhang JZ. Induction of CD4+CD25+ regulatory T cells by copolymer-I through activation of transcription factor Foxp3. Proc Natl Acad Sci USA. (2005) 102:6449–54. doi: 10.1073/pnas.0502187102
203. Haas J, Korporal M, Balint B, Fritzsching B, Schwarz A, Wildemann B. Glatiramer acetate improves regulatory T-cell function by expansion of naive CD4+CD25+FOXP3+CD31+ T-cells in patients with multiple sclerosis. J Neuroimmunol. (2009) 216:113–7. doi: 10.1016/j.jneuroim.2009.06.011
204. Polman CH, O'Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, et al. A Randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. (2006) 354:899–910. doi: 10.1056/NEJMoa044397
205. Rudick RA, Stuart WH, Calabresi PA, Confavreux C, Galetta SL, Radue E-W, et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N Engl J Med. (2006) 354:911–23. doi: 10.1056/NEJMoa044396
206. Kleinewietfeld M, Starke M, Mitri D, Borsellino G, Battistini L, Rötzschke O, et al. CD49d provides access to “untouched” human Foxp3+ Treg free of contaminating effector cells. Blood. (2009) 113:827–36. doi: 10.1182/blood-2008-04-150524
207. Stenner M-P, Waschbisch A, Buck D, Doerck S, Einsele H, Toyka KV, et al. Effects of natalizumab treatment on Foxp3+ T regulatory cells. PLoS ONE. (2008) 3:e3319. doi: 10.1371/journal.pone.0003319
208. Cohen JA, Barkhof F, Comi G, Hartung H-P, Khatri BO, Montalban X, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. (2010) 362:402–15. doi: 10.1056/NEJMoa0907839
209. Kappos L, Radue E-W, O'Connor P, Polman C, Hohlfeld R, Calabresi P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. (2010) 362:387–401. doi: 10.1056/NEJMoa0909494
210. Liu G, Burns S, Huang G, Boyd K, Proia RL, Flavell RA, et al. The receptor S1P1 overrides regulatory T cell–mediated immune suppression through Akt-mTOR. Nat Immunol. (2009) 10:769–77. doi: 10.1038/ni.1743
211. Eken A, Duhen R, Singh AK, Fry M, Buckner JH, Kita M, et al. S1P1 deletion differentially affects TH17 and regulatory T cells. Sci Rep. (2017) 7:12905. doi: 10.1038/s41598-017-13376-2
212. Dominguez-Villar M, Raddassi K, Danielsen A, Guarnaccia J, Hafler DA. Fingolimod modulates T cell phenotype and regulatory T cell plasticity in vivo. J Autoimmun. (2018) 96:40–9. doi: 10.1016/j.jaut.2018.08.002
213. Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. (2012) 367:1098–107. doi: 10.1056/NEJMoa1114287
214. Linker RA, Lee D-H, Ryan S, van Dam AM, Conrad R, Bista P, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. (2011) 134:678–92. doi: 10.1093/brain/awq386
215. Diebold M, Sievers C, Bantug G, Sanderson N, Kappos L, Kuhle J, et al. Dimethyl fumarate influences innate and adaptive immunity in multiple sclerosis. J Autoimmun. (2018) 86:39–50. doi: 10.1016/j.jaut.2017.09.009
216. Cohen JA, Coles AJ, Arnold DL, Confavreux C, Fox EJ, Hartung H-P, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. (2012) 380:1819–28. doi: 10.1016/S0140-6736(12)61769-3
217. Coles AJ, Twyman CL, Arnold DL, Cohen JA, Confavreux C, Fox EJ, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. (2012) 380:1829–39. doi: 10.1016/S0140-6736(12)61768-1
218. Cox AL, Thompson S, Jones JL, Robertson VH, Hale G, Waldmann H, et al. Lymphocyte homeostasis following therapeutic lymphocyte depletion in multiple sclerosis. Eur J Immunol. (2005) 35:3332–42. doi: 10.1002/eji.200535075
219. Mercanti S, Rolla S, Cucci A, Bardina V, Cocco E, Vladic A, et al. Alemtuzumab long-term immunologic effect Treg suppressor function increases up to 24 months. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e194. doi: 10.1212/NXI.0000000000000194
220. Havari E, Turner MJ, Campos-Rivera J, Shankara N, Nguyen T, Roberts B, et al. Impact of alemtuzumab treatment on the survival and function of human regulatory T cells in vitro. Immunology. (2014) 141:123–31. doi: 10.1111/imm.12178
221. Carson D, Wasson D, Taetle R, Yu A. Specific toxicity of 2-chlorodeoxyadenosine toward resting and proliferating human lymphocytes. Blood. (1983) 62:737–43. doi: 10.1182/blood.V62.4.737.737
222. Piro LD, Carrera CJ, Carson DA, Beutler E. Lasting remissions in hairy-cell leukemia induced by a single infusion of 2-chlorodeoxyadenosine. N Engl J Med. (1990) 322:1117–21. doi: 10.1056/NEJM199004193221605
223. Giovannoni G, Comi G, Cook S, Rammohan K, Rieckmann P, Sørensen P, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. (2010) 362:416–26. doi: 10.1056/NEJMoa0902533
224. Mitosek-Szewczyk K, Tabarkiewicz J, Wilczynska B, Lobejko K, Berbecki J, Nastaj M, et al. Impact of cladribine therapy on changes in circulating dendritic cell subsets, T cells and B cells in patients with multiple sclerosis. J Neurol Sci. (2013) 332:35–40. doi: 10.1016/j.jns.2013.06.003
225. Irwin MR. Sleep and inflammation: partners in sickness and in health. Nat Rev Immunol. (2019) 19:702–15. doi: 10.1038/s41577-019-0190-z
226. Scheiermann C, Gibbs J, Ince L, Loudon A. Clocking in to immunity. Nat Rev Immunol. (2018) 18:423–37. doi: 10.1038/s41577-018-0008-4
227. Buenafe AC. Diurnal rhythms are altered in a mouse model of multiple sclerosis. J Neuroimmunol. (2012) 243:12–7. doi: 10.1016/j.jneuroim.2011.12.002
228. Druzd D, Matveeva O, Ince L, Harrison U, He W, Schmal C, et al. Lymphocyte circadian clocks control lymph node trafficking and adaptive immune responses. Immunity. (2017) 46:120–32. doi: 10.1016/j.immuni.2016.12.011
229. He W, Holtkamp S, Hergenhan S, Kraus K, de Juan A, Weber J, et al. Circadian expression of migratory factors establishes lineage-specific signatures that guide the homing of leukocyte subsets to tissues. Immunity. (2018) 49:1175–90.e7. doi: 10.1016/j.immuni.2018.10.007
230. Sutton CE, Finlay CM, Raverdeau M, Early JO, DeCourcey J, Zaslona Z, et al. Loss of the molecular clock in myeloid cells exacerbates T cell-mediated CNS autoimmune disease. Nat Commun. (2017) 8:1923. doi: 10.1038/s41467-017-02111-0
231. Bollinger T, Bollinger A, Skrum L, Dimitrov S, Lange T, Solbach W. Sleep-dependent activity of T cells and regulatory T cells. Clin Exp Immunol. (2009) 155:231–8. doi: 10.1111/j.1365-2249.2008.03822.x
232. Kiernozek E, Kowalik A, Markowska M, Kozlowska E, Drela N. Day/night changes of thymus-deriving natural regulatory T cell development and function. J Neuroimmunol. (2014) 274:102–10. doi: 10.1016/j.jneuroim.2014.07.002
233. Mizutani H, Tamagawa-Mineoka R, Minami Y, Yagita K, Katoh N. Constant light exposure impairs immune tolerance development in mice. J Dermatol Sci. (2017) 86:63–70. doi: 10.1016/j.jdermsci.2016.12.016
234. Yang G, Zhang H, Liu Y, Feng Y, Luo X-Q, Liu Z-Q, et al. Alternation of circadian clock modulates forkhead box protein-3 gene transcription in CD4+ T cells in the intestine. J Allergy Clin Immun. (2016) 138:1446–9.e10. doi: 10.1016/j.jaci.2016.04.035
235. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. (2014) 510:92–101. doi: 10.1038/nature13479
236. Chiurchiù V, Leuti A, Dalli J, Jacobsson A, Battistini L, Maccarrone M, et al. Proresolving lipid mediators resolvin D1, resolvin D2, and maresin 1 are critical in modulating T cell responses. Sci Transl Med. (2016) 8:353ra111. doi: 10.1126/scitranslmed.aaf7483
237. Baumjohann D, Ansel MK. MicroRNA-mediated regulation of T helper cell differentiation and plasticity. Nat Rev Immunol. (2013) 13:666–78. doi: 10.1038/nri3494
238. Chen GY, Satpathy AT, Chang HY. Gene regulation in the immune system by long noncoding RNAs. Nat Immunol. (2017) 18:962–72. doi: 10.1038/ni.3771
239. Inácio DP, Amado T, Silva-Santos B, Gomes AQ. Control of T cell effector functions by miRNAs. Cancer Lett. (2018) 427:63–73. doi: 10.1016/j.canlet.2018.04.011
240. Bhairavabhotla R, Kim YC, Glass DD, Escobar TM, Patel MC, Zahr R, et al. Transcriptome profiling of human FoxP3+ regulatory T cells. Hum Immunol. (2016) 77:201–13. doi: 10.1016/j.humimm.2015.12.004
241. Rouas R, Fayyad-Kazan H, Zein N, Lewalle P, Rothé F, Simion A, et al. Human natural Treg microRNA signature: role of microRNA-31 and microRNA-21 in FOXP3 expression. Eur J Immunol. (2009) 39:1608–18. doi: 10.1002/eji.200838509
242. Du C, Liu C, Kang J, Zhao G, Ye Z, Huang S, et al. MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol. (2009) 10:1252–9. doi: 10.1038/ni.1798
243. Liu C, Yang H, Shi W, Wang T, Ruan Q. MicroRNA-mediated regulation of T helper type 17/regulatory T-cell balance in autoimmune disease. Immunology. (2018) 155:427–34. doi: 10.1111/imm.12994
244. Roy S, Awasthi A. Emerging roles of noncoding RNAs in T cell differentiation and functions in autoimmune diseases. Int Rev Immunol. (2019) 1–14. doi: 10.1080/08830185.2019.1648454
245. Santis G, Ferracin M, Biondani A, Caniatti L, Tola M, Castellazzi M, et al. Altered miRNA expression in T regulatory cells in course of multiple sclerosis. J Neuroimmunol. (2010) 226:165–71. doi: 10.1016/j.jneuroim.2010.06.009
Keywords: neuroimmunity, multiple sclerosis, Treg-regulatory T cell, FoxP3, Treg heterogeneity, immune regulation
Citation: Sambucci M, Gargano F, Guerrera G, Battistini L and Borsellino G (2019) One, No One, and One Hundred Thousand: T Regulatory Cells' Multiple Identities in Neuroimmunity. Front. Immunol. 10:2947. doi: 10.3389/fimmu.2019.02947
Received: 04 October 2019; Accepted: 02 December 2019;
Published: 20 December 2019.
Edited by:
Margarita Dominguez-Villar, Imperial College London, United KingdomReviewed by:
Silvia Deaglio, University of Turin, ItalyCopyright © 2019 Sambucci, Gargano, Guerrera, Battistini and Borsellino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giovanna Borsellino, Zy5ib3JzZWxsaW5vQGhzYW50YWx1Y2lhLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.