 Laura Ducimetière
Laura Ducimetière Marijne Vermeer
Marijne Vermeer Sonia Tugues
Sonia Tugues
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 13 December 2019
Sec. Cancer Immunity and Immunotherapy
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02895
This article is part of the Research TopicInnate Lymphoid Cells in Cancer: Friends or Foes?View all 12 articles
The multifaceted roles of Innate Lymphoid Cells (ILC) have been widely interrogated in tumor immunity. Whereas, Natural Killer (NK) cells possess undisputable tumor-suppressive properties across multiple types of cancer, the other ILC family members can either promote or inhibit tumor growth depending on the environmental conditions. The differential effects of ILCs on tumor outcome have been attributed to the high degree of heterogeneity and plasticity within the ILC family members. However, it is now becoming clear that ILCs responses are shaped by their dynamic crosstalk with the different components of the tumor microenvironment (TME). In this review, we will give insights into the molecular and cellular players of the ILCs-TME interactions and we will discuss how we can use this knowledge to successfully harness the activity of ILCs for anticancer therapies.
In the past years, Innate Lymphoid Cells (ILCs) have emerged as crucial players in cancer growth and therapy. The ILC family members are classified into five groups namely Natural Killer (NK) cells, group 1 ILCs (ILC1s), group 2 ILCs (ILC2s), group 3 ILCs (ILC3s), and lymphoid-tissue inducer cells (LTis) (1, 2). Initially described in the 1970s, NK cells are the founding members of the ILCs family (3). They develop in the bone marrow from common lymphoid progenitors and follow a sequential maturation and differentiation process, which is regulated by a variety of transcription factors (4). The T-box protein 21 (T-BET) and Eomesodermin (EOMES), for instance, undertake non-redundant roles in this process through stabilizing distinct NK cell subsets during maturation (5). Once in tissues, NK cells are potentially capable of eliminating infected or transformed cells via several mechanisms including degranulation, death receptor ligation, or the production of inflammatory cytokines (e.g., IFNγ, TNFα) (6). The latter feature is shared with another member of the family, the ILC1s, which are phenotypically very similar to NK cells (7, 8). However, the two lineages have been shown to diverge early in ontogeny and to differ in terms of cytotoxic machinery and tropic properties (9). Thus, ILC1s are typically defined as tissue resident since they are not found in the blood or lymphoid tissues, but rather in organs such as the gut, the liver, the salivary glands or the reproductive tract (9). In the liver, for example, T-bet dependent ILC1s have been shown to contribute to immune responses against haptens and viral antigens (10). In contrast, the intestinal ILC1 subset rather controls microbial pathogens and contributes to chronic inflammation (11).
ILC2s are characterized by their ability to produce Th2 cytokines (IL-4, IL-5, IL-9, or IL-13) and therefore contribute to type 2 inflammation promoting pathological responses associated to asthma or allergy, but also conferring protection against helminths (12). ILC2s require the transcription factor GATA-3 for their development (13). In the mucosal tissues, where they typically reside, ILC2s can be activated by epithelial-derived alarmins (IL-33, IL-25, or TLSP), whose contribution to ILC2 activation depends on the tissue type as well as on the nature and magnitude of the pathological insult (14). Thus, whereas IL-33 is believed to play a crucial role in the context of allergic airway inflammation, IL-25 is particularly relevant for the amplification of type 2 immune responses in the gut (15, 16).
ILC3s comprise a heterogenous and plastic population. They are divided into two subsets based on the expression of the natural cytotoxicity receptor (NCR) NKp46 in mice and Nkp44 in humans namely NCR+ILC3s and NCR−ILC3s (17). Both subsets require the transcription factor RORγt for their development and represent a major source of the cytokine IL-22 (18), which regulates interactions with the commensal flora and controls mucosal infections at barrier sites (19). ILC3s can also take proinflammatory roles through the release of IL-17 or IFNγ, contributing to the progression of psoriasis and colitis, respectively (20, 21). The production of abundant amounts of IL-17 and IL-22 is also a defining feature of LTis, which functionally resemble the population of NCR−ILC3s (22). LTis, however, are derived from a developmental pathway starting in the embryo (23), where they engage in the formation of lymphoid tissues through the production of lymphotoxins (24).
The properties of ILCs have been widely investigated in the context of tumorigenesis. Due to their high cytotoxic capacity, NK cells are particularly suitable to eliminate tumor cells. Indeed, several preclinical studies have revealed a central role for NK cells in tumor control, especially in metastatic disease (25–30). ILC2s and ILC3s can also modulate antitumor responses, but their role rather depends on the environmental cues they encounter in their resident tissue. Thus, whereas IL-12-stimulated NCR+ILC3s were found to control primary melanoma growth (31), the growth of this tumor type in the lungs is modulated by an IL-5-producing subset of ILC2s (32). This contrasts with the protumorigenic role of ILCs described for other tumor models. For example, IL-13-producing ILC2s promote tumor growth in leukemia and breast cancer (33, 34), and IL-22-producing ILC3s do likewise in the gut (35, 36). Finally, the recruitment of RORγt+ILC3s to tumors mediated by CCL21 was able to promote lymph node metastasis by modulating the local chemokine milieu in the TME (37).
With the growing interest in harnessing ILC responses for immunotherapeutic strategies against cancer, it is important to better understand the multifaceted roles of ILCs in tumor development. Here, we will first discuss how ILCs migrate and expand in the tumor site. Further, we will review current knowledge on how ILCs communicate with the environment, including the interactions they establish with the tumor cells and the different components of the TME. Finally, we will discuss whether these interactions are beneficial or deleterious to tumor growth and invasion.
Parabiosis studies have shown that NK cells are a highly mobile subset that constantly circulate throughout the bloodstream and the lymphatic system, whereas the rest of the ILC family members are defined as tissue resident cells (38–40). Confirming the maintenance by local self-renewal within tissues, only very small numbers of ILCs can be found in healthy human and mouse circulation (41, 42). It has been shown that some ILC progenitors express the integrin α4β7 and the chemokine receptors CCR7, CCR9, or CXCR6, which may enable them to migrate to peripheral and lymphoid tissues (42–44). Also mature ILCs express several tissue homing receptors such as CXCR6, which promotes the accumulation of ILC3s in the gut (45) and provides survival signals to maintain ILC1s within the hepatic niche (46). Other markers of tissue residency for ILCs are the integrin CD49a and the early activation marker CD69, which are upregulated during ILC activation (47–49).
Within the TME, NK cells represent by far the most abundant innate lymphocyte subset identified (48). However, despite correlating with a better prognosis, NK cell homing is highly inefficient in most solid tumors (50, 51). There are a few exceptions including clear cell renal carcinoma, which harbors unusually high numbers of intratumoral NK cells (52, 53). The mechanisms leading to NK cell paucity in the TME are not well-studied, but what it is by now clear is that the majority of NK cells infiltrating tumor tissue belong to the mouse CD27high and the human CD56bright subsets, which are recruited to the tumor in a CXCR3-dependent manner (54, 55). Even though the immature CD56bright NK cell population has been traditionally considered as a “cytokine producer,” whether it can control tumors as efficiently as the mature CD56dim population is still a matter of debate. Due to their high motility, NK cells can also be recruited in strategic locations in order to prevent further cancer spread. As such, highly cytotoxic populations of NK cells from both CD56dim and CD56bright subsets have been found in tumor-draining lymph nodes of melanoma patients (56, 57). On the other hand, immunosuppressive mediators such as TGFβ might favor the retention of NK cells in the bone marrow through the upregulation of CXCR4 and the downregulation of CX3CR1 (58).
Despite their residency properties, a few ILCs have been reported to circulate in human blood. Thus, increased frequencies of ILC1s and ILC2s were found in patients with colorectal cancer (59) and with gastric cancer, respectively (60, 61). RORγt+ILC3s were also reported to migrate via the bloodstream toward the tumor site in response to CCL21 in mouse models of breast cancer (37) and in melanoma (62). Within the TME, ILC subsets other than NK cells are only found at extremely scarce numbers. In human lung cancer, a NCR+ population of ILC3s was found to accumulate at the edge of lymphoid structures in the tumor tissue (37, 62, 63). An enrichment of ILCs in tumors compared to healthy tissue has also been observed for ILC1s in gastrointestinal tumors (49) or ILC2s in gastric, breast and prostate cancer (34, 49, 60). Despite the presence of ILCs in the above-mentioned types of cancer, whether they contribute to the underlying pathology in humans is still a matter of debate. Also, whether the enrichment of ILCs results from newly recruited cells or from local in situ proliferation has not been thoroughly investigated. The latter phenomenon was however observed for ILC2s in IL-33-treated breast cancer (33), and for ILC1s in mouse mammary pre-cancerous lesions (64).
From all the ILC family members, NK cells show the highest cytolytic activity, while the primary role of other ILCs is to produce cytokines in response to different stimuli. In order to eliminate transformed cells, NK cells are equipped with a plethora of activating and inhibitory receptors, which need to be tightly regulated to determine whether a target cell will be killed or spared (65). Once activated, NK cells eliminate target cells via death receptors pathways (e.g., Fas/FasL) or through the release of cytotoxic granules at the immunological synapse (66). The usage of these two cytotoxic pathways appears to be tightly regulated. As such, whereas NK cells use the fast granule-mediated pathways for their first killing events, they switch to death receptors-mediated killing during the last encounters with the tumor cells (67). Despite possessing such an efficient cytotoxic machinery, NK cells from tumor-bearing mice or cancer patients are often functionally impaired and display low amounts of effector molecules such as granzyme B, IFNγ, or FasL (68). This is mostly due to the signals these cells receive from the TME, and especially from the surrounding tumor cells.
Within the TME, tumor cells are constantly exposed to stress conditions, which induce the upregulation of ligands for NK cell activating receptors (69). Although a priori this would favor NK cell-mediated immune surveillance, cancer cells have developed several mechanisms that allow them to evade immune recognition. Among those, we highlight the dysregulation of ligands that bind NKG2D, a major NK cell activating receptor critical for antitumor immunity (70). A commonly proposed mechanism for evading NK cell surveillance has to do with the shedding of the NKG2D ligands MICA and MICB from the cell membrane, leading to soluble forms that promote the internalization and posterior degradation of the receptor (71–73). This was however challenged in a study performed in murine tumor models, which reported that the soluble high affinity NKG2D ligand MULT-1 actually caused NK cell activation and tumor rejection (74). Irrespective of whether NKG2D ligands are soluble or membrane-bound, what is clear by now is that it is their chronic engagement which causes the desensitization of the NK cell receptor as well as related signaling pathways (75). Moreover, although tumor cells represent the main source of ligands for activating receptors, the induction of NKG2D ligands on myeloid cells and endothelial cells has also been shown to contribute to impaired NK antitumor responses (76, 77). Finally, other ILC family members such as intestinal ILC1s and ILC3s can also express NKG2D on the cell surface (78). Whether this receptor is able to modulate the activity of these cells in the TME is however not known.
Besides desensitizing NKG2D, tumor cells use additional mechanisms to evade NK cell surveillance including the secretion of immunosuppressive molecules such as TGFβ, IL-10, prostaglandin E2 (PGE2) or indoleamine 2,3-dioxygense (IDO) (79, 80). The production of these factors is not restricted to cancer cells, and a variety of cell types populating the TME can also contribute to the immunosuppressive pool leading to impaired NK cell function. Nevertheless, TGFβ and PGE2 are able to shape NK cell activity directly via the inhibition of activating receptors (79–81), or indirectly through the recruitment of immunosuppressive cells types such as myeloid-derived suppressor cells (MDSCs) or regulatory T cells (Tregs) (82, 83).
ILCs have a remarkable plasticity allowing them to acquire features of another ILC population in order to adapt to changes in the tissue microenvironment. In tumors, ILC plasticity was suggested as a mechanism by Gao et al., who reported a TGFβ-dependent conversion of NK cells into “ILC1-like” cells in a mouse model of chemically induced sarcoma (84). This conversion, which is characterized by the upregulation of the integrin CD49a and the downregulation of Eomes, appears to be detrimental for tumor control (84). A similar CD49ahigh ILC1-derived subset with a tissue-residency phenotype was however found to exert cytotoxicity in oncogene-induced murine tumor models (64). Given the overlapping phenotypes between NK cells and ILC1s (85), it is difficult to postulate whether one subset really converts into the other or if cells rather evolve on a continuum. A complete transition seems unlikely, since ILC1s and NK cells lineages are believed to separate early during the differentiation process (78, 86).
The dependence of NK cell into ILC1 conversion on TGFβ supports increasing evidence that this cytokine does not only induce NK cell dysfunction, but also plays a crucial role in regulating ILC plasticity. Interestingly, TGFβ-imprinting is essential for the differentiation of the ILC1s residing in murine salivary glands via suppression of Eomes and the upregulation of CD49a (87). In humans, TGFβ was also described to enable the transition between mature CD16+ peripheral blood NK cells into a CD16−CD9+ phenotype that resembles a population of decidual NK cells (88). But TGFβ is not the sole factor reported to induce ILC plasticity. In fact, the proinflammatory cytokine IL-12 was shown to induce the differentiation of ILC2s into IFNγ-producing ILC1s, a process that was reversed by IL-4 (89, 90). Further, IL-12 mediates the conversion of ILC3s into type 1-like ILCs in a variety of pathological conditions (31, 91–93). The so-called “ex-ILC3s” were found to display cytotoxic activity in humans (93) and to effectively suppress tumor growth in a mouse model of melanoma (31). In the context of intestinal inflammation, the ILC3 to ILC1 plasticity was reversible in the presence of IL-23 (92). Together, these results reinforce the notion that ILCs are highly plastic cells which fine-tune immune responses to adapt to the changing environment.
The wide number of events that take place in the TME to evade ILC surveillance have been summarized in Figure 1.
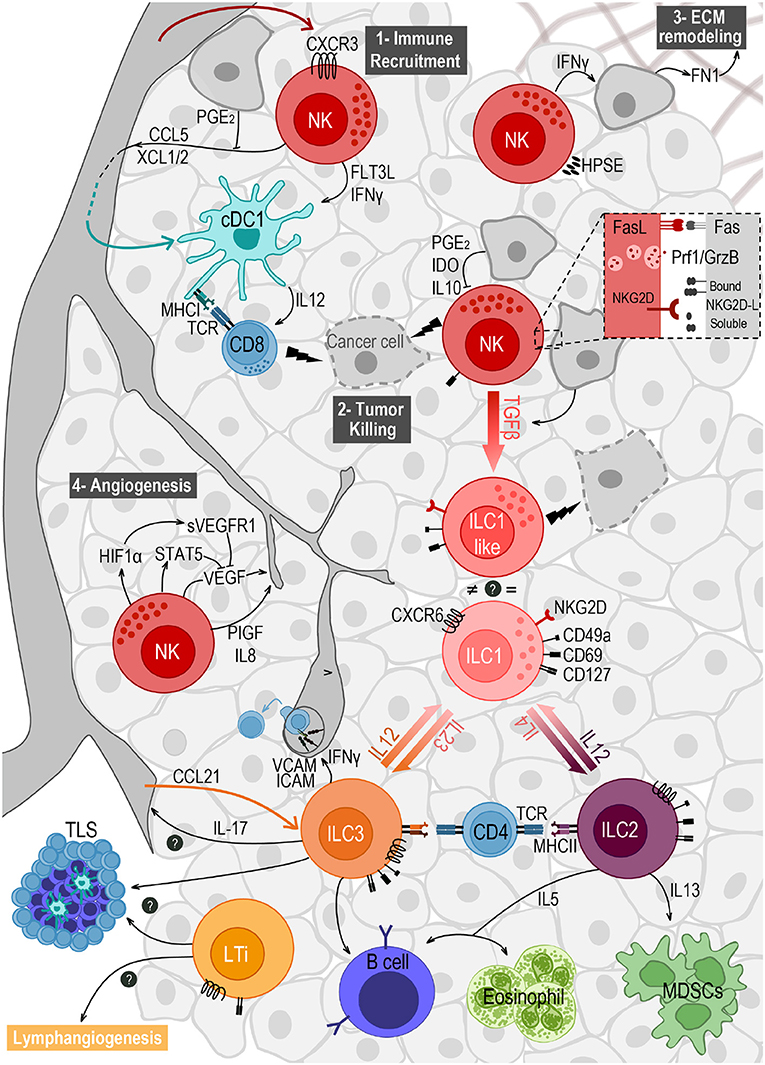
Figure 1. Crosstalk of ILCs and the different components of the tumor microenvironment. NK cells are the main ILC subset found in the TME, after migrating from the blood in a CXCR3-dependent way. They play important roles in (1) immune recruitment, (2) tumor cell killing, (3) extracellular matrix (ECM) remodeling, and (4) angiogenesis. (1) NK cells recruit cDC1s to the TME by secreting CCL5 and XCL1/2, and promote their survival and maturation through FLT3L and IFNγ. (2) NK cell mediated killing is mostly achieved by the engagement of death receptors (e.g., Fas/FasL) and by the release of cytotoxic granules containing perforin (Prf1) and granzymes (GrzB). This response can be triggered by the expression of stress markers on tumor cells, recognized by the receptor NKG2D. Ligands to this receptor (NKG2D-L) are membrane-bound but can also be shed and released in soluble form. (3) NK cells express the enzyme heparanase (HPSE) to degrade heparin sulfate proteoglycans, allowing them to migrate in the tumor tissue. NK cells' secretion of IFNγ induces the production of fibronectin 1 (FN1) by tumor cells, further remodeling the ECM. (4) NK cells modulate angiogenesis by releasing VEGF, PIGF and IL-8. VEGF secretion can be repressed by STAT5 or by soluble VEGF receptor (sVEGFR1) induced as a response to hypoxia. Tumor cells' secretion of TGFβ induces the conversion of NK cells into ILC1-like cells expressing CD49a and CD69, and exhibiting anti-tumor cytotoxic activity. Plasticity within the ILC family also includes the conversion from ILC2 to ILC1, and from ILC3 to ILC1, both induced by IL-12 and reversed by IL-4 and IL-23, respectively. ILC2s have a pro-tumorigenic role via secretion of IL-13 driving the expansion of myeloid-derived suppressor cells (MDSCs), and an anti-tumorigenic role through IL-5 mediated recruitment of eosinophils and activation of B cells. ILC3 can also stimulate antibody production by B cells, and can favor leukocyte recruitment to the TME when sensing IL-12, through IFNγ-mediated upregulation of ICAM-1 and VCAM-1. They may also play a role in angiogenesis through the production of IL-17. NCR+ILC3s and LTis accumulate in tertiary lymphoid structures (TLS) and may also promote lymphangiogenesis. Finally, both ILC2 and ILC3 express MHC class II and were able to prime CD4 T cells in vitro.
Angiogenesis, the formation of new blood vessels from pre-existing ones, is needed to satisfy the increasing demand of oxygen and nutrients of the growing tumor. This process is supported by several immune cell types via the production of pro-angiogenic growth factors (94, 95). NK cells, for instance, were the first ILC subset reported to modulate tumor vascularization. Hence, a population of CD56brightCD16− NK cells was shown to produce high amounts of the proangiogenic molecules VEGF, PlGF, or IL-8, leading to the formation of capillary-like structure in patients with NSCLC, melanoma, breast and colon carcinoma (96, 97). Interestingly, this population is reminiscent of a CD56brightCD16− subset of decidual NK cells, which may be involved in the tissue remodeling process associated with angiogenesis during embryonic development (98).
NK cells are able to regulate the production of the proangiogenic factor VEGF through various mechanisms. Firstly, the expression of VEGF can be repressed by the transcription STAT-5, leading to inhibition of angiogenesis and tumor growth (99). Since STAT-5 is required for NK cell cytotoxicity, it was proposed that cytokines that signal through this transcription factor (e.g., IL-2 and IL-15) may regulate tumor growth by promoting the conversion from angiogenic to cytotoxic NK cells (99). NK cells can also regulate their own production of VEGF when adapting to hypoxia (100). Thus, the induction of HIF-1α on NK cells induces the upregulation of the soluble receptor VEGFR-1 (sVEGFR-1), which sequesters VEGF leading to the formation of more functional vessels that induce tumor growth (100). Further, ILCs can induce changes in the tumor vasculature through the modulation of adhesion molecules (101, 102). A tumor-suppressive subset of IL-12-driven NKp46+ ILC3s promoted leukocyte recruitment through the induction of the adhesion molecules ICAM and VCAM on the tumor vessels (102), similarly to what was observed by tumor-infiltrating NCR+ILC3s in NSCLC tissues (63). ILC3s producing IL-17 may also play a role in regulating the tumor vasculature. IL-17 induces blood vessel permeability in pulmonary endothelial cells, thus leading to metastatic growth (103). Further, IL-17 signals through stromal cells to induce a variety of proangiogenic factors (e.g., VEGF, TGFβ, or IL-8) (104, 105). Finally LTis may also play a role in promoting lymphatic vessel growth, which actively participates in metastatic tumor dissemination (106). LTis interact with Mesenchymal Stem Cells (MSCs), which produce pro-lymphoangiogenic factors such as VEGF-C (107). Although the LTi-MSC crosstalk has been proposed to mediate lymph node metastasis in breast cancer (37), the involvement of the lymphatic vasculature in this setting remains unknown at this time.
Not only the vasculature, but also the extracellular matrix (ECM) is modified during the course of cancer progression. The ECM is a complex network of proteoglycans and fibrous proteins that support the surrounding cells and provide molecular cues for cell migration and differentiation (108). During cancer progression, the deregulation of the ECM promotes invasion, angiogenesis and facilitates immune cell infiltration (109). It has been shown that NK cells can modulate the ECM through the secretion of fibronectin 1 (FN1), leading to structural changes in the primary tumor and decreased metastasis (110). In addition, NK cells can facilitate their own migration through the ECM thanks to the expression of heparanase, an enzyme known to degrade heparin sulfate proteoglycans (HSPGs) (111). This raises concerns about the use of EMC inhibitors to block tumor cell invasion, since it may be detrimental for a proper migration of NK cells and possibly other subsets of immune cells.
Taken together, these reports highlight the importance of ILCs in modulating the tumor vasculature and the remodeling of the ECM (Figure 1), which could be exploited for immunotherapeutic purposes. Further work will have to address specific contributions of the distinct ILC subsets to this process. For instance, whether and how ILC2-signature cytokines regulate the angiogenic process has yet to be studied.
ILCs establish continuous interactions with a wide variety of cells within the TME. As such, understanding the nature of this crosstalk is crucial to unleash the full potential of ILC responses against developing tumors. Defined NK cell interactions in the cancer context include the interplay with DCs, the main sentinels of the innate immune system (112, 113). DCs can support NK cell responses through the secretion of several proinflammatory cytokines (IL-12, IL-15, IL-18, and Type I Interferon) (114). NK cells can in turn trigger DC maturation via the production of IFNs and Tumor Necrosis Factor (TNF) (115, 116). Within the TME, NK cells promote the recruitment of cDC1s, the DC subset capable of priming tumor-specific CD8 T cells (117). This is mediated through the secretion of CCL5 and XCL1/2 by intratumoral NK cells, and antagonized by PGE2 produced by the tumor cells (113). Apart from promoting the recruitment of DCs, NK cells can also prime and ensure their expansion. Thus, NK cells activated by MHC Class I low tumor cells can prime DCs to produce IL-12 and to induce protective CD8 T cell responses (118). Further, they appear to be the main source of the cytokine fms-like tyrosine kinase 3 ligand (FLT3L), a survival factor for DCs (112, 119). In contrast, the use of less immunogenic tumor cells led to the inhibition of DC activation by NK cells, which was mediated by the (TNF)-related apoptosis-inducing ligand (TRAIL) (120). This controversy was not observed in the human disease, where a high expression of NK cell and cDC1 signatures correlated with better prognosis and response to immunotherapy in a wide array of cancers (112, 113).
NK cells are not the only ILC subset that is able to shape myeloid cell responses. ILC2s, for instance, have been shown to either limit anti-tumor responses by triggering the expansion of MDSCs via secretion of IL-13 (34), or to enhance anti-cancer immunity by cooperating with DCs or eosinophils in the lung in a IL-5-dependent manner (32, 121). Also the crosstalk between ILC1s, ILC3s and myeloid cells has been shown to promote chronic inflammation leading to tumor initiation. First, ILC1s and “ex-ILC3s” have been found to accumulate in chronically inflamed guts in response to myeloid-derived cytokines such as IL-12 or IL-15 (47, 91). In this scenario, these two ILC subsets secrete high amounts of IFNγ, which engages neutrophils and macrophages to cause tissue injury (47, 122). Further, group 3 ILCs are particularly responsive to IL-23, a key pathogenic inducer of chronic intestinal inflammation (123). IL-23, which is primarily produced by cells of the myeloid lineage, induce the production of GM-CSF, IL-17 or IL-22 by ILC3s (20, 124, 125). Whereas GM-CSF feeds back on the myeloid cells to promote tissue damage and colitis (20, 125), IL-17 and IL-22 limit inflammation by maintaining the integrity of the epithelial barrier (126, 127). This contrasts with the protumorigenic role that both IL-22 and IL-17 exert in colorectal cancer (105, 128), where they were shown to have pro-proliferative and pro-angiogenic functions, respectively (105, 128).
In humans, the levels of IL-17 appear to be upregulated in colorectal cancer patients, where they associate to poor prognosis (35, 129, 130). Further studies will be required to determine the contribution of ILC3s to the total pool of IL-17 or IL-22-producing cells. Nevertheless, blocking the ILC-myeloid axis in tumors of intestinal origin arises as a promising approach and may represent a promising anti-cancer therapeutic strategy.
ILCs may also directly modulate the quality of T cell responses without prior DC crosstalk. NK cell-secreted IFNγ, for example, was shown to promote Th1 polarization in the lymph nodes in mouse models of infection (131, 132). A close cooperation between NK cells and T cells has also been shown in established lung carcinoma, where the stimulation of NK cells induced the recruitment of highly active T cells, leading to a more efficient tumor control (133). Also the engagement of NK cells with the TNF superfamily member LIGHT was found to trigger CD8 T responses at the tumor site (134). ILC2s can also modulate adaptive immune responses. The IL-33/ST2 signaling pathway, which drive ILC2 activation, shape an immunosuppressive microenvironment during intestinal tumorigenesis dominated by regulatory T cells (Tregs) (135). A possible mechanisms by which ILC2s might shape the Treg phenotype is through production of AREG, an EGF-like growth factor that enhances regulatory T cell functions (136), or via the production of Arginase 1 (Arg1), which inhibits T cell activation (137).
Upon activation, both ILC2s and ILC3s were able to upregulate the expression of MHCII molecules (138–142). In the case of ILC3s, this was accompanied by high levels of costimulatory molecules and the capacity to process antigens, thus promoting in vitro CD4 T cell priming (141). Whether this priming can also take place in the tumor setting is currently now known. Interestingly enough, the population of NCR+ILC3s described by Carrega et al. in human tumors was found to be located at the edge of tertiary lymphoid structures, an ectopic hub for acquired immune responses (63). Not only T cells, but also B cell responses can be regulated by ILCs. ILC2s, for instance, can modulate B cell function and antibody production through the production of IL-5 and expression of CD40 ligands (143, 144). Also, a population of splenic RORγt+ ILC3s located in the marginal zone was shown to help B cells for antibody production (145).
Collectively, the above-mentioned studies demonstrate that ILCs are poised to interact with other immune cells within the TME, and thereby modulate both innate and adaptive immune responses against tumors (summarized in Figure 1).
Manipulating ILC responses has emerged as an attractive therapeutic strategy against cancer. In principle, NK cells are the best-suited members of the ILC family to exploit for therapeutic purposes, due to their indisputable cytotoxic properties. In the past years, however, other ILC subsets endowed with either pro- or anti-tumor activity have gained increasing attention as important modulators of the TME. Irrespective of the subset, ILC therapies face a major obstacle, namely the poor capacity to infiltrate and to survive in the hostile tumor bed (50, 51). To date, we still miss a complete picture of the mechanisms regulating ILC migration into tumors. This would be of high interest not only to enhance the number of endogenous ILCs that reach the tumor, but also to increase the efficacy of cell transfer therapies. The latter includes, for instance, the utilization of NK cells engineered to express chimeric antigen receptor (CAR) specific for tumor antigens, which arises as a safe off-the-shelf therapy against refractory malignancies (146).
Once ILCs reach the tumor site, they encounter a hostile microenvironment, which imposes several limitations to dampen the activity of ILCs. Although tumor cells are the key drivers of NK cell dysfunction, other immunosuppressive cells populating the TME can significantly contribute to this process. The TME is characterized by a high degree of intra- and inter-tumor heterogeneity, which challenges the identification of targetable factors aimed at restoring tumor surveillance by ILCs. Nevertheless, a better understanding of the mechanisms that ILCs utilize to communicate with the TME will be key to effectively manipulate these cells for site-specific anticancer therapies. This can be achieved using multi-omics approaches that allow for the integration of data from diverse platforms, including single-cell transcriptomics, cytometry by time-of-flight (CyTOF) or multiplexed tissue imaging. A detailed and personalized multi-omics profile of the TME will be crucial for the design of novel approaches for cancer immunotherapy in the era of precision medicine.
It is important to point out that most of the studies directed to investigate ILCs-TME interactions relies on the use of non-specific strategies to deplete ILC subsets (e.g., blocking antibodies against asialo GM1, CD25, or CD90.2). Although this is being gradually substituted by genetic tools that allow for selective ablation and mapping of ILCs (147), the high plasticity observed within the different ILC family members complicates our ability to track and target these cells in the TME. The matter is further complicated for ILC1s, due to the lack of ILC1-knockout mice or antibodies that specifically deplete this ILC population. This calls for caution when interpreting the effects of the ILC1 population on both physiological and pathological conditions.
Clearly, as we only now start to understand the complex biology of the ILC family members, it is time to study the power of these cells not only from the direct effects they exert on cancer cells, but also from their ability to communicate with the TME. This will provide valuable insights into how to effectively manipulate ILCs for immune-mediated anticancer therapies.
All authors participated in the intellectual conception of the review, drafts, and final approval of the manuscript.
Work in the authors' lab was supported by the UBS Promedica Stiftung, the Swiss Cancer League (KFS-4431-02-2018) (ST), the Research Talent Development Fund from the University of Zurich and a PRIMA grant from the Swiss National Science Foundation (PR00P3_179775) (ST).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Klose CSN, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol. (2016) 17:765–74. doi: 10.1038/ni.3489
2. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Leading edge review innate lymphoid cells: 10 years on. Cell. (2018) 174:1054–66. doi: 10.1016/j.cell.2018.07.017
3. Herberman RB, Nunn ME, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic acid allogeneic tumors. I Distribution of reactivity and specificity. Int J Cancer. (1975) 16:216–29. doi: 10.1002/ijc.2910160204
4. Goh W, Huntington ND. Regulation of murine natural killer cell development. Front Immunol. (2017) 8:130. doi: 10.3389/fimmu.2017.00130
5. Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, et al. The transcription factors t-bet and eomes control key checkpoints of natural killer cell maturation. Immunity. (2012) 36:55–67. doi: 10.1016/j.immuni.2011.11.016
6. Abel AM, Yang C, Thakar MS, Malarkannan S. Natural killer cells: development, maturation, and clinical utilization. Front Immunol. (2018) 9:1869. doi: 10.3389/fimmu.2018.01869
7. Robinette ML, Fuchs A, Cortez VS, Lee JS, Wang Y, Durum SK, et al. Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat Immunol. (2015) 16:306–17. doi: 10.1038/ni.3094
8. Bernink JH, Mjösberg J, Spits H. Human ILC1: to be or not to be. Immunity. (2017) 46:756–7. doi: 10.1016/j.immuni.2017.05.001
9. Fuchs A. ILC1s in tissue inflammation and infection. Front Immunol. (2016) 7:104. doi: 10.3389/fimmu.2016.00104
10. Peng H, Jiang X, Chen Y, Sojka DK, Wei H, Gao X, et al. Liver-resident NK cells confer adaptive immunity in skin-contact inflammation. J Clin Invest. (2013) 123:1444–56. doi: 10.1172/JCI66381
11. Spits H, Bernink JH, Lanier L. NK cells and type 1 innate lymphoid cells: partners in host defense. Nat Immunol. (2016) 17:758–64. doi: 10.1038/ni.3482
12. Scanlon ST, McKenzie ANJ. Type 2 innate lymphoid cells: new players in asthma and allergy. Curr Opin Immunol. (2012) 24:707–12. doi: 10.1016/j.coi.2012.08.009
13. Yagi R, Zhong C, Northrup DL, Yu F, Bouladoux N, Spencer S, et al. The transcription factor GATA3 is critical for the development of all IL-7Rα-expressing innate lymphoid cells. Immunity. (2014) 40:378–88. doi: 10.1016/j.immuni.2014.01.012
14. Bouchery T, Le Gros G, Harris N. ILC2S—trailblazers in the host response against intestinal helminths. Front Immunol. (2019) 10:623. doi: 10.3389/fimmu.2019.00623
15. Kim HY, Chang Y-J, Subramanian S, Lee H-H, Albacker LA, Matangkasombut P, et al. Innate lymphoid cells responding to IL-33 mediate airway hyperreactivity independently of adaptive immunity. J Allergy Clin Immunol. (2012) 129:216–7.e1-6. doi: 10.1016/j.jaci.2011.10.036
16. von Moltke J, Ji M, Liang H-E, Locksley RM. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature. (2016) 529:221–5. doi: 10.1038/nature16161
17. Montaldo E, Juelke K, Romagnani C. Group 3 innate lymphoid cells (ILC3s): origin, differentiation, and plasticity in humans and mice. Eur J Immunol. (2015) 45:2171–82. doi: 10.1002/eji.201545598
18. Sanos SL, Bui VL, Mortha A, Oberle K, Heners C, Johner C, et al. RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nat Immunol. (2009) 10:83–91. doi: 10.1038/ni.1684
19. Melo-Gonzalez F, Hepworth MR. Functional and phenotypic heterogeneity of group 3 innate lymphoid cells. Immunology. (2017) 150:265–75. doi: 10.1111/imm.12697
20. Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. (2010) 464:1371–5. doi: 10.1038/nature08949
21. Villanova F, Flutter B, Tosi I, Grys K, Sreeneebus H, Perera GK, et al. Characterization of innate lymphoid cells in human skin and blood demonstrates increase of NKp44+ ILC3 in psoriasis. J Invest Dermatol. (2014) 134:984–91. doi: 10.1038/jid.2013.477
22. Takatori H, Kanno Y, Watford WT, Tato CM, Weiss G, Ivanov II, et al. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J Exp Med. (2009) 206:35–41. doi: 10.1084/jem.20072713
23. van de Pavert SA, Vivier E. Differentiation and function of group 3 innate lymphoid cells, from embryo to adult. Int Immunol. (2016) 28:35–42. doi: 10.1093/intimm/dxv052
24. De Togni P, Goellner J, Ruddle NH, Streeter PR, Fick A, Mariathasan S, et al. Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science. (1994) 264:703–7. doi: 10.1126/science.8171322
25. Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med. (2000) 6:443–6. doi: 10.1038/74704
26. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell aloreactivity in mismatched hematopoietic transplants. Science. (2002) 295:2097–100. doi: 10.1126/science.1068440
27. Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. (2003) 21:3940–7. doi: 10.1200/JCO.2003.05.013
28. Ohs I, van den Broek M, Nussbaum K, Münz C, Arnold SJ, Quezada SA, et al. Interleukin-12 bypasses common gamma-chain signalling in emergency natural killer cell lymphopoiesis. Nat Commun. (2016) 7:13708. doi: 10.1038/ncomms13708
29. Ohs I, Ducimetière L, Marinho J, Kulig P, Becher B, Tugues S. Restoration of natural killer cell antimetastatic activity by IL12 and checkpoint blockade. Cancer Res. (2017) 77:7059–71. doi: 10.1158/0008-5472.CAN-17-1032
30. Krasnova Y, Putz EM, Smyth MJ, Souza-Fonseca-Guimaraes F. Bench to bedside: NK cells and control of metastasis. Clin Immunol. (2017) 177:50–9. doi: 10.1016/j.clim.2015.10.001
31. Nussbaum K, Burkhard SH, Ohs I, Mair F, Klose CSN, Arnold SJ, et al. Tissue microenvironment dictates the fate and tumor-suppressive function of type 3 ILCs. J Exp Med. (2017) 214:2331–47. doi: 10.1084/jem.20162031
32. Ikutani M, Yanagibashi T, Ogasawara M, Tsuneyama K, Yamamoto S, Hattori Y, et al. Identification of innate IL-5–producing cells and their role in lung eosinophil regulation and antitumor immunity. J Immunol. (2012) 188:703–13. doi: 10.4049/jimmunol.1101270
33. Jovanovic IP, Pejnovic NN, Radosavljevic GD, Pantic JM, Milovanovic MZ, Arsenijevic NN, et al. Interleukin-33/ST2 axis promotes breast cancer growth and metastases by facilitating intratumoral accumulation of immunosuppressive and innate lymphoid cells. Int J Cancer. (2014) 134:1669–82. doi: 10.1002/ijc.28481
34. Trabanelli S, Chevalier MF, Martinez-Usatorre A, Gomez-Cadena A, Salomé B, Lecciso M, et al. Tumour-derived PGD2 and NKp30-B7H6 engagement drives an immunosuppressive ILC2-MDSC axis. Nat Commun. (2017) 8:593. doi: 10.1038/s41467-017-00678-2
35. Kirchberger S, Royston DJ, Boulard O, Thornton E, Franchini F, Szabady RL, et al. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J Exp Med. (2013) 210:917–31. doi: 10.1084/jem.20122308
36. Chan IH, Jain R, Tessmer MS, Gorman D, Mangadu R, Sathe M, et al. Interleukin-23 is sufficient to induce rapid de novo gut tumorigenesis, independent of carcinogens, through activation of innate lymphoid cells. Mucosal Immunol. (2014) 7:842–56. doi: 10.1038/mi.2013.101
37. Irshad S, Flores-Borja F, Lawler K, Monypenny J, Evans R, Male V, et al. RORγt+ innate lymphoid cells promote lymph node metastasis of breast cancers. Cancer Res. (2017) 77:1083–96. doi: 10.1158/0008-5472.CAN-16-0598
38. Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science. (2015) 350:981–5. doi: 10.1126/science.aac9593
39. Moro K, Kabata H, Tanabe M, Koga S, Takeno N, Mochizuki M, et al. Interferon and IL-27 antagonize the function of group 2 innate lymphoid cells and type 2 innate immune responses. Nat Immunol. (2016) 17:76–86. doi: 10.1038/ni.3309
40. Schneider C, Lee J, Koga S, Ricardo-Gonzalez RR, Nussbaum JC, Smith LK, et al. Tissue-resident group 2 innate lymphoid cells differentiate by layered ontogeny and in situ perinatal priming. Immunity. (2019) 50:1425–38.e5. doi: 10.1016/j.immuni.2019.04.019
41. Dutton EE, Gajdasik DW, Willis C, Fiancette R, Bishop EL, Camelo A, et al. Peripheral lymph nodes contain migratory and resident innate lymphoid cell populations. Sci Immunol. (2019) 4:eaau8082. doi: 10.1126/sciimmunol.aau8082
42. Lim AI, Li Y, Lopez-Lastra S, Amit I, Yssel H, Di Santo JP. Systemic human ILC precursors provide a substrate for tissue ILC differentiation. Cell. (2017) 168:1086–100.e10. doi: 10.1016/j.cell.2017.02.021
43. Freud AG, Becknell B, Roychowdhury S, Mao HC, Ferketich AK, Nuovo GJ, et al. A human CD34(+) subset resides in lymph nodes and differentiates into CD56bright natural killer cells. Immunity. (2005) 22:295–304. doi: 10.1016/j.immuni.2005.01.013
44. Kim CH, Hashimoto-Hill S, Kim M. Migration and tissue tropism of innate lymphoid cells. Trends Immunol. (2016) 23:9–17. doi: 10.1016/j.it.2015.11.003
45. Satoh-Takayama N, Serafini N, Verrier T, Rekiki A, Renauld JC, Frankel G, et al. The chemokine receptor CXCR6 controls the functional topography of interleukin-22 producing intestinal innate lymphoid cells. Immunity. (2014) 41:776–88. doi: 10.1016/j.immuni.2014.10.007
46. Paust S, Gill HS, Wang BZ, Flynn MP, Moseman EA, Senman B, et al. Critical role for the chemokine receptor CXCR6 in NK cell-mediated antigen-specific memory of haptens and viruses. Nat Immunol. (2010) 11:1127–35. doi: 10.1038/ni.1953
47. Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of il-12- and il-15-responsive ifn-γ-producing cells. Immunity. (2013) 38:769–81. doi: 10.1016/j.immuni.2013.02.010
48. Simoni Y, Fehlings M, Kløverpris HN, McGovern N, Koo SL, Loh CY, et al. Human innate lymphoid cell subsets possess tissue-type based heterogeneity in phenotype and frequency. Immunity. (2017) 46:148–61. doi: 10.1016/j.immuni.2016.11.005
49. Salimi M, Wang R, Yao X, Li X, Wang X, Hu Y, et al. Activated innate lymphoid cell populations accumulate in human tumour tissues. BMC Cancer. (2018) 18:341. doi: 10.1186/s12885-018-4262-4
50. Halama N, Braun M, Kahlert C, Spille A, Quack C, Rahbari N, et al. Natural killer cells are scarce in colorectal carcinoma tissue despite high levels of chemokines and cytokines. Clin Cancer Res. (2011) 17:678–89. doi: 10.1158/1078-0432.CCR-10-2173
51. Castriconi R, Carrega P, Dondero A, Bellora F, Casu B, Regis S, et al. Molecular mechanisms directing migration and retention of natural killer cells in human tissues. Front Immunol. (2018) 9:2324. doi: 10.3389/fimmu.2018.02324
52. Schleypen JS, Von Geldern M, Weiss EH, Kotzias N, Rohrmann K, Schendel DJ, et al. Renal cell carcinoma-infiltrating natural killer cells express differential repertoires of activating and inhibitory receptors and are inhibited by specific HLA class I allotypes. Int J cancer. (2003) 106:905–12. doi: 10.1002/ijc.11321
53. Eckl J, Buchner A, Prinz PU, Riesenberg R, Siegert SI, Kammerer R, et al. Transcript signature predicts tissue NK cell content and defines renal cell carcinoma subgroups independent of TNM staging. J Mol Med. (2012) 90:55–66. doi: 10.1007/s00109-011-0806-7
54. Wendel M, Galani IE, Suri-Payer E, Cerwenka A. Natural killer cell accumulation in tumors is dependent on IFN-gamma and CXCR3 ligands. Cancer Res. (2008) 68:8437–45. doi: 10.1158/0008-5472.CAN-08-1440
55. Habif G, Crinier A, André P, Vivier E, Narni-Mancinelli E. Targeting natural killer cells in solid tumors. Cell Mol Immunol. (2019) 16:415–22. doi: 10.1038/s41423-019-0224-2
56. Ali TH, Pisanti S, Ciaglia E, Mortarini R, Anichini A, Garofalo C, et al. Enrichment of CD56(dim)KIR + CD57 + highly cytotoxic NK cells in tumour-infiltrated lymph nodes of melanoma patients. Nat Commun. (2014) 5:5639. doi: 10.1038/ncomms6639
57. Messaoudene M, Fregni G, Fourmentraux-Neves E, Chanal J, Maubec E, Mazouz-Dorval S, et al. Mature cytotoxic CD56bright/CD16+ natural killer cells can infiltrate lymph nodes adjacent to metastatic melanoma. Cancer Res. (2014) 74:81–92. doi: 10.1158/0008-5472.CAN-13-1303
58. Castriconi R, Dondero A, Bellora F, Moretta L, Castellano A, Locatelli F, et al. Neuroblastoma-derived TGF-β1 modulates the chemokine receptor repertoire of human resting NK cells. J Immunol. (2013) 190:5321–8. doi: 10.4049/jimmunol.1202693
59. Loyon R, Jary M, Salomé B, Gomez-Cadena A, Galaine J, Kroemer M, et al. Peripheral innate lymphoid cells are increased in first line metastatic colorectal carcinoma patients: a negative correlation with Th1 immune responses. Front Immunol. (2019) 10:2121. doi: 10.3389/fimmu.2019.02121
60. Bie Q, Zhang P, Su Z, Zheng D, Ying X, Wu Y, et al. Polarization of ILC2s in peripheral blood might contribute to immunosuppressive microenvironment in patients with gastric cancer. J Immunol Res. (2014) 2014:923135. doi: 10.1155/2014/923135
61. De Weerdt I, Van Hoeven V, Marius Munneke J, Endstra S, Hofland T, Hazenberg MD, et al. Innate lymphoid cells are expanded and functionally altered in chronic lymphocytic leukemia. Haematologica. (2016) 101:e461–4. doi: 10.3324/haematol.2016.144725
62. Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science. (2010) 328:749–52. doi: 10.1126/science.1185837
63. Carrega P, Loiacono F, Di Carlo E, Scaramuccia A, Mora M, Conte R, et al. NCR(+)ILC3 concentrate in human lung cancer and associate with intratumoral lymphoid structures. Nat Commun. (2015) 6:8280. doi: 10.1038/ncomms9280
64. Dadi S, Chhangawala S, Whitlock BM, Franklin RA, Luo CT, Oh SA, et al. Cancer immunosurveillance by tissue-resident innate lymphoid cells and innate-like T cells. Cell. (2016) 164:365–77. doi: 10.1016/j.cell.2016.01.002
65. Martinet L, Smyth MJ. Balancing natural killer cell activation through paired receptors. Nat Rev Immunol. (2015) 15:243–54. doi: 10.1038/nri3799
66. Smyth MJ, Cretney E, Kelly JM, Westwood JA, Street SEA, Yagita H, et al. Activation of NK cell cytotoxicity. Mol Immunol. (2005) 42:501–10. doi: 10.1016/j.molimm.2004.07.034
67. Prager I, Liesche C, van Ooijen H, Urlaub D, Verron Q, Sandström N, et al. NK cells switch from granzyme B to death receptor-mediated cytotoxicity during serial killing. J Exp Med. (2019) 216:2113–27. doi: 10.1084/jem.20181454
69. Schmiedel D, Mandelboim O. NKG2D Ligands-critical targets for cancer immune escape and therapy. Front Immunol. (2018) 9:2040. doi: 10.3389/fimmu.2018.02040
70. Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity. (2008) 28:571–80. doi: 10.1016/j.immuni.2008.02.016
71. Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature(2002) 419:734–8. doi: 10.1038/nature01112
72. Salih HR, Rammensee H-G, Steinle A. Cutting edge: down-regulation of MICA on human tumors by proteolytic shedding. J Immunol. (2002) 169:4098–102. doi: 10.4049/jimmunol.169.8.4098
73. Liu G, Lu S, Wang X, Page ST, Higano CS, Plymate SR, et al. Perturbation of NK cell peripheral homeostasis accelerates prostate carcinoma metastasis. J Clin Invest. (2013) 123:4410–22. doi: 10.1172/JCI69369
74. Deng W, Gowen BG, Zhang L, Wang L, Lau S, Iannello A, et al. Antitumor immunity. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science. (2015) 348:136–9. doi: 10.1126/science.1258867
75. Coudert JD, Scarpellino L, Gros F, Vivier E, Held W. Sustained NKG2D engagement induces cross-tolerance of multiple distinct NK cell activation pathways. Blood. (2008) 111:3571–8. doi: 10.1182/blood-2007-07-100057
76. Crane CA, Austgen K, Haberthur K, Hofmann C, Moyes KW, Avanesyan L, et al. Immune evasion mediated by tumor-derived lactate dehydrogenase induction of NKG2D ligands on myeloid cells in glioblastoma patients. Proc Natl Acad Sci USA. (2014) 111:12823–8. doi: 10.1073/pnas.1413933111
77. Thompson TW, Kim AB, Li PJ, Wang J, Jackson BT, Huang KTH, et al. Endothelial cells express NKG2D ligands and desensitize antitumor NK responses. Elife. (2017) 6:e30881. doi: 10.7554/eLife.30881
78. KloseCSN, Flach M, Möhle L, Rogell L, Hoyler T, Ebert K, et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell. (2014) 157:340–56. doi: 10.1016/j.cell.2014.03.030
79. Holt D, Ma X, Kundu N, Fulton A. Prostaglandin E(2) (PGE (2)) suppresses natural killer cell function primarily through the PGE(2) receptor EP4. Cancer Immunol Immunother. (2011) 60:1577–86. doi: 10.1007/s00262-011-1064-9
80. Viel S, Marçais A, Guimaraes FSF, Loftus R, Rabilloud J, Grau M, et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci Signal. (2016) 9:ra19. doi: 10.1126/scisignal.aad1884
81. Castriconi R, Cantoni C, Chiesa M, Della Vitale M, Marcenaro E, Conte R, et al. Transforming growth factor β1 inhibits expression of NKP30 and NKG2d receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci USA. (2003) 100:4120–5. doi: 10.1073/pnas.0730640100
82. Ghiringhelli F, Ménard C, Terme M, Flament C, Taieb J, Chaput N, et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J Exp Med. (2005) 202:1075–85. doi: 10.1084/jem.20051511
83. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-β1. J Immunol. (2009) 182:240–9. doi: 10.4049/jimmunol.182.1.240
84. Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat Immunol. (2017) 18:1004–15. doi: 10.1038/ni.3800
85. Riggan L, Freud AG, O'Sullivan TE. True detective: unraveling group 1 innate lymphocyte heterogeneity. Trends Immunol. (2019) 51:609–24. doi: 10.1016/j.it.2019.08.005
86. Constantinides MG, McDonald BD, Verhoef PA, Bendelac A. A committed precursor to innate lymphoid cells. Nature. (2014) 508:397–401. doi: 10.1038/nature13047
87. Cortez VS, Cervantes-Barragan L, Robinette ML, Bando JK, Wang Y, Geiger TL, et al. Transforming growth factor-β signaling guides the differentiation of innate lymphoid cells in salivary glands. Immunity. (2016) 44:1127–39. doi: 10.1016/j.immuni.2016.03.007
88. Keskin DB, Allan DSJ, Rybalov B, Andzelm MM, Stern JNH, Kopcow HD, et al. TGFβ promotes conversion of CD16+ peripheral blood NK cells into CD16- NK cells with similarities to decidual NK cells. Proc Natl Acad Sci USA. (2007) 104:3378–83. doi: 10.1073/pnas.0611098104
89. Bal SM, Bernink JH, Nagasawa M, Groot J, Shikhagaie MM, Golebski K, et al. IL-1β, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat Immunol. (2016) 17:636–45. doi: 10.1038/ni.3444
90. Ohne Y, Silver JS, Thompson-Snipes LA, Collet MA, Blanck JP, Cantarel BL, et al. IL-1 is a critical regulator of group 2 innate lymphoid cell function and plasticity. Nat Immunol. (2016) 17:646–55. doi: 10.1038/ni.3447
91. Bernink JH, Peters CP, Munneke M, Te Velde AA, Meijer SL, Weijer K, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol. (2013) 14:221–9. doi: 10.1038/ni.2534
92. Bernink JH, Krabbendam L, Germar K, de Jong E, Gronke K, Kofoed-Nielsen M, et al. Interleukin-12 and−23 control plasticity Of Cd127+ group 1 and group 3 innate lymphoid cells in the intestinal lamina propria. Immunity. (2015) 43:146–60. doi: 10.1016/j.immuni.2015.06.019
93. Raykova A, Carrega P, Lehmann FM, Ivanek R, Landtwing V, Quast I, et al. Interleukins 12 and 15 induce cytotoxicity and early NK-cell differentiation in type 3 innate lymphoid cells. Blood Adv. (2017) 1:2679–91. doi: 10.1182/bloodadvances.2017008839
94. Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med. (2011) 17:1359–70. doi: 10.1038/nm.2537
95. Bruno A, Pagani A, Pulze L, Albini A, Dallaglio K, Noonan M, et al. Orchestration of angiogenesis by immune cells. Front Oncol. (2014) 21:309–22. doi: 10.3389/fonc.2014.00131
96. Bruno A, Focaccetti C, Pagani A, Imperatori AS, Spagnoletti M, Rotolo N, et al. The proangiogenic phenotype of natural killer cells in patients with non-small cell lung cancer. Neoplasia. (2013) 15:133–42. doi: 10.1593/neo.121758
97. Levi I, Amsalem H, Nissan A, Darash-Yahana M, Peretz T, Mandelboim O, et al. Characterization of tumor infiltrating natural killer cell subset. Oncotarget. (2015) 6:13835–43. doi: 10.18632/oncotarget.3453
98. Hanna J, Hamani Y, Prus D, Daniel L. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat Med. (2006) 12:1065–74. doi: 10.1038/nm1452
99. Gotthardt D, Putz EM, Grundschober E, Prchal-Murphy M, Straka E, Kudweis P, et al. STAT5 is a key regulator in NK cells and acts as a molecular switch from tumor surveillance to tumor promotion. Cancer Discov. (2016) 6:414–29. doi: 10.1158/2159-8290.CD-15-0732
100. Krzywinska E, Kantari-Mimoun C, Kerdiles Y, Sobecki M, Isagawa T, Gotthardt D, et al. Loss of HIF-1α in natural killer cells inhibits tumour growth by stimulating non-productive angiogenesis. Nat Commun. (2017) 8:1597. doi: 10.1038/s41467-017-01599-w
101. Sgadari C, Angiolillo AL, Tosato G. Inhibition of angiogenesis by interleukin-12 is mediated by the interferon-inducible protein 10. Blood. (1996) 87:3877–82. doi: 10.1182/blood.V87.9.3877.bloodjournal8793877
102. Eisenring M, Vom Berg J, Kristiansen G, Saller E, Becher B. IL-12 initiates tumor rejection via lymphoid tissue-inducer cells bearing the natural cytotoxicity receptor NKp4. Nat Immunol. (2010) 11:1030–8. doi: 10.1038/ni.1947
103. Kulig P, Burkhard S, Mikita-Geoffroy J, Croxford AL, Hövelmeyer N, Gyülvészi G, et al. IL17A-mediated endothelial breach promotes metastasis formation. Cancer Immunol Res. (2016) 4:26–32. doi: 10.1158/2326-6066.CIR-15-0154
104. Numasaki M, Fukushi J, Ono M, Narula SK, Zavodny PJ, Kudo T, et al. Interleukin-17 promotes angiogenesis and tumor growth. Blood. (2003) 101:2620–7. doi: 10.1182/blood-2002-05-1461
105. Murugaiyan G, Mittal A, Lopez-Diego R, Maier LM, Anderson DE, Weiner HL. IL-27 is a key regulator of IL-10 and IL-17 production by human CD4 + T cells. J Immunol. (2009) 183:2435–43. doi: 10.4049/jimmunol.0900568
106. Dieterich LC, Detmar M. Tumor lymphangiogenesis and new drug development. Adv Drug Deliv Rev. (2016) 99(Pt B):148–60. doi: 10.1016/j.addr.2015.12.011
107. Robering JW, Weigand A, Pfuhlmann R, Horch RE, Beier JP, Boos AM. Mesenchymal stem cells promote lymphangiogenic properties of lymphatic endothelial cells. J Cell Mol Med. (2018) 22:3740–50. doi: 10.1111/jcmm.13590
108. Pickup MW, Mouw JK, Weaver VM. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. (2014) 15:1243–53. doi: 10.15252/embr.201439246
109. Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. (2012) 196:395–406. doi: 10.1083/jcb.201102147
110. Glasner A, Levi A, Enk J, Isaacson B, Viukov S, Orlanski S, et al. NKp46 receptor-mediated interferon-γ production by natural killer cells increases fibronectin 1 to alter tumor architecture and control metastasis. Immunity. (2018) 48:107–19.e4. doi: 10.1016/j.immuni.2017.12.007
111. Putz EM, Mayfosh AJ, Kos K, Barkauskas DS, Nakamura K, Town L, et al. NK cell heparanase controls tumor invasion and immune surveillance. J Clin Invest. (2017) 127:2777–88. doi: 10.1172/JCI92958
112. Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, et al. A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nat Med. (2018) 24:1178–91. doi: 10.1038/s41591-018-0085-8
113. Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. (2018) 172:1022–37.e14. doi: 10.1016/j.cell.2018.01.004
114. Ferlazzo G, Pack M, Thomas D, Paludan C, Schmid D, Strowig T, et al. Distinct roles of IL-12 and IL-15 in human natural killer cell activation by dendritic cells from secondary lymphoid organs. Proc Natl Acad Sci USA. (2004) 101:16606–11. doi: 10.1073/pnas.0407522101
115. Gerosa F, Gobbi A, Zorzi P, Burg S, Briere F, Carra G, et al. The reciprocal interaction of NK cells with plasmacytoid or myeloid dendritic cells profoundly affects innate resistance functions. J Immunol. (2005) 174:727–34. doi: 10.4049/jimmunol.174.2.727
116. Vitale M, Della Chiesa M, Carlomagno S, Pende D, Aricò M, Moretta L, et al. NK-dependent DC maturation is mediated by TNFα and IFNγ released upon engagement of the NKp30 triggering receptor. Blood. (2005) 106:566–71. doi: 10.1182/blood-2004-10-4035
117. Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell. (2014) 26:638–52. doi: 10.1016/j.ccell.2014.09.007
118. Mocikat R, Braumüller H, Gumy A, Egeter O, Ziegler H, Reusch U, et al. Natural killer cells activated by MHC class I Low targets prime dendritic cells to induce protective CD8 T cell responses. Immunity. (2003) 19:561–9. doi: 10.1016/S1074-7613(03)00264-4
119. Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, et al. Expansion and activation of CD103+ dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity. (2016) 44:924–38. doi: 10.1016/j.immuni.2016.03.012
120. Iyori M, Zhang T, Pantel H, Gagne BA, Sentman CL. TRAIL/DR5 plays a critical role in NK cell-mediated negative regulation of dendritic cell cross-priming of T cells. J Immunol. (2011) 187:3087–95. doi: 10.4049/jimmunol.1003879
121. Saranchova I, Han J, Huang H, Fenninger F, Choi KB, Munro L, et al. Discovery of a metastatic immune escape mechanism initiated by the loss of expression of the tumour biomarker interleukin-33. Sci Rep. (2016) 6:30555. doi: 10.1038/srep30555
122. Vonarbourg C, Mortha A, Bui VL, Hernandez PP, Kiss EA, Hoyler T, et al. Regulated expression of nuclear receptor RORγt confers distinct functional fates to NK cell receptor-expressing RORγt+ innate lymphocytes. Immunity. (2010) 33:736–51. doi: 10.1016/j.immuni.2010.10.017
123. Ahern PP, Izcue A, Maloy KJ, Powrie F. The interleukin-23 axis in intestinal inflammation. Immunol Rev. (2008) 226:147–59. doi: 10.1111/j.1600-065X.2008.00705.x
124. Pearson C, Thornton EE, McKenzie B, Schaupp AL, Huskens N, Griseri T, et al. ILC3 GM-CSF production and mobilisation orchestrate acute intestinal inflammation. Elife. (2016) 5:e10066. doi: 10.7554/eLife.10066
125. Griseri T, Arnold IC, Pearson C, Krausgruber T, Schiering C, Franchini F, et al. Granulocyte macrophage colony-stimulating factor-activated eosinophils promote interleukin-23 driven chronic colitis. Immunity. (2015) 43:187–99. doi: 10.1016/j.immuni.2015.07.008
126. O'Connor W, Kamanaka M, Booth CJ, Town T, Nakae S, Iwakura Y, et al. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat Immunol. (2009) 10:603–9. doi: 10.1038/ni.1736
127. Maxwell JR, Zhang Y, Brown WA, Smith CL, Byrne FR, Fiorino M, et al. Differential roles for interleukin-23 and interleukin-17 in intestinal immunoregulation. Immunity. (2015) 43:739–50. doi: 10.1016/j.immuni.2015.08.019
128. Hernandez P, Gronke K, Diefenbach A. A catch-22: Interleukin-22 and cancer. Eur J Immunol. (2018) 48:15–31. doi: 10.1002/eji.201747183
129. Liu J, Duan Y, Cheng X, Chen X, Xie W, Long H, et al. IL-17 is associated with poor prognosis and promotes angiogenesis via stimulating VEGF production of cancer cells in colorectal carcinoma. Biochem Biophys Res Commun. (2011) 407:348–54. doi: 10.1016/j.bbrc.2011.03.021
130. Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, Bindea G, et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, Th2, Treg, Th17) in patients with colorectal cancer. Cancer Res. (2011) 71:1263–71. doi: 10.1158/0008-5472.CAN-10-2907
131. Martín-Fontecha A, Thomsen LL, Brett S, Gerard C, Lipp M, Lanzavecchia A, et al. Induced recruitment of NK cells to lymph nodes provides IFN-γ for TH1 priming. Nat Immunol. (2004) 5:1260-5. doi: 10.1038/ni1138
132. Paul S, Kulkarni N, Shilpi Lal G. Intratumoral natural killer cells show reduced effector and cytolytic properties and control the differentiation of effector Th1 cells. Oncoimmunology. (2016) 5:e1235106. doi: 10.1080/2162402X.2016.1235106
133. Schmidt L, Eskiocak B, Kohn R, Dang C, Joshi NS, DuPage M, et al. Enhanced adaptive immune responses in lung adenocarcinoma through natural killer cell stimulation. Proc Natl Acad Sci USA. (2019) 116:17460–9. doi: 10.1073/pnas.1904253116
134. Fan Z, Yu P, Wang Y, Wang Y, Fu ML, Liu W, et al. NK-cell activation by LIGHT triggers tumor-specific CD8 + T-cell immunity to reject established tumors. Blood. (2006) 107:1342–51. doi: 10.1182/blood-2005-08-3485
135. Pastille E, Wasmer MH, Adamczyk A, Vu VP, Mager LF, Phuong NNT, et al. The IL-33/ST2 pathway shapes the regulatory T cell phenotype to promote intestinal cancer. Mucosal Immunol. (2019) 12:990–1003. doi: 10.1038/s41385-019-0176-y
136. Zaiss DMW, Gause WC, Osborne LC, Artis D. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity. (2015) 42:216–26. doi: 10.1016/j.immuni.2015.01.020
137. Bando JK, Nussbaum JC, Liang H-E, Locksley RM. Type 2 innate lymphoid cells constitutively express arginase-I in the naïve and inflamed lung. J Leukoc Biol. (2013) 94:877–84. doi: 10.1189/jlb.0213084
138. Hepworth MR, Monticelli LA, Fung TC, Ziegler CGK, Grunberg S, Sinha R, et al. Innate lymphoid cells regulate CD4 + T-cell responses to intestinal commensal bacteria. Nature. (2013) 498:113–7. doi: 10.1038/nature12240
139. Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4+ T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity. (2014) 41:283–95. doi: 10.1016/j.immuni.2014.06.016
140. Rusakiewicz S, Perier A, Semeraro M, Pitt JM, von Strandmann EP, Reiners KS, et al. NKp30 isoforms and NKp30 ligands are predictive biomarkers of response to imatinib mesylate in metastatic GIST patients. Oncoimmunology. (2016) 6:e1137418. doi: 10.1080/2162402X.2015.1137418
141. Von Burg N, Chappaz S, Baerenwaldt A, Horvath E, Bose Dasgupta S, Ashok D, et al. Activated group 3 innate lymphoid cells promote T-cell-mediated immune responses. Proc Natl Acad Sci USA. (2014) 111:12835–40. doi: 10.1073/pnas.1406908111
142. Saranchova I, Han J, Zaman R, Arora H, Huang H, Fenninger F, et al. Type 2 innate lymphocytes actuate immunity against tumours and limit cancer metastasis. Sci Rep. (2018) 8:2924. doi: 10.1038/s41598-018-20608-6
143. Cruz-Migoni S, Caamaño J. Fat-associated lymphoid clusters in inflammation and immunity. Front Immunol. (2016) 7:612. doi: 10.3389/fimmu.2016.00612
144. Maggi L, Montaini G, Mazzoni A, Rossettini B, Capone M, Rossi MC, et al. Human circulating group 2 innate lymphoid cells can express CD154 and promote IgE production. J Allergy Clin Immunol. (2017) 139:964–76.e4. doi: 10.1016/j.jaci.2016.06.032
145. Magri G, Miyajima M, Bascones S, Mortha A, Puga I, Cassis L, et al. Innate lymphoid cells integrate stromal and immunological signals to enhance antibody production by splenic marginal zone B cells. Nat Immunol. (2014) 15:354–64. doi: 10.1038/ni.2830
146. Daher M, Rezvani K. Next generation natural killer cells for cancer immunotherapy: the promise of genetic engineering. Curr Opin Immunol. (2018) 51:146–53. doi: 10.1016/j.coi.2018.03.013
Keywords: innate lymphoid cells, tumor microenvironment, crosstalk, immune evasion, immune modulation
Citation: Ducimetière L, Vermeer M and Tugues S (2019) The Interplay Between Innate Lymphoid Cells and the Tumor Microenvironment. Front. Immunol. 10:2895. doi: 10.3389/fimmu.2019.02895
Received: 18 October 2019; Accepted: 26 November 2019;
Published: 13 December 2019.
Edited by:
Nicolas Jacquelot, Walter and Eliza Hall Institute of Medical Research, AustraliaReviewed by:
Aharon Freud, The Ohio State University, United StatesCopyright © 2019 Ducimetière, Vermeer and Tugues. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sonia Tugues, dHVndWVzQGltbXVub2xvZ3kudXpoLmNo
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.