 Olivia I. Coleman1
Olivia I. Coleman1 Dirk Haller1,2*
Dirk Haller1,2*- 1Chair of Nutrition and Immunology, Technical University of Munich, Munich, Germany
- 2ZIEL – Institute for Food & Health, Technical University of Munich, Munich, Germany
An imbalance in the correct protein folding milieu of the endoplasmic reticulum (ER) can cause ER stress, which leads to the activation of the unfolded protein response (UPR). The UPR constitutes a highly conserved and intricately regulated group of pathways that serve to restore ER homeostasis through adaptation or apoptosis. Numerous studies over the last decade have shown that the UPR plays a critical role in shaping immunity and inflammation, resulting in the recognition of the UPR as a key player in pathological processes including complex inflammatory, autoimmune and neoplastic diseases. The intestinal epithelium, with its many highly secretory cells, forms an important barrier and messenger between the luminal environment and the host immune system. It is not surprising, that numerous studies have associated ER stress and the UPR with intestinal diseases such as inflammatory bowel disease (IBD) and colorectal cancer (CRC). In this review, we discuss our current understanding of the roles of ER stress and the UPR in shaping immune responses and maintaining tissue homeostasis. Furthermore, the role played by the UPR in disease, with emphasis on IBD and CRC, is described here. As a key player in immunity and inflammation, the UPR has been increasingly recognized as an important pharmacological target in the development of therapeutic strategies for immune-mediated pathologies. We summarize available strategies targeting the UPR and their therapeutic implications. Understanding the balance between homeostasis and pathophysiology, as well as means of manipulating this balance, provides an important avenue for future research.
The Endoplasmic Reticulum and Its Intricately Regulated Unfolded Protein Response
In mammalian cells, the extensive tubular-reticular network known as the endoplasmic reticulum (ER) forms a crucial site for maintaining calcium homeostasis, cholesterol production and lipid synthesis, and most importantly acts as a gatekeeper for synthesizing and folding secreted and transmembrane proteins (1). ER protein-folding can be disrupted by envrionmental, physiological and pathological factors, resulting in ER stress. Changes in calcium homeostasis, an altered redox status, energy deficiency, lipid overload or the accumulation of unfolded or misfolded proteins are examples of conditions that can disrupt the ER protein-folding environment (2, 3). Furthermore, perturbations in membrane fluidity through cellular lipid composition can cause lipotoxic ER stress (4, 5). The correct folding and post-translational modifications of proteins are necessary in order to maintain proteostasis within a cell, making it essential for the ER to have a rigorous quality control system should the ER environment become compromised. To this end, ER-associated degradation (ERAD) removes and subsequently degrades unfolded or misfolded proteins (6), and the ER unfolded protein response (UPR) serves to restore normal functioning of the cell (adaptation) or gears toward cell death (apoptosis) in case of irreversible disruption (2).
The UPR forms a conserved group of intracellular signaling pathways that primarily aim to restore ER homeostasis in response to ER stress caused by the accumulation of unfolded or misfolded proteins (7). The UPR consist of three membrane-bound signal transducers, namely PKR-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1) and Activating transcription factor 6 (ATF6) (8, 9). Under conditions of homeostasis, the luminal domains of these three signal transducers are bound to the chaperone Glucose regulated protein 78 (Grp78; also known as BiP and HSPA5) (10). Upon ER stress, Grp78 is translocated to the unfolded or misfolded proteins in the ER and thereby allows activation of the UPR signal transducers and subsequent downstream signaling (2, 11). Dissociation of Grp78 from the type-I transmembrane protein PERK activates its oligomerization and autophosphorylation in the cytosolic kinase domain, to allow PERK to phosphorylate the alpha subunit of translation initiation factor 2 (eIF2α) to indirectly inactivate the latter and inhibit mRNA translation (2). This leads to the inhibition of global protein synthesis, but specifically favors translation of mRNAs with short open reading frames such asactivating transcription factor 4 (ATF4), whose translation is induced (12). IRE1 is also a type-I transmembrane protein, which oligomerizes and subsequently autophosphorylates in the kinase domain to trigger its ribonuclease activity. This leads to unconventional splicing of Xbox-binding protein 1 (Xbp1) that binds to the UPR element (UPRE) to induce the transcription of UPR-related genes (13, 14). Upon release from Grp78, the type-II transmembrane protein ATF6 is transported to the Golgi where it is proteolytically cleaved via the site 1 and site 2 proteases (S1P and S2P) (15–17). Subsequently, the amino-terminal of ATF6 (nATF6) is translocated to the nucleus to bind to the ER stress response element (ERSE) for the transcription of genes encoding XBP1, ERAD components, and ER chaperones (13, 14, 18). Mechanistically, and unlike PERK and IRE1α, ATF6 is not dimerized and may perhaps be less affected by ER membrane changes such as reduced membrane fluidity.
In addition to the UPR being a response system that restores proteostasis, it fulfills an important signaling function that plays an anticipatory role in cells that have a higher protein folding demand and require an increased protein-folding capacity, in the absence of ER stress (12). Triggers that can activate this signaling response of the UPR include differentiating cells, such as maturing immune cells (19, 20), and hormones, such as epidermal groth factor (EGF) and vascular endothelial growth factor (VEGF) (21, 22). Thus, while the primary purpose of the UPR is to enhance protein degradation, reduce protein synthesis, increase ER protein folding capacity and upregulate chaperones required for protein folding, it has become increasingly clear that the UPR plays a crucial role in tissue homeostasis. The context-dependent functionality of the UPR, which tailors the output according to the cellular stimulus, render it an important gatekeeper of cellular physiology.
ER stress occurs under both physiological and pathological conditions, and has been associated with immune and inflammatory diseases (23), viral infection (24), cardiovascular diseases (25), diabetes (26), cancer (27), cerebral ischemia (28), neurodegenerative diseases (29), as well as mental disorders (30, 31). What remains to be elucidated, however, is whether the UPR plays a causal role in these pathologies or merely presents a consequence of the respective disease. Experimental models of the UPR provide a useful tool for unraveling the timely and mechanistic involvement of the UPR in disease development of the host. Indeed, to date there are a number of UPR-related mouse models that display disease phenotypes and provide insights into the role of this complex signaling pathway in physiology and pathology. Table 1 provides an overview of UPR-related mouse models and their associated disease phenotypes. From this group of mouse models, with targeted UPR pathway proteins, it is evident that the UPR plays a critical role in a diverse range of pathologies. Findings from such mouse models will be discussed in more detail in the following sections. In light of the complexity of the canonical UPR and its regulation, many of the in vitro cell culture studies on ER stress are difficult to translate into the in vivo situation. UPR-related mouse models are therefore indispensable to gain mechanistic insights into the role of the UPR in human disease.
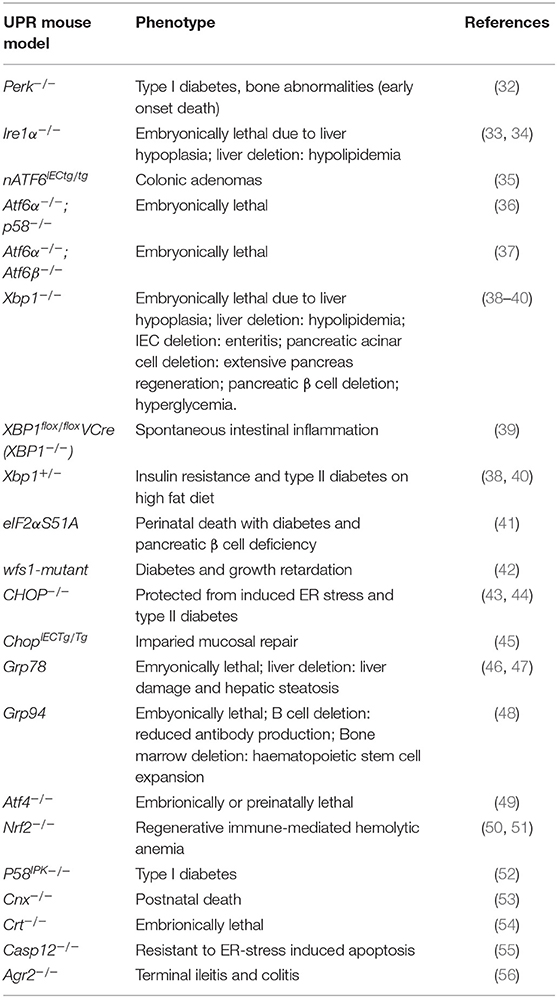
Table 1. UPR-related mouse models and their associated disease phenotypes.
UPR in Immune Cells and Immune Barrier Function
The UPR plays a critical role in the development of immune cells, with numerous studies highlighting its involvement in physiological immune processes (57). Discussed here, and summarized in Figure 1, is our understanding of the roles of ER stress and the UPR in immune responses that lead to immune activation, differentiation, and cytokine expression in immune cells. The UPR regulates cytokine production on multiple levels, extending from pattern recognition receptor (PRR) sensing to inflammatory signaling and cytokine transcription factor activation. UPR-PRR synergy that strengthens the immune response has been described by several groups and summarized in Claudio et al. (58) and Smith (59).
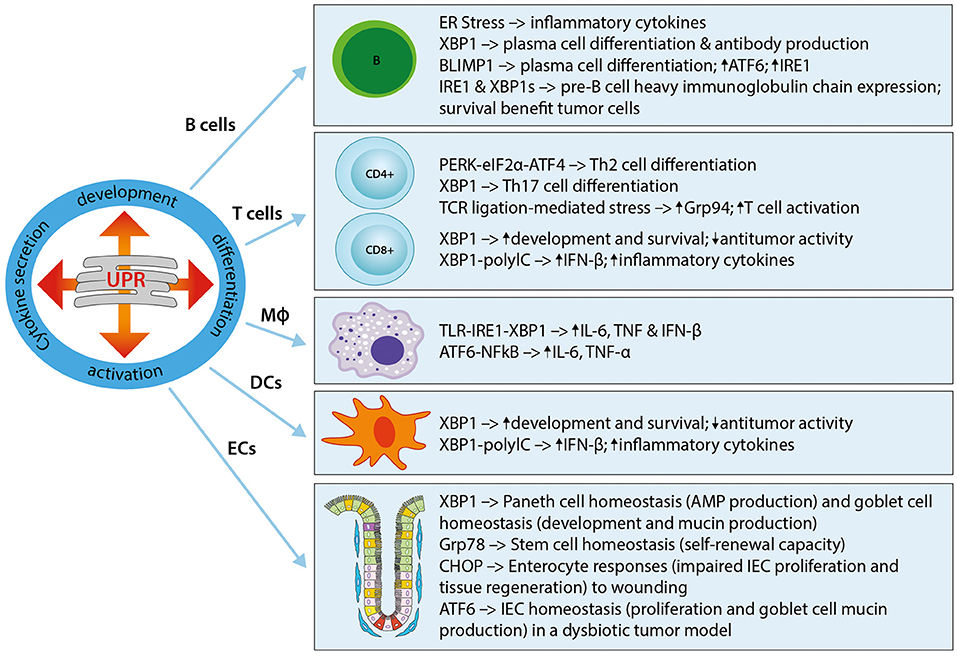
Figure 1. The UPR in immune cells and immune barrier function. The UPR plays a critical role in the development, differentiation, activation, and cytokine secretion of immune cells. Shown here are the effects of different UPR components on the main immune cell types, namely B cells, T cells (CD4+ and CD8+), macrophages (MΦ), and dendritic cells (DCs), as well as epithelial cells (ECs). Th2, T helper type 2; Th17, T helper type 17; Grp94, Glucose regulated protein 94; IL-6, Interleukin-6; IFN-β, Interferon-β; TNF, Tumor necrosis factor; TLR, Toll-like receptor; NFκB, Nuclear factor “kappa-light-chain-enhancer” of activated B-cells; ECs, epithelial cells; IEC, intestinal epithelial cell; AMP, antimicrobial peptide.
UPR in Immune Cells
An essential role for the UPR in B cells was first described in studies that showed UPR activation in the differentiation of B cells into plasma cells, and a requirement for the induction of Xbp1 for this process (38, 60–63). Further work revealed that the induction of XBP1is a differentiation-dependent event rather a response to increased immunoglobulin secretion (19, 20). XBP1 induces ER expansion in plasma cells, allowing for high immunoglobulin synthesis, and its deficiency abrogates immunoglobulin secretion by activated B cells through IRE1α hyperactivation (60, 62, 64, 65). Plasma cell differentiation is in part regulated by the transcription factor B lymphocyte-induced maturation protein 1 (Blimp-1) (62). Blimp-1 deficient B cells cannot activate transcription of plasma cell-related genes, including XBP1 (62). XBP1 is downstream of Blimp-1, as demonstrated in XBP1-deficient mice that resulted in normal Blimp-1 induction (62). Furthermore, Blimp-1 was shown to transcriptionally regulate ATF6 and IRE1 (66). XBP1 and also IRE1 are required during the pre-B cell stage during which immunoglobulin heavy chains are expressed for the first time, and XBP1 provides a survival benefit for tumor cells in pre-B acute lymphoblastic leukemia (33, 67).
In T cells, the UPR seems to play a role during cell differentiation. For example, the PERK-eIF2α-ATF4 axis has been implicated in Th2 cell differentiation, resulting in the upregulation of UPR genes (68). Similarly, XBP1 was shown to play a role in Th17 cell differentiation in response to inflammatory and autoimmune diseases (69, 70). Evidence for the important role played by the ER stress response in T cell activation was recently shown in a study where the ER molecular chaperone Grp94 was induced in CD4+ T cells following T cell receptor-ligation mediated ER stress (71). In turn, Grp94 deletion resulted in an activation defect. In CD8+ T cells, the IRE1α-XBP1 pathway activated upon acute infection was shown to be vital for effector T cell differentiation through increased expression of killer cell lectin-like receptor G1 (KLGR1) (72).
Both the development and the survival of antigen-presenting Dendritic cells (DCs) is driven by XBP1, with XBP1-deficiency resulting in reduced numbers of conventional and plasmacytoid DCs and increased apoptosis (73, 74). Interestingly, a recent study could also show a role for XBP1 in the suppression of antitumor immunity through the promotion of lipid accumulation and impaired antigen presentation (75). Further evidence for an important role of ER stress in DCs is shown by its ability to induce IFN-β production and IL-23 expression (74, 76). In DCs stimulated with the toll-like receptor (TLR) agonist polyinosinic:polycytidylic acid (PolyIC), silencing of XBP1 was shown to inhibit IFN-β production, whereas overexpression of XBP1 augmented inflammatory responses (74). TLR agonist stimulation of DCs under ER stress enhanced IL-23p19 expression, a target of the ER stress-induced transcription factor C/EBP homologous protein (CHOP), by stimulating the enhanced binding of CHOP to its promoter (76). In line with this, knockdown of CHOP reduced the expression of IL-23 in vitro (76). In phagocytic macrophages, the IRE1α-XBP1 ER stress axis is crucially involved in macrophage cytokine (IL-6, TNF, and IFN-β) responses to toll-like receptor (TLR) ligation in a pathway that involves TNF receptor-associated factor 6 (TRAF6) and the NAPDH oxidase-2 (NOX2) (77, 78). Furthermore, the IRE1α-XBP1 axis has also been implicated in the regulation of inflammatory cytokine (IL1-β) production via the activation of glycogen synthase kinase 3β (GSK3β) (79). A role for ATF6 in macrophages was demonstrated in a study of liver ischemia perfusion injury by Rao et al. in which prolonged ischemia activated the ATF6 arm of the UPR and subsequent pro-inflammatory cytokine production (TNF-α and IL-6) (80). It is important to note that the context-specific response and control of the individual UPR pathways is vital for the required immune response. For example, the survival of macrophages during an immune response is facilitated through the suppression of CHOP, downstream of ATF4 in the PERK pathway of the ER stress response (77, 81).
UPR in Immune Barrier Function
In addition to the traditionally classified immune cells described above, epithelial cells lining mucosal surfaces, such as intestinal, gastric, and pulmonary surfaces, are further regulators of innate and adaptive immune responses. As the largest barrier between the host and the external environment, the gastrointestinal tract, with its enteroendocrine, absorptive, Paneth cells, and goblet cells, is particularly dependent on correct cellular functioning to maintain a state of intestinal homeostasis. Mucus-producing goblet cells, immunoglobulin-, chemokine-, and cytokine-secreting absorptive enterocytes, as well as anti-microbial peptide-producing Paneth cells have been shown to be particularly dependent on the UPR. For example, specific deletion of XBP1 in intestinal epithelial cells (IECs) showed a decreased antimicrobial function in Paneth cells, with loss of their characteristic granules, a significant reduction in goblet cells, increased epithelial apoptosis and the development of intestinal enteritis via IRE1/XBP1 signaling, which was reversible under germ-free conditions (39, 82). Adolph et al. showed that the development of intestinal inflammation is promoted by stressed Paneth cells, as specific deletion of XBP1 in Paneth cells is sufficient to induce small intestinal enteritis (83). A study provided first evidence that intestinal ischemia/reperfusion induces UPR activation in the human small intestine, particularly in Paneth cells, and demonstrated subsequent induction of apoptosis in Paneth cells (84). Here, ER stress-induced Paneth cell apoptosis was shown to contribute to intesinal ischemia/reperfusion-induced bacterial translocation and systemic inflammation. With regard to epithelial stem cells, the UPR causes loss of self-renewal capacity in cells with ER stress (85, 86). Heijmans et al. showed that an activated UPR in crypt base columnar cells antagonizes stem cell properties and proliferation via stem cell-specific depletion of the ER chaperone Grp78 (87). More recently, they were able to show that heterozygosity of Grp78 in the intestinal epithelium compromises epithelial regeneration capacity and protects against adenoma formation (88). A similar mechanism to the UPR has been described in mitochondria and is termed the mitochondrial UPR (mtUPR) (89–91). Our lab showed that loss of the mitochondrial chaperone HSP60 activates the mtUPR resulting in mitochondrial dysfunction (92). In these mice, HSP60-deficiency causes a loss of stemness and cell proliferation in intestinal crypts. HSP60 deficiency in IECs triggered the paracrine release of Wnt-related signals associated with hyperproliferation of residual stem cells that escaped Hsp60 deletion, demonstrating a fundamental role of mitochondrial function in the control of intestinal stem cell homeostasis. Under conditions of chronic inflammation, where this homeostasis is constantly challenged, this mechanism may contribute to inflammation associated tumorigenesis. In a recent study using mice in which Apc (Adenomatous polyposis coli; the most frequent initial gene mutation in CRC) and the ER stress chaperone Grp78 were deleted in IECs, it could be shown that ER stress signaling results in a rapid loss of Apc mutated stem cells and self-renewal capacity through interfering with Wnt signaling (93). The ER UPR and its role in intestinal pathologies will be one focus of this review, and is discussed in detail in subsequent sections.
Taken together, it has become clear that ER stress and the UPR signaling pathways play a pivotal role in shaping immune cell development and responses in order to mount an adequate immune response. Furthermore, IEC secretory cell function and the UPR play an important role in the maintenance of homeostasis and the resolution of inflammatory conditions. It is therefore not surprising that the UPR with its associated inflammatory pathways is also a key player in pathologies, including complex inflammatory, autoimmune, and neoplastic diseases.
UPR and Intestinal Disease
ER stress and UPR activation critically impact the regulation of intestinal epithelial stem cell differentiation (87, 92), the development of chronic intestinal inflammation (39, 94, 95), and the pathogenesis of intestinal tumorigenesis (35). Understandably, a dysfunction of IECs, particularly highly secretory cells such as goblet cells, associates ER stress and the UPR with numerous gastrointestinal disorders such as inflammatory bowel disease (IBD), celiac disease, as well as cancer, including colorectal cancer (CRC) (96–98). With its multifaceted possibilities of physiological outcomes, understanding the role of the UPR in IBD and CRC will open up new avenues for treatments of these debilitating and life-threatening diseases.
UPR in IBD
IBD refers to a group of multifactorial, immunologically-mediated chronic inflammatory diseases, of which Crohn's disease (CD) and ulcerative colitis (UC) represent the two major forms of disease. IBD can be debilitating and may lead to life-threatening complications, and its incidence and prevalence are increasing worldwide. The onset of IBD is suggested to result from genetic susceptibility, immune dysregulation, the intestinal microbiota, and environmental factors such as diet (99–102). Genome wide association studies (GWAS) over the past decade have identified numerous susceptibility loci for CD and UC, the latest of which links 241 susceptibility loci to IBD (103). Many of these susceptibility loci encode proteins with important roles in proteostasis. Among the ER-relevant genes identified here are Orosmucoid-like 3 (ORMDL3) (104, 105), anterior gradient 2 (AGR2) (106), and XBP1 (39). ORMDL3 has long been recognized as a key UPR inducer by disturbing endoplasmic calcium homeostasis (107). ORMDL3 was also shown to selectively activate the ATF6 arm of the UPR in lung epithelia (108, 109). The precise mechanism by which genetic abnormalities in ORMDL3 contribute to IBD is to date not understood, but it was shown to protect against apoptosis (110). The protein disulfide isomerase AGR2 is highly expressed in secretory cells, and mice deficient in AGR2 develop terminal ileitis and colitis displaying Paneth cell hypertrophy and a loss of mucin-filled goblet cells along with UPR activation (Grp78 increase) (56, 111). Similar to AGR2 deficiency, XBP1 deletion in IECs particularly affects secretory cells, with a loss of Paneth cells and mucin-filled goblet cells (39). IEC-specific XBP1 deletion resulted in ER stress (IRE1-XBP1 axis), spontaneous inflammation and an increased susceptibility to dextran-sodium sulfate (DSS)-induced colitis (39). ATG16L1T300A is a major risk polymorphism in CD (112). Abnormalities in the secretory pathway of Paneth cells are a consequence of hypomorphic ATG16L1 (83, 113), and an IEC-specific deletion in mice (Atg16l1ΔIEC) demonstrates that the observed spontaneous transmural ileitis is driven by IRE1α, which accumulates in Paneth cells (114).
UC is characterized by depleted goblet cells and a reduced mucus layer (115, 116). Further evidence for a role of the UPR in goblet cells was provided by two strains of mice with distinct, non-complementing missense mutations in the major secreted intestinal mucin Muc2 (Winnie and Eeyore mice), which develop an UC-like phenotype (117). Goblet cells in these mice display evidence of ER stress and activation of the UPR, associating mucin misfolding and ER stress with the initiation of colitis in mice. Cytokines can either exacerbate or suppress ER stress and protein production in secretory cells. For example, IL-10 can act directly on goblet cells in the colon to reduce protein misfolding and ER stress and promote mucus barrier function (118). Furthermore, IL-22 was identified as a suppressor of ER stress, and was shown to reverse high-fat diet-induced intestinal epithelial stress and loss of mucosal barrier integrity (119, 120). Immunoglobulin A (IgA) is the major secreted immunoglobulin isotype found at mucosal surfaces. As discussed in the previous section, studies in mice with an IEC-specific deletion in XBP1 have shown that IEC-associated ER stress can serve as a nidus for spontaneous microbiota-dependent ileitis (39, 83). It was recently shown that IEC-associated ER stress induces the expansion and activation of peritoneal B1b cells, resulting in increased lamina propria and luminal IgA to induce a barrier-protective T cell-independent IgA response (121). This mechanism presents a beneficial self-contained host-derived response that occurs independently of the microbiota and inflammation.
Numerous models with perturbations in the UPR pathways (ATF6, p58IPK, IRE-1, CHOP, OASIS, and S1P) do not show spontaneous phenotypes but display increased susceptibility to DSS-induced colitis (45, 94, 122–124). Treton et al. reported that the coordinated expression of all three branches of the UPR is impaired in UC patient mucosa, and that a defective integrated stress response in these mucosa samples led to reduced ATF4 and CHOP transcripts and protein levels (125). Their findings demonstrate that inappropriate ER stress renders UC mucosa highly susceptible to pathological changes in the microenvironment and may present an in vivo signature for the susceptibility of unaffected UC mucosa to inflammation. Similarly, we observed a downregulation of CHOP mRNA and protein expression in mouse models of T-cell-mediated and bacteria-driven colitis (45). A further study conducted in our own group provided evidence of UPR activation (increased Grp78 expression) in IECs from IBD patients and a mouse model of intestinal colitis (IL-10−/−) (95). The finding that IL-10-mediated p38 signaling inhibited TNF-induced recruitment of ATF6 to the Grp78 promoter provides a plausible explanation for colitis development in IL10−/− mice. Traditionally, Grp78 is regarded as a luminal ER chaperone, however numerous studies have established that, under cell stress conditions and in specific cell types, it can be found in other locations including the cell surface and the cytosol (126–128). This extends the functions of Grp78 beyond its traditional protein folding and processing role, to affect cell growth and signaling. In addition to a role of the UPR in IBD, mtUPR has been implicated in disease pathogenesis. Our study using double-stranded-RNA-activated protein kinase (PKR) knockout mice demonstrated that the highly selective mtUPR pathway employs PKR to recruit signaling molecules associated with the disease-relevant UPR signaling cascade, namely eIF2α and transcription factor activator protein-1 (AP1/cJun) (129). The observed eIF2α phosphorylation and AP1/cJun activation were dependent on activities of the mitochondrial protease ClpP and the cytoplasmic kinase PKR. The induction of mtUPR and PKR expression could be observed in murine IECs as well as patients with IBD, indicating that PKR may link mitochondrial stress to intestinal inflammation.
Taken together, there is substantial evidence from mouse models, patient data and GWAS studies, which demonstrate a role of the UPR (and mtUPR) in intestinal inflammation.
UPR in CRC
Several studies suggest a complex relationship between ER stress and tumorigenesis due to the multifaceted outcomes of UPR activation, either by promoting pro-oncogenic adaptation and cellular survival or by acquiring pro-apoptotic tumor suppression (130, 131). Both cell extrinsic [hypoxia (132, 133), nutrient deprivation (134, 135), and acidosis (136, 137)] and cell intrinsic [oncogene activation (138–142), loss of tumor suppressor genes (143), chromosomal abnormalities (144)] factors can influence the tumor environment and therefore UPR signaling, to either mount a tumor-survival response (facilitating tumorigenesis) or an anti-tumor response (suppressing tumorigenesis). Sublethal tolerable levels of UPR activation allow adaptation to cell stress and sustain mechanisms of tumor progression, mostly through links between the ER stress response and fundamental biological processes, such as autophagy and ER-mitochondrial crosstalk (145).
Colorectal cancer (CRC) is one of the leading causes of death in the western society, being ranked third most lethal neoplasia in the United States in both men and women (146). As a key regulator of all UPR pathways, GRP78 constitutes a major marker for UPR signaling, and its enhanced expression correlates with the growth, invasion, and metastasis of tumors (147). Elevated levels of GRP78 could be observed in CRC cell lines and CRC patient tissue, with inhibition of GRP78 evoking enhanced sensitivity of CRC cells to chemotherapeutic agents (148). The selective contribution of the IRE1 pathway to an anticancer immune response in mice was demonstrated in three independent mouse cancer models fed a low-protein diet (149). Interestingly, an Xbp1-deficient epithelium results in an over-activation of IRE1α which drives the regenerative intestinal stem cell (ISC) expansion upon pathological ER stress, but is not involved in homeostatic ISC regulation (150). IRE1 signaling was also shown to induce vascular endothelial growth factor-A (VEGF-A), IL-β, and IL-6 during the process of CRC angiogenesis (151). Similar to IRE1, activation of the PERK pathway has also been shown to play a vital role in CRC initiation, progression and angiogenesis (152, 153). With respect to the causal role of the UPR effector ATF6 in tumor biology, very little is known, although its downstream target gene Grp78 is frequently found to be overexpressed (154). A missense polymorphism in ATF6 is associated with susceptibility to hepatocellular carcinoma (155). ATF6 mRNA expression positively correlates with CRC primary tumors and the likelihood of metastasis and relapse (156, 157). ATF6 was recently proposed as a marker for early dysplastic changes both in ulcerative colitis (UC)-associated and non-UC-associated CRC (158). In our newly generated transgenic mouse model expressing the active form of ATF6 in IECs (nATF6IEC), we observed spontaneous colorectal tumorigenesis through the induction of intestinal dysbiosis and innate immune response in the absence of early inflammation (35). Using germ-free mice, we showed that ATF6-activated UPR in the epithelium requires the presence of intestinal microorganisms for tumor formation. Our analysis of CRC patients in The Cancer Genome Atlas dataset identified aberrant ATF6 as a clinically relevant UPR mediator (35). Furthermore, our clinical results identified approximately 11% of CRC patients from all tumor stages who overexpressed ATF6, and linked increased ATF6 levels in tumors of a subset of CRC patients with increased risk of post-operative disease relapse, supporting our hypothesis that ATF6 represents a novel and clinically relevant tumor risk gene defining a subgroup of CRC patients.
The above findings demonstrate a clear involvement of the UPR in the different stages of CRC pathogenesis, however, it remains largely unclear how ER stress and the UPR promote survival of cancer cells. Cancer cells that are undergoing ER stress can actively modulate immune cell function through transmissible ER stress. Induced ER stress in cancer cells was shown to cause the upregulation of UPR genes and pro-inflammatory cytokines in responder macrophages (159). The same group showed that cell-extrinsic effects of tumor ER stress imprint myeloid DCs and impair CD8+ T cell priming (160). Further evidence for the modulation of immune cells through ER stress was provided by Lee et al. who showed that ER stress in tumor-bearing mice accelerated cancer progression and the immunosuppressive capacity of myeloid-derived suppressor cells (MDSCs) (161). It seems to be the magnitude of ER stress, which defines whether an immunosuppressive or immunogenic response is mounted.
As summarized in this section and in Figure 2, research over the past decade has provided much insight into the critical role played by the UPR in IBD and CRC. Fully elucidating the mechanisms by which the UPR promotes or prevents the progression of diseases such as IBD and CRC will pave the way for novel therapeutic approaches.
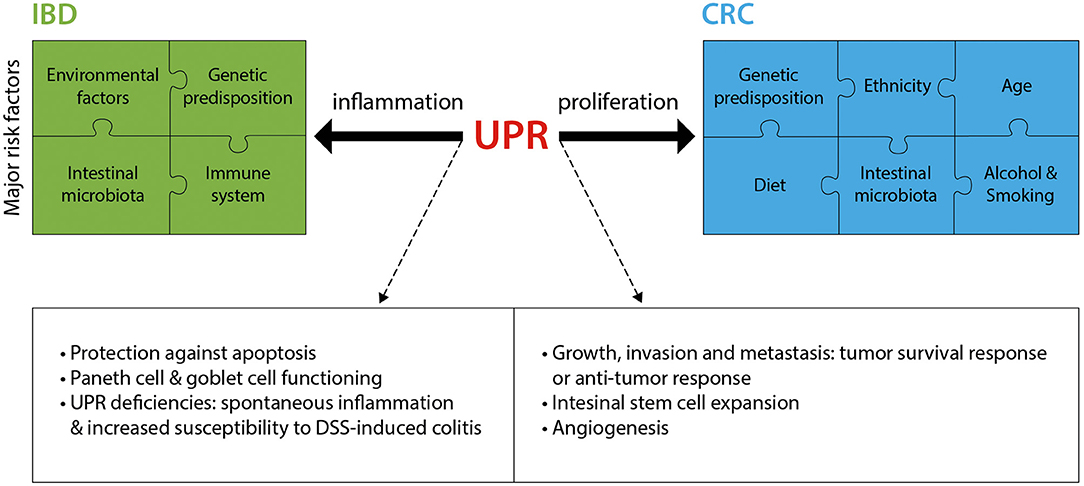
Figure 2. The UPR in IBD and CRC. IBD and CRC constitute complex diseases with numerous major risk factors contributing to disease pathology. The UPR significantly contributes to these two intestinal pathologies, mostly through an involvement in inflammation (IBD) and proliferation (CRC). Listed are the main known mechanisms by which the UPR is implicated in IBD and CRC. DSS, Dextran sodium sulfate.
UPR as a Pharmacological Target With Therapeutic Implications
As a key player in immunity and inflammation, the UPR has been increasingly recognized as an important pharmacological target, providing promising hope in the development of personalized therapeutic strategies for immune-mediated pathologies. To date there are several therapeutic opportunities involving the UPR. For descriptive and comparative purposes, these have been compiled in Table 2. Chemical chaperones are a group of low-molecular mass compounds that improve ER function. The most studied chemical chaperones are Tauro-ursodeoxycholic acid (TUDCA) and 4-phenyl butyrate (PBA) that, among other functions, were shown to reduce ER stress in intestinal epithelial cell lines (194), and in the intestinal epithelium where they decreased the severity to DSS-induced colitis in mice (162). In a further example, the clinically relevant Food and Drug Adnministration (FDA)-approved proteasome inhibitor Bortezomib sensitizes multiple myeloma cells to apoptosis through UPR induction (165). Increasing our molecular understanding of the intricate signaling pathways and their effects on the immune system is therefore indispensable to specifically and successfully target individual components of the UPR. Interestingly, a study by Wielenga et al. reported that ER stress-induced activation of the UPR forces colon cancer stem cells to differentiate, resulting in their enhanced sensitivity to chemotherapy in vitro and in vivo (195). These findings suggest that agents that induce the activation of the UPR may be used to specifically increase sensitivity of colon cancer stem cells to the effects of conventional chemotherapy. In light of the multitude of possible functions, responses and effects of the UPR, the therapeutic implications for inflammatory disorders and antitumor strategies in cancer may well be limitless. At the same time, however, it is indispensable to consider possible side effects when targeting the UPR, particularly with broad targeting approaches. This becomes evident with examples such as the receptor tyrosine kinase inhibitor sunitinib, which is FDA-approved for the treatment of renal cell carcinoma and gastrointestinal stromal tumors, but has also been shown to have negative effects on the anti-viral immune response (167).

Table 2. The UPR as a pharmacological target with therapeutic implications.
Concluding Remarks
Although our knowledge of the impact of ER stress and the UPR on immune responses requires much more insight and understanding, a growing body of new studies recognize the UPR as a fundamental mediator in cellular physiology and therefore also in the pathogenesis of inflammatory disorders, autoimmune responses, metabolic diseases, and tumorigenesis. Through its regulation of numerous cell-specific functions, the UPR is associated not only with the restoration of homeostasis, but also causally contributes to pathological processes. UPR signaling induces inflammatory responses, as well as inducing and controlling immune cell functions, making it an attractive research target with therapeutic implications for chronic immune-mediated diseases.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the German Research Foundation (DFG; Collaborative Research Center CRC1335; P11) and the Technical University of Munich (TUM) in the framework of the Open Access Publishing Program.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Braakman I, Bulleid NJ. Protein folding and modification in the mammalian endoplasmic reticulum. Annu Rev Biochem. (2011) 80:71–99. doi: 10.1146/annurev-biochem-062209-093836
2. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. (2007) 8:519–29. doi: 10.1038/nrm2199
3. Byrd AE, Brewer JW. Intricately regulated: a cellular toolbox for fine-tuning XBP1 expression and activity. Cells. (2012) 1:738–53. doi: 10.3390/cells1040738
4. Feng B, Yao PM, Li Y, Devlin CM, Zhang D, Harding HP, et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol. (2003) 5:781–92. doi: 10.1038/ncb1035
5. Cunha DA, Hekerman P, Ladriere L, Bazarra-Castro A, Ortis F, Wakeham MC, et al. Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J Cell Sci. (2008) 121:2308–18. doi: 10.1242/jcs.026062
6. Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. (2006) 125:443–451. doi: 10.1016/j.cell.2006.04.014
7. Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. (2005) 74:739–89. doi: 10.1146/annurev.biochem.73.011303.074134
8. Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol. (2004) 14:20–8. doi: 10.1016/j.tcb.2003.11.001
9. Lee AS. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods. (2005) 35:373–81. doi: 10.1016/j.ymeth.2004.10.010
10. Pincus D, Chevalier MW, Aragon T, van Anken E, Vidal SE, El-Samad H, et al. BiP binding to the ER-stress sensor Ire1 tunes the homeostatic behavior of the unfolded protein response. PLoS Biol. (2010) 8:e1000415. doi: 10.1371/journal.pbio.1000415
11. Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. (2000) 2:326–32. doi: 10.1038/35014014
12. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. (2011) 334:1081–6. doi: 10.1126/science.1209038
13. Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. (2001) 107:881–91. doi: 10.1016/S0092-8674(01)00611-0
14. Yamamoto K, Yoshida H, Kokame K, Kaufman RJ, Mori K. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J Biochem. (2004) 136:343–50. doi: 10.1093/jb/mvh122
15. Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. (2000) 6:1355–64. doi: 10.1016/S1097-2765(00)00133-7
16. Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. (2002) 3:99–111. doi: 10.1016/S1534-5807(02)00203-4
17. Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. (1999) 10:3787–99. doi: 10.1091/mbc.10.11.3787
18. Chen X, Karnovsky A, Sans MD, Andrews PC, Williams JA. Molecular characterization of the endoplasmic reticulum: insights from proteomic studies. Proteomics. (2010) 10:4040–52. doi: 10.1002/pmic.201000234
19. Hu CC, Dougan SK, McGehee AM, Love JC, Ploegh HL. XBP-1 regulates signal transduction, transcription factors and bone marrow colonization in B cells. EMBO J. (2009) 28:1624–36. doi: 10.1038/emboj.2009.117
20. van Anken E, Romijn EP, Maggioni C, Mezghrani A, Sitia R, Braakman I, et al. Sequential waves of functionally related proteins are expressed when B cells prepare for antibody secretion. Immunity. (2003) 18:243–53. doi: 10.1016/S1074-7613(03)00024-4
21. Yu L, Andruska N, Zheng X, Shapiro DJ. Anticipatory activation of the unfolded protein response by epidermal growth factor is required for immediate early gene expression and cell proliferation. Mol Cell Endocrinol. (2016) 422:31–41. doi: 10.1016/j.mce.2015.11.005
22. Karali E, Bellou S, Stellas D, Klinakis A, Murphy C, Fotsis T. VEGF Signals through ATF6 and PERK to promote endothelial cell survival and angiogenesis in the absence of ER stress. Mol Cell. (2014) 54:559–72. doi: 10.1016/j.molcel.2014.03.022
23. Brewer JW, Hendershot LM. Building an antibody factory: a job for the unfolded protein response. Nat Immunol. (2005) 6:23–9. doi: 10.1038/ni1149
24. He B. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ. (2006) 13:393–403. doi: 10.1038/sj.cdd.4401833
25. Vasa-Nicotera M. The new kid on the block: the unfolded protein response in the pathogenesis of atherosclerosis. Cell Death Differ. (2004) 11(Suppl. 1):S10–1. doi: 10.1038/sj.cdd.4401468
26. Harding HP, Ron D. Endoplasmic reticulum stress and the development of diabetes: a review. Diabetes. (2002) 51(Suppl. 3):S455–61. doi: 10.2337/diabetes.51.2007.S455
27. Ma Y, Hendershot LM. The role of the unfolded protein response in tumour development: friend or foe? Nat Rev Cancer. (2004) 4:966–77. doi: 10.1038/nrc1505
28. DeGracia DJ, Montie HL. Cerebral ischemia and the unfolded protein response. J Neurochem. (2004) 91:1–8. doi: 10.1111/j.1471-4159.2004.02703.x
29. Lindholm D, Wootz H, Korhonen L. ER stress and neurodegenerative diseases. Cell Death Differ. (2006) 13:385–92. doi: 10.1038/sj.cdd.4401778
30. Kakiuchi C, Iwamoto K, Ishiwata M, Bundo M, Kasahara T, Kusumi I, et al. Impaired feedback regulation of XBP1 as a genetic risk factor for bipolar disorder. Nat Genet. (2003) 35:171–5. doi: 10.1038/ng1235
31. Cichon S, Buervenich S, Kirov G, Akula N, Dimitrova A, Green E, et al. Lack of support for a genetic association of the XBP1 promoter polymorphism with bipolar disorder in probands of European origin. Nat Genet. (2004) 36:783–4; author reply: 784–5. doi: 10.1038/ng0804-783
32. Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, et al. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol Cell. (2001) 7:1153–63. doi: 10.1016/S1097-2765(01)00264-7
33. Zhang K, Wong HN, Song B, Miller CN, Scheuner D, Kaufman RJ. The unfolded protein response sensor IRE1alpha is required at 2 distinct steps in B cell lymphopoiesis. J Clin Invest. (2005) 115:268–81. doi: 10.1172/JCI200521848
34. Zhang K, Wang S, Malhotra J, Hassler JR, Back SH, Wang G, et al. The unfolded protein response transducer IRE1α prevents ER stress-induced hepatic steatosis. EMBO J. (2011) 30:1357–75. doi: 10.1038/emboj.2011.52
35. Coleman OI, Lobner EM, Bierwirth S, Sorbie A, Waldschmitt N, Rath E, et al. Activated ATF6 induces intestinal dysbiosis and innate immune response to promote colorectal tumorigenesis. Gastroenterology. (2018) 155:1539–52.e12. doi: 10.1053/j.gastro.2018.07.028
36. Gomez JA, Tyra HM, DeZwaan-McCabe D, Olivier AK, Rutkowski DT. Synthetic embryonic lethality upon deletion of the ER cochaperone p58(IPK) and the ER stress sensor ATF6α. Biochem Biophys Res Commun. (2014) 443:115–9. doi: 10.1016/j.bbrc.2013.11.060
37. Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell. (2007) 13:365–76. doi: 10.1016/j.devcel.2007.07.018
38. Reimold AM, Etkin A, Clauss I, Perkins A, Friend DS, Zhang J, et al. An essential role in liver development for transcription factor XBP-1. Genes Dev. (2000) 14:152–7.
39. Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. (2008) 134:743–56. doi: 10.1016/j.cell.2008.07.021
40. Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. (2004) 306:457–61. doi: 10.1126/science.1103160
41. Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, et al. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. (2001) 7:1165–76. doi: 10.1016/S1097-2765(01)00265-9
42. Ishihara H, Takeda S, Tamura A, Takahashi R, Yamaguchi S, Takei D, et al. Disruption of the WFS1 gene in mice causes progressive beta-cell loss and impaired stimulus-secretion coupling in insulin secretion. Hum Mol Genet. (2004) 13:1159–70. doi: 10.1093/hmg/ddh125
43. Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. (2004) 18:3066–77. doi: 10.1101/gad.1250704
44. Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, et al. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest. (2002) 109:525–32. doi: 10.1172/JCI0214550
45. Waldschmitt N, Berger E, Rath E, Sartor RB, Weigmann B, Heikenwalder M, et al. C/EBP homologous protein inhibits tissue repair in response to gut injury and is inversely regulated with chronic inflammation. Mucosal Immunol. (2014) 7:1452–66. doi: 10.1038/mi.2014.34
46. Luo S, Mao C, Lee B, Lee AS. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol Cell Biol. (2006) 26:5688–97. doi: 10.1128/MCB.00779-06
47. Ye R, Jung DY, Jun JY, Li J, Luo S, Ko HJ, et al. Grp78 heterozygosity promotes adaptive unfolded protein response and attenuates diet-induced obesity and insulin resistance. Diabetes. (2010) 59:6–16. doi: 10.2337/db09-0755
48. Mao C, Wang M, Luo B, Wey S, Dong D, Wesselschmidt R, et al. Targeted mutation of the mouse Grp94 gene disrupts development and perturbs endoplasmic reticulum stress signaling. PLoS ONE. (2010) 5:e10852. doi: 10.1371/journal.pone.0010852
49. Tanaka T, Tsujimura T, Takeda K, Sugihara A, Maekawa A, Terada N, et al. Targeted disruption of ATF4 discloses its essential role in the formation of eye lens fibres. Genes Cells. (1998) 3:801–10. doi: 10.1046/j.1365-2443.1998.00230.x
50. Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci USA. (1996) 93:13943–8. doi: 10.1073/pnas.93.24.13943
51. Lee JM, Chan K, Kan YW, Johnson JA. Targeted disruption of Nrf2 causes regenerative immune-mediated hemolytic anemia. Proc Natl Acad Sci USA. (2004) 101:9751–6. doi: 10.1073/pnas.0403620101
52. Ladiges WC, Knoblaugh SE, Morton JF, Korth MJ, Sopher BL, Baskin CR, et al. Pancreatic beta-cell failure and diabetes in mice with a deletion mutation of the endoplasmic reticulum molecular chaperone gene P58IPK. Diabetes. (2005) 54:1074–81. doi: 10.2337/diabetes.54.4.1074
53. Denzel A, Molinari M, Trigueros C, Martin JE, Velmurgan S, Brown S, et al. Early postnatal death and motor disorders in mice congenitally deficient in calnexin expression. Mol Cell Biol. (2002) 22:7398–404. doi: 10.1128/MCB.22.21.7398-7404.2002
54. Mesaeli N, Nakamura K, Zvaritch E, Dickie P, Dziak E, Krause KH, et al. Calreticulin is essential for cardiac development. J Cell Biol. (1999) 144:857–68. doi: 10.1083/jcb.144.5.857
55. Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. (2000) 403:98–103. doi: 10.1038/47513
56. Zhao F, Edwards R, Dizon D, Afrasiabi K, Mastroianni JR, Geyfman M, et al. Disruption of Paneth and goblet cell homeostasis and increased endoplasmic reticulum stress in Agr2-/- mice. Dev Biol. (2010) 338:270–9. doi: 10.1016/j.ydbio.2009.12.008
57. Rutkowski DT, Hegde RS. Regulation of basal cellular physiology by the homeostatic unfolded protein response. J Cell Biol. (2010) 189:783–94. doi: 10.1083/jcb.201003138
58. Claudio N, Dalet A, Gatti E, Pierre P. Mapping the crossroads of immune activation and cellular stress response pathways. EMBO J. (2013) 32:1214–24. doi: 10.1038/emboj.2013.80
59. Smith JA. A new paradigm: innate immune sensing of viruses via the unfolded protein response. Front Microbiol. (2014) 5:222. doi: 10.3389/fmicb.2014.00222
60. Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature. (2001) 412:300–7. doi: 10.1038/35085509
61. Gass JN, Gifford NM, Brewer JW. Activation of an unfolded protein response during differentiation of antibody-secreting B cells. J Biol Chem. (2002) 277:49047–54. doi: 10.1074/jbc.M205011200
62. Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. (2004) 21:81–93. doi: 10.1016/j.immuni.2004.06.010
63. Todd DJ, McHeyzer-Williams LJ, Kowal C, Lee AH, Volpe BT, Diamond B, et al. XBP1 governs late events in plasma cell differentiation and is not required for antigen-specific memory B cell development. J Exp Med. (2009) 206:2151–9. doi: 10.1084/jem.20090738
64. Benhamron S, Hadar R, Iwawaky T, So JS, Lee AH, Tirosh B. Regulated IRE1-dependent decay participates in curtailing immunoglobulin secretion from plasma cells. Eur J Immunol. (2014) 44:867–76. doi: 10.1002/eji.201343953
65. Taubenheim N, Tarlinton DM, Crawford S, Corcoran LM, Hodgkin PD, Nutt SL. High rate of antibody secretion is not integral to plasma cell differentiation as revealed by XBP-1 deficiency. J Immunol. (2012) 189:3328–38. doi: 10.4049/jimmunol.1201042
66. Tellier J, Shi W, Minnich M, Liao Y, Crawford S, Smyth GK, et al. Blimp-1 controls plasma cell function through the regulation of immunoglobulin secretion and the unfolded protein response. Nat Immunol. (2016) 17:323–30. doi: 10.1038/ni.3348
67. Masouleh BK, Geng HM, Hurtz C, Chan LN, Logan AC, Chang MS, et al. Mechanistic rationale for targeting the unfolded protein response in pre-B acute lymphoblastic leukemia. Proc Natl Acad Sci USA. (2014) 111:E2219–28. doi: 10.1073/pnas.1400958111
68. Scheu S, Stetson DB, Reinhardt RL, Leber JH, Mohrs M, Locksley RM. Activation of the integrated stress response during T helper cell differentiation. Nat Immunol. (2006) 7:644–51. doi: 10.1038/ni1338
69. Brucklacher-Waldert V, Ferreira C, Stebegg M, Fesneau O, Innocentin S, Marie JC, et al. Cellular stress in the context of an inflammatory environment supports TGF-beta-independent T helper-17 differentiation. Cell Rep. (2017) 19:2357–70. doi: 10.1016/j.celrep.2017.05.052
70. Stadhouders R, Lubberts E, Hendriks RW. A cellular and molecular view of T helper 17 cell plasticity in autoimmunity. J Autoimmun. (2018) 87:1–15. doi: 10.1016/j.jaut.2017.12.007
71. Thaxton JE, Wallace C, Riesenberg B, Zhang Y, Paulos CM, Beeson CC, et al. Modulation of endoplasmic reticulum stress controls CD4(+) T-cell activation and antitumor function. Cancer Immunol Res. (2017) 5:666–75. doi: 10.1158/2326-6066.CIR-17-0081
72. Kamimura D, Bevan MJ. Endoplasmic reticulum stress regulator XBP-1 contributes to effector CD8+ T cell differentiation during acute infection. J Immunol. (2008) 181:5433–41. doi: 10.4049/jimmunol.181.8.5433
73. Iwakoshi NN, Pypaert M, Glimcher LH. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J Exp Med. (2007) 204:2267–75. doi: 10.1084/jem.20070525
74. Hu F, Yu X, Wang H, Zuo D, Guo C, Yi H, et al. ER stress and its regulator X-box-binding protein-1 enhance polyIC-induced innate immune response in dendritic cells. Eur J Immunol. (2011) 41:1086–97. doi: 10.1002/eji.201040831
75. Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, et al. ER Stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell. (2015) 161:1527–38. doi: 10.1016/j.cell.2015.05.025
76. Goodall JC, Wu C, Zhang Y, McNeill L, Ellis L, Saudek V, et al. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc Natl Acad Sci USA. (2010) 107:17698–703. doi: 10.1073/pnas.1011736107
77. Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. (2010) 11:411–8. doi: 10.1038/ni.1857
78. Smith JA, Turner MJ, DeLay ML, Klenk EI, Sowders DP, Colbert RA. Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic IFN-beta induction via X-box binding protein 1. Eur J Immunol. (2008) 38:1194–203. doi: 10.1002/eji.200737882
79. Kim S, Joe Y, Kim HJ, Kim YS, Jeong SO, Pae HO, et al. Endoplasmic reticulum stress-induced IRE1α activation mediates cross-talk of GSK-3β and XBP-1 to regulate inflammatory cytokine production. J Immunol. (2015) 194:4498–506. doi: 10.4049/jimmunol.1401399
80. Rao J, Yue S, Fu Y, Zhu J, Wang X, Busuttil RW, et al. ATF6 mediates a pro-inflammatory synergy between ER stress and TLR activation in the pathogenesis of liver ischemia-reperfusion injury. Am J Transplant. (2014) 14:1552–61. doi: 10.1111/ajt.12711
81. Woo CW, Cui D, Arellano J, Dorweiler B, Harding H, Fitzgerald KA, et al. Adaptive suppression of the ATF4-CHOP branch of the unfolded protein response by toll-like receptor signalling. Nat Cell Biol. (2009) 11:1473–80. doi: 10.1038/ncb1996
82. Clevers HC, Bevins CL. Paneth cells: maestros of the small intestinal crypts. Annu Rev Physiol. (2013) 75:289–311. doi: 10.1146/annurev-physiol-030212-183744
83. Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Bock J, Martinez-Naves E, et al. Paneth cells as a site of origin for intestinal inflammation. Nature. (2013) 503:272–6. doi: 10.1038/nature12599
84. Grootjans J, Hodin CM, de Haan JJ, Derikx JP, Rouschop KM, Verheyen FK, et al. Level of activation of the unfolded protein response correlates with Paneth cell apoptosis in human small intestine exposed to ischemia/reperfusion. Gastroenterology. (2011) 140:529–39.e3. doi: 10.1053/j.gastro.2010.10.040
85. Rosekrans SL, Heijmans J, Buller NV, Westerlund J, Lee AS, Muncan V, et al. ER stress induces epithelial differentiation in the mouse oesophagus. Gut. (2015) 64:195–202. doi: 10.1136/gutjnl-2013-306347
86. van Galen P, Kreso A, Mbong N, Kent DG, Fitzmaurice T, Chambers JE, et al. The unfolded protein response governs integrity of the haematopoietic stem-cell pool during stress. Nature. (2014) 510:268–72. doi: 10.1038/nature13228
87. Heijmans J, van Lidth de Jeude JF, Koo BK, Rosekrans SL, Wielenga MC, van de Wetering M, et al. ER stress causes rapid loss of intestinal epithelial stemness through activation of the unfolded protein response. Cell Rep. (2013) 3:1128–39. doi: 10.1016/j.celrep.2013.02.031
88. van Lidth de Jeude JF, Spaan CN, Meijer BJ, Smit WL, Soeratram TTD, Wielenga MCB, et al. Heterozygosity of chaperone Grp78 reduces intestinal stem cell regeneration potential and protects against adenoma formation. Cancer Res. (2018) 78:6098–106. doi: 10.1158/0008-5472.CAN-17-3600
89. Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J. (2002) 21:4411–9. doi: 10.1093/emboj/cdf445
90. Aldridge JE, Horibe T, Hoogenraad NJ. Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS ONE. (2007) 2:e874. doi: 10.1371/journal.pone.0000874
91. Rath E, Moschetta A, Haller D. Mitochondrial function - gatekeeper of intestinal epithelial cell homeostasis. Nat Rev Gastroenterol Hepatol. (2018) 15:497–516. doi: 10.1038/s41575-018-0021-x
92. Berger E, Rath E, Yuan D, Waldschmitt N, Khaloian S, Allgauer M, et al. Mitochondrial function controls intestinal epithelial stemness and proliferation. Nat Commun. (2016) 7:13171. doi: 10.1038/ncomms13171
93. van Lidth de Jeude JF, Meijer BJ, Wielenga MCB, Spaan CN, Baan B, Rosekrans SL, et al. Induction of endoplasmic reticulum stress by deletion of Grp78 depletes Apc mutant intestinal epithelial stem cells. Oncogene. (2017) 36:3397–405. doi: 10.1038/onc.2016.326
94. Bertolotti A, Wang X, Novoa I, Jungreis R, Schlessinger K, Cho JH, et al. Increased sensitivity to dextran sodium sulfate colitis in IRE1beta-deficient mice. J Clin Invest. (2001) 107:585–93. doi: 10.1172/JCI11476
95. Shkoda A, Ruiz PA, Daniel H, Kim SC, Rogler G, Sartor RB, et al. Interleukin-10 blocked endoplasmic reticulum stress in intestinal epithelial cells: impact on chronic inflammation. Gastroenterology. (2007) 132:190–207. doi: 10.1053/j.gastro.2006.10.030
96. Pott J, Hornef M. Innate immune signalling at the intestinal epithelium in homeostasis and disease. EMBO Rep. (2012) 13:684–98. doi: 10.1038/embor.2012.96
97. Catalioto RM, Maggi CA, Giuliani S. Intestinal epithelial barrier dysfunction in disease and possible therapeutical interventions. Curr Med Chem. (2011) 18:398–426. doi: 10.2174/092986711794839179
98. Okumura R, Takeda K. Roles of intestinal epithelial cells in the maintenance of gut homeostasis. Exp Mol Med. (2017) 49:e338. doi: 10.1038/emm.2017.20
99. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. (2012) 491:119–24. doi: 10.1038/nature11582
100. Morgan XC, Kabakchiev B, Waldron L, Tyler AD, Tickle TL, Milgrom R, et al. Associations between host gene expression, the mucosal microbiome, and clinical outcome in the pelvic pouch of patients with inflammatory bowel disease. Genome Biol. (2015) 16:67. doi: 10.1186/s13059-015-0637-x
101. Sartor RB, Wu GD. Roles for intestinal bacteria, viruses, and fungi in pathogenesis of inflammatory bowel diseases and therapeutic approaches. Gastroenterology. (2017) 152:327–39.e4. doi: 10.1053/j.gastro.2016.10.012
102. Kaplan GG. IBD Global variations in environmental risk factors for IBD. Nat Rev Gastroenterol Hepatol. (2014) 11:708–9. doi: 10.1038/nrgastro.2014.182
103. de Lange KM, Moutsianas L, Lee JC, Lamb CA, Luo Y, Kennedy NA, et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet. (2017) 49:256–61. doi: 10.1038/ng.3760
104. Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. (2008) 40:955–62. doi: 10.1038/ng.175
105. McGovern DP, Gardet A, Torkvist L, Goyette P, Essers J, Taylor KD, et al. Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat Genet. (2010) 42:332–7. doi: 10.1038/ng.549
106. Zheng W, Rosenstiel P, Huse K, Sina C, Valentonyte R, Mah N, et al. Evaluation of AGR2 and AGR3 as candidate genes for inflammatory bowel disease. Genes Immun. (2006) 7:11–8. doi: 10.1038/sj.gene.6364263
107. Cantero-Recasens G, Fandos C, Rubio-Moscardo F, Valverde MA, Vicente R. The asthma-associated ORMDL3 gene product regulates endoplasmic reticulum-mediated calcium signaling and cellular stress. Hum Mol Genet. (2010) 19:111–21. doi: 10.1093/hmg/ddp471
108. Miller M, Rosenthal P, Beppu A, Mueller JL, Hoffman HM, Tam AB, et al. ORMDL3 transgenic mice have increased airway remodeling and airway responsiveness characteristic of asthma. J Immunol. (2014) 192:3475–87. doi: 10.4049/jimmunol.1303047
109. Miller M, Tam AB, Cho JY, Doherty TA, Pham A, Khorram N, et al. ORMDL3 is an inducible lung epithelial gene regulating metalloproteases, chemokines, OAS, and ATF6. Proc Natl Acad Sci USA. (2012) 109:16648–53. doi: 10.1073/pnas.1204151109
110. Kiefer K, Carreras-Sureda A, Garcia-Lopez R, Rubio-Moscardo F, Casas J, Fabrias G, et al. Coordinated regulation of the orosomucoid-like gene family expression controls de novo ceramide synthesis in mammalian cells. J Biol Chem. (2015) 290:2822–30. doi: 10.1074/jbc.M114.595116
111. Park SW, Zhen G, Verhaeghe C, Nakagami Y, Nguyenvu LT, Barczak AJ, et al. The protein disulfide isomerase AGR2 is essential for production of intestinal mucus. Proc Natl Acad Sci USA. (2009) 106:6950–5. doi: 10.1073/pnas.0808722106
112. Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. (2007) 39:207–11. doi: 10.1038/ng1954
113. Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. (2008) 456:259–63. doi: 10.1038/nature07416
114. Tschurtschenthaler M, Adolph TE, Ashcroft JW, Niederreiter L, Bharti R, Saveljeva S, et al. Defective ATG16L1-mediated removal of IRE1α drives Crohn's disease-like ileitis. J Exp Med. (2017) 214:401–22. doi: 10.1084/jem.20160791
115. Tytgat KMAJ, vanderWal JWG, Einerhand AWC, Buller HA, Dekker J. Quantitative analysis of MUC2 synthesis in ulcerative colitis. Biochem Biophys Res Commun. (1996) 224:397–405. doi: 10.1006/bbrc.1996.1039
116. Van Klinken BJW, Van der Wal JWG, Einerhand AWC, Buller HA, Dekker J. Sulphation and secretion of the predominant secretory human colonic mucin MUC2 in ulcerative colitis. Gut. (1999) 44:387–93. doi: 10.1136/gut.44.3.387
117. Heazlewood CK, Cook MC, Eri R, Price GR, Tauro SB, Taupin D, et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. (2008) 5:e54. doi: 10.1371/journal.pmed.0050054
118. Hasnain SZ, Tauro S, Das I, Tong H, Chen AC, Jeffery PL, et al. IL-10 promotes production of intestinal mucus by suppressing protein misfolding and endoplasmic reticulum stress in goblet cells. Gastroenterology. (2013) 144:357–68.e9. doi: 10.1053/j.gastro.2012.10.043
119. Hasnain SZ, Borg DJ, Harcourt BE, Tong H, Sheng YH, Ng CP, et al. Glycemic control in diabetes is restored by therapeutic manipulation of cytokines that regulate beta cell stress. Nat Med. (2014) 20:1417–26. doi: 10.1038/nm.3705
120. Gulhane M, Murray L, Lourie R, Tong H, Sheng YH, Wang R, et al. High fat diets induce colonic epithelial cell stress and inflammation that is reversed by IL-22. Sci Rep. (2016) 6:28990. doi: 10.1038/srep28990
121. Grootjans J, Krupka N, Hosomi S, Matute JD, Hanley T, Saveljeva S, et al. Epithelial endoplasmic reticulum stress orchestrates a protective IgA response. Science. (2019) 363:993–8. doi: 10.1126/science.aat7186
122. Hino K, Saito A, Asada R, Kanemoto S, Imaizumi K. Increased susceptibility to dextran sulfate sodium-induced colitis in the endoplasmic reticulum stress transducer OASIS deficient mice. PLoS ONE. (2014) 9:e88048. doi: 10.1371/journal.pone.0088048
123. Asada R, Saito A, Kawasaki N, Kanemoto S, Iwamoto H, Oki M, et al. The endoplasmic reticulum stress transducer OASIS is involved in the terminal differentiation of goblet cells in the large intestine. J Biol Chem. (2012) 287:8144–53. doi: 10.1074/jbc.M111.332593
124. Brandl K, Rutschmann S, Li X, Du X, Xiao N, Schnabl B, et al. Enhanced sensitivity to DSS colitis caused by a hypomorphic Mbtps1 mutation disrupting the ATF6-driven unfolded protein response. Proc Natl Acad Sci USA. (2009) 106:3300–5. doi: 10.1073/pnas.0813036106
125. Treton X, Pedruzzi E, Cazals-Hatem D, Grodet A, Panis Y, Groyer A, et al. Altered endoplasmic reticulum stress affects translation in inactive colon tissue from patients with ulcerative colitis. Gastroenterology. (2011) 141:1024–35. doi: 10.1053/j.gastro.2011.05.033
126. Kern J, Untergasser G, Zenzmaier C, Sarg B, Gastl G, Gunsilius E, et al. GRP-78 secreted by tumor cells blocks the antiangiogenic activity of bortezomib. Blood. (2009) 114:3960–7. doi: 10.1182/blood-2009-03-209668
127. Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem J. (2011) 434:181–8. doi: 10.1042/BJ20101569
128. Ouyang YB, Xu LJ, Emery JF, Lee AS, Giffard RG. Overexpressing GRP78 influences Ca2+ handling and function of mitochondria in astrocytes after ischemia-like stress. Mitochondrion. (2011) 11:279–86. doi: 10.1016/j.mito.2010.10.007
129. Rath E, Berger E, Messlik A, Nunes T, Liu B, Kim SC, et al. Induction of dsRNA-activated protein kinase links mitochondrial unfolded protein response to the pathogenesis of intestinal inflammation. Gut. (2012) 61:1269–78. doi: 10.1136/gutjnl-2011-300767
130. Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, et al. IRE1 signaling affects cell fate during the unfolded protein response. Science. (2007) 318:944–9. doi: 10.1126/science.1146361
131. Tay KH, Luan Q, Croft A, Jiang CC, Jin L, Zhang XD, et al. Sustained IRE1 and ATF6 signaling is important for survival of melanoma cells undergoing ER stress. Cell Signal. (2014) 26:287–94. doi: 10.1016/j.cellsig.2013.11.008
132. Gutierrez T, Simmen T. Endoplasmic reticulum chaperones and oxidoreductases: critical regulators of tumor cell survival and immunorecognition. Front Oncol. (2014) 4:291. doi: 10.3389/fonc.2014.00291
133. Koritzinsky M, Levitin F, van den Beucken T, Rumantir RA, Harding NJ, Chu KC, et al. Two phases of disulfide bond formation have differing requirements for oxygen. J Cell Biol. (2013) 203:615–27. doi: 10.1083/jcb.201307185
134. Wek RC, Cavener DR. Translational control and the unfolded protein response. Antioxid Redox Signal. (2007) 9:2357–71. doi: 10.1089/ars.2007.1764
135. Zhang P, McGrath BC, Reinert J, Olsen DS, Lei L, Gill S, et al. The GCN2 eIF2alpha kinase is required for adaptation to amino acid deprivation in mice. Mol Cell Biol. (2002) 22:6681–8. doi: 10.1128/MCB.22.19.6681-6688.2002
136. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
137. Tang CH, Ranatunga S, Kriss CL, Cubitt CL, Tao J, Pinilla-Ibarz JA, et al. Inhibition of ER stress-associated IRE-1/XBP-1 pathway reduces leukemic cell survival. J Clin Invest. (2014) 124:2585–98. doi: 10.1172/JCI73448
138. Hart LS, Cunningham JT, Datta T, Dey S, Tameire F, Lehman SL, et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J Clin Invest. (2012) 122:4621–34. doi: 10.1172/JCI62973
139. Denoyelle C, Abou-Rjaily G, Bezrookove V, Verhaegen M, Johnson TM, Fullen DR, et al. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat Cell Biol. (2006) 8:1053–63. doi: 10.1038/ncb1471
140. Corazzari M, Rapino F, Ciccosanti F, Giglio P, Antonioli M, Conti B, et al. Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ. (2015) 22:946–58. doi: 10.1038/cdd.2014.183
141. Nam S, Chang HR, Jung HR, Gim Y, Kim NY, Grailhe R, et al. A pathway-based approach for identifying biomarkers of tumor progression to trastuzumab-resistant breast cancer. Cancer Lett. (2015) 356:880–90. doi: 10.1016/j.canlet.2014.10.038
142. Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol. (2011) 9:16–32. doi: 10.1038/nrclinonc.2011.177
143. Ozcan U, Ozcan L, Yilmaz E, Duvel K, Sahin M, Manning BD, et al. Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Mol Cell. (2008) 29:541–51. doi: 10.1016/j.molcel.2007.12.023
144. Oromendia AB, Amon A. Aneuploidy: implications for protein homeostasis and disease. Dis Model Mech. (2014) 7:15–20. doi: 10.1242/dmm.013391
145. Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer. (2014) 14:581–97. doi: 10.1038/nrc3800
146. Siegel R, Desantis C, Jemal A. Colorectal cancer statistics, 2014. CA Cancer J Clin. (2014) 64:104–17. doi: 10.3322/caac.21220
147. Lee AS. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer. (2014) 14:263–76. doi: 10.1038/nrc3701
148. Mhaidat NM, Alzoubi KH, Khabour OF, Banihani MN, Al-Balas QA, Swaidan S. GRP78 regulates sensitivity of human colorectal cancer cells to DNA targeting agents. Cytotechnology. (2016) 68:459–67. doi: 10.1007/s10616-014-9799-8
149. Rubio-Patino C, Bossowski JP, De Donatis GM, Mondragon L, Villa E, Aira LE, et al. Low-protein diet induces IRE1α-dependent anticancer immunosurveillance. Cell Metab. (2018) 27:828–42.e7. doi: 10.1016/j.cmet.2018.02.009
150. Niederreiter L, Fritz TM, Adolph TE, Krismer AM, Offner FA, Tschurtschenthaler M, et al. ER stress transcription factor Xbp1 suppresses intestinal tumorigenesis and directs intestinal stem cells. J Exp Med. (2013) 210:2041–56. doi: 10.1084/jem.20122341
151. Drogat B, Auguste P, Nguyen DT, Bouchecareilh M, Pineau R, Nalbantoglu J, et al. IRE1 signaling is essential for ischemia-induced vascular endothelial growth factor-a expression and contributes to angiogenesis and tumor growth in vivo. Cancer Res. (2007) 67:6700–07. doi: 10.1158/0008-5472.CAN-06-3235
152. Blais JD, Addison CL, Edge R, Falls T, Zhao H, Wary K, et al. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol Cell Biol. (2006) 26:9517–32. doi: 10.1128/MCB.01145-06
153. Wang YG, Alam GN, Ning Y, Visioli F, Dong ZH, Nor JE, et al. The unfolded protein response induces the angiogenic switch in human tumor cells through the PERK/ATF4 pathway. Cancer Res. (2012) 72:5396–406. doi: 10.1158/0008-5472.CAN-12-0474
154. Li Z, Li Z. Glucose regulated protein 78: a critical link between tumor microenvironment and cancer hallmarks. Biochim Biophys Acta. (2012) 1826:13–22. doi: 10.1016/j.bbcan.2012.02.001
155. Wu X, Xin Z, Zhang W, Zheng S, Wu J, Chen K, et al. A missense polymorphism in ATF6 gene is associated with susceptibility to hepatocellular carcinoma probably by altering ATF6 level. Int J Cancer. (2014) 135:61–8. doi: 10.1002/ijc.28649
156. Lin YH, Friederichs J, Black MA, Mages J, Rosenberg R, Guilford PJ, et al. Multiple gene expression classifiers from different array platforms predict poor prognosis of colorectal cancer. Clin Cancer Res. (2007) 13:498–507. doi: 10.1158/1078-0432.CCR-05-2734
157. Schewe DM, Aguirre-Ghiso JA. ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc Natl Acad Sci USA. (2008) 105:10519–24. doi: 10.1073/pnas.0800939105
158. Hanaoka M, Ishikawa T, Ishiguro M, Tokura M, Yamauchi S, Kikuchi A, et al. Expression of ATF6 as a marker of pre-cancerous atypical change in ulcerative colitis-associated colorectal cancer: a potential role in the management of dysplasia. J Gastroenterol. (2017) 53:631–41. doi: 10.1007/s00535-017-1387-1
159. Mahadevan NR, Rodvold J, Sepulveda H, Rossi S, Drew AF, Zanetti M. Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc Natl Acad Sci USA. (2011) 108:6561–6. doi: 10.1073/pnas.1008942108
160. Mahadevan NR, Anufreichik V, Rodvold JJ, Chiu KT, Sepulveda H, Zanetti M. Cell-extrinsic effects of tumor ER stress imprint myeloid dendritic cells and impair CD8(+) T cell priming. PLoS ONE. (2012) 7:e51845. doi: 10.1371/journal.pone.0051845
161. Lee BR, Chang SY, Hong EH, Kwon BE, Kim HM, Kim YJ, et al. Elevated endoplasmic reticulum stress reinforced immunosuppression in the tumor microenvironment via myeloid-derived suppressor cells. Oncotarget. (2014) 5:12331–45. doi: 10.18632/oncotarget.2589
162. Cao SS, Zimmermann EM, Chuang BM, Song B, Nwokoye A, Wilkinson JE, et al. The unfolded protein response and chemical chaperones reduce protein misfolding and colitis in mice. Gastroenterology. (2013) 144:989–1000.e6. doi: 10.1053/j.gastro.2013.01.023
163. Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. (2006) 313:1137–40. doi: 10.1126/science.1128294
164. Kim HJ, Jeong JS, Kim SR, Park SY, Chae HJ, Lee YC. Inhibition of endoplasmic reticulum stress alleviates lipopolysaccharide-induced lung inflammation through modulation of NF-kappa B/HIF-1α signaling pathway. Sci Rep. (2013) 3:1142. doi: 10.1038/srep01142
165. Mujtaba T, Dou QP. Advances in the understanding of mechanisms and therapeutic use of bortezomib. Discov Med. (2011) 12:471–80.
166. Ali MMU, Bagratuni T, Davenport EL, Nowak PR, Silva-Santisteban MC, Hardcastle A, et al. Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. Embo J. (2011) 30:894–905. doi: 10.1038/emboj.2011.18
167. Jha BK, Polyakova I, Kessler P, Dong B, Dickerman B, Sen GC, et al. Inhibition of RNase L and RNA-dependent protein kinase (PKR) by sunitinib impairs antiviral innate immunity. J Biol Chem. (2011) 286:26319–26. doi: 10.1074/jbc.M111.253443
168. Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood. (2011) 117:1311–4. doi: 10.1182/blood-2010-08-303099
169. Mimura N, Fulciniti M, Gorgun G, Tai T, Cirstea D, Santo L, et al. Blockade of XBP1 splicing by inhibition of IRE1α is a promising therapeutic option in multiple myeloma. Blood. (2012) 119:5772–81. doi: 10.1182/blood-2011-07-366633
170. Zhao N, Cao J, Xu LY, Tang QZ, Dobrolecki LE, Lv XD, et al. Pharmacological targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven breast cancer. J Clin Invest. (2018) 128:1283–99. doi: 10.1172/JCI95873
171. Ghosh R, Wang L, Wang ES, Perera BGK, Igbaria A, Morita S, et al. Allosteric inhibition of the IRE1α RNase preserves cell viability and function during endoplasmic reticulum stress. Cell. (2014) 158:534–48. doi: 10.1016/j.cell.2014.07.002
172. Sheng X, Nenseth HZ, Qu S, Kuzu OF, Frahnow T, Simon L, et al. IRE1α-XBP1s pathway promotes prostate cancer by activating c-MYC signaling. Nat Commun. (2019) 10:323. doi: 10.1038/s41467-018-08152-3
173. Morita S, Villalta SA, Feldman HC, Register AC, Rosenthal W, Hoffmann-Petersen IT, et al. Targeting ABL-IRE1α signaling spares ER- stressed pancreatic β cells to reverse autoimmune diabetes. Cell Metab. (2017) 25:883–97.e8. doi: 10.1016/j.cmet.2017.04.026
174. Ming J, Ruan SN, Wang MY, Ye D, Fan NN, Meng QY, et al. A novel chemical, STF-083010, reverses tamoxifen-related drug resistance in breast cancer by inhibiting IRE1/XBP1. Oncotarget. (2015) 6:40692–703. doi: 10.18632/oncotarget.5827
175. Thamsen M, Ghosh R, Auyeung VC, Brumwell A, Chapman HA, Backes BJ, et al. Small molecule inhibition of IRE1α kinase/RNase has anti-fibrotic effects in the lung. PLoS ONE. (2019) 14:e0209824. doi: 10.1371/journal.pone.0209824
176. Atkins C, Liu Q, Minthorn E, Zhang SY, Figueroa DJ, Moss K, et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. (2013) 73:1993–2002. doi: 10.1158/0008-5472.CAN-12-3109
177. Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-p yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem. (2012) 55:7193–207. doi: 10.1021/jm300713s
178. Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med. (2013) 5:206ra138. doi: 10.1126/scitranslmed.3006767
179. Radford H, Moreno JA, Verity N, Halliday M, Mallucci GR. PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. (2015) 130:633–42. doi: 10.1007/s00401-015-1487-z
180. Mercado G, Castillo V, Soto P, Lopez N, Axten JM, Sardi SP, et al. Targeting PERK signaling with the small molecule GSK2606414 prevents neurodegeneration in a model of Parkinson's disease. Neurobiol Dis. (2018) 112:136–48. doi: 10.1016/j.nbd.2018.01.004
181. Grande V, Ornaghi F, Comerio L, Restelli E, Masone A, Corbelli A, et al. PERK inhibition delays neurodegeneration and improves motor function in a mouse model of Marinesco-Sjogren syndrome. Hum Mol Genet. (2018) 27:2477–89. doi: 10.1093/hmg/ddy152
182. Ounallah-Saad H, Sharma V, Edry E, Rosenblum K. Genetic or pharmacological reduction of PERK enhances cortical-dependent taste learning. J Neurosci. (2014) 34:14624–32. doi: 10.1523/JNEUROSCI.2117-14.2014
183. Kim MJ, Min SH, Shin SY, Kim MN, Lee H, Jang JY, et al. Attenuation of PERK enhances glucose-stimulated insulin secretion in islets. J Endocrinol. (2018) 236:125–36. doi: 10.1530/JOE-17-0497
184. Blackwood EA, Azizi K, Thuerauf DJ, Paxman RJ, Plate L, Kelly JW, et al. Pharmacologic ATF6 activation confers global protection in widespread disease models by reprograming cellular proteostasis. Nat Commun. (2019) 10:187. doi: 10.1038/s41467-018-08129-2
185. Colla E, Coune P, Liu Y, Pletnikova O, Troncoso JC, Iwatsubo T, et al. Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J Neurosci. (2012) 32:3306–20. doi: 10.1523/JNEUROSCI.5367-11.2012
186. Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, et al. Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature. (2012) 485:507–11. doi: 10.1038/nature11058
187. Rubovitch V, Barak S, Rachmany L, Goldstein RB, Zilberstein Y, Pick CG. The neuroprotective effect of salubrinal in a mouse model of traumatic brain injury. Neuromolecular Med. (2015) 17:58–70. doi: 10.1007/s12017-015-8340-3
188. Ohri SS, Hetman M, Whittemore SR. Restoring endoplasmic reticulum homeostasis improves functional recovery after spinal cord injury. Neurobiol Dis. (2013) 58:29–37. doi: 10.1016/j.nbd.2013.04.021
189. Way SW, Podojil JR, Clayton BL, Zaremba A, Collins TL, Kunjamma RB, et al. Pharmaceutical integrated stress response enhancement protects oligodendrocytes and provides a potential multiple sclerosis therapeutic. Nat Commun. (2015) 6:6532. doi: 10.1038/ncomms7532
190. Vieira FG, Ping Q, Moreno AJ, Kidd JD, Thompson K, Jiang B, et al. Guanabenz Treatment accelerates disease in a mutant SOD1 mouse model of ALS. PLoS ONE. (2015) 10:e0135570. doi: 10.1371/journal.pone.0135570
191. Wang L, Popko B, Tixier E, Roos RP. Guanabenz, which enhances the unfolded protein response, ameliorates mutant SOD1-induced amyotrophic lateral sclerosis. Neurobiol Dis. (2014) 71:317–24. doi: 10.1016/j.nbd.2014.08.010
192. Jiang HQ, Ren M, Jiang HZ, Wang J, Zhang J, Yin X, et al. Guanabenz delays the onset of disease symptoms, extends lifespan, improves motor performance and attenuates motor neuron loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neuroscience. (2014) 277:132–8. doi: 10.1016/j.neuroscience.2014.03.047
193. Das I, Krzyzosiak A, Schneider K, Wrabetz L, D'Antonio M, Barry N, et al. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science. (2015) 348:239–42. doi: 10.1126/science.aaa4484
194. Berger E, Haller D. Structure-function analysis of the tertiary bile acid TUDCA for the resolution of endoplasmic reticulum stress in intestinal epithelial cells. Biochem Biophys Res Commun. (2011) 409:610–5. doi: 10.1016/j.bbrc.2011.05.043
Keywords: UPR, immunity, tissue homeostasis, IBD, CRC
Citation: Coleman OI and Haller D (2019) ER Stress and the UPR in Shaping Intestinal Tissue Homeostasis and Immunity. Front. Immunol. 10:2825. doi: 10.3389/fimmu.2019.02825
Received: 30 September 2019; Accepted: 18 November 2019;
Published: 04 December 2019.
Edited by:
Cláudia Pereira, University of Coimbra, PortugalReviewed by:
Joep Grootjans, University of Amsterdam, NetherlandsJudith Smith, University of Wisconsin-Madison, United States
Copyright © 2019 Coleman and Haller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dirk Haller, ZGlyay5oYWxsZXJAdHVtLmRl