 Andreu Garcia-Vilanova
Andreu Garcia-Vilanova John Chan2,3
John Chan2,3 Jordi B. Torrelles
Jordi B. Torrelles
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 18 December 2019
Sec. Vaccines and Molecular Therapeutics
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02909
This article is part of the Research TopicMycobacterial Glycolipids – Role in Immunomodulation and Targets for Vaccine DevelopmentView all 7 articles
The Mycobacterium tuberculosis cell envelope has been evolving over time to make the bacterium transmissible and adaptable to the human host. In this context, the M. tuberculosis cell envelope contains a peripheral barrier full of lipids, some of them unique, which confer M. tuberculosis with a unique shield against the different host environments that the bacterium will encounter at the different stages of infection. This lipid barrier is mainly composed of glycolipids that can be characterized by three different subsets: trehalose-containing, mannose-containing, and 6-deoxy-pyranose-containing glycolipids. In this review, we explore the roles of these cell envelope glycolipids in M. tuberculosis virulence and pathogenesis, drug resistance, and further, how these glycolipids may dictate the M. tuberculosis cell envelope evolution from ancient to modern strains. Finally, we address how these M. tuberculosis cell envelope glycolipids are impacted by the host lung alveolar environment, their role in vaccination and masking host immunity, and subsequently the impact of these glycolipids in shaping how M. tuberculosis interacts with host cells, manipulating their immune response to favor the establishment of an infection.
Currently, in the twenty-first century, one person is infected with Mycobacterium tuberculosis (M. tuberculosis) every 6 s, and one person dies of tuberculosis (TB) every 21 s. Multi-, extensively, extremely (also known as total) drug-resistant (MDR/XDR/XXDR/TDR) M. tuberculosis strains are emerging worldwide as a threat to public health and TB control, raising concerns of a future epidemic of virtually untreatable TB (1–5). It is estimated that drug-resistant TB will kill 75 million people and cost the global economy $16.7 trillion over the next 35 years (6).
Most current and proposed therapies target the M. tuberculosis cell envelope, a complex and dynamic structure. Despite this, little is known regarding the role of M. tuberculosis cell envelope lipids, and specially glycolipids, in the establishment and maintenance of the infection and in the development of active TB disease. During its path to infection and disease outcome, M. tuberculosis gets in contact with different host environments (7), where a critical gap in our knowledge is defining the changes in the M. tuberculosis cell envelope structures during the course of pulmonary infection, as well as how these changes determine the M. tuberculosis infection outcome.
Little is also known about the cell envelope composition of MDR/XDR/XXDR/TDR M. tuberculosis strains. For these strains, it is still unknown what bacterial and host factors are involved in the induction of their overwhelming tissue damaging, driving to the collapse of the lung merely months after active TB is diagnosed. The M. tuberculosis cell envelope, in its majority (~80%), comprises carbohydrates and lipids readily exposed to the host immune system and thus being instrumental in determining the M. tuberculosis pathway of infection and the host inflammatory response that defines the TB disease outcome. In this context, structural-biological function relationships for the majority of the M. tuberculosis cell envelope glycolipids are still being elucidated. Mainly, M. tuberculosis glycolipids are composed of two carbohydrates, trehalose (a glucose disaccharide) and mannose (Figure 1). Related to the M. tuberculosis cell envelope trehalose glycolipid content, it is plausible that trehalose synthesis and degradation play a role in the process of reciprocal carbon mobilization across the M. tuberculosis cell envelope, especially during infection, where the regulation of trehalose containing lipids on the M. tuberculosis cell envelope may be modulated by many environmental and physiological factors that affect both M. tuberculosis and the host cell (8, 9). In regard to mannose-containing glycolipids and lipoglycans, studies have focused on showing how M. tuberculosis mimics the host by containing cell envelope components (e.g., mannose-containing molecules) whose terminal epitopes closely resemble mammalian glycoforms such as mannoproteins (Figure 2). Indeed, studies have shown that M. tuberculosis uses this host resemblance to its advantage, gaining entrance and establishing a unique intracellular niche within the host (10, 11).
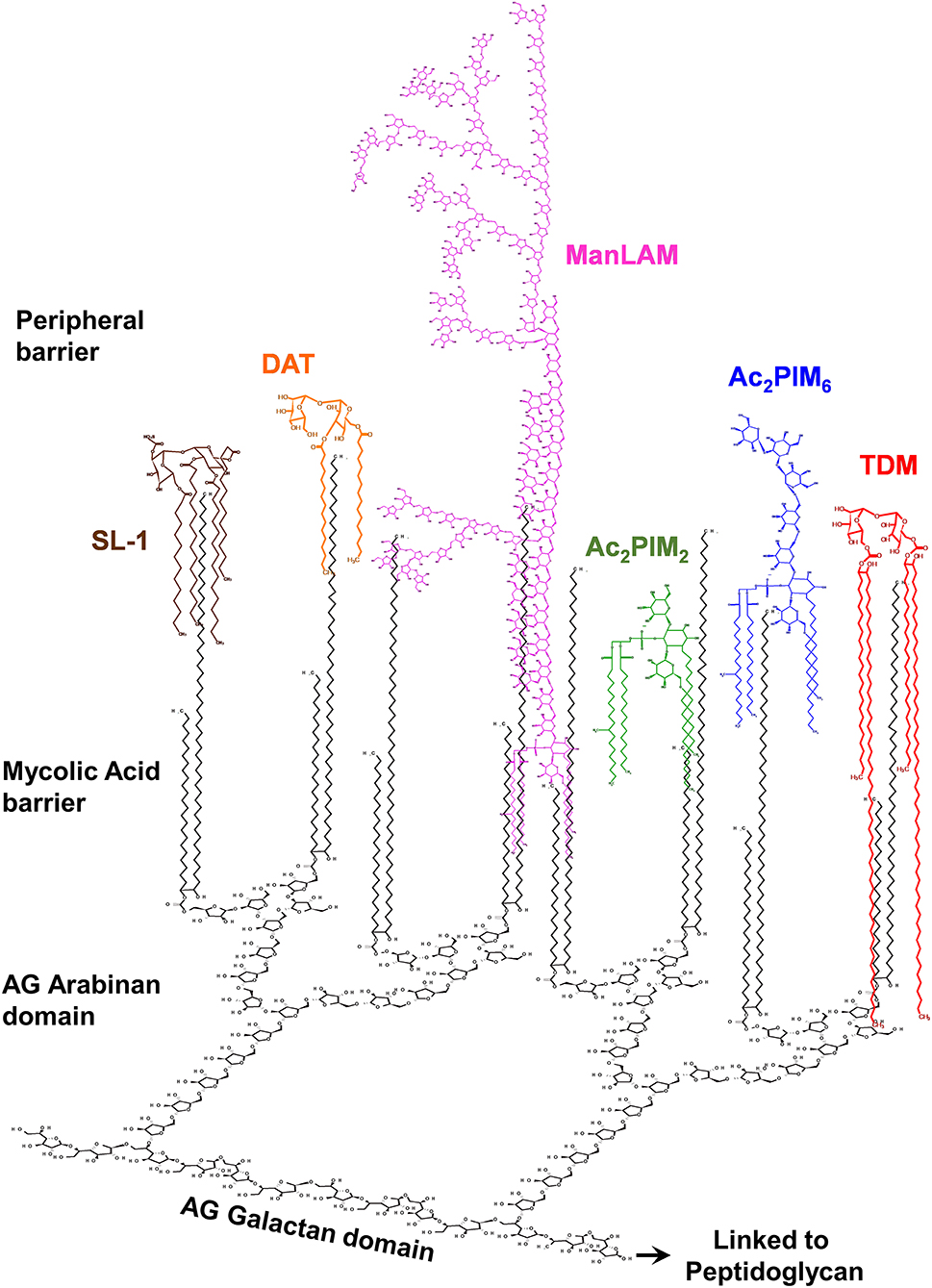
Figure 1. The M. tuberculosis complex cell envelope with emphasis on surface glycolipids. The M. tuberculosis cell envelope is composed of a covalently linked core called mycolic-acids-arabinogalactan-peptidoglycan (mAGP) complex. On the top of this core is described to be a peripheral lipid barrier, where lipids intercalate among the mycolic acids of mAGP. Mycolic acids are perpendicular to the plasma membrane. Here the M. tuberculosis cell envelope surface location of mannose-containing glycolipids (phosphatidyl-myo-inositol mannosides, or PIMs) and lipoglycans (e.g., the mannose capped lipoarabinomannan, or ManLAM), as well as trehalose containing lipids [e.g., trehalose-dimycolate (TDM), sulfolipid-1 (SL-1), and diacyl-trehalose (DAT)], is depicted. The trehalose containing lipooligoscharides (LOSs), phthienoates and hydroxyphthienoates, 6-deoxy-pyranose-lipids (phenolic glycolipids, or PGLs), and mannose-containing mannosyl-β-1–phosphomycoketides (MPM) are not depicted. Outer material components (i.e., α-glucan, mannan, arabinomannan, and xylan) are also not shown. Relative number of molecules and size are not accurately depicted, reflecting published experimental data.
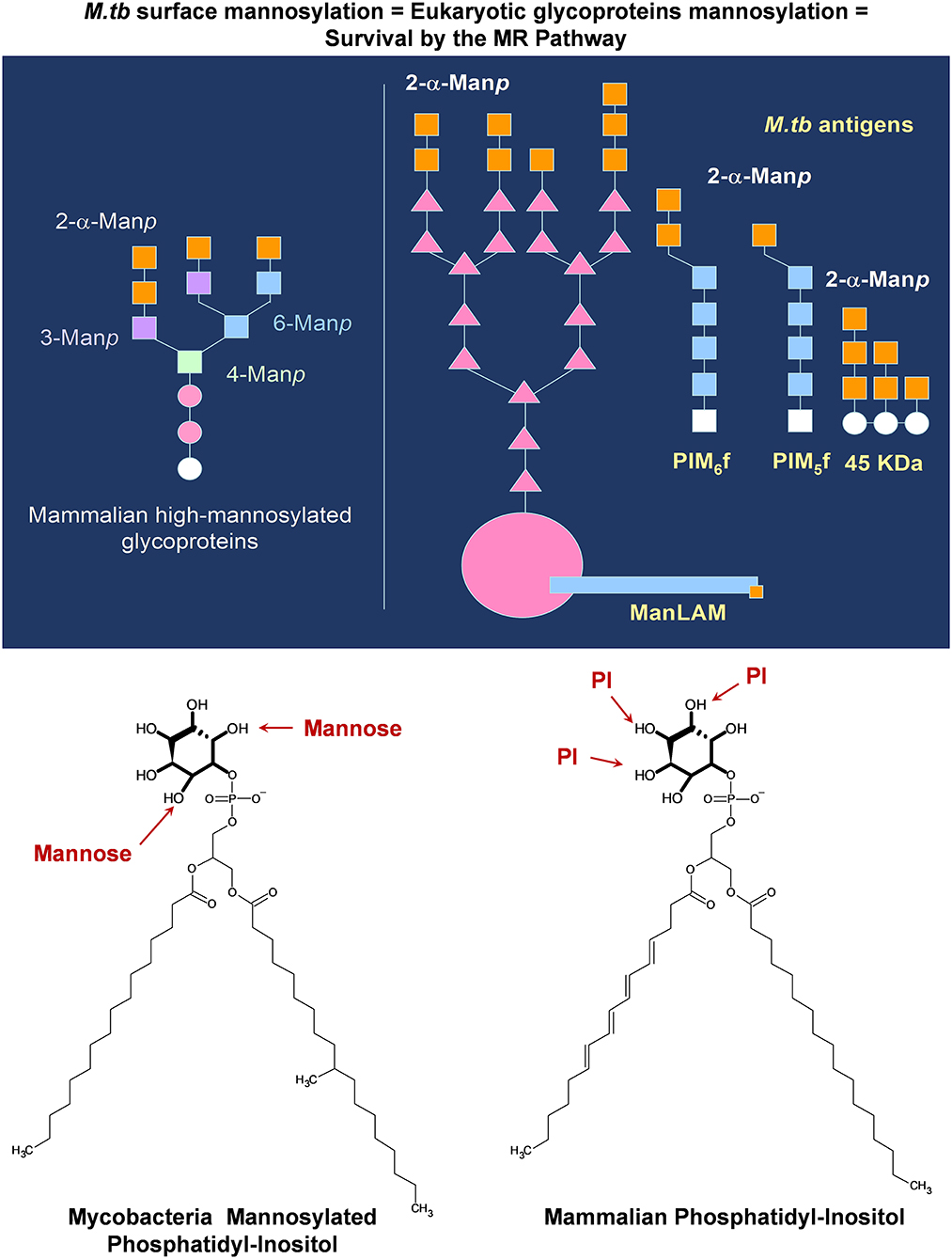
Figure 2. M. tuberculosis complex mannose-containing lipids mimic mammalian structures allowing the bacterium to gain entry into host cells and thus potentially bypassing the host immune response. M. tuberculosis mannose-containing glycolipids (PIMs) and lipoglycans [ManLAM and lipomannan (LM), and some mannose containing proteins] contain α-2-mannose in their non-reducing termini, mimicking non-siliated mammalian glycoproteins in circulation. These non-siliated mammalian proteins can trigger inflammation, and thus, the host contains a series of homeostatic receptors, such as the mannose receptor (MR), to remove them out from circulation. M. tuberculosis can take advantage of this host clearance system by increasing its mannose surface, allowing the M. tuberculosis bacterium to interact with the homeostatic MR (among others), gaining entry and surviving within host cells. Moreover, these same glycolipids and lipoglycans contain a GPI-anchor of similar structure to mamalian phosphatidyl-inositol, which intercalates within vesicular membranes, regulating vesicular trafficking and fusion.
Many M. tuberculosis-host factors are involved in this first encounter interface at the initial stages of the infection. First, the constitution of the M. tuberculosis cell envelope, which is strain-dependent, from the worldwide used laboratory strains (M. tuberculosis H37Ra, M. tuberculosis H37Rv, and M. tuberculosis Erdman), to the highly transmissible CDC1551, to the hypervirulent HN878, and to several relevant drug-resistant M. tuberculosis clinical isolates in endemic TB areas (12–16). We will discuss their cell envelope glycolipid constitution in relation to their infection outcome. Second, we will focus on the alveolar space and its predominant cells, the alveolar macrophage (AM) and the alveolar epithelial cell, and how these alveolar cells act as a niche for M. tuberculosis and discuss their contribution to control or favor the infection. We will also discuss our results in an area frequently bypassed, the host lung alveolar environment and its impact on the M. tuberculosis cell envelope, addressing how the lung environment can determine and change the nature of the M. tuberculosis cell envelope during infection and how these changes on the M. tuberculosis cell envelope may determine/contribute to the pathway of infection and disease outcome. Importantly, we will also discuss our recent findings on the potential role of the mycobacterial glycolipids in masking the generation of the host protective immune response against M. tuberculosis infection and TB disease.
The M. tuberculosis complex includes M. tuberculosis, M. africanum, M. bovis, and the Bacillus Calmette–Guérin (BCG) strain, M. canetti, M. caprae, M. microti, M. mungi, M. orygis, M. pinnipedii, and M. suricattae (with M. dassie being not yet included). The M. tuberculosis complex cell envelope has a unique, complex, thick, and waxy cell envelope, with properties, such as low permeability and intrinsic resistance to entry of many hydrophobic antibiotics and resistance to harsh environments which, distinguish M. tuberculosis from other pathogenic bacteria (17, 18). This envelope comprises an array of lipids with diverse polarity, such as glycolipids, aminolipids, phospholipids, phosphoglycolipids, sulfolipids, phenolic-glycolipids, and other lipids; all of them exhibit important roles in M. tuberculosis pathogenicity and are an important target, due to their alleged essentiality and proved immunogenicity, for new drug treatments and diagnostics. Most, if not all, M. tuberculosis cell envelope components have been shown to provide M. tuberculosis with some advantage for establishing an infection or survival in the host, for inhibiting bacteriostatic components of the host lung mucosa, or by allowing it to survive harsh intracellular environments, adapting its metabolism to the host environment. Thus, the cell envelope of M. tuberculosis, per se, may contain all of the elements associated with active TB disease, including elements driving tissue caseation, hypersensitization, cytotoxicity, and the antigens triggering or dampening humoral and adaptive immunity (19). However, although obvious, cell envelope compositional differences exist among M. tuberculosis complex strains and non-pathogenic mycobacteria; a detailed electron microscopy study has not yet identified any special and unique features on the M. tuberculosis complex strains.
The M. tuberculosis cell envelope is defined in five distinct compartments, the plasma membrane, a periplasmic space, the cell envelope core, the peripheral lipid layer, and around it, the outer material. All these parts are thought to provide structural, mechanical, and metabolic support, as well as osmotic protection and transport and communication with host environments surrounding the bacillus during different stages of infection. Here, we will discuss M. tuberculosis cell envelope, focusing on the M. tuberculosis peripheral lipid layer constituents and their role in M. tuberculosis pathogenesis.
The interest of studying the tubercle bacillus lipoids remotes to more than 90 years ago by Anderson et al. in 1929 stating: “The crude wax obtained from the chloroform extract of the human type of tubercle bacillus, Strain H-37, yielded on purification, two fractions; viz., a white solid substance designated as purified wax and a soft yellowish brown salve-like mass which was called soft wax and it possessed a slight but agreeable perfume-like odor” (20). Since then, a fascinating diversity of lipid structures and their biological activities have been and are still being described. In this context, glycolipids are one of the major M. tuberculosis cell envelope constituents, defined by their polarity, cytotoxicity, and/or immunological properties (17) (Figure 1). These mainly comprise the acyl trehaloses family sharing a common α-D-Glcp(1→ 1′)-α-D-Glcp unit and are the class of M. tuberculosis lipids most extensively studied by lipidologists and mycobacteriologists. These specifically include di-, tri-, and penta-acyltrehaloses (DATs/TATs/PATs); mono- and di-mycolyl trehaloses (TMM/TDM); and tetraacyl-trehalose sulfo(glyco)lipids (SLs) (21), and the lesser studied to date, oligosaccharides containing lipids [lipooligosaccharides (LOSs)] (Figure 3). Another class of M. tuberculosis glycolipids of important relevance is the phosphatidyl-myo-inositol mannose derivatives (PIMs). Some of them are precursors of two major lipoglycans on the M. tuberculosis cell surface, the lipomannan, and the mannose capped lipoarabinomannan (ManLAM) (Figure 3). Lastly, and no less important, some M. tuberculosis strains also contain phenolic glycolipids (PGLs) (Figure 3). All these glycolipids are thought to perform filler roles in completing the outer leaflet of the asymmetric peripheral lipid layer on the M. tuberculosis cell envelope (17). The importance of these lipids in M. tuberculosis infection and pathogenesis is described in the sections below and summarized in Table 1 and Figure 4.
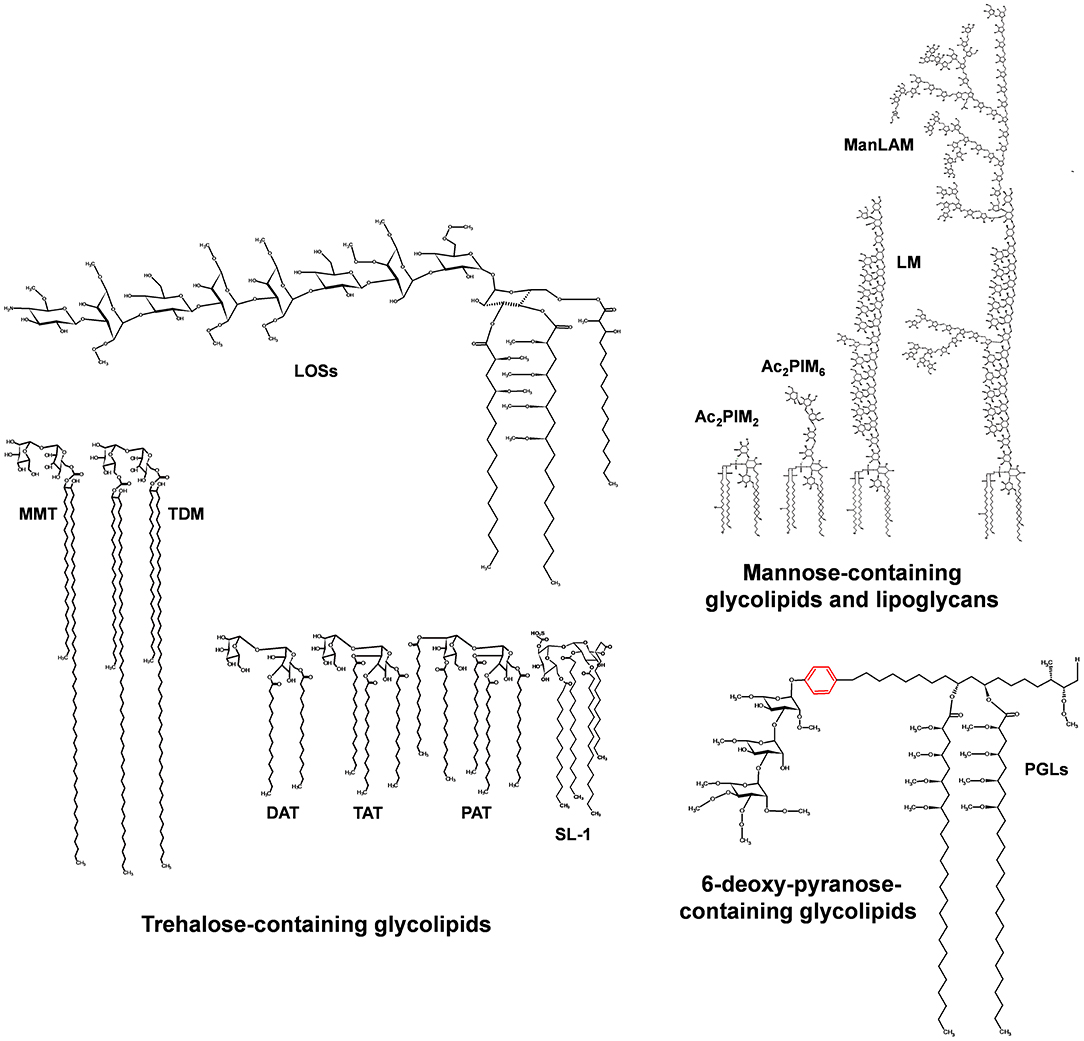
Figure 3. Major glycolipids of the M. tuberculosis cell envelope surface. Three groups of glycolipids can be found on the M. tuberculosis cell envelope: (i) trehalose containing lipids, which involve di-, tri-, and penta-acylated trehaloses (DAT, TAT, PAT); trehalose mono- and di-mycolate (TMM, TDM); sulfolipids (SL-1); and lipooligosaccharides (LOSs); (ii) mannose-containing lipids, which involve the phosphatidyl-myo-inositol mannosides of different lengths (1 to 6 mannoses) and acylation patterns 1 to 4 fatty acid of different nature, being the most common palmitic (16:0) and tuberculostearic acid (TBST); and (iii) 6-deoxy-pyranose containing lipids, mainly phenolic glycolipids (PGLs). LOS and PGL types are restricted to a few strains of the M. tuberculosis complex.
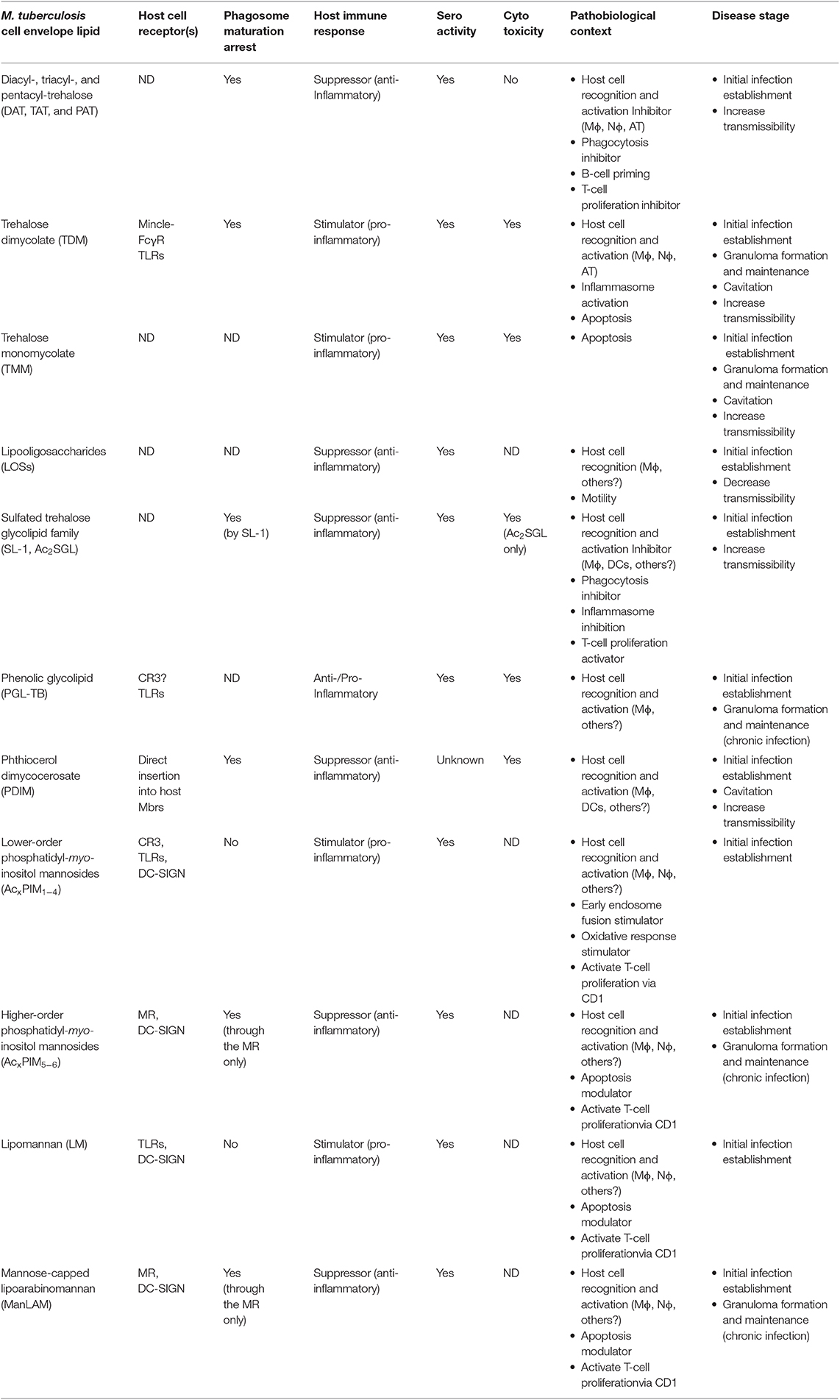
Table 1. M. tuberculosis cell wall glycolipids and lipoglycans and their interaction with the host.
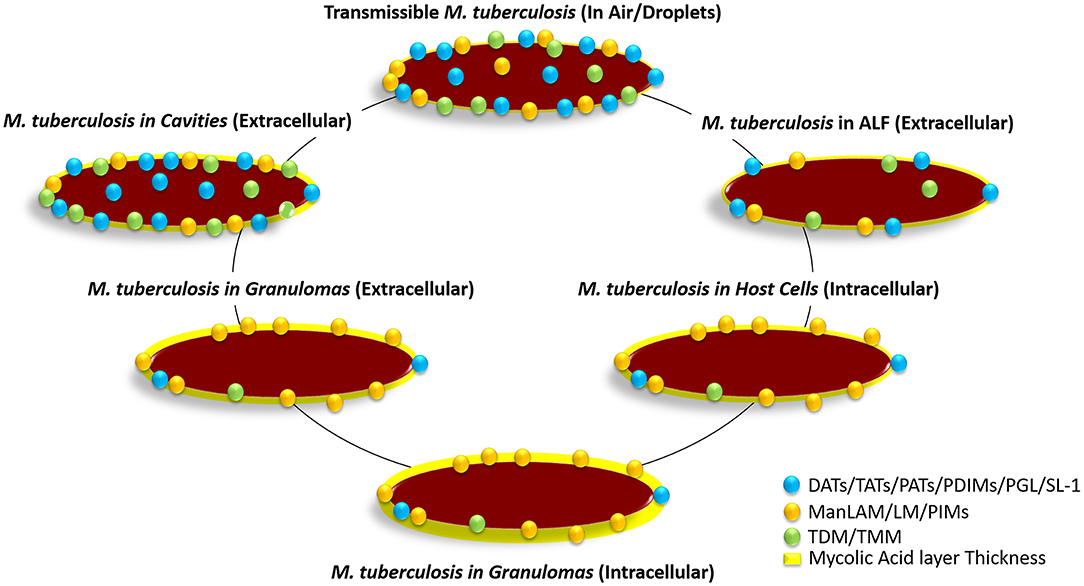
Figure 4. Hypothetical distribution of glycolipids on the M. tuberculosis bacterium cell envelope surface at the different stages of infection. The transmissible bacterium is thought to contain a large number of glycolipids (DAT, TAT, PAT, SL-1, TMM, TDM, PDIMS, PIMs, etc.) on the cell envelope driving surface hydrophobicity, which in turn favors its transmission through the air. Upon infection and after reaching the alveolar space, M. tuberculosis gets in contact with alveolar lining fluid (ALF; hypophase of the lung mucosa). ALF contains an array of homeostatic hydrolases that significantly alter the M. tuberculosis cell envelope surface removing glycolipids and lipoglycans, and thus somehow altering its cell surface hydrophobicity and determining how the bacterium will interact with alveolar host cells. After phagocytosis by AMs, the bacterium is shown to hyperproduce PIMs, as well as switching to metabolic networks such as beta-oxidation, glyoxylate shunt, and the reverse methylcitrate cycle. This metabolic switch within host cells allows the bacterium to break down own and host captured long-chain fatty acids and cholesterol to generate acetyl-CoA and propionyl-CoA, increasing its production of mycolic acids in detriment of producing glycolipids. This bacterium metabolic state is thought to be maintained at latency stages when the bacterium remains intracellular within granulomas. Upon reactivation, the bacterium metabolic state changes again increasing glycolipid production in detriment of producing mycolic acids, restoring the bacterium surface hydrophobicity, while remaining extracellular within granulomas. This bacterial surface hydrophobicity gets further accentuated when M. tuberculosis escapes disrupted granulomas becoming extracellular within cavities, where host-driven hydrolytic destruction of tissues enhances M. tuberculosis escape to the blood stream and airways becoming transmissible again closing the cycle. Relative number of molecules and size are not accurately depicted reflecting published experimental data.
Diacyl- (DAT; a 2, 3-diacyltrehalose), triacyl-trehalose (TAT; a 2, 3, 6-triacyltrehalose), and pentaacyl-trehalose (PAT; a 2, 3, 6, 2′, 4′-pentaacyltrehalose) contain polymethylated acyl groups (22). Recent studies unveiled that DAT family alone isolated from the M. tuberculosis cell envelope contains more than 30 molecular species, with each species containing up to six isomeric structures (23).
Their purpose and function in the M. tuberculosis cell envelope are still unclear. Some studies related DAT functions to its capacity to negatively regulate the host pro-inflammatory response induced by M. tuberculosis during infection in a dose-dependent manner (24). Indeed, DATs do not seem to participate in the receptor-mediated phagocytosis process of M. tuberculosis, do not induce host cytotoxicity, but trigger IL-10 production and are capable of inhibiting T-cell proliferation (25), supporting their role in potentially being used by M. tuberculosis to offset the host immune response during initial stages of the infection (24) (Table 1). Moreover, other studies indicate the capacity of DATs acting together with PAT (the major polyacyltrehalose of M. tuberculosis containing four mycolipenoyl and one fully saturated palmitoyl) in the maintenance of the M. tuberculosis cell envelope, as well as in pathogenesis promoting the M. tuberculosis survival within host cells (26) and being antigenic (27). In this context, several studies emphasize the potential primary role of DATs in regulating the composition of the M. tuberculosis cell envelope, serving as anchors of the M. tuberculosis outer material, which blocks M. tuberculosis recognition and phagocytosis by host cells (28, 29). In this regard, M. tuberculosis DAT mutants have altered cell envelope surface and have enhanced phagocytosis by host cells, but their absence from the M. tuberculosis cell envelope does not alter the bacterium capacity to replicate and survive in vivo using the mouse model (28, 30). Conversely, other studies have shown that M. tuberculosis mutants lacking DATs and sulfoglycolipids cannot block phagosome maturation, thus revealing the importance of these molecules in the M. tuberculosis pathogenesis (31). Currently, the contribution of M. tuberculosis acytrehaloses to pathogenesis is still understudied, and the main use of these M. tuberculosis glycolipids is still for serodiagnosis (32–36).
Other trehalose-containing family of lipids characterized by their long acyl groups is the mycolyl-containing trehalose lipids (37). These are widely distributed and considered the most abundant, non-covalently associated M. tuberculosis cell envelope lipids, providing M. tuberculosis with a waxy, impermeable appearance. Both TDM (6,6′-dimycoloyl-α-D-trehalose) and TMM (6-monomycoloyl-α-D-trehalose) have their mycolates oriented toward the cell plasma membrane, presenting their trehalose moiety to the external environment.
TDM (or cord factor) was first obtained after a petroleum ether extraction from cells of a virulent strain of M. tuberculosis (38). After the extraction, M. tuberculosis presented a drastic disorganization of the typical cellular cords formed in a culture, and the resulting petroleum ether organic extract was toxic in mice. This toxicity was later shown to be related to TDM (39). Subsequently, further studies indicated that TDM toxicity is linked to the regulation of tissue-specific nicotinamide adenine dinuclease (NAD) activity by TDM-induced NADse, decreasing tissue-specific NAD levels by blocking the electron flow along the mitochondrial respiratory chain and thus oxidative phosphorylation (40–44). M. tuberculosis TDM has two mycolic acids (or mycolates). Each TDM mycolate has two acyl-chains, a long β-hydroxy chain consisting of 50 to 60 carbons, and a shorter α-alkyl chain typically with 24 carbons (45). Thus, each mycolic acid of TDM is reported to contain between 74 and 90 carbons (45–47). The length and ester-linkages of the mycolates on TDM molecules define their toxicity; a longer mycolate shows higher toxicity (48, 49). Related to the M. tuberculosis cell envelope, TDM seems to interlock with ManLAM, which is also found on the surface of M. tuberculosis, to form an asymmetrical bilayer and thus directly participates in the maintenance of the M. tuberculosis cell envelope integrity (37). Studies removing TDM from the bacterium surface further indicated the importance of TDM in the M. tuberculosis-host relationship that takes place in infections in vitro and in vivo (50, 51). In this regard, M. tuberculosis is shown to produce large quantities of TDM during infection and when inside of the phagosome (52). TDM also seems to interlock host fibrinogen, the latter then acting as a cofactor for TDM biological effects (53).
M. tuberculosis TDM's biological properties have been described to rely on the whole molecule (54, 55), and these include (Table 1): interacting with Mincle (macrophage-inducible C-type lectin) (56) on the macrophage surface, driving M. tuberculosis-host recognition, and interacting with Mincle working together with the Fcγ-receptor transmembrane segment inducing Th1 (e.g., TNF, IL-12), Th2 (IL-6), and inflammasome (IL-1β) pro-inflammation mediators (57–59); actively participating in driving mycobacterial phagosome maturation arrest (55, 60) after phagocytosis of virulent strains of M. tuberculosis, allowing the bacillus to survive within the phagocyte; indirectly participating via its recognition by Mincle in the influx of neutrophils to the infection site (61); inducing host cell apoptosis (62–64); participating in the induction and vascularization of lung granulomas (65); being antigenic and having antitumoral activity (63, 66); having adjuvant properties being able to generate optimal antibody responses (67); inducing host cachexia, a typical symptom in active TB disease (68); and being able to trigger non-specific host immune responses against both parasitic and bacterial infections (63, 69–72). TDM is also shown to undergo cyclopropane modifications on its mycolates (73), driving a TDM-dependent reprogramming of the macrophage global gene expression during infection in vitro and in vivo (74).
TMM is known to be an essential precursor for the biosynthesis of TDM and the mycolic acid layer covalently linked to the arabinogalactan-peptidoglycan (mAGP) core of the M. tuberculosis cell envelope (75). Although TDM has been largely studied in its close relation with M. tuberculosis pathogenesis, this is not the case for TMM. TMM is thought to be involved in the maintenance of the structure and function of the M. tuberculosis cell envelope, working together with arabinogalactan mycolate, TDM (76), or phosphatidyl-ethanolamine, the latter shown to participate in the TMM translocation from the plasma membrane to M. tuberculosis cell envelope core (77). In this context, TMM is shown to be used by M. tuberculosis to transfer mycolic acids toward molecules like the envelope-linked arabinogalactan. In agreement with this fact, the known secreted immunogenic Ag 85 complex has been identified as a trehalose mycolyltransferase (78, 79).
As TDM, TMM has similar biological properties, also inducing lethal host cytotoxicity and host cell apoptosis; has granulomagenic activity; has adjuvant activity; and is capable of stimulating TNF via activation of the protein kinase C pathway (10, 80–84) (Table 1).
Another family of trehalose-based lipids present on the M. tuberculosis cell envelope is the LOSs. LOSs in the M. tuberculosis complex are understudied, produced by some strains (e.g., M. canettii) (85), and absent in most clinical isolates of M. tuberculosis as well as laboratory strains such as H37Rv, H37Ra, Erdman (86). The majority of their biological attributes are derived from studies performed in other mycobacterial species such as Mycobacterium kansasii, Mycobacterium smegmatis, and Mycobacterium marinum among others (85–89). LOSs are considered to be located on the surface of the mycobacterial cell envelope (90–92), but their contribution in shaping it is still unclear (93). However, the typical smooth and glossy colony formation observed in the M. tuberculosis complex strain canettii is speculated to be directly linked to the heavy presence of LOSs in this strain cell envelope (85, 94, 95).
All described LOSs share one trehalose with at least one or two long poly-methylated branched fatty acid chains that can be saturated or unsaturated, and located in both glucose residues of the trehalose end of the polymer (90). Depending on the mycobacterial species, a monosaccharide, or more frequently, an oligosaccharide (2 to 6 carbohydrate residues) is also present linked either at C3, C4, or C6 of the trehalose end (89, 96, 97). LOSs from different mycobacteria present different grade of variability in this oligosaccharide, the most frequent one being a tetraglucose structure [D-Glcp(β1→ 3)-D-Glcp-(β1→ 4)-D-Glcp-(α1→ 1α)-D-Glcp], which includes the trehalose (in bold). Pyruvic acid residues (carboxyethylidene) have also been described giving an anionic character to these molecules.
Related to LOSs biological properties, these may play an important role in regulating bacterial transmission, motility, and biofilm formation; are immunogenic and also phage receptors; and may play a role in the establishment of the infection (86, 98) (Table 1). LOSs are mainly used for serotyping, as LOS-specific antibodies are raised against the immunodominant LOS oligosaccharide. LOSs are also shown to directly or indirectly participate in the mycobacterial infection of host macrophages (86). In this regard, their role in M. tuberculosis virulence is unclear. Some studies suggest LOSs being attenuated/masking factors such as DATs, where mycobacterial species containing LOS are readily cleared in vivo; conversely, mycobacterial strains lacking LOSs cause chronic and systemic infections in vivo (92, 99).
One role of LOSs is to participate in reducing the hydrophobicity of the mycobacterial cell envelope. Studies suggest that M. tuberculosis complex strain canettii is an ancestor strain, mainly isolated in the horn of Africa, containing abundant LOSs (100). However, as evolution goes, modern M. tuberculosis complex strains became more hydrophobic, replacing LOSs in their cell envelope in detriment of adding more apolar lipids. This change of polarity/hydrophobicity is thought to provide critical advantages to the M. tuberculosis complex, increasing their capability for aerosol transmission (101), affecting their virulence and pathogenicity (100, 102).
As in the case of the trehalose containing DATs, TATs, PATs, TDMs, and TMMs, sulfated trehaloses (or sulfolipids, designated by SL) (103) are also present in virulent strains of M. tuberculosis, specifically SL-1 (104, 105). Sulfate derivatives are rare in nature, where the degree of sulfation could directly change the binding properties directing extracellular interactions between biomolecules from both, M. tuberculosis and host cells (106). Concretely, M. tuberculosis SLs consists of a trehalose-2-sulfate disaccharide that can be acylated by two [defining the diacylated sulfoglycolipid or Ac2SGL (107)] to four [defining SL-1 (108)] very long (up to C64) saturated and unsaturated, highly branched fatty acids (109, 110).
Like TDM, SL-1 [and potentially Ac2SGL (107)] is involved in the establishment of the infection. SL-1 seems to induce phagosome maturation arrest in macrophages (111); is cytotoxic in vitro (112, 113); inhibits macrophage priming/activation (114, 115), phagocytosis, and the inflammasome (115); induces superoxide release in activated neutrophils and monocytes; and are immunogenic with potential for serodiagnosis (33, 116–118) (Table 1). Specifically for Ac2SGL (107), this can stimulate and proliferate CD1b-restricted T cells (119), and stimulate the production of Th1 cytokines by CD8+ T cells (119). However, in vitro and in vivo studies, the latter using different animal models, dispute the role of SL-1 in M. tuberculosis virulence and pathogenesis (114–117, 120–122). SL-1 has been shown acting together with TDM, damaging the mitochondrial structures in the host cell in vitro; however, SL-1 alone intraperitoneally injected in mice did not induce cytotoxicity. M. tuberculosis mutants (pks2) lacking both SL-1 and its precursor Ac2SGL indicate that both are not required for M. tuberculosis to grow and cause pathogenesis in vivo. Conversely, an M. tuberculosis mutant (mmpl8) only lacking SL-1 but accumulating Ac2SGL were shown to be attenuated in vivo, indicating that the SL-1 precursor Ac2SGL is indeed immunogenically participating in M. tuberculosis virulence (108). In this regard, it has been suggested that as Ac2SGL is a precursor of SL-1 lacking two acyl groups (phthioceranic acid and hydroxyphthioceranic acid), it could be thus plausible that M. tuberculosis uses the ratio of SL-1/Ac2SGL in its cell envelope to modulate the host immune response to its advantage (119, 123).
Lesser-studied M. tuberculosis trehalose-containing lipids are the phthienoates (or mycolipenates) and hydroxyphthienoates (or mycolipanolates). These consist of four species with different polarities (124). The less polar species have acyl substituents mainly straight-chain 16:0 and 18:0 acids and 2,4,6-trimethyl tetracos-2-enoic (C27 phthienoic or mycolipenic) acid, or a 3-hydroxy-2,4,6-trimethyltetracosanoic (C27-hydroxyphthienoic or mycolipanolic) acid. The polar pair species mainly have straight-chain 16:0 and 18:0, C27-mycolipanolic acid, minor proportions of C25- and C27-mycolipenic acids, and major proportions of 2,4-dimethyldocosanoic acid (124, 125). Their relative contribution to the M. tuberculosis pathogenesis remains uncertain, but their function in maintaining the integrity of the cell envelope is stipulated (124).
Other M. tuberculosis glycolipids containing oligosaccharides are the PGLs. PGLs are considered a glycosylated form of phthiocerol dimycocerosates (PDIM) (126). These glycolipids have been mainly studied in Mycobacterium leprae (triglycosylated PGL-I) (127–129) and Mycobacterium marinum (monoglycosylated PGL), being used for diagnostic testing in leprosy patients (130). The exact structure of PGL is species-specific, and its biological properties are starting to be elucidated. In this regard, while in leprosy, M. leprae PGL-1 subverts tissue macrophages to produce neurotoxic radical nitrogen species (e.g., nitric oxide) leading to nerve damage (131). Along this line, PGL-1 has also been shown to be effective oxygen radicals scavengers (132). In M. marinum infection, PGL drives phagosome maturation arrest (133), and allows the bacterium to escape from hostile tissue-resident macrophages during the initial stages of the infection (131, 134, 135). By doing so, M. marinum uses PGL to infect resident macrophages and induce the secretion of the chemokine (C-C motif) ligand 2 (CCL2) attracting non-bactericidal monocytes, which will be subsequently infected by M. marinum and used as Trojan horses to cause a systemic infection (134–136).
As LOSs, PGLs are not present in all M. tuberculosis complex strains. These have been found in abundance in the M. tuberculosis complex canettii strain (PGL-Tb) (137) and in same M. tuberculosis clinical isolates of the W-Beijing family. Due to the different amounts of PGLs produced by different M. tuberculosis strains (138), their use for M. tuberculosis serodiagnostic did not prove to be useful as TB patients presented significant variations in their response to this antigen (85). Interestingly, the hypervirulent phenotype observed in several strains of M. tuberculosis (e.g., the Beijing strain HN878) has been associated with the presence of PGL and triglycerides in their cell envelope (139, 140).
Contrary to the debatable PGL hypervirulence effects where M. tuberculosis PGL-deficient strains induce pro-inflammation (141), recent studies, using a necrotic M. tuberculosis mouse model, show that loss of M. tuberculosis PGLs decreases the Th17 response (e.g., IL-17A), where IL-17A production seems critical to limit the development of hypoxic necrotic granulomas and reduces disease severity in TB (142). Thus, indirectly, M. tuberculosis PGL may favor M. tuberculosis infection reducing tissue damage. This concept is supported by other studies analyzing multiple clinical isolates, where the clinical spectrum of M. tuberculosis infection resulting in active TB or latent infection could be dictated not only by the host but also by the amounts and ratios of surface-exposed glycolipids defined by the strain genotype (134, 143, 144). Thus, serum opsonized M. tuberculosis clinical isolates expressing abundant PGL on their surface had reduced phagocytosis by macrophages, but the ingested M. tuberculosis bacilli replicated at faster rate (144). This was hypothetically attributed to the opsonization of M. tuberculosis surface-exposed PGL by host-soluble complement component 3 (C3) and the subsequent use by M. tuberculosis of the macrophage phagocytic complement receptor 3 (CR3) to infect host cells (144). Later studies confirmed that the sugar moiety of PGL is unique in its capacity to bind the lectin domain of CR3 for efficient infection of human macrophages, and M. tuberculosis PGLs are able to inhibit Toll-like receptor 2-triggered NF-κB activation and thus the pro-inflammatory response, thus assisting M. tuberculosis to subvert the host immune response (145) (Table 1). This capacity to dampen the host response is further corroborated by studies using M. tuberculosis PGL analogs, where these inhibited the production of Th1 (TNF), Th2 (IL-6), and the inflammasome (IL1-β)-related responses in a Toll-like receptor 2-mediated manner (143). Other supporting studies using recombinant BCG expressing PGL-1 show that this strain can exploit the host CR3, promoting bacterial uptake but at the same time inhibiting pro-inflammatory responses (e.g., TNF) (146). Overall, the direct interaction of PGL with host cell receptors is still unclear. Studies focused in determining which host receptors could recognize and bind M. tuberculosis complex BSA-conjugated glycans indicate that dendritic cell-specific ICAM-3-grabbing non-integrin (DC-SIGN or CD209), the DC-SIGN homolog DC-SIGNR (for DC-SIGN related), Langerin (CD207, C-type lectin receptor on Langerhans cells), the mannose receptor (MR or CD206), galactose receptor, dendritic cell C-type lectin 2 (Dectin-2), macrophage-inducible Ca2+-dependent lectin receptor (Mincle or CLEC4E), and BDCA-2 (CLEC4C or CD303) had low affinity for the rhamnose (6-deoxy-L-mannose) and fucose (6-deoxy-L-galactose) found in PGLs, indicating that PGL could be targeted by other, yet to be discovered, host C-type lectins (147).
Interestingly, studies also highlighted the role of PGL in BCG efficacy, where a mutant BCG (deleting fadD28) lacking both PDIMs and PGLs not only made BCG more attenuated in mice but also reduced its efficacy as a vaccine against M. tuberculosis infection (148).
This family involves phosphatidyl-myo-inositol (PI) glycosylated derivatives known as PIMs and the predominant mannosyl-β-1-phosphomycoketides (MPM). PIMs are important lipids in the cell envelope of M. tuberculosis, both as key cell envelope constituents involved in metabolic processes and as essential participants in M. tuberculosis-host interactions. PI is an acidic (anionic) phospholipid consisting of a phosphatidic acid linked via a phosphate group to myo-D-inositol. PIMs are considered, together with phosphatidyl-ethanolamine, the major phospholipid components of the M. tuberculosis cell envelope (75). There are found on the M. tuberculosis cell envelope as a mixture of compounds differing from each other by their number of mannose and fatty acid residues. The basic structure of PIM1 consists of a mannosyl unit attached to position C-2 of the myo-inositol of a PI anchor. Position C-6 of myo-inositol can be further substituted by an α-D-mannosyl or a linked di- or trimannosyl unit, giving PIM2, PIM3, and PIM4, respectively. The terminal mannose residue of PIM4 can be further substituted at position C-2 by a α-D-mannosyl leading to PIM5, which can also be further substituted at the same position leading to PIM6, the higher PIM encountered in mycobacteria to date (10). PIMs are shown to be located both in the M. tuberculosis plasma membrane and on the cell envelope surface (149). PIMs are intrinsically highly heterogeneous not only due to their mannose content but also due to the number and nature of their acyl groups. PIMs can be mono- to tetra-acylated, where the basic PIM is the diacylated form (150) esterified by palmitic (C16) and tuberculostearic (TBST or 10-methyl-octadecanoic) acids on C-1 and C-2 position of the glycerol, with a third fatty acyl substituent on the C-6 position of the (1→ 2) linked mannose to the inositol and with a fourth fatty acyl on the C-3 of the myo-inositol (150, 151). To simplify, PIMs can be grouped into lower- and higher-order PIMs depending on their number of mannoses, where lower-order PIMs contain one to four mannoses and higher-order PIMs contain five to six mannoses (152). The most common PIMs found in M. tuberculosis complex are AcPIM2 and Ac1PIM2 (di- and triacylated PIM2) and AcPIM6 and Ac1PIM6 (di-and triacylated PIM6) (150). All lower-order PIMs have a terminal α(1→ 6)-mannose, are exposed on the cell surface, and directly participate in the establishment of the infection, playing a role in macrophage recognition and phagocytosis process through association with the non-opsonic domain of CR3 (153). Lower-order PIMs also participate in facilitating trafficking processes within the phagocyte by promoting early endosomal fusion with M. tuberculosis containing phagosomes, thus providing nutrients to the bacterium (154) (Table 1).
In contrast, all higher-order PIMs have a terminal α(1→ 2)-mannose. In the case of AcxPIM5, it is a single α(1→ 2)-mannose, and in the case of AcxPIM6, it is an α(1→ 2)-dimannoside similar to the mannose caps of mannose-capped lipoarabinomannan [ManLAM, reviewed in Turner and Torrelles (155)]. Only tri-acylated forms of higher-order PIMs are shown to interact with the MR and interfering with trafficking pathways resulting in phagosome maturation arrest (152). Conversely, all M. tuberculosis PIMs seem to interact with DC-SIGN (152), although differences in DC-SIGN PIM recognition specificity may be species-dependent (156). Further studies have also shown that cytosolic soluble CD1e is involved in AcPIM6 processing and presentation via CD1 with subsequent T-cell activation (157) (Table 1).
Overall, without discerning the type of PIMs used, these have been shown to participate in host cell recognition, phagocytosis, oxidant and cytokine production, regulating intracellular vesicular trafficking, regulating signaling pathways, apoptosis, and antigen presentation (158–160) (Table 1). However, as pointed out, these intrinsic PIM properties, apart of being species-specific and depending on their heterogeneous nature, also may be derived from differences in the protocols used for PIM isolation and for in vitro experiments including immune cell types and procedures to generate them (160, 161).
To date, the role and metabolism of PIMs during M. tuberculosis infection and pathogenesis is still unclear, as different PIMs are recognized by different receptors triggering different host responses and outcomes. PIMs are downregulated in stationary phase (162), but it has been observed that the production of PIMs by M. tuberculosis is enhanced during infection in primary human macrophages (163). The latter result indicates that M. tuberculosis can adapt to the environment during infection by modulating its cell envelope composition (164–168). That PIM expression levels during early stages of infection may be in part directly or indirectly involved in bacterial growth, virulence, and entry into persistence is still unclear; however, studies show that a PIM-hyperproducing M. tuberculosis strain (ΔRv2623) is hypervirulent in vivo being unable to establish a chronic infection (163), while a PIM-hypoproducing M. tuberculosis strain (ΔRv1747) is attenuated in vivo in mice (169) (Table 1).
Another M. tuberculosis mannose-containing glycolipid is MPM. This glycolipid consists of a mannosyl-β-1–phosphoisoprenoid with a C32 4,8,12,16,20-pentamethylpentacosyl chain (170) and has been shown to activate CD1c-restricted T cells (171).
There are no doubts that the cell envelope of M. tuberculosis has evolved over the years to become the perfect shield for this pathogen, from the perspective of its hydrophobicity, transmissibility, adaptation to the host, drug resistance, virulence, and pathogenesis.
Several studies have discussed at large how M. tuberculosis has evolved increasing the hydrophobicity of its cell envelope to its advantage. In this regard, the role of the cell envelope lipids in the transition of environmental mycobacteria becoming highly transmissible, pathogenic and host-dependent is pointed out (102). The possibility that ancient M. tuberculosis complex strains such as M. canettii, mainly isolated in the horn of Africa (172, 173), have evolved has been explored, that is, changing their metabolism and increasing their cell envelope hydrophobicity as they evolved to the modern M. tuberculosis complex strains, e.g., changing their cell envelope's outmost exposed lipid polarity and thus evolving into a more hydrophobic organism. This was achieved by stopping the production of polar lipids such as LOSs or PGLs (which currently are just described in a subset of M. tuberculosis complex modern strains, Beijing clinical isolates), reducing their polarity (e.g., by losing sugars or adding acyl chains in their cell envelope glycolipids, e.g., DAT, TAT, PAT, SLs, TDM, TMM), and increasing the presence of apolar lipids in their cell envelopes [e.g., dimycocerosates of the phthiocerol family (PDIMs)] (102). Thus, cell envelope hydrophobicity could be linked to a simplified cell envelope, efficiency in transmissibility, and increased pathogenesis [extended review in Jankute et al. (102)]. Conversely, other studies shuffle the possibility that modern M. tuberculosis complex strains are adapting to the host by decreasing the hydrophobicity of their cell envelopes by adding mannose-containing components on their cell envelope, mimicking mammalian glycoproteins (Figure 2), and thus deceiving the host immune response during infection (10).
Looking at both scenarios and at the properties described for the glycolipids present in the cell envelope of M. tuberculosis complex, these two possibilities seem compatible, but potentially with different outcomes. Increasing hydrophobicity allows M. tuberculosis to be highly transmissible, and this could be the case for CDC1551, an M. tuberculosis strain shown to have this highly transmissible property (174). Increasing the hydrophobicity of M. tuberculosis can also trigger increased interactions of M. tuberculosis bacilli with signaling receptors that drive pro-inflammation and cellular trafficking to the infection site, and depending on the metabolic status of the host and the bacterium at this initial encounter, could result in active TB disease. This is supported by the role of triglycerides in driving hypervirulence in place of the initially described PGL (139, 140), which seems now to be involved in attenuating the host immune response (142).
Conversely, the other scenario is decreasing hydrophobicity by M. tuberculosis adding mannose-containing biomolecules in its cell envelope, such as the glycolipids PIMs, and their biosynthetically related lipoglycans and their derivatives [lipomannan, ManLAM, mannan, and arabinomannan (10)] (Table 1). M. tuberculosis complex strains with high mannose content in their cell envelope could be less effectively transmissible; however, when infecting the host, these trigger an anti-inflammatory response by interacting their mannose-containing glycolipids with homeostatic receptors of phagocytes [e.g., the MR (10, 175)], confusing the host cell. This scenario can result in a permissive intracellular environment for M. tuberculosis to permanently stay, without being detected, allowing host cells to control the infection, ultimately driving to the latency state. This is supported by the role of other glycolipids present in modern M. tuberculosis complex strains, such as DATs, which are shown dampening the host immune response (24).
The fact is that M. tuberculosis-host cell interactions, infection, and potential outcomes may also depend on the M. tuberculosis metabolic status prior to and during infection (Figure 4). M. tuberculosis overproduces PIMs during in vitro infections in macrophages (163). M. tuberculosis Ac1PIM6, as well as ManLAM, interacts with the MR defining a pathway of intracellular survival for M. tuberculosis and at the same time dampens the host immune response potentially benefiting M. tuberculosis adaption to the host (152, 176). However, a third unexplored possibility is that the cell envelope of M. tuberculosis shaping directly depends on the host lung environment, during the early stages of infection (in the alveolar space) or in later stages of infection, in cavities, when transmissible bacilli are shaped to be transmitted. In both cases, M. tuberculosis remains extracellular. In the initial stages, when infection occurs, M. tuberculosis is deposited on the alveolar space of the lung, where it will find the human lung mucosa before encountering any host cell [e.g., AMs, alveolar epithelial cells (7)]. In this scenario, the alveolar epithelium is covered by lung mucosal fluid, which is composed of two phases, a lipid layer called surfactant (mainly composed of phospholipids) and an hypophase called alveolar lining fluid (or ALF). Host cells are thought to be immersed and operating in the ALF. ALF is thought to trap any particle that reaches the alveolar space. ALF has been described to contain an array of hydrolytic enzymes [hydrolases (164)], whose main function is to assist host cells in recycling ALF every 18 h (177). Studies have demonstrated that the nature of these host hydrolases impacts the M. tuberculosis cell envelope as it has been described, where 15-min exposure of M. tuberculosis to human ALF reduces the cell surface presence of ManLAM and TDM by ~70 and ~40%, respectively (164). Other affected lipids significantly reduced from the M. tuberculosis cell envelope upon contact with human ALF are triglycerides, PDIMs, SLs, PIMs, and at lower degree DATs (personal communication). The impact of these human ALF modifications on the M. tuberculosis cell envelope results in a better control of the infection by human phagocytes (macrophages and neutrophils) by increasing M. tuberculosis-containing phagosome-lysosome fusion and acidification events (164, 178). Thus, the repertoire of lipids exposed on the M. tuberculosis cell envelope surface during the infection initial stages and prior to the first contact with host cells may in fact depend on the host ALF status, e.g., activity of the ALF hydrolases shaping the cell envelope.
Nevertheless, these M. tuberculosis cell envelope glycolipid modifications also result in the release of M. tuberculosis cell envelope fragments to the milieu (164). Their implications in M. tuberculosis pathogenesis are starting to be elucidated, but studies have shown that these fragments further assist macrophages in controlling M. tuberculosis infection by further increasing phagosome-lysosome fusion events in an IL-10-dependent manner (179), as well as drive neutrophils to control M. tuberculosis infection better by increasing their oxidative response (180).
The fact is that the status of the host lung environment could somehow “normalize the cell envelope of M. tuberculosis,” as well as redirecting M. tuberculosis adaptation to the host. Furthermore, M. tuberculosis exposure to ALF alters the cell envelope but also differentially drives how other innate soluble components described in the lung mucosa will interact with M. tuberculosis and drive infection and potentially disease outcome. In this context, TB comorbidities such as aging (181, 182), HIV, diabetes, smoking, etc., may impact the status of ALF, its hydrolases, and other soluble innate components functionality (182), and thus, the M. tuberculosis cell envelope may be differentially impacted depending on the status of the human ALF at the moment of the establishment of the infection.
This scenario has been tested in studies where the BCG vaccine has been exposed to human ALF and subsequently used to vaccinate mice showing that ALF-exposed BCG has an altered cell envelope and increased protection against M. tuberculosis challenge (183). Based on this study where ALF modified the BCG cell envelope and drove improved protection, studies delipidating BCG mostly removing apolar lipids showed the ability of this delipidated BCG to be delivered directly into the lung, minimizing tissue inflammation and to further increase protection against M. tuberculosis infection and significantly reducing tissue damage, the hallmark of TB disease (184). All these studies put in trial the role of the M. tuberculosis glycolipids during infection, how M. tuberculosis metabolically adapts to the lung environment, how M. tuberculosis uses its cell envelope glycolipids to mask its presence to host cells or to confuse them by mimicking mammalian structures, and how M. tuberculosis reconstitutes and reorganizes its cell envelope during infection and in the different environments that may encounter within the host. These environments could be extracellular at the earlier stages of infection, after escaping the infected cell via necrosis, or in cavities, where hypothetically M. tuberculosis bacilli could be exposed to lung tissue hydrolases causative of cavities, and thus, these hydrolases could shape bacteria that escape to the airways and are ready to be transmitted. These environments could also be intracellular within host cells in the alveolar space (e.g., AMs and alveolar epithelial cells) or within the interstitium in granuloma structures.
When it comes to drug resistance, limited information exists about the exact glycolipid composition and their role conferring the drug resistance phenotype in multi- (MDR), extensive- (XDR), and extreme- (XXDR) drug-resistant M. tuberculosis strains. In this regard, studies investigating M. tuberculosis strains containing rifampicin drug resistance-related mutations show that these strains change their cell envelope lipid composition by overexpressing PGL biosynthesis structurally related PDIMs influencing the macrophage metabolism and response during infection (16). Other studies looking at the role of tesA (a type II thioesterase) in M. marinum indicated a potential link between increased drug susceptibility and the lack of PDIM and PGLs on the bacterium cell envelope (185). Additional studies found that mycolic acid cyclopropanation is essential for M. tuberculosis viability, drug resistance, and cell envelope integrity (186). Overall, how glycolipids participate in the development of drug resistance directly or indirectly rearranging the M. tuberculosis cell envelope is still uncertain. Studies performed using transmission electron and atomic force microscopy techniques dig into this question showing that MDR-, XDR-, and XXDR-M. tuberculosis strains have thicker cell envelope and irregular cell surface with tubular extensions than susceptible strains (187, 188). As some XDR- and XXDR-M. tuberculosis strains are related to the Beijing family (189), which are shown to have their cell envelope overpopulated with triglycerides, it is plausible to question any relationship between the abundance of a specific hydrophobic lipid on the M. tuberculosis cell envelope and drug resistance (190). Many of these questions remain unanswered.
At the end, studies have been focused on depicting the role of purified glycolipids on M. tuberculosis-host cell interactions and pathogenesis, using purified lipids, embedded into vehicles (coated in beads or nanoparticles), or using M. tuberculosis glycolipid(s)-depleted mutants. This conferred us an enormous knowledge about their role in M. tuberculosis cell envelope rearrangements, adaptation, metabolism, catabolism, virulence, and pathogenesis. However, it is still unknown how M. tuberculosis uses these lipids during the different stages of infection, how the host shapes and alters them, and how the status of the host with active TB and M. tuberculosis itself works together within the cavities to define the successful transmissible form of M. tuberculosis.
In this context, persistence of M. tuberculosis within host cells and in granulomas depends on the bacterial use of lipid metabolic networks such as beta-oxidation, glyoxylate shunt, and the reverse methylcitrate cycle, to break down own and host captured long-chain fatty acids and cholesterol to generate acetyl-CoA and propionyl-CoA (191, 192). M. tuberculosis could regulate acetyl-CoA and propionyl-CoA production depending of the status of the host. Where during initial infection stages and granuloma establishment and maintenance (chronic infection), M. tuberculosis converts acetyl-CoA into mycolic acids to thicken its cell envelope and to use them as energy storage units in detriment of producing SL-1, DAT, and PAT, among others (47, 193, 194). Conversely, during reactivation leading to cavity formation, M. tuberculosis could further convert acetyl-CoA into mycolic acids, further increasing their production (which in turn are used to produce TDM and TMM), and convert propionyl-CoA and methyl malonyl-CoA into Ac2SG, SL-1, PAT, and PDIM (methyl-CoA-lipids) (14, 195, 196), increasing its cell envelope hydrophobicity, which will enhance its host to host transmissibility (Table 1, Figure 4).
In vitro and in vivo studies using mycobacterial lipids and lipoglycans have shown that these induce both humoral and CD1-restricted T cell-mediated responses in the context of BCG vaccination (197–200). In this context, the CD1 locus encodes five related proteins (CD1a–e). CD1a–c are shown to present antigens to T cells. Concretely, CD1a is shown to present lipopeptides (201); CD1b presents mycobacterial cell envelope lipids and lipoglycans, such as mycolic acids, glucose monomycolate, glycerol monomycolate, SLs, LAM, and PIMs, among others (119, 202–205); and CD1c presents isoprenoid phosphoglycolipids (206). CD1d presents antigens to NKT cells, with the sponge-derived α-galactosyl-ceramide being the major antigen described (207). CD1e is located intracellularly and shown to present metabolized PIM6 (157). Overall these data indicate that mycobacterial lipids could directly participate in generating long-standing protective immune responses via CD1-restricted T cells during M. tuberculosis infection. The role of these lipid-restricted CD1 T-cell populations in generating a protective immune response against M. tuberculosis infection has been debatable due to the lack of human homologs of CD1a, CD1b, and CD1c in mice, but studies using human CD1a–c transgenic (hCD1Tg) mice (208) concluded that, contrary to what has been observed in NK T cells, CD1-restricted T cells in reality exhibit delayed primary responses and more rapid secondary responses, similar to conventional T cells, indicating that these lipid-restricted CD1 T-cell populations in reality participate in adaptive immune responses upon mycobacterial infection and thus embrace the use of mycobacterial lipids to target these CD1-restricted T cells for vaccine strategies.
Conversely, for several years, the use of adjuvants in TB vaccination strategies relied on liposomes and oil extracts (209–211). Current vaccine strategies evolved the adjuvant field by targeting the stimulation of pattern recognition receptors, such as Toll-like receptors (212). These new generations of adjuvants include cationic lipids, micro-/nanoparticles, cytokines, toxin derivatives, CpG-containing DNA-based molecules, antimicrobial peptides/proteins, and mycobacterial proteins conjugates and lipids (209). These adjuvants in combination with the current BCG vaccine or with vaccine subunits are shown to be successful in boosting vaccine efficacies (209, 210, 213–215). In this context, there are ongoing vaccination strategies using mycobacterial lipids (or their analogs) as adjuvants driving better and prolonged protection. As an example, the combination of the Hybrid 1(H1) subunit vaccine with the liposomal adjuvant CAF01 [composed of a cationic liposome vehicle (dimethyldioctadecyl-ammonium) and the synthetic TDM analog, trehalose-6,6-dibehenate or TDB] (216–218). This novel vaccine seems to be well tolerated and able of generating long-lasting protective T-cell responses (216).
Having said this, we need to account that M. tuberculosis is a master manipulator of host immune responses, being able to passively (directed by the host) or actively (directed by the bacterium itself) change its metabolism and cell envelope lipid composition, adapting to the different adverse host environments (e.g., ALF, AMs, granuloma) during the course of infection (10, 219–222). Thus, mycobacterial lipids may generate or mask a protective host immune response against infection depending on the human lung environment and thus their debatable potential application in vaccine strategies, which has been reviewed by us in detail elsewhere (223, 224).
Overall, a comprehensive study of the M. tuberculosis cell envelope lipid constitution during infection could be critical to develop novel vaccine strategies. Our in vitro data demonstrate that M. tuberculosis, upon contact with the lung mucosa from healthy adult individuals, is temporally depleted of surface exposed lipids resulting in a better control of the infection by alveolar resident cells (164, 178–180, 225). Our in vivo data also show that this temporal depletion of lipids on the M. tuberculosis surface results in a better control of M. tuberculosis growth in the lung of infected mice over time (personal communication). However, this is not observed when M. tuberculosis is upon contact with the lung mucosa of healthy elderly individuals (182) or upon contact with the lung mucosa of HIV+ individuals (personal communication). Instead, in both of these cases, M. tuberculosis contact with the lung mucosa of the elderly and HIV+ populations accentuates its intracellular growth in AMs (in vitro) and in mice (in vivo) (182) (personal communication). These results point out the importance of the status of the human lung mucosa in altering the M. tuberculosis cell envelope lipid composition (164), resulting in a successful immune response initially protective against M. tuberculosis infection and intracellular growth. Subsequently, when inside human macrophages, M. tuberculosis seems to overproduce specific lipids, e.g. PIMs (163). Thus, the host-directed temporal removal of lipids and the bacterial overproduction of lipids within host cells may be important to understand which lipids (their presence and/or absence) are important to generate a prolonged and protective immune response, which may improve current vaccination strategies.
M. tuberculosis is proven to expend energy elaborating a great variety of glycolipids of rather unusual structure, both in the inner cell envelope and exposed on its cell surface. These include mainly three groups of glycolipids, trehalose-containing lipids [e.g., acylglucosides (DAT, TAT, PAT), sulfatides (SLs), and LOSs], the 6-deoxy-pyranose containing phenolic glycolipids (e.g., PGLs), and the ubiquitous mannose-containing glycolipids (e.g., PIMs). Some of these glycolipids are described as M. tuberculosis virulence factors being recognized by specific host cell receptors (or directly by their interaction with membranes) and thus helping the bacillus to survive within host cells. Therefore, enzymes involved in glycolipids biosynthesis have been and are extensively studied as these may represent potential drug targets (226). In this context, the role of the M. tuberculosis cell envelope lipids during dormancy, nutrient starvation, hypoxia, and oxidative stress has been reported using the in vitro Wayne's model, pointing out that under these conditions M. tuberculosis adapts its proteome and lipidome (222, 227–231).
Attempts to define glycolipids biological function by genetically manipulating M. tuberculosis to obtain glycolipid deficient mutants are somehow providing us if these glycolipids are important for M. tuberculosis infection and pathogenesis; however, these studies may bypass the impact of M. tuberculosis cell envelope rearrangements and the host environment impact on these glycolipids or new rearrangements due to the mutation. Thus, the production of mutants or conditional mutants that provide different levels of production of these glycolipids in the M. tuberculosis cell envelope at different stages of the infection may provide us more accurate information about the role of these glycolipids during infection and their real implication in pathogenesis, in place of studying mutants lacking a specific glycolipid.
AG-V, JC, and JT made substantial contributions to the conception, writing, and editing of this review.
This work has been partially supported by NIH/NIA (P01 AG-051428) and the Robert J. Kleberg and Helen C. Kleberg Foundation to JT, and by NIH/NIAID (R01 AI 146340) to JC and JT.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. (CDC) CfDCaP. Emergence of Mycobacterium tuberculosis With Extensive Resistance to Second-Line Drugs–Worldwide, 2000-2004 MMWR Morb Mortal Wkly Rep (2006). p. 301–5.
2. Gandhi NR, Moll A, Sturm AW, Pawinski R, Govender T, Lalloo U, et al. Extensively drug-resistant tuberculosis as a cause of death in patients co-infected with tuberculosis and HIV in a rural area of South Africa. Lancet. (2006) 368:1575–80. doi: 10.1016/S0140-6736(06)69573-1
3. Basu S, Friedland GH, Medlock J, Andrews JR, Shah NS, Gandhi NR, et al. Averting epidemics of extensively drug-resistant tuberculosis. Proc Natl Acad Sci USA. (2009) 106:7672–7. doi: 10.1073/pnas.0812472106
4. Andrews JR, Shah NS, Weissman D, Moll AP, Friedland G, Gandhi NR. Predictors of multidrug- and extensively drug-resistant tuberculosis in a high HIV prevalence community. PLoS ONE. (2010) 5:e15735. doi: 10.1371/journal.pone.0015735
5. Gandhi NR, Shah NS, Andrews JR, Vella V, Moll AP, Scott M, et al. HIV coinfection in multidrug- and extensively drug-resistant tuberculosis results in high early mortality. Am J Respir Crit Care Med. (2010) 181:80–6. doi: 10.1164/rccm.200907-0989OC
6. WHO. Global Tuberculosis Report 2018. Geneva: WHO Organization, (2018). Available online at: https://www.who.int/tb/publications/global_report/en/.
7. Torrelles JB, Schlesinger LS. Integrating lung physiology, immunology, and tuberculosis. Trends Microbiol. (2017) 25:688–97. doi: 10.1016/j.tim.2017.03.007
8. Avigad G, Dey PM. 4- carbohydrate metabolism: storage carbohydrates. In: Dey PM, Harborne JB, editors. Plant Biochemistry. London: Academic Press (1997). p. 143–204. doi: 10.1016/B978-012214674-9/50005-9
9. Kalscheuer R, Weinrick B, Veeraraghavan U, Besra GS, Jacobs WR Jr. Trehalose-recycling ABC transporter LpqY-SugA-SugB-SugC is essential for virulence of Mycobacterium tuberculosis. Proc Natl Acad Sci USA. (2010) 107:21761–6. doi: 10.1073/pnas.1014642108
10. Torrelles JB, Schlesinger LS. Diversity in Mycobacterium tuberculosis mannosylated cell wall determinants impacts adaptation to the host. Tuberculosis. (2010) 90:84–93. doi: 10.1016/j.tube.2010.02.003
11. Dobos KM, Khoo KH, Swiderek KM, Brennan PJ, Belisle JT. Definition of the full extent of glycosylation of the 45- kilodalton glycoprotein of Mycobacterium tuberculosis. J Bacteriol. (1996) 178:2498–506. doi: 10.1128/jb.178.9.2498-2506.1996
12. Gago G, Diacovich L, Gramajo H. Lipid metabolism and its implication in mycobacteria-host interaction. Curr Opin Microbiol. (2018) 41:36–42. doi: 10.1016/j.mib.2017.11.020
13. Velayati AA, Farnia P, Hoffner S. Drug-resistant Mycobacterium tuberculosis: Epidemiology and role of morphological alterations. J Glob Antimicrob Resist. (2018) 12:192–6. doi: 10.1016/j.jgar.2017.10.006
14. Ghazaei C. Mycobacterium tuberculosis and lipids: insights into molecular mechanisms from persistence to virulence. J Res Med Sci. (2018) 23:63. doi: 10.4103/jrms.JRMS_904_17
15. Chakraborty P, Kulkarni S, Rajan R, Sainis K. Drug resistant clinical isolates of Mycobacterium tuberculosis from different genotypes exhibit differential host responses in THP-1 cells. PLoS ONE. (2013) 8:e62966. doi: 10.1371/journal.pone.0062966
16. Howard NC, Marin ND, Ahmed M, Rosa BA, Martin J, Bambouskova M, et al. Mycobacterium tuberculosis carrying a rifampicin drug resistance mutation reprograms macrophage metabolism through cell wall lipid changes. Nat Microbiol. (2018) 3:1099–108. doi: 10.1038/s41564-018-0245-0
17. Brennan PJ, Nikaido H. The envelope of mycobacteria. Annu Rev Biochem. (1995) 64:29–63. doi: 10.1146/annurev.bi.64.070195.000333
18. Jarlier V, Nikaido H. Mycobacterial cell wall: structure and role in natural resistance to antibiotics. FEMS Microbiol Lett. (1994) 123:11–8. doi: 10.1016/0378-1097(94)90267-4
19. Brennan PJ. Mycobacterium and other actinomycetes. In: Ratledge C, Wilkinson VL, editors. Microbial Lipid. London: Acad. Press. (1988). p. 203–98.
20. Anderson RJ. Chemical Investigation of biologically active lipoids of tubercle bacilli. Proc Natl Acad Sci USA. (1929) 15:628–33. doi: 10.1073/pnas.15.8.628
21. Rhoades ER, Streeter C, Turk J, Hsu FF. Characterization of sulfolipids of Mycobacterium tuberculosis H37Rv by multiple-stage linear ion-trap high-resolution mass spectrometry with electrospray ionization reveals that the family of sulfolipid II predominates. Biochemistry. (2011) 50:9135–47. doi: 10.1021/bi2012178
22. Gautier N, López Marín LM, Lanéelle M-A, Daffé M. Structure of mycoside F, a family of trehalose-containing glycolipids of Mycobacterium fortuitum. FEMS Microbiol Lett. (1992) 98:81–8. doi: 10.1016/0378-1097(92)90136-C
23. Frankfater C, Abramovitch RB, Purdy GE, Turk J, Legentil L, Lemiegre L, et al. Multiple-stage precursor ion separation and high resolution mass spectrometry toward structural characterization of 2,3-diacyltrehalose family from Mycobacterium tuberculosis. Separations. (2019) 6:4. doi: 10.3390/separations6010004
24. Lee KS, Dubey VS, Kolattukudy PE, Song CH, Shin AR, Jung SB, et al. Diacyltrehalose of Mycobacterium tuberculosis inhibits lipopolysaccharide- and mycobacteria-induced proinflammatory cytokine production in human monocytic cells. FEMS Microbiol Lett. (2007) 267:121–8. doi: 10.1111/j.1574-6968.2006.00553.x
25. Saavedra R, Segura E, Leyva R, Esparza LA, Lopez-Marin LM. Mycobacterial di-O-acyl-trehalose inhibits mitogen- and antigen-induced proliferation of murine T cells in vitro. Clin Diagn Lab Immunol. (2001) 8:1081–8. doi: 10.1128/CDLI.8.6.1-91-1088.2001
26. Bailo R, Bhatt A, Ainsa JA. Lipid transport in Mycobacterium tuberculosis and its implications in virulence and drug development. Biochem Pharmacol. (2015) 96:159–67. doi: 10.1016/j.bcp.2015.05.001
27. Papa F, Cruaud P, David HL. Antigenicity and specificity of selected glycolipid fractions from Mycobacterium tuberculosis. Res Microbiol. (1989) 140:569–78. doi: 10.1016/0923-2508(89)90089-2
28. Rousseau C, Neyrolles O, Bordat Y, Giroux S, Sirakova TD, Prevost MC, et al. Deficiency in mycolipenate- and mycosanoate-derived acyltrehaloses enhances early interactions of Mycobacterium tuberculosis with host cells. Cell Microbiol. (2003) 5:405–15. doi: 10.1046/j.1462-5822.2003.00289.x
29. Hatzios SK, Schelle MW, Holsclaw CM, Behrens CR, Botyanszki Z, Lin FL, et al. PapA3 is an acyltransferase required for polyacyltrehalose biosynthesis in Mycobacterium tuberculosis. J Biol Chem. (2009) 284:12745–51. doi: 10.1074/jbc.M809088200
30. Dubey VS, Sirakova TD, Kolattukudy PE. Disruption of msl3 abolishes the synthesis of mycolipanoic and mycolipenic acids required for polyacyltrehalose synthesis in Mycobacterium tuberculosis H37Rv and causes cell aggregation. Mol Microbiol. (2002) 45:1451–9. doi: 10.1046/j.1365-2958.2002.03119.x
31. Brodin P, Poquet Y, Levillain F, Peguillet I, Larrouy-Maumus G, Gilleron M, et al. High content phenotypic cell-based visual screen identifies Mycobacterium tuberculosis acyltrehalose-containing glycolipids involved in phagosome remodeling. PLoS Pathog. (2010) 6:e1001100. doi: 10.1371/journal.ppat.1001100
32. Muñoz M, Luquin M, García-Barcelo M, Julián E, Ausina V, Lanéelle MA. Distribution of surface-exposed antigenic glycolipids in recent clinical isolates off Mycobacterium tuberculosis. Res Microbiol. (1997) 148:405–12. doi: 10.1016/S0923-2508(97)83871-5
33. Julian E, Matas L, Perez A, Alcaide J, Laneelle MA, Luquin M. Serodiagnosis of tuberculosis: comparison of immunoglobulin A (IgA) response to sulfolipid I with IgG and IgM responses to 2,3-diacyltrehalose, 2,3,6-triacyltrehalose, and cord factor antigens. J Clin Microbiol. (2002) 40:3782–8. doi: 10.1128/JCM.40.10.3782-3788.2002
34. Sempere M, Ariza M, Sanchez-Monton T, Rodriguez-Gonzalez T, Martin-Luengo F, Valero-Guillen PL. Evaluacion de aciltrehalosas de Mycobacterium fortuitum en el diagnostico serologico de tuberculosis pulmonar. Enferm Infecc Microbiol Clin. (1995) 13:292–6.
35. Ridell M, Wallerstrom G, Minnikin DE, Bolton RC, Magnusson M. A comparative serological study of antigenic glycolipids from Mycobacterium tuberculosis. Tubercle Lung Dis. (1992) 73:101–5. doi: 10.1016/0962-8479(92)90063-P
36. Tortola MT, Laneelle MA, Martin-Casabona N. Comparison of two 2,3-diacyl trehalose antigens from Mycobacterium tuberculosis and Mycobacterium fortuitum for serology in tuberculosis patients. Clin Diagn Lab Immunol. (1996) 3:563–6.
37. Hunter RL, Olsen MR, Jagannath C, Actor JK. Multiple roles of cord factor in the pathogenesis of primary, secondary, and cavitary tuberculosis, including a revised description of the pathology of secondary disease. Ann Clin Lab Sci. (2006) 36:371–86. Available online at: http://www.annclinlabsci.org/content/36/4/371.full.pdf+html
38. Bloch H. Studies on the virulence of tubercle bacilli: isolation and biological properties of a constituent of virulent organisms. J Exp Med. (1950) 91:197–219. doi: 10.1084/jem.91.2.197
39. Noll H, Bloch H, Asselineau J, Lederer E. The chemical structure of the cord factor of Mycobacterium tuberculosis. Biochim. Biophys. Acta. (1956) 20:299–309. doi: 10.1016/0006-3002(56)90289-X
40. Mdluli K, Slayden RA, Zhu Y, Ramaswamy S, Pan X, Mead D, et al. Inhibition of a Mycobacterium tuberculosis β-ketoacyl ACP synthase by isoniazid. Science. (1998) 280:1607–10. doi: 10.1126/science.280.5369.1607
41. Artman M, Bekierkunst A, Goldenberg I. Tissue methabolism in infection: Biochemical changes in mice treated with cord factor. Arch Biochem Biophys. (1964) 105:80–5. doi: 10.1016/0003-9861(64)90237-1
42. Yuan Y, Zhu Y, Crane DD, Barry CEI. The effect of oxygenated mycolic acid composition on cell wall function and macrophage growth in Mycobacterium tuberculosis. Mol Microbiol. (1998) 29:1449–58. doi: 10.1046/j.1365-2958.1998.01026.x
43. Kordulakova J, Gilleron M, Puzo G, Brennan PJ, Gicquel B, Mikusova K, et al. Identification of the required acyltransferase step in the biosynthesis of the phosphatidylinositol mannosides of mycobacterium species. J Biol Chem. (2003) 278:36285–95. doi: 10.1074/jbc.M303639200
44. Rajni, Rao N, Meena LS. Biosynthesis and virulent behavior of lipids produced by Mycobacterium tuberculosis: LAM and cord factor: an overview. Biotechnol Res Int. (2011) 2011:274693. doi: 10.4061/2011/274693
45. Actor JK. Trehalose dimycolate (cord factor) as a contributing factor to tuberculosis pathogenesis. In: Cirillo JD, Kong Y, editors. Tuberculosis Host-Pathogen Interactions. Cham: Springer International Publishing. (2019). p. 43–61. doi: 10.1007/978-3-030-25381-3_3
46. Marrakchi H, Laneelle MA, Daffe M. Mycolic acids: structures, biosynthesis, and beyond. Chem Biol. (2014) 21:67–85. doi: 10.1016/j.chembiol.2013.11.011
47. Takayama KCW, Besra GS. Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin Microbiol Rev. (2005) 18:81–101. doi: 10.1128/CMR.18.1.81-101.2005
48. Fujita Y, Okamoto Y, Uenishi Y, Sunagawa M, Uchiyama T, Yano I. Molecular and supra-molecular structure related differences in toxicity and granulomatogenic activity of mycobacterial cord factor in mice. Microb Pathog. (2007) 43:10–21. doi: 10.1016/j.micpath.2007.02.006
49. Kato M. Action of a toxic glycolipid of Corynebacterium diphtheriae on mitochondrial structure and function. J Bacteriol. (1970) 101:709–16.
50. Silva CL, Ekizlerian SM, Fazioli RA. Role of cord factor in the modulation of infection caused by mycobacteria. Am J Pathol. (1985) 118:238–47.
51. Indrigo J, Hunter RL Jr, Actor JK. Influence of trehalose 6,6'-dimycolate (TDM) during mycobacterial infection of bone marrow macrophages. Microbiology. (2002) 148(Pt 7):1991–8. doi: 10.1099/00221287-148-7-1991
52. Fischer K, Chatterjee D, Torrelles J, Brennan PJ, Kaufmann SHE, Schaible UE. Mycobacterial lysocardiolipin is exported from phagosomes upon cleavage of cardiolipin by a macrophage-derived lysosomal phospholipase A2. J Immunol. (2001) 167:2187–92. doi: 10.4049/jimmunol.167.4.2187
53. Sakamoto K, Geisel RE, Kim MJ, Wyatt BT, Sellers LB, Smiley ST, et al. Fibrinogen regulates the cytotoxicity of mycobacterial trehalose dimycolate but is not required for cell recruitment, cytokine response, or control of mycobacterial infection. Infect Immun. (2010) 78:1004–11. doi: 10.1128/IAI.00451-09
54. Hunter RL, Olsen M, Jagannath C, Actor JK. Trehalose 6,6'-dimycolate and lipid in the pathogenesis of caseating granulomas of tuberculosis in mice. Am J Pathol. (2006) 168:1249–61. doi: 10.2353/ajpath.2006.050848
55. Spargo BJ, Crowe LM, Ioneda T, Beaman BL, Crowe JH. Cord factor (a,a-trehalose 6,6'-dimycolate) inhibits fusion between phospholipid vesicles. Proc Natl Acad Sci USA. (1991) 88:737–40. doi: 10.1073/pnas.88.3.737
56. Yamasaki S, Ishikawa E, Sakuma M, Hara H, Ogata K, Saito T. Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat Immunol. (2008) 9:1179–88. doi: 10.1038/ni.1651
57. Ishikawa E, Ishikawa T, Morita YS, Toyonaga K, Yamada H, Takeuchi O, et al. Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J Exp Med. (2009) 206:2879–88. doi: 10.1084/jem.20091750
58. Schoenen H, Bodendorfer B, Hitchens K, Manzanero S, Werninghaus K, Nimmerjahn F, et al. Cutting edge: mincle is essential for recognition and adjuvanticity of the mycobacterial cord factor and its synthetic analog trehalose-dibehenate. J Immunol. (2010) 184:2756–60. doi: 10.4049/jimmunol.0904013
59. Welsh KJ, Abbott AN, Hwang SA, Indrigo J, Armitige LY, Blackburn MR, et al. A role for tumour necrosis factor-alpha, complement C5 and interleukin-6 in the initiation and development of the mycobacterial cord factor trehalose 6,6′-dimycolate induced granulomatous response. Microbiology. (2008) 154:1813–24. doi: 10.1099/mic.0.2008/016923-0
60. Indrigo J, Hunter RL Jr, Actor JK. Cord factor trehalose 6,6′-dimycolate (TDM) mediates trafficking events during mycobacterial infection of murine macrophages. Microbiology. (2003) 149:2049–59. doi: 10.1099/mic.0.26226-0
61. Lee WB, Kang JS, Yan JJ, Lee MS, Jeon BY, Cho SN, et al. Neutrophils promote mycobacterial trehalose dimycolate-induced lung inflammation via the Mincle pathway. PLoS Pathog. (2012) 8:e1002614. doi: 10.1371/journal.ppat.1002614
62. Ozeki Y, Kaneda K, Fujiwara N, Morimoto M, Oka S, Yano I. In vivo induction of apoptosis in the thymus by administration of mycobacterial cord factor (trehalose 6,6′-dimycolate). Infect Immun. (1997) 65:1793–9.
63. Bekierkunst A. Acute granulomatous response produced in mice by trehalose-6,6-dimycolate. J Bacteriol. (1968) 96:958–61.
64. Sakaguchi I, Ikeda N, Nakayama M, Kato Y, Yano I, Kaneda K. Trehalose 6,6'-dimycolate (Cord factor) enhances neovascularization through vascular endothelial growth factor production by neutrophils and macrophages. Infect Immun. (2000) 68:2043–52. doi: 10.1128/IAI.68.4.2043-2052.2000
65. Werninghaus K, Babiak A, Gross O, Holscher C, Dietrich H, Agger EM, et al. Adjuvanticity of a synthetic cord factor analogue for subunit Mycobacterium tuberculosis vaccination requires FcRgamma-Syk-Card9-dependent innate immune activation. J Exp Med. (2009) 206:89–97. doi: 10.1084/jem.20081445
66. Bekierkunst A, Levij IS, Yarkoni E. Suppression of urethan-induced lung adenomas in mice treated with trehalose-6,6-dimycolate (cord factor) and living bacillus Calmette Guerin. Science. (1971) 174:1240–2. doi: 10.1126/science.174.4015.1240
67. Bekierkunst A, Yarkoni E, Flechner I, Morecki S, Vilkas E, Lederer E. Immune response to sheep red blood cells in mice pretreated with mycobacterial fractions. Infect Immun. (1971) 4:256–63.
68. Silva CL, Faccioli LH. Tumor necrosis factor (cachectin) mediates induction of cachexia by cord factor from mycobacteria. Infect Immun. (1988) 56:3067–71.
69. Parant M, Parant F, Chedid L, Drapier JC, Petit JF, Wietzerbin J, et al. Enhancement of nonspecific immunity to bacterial infection by cord factor (6,6'-trehalose dimycolate). J Infect Dis. (1977) 135:771–7. doi: 10.1093/infdis/135.5.771
70. Parant M, Audibert F, Parant F, Chedid L, Soler E, Polonsky J, et al. Nonspecific immunostimulant activities of synthetic trehalose-6,6'-diesters (lower homologs of cord factor). Infect Immun. (1978) 20:12–9.
71. Granger DL, Yamamoto KI, Ribi E. Delayed hypersensitivity and granulomatous response after immunization with protein antigens associated with a mycobacterial glycolipid and oil droplets. J Immunol. (1976) 116:482–8.
72. Yarkoni E, Bekierkunst A. Nonspecific resistance against infection with Salmonella typhi and Salmonella typhimurium induced in mice by cord factor (trehalose-6,6'-dimycolate) and its analogues. Infect Immun. (1976) 14:1125–9.
73. Rao V, Fujiwara N, Porcelli SA, Glickman MS. Mycobacterium tuberculosis controls host innate immune activation through cyclopropane modification of a glycolipid effector molecule. J Exp Med. (2005) 201:535–43. doi: 10.1084/jem.20041668
74. Sakamoto K, Kim MJ, Rhoades ER, Allavena RE, Ehrt S, Wainwright HC, et al. Mycobacterial trehalose dimycolate reprograms macrophage global gene expression and activates matrix metalloproteinases. Infect Immun. (2013) 81:764–76. doi: 10.1128/IAI.00906-12
75. Crick DC, Brennan PJ, McNeil MR. The cell wall of Mycobacterium tuberculosis. In: Rom WM, Garay SM, editors. Tuberculosis, 2nd ed. Philadelphia: Lippincott Williams and Wilkins (2003).
76. Walker RW, Prome JC, Lacave CS. Biosynthesis of mycolic acids. Formation of a C32 beta-keto ester from palmitic acid in a cell-free system of Corynebacterium diphtheriae. Biochim Biophys Acta. (1973) 326:52–62. doi: 10.1016/0005-2760(73)90027-1
77. Su C-C, Klenotic PA, Bolla JR, Purdy GE, Robinson CV, Yu EW. MmpL3 is a lipid transporter that binds trehalose monomycolate and phosphatidylethanolamine. Proc Natl Acad Sci USA. (2019) 116:11241–6. doi: 10.1073/pnas.1901346116
78. Sathyamoorthy N, Takayama K. Purification and characterization of a novel mycolic acid exchange enzyme from Mycobacterium smegmatis. J Biol Chem. (1987) 262:13417–23.
79. Belisle JT, Vissa VD, Sievert T, Takayama K, Brennan PJ, Besra GS. Role of the major antigen of Mycobacterium tuberculosis in cell wall biogenesis. Science. (1997) 276:1420–2. doi: 10.1126/science.276.5317.1420
80. Numata F, Nishimura K, Ishida H, Ukei S, Tone Y, Ishihara C, et al. Lethal and adjuvant activities of cord factor (trehalose-6,6'-dimycolate) and synthetic analogs in mice. Chem Pharm Bull. (1985) 33:4544–55. doi: 10.1248/cpb.33.4544
81. Sueoka E, Nishiwaki S, Okabe S, Iida N, Suganuma M, Yano I, et al. Activation of protein kinase C by mycobacterial cord factor, trehalose 6-monomycolate, resulting in tumor necrosis factor-α release in mouse lung tissues. Jpn J Cancer Res. (1995) 86:749–55. doi: 10.1111/j.1349-7006.1995.tb02464.x
82. Rhoades ER, Geisel RE, Butcher BA, McDonough S, Russell DG. Cell wall lipids from Mycobacterium bovis BCG are inflammatory when inoculated within a gel matrix: characterization of a new model of the granulomatous response to mycobacterial components. Tuberculosis. (2005) 85:159–76. doi: 10.1016/j.tube.2004.10.001
83. Lemaire G, Tenu JP, Petit JF, Lederer E. Natural and synthetic trehalose diesters as immunomodulators. Med Res Rev. (1986) 6:243–74. doi: 10.1002/med.2610060302
84. Fiolek TJ, Banahene N, Kavunja HW, Holmes NJ, Rylski AK, Pohane AA, et al. Engineering the mycomembrane of live mycobacteria with an expanded set of trehalose monomycolate analogues. Chembiochem. (2019) 20:1282–91. doi: 10.1002/cbic.201800687
85. Daffe M, McNeil M, Brennan PJ. Novel type-specific lipooligosaccharides from Mycobacterium tuberculosis. Biochemistry. (1991) 30:378–88. doi: 10.1021/bi00216a011
86. Ren H, Dover LG, Islam ST, Alexander DC, Chen JM, Besra GS, et al. Identification of the lipooligosaccharide biosynthetic gene cluster from Mycobacterium marinum. Mol Microbiol. (2007) 63:1345–59. doi: 10.1111/j.1365-2958.2007.05603.x
87. Hunter SW, Murphy RC, Clay K, Goren MB, Brennan PJ. Trehalose-containing lipooligosaccharides. A new class of species-specific antigens from Mycobacterium. J Biol Chem. (1983) 258:10481–7.
88. Saadat S, Ballou CE. Pyruvylated glycolipids from Mycobacterium smegmatis. Structures of two oligosaccharide components. J Biol Chem. (1983) 258:1813–8.
89. Daffé M. Lemassu A. Glycobiology of the mycobacterial surface: structures and biological activities of the cell envelope glycoconjugates. In: Doyle RJ, editor. Glycomicrobiology. Boston, MA: Springer. (2000).
90. Etienne G, Malaga W, Laval F, Lemassu A, Guilhot C, Daffe M. Identification of the polyketide synthase involved in the biosynthesis of the surface-exposed lipooligosaccharides in mycobacteria. J Bacteriol. (2009) 191:2613–21. doi: 10.1128/JB.01235-08
91. Ortalo-Magne A, Lemassu A, Laneelle M-A, Bardou F, Silve G, Gounon P, et al. Identification of the surface-exposed lipids on the cell envelopes of Mycobacterium tuberculosis and other mycobacterial species. J Bacteriol. (1996) 178:456–61. doi: 10.1128/jb.178.2.456-461.1996
92. Belisle JT, Brennan PJ. Chemical basis of rough and smooth variation in mycobacteria. J Bacteriol. (1989) 171:3465–70. doi: 10.1128/jb.171.6.3465-3470.1989
93. Lemassu A, Lévy-Frébault VV, Lanéelle M-A, Daffé M. Lack of correlation between colony morphology and lipooligosaccharide content in the Mycobacterium tuberculosis complex. J Gen Microbiol. (1992) 138:1535–41. doi: 10.1099/00221287-138-7-1535
94. van Soolingen D, Hoogenboezem T, de Haas PE, Hermans PW, Koedam MA, Teppema KS, et al. A novel pathogenic taxon of the Mycobacterium tuberculosis complex, Canetti: characterization of an exceptional isolate from Africa. Int J Syst Bacteriol. (1997) 47:1236–45. doi: 10.1099/00207713-47-4-1236
95. Pfyffer GE, Auckenthaler R, van Embden JD, van Soolingen D. Mycobacterium canettii, the smooth variant of M. tuberculosis, isolated from a Swiss patient exposed in Africa. Emerg Infect Dis. (1998) 4:631–4. doi: 10.3201/eid0404.980414
96. Gilleron M, Vercauteren J, Puzo G. Lipo-oligosaccharidic antigen from Mycobacterium gastri. Complete structure of a novel C4-branched 3,6-dideoxy-β- xylo-hexopyranose. Biochemistry. (1994) 33:1930–7. doi: 10.1021/bi00173a041
97. Hunter SW, Jardine I, Yanagihara DL, Brennan PJ. Trehalose-containing lipooligosaccharides from mycobacteria: structures of the oligosaccharide segments and recognition of a unique N-acylkanosamine-containing epitope. Biochemistry. (1985) 24:2798–805. doi: 10.1021/bi00332a030
98. Besra GS, Chatterjee D. Lipids and carbohydrates of Mycobacterium tuberculosis. In: Bloom BR, editor. Tuberculosis: Pathogenesis, Protection and Control. Washington, DC: American Society for Microbiology. (1994). p. 285–306. doi: 10.1128/9781555818357.ch20
99. Collins FM, Cunningham DS. Systemic Mycobacterium kansasii infection and regulation of the alloantigenic response. Infect Immun. (1981) 32:614–24.
100. Minnikin DE, Lee OYC, Wu HHT, Besra GS, Bhatt A, Nataraj V, et al. Ancient mycobacterial lipids: key reference biomarkers in charting the evolution of tuberculosis. Tuberculosis. (2015) 95(Suppl. 1):S133–9. doi: 10.1016/j.tube.2015.02.009
101. Falkinham JO 3rd. Mycobacterial aerosols and respiratory disease. Emerg Infect Dis. (2003) 9:763–7. doi: 10.3201/eid0907.020415
102. Jankute M, Nataraj V, Lee OYC, Wu HHT, Ridell M, Garton NJ, et al. The role of hydrophobicity in tuberculosis evolution and pathogenicity. Sci Rep. (2017) 7:1315. doi: 10.1038/s41598-017-01501-0
103. Middlebrook G, Coleman CM, Schaefer WB. Sulfolipid from virulent Tubercle bacilli. Proc Natl Acad Sci USA. (1959) 45:1801–4. doi: 10.1073/pnas.45.12.1801
104. Goren MB. Sulfolipid I of Mycobacterium tuberculosis, strain H37Rv. II. Structural studies. Biochim Biophys Acta. (1970) 210:127–38. doi: 10.1016/0005-2760(70)90068-8
105. Goren MB. Sulfolipid I of Mycobacterium tuberculosis, strain H37Rv. I. Purification and properties. Biochim Biophys Acta. (1970) 210:116–26. doi: 10.1016/0005-2760(70)90067-6
106. Mougous JD, Petzold CJ, Senaratne RH, Lee DH, Akey DL, Lin FL, et al. Identification, function and structure of the mycobacterial sulfotransferase that initiates sulfolipid-1 biosynthesis. Nat Struct Mol Biol. (2004) 11:721–9. doi: 10.1038/nsmb802
107. Kumar P, Schelle MW, Jain M, Lin FL, Petzold CJ, Leavell MD, et al. PapA1 and PapA2 are acyltransferases essential for the biosynthesis of the Mycobacterium tuberculosis virulence factor sulfolipid-1. Proc Natl Acad Sci USA. (2007) 104:11221–6. doi: 10.1073/pnas.0611649104
108. Converse SE, Mougous JD, Leavell MD, Leary JA, Bertozzi CR, Cox JS. MmpL8 is required for sulfolipid-1 biosynthesis and Mycobacterium tuberculosis virulence. Proc Natl Acad Sci USA. (2003) 100:6121–6. doi: 10.1073/pnas.1030024100
109. Goren MB, Mor N. Influence of phagosomal contents on the apparent inhibition of phagosome-lysosome fusion mediated by polyanionic substances in mouse peritoneal macrophages. Biochem Cell Biol. (1990) 68:24–32. doi: 10.1139/o90-004
110. Leigh CD, Bertozzi CR. Synthetic studies toward Mycobacterium tuberculosis sulfolipid-I. J Org Chem. (2008) 73:1008–17. doi: 10.1021/jo702032c
111. Goren MB, Hart PD, Young MR, Armstrong JA. Prevention of phagosome-lysosome fusion in cultured macrophages by sulfatides of Mycobacterium tuberculosis. Proc Natl Acad Sci USA. (1976) 73:2510–4. doi: 10.1073/pnas.73.7.2510
112. Kato M, Goren MB. Enhancement of cord factor-toxicity by sulfolipid. Jpn J Med Sci Biol. (1974) 27:120–4.
113. Kato M, Goren MB. Synergistic action of cord factor and mycobacterial sulfatides on mitochondria. Infect Immun. (1974) 10:733–41.
114. Brozna JP, Horan M, Rademacher JM, Pabst KA, Pabst MJ. Monocyte responses to sulfatide from Mycobacterium tuberculosis: inhibition of priming for enhanced release of superoxide, associated with increased secretion of interleukin-1 and tumor necrosis factor alpha, and altered protein phosphorylation. Infect Immun. (1991) 59:2542–8.
115. Pabst JJ, Gross JM, Prozna JP, Goren MB. Inhibition of macrophage priming by sulfatide from Mycobacterium tuberculosis. J Immunol. (1988) 140:634–40.
116. Zhang L, English D, Andersen BR. Activation of human neutrophils by Mycobacterium tuberculosis-derived sulfolipid-1. J Immunol. (1991) 146:2730–6.
117. Zhang L, Goren MB, Holzer TJ, Andersen BR. Effect of Mycobacterium tuberculosis-derived sulfolipid I on human phagocytic cells. Infect Immun. (1988) 56:2876–83.
118. Julian E, Matas L, Alcaide J, Luquin M. Comparison of antibody responses to a potential combination of specific glycolipids and proteins for test sensitivity improvement in tuberculosis serodiagnosis. Clin Diagnos Lab Immunol. (2004) 11:70–6. doi: 10.1128/CDLI.11.1.70-76.2004
119. Gilleron M, Stenger S, Mazorra Z, Wittke F, Mariotti S, Bohmer G, et al. Diacylated sulfoglycolipids are novel mycobacterial antigens stimulating CD1-restricted T cells during infection with Mycobacterium tuberculosis. J Exp Med. (2004) 199:649–59. doi: 10.1084/jem.20031097
120. Gangadharam PR, Cohn ML, Middlebrook G. Infectivity, Pathogenicity and sulpholipid fraction of some Indian and British strains of Tubercle bacilli. Tubercle. (1963) 44:452–5. doi: 10.1016/S0041-3879(63)80087-2
121. Goren MB, Brokl O, Schaefer WB. Lipids of putative relevance to virulence in Mycobacterium tuberculosis: correlation of virulence with elaboration of sulfatides and strongly acidic lipids. Infect Immun. (1974) 9:142–9.
122. Rousseau C, Sirakova TD, Dubey VS, Bordat Y, Kolattukudy PE, Gicquel B, et al. Virulence attenuation of two Mas-like polyketide synthase mutants of Mycobacterium tuberculosis. Microbiology. (2003) 149:1837–47. doi: 10.1099/mic.0.26278-0
123. Queiroz A, Riley LW. Bacterial immunostat: Mycobacterium tuberculosis lipids and their role in the host immune response. Rev Soc Bras Med Trop. (2017) 50:9–18. doi: 10.1590/0037-8682-0230-2016
124. Minnikin DE, Dobson G, Sesardic D, Ridell M. Mycolipenates and mycolipanolates of trehalose from Mycobacterium tuberculosis. J Gen Microbiol. (1985) 131:1369–74. doi: 10.1099/00221287-131-6-1369
125. Daffe M, Lacave C, Laneelle MA, Gillois M, Laneelle G. Polyphthienoyl trehalose, glycolipids specific for virulent strains of the tubercle bacillus. Eur J Biochem. (1988) 172:579–84. doi: 10.1111/j.1432-1033.1988.tb13928.x
126. Arbues A, Lugo-Villarino G, Neyrolles O, Guilhot C, Astarie-Dequeker C. Playing hide-and-seek with host macrophages through the use of mycobacterial cell envelope phthiocerol dimycocerosates and phenolic glycolipids. Front Cell Infect Microbiol. (2014) 4:173. doi: 10.3389/fcimb.2014.00173
127. Hunter SW, Brennan PJ. Further specific extracellular phenolic glycolipid antigens and a related diacylpthiocerol from Mycobacterium leprae. J Biol Chem. (1983) 258:7556–62.
128. Hunter SW, Fujiwara T, Brennan PJ. Structure and antigenicity of the major specific glycolipid antigen of Mycobacterium leprae. J Biol Chem. (1983) 257:15072–8.
129. Hunter SW, Brennan PJ. A novel phenolic glycolipid from Mycobacterium leprae possibly involved in immunogenicity and pathogenicity. J Bacteriol. (1981) 147:728–35.
130. Cho S-N, Yanagihara DL, Hunter SW, Gelber RH, Brennan PJ. Serological specificity of phenolic glycolipid 1 from Mycobacterium leprae and use in serodiagnosis of leprosy. Infect Immun. (1983) 41:1077–83.
131. Madigan CA, Cambier CJ, Kelly-Scumpia KM, Scumpia PO, Cheng T-Y, Zailaa J, et al. A macrophage response to Mycobacterium leprae phenolic glycolipid initiates nerve damage in leprosy. Cell. (2017) 170:973–85.e10. doi: 10.1016/j.cell.2017.07.030
132. Chan J, Fujiwara T, Brennan P, McNeil M, Turco SJ, Sibille J-C, et al. Microbial glycolipids: possible virulence factors that scavenge oxygen radicals. Proc Natl Acad Sci USA. (1989) 86:2453–7. doi: 10.1073/pnas.86.7.2453
133. Robinson N, Kolter T, Wolke M, Rybniker J, Hartmann P, Plum G. Mycobacterial phenolic glycolipid inhibits phagosome maturation and subverts the pro-inflammatory cytokine response. Traffic. (2008) 9:1936–47. doi: 10.1111/j.1600-0854.2008.00804.x
134. Cambier CJ, O'Leary SM, O'Sullivan MP, Keane J, Ramakrishnan L. Phenolic glycolipid facilitates mycobacterial escape from microbicidal tissue-resident macrophages. Immunity. (2017) 47:552–65.e4. doi: 10.1016/j.immuni.2017.08.003
135. Singh AK, Bishai WR. Partners in crime: phenolic glycolipids and macrophages. Trends Mol Med. (2017) 23:981–3. doi: 10.1016/j.molmed.2017.09.003
136. Bolz M, Ernst JD. Fishing for answers in human mycobacterial infections. Immunity. (2017) 47:395–7. doi: 10.1016/j.immuni.2017.09.005
137. Daffe M, Lacave C, Laneelle M, Laneelle G. Structure of the major triglycosyl phenol-phthiocerol of Mycobacterium tuberculosis (strain Canetti). Eur J Biochem. (1987) 167:155–60. doi: 10.1111/j.1432-1033.1987.tb13317.x
138. Cho S-N, Shin J-S, Daffe M, Chong Y, Kim S-K, Kim J-D. Production of monoclonal antibody to a phenolic glycolipid of Mycobacterium tuberculosis and its use in detection of the antigen in clinical isolates. J Clin Microbiol. (1992) 30:3065–9.
139. Reed MB, Domenech P, Manca C, Su H, Barczak AK, Kreiswirth BN, et al. A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature. (2004) 431:84–7. doi: 10.1038/nature02837
140. Reed MB, Gagneux S, Deriemer K, Small PM, Barry CE III. The W-Beijing lineage of Mycobacterium tuberculosis overproduces triglycerides and has the DosR dormancy regulon constitutively upregulated. J Bacteriol. (2007) 189:2583–9. doi: 10.1128/JB.01670-06
141. Sinsimer D, Huet G, Manca C, Tsenova L, Koo MS, Kurepina N, et al. The phenolic glycolipid of Mycobacterium tuberculosis differentially modulates the early host cytokine response but does not in itself confer hypervirulence. Infect Immun. (2008) 76:3027–36. doi: 10.1128/IAI.01663-07
142. Domingo-Gonzalez R, Das S, Griffiths KL, Ahmed M, Bambouskova M, Gopal R, et al. Interleukin-17 limits hypoxia-inducible factor 1alpha and development of hypoxic granulomas during tuberculosis. JCI Insight. (2017) 2:92973. doi: 10.1172/jci.insight.92973
143. Elsaidi HRH, Lowary TL. Inhibition of cytokine release by Mycobacterium tuberculosis phenolic glycolipid analogues. Chembiochem. (2014) 15:1176–82. doi: 10.1002/cbic.201402001
144. Torrelles JB, Knaup R, Kolareth A, Slepushkina T, Kaufman TM, Kang PB, et al. Identification of Mycobacterium tuberculosis clinical isolates with altered phagocytosis by human macrophages due to a truncated lipoarabinomannan. J Biol Chem. (2008) 283:31417–28. doi: 10.1074/jbc.M806350200
145. Arbues A, Malaga W, Constant P, Guilhot C, Prandi J, Astarie-Dequeker C. Trisaccharides of phenolic glycolipids confer advantages to pathogenic mycobacteria through manipulation of host-cell pattern-recognition receptors. ACS Chem Biol. (2016) 11:2865–75. doi: 10.1021/acschembio.6b00568
146. Tabouret G, Astarie-Dequeker C, Demangel C, Malaga W, Constant P, Ray A, et al. Mycobacterium leprae phenolglycolipid-1 expressed by engineered M. bovis BCG modulates early interaction with human phagocytes. PLoS Pathogens. (2010) 6:e1001159. doi: 10.1371/journal.ppat.1001159
147. Zheng RB, Jegouzo SAF, Joe M, Bai Y, Tran H-A, Shen K, et al. Insights into interactions of mycobacteria with the host innate immune system from a novel array of synthetic Mycobacterial glycans. ACS Chem Biol. (2017) 12:2990–3002. doi: 10.1021/acschembio.7b00797
148. Tran V, Ahn SK, Ng M, Li M, Liu J. Loss of lipid virulence factors reduces the efficacy of the BCG vaccine. Sci Rep. (2016) 6:29076. doi: 10.1038/srep29076
149. Ortalo-Magne A, Andersen AB, Daffe M. The outermost capsular arabinomannans and other mannoconjugates of virulent and avirulent tubercle bacilli. Microbiology. (1996) 142:927–35. doi: 10.1099/00221287-142-4-927
150. Khoo K-H, Dell A, Morris HR, Brennan PJ, Chatterjee D. Structural definition of acylated phosphatidylinositol mannosides from Mycobacterium tuberculosis: definition of a common anchor for lipomannan and lipoarabinomannan. Glycobiology. (1995) 5:117–27. doi: 10.1093/glycob/5.1.117
151. Gilleron M, Nigou J, Cahuzac B, Puzo G. Structural study of the lipomannans from Mycobacterium bovis BCG: characterisation of multiacylated forms of the phosphatidyl-myo-inositol anchor. J Mol Biol. (1999) 285:2147–60. doi: 10.1006/jmbi.1998.2438
152. Torrelles JB, Azad AK, Schlesinger LS. Fine discrimination in the recognition of individual species of phosphatidyl-myo-inositol mannosides from Mycobacterium tuberculosis by C-type lectin pattern recognition receptors. J Immunol. (2006) 177:1805–16. doi: 10.4049/jimmunol.177.3.1805
153. Villeneuve C, Gilleron M, Maridonneau-Parini I, Daffe M, Astarie-Dequeker C, Etienne G. Mycobacteria use their surface-exposed glycolipids to infect human macrophages through a receptor-dependent process. J Lipid Res. (2005) 46:475–83. doi: 10.1194/jlr.M400308-JLR200
154. Vergne I, Fratti RA, Hill PJ, Chua J, Belisle J, Deretic V. Mycobacterium tuberculosis phagosome maturation arrest: mycobacterial phosphatidylinositol analog phosphatidylinositol mannoside stimulates early endosomal fusion. Mol Biol Cell. (2004) 15:751–60. doi: 10.1091/mbc.e03-05-0307
155. Turner J, Torrelles JB. Mannose-capped lipoarabinomannan in Mycobacterium tuberculosis pathogenesis. Pathog Dis. (2018) 76:fty026. doi: 10.1093/femspd/fty026
156. Driessen NN, Ummels R, Maaskant JJ, Gurcha SS, Besra GS, Ainge GD, et al. Role of phosphatidylinositol mannosides in the interaction between mycobacteria and DC-SIGN. Infect Immun. (2009) 77:4538–47. doi: 10.1128/IAI.01256-08
157. dlS H, Mariotti S, Angenieux C, Gilleron M, Garcia-Alles LF, Malm D, et al. Assistance of microbial glycolipid antigen processing by CD1e. Science. (2005) 310:1321–4. doi: 10.1126/science.1115301
158. Gilleron M, Quesniaux VF, Puzo G. Acylation state of the phosphatidylinositol hexamannosides from Mycobacterium bovis bacillus Calmette Guerin and Mycobacterium tuberculosis H37Rv and its implication in Toll-like receptor response. J Biol Chem. (2003) 278:29880–9. doi: 10.1074/jbc.M303446200
159. Jones BW, Means TK, Heldwein KA, Keen MA, Hill PJ, Belisle JT, et al. Different toll-like receptor agonists induce distinct macrophage responses. J Leukoc Biol. (2001) 69:1036–44. doi: 10.1189/jlb.69.6.1036
160. Mazurek J, Ignatowicz L, Kallenius G, Svenson SB, Pawlowski A, Hamasur B. Divergent effects of mycobacterial cell wall glycolipids on maturation and function of human monocyte-derived dendritic cells. PLoS ONE. (2012) 7:e42515. doi: 10.1371/journal.pone.0042515
161. Kallenius G, Correia-Neves M, Buteme H, Hamasur B, Svenson SB. Lipoarabinomannan, and its related glycolipids, induce divergent and opposing immune responses to Mycobacterium tuberculosis depending on structural diversity and experimental variations. Tuberculosis. (2016) 96:120–30. doi: 10.1016/j.tube.2015.09.005
162. Guerin ME, Kordulakova J, Alzari PM, Brennan PJ, Jackson M. Molecular basis of phosphatidyl-myo-inositol mannoside biosynthesis and regulation in mycobacteria. J Biol Chem. (2010) 285:33577–83. doi: 10.1074/jbc.R110.168328
163. Glass LN, Swapna G, Chavadi SS, Tufariello JM, Mi K, Drumm JE, et al. Mycobacterium tuberculosis universal stress protein Rv2623 interacts with the putative ATP binding cassette (ABC) transporter Rv1747 to regulate mycobacterial growth. PLoS Pathog. (2017) 13:e1006515. doi: 10.1371/journal.ppat.1006515
164. Arcos J, Sasindran SJ, Fujiwara N, Turner J, Schlesinger LS, Torrelles JB. Human lung hydrolases delineate Mycobacterium tuberculosis-macrophage interactions and the capacity to control infection. J Immunol. (2011) 187:372–81. doi: 10.4049/jimmunol.1100823
165. Guirado E, Mbawuike U, Keiser TL, Arcos J, Azad AK, Wang SH, et al. Characterization of host and microbial determinants in individuals with latent tuberculosis infection using a human granuloma model. MBio. (2015) 6:e02537–14. doi: 10.1128/mBio.02537-14
166. Keiser TL, Azad AK, Guirado E, Bonacci R, Schlesinger LS. Comparative transcriptional study of the putative mannose donor biosynthesis genes in virulent Mycobacterium tuberculosis and attenuated Mycobacterium bovis BCG strains. Infect Immun. (2011) 79:4668–73. doi: 10.1128/IAI.05635-11
167. Lavollay M, Arthur M, Fourgeaud M, Dubost L, Marie A, Veziris N, et al. The peptidoglycan of stationary-phase Mycobacterium tuberculosis predominantly contains cross-links generated by L,D-transpeptidation. J Bacteriol. (2008) 190:4360–6. doi: 10.1128/JB.00239-08
168. Singh A, Crossman DK, Mai D, Guidry L, Voskuil MI, Renfrow MB, et al. Mycobacterium tuberculosis WhiB3 maintains redox homeostasis by regulating virulence lipid anabolism to modulate macrophage response. PLoS Pathogens. (2009) 5:e1000545. doi: 10.1371/journal.ppat.1000545
169. Curry JM, Whalan R, Hunt DM, Gohil K, Strom M, Rickman L, et al. An ABC transporter containing a forkhead-associated domain interacts with a serine-threonine protein kinase and is required for growth of Mycobacterium tuberculosis in mice. Infect Immun. (2005) 73:4471–7. doi: 10.1128/IAI.73.8.4471-4477.2005
170. Jackson M, Stadthagen G, Gicquel B. Long-chain multiple methyl-branched fatty acid-containing lipids of Mycobacterium tuberculosis: biosynthesis, transport, regulation and biological activities. Tuberculosis. (2007) 87:78–86. doi: 10.1016/j.tube.2006.05.003
171. Matsunaga I, Bhatt A, Young DC, Cheng TY, Eyles SJ, Besra GS, et al. Mycobacterium tuberculosis pks12 produces a novel polyketide presented by CD1c to T cells. J Exp Med. (2004) 200:1559–69. doi: 10.1084/jem.20041429
172. Fabre M, Koeck J-L, Le Fleche P, Simon F, Herve V, Vergnaud G, et al. High genetic diversity revealed by variable-number tandem repeat genotyping and analysis of hsp65 gene polymorphism in a large collection of “Mycobacterium canettii” strains indicates that the M. tuberculosis complex is a recently emerged clone of “M. canettii”. J Clin Microbiol. (2004) 42:3248–55. doi: 10.1128/JCM.42.7.3248-3255.2004
173. Briquet A, Vong R, Roseau J-B, Javelle E, Cazes N, Riviere F, et al. Clinical features of Mycobacterium canettii infection: a retrospective study of 20 cases among French soldiers and relatives. Clin Infect Dis. (2019) 69:2003–10. doi: 10.1093/cid/ciz107
174. Valway SE, Sanchez MPC, Shinnick TF, Orme I, Agerton T, Hoy D, et al. An outbreak involving extensive transmission of a virulent strain of Mycobacerium tuberculosis. N Engl J Med. (1998) 338:633–9. doi: 10.1056/NEJM199803053381001
175. Schlesinger LS. Macrophage phagocytosis of virulent but not attenuated strains of Mycobacterium tuberculosis is mediated by mannose receptors in addition to complement receptors. J Immunol. (1993) 150:2920–30.
176. Kang PB, Azad AK, Torrelles JB, Kaufman TM, Beharka A, Tibesar E, et al. The human macrophage mannose receptor directs Mycobacterium tuberculosis lipoarabinomannan-mediated phagosome biogenesis. J Exp Med. (2005) 202:987–99. doi: 10.1084/jem.20051239
177. Notter RH. Lung Surfactants: Basic Science and Clinical Applications. New York, NY: Marcel Dekker (2000). p. 1–444. doi: 10.1201/9781482270426
178. Arcos J, Diangelo L, Scordo J, Sasindran J, Moliva J, Turner J, et al. Lung mucosa lining fluid modifies Mycobacterium tuberculosis to reprogram human neutrophil killing mechanisms. J Infect Dis. (2015) 212:948–58. doi: 10.1093/infdis/jiv146
179. Arcos J, Sasindran SJ, Moliva JI, Scordo JM, Sidiki S, Guo H, et al. Mycobacterium tuberculosis cell wall released fragments by the action of the human lung mucosa modulate macrophages to control infection in an IL-10-dependent manner. Mucosal Immunol. (2017) 10:1248–58. doi: 10.1038/mi.2016.115
180. Scordo JM, Arcos J, Kelley HV, Diangelo L, Sasindran SJ, Youngmin E, et al. Mycobacterium tuberculosis cell wall fragments released upon bacterial contact with the human lung mucosa alter the neutrophil response to infection. Front Immunol. (2017) 8:307. doi: 10.3389/fimmu.2017.00307
181. Moliva JI, Rajaram MV, Sidiki S, Sasindran SJ, Guirado E, Pan XJ, et al. Molecular composition of the alveolar lining fluid in the aging lung. Age. (2014) 36:9633. doi: 10.1007/s11357-014-9633-4
182. Moliva JI, Duncan MA, Olmo-Fontanez A, Akhter A, Arnett E, Scordo JM, et al. The lung mucosa environment in the elderly increases host susceptibility to Mycobacterium tuberculosis infection. J Infect Dis. (2019). doi: 10.1093/infdis/jiz138
183. Moliva JI, Hossfeld AP, Canan CH, Dwivedi V, Wewers MD, Beamer G, et al. Exposure to human alveolar lining fluid enhances Mycobacterium bovis BCG vaccine efficacy against Mycobacterium tuberculosis infection in a CD8(+) T-cell-dependent manner. Mucosal Immunol. (2018) 11:968–78. doi: 10.1038/mi.2017.80
184. Moliva JI, Hossfeld AP, Sidiki S, Canan CH, Dwivedi V, Beamer G, et al. Selective delipidation of Mycobacterium bovis BCG enables direct pulmonary vaccination and enhances protection against Mycobacterium tuberculosis. Mucosal Immunol. (2019) 12:805–15. doi: 10.1038/s41385-019-0148-2
185. Chavadi SS, Edupuganti UR, Vergnolle O, Fatima I, Singh SM, Soll CE, et al. Inactivation of tesA reduces cell wall lipid production and increases drug susceptibility in mycobacteria. J Biol Chem. (2011) 286:24616–25. doi: 10.1074/jbc.M111.247601
186. Barkan D, Liu Z, Sacchettini JC, Glickman MS. Mycolic acid cyclopropanation is essential for viability, drug resistance, and cell wall integrity of Mycobacterium tuberculosis. Chem Biol. (2009) 16:499–509. doi: 10.1016/j.chembiol.2009.04.001
187. Velayati AA, Farnia P, Ibrahim TA, Haroun RZ, Kuan HO, Ghanavi J, et al. Differences in cell wall thickness between resistant and nonresistant strains of Mycobacterium tuberculosis: using transmission electron microscopy. Chemotherapy. (2009) 55:303–7. doi: 10.1159/000226425
188. Velayati AA, Farnia P, Merza MA, Zhavnerko GK, Tabarsi P, Titov LP, et al. New insight into extremely drug-resistant tuberculosis: using atomic force microscopy. Eur Respir J. (2010) 36:1490–3. doi: 10.1183/09031936.00064510
189. Velayati AA, Masjedi MR, Farnia P, Tabarsi P, Ghanavi J, Ziazarifi AH, et al. Emergence of new forms of totally drug-resistant tuberculosis bacilli: super extensively drug-resistant tuberculosis or totally drug-resistant strains in iran. Chest. (2009) 136:420–5. doi: 10.1378/chest.08-2427
190. Torrelles JB. Broadening our view about the role of Mycobacterium tuberculosis cell envelope components during infection: a battle for survival. In: Cardona PJ, editor. Understanding Tuberculosis—Analyzing the Origin of Mycobacterium tuberculosis Pathogenicity. Rijeka, Croatia: Intech (2012). p. 1–46.
191. Jain M, Petzold CJ, Schelle MW, Leavell MD, Mougous JD, Bertozzi CR, et al. Lipidomics reveals control of Mycobacterium tuberculosis virulence lipids via metabolic coupling. Proc Natl Acad Sci USA. (2007) 104:5133–8. doi: 10.1073/pnas.0610634104
192. Serafini A, Tan L, Horswell S, Howell S, Greenwood DJ, Hunt DM, et al. Mycobacterium tuberculosis requires glyoxylate shunt and reverse methylcitrate cycle for lactate and pyruvate metabolism. Mol Microbiol. (2019) 112:1284–307. doi: 10.1111/mmi.14362
193. Sequeira PC, Senaratne RH, Riley LW. Inhibition of toll-like receptor 2 (TLR-2)-mediated response in human alveolar epithelial cells by mycolic acids and Mycobacterium tuberculosis mce1 operon mutant. Pathog Dis. (2014) 70:132–40. doi: 10.1111/2049-632X.12110
194. Dunphy KY, Senaratne RH, Masuzawa M, Kendall LV, Riley LW. Attenuation of Mycobacterium tuberculosis functionally disrupted in a fatty acyl-coenzyme A synthetase gene fadD5. J Infect Dis. (2010) 201:1232–9. doi: 10.1086/651452
195. Munoz-Elias EJ, Upton AM, Cherian J, McKinney JD. Role of the methylcitrate cycle in Mycobacterium tuberculosis metabolism, intracellular growth, and virulence. Mol Microbiol. (2006) 60:1109–22. doi: 10.1111/j.1365-2958.2006.05155.x
196. McKinney JD, Honer zu Bentrup K, Munoz-Elias EJ, Miczak A, Chen B, Chan WT, et al. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature. (2000) 406:735–8. doi: 10.1038/35021074
197. Moody DB, Reinhold BB, Guy MR, Beckman EM, Frederique DE, Furlong ST, et al. Structural requirements for glycolipid antigen recognition by CD1b-restricted T cells. Science. (1997) 278:283–6. doi: 10.1126/science.278.5336.283
198. Porcelli SA, Modlin RL. The CD1 system: antigen-presenting molecules for T cell recognition of lipids and glycolipids. Annu Rev Immunol. (1999) 17:297–329. doi: 10.1146/annurev.immunol.17.1.297
199. Kawashima T, Norose Y, Watanabe Y, Enomoto Y, Narazaki H, Watari E, et al. Cutting edge: major CD8 T cell response to live bacillus Calmette-Guerin is mediated by CD1 molecules. J Immunol. (2003) 170:5345–8. doi: 10.4049/jimmunol.170.11.5345
200. Watanabe Y, Watari E, Matsunaga I, Hiromatsu K, Dascher CC, Kawashima T, et al. BCG vaccine elicits both T-cell mediated and humoral immune responses directed against mycobacterial lipid components. Vaccine. (2006) 24:5700–7. doi: 10.1016/j.vaccine.2006.04.049
201. Moody DB, Young DC, Cheng TY, Rosat JP, Roura-Mir C, O'Connor PB, et al. T cell activation by lipopeptide antigens. Science. (2004) 303:527–31. doi: 10.1126/science.1089353
202. Beckman EV, Porcelli SA, Morita CT, Behar SM, Furlong ST, Brenner MB. Recognition of a lipid antigen by CD1-restricted alpha beta+ T cells. Nature. (1994) 372:691–4. doi: 10.1038/372691a0
203. Sieling PA, Chatterjee D, Porcelli SA, Prigozy TI, Mazzaccaro RJ, Soriano T, et al. CD1-restricted T cell recognition of microbial lipoglycan antigens. Science. (1995) 269:227–30. doi: 10.1126/science.7542404
204. Moody DB, Sugita M, Peters PJ, Brenner MB, Porcelli SA. The CD1-restricted T-cell response to mycobacteria. Res Immunol. (1996) 147:550–9. doi: 10.1016/S0923-2494(97)85221-2
205. Layre E, Collmann A, Bastian M, Mariotti S, Czaplicki J, Prandi J, et al. Mycolic acids constitute a scaffold for mycobacterial lipid antigens stimulating CD1-restricted T cells. Chem Biol. (2009) 16:82–92. doi: 10.1016/j.chembiol.2008.11.008
206. Moody DB, Ulrichs T, Muhlecker W, Young DC, Gurcha SS, Grant E, et al. CD1c-mediated T-cell recognition of isoprenoid glycolipids in Mycobacterium tuberculosis infection. Nature. (2000) 404:884–8. doi: 10.1038/35009119
207. Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol. (2007) 25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711
208. Felio K, Nguyen H, Dascher CC, Choi HJ, Li S, Zimmer MI, et al. CD1-restricted adaptive immune responses to Mycobacteria in human group 1 CD1 transgenic mice. J Exp Med. (2009) 206:2497–509. doi: 10.1084/jem.20090898
209. Moreno-Mendieta SA, Rocha-Zavaleta L, Rodriguez-Sanoja R. Adjuvants in tuberculosis vaccine development. FEMS Immunol Med Microbiol. (2010) 58:75–84. doi: 10.1111/j.1574-695X.2009.00629.x
210. Ribi E, Granger DL, Milner KC, Yamamoto K, Strain SM, Parker R, et al. Induction of resistance to tuberculosis in mice with defined components of mycobacteria and with some unrelated materials. Immunology. (1982) 46:297–305.
211. Andersen P. Effective vaccination of mice against Mycobacterium tuberculosis infection with a soluble mixture of secreted mycobacterial proteins. Infect Immun. (1994) 62:2536–44.
212. Ishikawa E, Mori D, Yamasaki S. Recognition of mycobacterial lipids by immune receptors. Trends Immunol. (2017) 38:66–76. doi: 10.1016/j.it.2016.10.009
213. Agger EM. Novel adjuvant formulations for delivery of anti-tuberculosis vaccine candidates. Adv Drug Deliv Rev. (2016) 102:73–82. doi: 10.1016/j.addr.2015.11.012
214. Lindblad EB, Elhay MJ, Silva R, Appelberg R, Andersen P. Adjuvant modulation of immune responses to tuberculosis subunit vaccines. Infect Immun. (1997) 65:623–9.
215. Hwang SA, Arora R, Kruzel ML, Actor JK. Lactoferrin enhances efficacy of the BCG vaccine: comparison between two inbred mice strains (C57BL/6 and BALB/c). Tuberculosis. (2009) 89(Suppl. 1):S49–54. doi: 10.1016/S1472-9792(09)70012-5
216. van Dissel JT, Joosten SA, Hoff ST, Soonawala D, Prins C, Hokey DA, et al. A novel liposomal adjuvant system, CAF01, promotes long-lived Mycobacterium tuberculosis-specific T-cell responses in human. Vaccine. (2014) 32:7098–107. doi: 10.1016/j.vaccine.2014.10.036
217. van Dissel JT, Arend SM, Prins C, Bang P, Tingskov PN, Lingnau K, et al. Ag85B-ESAT-6 adjuvanted with IC31 promotes strong and long-lived Mycobacterium tuberculosis specific T cell responses in naive human volunteers. Vaccine. (2010) 28:3571–81. doi: 10.1016/j.vaccine.2010.02.094
218. van Dissel JT, Soonawala D, Joosten SA, Prins C, Arend SM, Bang P, et al. Ag85B-ESAT-6 adjuvanted with IC31(R) promotes strong and long-lived Mycobacterium tuberculosis specific T cell responses in volunteers with previous BCG vaccination or tuberculosis infection. Vaccine. (2011) 29:2100–9. doi: 10.1016/j.vaccine.2010.12.135
219. Fozo EM, Rucks EA. The Making and Taking of Lipids: The role of bacterial lipid synthesis and the harnessing of host lipids in bacterial pathogenesis. Adv Microb Physiol. (2016) 69:51–155. doi: 10.1016/bs.ampbs.2016.07.001
220. Kaufmann SH, Cole ST, Mizrahi V, Rubin E, Nathan C. Mycobacterium tuberculosis and the host response. J Exp Med. (2005) 201:1693–7. doi: 10.1084/jem.20050842
221. Peterson EJ, Bailo R, Rothchild AC, Arrieta-Ortiz ML, Kaur A, Pan M, et al. Path-seq identifies an essential mycolate remodeling program for mycobacterial host adaptation. Mol Syst Biol. (2019) 15:e8584. doi: 10.15252/msb.20188584
222. Eoh H, Wang Z, Layre E, Rath P, Morris R, Branch Moody D, et al. Metabolic anticipation in Mycobacterium tuberculosis. Nat Microbiol. (2017) 2:17084. doi: 10.1038/nmicrobiol.2017.84
223. Moliva JI, Turner J, Torrelles JB. Prospects in Mycobacterium bovis Bacille Calmette et Guerin (BCG) vaccine diversity and delivery: why does BCG fail to protect against tuberculosis? Vaccine. (2015) 33:5035–41. doi: 10.1016/j.vaccine.2015.08.033
224. Moliva JI, Turner J, Torrelles JB. Immune responses to BCG vaccination: why do they fail to protect against Mycobacterium tuberculosis? Front Immunol. (2017) 8:407. doi: 10.3389/fimmu.2017.00407
225. Scordo JM, Olmo-Fontanez AM, Kelley HV, Sidiki S, Arcos J, Akhter A, et al. The human lung mucosa drives differential Mycobacterium tuberculosis infection outcome in the alveolar epithelium. Mucosal Immunol. (2019) 12:795–804. doi: 10.1038/s41385-019-0156-2
226. Kaur D, Guerin ME, Skovierova H, Brennan PJ, Jackson M. Chapter 2: Biogenesis of the cell wall and other glycoconjugates of Mycobacterium tuberculosis. Adv Appl Microbiol. (2009) 69:23–78. doi: 10.1016/S0065-2164(09)69002-X
227. Raghunandanan S, Jose L, Gopinath V, Kumar RA. Comparative label-free lipidomic analysis of Mycobacterium tuberculosis during dormancy and reactivation. Sci Rep. (2019) 9:3660. doi: 10.1038/s41598-019-40051-5
228. Schubert OT, Ludwig C, Kogadeeva M, Zimmermann M, Rosenberger G, Gengenbacher M, et al. Absolute proteome composition and dynamics during dormancy and resuscitation of Mycobacterium tuberculosis. Cell Host Microbe. (2015) 18:96–108. doi: 10.1016/j.chom.2015.06.001
229. Rodriguez JG, Hernandez AC, Helguera-Repetto C, Aguilar Ayala D, Guadarrama-Medina R, Anzola JM, et al. Global adaptation to a lipid environment triggers the dormancy-related phenotype of Mycobacterium tuberculosis. mBio. (2014) 5:e01125–14. doi: 10.1128/mBio.01125-14
230. Deb C, Lee CM, Dubey VS, Daniel J, Abomoelak B, Sirakova TD, et al. A novel in vitro multiple-stress dormancy model for Mycobacterium tuberculosis generates a lipid-loaded, drug-tolerant, dormant pathogen. PLoS ONE. (2009) 4:e6077. doi: 10.1371/journal.pone.0006077
Keywords: tuberculosis, Mycobacterium tuberculosis, cell envelope glycolipids, infectious diseases, vaccine strategies, immunomodulatory lipids, immune responses
Citation: Garcia-Vilanova A, Chan J and Torrelles JB (2019) Underestimated Manipulative Roles of Mycobacterium tuberculosis Cell Envelope Glycolipids During Infection. Front. Immunol. 10:2909. doi: 10.3389/fimmu.2019.02909
Received: 29 August 2019; Accepted: 27 November 2019;
Published: 18 December 2019.
Edited by:
Andrea Cooper, University of Leicester, United KingdomReviewed by:
Shigeto Yoshida, Kanazawa University, JapanCopyright © 2019 Garcia-Vilanova, Chan and Torrelles. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jordi B. Torrelles, anRvcnJlbGxlc0B0eGJpb21lZC5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.