
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 06 December 2019
Sec. Autoimmune and Autoinflammatory Disorders
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02814
 Amandine Charras1†
Amandine Charras1† Pinelopi Arvaniti2,3,4†
Pinelopi Arvaniti2,3,4† Christelle Le Dantec1
Christelle Le Dantec1 George N. Dalekos3,4Kaliopi Zachou3,4
George N. Dalekos3,4Kaliopi Zachou3,4 Anne Bordron1‡
Anne Bordron1‡ Yves Renaudineau1,2*‡
Yves Renaudineau1,2*‡Primary Sjögren's syndrome (SjS) is a complex autoimmune epithelitis, with few treatment options, but the use of Janus kinase (JAK) inhibitors is promising because suppression of the JAK/signal transducer and activator of transcription (STAT) pathway improves sicca manifestations. Playing a primary and pathogenic role in disease development, the oxidative stress response is upregulated in activated salivary gland epithelial cells (SGECs) from patients with SjS. Therefore, the aim of this study was to investigate whether JAK inhibitors would suppress SGEC activation in response to an oxidative stress. For this purpose, the human salivary gland (HSG) cell line was used, and cells were treated with the reactive oxygen species (ROS) inducer hydrogen peroxide (H2O2) or with interferons (IFN Type I and Type II), used as positive controls, to mimic activated SGECs as observed in SjS patients. Afterward, the levels of the intracellular adhesion molecule-1 (ICAM-1) and the regulatory programmed-death ligand-1 (PD-L1) were measured by real-time PCR and flow cytometry, and the STAT1/3 phosphorylation status was assessed by Western blotting. Using the HSG cell line, our results showed that both ICAM-1 and PD-L1 are induced by ROS through pSTAT3, and that this activation pathway is reversed by the use of JAK inhibitors, AG490 and ruxolitinib, as well as by N-acetylcysteine, which is a direct inhibitor of ROS. These findings open new perspectives regarding the pathogenesis and therapeutic possibilities for SjS.
Primary Sjögren's syndrome (SjS) is a chronic autoimmune epithelitis affecting mainly women, which is characterized by lymphocytic infiltration of lachrymal and salivary glands, resulting in progressive loss of their secretory function. Patients suffer from xerophthalmia and xerostomia, also known as sicca syndrome, while systemic manifestations and multiple organ involvement are not infrequent and represent a worse clinical course of the disease, with high risk for the development of B-cell lymphoma (1–4). Although both genetic and epigenetic studies have opened new perspectives for understanding this complex disease, the etiology of SjS remains obscure (5). Histological data from salivary glands have shown activation of oxidative stress in salivary gland epithelial cells (SGECs) associated with infiltration by activated CD4(+) and CD8(+) T lymphocytes (LT) (6). Along with the progression of the disease, dendritic cells and B lymphocytes are also recruited, leading to local production of interferon (IFN) type I and type II and of circulating autoantibodies, such as anti-sicca syndrome type A (SSA/Ro) and type B (SSB/La) (7–11).
A multitude of soluble factors and cell surface molecules are upregulated in SGECs during SjS development, including both positive and negative regulators of autoimmune responses and pathologies. Among them, the intracellular adhesion molecule-1 (ICAM-1) seems to play an important role in SjS pathogenesis. The interaction of SGECs, which express ICAM-1, with immune cells through their ligands, leads to the retention of infiltrating lymphocytes. ICAM-1 upregulation in SjS was correlated not only with the lymphocytic infiltration but also with disease activity and severity (12–14). Moreover, studies in animal models have shown that blocking ICAM-1 may result in attenuation of salivary gland inflammation in early stages of the disease (15). Another example is programmed-death ligand-1 (PD-L1), a transmembrane protein, that presents a dual function. On one hand, PD-L1 is associated with epithelial cell survival and resistance to IFN-mediated apoptosis (16); on the other hand, PD-L1 downregulates various immune responses by engaging the co-inhibitory receptor programmed death-1 (PD-1) protein. When activated by its ligand, PD-1 suppresses T-cell activation and IFNγ production by TH1 cells. PD-L1 is expressed constitutively in myeloid cells and can be induced in many cell types, including epithelial cells, after exposure to pro-inflammatory stimuli. It has been shown that PD-L1 expression is elevated in SGECs of SjS patients, suggesting a potential protective role for the epithelial cells as well as an immunosuppressive role for infiltrating T cells in this disease (17, 18). As a consequence, blocking the interaction between PD-L1 and PD-1 represents an emerging therapeutic target for both autoimmune and malignant diseases (19, 20).
Reactive oxygen species (ROS) have been implicated in the induction of inflammation and tissue damage in the field of SjS (21) and other autoimmune diseases (22–25). The role of ROS in the activation of SGECs during SjS is supported by recent studies (6). First, ROS were proved to increase ICAM-1 in epithelial cell lines and to increase its ability to bind neutrophils (26), whereas treatment with N-acetylcysteine (NAC), a ROS scavenger, was able to block ICAM-1 expression in myeloid cell lines (27). ICAM-1 blockade with selective anti-ICAM-1 monoclonal antibodies prevented ROS production in epithelial cells (28). Second, the role of ROS on PD-L1 expression is also suggested as recent studies have shown an increase of PD-L1 after treatment with ROS inducers in macrophages (29). Moreover, a link between ROS and PD-L1 upregulation was described in non-small epithelial cell lung carcinoma and was correlated with cisplatin resistance (30). According to current literature, IFNs have been proven to be the main regulators of both ICAM-1 and PD-L1 expression in epithelial cells, including SGECs through the activation of the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway and subsequent induction of IFN gamma activated site (GAS) and IFN response element (IRE) (31–34). Our hypothesis is that part of this activation may be related to the oxidative stress.
Based on the hypothesis mentioned above and our recent study showing an interplay between ROS and the JAK/STAT pathway to control epigenetic actors (35), the aim of our study was to investigate the role of ROS on the expression of ICAM-1 and PD-L1, as well as to elucidate the potential reversible effect of JAK inhibitors and NAC in this process, a finding that could lead to the development of a novel therapeutic target in this still incurable disease.
The human salivary gland (HSG) cell line, commonly used as an in vitro model to study salivary gland pathways, was used (36–39). Cells were suspended in Dulbecco's modified Eagle's medium (DMEM; Lonza Inc., Allendale, NJ), supplemented with fetal bovine serum (Eurobio, Les Ulis, France), with 2 mM L-glutamine, 250 mg/ml amphotericin B, and penicillin/streptomycin, at 37°C and with 5% CO2. Trypsin was used to collect cells, and cells were cultured afterward, unless otherwise specified, in the presence of 25 ng/ml type I IFNα (500 U/ml, ImmunoTools, Friesoythe, Germany); 25 ng/ml type II IFNγ (500 U/ml, ImmunoTools); 150 μM hydrogen peroxide (H2O2); 40 μM AG490, a JAK2/epidermal growth factor receptor (EGFR) inhibitor (Sigma, St. Louis, USA); 100 nM ruxolitinib, a JAK1/2 inhibitor (Jakavi, Novartis, Basel, Switzerland); and 50 mM NAC (Hidonac®, injecting solution 5 g/ml), a membrane-penetrating antioxidant and ROS modulator for 48 h.
For quantitative PCR, total RNA was extracted from the cells using the RNAble® (Eurobio) according to the manufacturer's instructions. The purity and quantity of the RNA were measured by determining the ratio of absorbance at 260 and 280 nm (NanoDrop® 1000, Latech). Next, 2 μg of total RNA was converted to cDNA with the Superscript II (ThermoFisher, Waltham, USA) according to the manufacturer's instructions and stored at −20°C. The master mix contained 3 μl of cDNA at dilution 1:50 (12 ng of cDNA), 1 × Power SYBR Green PCR Master Mix or 1 × Taqman assay kit (Applied Biosystems, Foster City, CA) and 250 nM of each primer. For the SYBR Green PCR: ICAM-1 (primer sense 5′-GCCGGCCAGCTTATACACAA-3′; reverse 5′-TGGCCACGTCCAGTTTCC-3′) and GAPDH (primer sense 5′-TGCCCTCAACGACCACTTT-3′; reverse 5′-GGTCCAGGGGTCTTACTCCTT-3′; For the Taqman assays: Hs00204257 (PD-L1) and Hs02758991 (GAPDH). The relative quantification of gene expression was calculated using the formula of 2-ΔΔCT with the use of GAPDH as an internal control, and results were expressed relative to the baseline.
For cell surface marker determination, HSG cells were cultured for 48 h in the presence of H2O2, IFNα, or IFNγ both with and without the presence of AG490, ruxolitinib, and NAC. Afterward, the effect on ICAM-1 and PD-L1 plasma membrane expression was evaluated using ICAM-1 (CD54)-FITC (Beckman Coulter, Brea, CA) and PD-L1 (CD274)-PE (Thermo Fisher Scientific, Waltham, US) anti-mouse antibodies.
For measurement of the oxidative stress, the cell-permeant 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA, Carlsbad, CA) was used as an indicator for ROS in cells. Upon cleavage of the acetate groups by intracellular esterases and oxidation, the nonfluorescent H2DCFDA is converted to the highly fluorescent 2′,7′-dichlorofluorescein (DCF). Thus, the florescence level reflects the ROS generation in the cell. After cell collection, cells were washed with PBS, stained with 10 μg of H2DCFDA at 37°C, washed with PBS, and then fluorescence was monitored by flow cytometry. A treatment with 250 μM of H2O2 was used as a positive marker. For the evaluation of cell apoptosis, cells were recovered, washed, and stained for 15 min with FITC-conjugated annexin-V (AV)/propidium iodide (PI) according to the Beckman-Coulter apoptosis kit protocol.
A whole-protein extraction was performed by the use of a cellular lysis buffer containing 5 M sodium chloride (NaCl), 1 M Tris hydrochloride (Tris-HCl), 100 mM sodium fluoride (NaF), and 0.1 M ethylene diamine tetraacetic acid (EDTA), enriched with a cocktail of protease inhibitors. The quantity of extracted proteins was estimated with the MicroBCA Assay Protein Quantification kit (Interchim, San Diego, CA). The amounts of pSTAT1 (Y701), pSTAT3 (Y705), STAT1, and beta-actin were evaluated by WB using specific anti-mouse primary antibodies in the BD Pharmingen kit (BD Biosciences, France) and followed with the appropriate horseradish peroxidase (HRP)-conjugated secondary anti-mouse or anti-rabbit IgG antibodies according to manufacturer's instructions (anti-mouse biotinylated 1/5,000; streptavidin HRP 1/500) (GE Health Care Limited, UK). Signals were visualized by Luminata Forte Western HRP Substrate (EMD Millipore, Billerica, USA).
The results are expressed as arithmetic means with standard error of the mean (SEM), and differences among groups were analyzed by one-way ANOVA, and the Tukey's test was used for post hoc multiple comparisons test using GraphPad-Prism 7.0 software (La Jolla, CA, USA). Significance was assessed as p < 0.05.
To evaluate the effect of ROS mediated by H2O2 on the expression of ICAM-1 and PD-L1 compared to the effect of IFN type I (IFNα) or IFN type II (IFNγ), HSG cells were cultured for 48 h under treatment with H2O2 (25–150 μM), IFNα (25 ng/ml), or IFNγ (25 ng/ml). Then, the levels of ICAM-1 and PD-L1 were tested for transcriptional expression by real-time PCR, and for protein synthesis by flow cytometry, and mean fluorescence intensity (MFI) was reported for each treatment (Figures 1A–F). Regarding ROS activation mediated by H2O2, the expression levels of ICAM-1 and PD-L1 were increased in a dose–response relationship. At optimal concentration (150 μM H2O2), an increase for both ICAM-1 and PD-L1 was observed by real-time PCR and the induction confirmed by FACS. As expected, the positive controls IFNα and IFNγ strongly increased ICAM-1 and PD-L1, respectively. To exclude that IFNα/γ could also induce ROS production, cells were incubated for 6–48 h with IFNα, IFNγ, and H2O2, and the production of ROS was followed by flow cytometry (Figure 2A). Results revealed that only H2O2 induces ROS production in HSG cells with a maximal effect observed at 48 h, whereas no effect was observed for IFNα and IFNγ.
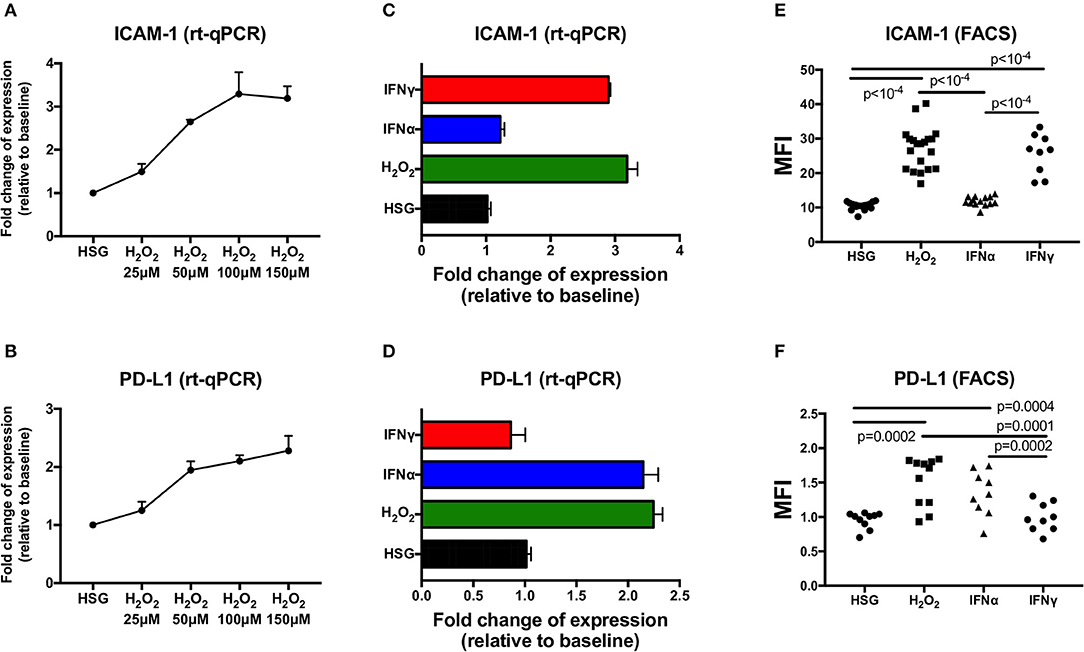
Figure 1. Expression of ICAM-1 and PD-L1 measured by real-time PCR and flow cytometry in the human salivary gland (HSG) cell line after 48 h of treatment with H2O2, IFNα, and IFNγ, respectively. (A,B) H2O2 dose effect on ICAM-1 and PD-L1 mRA expression (n = 3). (C,D) HSG (no treatment), H2O2 (150 μM), IFNα, and IFNγ effect on ICAM-1 and PD-L1 mRA expression (n = 3). (E,F) No treatment (n = 17 and 10, respectively), H2O2 (n = 20 and 11), IFNα (n = 14 and 9), and IFNγ (n = 9 both) effect on cell surface ICAM-1 and PD-L1 expression as evaluated by flow cytometry. MFI, mean fluorescence intensity; ICAM-1, intracellular adhesion molecule-1; PD-L1, programmed death ligand 1.
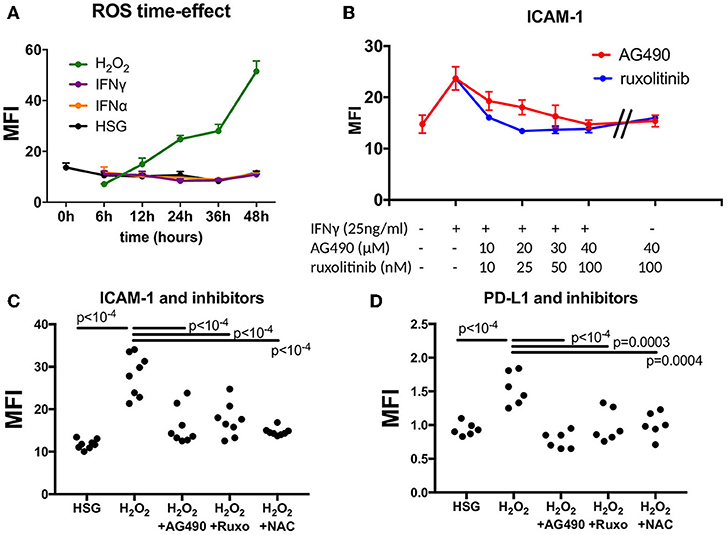
Figure 2. Effect of JAK inhibitors and NAC on ICAM-1 and PD-L1 induction mediated by IFNγ and H2O2. (A) Measurement of ROS in a time effect by flow cytometry after treatment with H2O2, IFNα, and IFNγ (n = 3). (B) Dose effect of the JAK inhibitors AG490 and ruxotinib to reverse ICAM-1 induction mediated by IFNγ. (C) AG490, ruxolitinib, and NAC reverse H2O2-mediated induction of ICAM-1 (n = 8). (D) AG490, ruxolitinib, and NAC reverse H2O2-mediated induction of PD-L1 (n = 6). ICAM-1, intracellular adhesion molecule-1; NAC, N-acetylcysteine; PD-L1, programmed death ligand 1; ROS, reactive oxygen species; H2O2, hydrogen peroxide.
Next, we tested whether ICAM-1 and PD-L1 induction mediated by H2O2 was reversed by JAK inhibitors and NAC. At first, we used increasing concentrations of AG490 and ruxolitinib in the presence of IFNγ (25 ng/ml) and estimated the optimal concentration achieving ICAM-1 reduction, which was observed at 40 μM of AG490 and at 100 nM of ruxolitinib (Figure 2B). Similarly, for NAC, we used increasing concentrations of NAC in the presence of 150 μM of H2O2, and the optimal effect on ROS reduction was achieved at 50 mM (data not shown). When cells were exposed to JAK inhibitors and NAC at optimal concentrations, no effects on ICAM-1 and PD-L1 were observed. (Figure 2B and data not shown) In all conditions, apoptosis was not significantly increased (<5%) as evaluated by annexin V and propidium iodide staining (data not shown).
Afterward, cells were treated with 150 μM of H2O2 in the presence of 40 μM of AG490, 100 nM of ruxolotinib, and 50 mM of NAC for 48 h, and MFI was calculated by flow cytometry (Figures 2C,D). Results revealed a significant decrease of both ICAM-1 and PD-L1 after treatment with H2O2 in the presence of AG490, ruxolitinib, and NAC. We concluded from these experiments that ROS were effective in controlling ICAM-1 and PD-L1 expression in the HSG cell line through the JAK/STAT pathway.
To test our hypothesis that the H2O2 effect on ICAM-1 and PD-L1 involved a JAK/STAT pathway, we further tested STAT1 (Y701) and STAT3 (Y705) phosphorylation (p) status in HSG after incubation with increasing concentrations of H2O2. Afterward, whole-cell extracts were isolated, and the pSTAT1 and pSTAT3 status was assessed by Western blot. H2O2 treatment was effective in increasing pSTAT3, with the optimal effect reported at 150 μM, whereas no effects were reported for STAT1 and pSTAT1 (Figure 3A). In addition to this, we used, as in previous experiments, the two JAK inhibitors (AG490 40 μM and ruxolitinib 100 nM) as well as NAC (50 mM) to test their effect on pSTAT3 induction mediated by H2O2. We showed that both JAK inhibitors and NAC reverse pSTAT3 activation (Figures 3B,C).
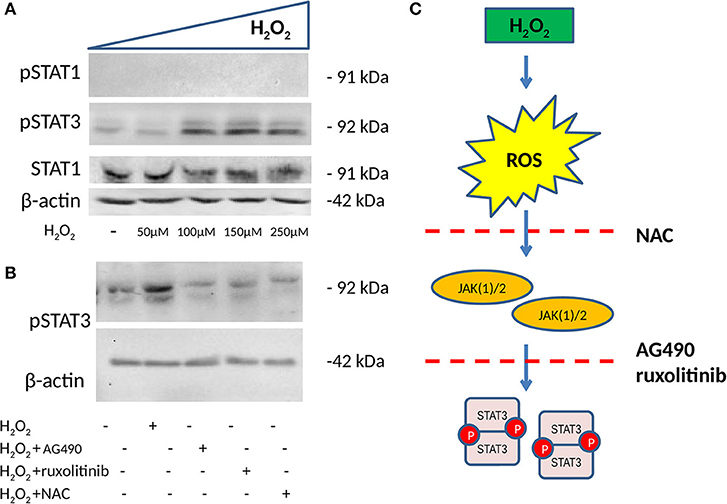
Figure 3. Phosphorylation status of pSTAT1 Y701 and pSTAT3 Y705 measured by WB (n = 3 each). (A) WB showing the expression of pSTAT1, pSTAT3, and total STAT1 after 48 h of treatment with increasing concentrations of H2O2. H2O2 increases pSTAT3 with the optimal effect observed at 150 μM, whereas no effect is observed on the expression of pSTAT1. (B) WB showing expression of pSTAT3 after 48 h of treatment with 150 μM of H2O2 with and without two JAK(1)/2 inhibitors, AG490 40 μM, a JAK2 inhibitor, ruxolitinib 100 nM a JAK1/2 inhibitor and NAC 50 mM. Both JAK1/2 inhibitors and NAC reverse H2O2-mediated induction of pSTAT3. (C) ROS induction mediated by H2O2 phosphorylates STAT3, a process that is reversed by NAC and JAK(1)/2 inhibitors. ROS, reactive oxygen species; H2O2, hydrogen peroxide; JAK, Janus kinase; STAT, signal transducer and activator of transcription; ICAM-1, intracellular adhesion molecule-1; PD-L1, programmed death ligand 1; WB, Western blot.
Our study extends the initial observations that oxidative stress and ROS are implicated in inflammation and tissue damage in SjS (6, 21, 40), as we demonstrate that ROS are effective in inducing ICAM-1 and PD-L1 expression in the HSG cell line through phosphorylation of STAT3. In addition and in agreement with in vivo studies supporting the involvement of JAK inhibitors and NAC in SjS (41), we have reported that the effects of H2O2 on ICAM-1 and PD-L1 can be reversed not only by JAK inhibitors but also by NAC, which is a direct ROS inhibitor. These findings open new perspectives in understanding SjS pathogenesis and the subsequent development of new therapeutic targets based on an in vitro cellular model using H2O2-activated HSG cells.
Studies on SGECs have shown upregulation of both ICAM-1 and PD-L1 in SjS patients (12–14, 17, 18), and until recently, their expression was related to type I and type II IFN signaling through the implication of the JAK-STAT pathway (22–24, 27, 28). However, recently published data, mainly in the field of immune cells and carcinogenesis, support the role of ROS on ICAM-1 and PD-L1 expression (26–29). In our study, we revealed a significant induction of both ICAM-1 and PD-L1 in HSG cells after treatment with H2O2 compared to the expected induction mediated by IFNs. As a consequence, part of the IFN signature described in SjS may be, in fact, related to the oxidative stress, and this is supported by a recent study using an epigenetic approach (40, 42).
In addition to this, we also demonstrated that JAK inhibitors and NAC reverse ICAM-1 and PD-L1 induction mediated by H2O2. Regarding the JAK target and based on the fact that ruxolitinib is a specific JAK1/2 inhibitor with very low affinity for non-JAK targets and that AG490 selectively inhibits JAK2 and EGFR, this supports a key role played by JAK2. Such hypothesis regarding the role of JAK2 needs confirmation with another approach. The effect of JAK inhibitors on regulating the expression of ICAM-1 and PD-L1 mediated by IFN has been studied, and it was shown that JAK inhibitors may affect the progression of autoimmune or malignant diseases by inhibiting ICAM-1 expression (43, 44) and that they could act beneficially in malignant diseases by blocking PD-L1 and reinforcing in this way the effect of antineoplastic drugs on tumor cells (16, 45, 46). Moreover, our findings are in agreement with previous reports in which the expression of ICAM-1 induced by 12-O-tetradecanoyl-phorbol-13-acetate (TPA) was linked to the generation of ROS and was reversed after treatment with NAC, a ROS scavenger (28), but also with the fact that PD-L1 was upregulated in macrophages when treated with ROS inducers (29). NAC has been proved to have a therapeutic effect in SjS mainly by attenuating ocular symptoms but is also believed to act as an anti-inflammatory agent by blocking IL-8 and ICAM-1 expression in epithelial cells, as has been described in the field of obstructive pulmonary disease (47, 48).
To investigate the potential implication of JAK/STAT pathway in ICAM-1 and PD-L1 upregulation by ROS, we investigated the phosphorylation status of pSTAT3 and not only demonstrated induction of pSTAT3 mediated by H2O2, whereas no effect was observed for pSTAT1, but also that JAK inhibitors and NAC reverse pSTAT3 induction mediated by ROS. Our findings are in agreement with previous studies where it was shown that ROS activate pSTAT3 in the field of carcinogenesis and in human T cells (25, 49) and that NAC reduces pSTAT3 expression and protein phosphorylation in animal models (50). Intracellular mechanisms linking ROS with STAT3 activation are poorly understood and may involve either intrinsic factors because the mitochondria have the capacity to interact with STAT3 or extrinsic factors through an autocrine loop with cytokines, such as IL-6 (51, 52). As a result, our findings suggest that oxidative stress mediates, through a direct or indirect mechanism, the phosphorylation of STAT3 (Y705) and the expression of ICAM-1 and PD-L1 in SGECs, a process that is reversed by JAK/STAT and ROS inhibitors.
The role of ICAM-1 and PD-L1 signaling blockade on SjS development has been described previously in animal models. Roescher et al. described that blocking ICAM-1 in non-obese diabetic mice at early stages of the disease leads to a moderate reduction of inflammation but may cause adverse events once the inflammation is established (15). In the same animal model, Zhou et al. described that upregulation of PD-L1 impedes TH1 and B-cell recruitment by reducing IFNγ production, acting in this way as a negative feedback of the disease, whereas PD-L1 blockade accelerates salivary gland dysfunction (17). However, the inhibition of PD-L1 in this manner represents a much more complex mechanism because PD-L1's functions are not restricted to a critical role in maintaining self-tolerance by blocking T-cell activation through binding PD-1 (53, 54). In autoimmune diseases, autoreactive T cells must be continually kept in check through inhibitory receptor engagement, but in SjS, strong CD8(+) activation is observed, which supports an ineffective PD-L1 pathway in such cases (55, 56). As a result, early signaling through PD-L1 would be insufficient to maintain peripheral tolerance; therefore, tissue expression of PD-L1 would be required to prevent epithelial destruction. This model explains why PD-L1 expression increases along with inflammation (55–58). On the other hand, the elevation of intracellular PD-L1 modulates epithelial cells' metabolic pathways (i) by reducing sensitivity of expressing cells to T-cell toxicity, (ii) by enhancing cell survival through an induction of AkT/mTOR pathway, and (iii) by neutralizing the proapoptotic effect of the IFN pathway (58–60). This means that co-inhibitory or co-stimulatory signals provided by PD-L1 may depend upon timing and locale, the type of cells involved, and the relative levels of PD-L1 molecules (53, 61, 62). Indeed, PD-L1 blockade in cancer cells restores their sensitivity to T-cell toxicity as well as IFN-mediated apoptotic effect (63, 64).
Taking into consideration the abovementioned data and combining them with our findings as described in Figure 4, we can speculate that oxidative stress in salivary glands of patients with SjS leads to an upregulation of ICAM-1 and PD-L1 through the implication of a ROS/JAK(1/)2/STAT3 pathway, which acts synergistically with IFN/JAK/STAT-mediated effect on ICAM-1 and PD-L1 expression, initiating in this manner the adhesion and infiltration of activated T cells. PD-L1 is activated along with initiation of inflammation as a negative feedback, but during the development of the disease, there is an inability to control tissue damage and on the contrary contributes to SGEC activation and increased survival and subsequently to disease perpetuation. The administration of JAK inhibitors and NAC reverses ICAM-1 and PD-L1 induction mediated by ROS and strengthens their ability to reduce ICAM-1 and PD-L1 expression mediated by IFNs, a fact that could act beneficially if administered early in disease development.
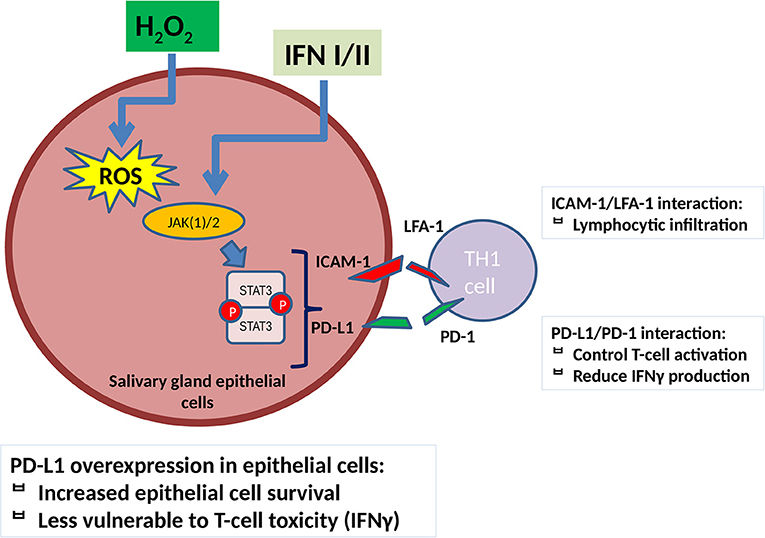
Figure 4. ROS contribute to ICAM-1 and PD-L1 induction in salivary gland epithelial cells through a JAK(1/)2/STAT3 pathway. ICAM-1 upregulation leads to SGEC activation and induces lymphocytic infiltration. PD-L1 reduces T-cell activation and IFNγ production on one hand but, on the other hand, contributes to SGECs activation and disease perpetuation as it increases salivary gland epithelial cell survival and reduces pro-inflammatory TH1 cell-mediated activation and toxicity. JAK(1/)2 inhibitors, AG490 and ruxolitinib, as well as NAC reverse this process as described in Figure 3. IFN(α/γ), interferon α/γ, ICAM-1, intracellular adhesion molecule-1; PD-L1, programmed death ligand 1; TH1, T helper 1 lymphocyte; ROS, reactive oxygen species; H2O2, hydrogen peroxide; TYK, tyrosine kinase; JAK, janus kinase; STAT, signal transducer and activator of transcription.
Adhesion molecules and cell surface immunomodulatory factors are suspected of having an important role in SjS pathogenesis. In our study, we demonstrated in the HSG cell line that ROS induce expression of ICAM-1 and PD-L1 through activation of STAT3. JAK inhibitors and NAC reverse this process, reducing ICAM-1 and PD-L1 expression mediated by ROS. The effect of JAK inhibitors and NAC on ICAM-1 and PD-L1 production could act beneficially in SjS, reducing activation of SGECs and subsequently reducing inflammation and lymphocytic infiltration.
The datasets generated for this study are available on request to the corresponding author.
AC, AB, and YR designed the study. AC, PA, and CL performed the research. AC, PA, GD, KZ, AB, and YR analyzed the data. YR and PA prepared the initial draft. The final manuscript was read and approved by all authors.
This study was supported by research funding from the Association Française de Gougerot Sjögren. PA was supported by a research grant from the Federation for the Development of Internal Medicine in Europe (FDIME).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are thankful to Dr. Wesley H. Brooks (University of South Florida, USA) for editorial assistance and to Servier Medical for providing free art for the figures.
SjS, primary Sjögren's syndrome; ROS, reactive oxygen species; IFNα, interferon α; IFNγ, interferon γ; SGECs, salivary gland epithelial cells; LT, T-lymphocytes; ICAM-1, intracellular adhesion molecule-1; PD-L1, programmed death ligand-1; JAK, Janus kinase; STAT, signal transducer and activator of transcription; WB, Western blotting; PD-1, programmed death-1; GAS element, IFN-gamma activated site; MFI, mean fluorescence intensity.
1. Mavragani CP, Moutsopoulos HM. Sjogren's syndrome. Annu Rev Pathol. (2014) 9:273–85. doi: 10.1146/annurev-pathol-012513-104728
2. Ramos-Casals M, Brito-Zerón P, Sisó-Almirall A, Bosch X. Primary sjogren syndrome. BMJ. (2014) 344:3821. doi: 10.1136/bmj.e3821
3. Ramos-Casals M, Brito-Zerón P, Kostov B, Sisó-Almirall A, Bosch X, Buss D, et al. Google-driven search for big data in autoimmune geoepidemiology: analysis of 394,827 patients with systemic autoimmune diseases. Autoimmun Rev. (2015) 14:670–9. doi: 10.1016/j.autrev.2015.03.008
4. Bordron A, Charras A, Le Dantec C, Renaudineau Y. Épigénome et syndrome de Gougerot-Sjögren. Rev Médecine Interne. (2018) 39:346–51. doi: 10.1016/j.revmed.2017.09.003
5. Renaudineau Y, Ballestar E. Epigenetics: DNA methylation signatures in Sjögren syndrome. Nat Rev Rheumatol. (2016) 12:565–6. doi: 10.1038/nrrheum.2016.144
6. Pagano G, Castello G, Pallardó FV. Sjögren's syndrome-associated oxidative stress and mitochondrial dysfunction: prospects for chemoprevention trials. Free Radic Res. (2013) 47:71–3. doi: 10.3109/10715762.2012.748904
7. Christodoulou MI, Kapsogeorgou EK, Moutsopoulos HM. Characteristics of the minor salivary gland infiltrates in Sjogren's syndrome. J Autoimmun. (2010) 34:400–7. doi: 10.1016/j.jaut.2009.10.004
8. Mingueneau M, Boudaoud S, Haskett S, Reynolds TL, Nocturne G, Norton E, et al. Cytometry by time-of-flight immunophenotyping identifies a blood Sjogren's signature correlating with disease activity and glandular inflammation. J Allergy Clin Immunol. (2016) 137:1809–21. doi: 10.1016/j.jaci.2016.01.024
9. Kapsogeorgou EK, Christodoulou MI, Panagiotakos DB, Paikos S, Tassidou A, Tzioufas AG, et al. Minor salivary gland inflammatory lesions in Sjogren syndrome: do they evolve? J Rheumatol. (2013) 40:1566–71. doi: 10.3899/jrheum.130256
10. Ding J, Zhang W, Haskett S, Pellerin A, Xu S, Petersen B, et al. BAFFoverexpression increases lymphocytic infiltration in Sjogren's target tissue, but only inefficiently promotes ectopic B-cell differentiation. Clin Immunol. (2016) 169:69–79. doi: 10.1016/j.clim.2016.06.007
11. Capaldo C, Carvajal Alegria G, Cornec D, Jousse-Joulin S, Devauchelle-Pensec V, Renaudineau Y. The active immunological profile in patients with primary Sjögren's syndrome is restricted to typically encountered autoantibodies. Clin Exp Rheumatol. (2016) 34:722.
12. Saito I, Terauchi K, Shimuta M, Nishiimura S, Yoshino K, Takeuchi T, et al. Expression of cell adhesion molecules in the salivary and lacrimal glands of sjogren's syndrome. J Clin Lab Anal. (1993) 7:180–7 doi: 10.1002/jcla.1860070309
13. Aziz KE, McCluskey PJ, Wakefield D. Pattern of adhesion molecule expression in labial salivary glands from patients with primary Sjogren's syndrome. Ocul Immunol Inflamm. (1995) 3:221–36. doi: 10.3109/09273949509069116.
14. Błochowiak KJ, Olewicz-Gawlik A, Trzybulska D, Nowak-Gabryel M, Kociecki J, Witmanowski H, et al. Serum ICAM-1, VCAM-1 and E-selectin levels in patients with primary and secondary Sjögren's syndrome. Adv Clin Exp Med. (2017) 26:835–42. doi: 10.17219/acem/61434
15. Roescher N, Vosters JL, Yin H, Illei GG, Tak PP, Chiorini JA. Effect of soluble ICAM-1 on a Sjogren's Syndrome-like phenotype in NOD mice is disease stage dependent. PLoS ONE. (2011) 5:19962. doi: 10.1371/journal.pone.0019962
16. Marcucci F, Rumio C, Corti A. Tumor cell-associated immune checkpoint molecules - Drivers of malignancy and stemness. Biochim Biophys Acta Rev Cancer. (2017) 1868:571–83. doi: 10.1016/j.bbcan.2017.10.006
17. Zhou J, Jin J, Kawai T, Yu Q. Endogenous programmed death ligand-1 restrains the development and onset of Sjögren's syndrome in non-obese diabetic mice. Sci Rep. (2016) 6:39105. doi: 10.1038/srep39105
18. Kobayashi M, Kawano S, Hatachi S, Kurimoto C, Okazaki T, Iwai Y, et al. Enhanced expression of programmed death-1. (PD-1)/PD-L1 in salivary glands of patients with Sjogren's syndrome. J Rheumatol. (2005) 32:2156–63.
19. Doi T, Ishikawa T, Okayama T, Oka K, Mizushima K, Yasuda T, et al. The JAK/STAT pathway is involved in the upregulation of PD-L1 expression in pancreatic cancer cell lines. Oncol Rep. (2017) 37:1545–54. doi: 10.3892/or.2017.5399
20. Leonardi GC, Gainor JF, Altan M. Safety of programmed death−1 pathway inhibitors among patients with Non–small-cell lung cancer and preexisting autoimmune disorders. J Clin Oncol. (2018) 36:1905–12. doi: 10.1200/JCO.2017.77.0305
21. Ryo K, Yamada H, Nakagawa Y, Tai Y, Obara K, Inoue H, et al. Possible involvement of oxidative stress in salivary gland of patients with Sjogren's syndrome. Pathobiology. (2006) 73:252–60. doi: 10.1159/000098211
22. Smallwood MJ, Nissim A, Knight AR, Whiteman M, Haigh R, Winyard PG. Oxidative stress in autoimmune rheumatic diseases. Free Radic Biol Med. (2018) 125:3–14. doi: 10.1016/j.freeradbiomed.2018.05.086
23. Dalmazi G, Hirshberg J, Lyle D, Freij JB, Caturegli P. Reactive oxygen species in organ-specific autoimmunity. Autoimmun Highlights. (2016) 7:11. doi: 10.1007/s13317-016-0083-0
24. Kaffe ET, Rigopoulou EI, Koukoulis GK, Dalekos GN, Moulas AN. Oxidative stress and antioxidant status in patients with autoimmune liver diseases. Redox Rep. (2015) 20:33–41. doi: 10.1179/1351000214Y.0000000101
25. Simon AR, Rai U, Fanburg BL, Cochran BH. Activation of the JAK-STAT pathway by reactive oxygen species. Am J Physiol. (1998) 275:1640–52. doi: 10.1152/ajpcell.1998.275.6.C1640
26. Sellak H, Franzini E, Hakim J, Pasquier C. Reactive oxygen species rapidly increase endothelial ICAM-1 ability to bind neutrophils without detectable upregulation. Blood. (1994) 83:2669–77. doi: 10.1182/blood.V83.9.2669.2669
27. Wang Q, Doerschuk CM. Neutrophil-induced changes in the biomechanical properties of endothelial cells: roles of ICAM-1 and reactive oxygen species. J Immunol. (2000) 164:6487–94. doi: 10.4049/jimmunol.164.12.6487
28. Traore K, Sharma RB, Burek CL, Trush MA. Role of ROS and MAPK in TPA-induced ICAM-1 expression in the myeloid ML-1 cell line. J Cell Biochem. (2007) 100:1010–21. doi: 10.1002/jcb.21101
29. Roux C, Jafari SM, Shinde R, Duncan G, Cescon DW, Silvester J, et al. Reactive oxygen species modulate macrophage immunosuppressive phenotype through the up-regulation of PD-L1. Proc Natl Acad Sci USA. (2019) 15:201819473. doi: 10.1073/pnas.1819473116
30. Wangpaichitr M, Kandemir H, Li YY, Wu C, Nguyen D, Feun LG, et al. Relationship of metabolic alterations and PD-L1 expression in cisplatin resistant lung cancer cell. Dev Biol. (2017) 6:183. doi: 10.4172/2168-9296
31. Wu AJ, Kurrasch RH, Katz J, Fox PC, Baum BJ, Atkinson JC. Effect of tumor necrosis factor-alpha and interferon-gamma on the growth of a human salivary gland cell line. J Cell Physiol. (1994) 161:217–26. doi: 10.1002/jcp.1041610205
32. Chang YJ, Holtzman MJ, Chen CC. Interferon-gamma-induced epithelial ICAM-1 expression and monocyte adhesion. Involvement of protein kinase C-dependent c-Src tyrosine kinase activation pathway. J Biol Chem. (2002) 277:7118–26. doi: 10.1074/jbc.M109924200
33. Garcia-Diaz A, Sanghoon Shin D, Moreno HB, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. (2017) 19:1189–201. doi: 10.1016/j.celrep.2017.04.031
34. Mimura K, Teh JL, Okayama H, Shiraishi K, Kua LF, Koh V, et al. PD-L1 expression is mainly regulated by interferon gamma associated with JAK-STAT pathway in gastric cancer. Cancer Sci. (2018) 109:43–53. doi: 10.1111/cas.13424
35. Charras A, Arvaniti P, Le Dantec C, Arleevskaya MI, Zachou K, Dalekos GN, et al. JAK inhibitors suppress innate epigenetic reprogramming: a promise for patients with Sjögren's syndrome. Clin Rev Allergy Immunol. (2019). doi: 10.1007/s12016-019-08743-y. [Epub ahead of print].
36. Nelson J, Manzella K, Baker OJ. Current cell models for bioengineering a salivary gland: a mini-review of emerging technologies. Oral Dis. (2013) 19:236–44. doi: 10.1111/j.1601-0825.2012.01958.x
37. Konsta OD, Charras A, Le Dantec C, Kapsogeorgeou E, Bordron A, Brooks WH, et al. Epigenetic modifications in salivary glands from patients with Sjögren's syndrome affect cytokeratin 19 expression. Bull Group Int Rech Sci Stomatol Odontol. (2016) 53:e01.
38. Thabet Y, Le Dantec C, Ghedira I, Devauchelle V, Cornec D, Pers JO, et al. Epigenetic dysregulation in salivary glands from patients with primary Sjogren's syndrome may be ascribed to infiltrating B cells. J Autoimmun. (2013) 41:175–81. doi: 10.1016/j.jaut.2013.02.002
39. Konsta OD, Le Dantec C, Charras A, Cornec D, Kapsogeorgou EK, Tzioufas AG, et al. Defective DNA methylation in salivary gland epithelial acini from patients with Sjogren's syndrome is associated with SSB gene expression, anti-SSB/LA detection, and lymphocyte infiltration. J Autoimmun. (2016) 68:30–8. doi: 10.1016/j.jaut.2015.12.002
40. Cole MB, Quach H, Quach D, Baker A, Taylor KE, Barcellos LF, et al. Epigenetic signatures of salivary gland inflammation in sjögren's syndrome. Arthritis Rheumatol. (2016) 68:2936–44. doi: 10.1002/art.39792
41. Lee J, Lee J, Kwok SK, Baek S, Jang SG, Hong SM, et al. JAK-1 inhibition suppresses interferon-induced BAFF production in human salivary gland: potential therapeutic strategy for primary sjögren's syndrome. Arthritis Rheumatol. (2018) 70:2057–66. doi: 10.1002/art.40589
42. Charras A, Konsta OD, Le Dantec C, Bagacean C, Kapsogeorgou EK, Tzioufas AG, et al. Cell-specific epigenome-wide DNA methylation profile in long-term cultured minor salivary gland epithelial cells from patients with Sjögren's syndrome. Ann Rheum Dis. (2017) 76:625–28. doi: 10.1136/annrheumdis-2016-210167
43. Duzagac F, Inan S, Ela Simsek F, Acikgoz E, Guven U, Khan SA, et al. JAK/STAT pathway interacts with intercellular cell adhesion molecule. (ICAM) and vascular cell adhesion molecule. (VCAM) while prostate cancer stem cells form tumor spheroids. J Buon. (2015) 20:1250–7.
44. Jin W, Huang W, Chen L, Jin M, Wang Q, Gao Z, et al. Topical application of JAK1/JAK2 inhibitor momelotinib exhibits significant anti-inflammatory responses in DNCB-induced atopic dermatitis model mice. Int J Mol Sci. (2018) 19:3973. doi: 10.3390/ijms19123973
45. Kuol N, Stojanovska L, Nurgali K, Apostolopoulos V. PD-1/PD-L1 in disease Immunotherapy. (2018) 10:149–60. doi: 10.2217/imt-2017-0120
46. Luo N, Formisano L, Gonzalez-Ericsson PI, et al. Melanoma response to anti-PD-L1 immunotherapy requires JAK1 signaling, but not JAK2. Oncoimmunology. (2018) 7:1438106. doi: 10.1080/2162402X.2018.1438106
47. Walters MT, Rubin CE, Keightley SJ, Ward CD, Cawley MI. A double-blind, cross-over, study of oral N-acetylcysteine in Sjögren's syndrome. Scand J Rheumatol Suppl. (1986) 61:253–8.
48. Radomska-Leśniewska DM, Sadowska AM, Van Overveld FJ, Demkow U, Zielinski J, De Backer WA. Influence of N-acetylcysteine on ICAM-1 expression and IL-8 release from endothelial and epithelial cells. J Physiol Pharmacol. (2006) 57:325–34.
49. Carballo M, Conde M, El Bekay R, Martín-Nieto J, Camacho MJ, Monteseirín J, et al. Oxidative stress triggers STAT3 tyrosine phosphorylation and nuclear translocation in human lymphocytes. J Biol Chem. (1999) 274:17580–6. doi: 10.1074/jbc.274.25.17580
50. Al-Samhari MM, Al-Rasheed NM, Al-Rejaie S, Al-Rasheed NM, Hasan IH, Mahmoud AM, et al. Possible involvement of the JAK/STAT signaling pathway in N-acetylcysteine-mediated antidepressant-like effects. Exp Biol Med. (2016) 241:509–18. doi: 10.1177/1535370215619707
51. Cheng X, Peuckert C, Wölfl S. Essential role of mitochondrial Stat3 in p38(MAPK) mediated apoptosis under oxidative stress. Sci Rep. (2017) 7:15388. doi: 10.1038/s41598-017-15342-4
52. Wu WC, Hu DN, Gao HX, Chen M, Wang D, Rosen R, et al. Subtoxic levels hydrogen peroxide-induced production of interleukin-6 by retinal pigment epithelial cells. Mol Vis. (2010) 16:1864–73.
53. Dai S, Jia R, Zhang X, Fang Q, Huang L. The PD-1/PD-Ls pathway and autoimmune diseases. Cell Immunol. (2004) 290:72–9. doi: 10.1016/j.cellimm.2014.05.006
54. Fife BT, Pauken KE. The role of the PD-1 pathway in autoimmunity and peripheral tolerance. Ann N YAcad Sci. (2011) 1217:45–59. doi: 10.1111/j.1749-6632.2010.05919.x
55. Mezache L, Magro C, Hofmeister C, Pichiorri F, Sborov D, Nuovo GJ. Modulation of PD-L1 and CD8 activity in idiopathic and infectious chronic inflammatory conditions. Appl Immunohistochem Mol Morphol. (2017) 25:100–9. doi: 10.1097/PAI.0000000000000298
56. Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T. Establishment of NOD-Pdcd1-/- mice as an efficient animal model of type I diabetes. Proc Natl Acad Sci USA. (2005) 102:11823–8. doi: 10.1073/pnas.0505497102
57. Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol. (2002) 169:5538–45. doi: 10.4049/jimmunol.169.10.5538
58. Kinter AL, Godbout EJ, McNally JP, Sereti I, Roby GA, O'Shea MA, et al. The common gamma-chain cytokines IL-2, IL-7, IL-15, and IL-21 induce the expression of programmed death-1 and its ligands. J Immunol. (2008) 181:6738–46. doi: 10.4049/jimmunol.181.10.6738
59. Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, et al. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by Tcell receptor. (TCR) transgenic CD8+T cells. Cancer Res. (2004) 64:1140–5. doi: 10.1158/0008-5472.CAN-03-3259
60. Clark CA, Gupta HB, Sareddy G, Pandeswara S, Lao S, Yuan B, et al. Tumor-Intrinsic PD-L1 signals regulate cell growth, pathogenesis, and autophagy in ovarian cancer and melanoma. Cancer Res. (2016) 76:6964–74. doi: 10.1158/0008-5472.CAN-16-0258
61. Yadav D, Hill N, Yagita H, Azuma M, Sarvetnick N. Altered availability of PD- 1/PD ligands is associated with the failure to control autoimmunity in NOD mice. Cell Immunol. (2009) 258:161–71. doi: 10.1016/j.cellimm.2009.04.006
62. Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. (2006) 203:883–95. doi: 10.1084/jem.20051776
63. Escors D, Gato-Cañas M, Zuazo M, Arasanz H, García-Granda MJ, Vera R, et al. The intracellular signalosome of PD-L1 in cancer cells. Signal Transduct Target Ther. (2018) 3:26. doi: 10.1038/s41392-018-0022-9
Keywords: primary sjögren's syndrome, ROS, JAK/STAT pathway, ICAM-1, PD-L1
Citation: Charras A, Arvaniti P, Le Dantec C, Dalekos GN, Zachou K, Bordron A and Renaudineau Y (2019) JAK Inhibitors and Oxidative Stress Control. Front. Immunol. 10:2814. doi: 10.3389/fimmu.2019.02814
Received: 12 May 2019; Accepted: 15 November 2019;
Published: 06 December 2019.
Edited by:
Ann Marie Reed, School of Medicine, Duke University, United StatesReviewed by:
Arian Dominic John Laurence, University College London Hospitals NHS Foundation Trust, United KingdomCopyright © 2019 Charras, Arvaniti, Le Dantec, Dalekos, Zachou, Bordron and Renaudineau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yves Renaudineau, eXZlcy5yZW5hdWRpbmVhdUB1bml2LWJyZXN0LmZy
†These authors have contributed equally to this work
‡These authors share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.