 Chukwunonso Onyilagha
Chukwunonso Onyilagha Jude Ezeh Uzonna
Jude Ezeh Uzonna
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 22 November 2019
Sec. Microbial Immunology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02738
This article is part of the Research Topic Regulation of Immunity to Parasitic Infections Endemic to Africa View all 17 articles
Parasites, including African trypanosomes, utilize several immune evasion strategies to ensure their survival and completion of their life cycles within their hosts. The defense factors activated by the host to resolve inflammation and restore homeostasis during active infection could be exploited and/or manipulated by the parasites in an attempt to ensure their survival and propagation. This often results in the parasites evading the host immune responses as well as the host sustaining some self-inflicted collateral tissue damage. During infection with African trypanosomes, both effector and suppressor cells are activated and the balance between these opposing arms of immunity determines susceptibility or resistance of infected host to the parasites. Immune evasion by the parasites could be directly related to parasite factors, (e.g., antigenic variation), or indirectly through the induction of suppressor cells following infection. Several cell types, including suppressive macrophages, myeloid-derived suppressor cells (MDSCs), and regulatory T cells have been shown to contribute to immunosuppression in African trypanosomiasis. In this review, we discuss the key factors that contribute to immunity and immunosuppression during T. congolense infection, and how these factors could aid immune evasion by African trypanosomes. Understanding the regulatory mechanisms that influence resistance and/or susceptibility during African trypanosomiasis could be beneficial in designing effective vaccination and therapeutic strategies against the disease.
African trypanosomiasis is a disease caused by extracellular hemoprotozoan parasites that belong to the genus Trypanosoma. Trypanosomes are unicellular parasites that are equipped with flagella which help with their movement (1). The disease is associated with serious health and economic problems in the affected countries, and can be fatal if not properly treated (2, 3). The human form of the disease is caused by Trypanosoma brucei rhodesiense and Trypanosoma brucei gambiense while the animal form of the disease is mostly caused by Trypanosoma congolense, Trypanosoma vivax, and Trypanosoma brucei brucei. Trypanosomiasis is a vector borne disease. The transmission of the parasites from one host to another occurs during a blood meal by several species of tsetse flies belonging to the genus Glossina. For both human and animal African trypanosomiasis, Glossina morsitans, Glossina tachinoides, Glossina fuscipes, Glossina pallidipes, Glossina swynnertoni, and Glossina palpalis are some of the important vectors responsible for transmission due to their wide distribution in countries where the disease is endemic (4), and their presence is often used as a key predictor of the disease.
Although it has been estimated that 65 million people living in 36 countries in sub-Saharan Africa are at risk of contracting the disease, the number of reported cases per year dramatically reduced (~10,000 new cases annually) in 2009 due to increasing efforts to combat the disease (5–7). In 2018, the number of reported cases further reduced to 977, an almost 90% decline in 10 years (8). However, the real number of cases may be grossly underestimated because the disease is mostly found in rural communities in the endemic areas, and it has been estimated that only about 10% of the affected people living in these areas are accounted for (9, 10). In other words, the majority of the cases remain either undiagnosed or unreported, suggesting that the disease impact and statistics could be worse than currently believed.
Human African trypanosomiasis affects both the young and old especially those that engage in farm-related activities in the endemic rural areas, although few reported cases have also been reported in urban areas (11). This is most likely related to favorable environmental conditions in rural areas that favor breeding of the insect vector. Woody vegetations are known to support tsetse fly abundance as the flies tend to rest on tree trunks during the hot humid day. Efforts toward eliminating the disease were almost successful in the 1960s, but because of political instabilities and accompanying poor surveillance, the eradication process was disrupted, which allowed the disease to re-emerge (12–15). Economically, the threat posed by African trypanosomiasis to animals is much more than that posed to humans (16). The disease has been linked to severe food and economic loss in the affected regions (2, 3).
Natural infection with African trypanosomes starts with intradermal injection of parasites along with tsetse saliva into the mammalian host by an infected tsetse fly. Following the introduction of the parasite into the dermis, the parasites undergo several transformations at this site with associated inflammatory response resulting in the development of chancre (1). These events precede the entry of the parasites into the blood stream of the infected host. Human African Trypanosomiasis usually develops from hemolymphatic phase to meningo-encephalitic phase; the meningo-encephalitic stage is one of the hallmarks of the disease and is often associated with severe alteration in the sleep-wake cycle (17). Cerebral infections with African trypanosomes in animals have also been documented; while T. congolense and T. vivax rarely invade the central nervous system (CNS), T. brucei brucei has been recovered from cerebrospinal fluid (CSF) and are able to cause central nervous system (CNS) impairment (18). Although there has been a report of T. congolense being found in the CSF, this was speculated to be aided by a mixed infection with other species (19). The tsetse fly saliva has been proposed to enable the blood-feeding process, promotes parasite transmission, and possesses powerful immunomodulatory properties including skewing T helper cell responses and anti-/proinflammatory properties (20–23). In contrast to this, the overwhelming majority of experimental infection studies utilize the intraperitoneal infection route, which involves inoculation of blood stream forms directly into the peritoneal cavity. It is conceivable that the use of intradermal route in experimental African trypanosome infection would capture some of the series of early events that occur during a natural infection (24). Therefore, it is likely that the intraperitoneal route of infection (as is used during most experimental infections) may not clearly represent the early immune response that occurs during natural infection when the vector bites their human or animal hosts (24). Indeed, it has been shown that intradermal infection of experimental animals is able to induce the activation of some immune mediators that are distinct from those seen during intraperitoneal infection (24, 25). Therefore, the use both the intradermal and intraperitoneal infection routes of infection (when possible) in experimental African trypanosomiasis would be helpful in comparing the immune responses due to different infection routes. This would enable us to further identify the missing links that could help in the understanding of the host-parasite interactions that regulate disease outcome.
Both the innate and adaptive components of the immune system play crucial roles in resistance to African trypanosomiasis. Although African trypanosomes are free living in the bloodstreams of their mammalian host, and are therefore direct targets of antibody-mediated destruction, experimental animal models of infection show that a full component of the immune system (innate and adaptive) are critical for the development of optimal resistance to the infection.
Macrophages are one of the most important cells that contribute to innate immunity to African trypanosomiasis. They are capable of influencing the adaptive immune response directly through antigen presentation or indirectly by secreting many effector molecules including cytokines. During infection with African trypanosomes, classically-activated macrophages have been shown to contribute to parasite clearance via the process of phagocytosis (26), as well as through the production of proinflammatory cytokines and nitric oxide (27–31). Macaskill et al. conducted a study to specifically examine the roles played by antibody, macrophage activation, and complement in parasite clearance using trypanosomes labeled with [75Se]-methionine. Their results showed that clearance of parasites from circulation was mostly dependent on antibody-mediated phagocytosis by liver macrophages (32).
Both classical and alternative macrophage activation occur during African trypanosomiasis, and their effects vary depending on the timing of their activations. Enhanced survival of trypanosome-infected mice has been associated with the ability to switch from classically-activated macrophages (M1) at the early stages of infection to an alternatively-activated phenotype (M2) during the advanced stages of infection (33, 34). This switch from M1 to M2 macrophages is essential because if left unchecked, classically-activated macrophages produce excessive amounts of proinflammatory cytokines that induce immune hyperactivation, causing collateral tissue damage and death in trypanosome-infected animals (35, 36). Thus, the inability of mice to upregulate alternative macrophage activation while concomitantly downregulating classical activation during the advanced stage of T. congolense infection led to enhanced susceptibility to infection, which was associated with excessive production of proinflammatory cytokines and early death of infected mice (37).
Several studies have associated pathology—mostly anemia and cachexia—during African trypanosome infection with the over-activation of macrophages and the resulting release of harmful molecules such as TNF-α and nitric oxide (38, 39). However, the underlying mechanisms involved in this process are still not fully investigated. While TNF-α is directly linked to the development of anemia in T. brucei infection, it appears not to be involved in T. congolense infection. Instead a direct alteration of red blood cells has been linked. (38–42).
The roles of macrophages during intradermal infection have not been well-investigated. Wei et al. demonstrated that mice that are deficient in inducible nitric oxide synthase (iNOS) were susceptible to intradermal low dose T. congolense infection (24), and proposed that macrophages are at the center of innate control of primary intradermal infection. However, results from our lab have shown that depletion of macrophages before T. congolense infection did not alter the resistance of mice to primary intradermal infection (25). This observation suggests that host factors other than macrophages are responsible for mediating early resistance observed during intradermal infection. More studies are needed to fully determine the roles played by macrophages in enhanced resistance observed during intradermal infection.
Although African trypanosomes have different mechanisms to avoid lysis by complement, the activation of this important arm of the innate immune defense system is considered to be critical for mediating parasite clearance in infected animals. Both the classical and alternative pathways of complement system are activated during infection with African trypanosomes. Activation of the classical pathway is mediated by specific antibodies against the parasite and has been reported to contribute to parasite clearance via antibody-mediated lysis, opsonization and phagocytosis (43). The antibody-independent alternative pathway is usually activated during the early stages of infection when specific antibodies have not been formed, and has also been shown to contribute to parasite clearance (44).
Although phagocytosis of African trypanosomes by macrophages can occur in vitro in the absence of complement (45), the efficiency of parasite clearance and immune complex removal is greatly enhanced in the presence of complement (46). Indeed, it was demonstrated that depletion of the complement component C3 by treatment with cobra venom factor leads to significant reduction in hepatic uptake of opsonized trypanosomes (32). However, other studies showed that partial depletion of C3 did not affect either parasitemia control of T. brucei brucei or phagocytosis of T. brucei rhodesiense (47). Jarvinen and Dalmasso found that parasite control could not be attributed to the presence of C3, C5, or late acting complement factors (48). In addition, Jones and Hancock found no marked differences in the survival periods of C5-deficient and C5-sufficient mice infected with African trypanosomes (49). Collectively, these findings suggest that complement activation may be required but not critical for resistance to African trypanosomes. However, given that the cleavage products of C3 and C5 (C3a and C5a) have been reported to help in the initiation of inflammatory responses during infection (50), and that alternative and classical complement activation is higher in the relatively resistant C57BL/6 mice following T. congolense infection (51, 52), it is conceivable that complement activation may contribute to optimal resistance to certain species of African trypanosomes.
Both CD4+ (helper) and CD8+ (cytotoxic) T cells play a critical role in regulating the outcome of infection with African trypanosomes. CD4+ T cells contribute to resistance by producing cytokines that regulate other innate and adaptive immune cells, and by providing help to B cells to ensure efficient isotype class-switching and production of specific antibody responses to parasite antigens. CD4+ T helper cells provide signals that regulate B cell survival and differentiation into antibody producing cells (53). In support of this, Shi et al. showed that anti-parasite IgG2a production, as well as IFN-γ and IL-10 levels, was impaired in T. congolense-infected CD4+ T cell deficient mice (54). Following this observation, Tabel et al. proposed that future vaccines against African trypanosomiasis should be targeted toward encouraging the generation of T helper 1 cells (Th1) that would support B cells in class-switching from IgM to IgG2a during infection (55). Interestingly, it was found that CD4+ T cell deficient mice infected with T. congolense had significantly lower parasitemia and prolonged survival period compared with their WT mice, suggesting that CD4+ T cells might also contribute to disease pathogenesis and death in infected mice (54).
The role of CD8+ T cells has been controversial in African trypanosomiasis. Earlier studies suggested that polyclonal activation of CD8+ T cells by T. brucei brucei-derived T lymphocyte triggering factor (TLTF) leads to massive release of IFN-γ, which is responsible for profound immunosuppression and susceptibility to the infection (56, 57). However, a study by Wei and Tabel showed that the beneficial effect of anti-CD25 treatment in mice during T. congolense infection was lost upon depletion of CD8+ T cells (58), suggesting that CD8+ T cells may be playing a protective role during T. congolense infection. In another study that investigated the relative contributions of CD4+ and CD8+ T cells in T. brucei infection, Liu et al. (59) reported that IgG antibody synthesis was dependent on CD4+ T cells and not CD8+ T cells. In addition, they showed that infected CD8+ T cell-deficient mice had lower parasitemia and survived significantly longer than their WT counterpart mice. However, this enhanced survival was lost upon depletion of CD4+ T cells. Furthermore, cytokine (IFN-γ and IL-10) production during infection was also attributed to CD4+ but not CD8+ T cells (59). These observations indicate that CD8+ T cells mediate susceptibility while CD4+ T cells mediate protection during infection with T. brucei brucei.
Our understanding of the roles played by T cells has been simplified by the Th1 (proinflammatory properties) and Th2 (anti-inflammatory properties) paradigm. Overall, the control of parasites during infection with African trypanosomes is believed to be associated with a Th1 response during the early phase of infection and a switch to a Th2 phenotype in the advanced stage of infection (33, 34). In support this, T. congolense-infected TSLPR−/− mice with impaired Th2 response are highly susceptible to T. congolense infection during the chronic stage of the disease, and died significantly earlier than their WT controls (37). This susceptibility was associated with increased production of proinflammatory cytokines by CD4+ T cells from these mice, which was reversed upon treatment of the infected TSLPR−/− mice with anti-IFN-γ monoclonal antibody (37).
Because African trypanosomes are extracellular bloodstream parasites, they are constantly exposed to humoral immune factors and are direct target for antibody-mediated destruction. Indeed, B cell deficient mice are highly susceptible to African trypanosomes (60). In addition, passive transfer of variant-specific antibodies or B cells (but not T cells) to immunocompromised mice results in variant-specific protection (60, 61). Furthermore, the differential resistance observed in several strains of mice and cattle following infection with African trypanosomes has been attributed to differences in the production of parasite-specific antibodies (62–64). A study using mice that lack Bam32 further demonstrated the importance of strong B cell response for protection against infection with African trypanosomes (65). Bam32 is a B cell adaptor protein that plays a critical role in B cell activation (66), survival (67), and antigen presentation (68). Bam32 deficient mice on a relatively resistant background were more susceptible than their WT counterpart mice to T. congolense infection and showed impaired germinal center response as well as significantly low levels of parasite-specific IgG antibodies in the serum (65, 68).
B cells become activated upon encountering their cognate antigens, and this is followed by the initiation of germinal center formation with the help of follicular CD4+ T helper cells (Tfh) (69). Germinal centers are large areas in the secondary lymphoid organs where intense B activities such as proliferation, somatic hypermutation, selection, and class switch recombination take place, resulting in the production of various antibody isotypes with high antigen binding affinity (53). The requirement of B cells during infection with African trypanosomiasis centers on optimal activation, efficient germinal center (GC) formation, and production of strain-specific antibodies.
One of the hallmarks of African trypanosomiasis is excessive polyclonal activation of B cells leading to increased serum levels of trypanosome-specific and non-specific antibodies, including heterophilic and autoantibodies (70, 71). These observations led to the postulation that African trypanosomes possess molecules (VSG for instance) that could non-specifically activate B cells to produce antibodies (70). During infection with African trypanosomes, both T cell-dependent and T cell-independent antibody response are produced against the variant and invariant VSG molecules, cytoplasmic, and other nuclear parasite antigens (72). However, the overall quality (as assessed by binding affinity, isotype, and quantity) of the response is increased in the presence of T cells (72). For example, although IgM anti-VSG antibodies (which are mostly produced in a T cell-independent manner) can mediate parasite clearance, the different subclasses of IgG antibodies, whose production are T cell dependent, mediate a more effective parasite clearance in both mice and cattle compared to IgM (62, 73).
The profile and magnitude of cytokines produced during African trypanosomiasis play a critical role in determining susceptibility and resistance to the disease. Although the contributions of some cytokines in the pathogenesis of African trypanosomiasis have been demonstrated in different experimental settings, it is challenging to fully determine the precise role of specific cytokines in disease pathogenesis because of their pleiotropic activities (55). Studies have shown that infection with African trypanosomes leads to massive production of proinflammatory cytokines in the infected mammalian host. The initial inflammatory response during infection usually leads to the release of proinflammatory mediators like TNF-α, IL-1, IL-6, and NO by classically activated macrophages, and these have been shown to play important roles in mediating early protection during infection (26, 30, 74–78). Other cytokines like IL-12, MCP-1, IL-10, IFN-γ, and IL-4 have also been shown to mediate either pro-inflammatory or anti-inflammatory activities during infection (26, 54, 79–83).
Although the initial outburst of inflammatory cytokines is essential for resistance, it requires regulation to prevent collateral tissue damage. The switch from classically activated to alternatively activated macrophages later during infection by type II cytokines such as IL-4, IL13, and TSLP is critical for maintaining a Th2 type environment which have anti-inflammatory properties (1, 37). In fact, protection during infection with African trypanosomes is associated with the ability to switch from an early Th1 to Th2 response during the later stages of infection (33, 34, 37, 84). In line with this, the absence of TSLP signaling (which is a key cytokine that drives Th2 differentiation), in T. congolense-infected mice led to the inability to control more than two waves of parasitemia and early death compared to their wild-type counterpart mice (37). This inability to effectively control parasitemia was associated with overproduction of proinflammatory cytokines (including IFN-γ and TNF-α) and impaired activation of alternatively activated macrophages (37).
The effect of cytokines in the pathogenesis of African trypanosomiasis is complex and depends on the quantity and time of production during the infection (55). For example, while the production of IFN-γ and TNF-α are critical for protection during infection with African trypanosomes (85–88), their production in excessive amounts is detrimental and leads to susceptibility and death of infected mice (27, 79, 82). For instance, while IFN-γ-deficient mice are highly susceptible to T. brucei or T. congolense infection and fail to control the first wave of parasitemia (85, 87), neutralization of IFN-γ by antibody treatment during T. congolense infection is associated with enhanced resistance (lower parasitemia and prolonged survival) of the highly susceptible mice (79). Furthermore, acute death of infected relatively resistant mice following treatment with anti-IL-10 receptor antibody was completely abrogated by co-treatment with anti-IFN-γ mAb (82). These observations suggest that IFN-γ is a key mediator of death in these mice.
Another key cytokine that regulates the outcome of infection with African trypanosomes is IL-10. Due to its anti-inflammatory properties, IL-10 acts to downregulate excessive effector activities of both T cells and macrophages (82, 89), which are key cells that are involved in the production of inflammatory cytokines following infection with African trypanosomes. In T. congolense-infected cattle, reduced nitric oxide production and increased IL-10 and IL-4 mRNA levels were linked to protection (73, 90).
Adaptation mechanisms within the host are known to exist among bacteria, parasites, and viruses. Antigenic variation is one of the hallmarks of African trypanosomes and constitutes a major adaptation mechanism of evading their host immune response. In fact, it has been suggested that the inability to fully understand the mechanisms that regulate antigenic variation during infection with African trypanosomes is one of the major obstacles standing in the way of developing a vaccine for African trypanosomiasis (38).
Upon injection into the skin, the parasites grow and multiply in the bloodstream of their mammalian host. Because African trypanosomes are completely extracellular in nature, they are continuously exposed to the host's humoral immune defenses. Although clone-specific antibodies are effective in mediating parasite clearance (62, 85), the natural ability of the parasites to undergo antigenic variation during the course of infection renders the antibodies ineffective at mediating cure. The expression of new VSG allows the parasites to evade antibody-mediated destruction thus permitting them to grow and multiply, and requiring the host to initiate new antibody responses against the emerging new clones.
Bloodstream forms of African trypanosomes are covered with a densely packed protective coat comprising of over 107 copies of variant surface glycoprotein (VSG) molecules (1). These millions of identical glycoprotein molecules function to prevent complement-mediated lysis of the parasites (91, 92). They are attached to the surface of the plasma membrane of the parasite via the glycosylphosphatidylinositol (GPI) anchor (93). Although mostly membrane bound, soluble forms of VSG are released into the circulation upon cleavage of the GPI anchor by parasite-encoded phospholipase C (PLC) known as GPI-PLC (94). This process has been reported to trigger some inflammatory responses during infection (95).
Trypanosomes contain hundreds of VSG genes (constituting about 10% of the entire parasite genome) with only about 7% being fully functional (96). The transcription of VSG genes occurs at a telomere of the chromosomes containing the VSG expression sites (97). Because only one expression site can be active at any given time, only one of the VSG molecules is expressed on the surface coat of the parasite leading to identical display of surface coats (1). In addition, because antibody response is made only to this particular antigenic type that is being expressed, a switch in VSG expression would lead to the initiation of new antibody response, a condition that could subsequently pave the way for immune exhaustion due to the continuous need to mount immune response to numerous VSG-expressing clones. Trypanosomes are able to control VSG gene expression by turning off an active expression site (and turning on a previously silent expression site) and by rearrangement of the VSG genes mostly by reciprocal recombination and gene conversion (98–100). Efforts targeted at disrupting the VSG switching by the parasites could be a major step in designing effective disease control strategies.
Because of their extracellular life style, B cells play a critical role in clearance of African trypanosomes from the blood of infected hosts by producing parasite-specific antibodies. As a result, they have developed mechanisms to suppress and evade the host specific antibody responses. Trypanosomes are able to exploit the ability of B cells to produce antibodies and use this to induce excessive activation of antibody-producing cells. This often leads to an increase in plasma levels of specific and non-specific immunoglobulins in infected animals. This hypergammaglobulinemia, which results from extensive B cell expansion, was first reported as a major feature of infection with African trypanosomes (101). While specific antibodies against parasite antigens are produced in both T cell-dependent and independent pathways (72), the induction of non-specific (polyclonal) B cell activation in a T cell-independent manner represents a strategy through which the parasites regulate specific antibody responses that contributes to immune evasion (102). Although not clearly known, it has also been reported that experimental T. congolense infection causes B cell depletion in the bone marrow as well as the periphery, which in turn impact negatively on the specificity of anti-VSG immunoglobulins produced during infection (103). The resulting impaired B cell development and maturation makes it impossible for the optimal development of memory B cell subsets that are key when considering vaccination strategies (104).
Several trypanosomal moieties, including DNA and VSG have been shown to contribute to polyclonal B cell activation following infection with African trypanosomes. Trypanosomal DNAs are able to initiate TLR-9 signaling leading to non-specific B cell activation and production of poly-specific antibodies to VSG (102, 105). Indeed, autoantibodies against red blood cells, cardiolipids, nucleic acids, rheumatoid factors, and component of CNS myelin (the galactocerebrosides) have been detected in sera of infected animals (106–111). Furthermore, the overproduction of antibodies could result in over-engagement of the Fcγ receptors on phagocytes, which would eventually contribute to impaired opsonization and phagocytosis of parasites (102, 105). In addition, an increased in a sub-population of CD5 expressing B cells have also been documented, and these cells are believed to be responsible for the excessive production of immunoglobulins and autoantibodies during T. congolense infection in cattle (105).
African trypanosomes are able to take advantage of complement activation to evade the host immune response. Because the classical complement activation is critical for lysing parasites coated with VSG-specific antibodies, trypanosomes, in an effort to enhance their survival, are able to shed enormous VSG in the circulation which leads to the formation of immune complexes with antibodies. This “decoy mechanism” of shedding VSG prevents the deposition of membrane attack complex on the parasite cell membrane, thereby preventing complement-mediated lysis of the parasite. The continuous activation of the complement system often leads to a state of hypocomplementemia, which is a hallmark of infection with African trypanosomiasis (112). Also, due to their spatial and dense arrangement, the VSG is known to mask the binding of complement proteins on the plasma membrane leading to the inability to trigger alternative complement activation. This results in halting of the alternative pathway activation at the C3 convertase stage thereby preventing further activation of C5–C9 stages and subsequent formation of the membrane attack complex that usually initiate lysis of trypanosomes (113, 114).
One of the hallmarks of African trypanosomiasis is suppression of lymphocyte proliferation, and this has been reported to be one of the key factors that prevent parasite control in infected animals (115–117). Several cell types have been implicated in the pathogenesis of immunosuppression in African trypanosomiasis. Although suppressor macrophages and suppressor T cells were the earliest identified culprits (118–120), recent studies have clearly revealed additional roles played by regulatory T cells (25, 58, 121) and myeloid-derived suppressor cells (122). Schleifer and Mansfield (123) showed that macrophages suppress T cell responses through the production of reactive nitrogen intermediates and prostaglandins. Similarly, independent studies (using anti-CD25 mAb treatment) have shown that CD4+CD25+Foxp3+ T cells (Tregs) play a role in suppression of immune response to T. congolense (25, 58, 121). In these studies, immunity to the parasites (including control of parasitemia and survival) was enhanced in the absence of Tregs. Indeed, it was found that during intradermal infection with T. congolense, the parasites induces the expansion of regulatory T cells as a mechanism to induce immune suppression in order to evade host's immune response and enhance their survival (25).
Suppressor macrophages have also been shown to mediate suppression of host immune response as a means to enhance parasite survival. For example, trypanosomes are able to take advantage of the alternative macrophage activation to enhance their survival in the host. These parasites have been shown to preferentially induce alternative macrophage activation with the sole aim of upregulating host arginase, which has been shown to reduce the synthesis of trypanocidal nitrosylated compounds as well as upregulate L-ornithine production, a critical process in the synthesis of polyamines that is required for parasite growth (124, 125). Recently, we showed that T. congolense infection was associated with the induction of myeloid-derived suppressor cells, which suppressed T cell responses (proliferation and IFN-γ production) during T. congolense infection in an arginase-1-dependent mechanism (122). This suggests that targeting the arginase pathway during infection could limit parasite survival by increasing various effector responses directed against the parasites.
Two species of African trypanosomes, Trypanosoma brucei gambiense and Trypanosoma brucei rhodesiense, are able to infect their human host. This is due to the parasites' ability to manipulate and evade the effect of TLFs that are known to have anti-parasite activity. TLF comprises of two serum complexes, Trypanosome Lytic Factor 1 and 2 (TLF1 and TLF2) (126, 127). These complexes are made up of primate-specific protein haptoglobin-protein (HPR) and apolipoprotein L1 (APOL1) (128). It is known that HPR is responsible for targeting TLF1 to the parasites, while APOL1 is the trypanolytic toxin (129–131). In Trypanosoma brucei rhodesiense, resistance to APOL1 is related to the expression of an expression site-associated gene (ESAG) generally referred to as serum resistance-associated (SRA) gene (132, 133). SRA is able to bind to APOL1, and the resulting interaction leads to the inactivation of APOL1 via the inhibition of its membrane insertion, resulting in degradation by proteases (128, 134). In Trypanosoma brucei gambiense, resistance is conferred through T. b. gambiense-specific glycoprotein (TgsGP) (135). The deletion of the gene that encodes TgsGP renders the parasite susceptible to normal human serum (136, 137). TgsGP inhibits APOL1 toxicity though a hydrophobic β-sheet (136) that initiates membrane stiffening, which could inhibit APOL1 membrane insertion leading to APOL1 degradation by endosomal proteases (128).
African trypanosomes (specifically T. brucei) encodes a suppressive protein, Trypanosome-suppressive immunomodulating factor (TSIF) that has been shown to have immunosuppressive actions in vivo (138). Treatment of mice with recombinant TSIF resulted in the activation macrophages that predominantly produce TNF-α and NO, suggesting that this protein favors the classical activation of macrophages (138). TSIF has also been shown to significantly impair Th2-induced inflammation in allergic asthma model, which further strengthens its ability to preferentially induce a Th1 inflammatory condition (139). Although, classical macrophage activation could be important for trypanosome clearance during the early phase of infection, a switch to a more alternatively-activated macrophage phenotype toward the late stage infection is required to prevent excessive inflammation and death of infected mice (33, 34, 37, 84). By employing the use of TSIF, trypanosomes are able to manipulate the host immune system by sustaining the classical macrophage activation during infection thereby suppressing the development of alternatively activated macrophages (Th2 condition). This could lead to exacerbated inflammation and death of infected animals.
There is currently no vaccine against African trypanosomiasis and the current drugs for treatment of the disease are relatively ineffective. In addition to being toxic and expensive, the effectiveness of the current drugs is further hampered by increased risk of drug resistance and disease relapse (140–142). The development of a vaccine against this disease has been made virtually impossible due to antigenic variation and the difficulty in understanding the factors that regulate this and other important and highly complex host immune evasion mechanisms (38). Given that decades of research have failed to develop a vaccine against African trypanosomiasis, it is imperative that current research strategies have not yielded meaningful information regarding the induction and maintenance of effective and/or protective immune response to these parasites (143). Therefore, a change in strategy and our current way of thinking (including experimental designs and animal models) is needed. Studies focusing on halting various events that contribute to impaired immune response and immune evasion strategies by the parasites (Figure 1) could represent a major pathway toward disease control.
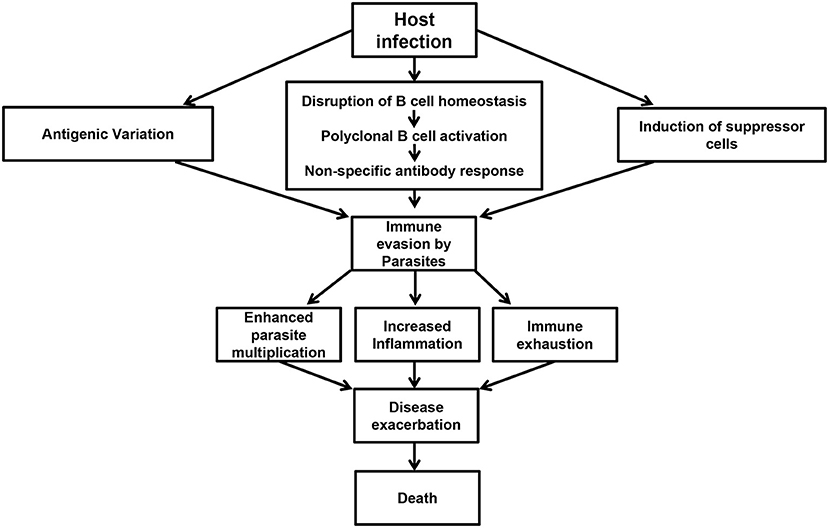
Figure 1. Events preceding immune evasion by parasites. Following infection, trypanosomes are able to undergo antigenic variation, disrupt B cell homeostasis, and initiate polyclonal B cell activation leading to the production of non-specific antibody responses. In addition, the infection is also associated with the induction of suppressor cells like Tregs and MDSCs. These events contribute to immune evasion by the parasites resulting in poor parasite control, increased inflammation, immune exhaustion, disease exacerbation, and death in untreated animals.
Because using the intraperitoneal route of infection does not mimic or represent the activities that take place during natural infection (which is initiated via intradermal inoculation by the tsetse fly), a switch to a more natural intradermal route of infection could be more beneficial in understanding key immune responses to these parasites, and could provide helpful information regarding vaccine development (25, 144). Furthermore, the causes of polyclonal B cell activation need to be further investigated since the parasite's ability to induce non-specific B cell responses to enhance their survival in the host still poses a great challenge in vaccine discovery. Although data showing the importance of optimal germinal center reaction during infection are beginning to appear (65), similar studies aimed at understanding other factors that regulate the parasite's ability to induce non-specific antibody responses could be beneficial. We showed that the ability of CD4+ T cells to proliferate and produce IFN-γ during T. congolense infection was inhibited by myeloid-derived suppressor cells in an arginase-1-dependent manner (122). This identifies arginase-1 as a potential target in African trypanosomiasis when considering treatment and vaccination strategies (144). It would be highly interesting to determine whether these exciting and novel mechanisms of immune regulation during T. congolense infections operate during infection with other African trypanosome species, particularly those that cause disease in humans.
In addition to the ongoing search for anti-parasite vaccine, adopting the anti-disease vaccine approach (145) could also serve as a great option to explore in African trypanosomiasis. This is based on the fact that trypanosome-associated factors could be targeted and nullified, which in turn would limit pathologies that are the major cause of disease. In line with this, cysteine peptidases have been identified as a potential candidate for anti-disease vaccine target (146) as they have been proposed to be likely involved in inducing alterations and pathologies that result in immunosuppression, anemia, and CNS disorder (147–150). Also, because the VSG GPI anchor induces very strong proinflammatory cytokine response that contributes to disease and pathologies (151, 152), it has been suggested that the GPI could be a promising target for anti-disease vaccine development (153). Indeed, it has been shown that GPI-based treatment alleviates trypanosomiasis-associated immunopathology, including anemia, weight loss, liver damage, inflammation, and proinflammatory cytokine production resulting in prolonged survival (154, 155). Although the efficacy of using synthetic GPI as vaccine is yet to be evaluated in infections caused by African trypanosomes, a study conducted in a mouse model of severe malaria using chemically-synthesized Plasmodium falciparum GPI glycan for immunization led to a significant reduction in early death and malaria-associated pathologies during challenge infection (153). This was associated with in vitro antibody (against GPI)-dependent neutralization of P. falciparum-induced inflammation (153). It would be interesting to investigate the impact of vaccination with synthetic GPI in the pathogenesis of experimental African trypanosomiasis, including the production of disease-enhancing proinflammatory cytokines.
CO: literature search and drafting. JU: provided corrections to the draft.
This work was supported by grant from The Natural Sciences and Engineering Research Council (NSERC) of Canada.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Baral TN. Immunobiology of African trypanosomes: need of alternative interventions. J Biomed Biotechnol. (2010) 2010:389153. doi: 10.1155/2010/389153
2. Kristjanson PM, Swallow BM, Rowlands GJ, Kruska RL, de Leeuw PN. Measuring the costs of African animal trypanosomosis, the potential benefits of control and returns to research. Agric Syst. (1999) 59:79–98. doi: 10.1016/S0308-521X(98)00086-9
3. Hursey BS. The programme against African trypanosomiasis: aims, objectives and achievements. Trends Parasitol. (2001) 17:2–3. doi: 10.1016/S1471-4922(00)01851-1
4. Kuzoe FA. Current situation of African trypanosomiasis. Acta Trop. (1993) 54:153–62. doi: 10.1016/0001-706X(93)90089-T
5. Simarro PP, Cecchi G, Paone M, Franco JR, Diarra A, Ruiz JA, et al. The Atlas of human African trypanosomiasis: a contribution to global mapping of neglected tropical diseases. Int J Health Geogr. (2010) 9:57. doi: 10.1186/1476-072X-9-57
6. Stuart K, Brun R, Croft S, Fairlamb A, Gurtler RE, McKerrow J, et al. Kinetoplastids: related protozoan pathogens, different diseases. J Clin Invest. (2008) 118:1301–10. doi: 10.1172/JCI33945
7. Brun R, Don R, Jacobs RT, Wang MZ, Barrett MP. Development of novel drugs for human African trypanosomiasis. Future Microbiol. (2011) 6:677–91. doi: 10.2217/fmb.11.44
8. WHO. Trypanosomiasis, Human African (Sleeping Sickness). (2019). Available online at: www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness) (accessed November 5, 2019).
9. WHO. African Trypanosomiasis (Sleeping Sickness). (2010). Available online at: www.who.int/mediacentre/factsheets/fs259/en/ (accessed December 13, 2010).
10. WHO. Control and Surveillance of African Trypanosomiasis. Report of a WHO Expert Committee. World Health Organisation (1998), p. 1–114.
11. Robays J, Ebeja Kadima A, Lutumba P, Miaka mia Bilenge C, Kande Betu Ku Mesu V, De Deken R, et al. Human African trypanosomiasis amongst urban residents in Kinshasa: a case-control study. Trop Med Int Health. (2004) 9:869–75. doi: 10.1111/j.1365-3156.2004.01266.x
12. Smith DH, Pepin J, Stich AH. Human African trypanosomiasis: an emerging public health crisis. Br Med Bull. (1998) 54:341–55. doi: 10.1093/oxfordjournals.bmb.a011692
13. Barrett MP. The fall and rise of sleeping sickness. Lancet. (1999) 353:1113–4. doi: 10.1016/S0140-6736(98)00416-4
14. Moore A, Richer M. Re-emergence of epidemic sleeping sickness in southern Sudan. Trop Med Int Health. (2001) 6:342–7. doi: 10.1046/j.1365-3156.2001.00714.x
15. Van Nieuwenhove S, Betu-Ku-Mesu VK, Diabakana PM, Declercq J, Bilenge CM. Sleeping sickness resurgence in the DRC: the past decade. Trop Med Int Health. (2001) 6:335–41. doi: 10.1046/j.1365-3156.2001.00731.x
16. Jordan AM. Tsetse flies as vectors of trypanosomes. Vet Parasitol. (1976) 2:143–52. doi: 10.1016/0304-4017(76)90059-5
17. Buguet A, Bisser S, Josenando T, Chapotot F, Cespuglio R. Sleep structure: a new diagnostic tool for stage determination in sleeping sickness. Acta Trop. (2005) 93:107–17. doi: 10.1016/j.actatropica.2004.10.001
18. Morrison WI, Murray M, Whitelaw DD, Sayer PD. Pathology of infection with Trypanosoma brucei: disease syndromes in dogs and cattle resulting from severe tissue damage. Contrib Microbiol Immunol. (1983) 7:103–19.
19. Haase M, Bernard S, Guidot G. Trypanosomiasis in Zebu cattle. Reappearance of Trypanosoma congolense in brain tissue after treatment with Berenil. Rev Elev Med Vet Pays Trop. (1981) 34:149–54.
20. Caljon G, De Ridder K, De Baetselier P, Coosemans M, Van Den Abbeele J. Identification of a tsetse fly salivary protein with dual inhibitory action on human platelet aggregation. PLoS One. (2010) 5:e9671. doi: 10.1371/journal.pone.0009671
21. Caljon G, Van Den Abbeele J, Sternberg JM, Coosemans M, De Baetselier P, Magez S. Tsetse fly saliva biases the immune response to Th2 and induces anti-vector antibodies that are a useful tool for exposure assessment. Int J Parasitol. (2006) 36:1025–35. doi: 10.1016/j.ijpara.2006.05.002
22. Caljon G, Van Den Abbeele J, Stijlemans B, Coosemans M, De Baetselier P, Magez S. Tsetse fly saliva accelerates the onset of Trypanosoma brucei infection in a mouse model associated with a reduced host inflammatory response. Infect Immun. (2006) 74:6324–30. doi: 10.1128/IAI.01046-06
23. Van Den Abbeele J, Caljon G, Dierick JF, Moens L, De Ridder K, Coosemans M. The Glossina morsitans tsetse fly saliva: general characteristics and identification of novel salivary proteins. Insect Biochem Mol Biol. (2007) 37:1075–85. doi: 10.1016/j.ibmb.2007.06.006
24. Wei G, Bull H, Zhou X, Tabel H. Intradermal infections of mice by low numbers of african trypanosomes are controlled by innate resistance but enhance susceptibility to reinfection. J Infect Dis. (2011) 203:418–29. doi: 10.1093/infdis/jiq051
25. Onyilagha C, Okwor I, Kuriakose S, Singh R, Uzonna J. Low-dose intradermal infection with trypanosoma congolense leads to expansion of regulatory T cells and enhanced susceptibility to reinfection. Infect Immun. (2014) 82:1074–83. doi: 10.1128/IAI.01028-13
26. Shi M, Wei G, Pan W, Tabel H. Trypanosoma congolense infections: antibody-mediated phagocytosis by Kupffer cells. J Leukoc Biol. (2004) 76:399–405. doi: 10.1189/jlb.1003500
27. Kaushik RS, Uzonna JE, Zhang Y, Gordon JR, Tabel H. Innate resistance to experimental African trypanosomiasis: differences in cytokine (TNF-alpha, IL-6, IL-10 and IL-12) production by bone marrow-derived macrophages from resistant and susceptible mice. Cytokine. (2000) 12:1024–34. doi: 10.1006/cyto.2000.0685
28. Shoda LK, Kegerreis KA, Suarez CE, Roditi I, Corral RS, Bertot GM, et al. DNA from protozoan parasites Babesia bovis, Trypanosoma cruzi, and T. brucei is mitogenic for B lymphocytes and stimulates macrophage expression of interleukin-12, tumor necrosis factor alpha, and nitric oxide. Infect Immun. (2001) 69:2162–71. doi: 10.1128/IAI.69.4.2162-2171.2001
29. Coller SP, Mansfield JM, Paulnock DM. Glycosylinositolphosphate soluble variant surface glycoprotein inhibits IFN-gamma-induced nitric oxide production via reduction in STAT1 phosphorylation in African trypanosomiasis. J Immunol. (2003) 171:1466–72. doi: 10.4049/jimmunol.171.3.1466
30. Kaushik RS, Uzonna JE, Gordon JR, Tabel H. Innate resistance to Trypanosoma congolense infections: differential production of nitric oxide by macrophages from susceptible BALB/c and resistant C57Bl/6 mice. Exp Parasitol. (1999) 92:131–43. doi: 10.1006/expr.1999.4408
31. Vincendeau P, Daulouede S, Veyret B, Darde ML, Bouteille B, Lemesre JL. Nitric oxide-mediated cytostatic activity on Trypanosoma brucei gambiense and Trypanosoma brucei brucei. Exp Parasitol. (1992) 75:353–60. doi: 10.1016/0014-4894(92)90220-5
32. Macaskill JA, Holmes PH, Whitelaw DD, McConnell I, Jennings FW, Urquhart GM. Immunological clearance of 75Se-labelled Trypanosoma brucei in mice. II. Mechanisms in immune animals. Immunology. (1980) 40:629–35.
33. Murata T, Taguchi J, Puri RK, Mohri H. Sharing of receptor subunits and signal transduction pathway between the IL-4 and IL-13 receptor system. Int J Hematol. (1999) 69:13–20.
34. Namangala B, de Baetselier P, Brijs L, Stijlemans B, Noel W, Pays E, et al. Attenuation of Trypanosoma brucei is associated with reduced immunosuppression and concomitant production of Th2 lymphokines. J Infect Dis. (2000) 181:1110–20. doi: 10.1086/315322
35. Baetselier PD, Namangala B, Noel W, Brys L, Pays E, Beschin A. Alternative versus classical macrophage activation during experimental African trypanosomosis. Int J Parasitol. (2001) 31:575–87. doi: 10.1016/S0020-7519(01)00170-9
36. Goerdt S, Orfanos CE. Other functions, other genes: alternative activation of antigen-presenting cells. Immunity. (1999) 10:137–42. doi: 10.1016/S1074-7613(00)80014-X
37. Onyilagha C, Singh R, Gounni AS, Uzonna JE. Thymic stromal lymphopoietin is critical for regulation of proinflammatory cytokine response and resistance to experimental Trypanosoma congolense infection. Front Immunol. (2017) 8:803. doi: 10.3389/fimmu.2017.00803
38. Magez S, Truyens C, Merimi M, Radwanska M, Stijlemans B, Brouckaert P, et al. P75 tumor necrosis factor-receptor shedding occurs as a protective host response during African trypanosomiasis. J Infect Dis. (2004) 189:527–39. doi: 10.1086/381151
39. Naessens J, Kitani H, Nakamura Y, Yagi Y, Sekikawa K, Iraqi F. TNF-alpha mediates the development of anaemia in a murine Trypanosoma brucei rhodesiense infection, but not the anaemia associated with a murine Trypanosoma congolense infection. Clin Exp Immunol. (2005) 139:405–10. doi: 10.1111/j.1365-2249.2004.02717.x
40. Darji A, Beschin A, Sileghem M, Heremans H, Brys L, De Baetselier P. In vitro simulation of immunosuppression caused by Trypanosoma brucei: active involvement of gamma interferon and tumor necrosis factor in the pathway of suppression. Infect Immun. (1996) 64:1937–43.
41. Magez S, Lucas R, Darji A, Songa EB, Hamers R, De Baetselier P. Murine tumour necrosis factor plays a protective role during the initial phase of the experimental infection with Trypanosoma brucei brucei. Parasite Immunol. (1993) 15:635–41. doi: 10.1111/j.1365-3024.1993.tb00577.x
42. Nok AJ, Balogun EO. A bloodstream Trypanosoma congolense sialidase could be involved in anemia during experimental trypanosomiasis. J Biochem. (2003) 133:725–30. doi: 10.1093/jb/mvg093
43. Mansfield JM, Levine RF, Dempsey WL, Wellhausen SR, Hansen CT. Lymphocyte function in experimental African trypanosomiasis. IV. Immunosuppression and suppressor cells in the athymic nu/nu mouse. Cell Immunol. (1981) 63:210–5. doi: 10.1016/0008-8749(81)90043-5
44. Musoke AJ, Barbet AF. Activation of complement by variant-specific surface antigen of trypanosoma brucei. Nature. (1977) 270:438–40. doi: 10.1038/270438a0
45. Ngaira JM, Nantulya VM, Musoke AJ, Hirumi K. Phagocytosis of antibody-sensitized Trypanosoma brucei in vitro by bovine peripheral blood monocytes. Immunology. (1983) 49:393–400.
46. Stevens DR, Moulton JE. Ultrastructural and immunological aspects of the phagocytosis of Trypanosoma brucei by mouse peritoneal macrophages. Infect Immun. (1978) 19:972–82.
47. Dempsey WL, Mansfield JM. Lymphocyte function in experimental African trypanosomiasis. V. Role of antibody and the mononuclear phagocyte system in variant-specific immunity. J Immunol. (1983) 130:405–11.
48. Jarvinen JA, Dalmasso AP. Trypanosoma musculi infections in normocomplementemic, C5-deficient, and C3-depleted mice. Infect Immun. (1977) 16:557–63.
49. Jones JF, Hancock GE. Trypanosomiasis in mice with naturally occurring immunodeficiencies. Infect Immun. (1983) 42:848–51.
50. Mekori YA, Metcalfe DD. Mast cells in innate immunity. Immunol Rev. (2000) 173:131–40. doi: 10.1034/j.1600-065X.2000.917305.x
51. Ogunremi O, Tabel H, Kremmer E, Wasiliu M. Differences in the activity of the alternative pathway of complement in BALB/c and C57Bl/6 mice. Exp Clin Immunogenet. (1993) 10:31–7.
52. Otesile EB, Lee M, Tabel H. Plasma levels of proteins of the alternative complement pathway in inbred mice that differ in resistance to Trypanosoma congolense infections. J Parasitol. (1991) 77:958–964. doi: 10.2307/3282750
53. Tellier J, Nutt SL. The unique features of follicular T cell subsets. Cell Mol Life Sci. (2013) 70:4771–84. doi: 10.1007/s00018-013-1420-3
54. Shi M, Wei G, Pan W, Tabel H. Experimental African trypanosomiasis: a subset of pathogenic, IFN-gamma-producing, MHC class II-restricted CD4+ T cells mediates early mortality in highly susceptible mice. J Immunol. (2006) 176:1724–32. doi: 10.4049/jimmunol.176.3.1724
55. Tabel H, Wei G, Shi M. T cells and immunopathogenesis of experimental African trypanosomiasis. Immunol Rev. (2008) 225:128–39. doi: 10.1111/j.1600-065X.2008.00675.x
56. Bakhiet M, Olsson T, Edlund C, Hojeberg B, Holmberg K, Lorentzen J, et al. A Trypanosoma brucei brucei-derived factor that triggers CD8+ lymphocytes to interferon-gamma secretion: purification, characterization and protective effects in vivo by treatment with a monoclonal antibody against the factor. Scand J Immunol. (1993) 37:165–78. doi: 10.1111/j.1365-3083.1993.tb01753.x
57. Bakhiet M, Olsson T, Ljungdahl A, Hojeberg B, Van Der Meide P, Kristensson K. Induction of interferon-gamma, transforming growth factor-beta, and interleukin-4 in mouse strains with different susceptibilities to Trypanosoma brucei brucei. J Interferon Cytokine Res. (1996) 16:427–33. doi: 10.1089/jir.1996.16.427
58. Wei G, Tabel H. Regulatory T cells prevent control of experimental African trypanosomiasis. J Immunol. (2008) 180:2514–21. doi: 10.4049/jimmunol.180.4.2514
59. Liu G, Sun D, Wu H, Zhang M, Huan H, Xu J, Zhang X, Zhou H, Shi M. Distinct contributions of CD4+ and CD8+ T cells to pathogenesis of Trypanosoma brucei infection in the context of gamma interferon and interleukin-10. Infect Immun. (2015) 83:2785–95. doi: 10.1128/IAI.00357-15
60. Campbell GH, Phillips SM. Adoptive transfer of variant-specific resistance to Trypanosoma rhodesiense with B lymphocytes and serum. Infect Immun. (1976) 14:1144–50.
61. Otesile EB, Tabel H. Enhanced resistance of highly susceptible Balb/c mice to infection with Trypanosoma congolense after infection and cure. J Parasitol. (1987) 73:947–53. doi: 10.2307/3282517
62. Uzonna JE, Kaushik RS, Gordon JR, Tabel H. Cytokines and antibody responses during Trypanosoma congolense infections in two inbred mouse strains that differ in resistance. Parasite Immunol. (1999) 21:57–71. doi: 10.1111/cge.1999.21.2.57
63. Williams DJ, Taylor K, Newson J, Gichuki B, Naessens J. The role of anti-variable surface glycoprotein antibody responses in bovine trypanotolerance. Parasite Immunol. (1996) 18:209–18. doi: 10.1046/j.1365-3024.1996.d01-76.x
64. Morrison WI, Murray M. The role of humoral immune responses in determining susceptibility of A/J and C57BL/6 mice to infection with Trypanosoma congolense. Parasite Immunol. (1985) 7:63–79. doi: 10.1111/j.1365-3024.1985.tb00479.x
65. Onyilagha C, Jia P, Jayachandran N, Hou S, Okwor I, Kuriakose S, et al. The B cell adaptor molecule Bam32 is critically important for optimal antibody response and resistance to Trypanosoma congolense infection in mice. PLoS Negl Trop Dis. (2015) 9:e0003716. doi: 10.1371/journal.pntd.0003716
66. Marshall AJ, Niiro H, Lerner CG, Yun TJ, Thomas S, Disteche CM, et al. A novel B lymphocyte-associated adaptor protein, Bam32, regulates antigen receptor signaling downstream of phosphatidylinositol 3-kinase. J Exp Med. (2000) 191:1319–32. doi: 10.1084/jem.191.8.1319
67. Niiro H, Maeda A, Kurosaki T, Clark EA. The B lymphocyte adaptor molecule of 32 kD (Bam32) regulates B cell antigen receptor signaling and cell survival. J Exp Med. (2002) 195:143–9. doi: 10.1084/jem.20011524
68. Al-Alwan M, Hou S, Zhang TT, Makondo K, Marshall AJ. Bam32/DAPP1 promotes B cell adhesion and formation of polarized conjugates with T cells. J Immunol. (2010) 184:6961–9. doi: 10.4049/jimmunol.0904176
69. Gatto D, Brink R. The germinal center reaction. J Allergy Clin Immunol. (2010) 126:898–907. doi: 10.1016/j.jaci.2010.09.007
70. Hudson KM, Byner C, Freeman J, Terry RJ. Immunodepression, high IgM levels and evasion of the immune response in murine trypanosomiasis. Nature. (1976) 264:256–58. doi: 10.1038/264256a0
71. Luckins AG, Mehlitz D. Observations on serum immunoglobulin levels in cattle infected with Trypanosoma brucei, T. vivax and T. congolense. Ann Trop Med Parasitol. (1976) 70:479–80. doi: 10.1080/00034983.1976.11687153
72. Reinitz DM, Mansfield JM. T-cell-independent and T-cell-dependent B-cell responses to exposed variant surface glycoprotein epitopes in trypanosome-infected mice. Infect Immun. (1990) 58:2337–42.
73. Mertens B, Taylor K, Muriuki C, Rocchi M. Cytokine mRNA profiles in trypanotolerant and trypanosusceptible cattle infected with the protozoan parasite Trypanosoma congolense: protective role for interleukin-4? J Interferon Cytokine Res. (1999) 19:59–65. doi: 10.1089/107999099314423
74. Duxbury RE, Sadun EH, Wellde BT, Anderson JS, Muriithi IE. Immunization of cattle with x-irradiated African trypanosomes. Trans R Soc Trop Med Hyg. (1972) 66:349–50. doi: 10.1016/0035-9203(72)90237-4
75. Mosser DM, Roberts JF. Trypanosoma brucei: recognition In vitro of two developmental forms by murine macrophages. Exp Parasitol. (1982) 54:310–6. doi: 10.1016/0014-4894(82)90040-6
76. Mabbott NA, Sutherland IA, Sternberg JM. Trypanosoma brucei is protected from the cytostatic effects of nitric oxide under in vivo conditions. Parasitol Res. (1994) 80:687–90. doi: 10.1007/BF00932954
77. Sternberg MJ, Mabbott NA. Nitric oxide-mediated suppression of T cell responses during Trypanosoma brucei infection: soluble trypanosome products and interferon-gamma are synergistic inducers of nitric oxide synthase. Eur J Immunol. (1996) 26:539–43. doi: 10.1002/eji.1830260306
78. Magez S, Geuskens M, Beschin A, del Favero H, Verschueren H, Lucas R, et al. Specific uptake of tumor necrosis factor-alpha is involved in growth control of Trypanosoma brucei. J Cell Biol. (1997) 137:715–27. doi: 10.1083/jcb.137.3.715
79. Uzonna JE, Kaushik RS, Gordon JR, Tabel H. Experimental murine Trypanosoma congolense infections. I. Administration of anti-IFN-gamma antibodies alters trypanosome-susceptible mice to a resistant-like phenotype. J Immunol. (1998) 161:5507–15.
80. Uzonna JE, Kaushik RS, Gordon JR, Tabel H. Immunoregulation in experimental murine Trypanosoma congolense infection: anti-IL-10 antibodies reverse trypanosome-mediated suppression of lymphocyte proliferation In vitro and moderately prolong the lifespan of genetically susceptible BALB/c mice. Parasite Immunol. (1998) 20:293–302. doi: 10.1046/j.1365-3024.1998.00156.x
81. Uzonna JE, Kaushik RS, Zhang Y, Gordon JR, Tabel H. Experimental murine Trypanosoma congolense infections>. II. Role of splenic adherent CD3+Thy1.2+ TCR-alpha beta- gamma delta- CD4+8- and CD3+Thy1.2+ TCR-alpha beta- gamma delta- CD4-8- cells in the production of IL-4, IL-10, and IFN-gamma and in trypanosome-elicited immunosuppression. J Immunol. (1998) 161:6189–97.
82. Shi M, Pan W, Tabel H. Experimental African trypanosomiasis: IFN-gamma mediates early mortality. Eur J Immunol. (2003) 33:108–18. doi: 10.1002/immu.200390013
83. Noel W, Hassanzadeh G, Raes G, Namangala B, Daems I, Brys L, et al. Infection stage-dependent modulation of macrophage activation in Trypanosoma congolense -resistant and -susceptible mice. Infect Immun. (2002) 70:6180–7. doi: 10.1128/IAI.70.11.6180-6187.2002
84. Namangala B, De Baetselier P, Beschin A. Both type-I and type-II responses contribute to murine trypanotolerance. J Vet Med Sci. (2009) 71:313–8. doi: 10.1292/jvms.71.313
85. Magez S, Radwanska M, Drennan M, Fick L, Baral TN, Brombacher F, et al. Interferon-gamma and nitric oxide in combination with antibodies are key protective host immune factors during trypanosoma congolense Tc13 Infections. J Infect Dis. (2006) 193:1575–83. doi: 10.1086/503808
86. Magez S, Radwanska M, Beschin A, Sekikawa K, De Baetselier P. Tumor necrosis factor alpha is a key mediator in the regulation of experimental Trypanosoma brucei infections. Infect Immun. (1999) 67:3128–32.
87. Hertz CJ, Filutowicz H, Mansfield JM. Resistance to the African trypanosomes is IFN-gamma dependent. J Immunol. (1998) 161:6775–83.
88. Schopf LR, Filutowicz H, Bi XJ, Mansfield JM. Interleukin-4-dependent immunoglobulin G1 isotype switch in the presence of a polarized antigen-specific Th1-cell response to the trypanosome variant surface glycoprotein. Infect Immun. (1998) 66:451–61.
89. Shi MQ, Wei GJ, Tabel H. Trypanosoma congolense infections: MHC class II-restricted immune responses mediate either protection or disease, depending on IL-10 function. Parasite Immunol. (2007) 29:107–11. doi: 10.1111/j.1365-3024.2006.00925.x
90. Taylor K, Mertens B, Lutje V, Saya R. Trypanosoma congolense infection of trypanotolerant N'Dama (Bos taurus) cattle is associated with decreased secretion of nitric oxide by interferon-gamma-activated monocytes and increased transcription of interleukin-10. Parasite Immunol. (1998) 20:421–9. doi: 10.1046/j.1365-3024.1998.00165.x
91. Cross GA. Identification, purification and properties of clone-specific glycoprotein antigens constituting the surface coat of Trypanosoma brucei. Parasitology. (1975) 71:393–417. doi: 10.1017/S003118200004717X
92. Pays E. The variant surface glycoprotein as a tool for adaptation in African trypanosomes. Microbes Infect. (2006) 8:930–7. doi: 10.1016/j.micinf.2005.10.002
93. Ferguson MA, Homans SW, Dwek RA, Rademacher TW. Glycosyl-phosphatidylinositol moiety that anchors Trypanosoma brucei variant surface glycoprotein to the membrane. Science. (1988) 239:753–9. doi: 10.1126/science.3340856
94. Carrington M, Carnall N, Crow MS, Gaud A, Redpath MB, Wasunna CL, et al. The properties and function of the glycosylphosphatidylinositol-phospholipase C in Trypanosoma brucei. Mol Biochem Parasitol. (1998) 91:153–64. doi: 10.1016/S0166-6851(97)00190-4
95. Magez S, Stijlemans B, Radwanska M, Pays E, Ferguson MA, De Baetselier P. The glycosyl-inositol-phosphate and dimyristoylglycerol moieties of the glycosylphosphatidylinositol anchor of the trypanosome variant-specific surface glycoprotein are distinct macrophage-activating factors. J Immunol. (1998) 160:1949–56.
96. Berriman M, Ghedin E, Hertz-Fowler C, Blandin G, Renauld H, Bartholomeu DC, et al. The genome of the African trypanosome Trypanosoma brucei. Science. (2005) 309:416–22. doi: 10.1126/science.1112642
97. Pays E, Lips S, Nolan D, Vanhamme L, Perez-Morga D. The VSG expression sites of Trypanosoma brucei: multipurpose tools for the adaptation of the parasite to mammalian hosts. Mol Biochem Parasitol. (2001) 114:1–16. doi: 10.1016/S0166-6851(01)00242-0
98. Borst P, Ulbert S. Control of VSG gene expression sites. Mol Biochem Parasitol. (2001) 114:17–27. doi: 10.1016/S0166-6851(01)00243-2
99. Pays E, Guyaux M, Aerts D, Van Meirvenne N, Steinert M. Telomeric reciprocal recombination as a possible mechanism for antigenic variation in trypanosomes. Nature. (1985) 316:562–4. doi: 10.1038/316562a0
100. Pays E, Van Assel S, Laurent M, Darville M, Vervoort T, Van Meirvenne N, et al. Gene conversion as a mechanism for antigenic variation in trypanosomes. Cell. (1983) 34:371–81. doi: 10.1016/0092-8674(83)90371-9
101. Murray M, Murray PK, Jennings FW, Fisher EW, Urquhart GM. The pathology of Trypanosoma brucei infection in the rat. Res Vet Sci. (1974) 16:77–84. doi: 10.1016/S0034-5288(18)33777-9
102. Stijlemans B, Radwanska M, De Trez C, Magez S. African trypanosomes undermine humoral responses and vaccine development: link with inflammatory responses? Front Immunol. (2017) 8:582. doi: 10.3389/fimmu.2017.00582
103. Obishakin E, de Trez C, Magez S. Chronic Trypanosoma congolense infections in mice cause a sustained disruption of the B-cell homeostasis in the bone marrow and spleen. Parasite Immunol. (2014) 36:187–98. doi: 10.1111/pim.12099
104. Radwanska M, Guirnalda P, De Trez C, Ryffel B, Black S, Magez S. Trypanosomiasis-induced B cell apoptosis results in loss of protective anti-parasite antibody responses and abolishment of vaccine-induced memory responses. PLoS Pathog. (2008) 4:e1000078. doi: 10.1371/journal.ppat.1000078
105. Drennan MB, Stijlemans B, Van den Abbeele J, Quesniaux VJ, Barkhuizen M, Brombacher F, et al. The induction of a type 1 immune response following a Trypanosoma brucei infection is MyD88 dependent. J Immunol. (2005) 175:2501–9. doi: 10.4049/jimmunol.175.4.2501
106. Kobayakawa T, Louis J, Izui S, Lambert PH. Autoimmune response to DNA, red blood cells, and thymocyte antigens in association with polyclonal antibody synthesis during experimental African trypanosomiasis. J Immunol. (1979) 122:296–301.
107. MacKenzie AR, Boreham PF. Autoimmunity in trypanosome infections. I. Tissue autoantibodies in Trypanosoma (Trypanozoon) brucei infections of the rabbit. Immunology. (1974) 26:1225–38.
108. Hunter CA, Jennings FW, Tierney JF, Murray M, Kennedy PG. Correlation of autoantibody titres with central nervous system pathology in experimental African trypanosomiasis. J Neuroimmunol. (1992) 41:143–8. doi: 10.1016/0165-5728(92)90064-R
109. Kazyumba G, Berney M, Brighouse G, Cruchaud A, Lambert PH. Expression of the B cell repertoire and autoantibodies in human African trypanosomiasis. Clin Exp Immunol. (1986) 65:10–8.
110. Jauberteau MO, Younes-Chennoufi AB, Amevigbe M, Bouteille B, Dumas M, Breton JC, et al. Galactocerebrosides are antigens for immunoglobulins in sera of an experimental model of trypanosomiasis in sheep. J Neurol Sci. (1991) 101:82–6. doi: 10.1016/0022-510X(91)90020-8
111. Amevigbe MD, Jauberteau-Marchan MO, Bouteille B, Doua F, Breton JC, Nicolas JA, et al. Human African trypanosomiasis: presence of antibodies to galactocerebrosides. Am J Trop Med Hyg. (1992) 47:652–62. doi: 10.4269/ajtmh.1992.47.652
112. Balber AE, Bangs JD, Jones SM, Proia RL. Inactivation or elimination of potentially trypanolytic, complement-activating immune complexes by pathogenic trypanosomes. Infect Immun. (1979) 24:617–27.
113. Devine DV, Falk RJ, Balber AE. Restriction of the alternative pathway of human complement by intact Trypanosoma brucei subsp. gambiense. Infect Immun. (1986) 52:223–9.
114. Ferrante A, Allison AC. Alternative pathway activation of complement by African trypanosomes lacking a glycoprotein coat. Parasite Immunol. (1983) 5:491–8. doi: 10.1111/j.1365-3024.1983.tb00763.x
115. Askonas BA, Corsini AC, Clayton CE, Ogilvie BM. Functional depletion of T- and B-memory cells and other lymphoid cell subpopulations-during trypanosomiasis. Immunology. (1979) 36:313–21.
116. Corsini AC, Clayton C, Askonas BA, Ogilvie BM. Suppressor cells and loss of B-cell potential in mice infected with Trypanosoma brucei. Clin Exp Immunol. (1977) 29:122–31.
117. Pearson TW, Roelants GE, Lundin LB, Mayor-Withey KS. Immune depression in trypanosome-infected mice. I. Depressed T lymphocyte responses. Eur J Immunol. (1978) 8:723–7. doi: 10.1002/eji.1830081010
118. Askonas BA. Macrophages as mediators of immunosuppression in murine African trypanosomiasis. Curr Top Microbiol Immunol. (1985) 117:119–27. doi: 10.1007/978-3-642-70538-0_6
119. Yamamoto K, Onodera M, Kato K, Kakinuma M, Kimura T, Richards FF. Involvement of suppressor cells induced with membrane fractions of trypanosomes in immunosuppression of trypanosomiasis. Parasite Immunol. (1985) 7:95–106. doi: 10.1111/j.1365-3024.1985.tb00062.x
120. Jayawardena AN, Waksman BH, Eardley DD. Activation of distinct helper and suppressor T cells in experimental trypanosomiasis. J Immunol. (1978) 121:622–8.
121. Okwor I, Onyilagha C, Kuriakose S, Mou Z, Jia P, Uzonna JE. Regulatory T cells enhance susceptibility to experimental Trypanosoma congolense infection independent of mouse genetic background. PLoS Negl Trop Dis. (2012) 6:e1761. doi: 10.1371/journal.pntd.0001761
122. Onyilagha C, Kuriakose S, Ikeogu N, Jia P, Uzonna J. Myeloid-derived suppressor cells contribute to susceptibility to Trypanosoma congolense infection by suppressing CD4(+) T cell proliferation and IFN-gamma production. J Immunol. (2018) 201:507–15. doi: 10.4049/jimmunol.1800180
123. Schleifer KW, Mansfield JM. Suppressor macrophages in African trypanosomiasis inhibit T cell proliferative responses by nitric oxide and prostaglandins. J Immunol. (1993) 151:5492–503.
124. Gobert AP, Daulouede S, Lepoivre M, Boucher JL, Bouteille B, Buguet A, et al. L-Arginine availability modulates local nitric oxide production and parasite killing in experimental trypanosomiasis. Infect Immun. (2000) 68:4653–7. doi: 10.1128/IAI.68.8.4653-4657.2000
125. Vincendeau P, Gobert AP, Daulouede S, Moynet D, Mossalayi MD. Arginases in parasitic diseases. Trends Parasitol. (2003) 19:9–12. doi: 10.1016/S1471-4922(02)00010-7
126. Hajduk SL, Moore DR, Vasudevacharya J, Siqueira H, Torri AF, Tytler EM, et al. Lysis of Trypanosoma brucei by a toxic subspecies of human high density lipoprotein. J Biol Chem. (1989) 264:5210–7.
127. Raper J, Fung R, Ghiso J, Nussenzweig V, Tomlinson S. Characterization of a novel trypanosome lytic factor from human serum. Infect Immun. (1999) 67:1910–6.
128. Pays E, Vanhollebeke B, Uzureau P, Lecordier L, Perez-Morga D. The molecular arms race between African trypanosomes and humans. Nat Rev Microbiol. (2014) 12:575–84. doi: 10.1038/nrmicro3298
129. Drain J, Bishop JR, Hajduk SL. Haptoglobin-related protein mediates trypanosome lytic factor binding to trypanosomes. J Biol Chem. (2001) 276:30254–60. doi: 10.1074/jbc.M010198200
130. Vanhollebeke B, Nielsen MJ, Watanabe Y, Truc P, Vanhamme L, Nakajima K, et al. Distinct roles of haptoglobin-related protein and apolipoprotein L-I in trypanolysis by human serum. Proc Natl Acad Sci U.S.A. (2007) 104:4118–23. doi: 10.1073/pnas.0609902104
131. Molina-Portela MP, Samanovic M, Raper J. Distinct roles of apolipoprotein components within the trypanosome lytic factor complex revealed in a novel transgenic mouse model. J Exp Med. (2008) 205:1721–8. doi: 10.1084/jem.20071463
132. Vanhamme L, Paturiaux-Hanocq F, Poelvoorde P, Nolan DP, Lins L, Van Den Abbeele J, et al. Apolipoprotein L-I is the trypanosome lytic factor of human serum. Nature. (2003) 422:83–7. doi: 10.1038/nature01461
133. Xong HV, Vanhamme L, Chamekh M, Chimfwembe CE, Van Den Abbeele J, Pays A, et al. A VSG expression site-associated gene confers resistance to human serum in Trypanosoma rhodesiense. Cell. (1998) 95:839–46. doi: 10.1016/S0092-8674(00)81706-7
134. Lecordier L, Vanhollebeke B, Poelvoorde P, Tebabi P, Paturiaux-Hanocq F, Andris F, et al. C-terminal mutants of apolipoprotein L-I efficiently kill both Trypanosoma brucei brucei and Trypanosoma brucei rhodesiense. PLoS Pathog. (2009) 5:e1000685. doi: 10.1371/journal.ppat.1000685
135. Berberof M, Perez-Morga D, Pays E. A receptor-like flagellar pocket glycoprotein specific to Trypanosoma brucei gambiense. Mol Biochem Parasitol. (2001) 113:127–38. doi: 10.1016/S0166-6851(01)00208-0
136. Uzureau P, Uzureau S, Lecordier L, Fontaine F, Tebabi P, Homble F, et al. Mechanism of Trypanosoma brucei gambiense resistance to human serum. Nature. (2013) 501:430–4. doi: 10.1038/nature12516
137. Capewell P, Clucas C, DeJesus E, Kieft R, Hajduk S, Veitch N, et al. The TgsGP gene is essential for resistance to human serum in Trypanosoma brucei gambiense. PLoS Pathog. (2013) 9:e1003686. doi: 10.1371/journal.ppat.1003686
138. Gomez-Rodriguez J, Stijlemans B, De Muylder G, Korf H, Brys L, Berberof M, et al. Identification of a parasitic immunomodulatory protein triggering the development of suppressive M1 macrophages during African trypanosomiasis. J Infect Dis. (2009) 200:1849–60. doi: 10.1086/648374
139. Umetsu DT, Dekruyff RH. Immune dysregulation in asthma. Curr Opin Immunol. (2006) 18:727–32. doi: 10.1016/j.coi.2006.09.007
140. Brown KN, Hill J, Holland AE. Anti-trypanosomal activity of certain phenyldiazoamino- and phenylazoamino-phenanthridinium compounds. Br J Pharmacol Chemother. (1961) 17:396–405. doi: 10.1111/j.1476-5381.1961.tb01125.x
141. Shapiro TA, Englund PT. Selective cleavage of kinetoplast DNA minicircles promoted by antitrypanosomal drugs. Proc Natl Acad Sci USA. (1990) 87:950–4. doi: 10.1073/pnas.87.3.950
142. Gonzalez VM, Perez JM, Alonso C. The berenil ligand directs the DNA binding of the cytotoxic drug Pt-berenil. J Inorg Biochem. (1997) 68:283–7. doi: 10.1016/S0162-0134(97)00111-6
143. Magez S, Caljon G, Tran T, Stijlemans B, Radwanska M. Current status of vaccination against African trypanosomiasis. Parasitology. (2010) 137:2017–27. doi: 10.1017/S0031182010000223
144. Tabel H, Wei G, Bull HJ. Immunosuppression: cause for failures of vaccines against African Trypanosomiases. PLoS Negl Trop Dis. (2013) 7:e2090. doi: 10.1371/journal.pntd.0002090
145. Playfair JH, Taverne J, Bate CA, de Souza JB. The malaria vaccine: anti-parasite or anti-disease? Immunol Today. (1990) 11:25–7. doi: 10.1016/0167-5699(90)90007-V
146. Antoine-Moussiaux N, Buscher P, Desmecht D. Host-parasite interactions in trypanosomiasis: on the way to an antidisease strategy. Infect Immun. (2009) 77:1276–84. doi: 10.1128/IAI.01185-08
147. Authie E, Boulange A, Muteti D, Lalmanach G, Gauthier F, Musoke AJ. Immunisation of cattle with cysteine proteinases of Trypanosoma congolense: targetting the disease rather than the parasite. Int J Parasitol. (2001) 31:1429–33. doi: 10.1016/S0020-7519(01)00266-1
148. Authie E, Muteti DK, Mbawa ZR, Lonsdale-Eccles JD, Webster P, Wells CW. Identification of a 33-kilodalton immunodominant antigen of Trypanosoma congolense as a cysteine protease. Mol Biochem Parasitol. (1992) 56:103–16. doi: 10.1016/0166-6851(92)90158-G
149. Fish WR, Nkhungulu ZM, Muriuki CW, Ndegwa DM, Lonsdale-Eccles JD, Steyaert J. Primary structure and partial characterization of a life-cycle-regulated cysteine protease from Trypanosoma (Nannomonas) congolense. Gene. (1995) 161:125–8. doi: 10.1016/0378-1119(95)00304-O
150. Nikolskaia OV, de ALAP, Kim YV, Lonsdale-Eccles JD, Fukuma T, Scharfstein J, et al. Blood-brain barrier traversal by African trypanosomes requires calcium signaling induced by parasite cysteine protease. J Clin Invest. (2006) 116:2739–47. doi: 10.1172/JCI27798
151. Ropert C, Almeida IC, Closel M, Travassos LR, Ferguson MA, Cohen P, et al. Requirement of mitogen-activated protein kinases and I kappa B phosphorylation for induction of proinflammatory cytokines synthesis by macrophages indicates functional similarity of receptors triggered by glycosylphosphatidylinositol anchors from parasitic protozoa and bacterial lipopolysaccharide. J Immunol. (2001) 166:3423–31. doi: 10.4049/jimmunol.166.5.3423
152. Almeida IC, Camargo MM, Procopio DO, Silva LS, Mehlert A, Travassos LR, et al. Highly purified glycosylphosphatidylinositols from Trypanosoma cruzi are potent proinflammatory agents. EMBO J. (2000) 19:1476–85. doi: 10.1093/emboj/19.7.1476
153. Schofield L, Hewitt MC, Evans K, Siomos MA, Seeberger PH. Synthetic GPI as a candidate anti-toxic vaccine in a model of malaria. Nature. (2002) 418:785–9. doi: 10.1038/nature00937
154. Stijlemans B, Baral TN, Guilliams M, Brys L, Korf J, Drennan M, et al. A glycosylphosphatidylinositol-based treatment alleviates trypanosomiasis-associated immunopathology. J Immunol. (2007) 179:4003–14. doi: 10.4049/jimmunol.179.6.4003
155. Stijlemans B, Vankrunkelsven A, Brys L, Raes G, Magez S, De Baetselier P. Scrutinizing the mechanisms underlying the induction of anemia of inflammation through GPI-mediated modulation of macrophage activation in a model of African trypanosomiasis. Microbes Infect. (2010) 12:389–99. doi: 10.1016/j.micinf.2010.02.006
Keywords: African trypanosomes, immunity, immunosuppression, immune evasion, resistance, susceptibility
Citation: Onyilagha C and Uzonna JE (2019) Host Immune Responses and Immune Evasion Strategies in African Trypanosomiasis. Front. Immunol. 10:2738. doi: 10.3389/fimmu.2019.02738
Received: 14 June 2019; Accepted: 08 November 2019;
Published: 22 November 2019.
Edited by:
Michael Harrison Hsieh, Children's National Health System, United StatesReviewed by:
Guy Caljon, University of Antwerp, BelgiumCopyright © 2019 Onyilagha and Uzonna. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jude Ezeh Uzonna, anVkZS51em9ubmFAdW1hbml0b2JhLmNh
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.