
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 15 November 2019
Sec. NK and Innate Lymphoid Cell Biology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02689
This article is part of the Research TopicTGF-β as a Key Regulator of NK and ILCs Development and FunctionsView all 5 articles
 Mariya Lazarova
Mariya Lazarova Alexander Steinle*
Alexander Steinle*Transforming growth factor-β (TGF-β) suppresses innate and adaptive immune responses via multiple mechanisms. TGF-β also importantly contributes to the formation of an immunosuppressive tumor microenvironment thereby promoting tumor growth. Amongst others, TGF-β impairs tumor recognition by cytotoxic lymphocytes via NKG2D. NKG2D is a homodimeric C-type lectin-like receptor expressed on virtually all human NK cells and cytotoxic T cells, and stimulates their effector functions upon engagement by NKG2D ligands (NKG2DL). While NKG2DL are mostly absent from healthy cells, their expression is induced by cellular stress and malignant transformation, and, accordingly, frequently detected on various tumor cells. Hence, the NKG2D axis is thought to play a decisive role in cancer immunosurveillance and, obviously, often is compromised in clinically apparent tumors. There is mounting evidence that TGF-β, produced by tumor cells and immune cells in the tumor microenvironment, plays a key role in blunting the NKG2D-mediated tumor surveillance. Here, we review the current knowledge on the impairment of NKG2D-mediated cancer immunity through TGF-β and discuss therapeutic approaches aiming at counteracting this major immune escape pathway. By reducing tumor-associated expression of NKG2DL and blinding cytotoxic lymphocytes through down-regulation of NKG2D, TGF-β is acting upon both sides of the NKG2D axis severely compromising NKG2D-mediated tumor rejection. Consequently, novel therapies targeting the TGF-β pathway are expected to reinvigorate NKG2D-mediated tumor elimination and thereby to improve the survival of cancer patients.
Transforming growth factor-β (TGF-β) is a potent suppressor of immune responses affecting many subsets of immune cells in various ways (1). For example, TGF-β impairs MHC class II expression (2, 3), thus potentially impairing priming of CD4 T cells, and suppresses the activity of cytotoxic lymphocytes by inhibiting the differentiation, proliferation, and effector functions of CD8 T cells and NK cells (1, 4). TGF-β also promotes the differentiation of suppressive immune cells subsets (5–7). In physiological settings, the TGF-β-mediated immune suppression is crucial for the establishment of immune tolerance and prevention of chronic inflammation, e.g., in the gastrointestinal tract (4, 8, 9), but in malignant disease TGF-β promotes immune escape, tumor progression and metastasis (4, 10–13). Importantly, there is emerging evidence that TGF-β also impairs immunorecognition of tumor cells by NK cells and cytotoxic T cells through down-regulation of activating immunoreceptors such as NKG2D. NKG2D ligation by stress-induced MHC class I-like glycoproteins on tumor cells transmits a potent stimulatory signal into cytotoxic lymphocytes and therefore promotes immunosurveillance of malignant cells (14, 15). Hence, evasion from NKG2D-mediated recognition is thought to represent a major mechanism allowing tumors to escape from tumor immunity. In this review, we specifically focus on the TGF-β-mediated impairment of immunorecognition through the NKG2D axis and its implications for tumor immunity and cancer therapies. The function and biology of TGF-β as well as of NKG2D will be summarized only briefly as both have extensively been reviewed elsewhere (4, 10, 16–18).
The three members of the human TGF-β family, TGF-β1,−2, and−3 are synthesized as precursor proteins containing an N-terminal latency-associated peptide (LAP) (~280–300 amino acids) followed by a shorter C-terminal polypeptide (112–114 amino acids) which represents the biologically active mature cytokine (19, 20). During the intracellular processing of this precursor protein, LAP is cleaved but remains associated with the TGF-β dimer forming an inactive latency complex that is sequestered into the extracellular matrix. Activation of TGF-β requires release from this latency complex (21). In addition, TGF-β can be present on the surface of regulatory T cells (Tregs), endothelial cells, platelets, macrophages and microglia in a membrane-associated form (22, 23). Mature TGF-β homodimers bind, with or without the assistance of the accessory receptor betaglycan (BG, also called TGF-β receptor III), first to homodimers of the TGF-β receptor II (TGF-βRII) which then phosphorylate TGF-β receptor I homodimers (TGF-βRI, ALK5) under formation of a hexameric complex of TGF-β, TGF-βRII, and TGF-βRI homodimers. Subsequently, TGF-βRI phosphorylates the cytoplasmic SMAD2 and SMAD3 proteins, which then, under association with SMAD4, transmigrate into the nucleus and exert transcriptional activity (4, 16). TGF-β receptors are expressed on virtually all immune cells. Of note, TGF-βRII expression was shown to decline in the course of mouse NK cell maturation (24).
TGF-β1 is the predominant TGF-β family member expressed by immune cells and suppresses innate and adaptive immune responses at multiple levels (4, 25). Amongst others, TGF-β has a prominent role in dampening T and NK cell responses: TGF-β impairs T cell proliferation and effector functions through down-regulation of IL-2 during T cell priming (26) and has been shown to induce cell cycle arrest and apoptosis of T cells (27–29). TGF-β directly inhibits the cytotoxic functions of CD8 T cells (30) and the differentiation of both Th1 and Th2 subsets by downregulation of their key transcription factors (31–35). Further, TGF-β downregulates the expression of MHC class II molecules via affecting CIITA expression (2, 3) thus impairing the capacity of antigen presenting cells (APC) for antigen presentation and CD4 T cell priming. TGF-β also inhibits the expansion, cytotoxicity, and cytokine production by NK cells (36–39). More recently, TGF-β was shown to block the IL-15-induced metabolic activity and proliferation of NK cells by inhibiting mTOR activity (24). In addition, TGF-β promotes conversion of NK cells into non-cytotoxic ILC1 in the tumor microenvironment (TME) thereby blunting tumor killing (40). TGF-β further promotes differentiation of Tregs (5, 6) and of myeloid derived suppressor cells (MDSC) (7). An eminent importance of TGF-β in affecting cancer immunosurveillance and efficacy of checkpoint blockade cancer therapy was recently highlighted by a series of studies on human cancer patients and of mouse tumor models: TGF-β produced by the TME was shown to restrict tumor infiltration by T cells and other cytotoxic lymphocytes and to block the acquisition of a Th1 effector phenotype (41–43). Inhibition of TGF-β activity not only facilitated T cell infiltration into central sites of the tumor, but also unleashed vigorous and efficient anti-tumor immunity, particularly in the course of checkpoint blockade (41–43). On the other hand, immunosuppression by TGF-β plays a central physiologic role in the establishment of immune tolerance and control of inflammation. Germline deletion of TGF-β1 in mice is lethal due to multi-organ inflammation (8, 9). Loss of TGF-β signaling, particularly in T cells, is associated with uncontrolled adaptive T cell responses and severe inflammatory disease (4, 44–46). In the persistent presence of antigen stimuli, e.g., in the gastrointestinal tract, TGF-β aids in suppression of immune responses in order to prevent chronic inflammation (4).
Loss of function mutations in the TGF-β receptors or in SMAD proteins are found in many tumors indicating a function as a tumor suppressor (4). TGF-β inhibits cell growth (4, 47–50), blocks the transition of pre-malignant cells to a more evasive phenotype and induces their apoptosis (51, 52). In contrast, there is also broad evidence suggesting that TGF-β supports tumorigenesis and invasiveness, and enables tumor growth by establishing an immunosuppressive and T cell excluding TME. For example, elevated TGF-β levels in the TME impair anti-tumor T cell responses (11–13, 53) with restricting T cell infiltration into the tumors as shown for mouse models of metastatic colorectal, urothelial and epithelial ovarian cancers (41–43). TGF-β is thought to function as a tumor suppressor at the early stages of tumor development, but with the progression of disease, cancer cells may decouple growth-inhibitory paracrine TGF-β signals by obstructing their TGF-β receptor signaling pathway and rather exploit the immunosilencing capacity of TGF-β to facilitate immune evasion and metastatic dissemination (4, 16).
NKG2D is a type II transmembrane glycoprotein comprising an extracellular C-type lectin-like domain, a transmembrane domain, and a short cytoplasmic portion without signaling motifs (54, 55). NKG2D glycoproteins form disulfide-linked homodimers with both monomers building a single ligand binding site (56). In humans, NKG2D homodimers associate with two pairs of DAP10 adaptor proteins through interaction of charged residues in the respective transmembrane domains. Formation of this hexameric complex is required for cell surface expression of NKG2D and signal transduction (55, 57). NKG2D is found on virtually all human NK cells and CD8 T cells, on most γδ T cells and iNKT cells, as well as on a few CD4 T cells (54, 58). Ligation of NKG2D activates cytotoxicity and cytokine production of NK cells and provides stimulatory signals for effector CD8 T cells (54, 59–63). NKG2D expression is enhanced through cytokines promoting NK and T cell survival and expansion such as IL-15 and IL-2 (62–64).
NKG2D ligands (NKG2DL) are stress-inducible membrane-bound proteins distantly related to MHC class I molecules. In human, there are two families of NKG2DL, the MIC family consisting of MICA and MICB, and the ULBP family consisting of ULBP1 through ULBP6 (14, 65, 66). All NKG2DL contain an ectodomain with an MHC class I-like α1α2-fold (14, 56, 67), but unlike MHC molecules NKG2DLs neither associate with β2-microglobulin, nor present antigenic peptides (54, 61). MICs contain an additional Ig-like α3 domain in their extracellular part that is absent from ULBPs (14, 56). Most MICs are single-pass transmembrane proteins, although there are also reports for GPI-anchored MICA variants (68, 69). ULBP1 through ULBP3 and ULBP6 are GPI-anchored, whereas ULBP4 and ULBP5 are inserted into the membrane with a single transmembrane domain (67, 70, 71).
While NKG2DLs are typically absent from the cell surface of healthy cells, NKG2DL transcripts are found in almost all human tissues (72), indicating a dominant control of NKG2DL expression at the post-transcriptional level. NKG2DL are surfacing on activated hematopoietic cells which may contribute to an NKG2D-mediated regulation of immune responses and may dampen T cell responses (73, 74), e.g., during the resolution of an infection (75, 76). NKG2DL are also found on many human tumor cell lines and primary human tumors (77), and are up-regulated during viral infections, particularly during infections with viruses of the herpesvirus family (78, 79). Such NKG2DL expression marks infected or malignant cells as “dangerous” for the immune system and facilitates their clearance through cytotoxic lymphocytes. NKG2DL on tumor cells enhance their susceptibility to NK cell killing (54, 80), protects against tumor initiation (81) promotes tumor rejection and/or reduce the tumor progression (82–85). Tumors utilize a variety of mechanisms to escape from NKG2D-mediated immunosurveillance: these mechanisms include the release of soluble NKG2DLs (sNKG2DL) either by proteolytic cleavage (71, 86–88) or by exosomal release of membrane-bound NKG2DLs (89, 90). Release of sNKG2DL reduces the density of NKG2DL on malignant cells and thereby impairs NKG2D-mediated recognition and elimination of tumor cells by cytotoxic lymphocytes (82–85). While some studies also report down-modulation of surface NKG2D on cytotoxic lymphocytes through sNKG2DL-mediated internalization (91, 92), other studies were unable to confirm these findings or attributed NKG2D down-modulation instead to TGF-β (82, 93, 94). Possibly, potent NKG2D down-modulation by TGF-β (see below) in serum samples of cancer patients containing both TGF-β and sNKG2DL may have led to some erroneous conclusions regarding sNKG2DL-mediated NKG2D down-modulation in previous studies (15, 92–94). Also, sera of tumor-free MICA-transgenic mice containing very high levels of sMICA did not affect NKG2D surface levels by splenic mouse NK cells (82). However, persistent exposure of NKG2D to membrane-bound MICA down-regulated surface NKG2D and reduced NK cell cytotoxicity in these MICA-transgenic mice as well as in other transgenic mouse models overexpressing NKG2DL (82, 95, 96). Hence, strong overexpression of NKG2DL may represent a strategy of tumor cells to blunt NKG2D-mediated immunosurveillance. In contrast to proteolytically shed monomeric sNKG2DL (i.e., most MICA variants, MICB, and ULBP2), exosomally released NKG2DL such as the prevalent MICA*08, ULBP1 or ULBP3 may down-modulate surface NKG2D through multivalency-based cross-linking (89, 90). Further escape mechanisms from NKG2D-mediated cancer immunosurveillance include down-regulation of NKG2DL through miRNAs (97, 98), epigenetic changes or transcriptional repression (99, 100), and TGF-β mediated signaling as outlined below. Intraindividual heterogeneity of malignant cells can also impair NKG2D-mediated tumor clearance: a recent study by Paczulla et al. showed that malignant cells of human acute myeloid leukemia (AML) patients are heterogeneous for NKG2DL expression with leukemic stem cells (LSC) being devoid of NKG2DL and therefore resistant to NK cell-mediated elimination (100). Poly-ADP-ribose polymerase 1 (PARP1) was shown to repress transcription of NKG2DL in LSC thereby enabling their escape from NKG2D-mediated immunosurveillance (100).
Soon after cloning of the TGF-β1 cDNA (101), TGF-β1 was shown to inhibit both the proliferation of T cells (102) and the anti-tumor cytotoxicity of NK cells (36). While it was demonstrated that TGF-β impairs effector functions of NK cells against target cells, the underlying mechanisms remained elusive until it was reported by Moretta and colleagues that TGF-β downregulates the surface expression of the activating NK receptors NKG2D and NKp30, thereby impairing NK cytolysis of tumor cell lines in vitro (103) (Figure 1). Obviously, this effect depends on the extent of expression of NKG2DL and ligands of NKp30 by the respective tumor cells. Subsequent studies confirmed and extended these observations (104, 105): TGF-β inhibits NKG2D-mediated lysis of target cells without altering the expression of perforin or Fas ligand, or without affecting NK cell viability, indicating that down-regulation of NKG2D is a major effect of TGF-β on NK cytolysis of tumor cells (105). A study on glioblastoma not only reported TGF-β-induced reduction of NKG2D expression on NK cells, but also on cytotoxic T lymphocytes (CTL). Decreased NKG2D expression resulted in the decreased cytolysis of NKG2DL positive targets by NK cells and a reduced NKG2D-mediated co-stimulation of CD8 T cells (104). The elevated TGF-β levels in sera of patients with lung and colorectal cancers were shown to down-regulate NKG2D on NK cells. Other studies linked increased tumor-associated TGF-β levels with the impairment of the function of NK cells and CTLs, and NKG2D down-regulation in various malignancies including Hodgkin lymphoma (106), gastric cancer (107) and head and neck squamous cell carcinoma (108, 109). Hence, impaired NKG2D expression may serve as a biomarker for TGF-β-compromised cytotoxic lymphocytes. TGF-β-mediated down-regulation of NKG2D and associated impaired NK cell functions were also reported in the context of infections with hepatitis B and C viruses (110, 111).
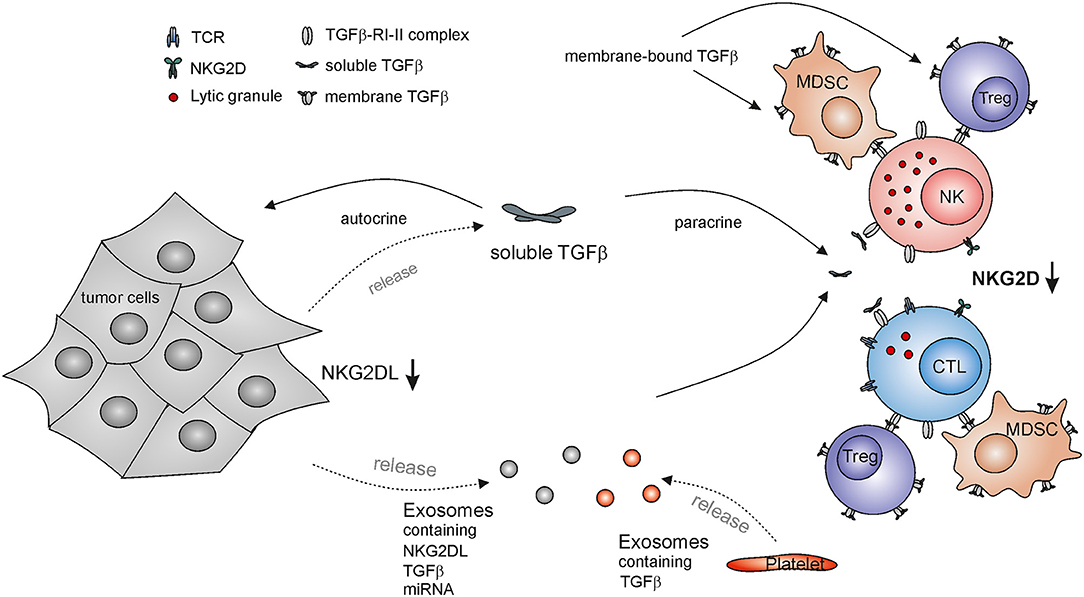
Figure 1. TGF-β-mediated escape from NKG2D-mediated tumor immunorecognition by cytotoxic lymphocytes. NKG2D down-regulation on cytotoxic lymphocytes impairs their immunosurveillance of NKG2DL-expressing malignant cells and subsequent tumor elimination. Tumor cells release both soluble TGF-β and TGF-β-containing exosomes locally and systemically acting on NK cells and cytotoxic T lymphocytes (CTL), thereby inducing downregulation of NKG2D. In addition, tumor-derived exosomes may contain NKG2DLs and miRNA with the capacity to down-regulate NKG2D surface expression. TGF-β also acts on tumor cells in an autocrine or paracrine manner thereby reducing NKG2DL expression and further subverting cancer immunosurveillance by the NKG2D-NKG2DL axis. Other major source of TGF-β are platelets as well as regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSCs) which also present membrane bound TGF-β.
Elevated TGF-β levels as detected in glioblastoma patients were also shown to affect the expression of NKG2DLs (104, 112): experimentally reduced TGF-β expression by glioma cells led to an increase of MICA, ULBP2, and ULBP4 transcripts and increased cell surface expression of MICA and ULBP2 as well as of a reduction of tumorigenicity in vivo (104, 112). Thus, tumor derived TGF-β can act in a paracrine fashion to decrease NKG2D expression on cytotoxic lymphocytes in the TME and in an autocrine manner to diminish tumor-associated NKG2DL expression thereby impairing the innate recognition and clearance of tumors (104). Hence, TGF-β-mediated repression of NKG2DL expression together with proteolytic shedding of NKG2DL has been suggested to facilitate the immune escape of glioma in the immune-privileged brain (112). However, there are also some reports that TGF-β treatment increases surface levels of NKG2DLs (113). The induction of cell surface expression of MICA and MICB upon culture with TGF-β was described for several human cell lines and appears at least partially dependent on mTOR signaling. In the case of HaCat cells, the increase in NKG2DL was associated with the TGF-β-induced epithelial-to-mesenchymal transition (113). These reports indicate that the regulation of NKG2DL expression by TGF-β may be dependent on the cell type and the context of the microenvironment.
TGF-β can be presented as a membrane bound form on the surface of several cell types (22, 23) and there is evidence that membrane-bound TGF-β can also regulate NKG2D expression. Surface-bound TGF-β presented by Tregs was found to decrease NKG2D expression on NK cells and this correlated with the inhibition of NK cell cytotoxicity (114). Adoptive transfer of Tregs in Treg-deficient mice resulted in a decreased NKG2D expression and NK cell cytotoxicity in vivo and reduced the anti-tumor effector functions of NK cells in an NKG2D-sensitive tumor model in a TGF-β dependent manner (114). Other reports confirmed that TGF-β produced by Tregs impairs NKG2D-mediated NK cell killing of target cells in vitro (115). Decreased NKG2D expression was also found on NK cells in murine models of liver and lung cancer and correlated with the frequency of MDSC. MDSC isolated from cancer-bearing mice were able to impair NK cells functions and NKG2D expression on NK cells in vitro, and after adoptive transfer in healthy mice, and depletion of MDSC from tumor-bearing mice restored the functionality and NKG2D expression on NK cells and delayed the tumor progression in vivo (116). The observed effects were also mediated through a membrane-bound TGF-β presented by MDSC, while NK cells deficient in TGF-β signaling were resistant to the MDSC-mediated effects (116). Exosomal secretion of NKG2DL can impair NKG2D expression on cytotoxic lymphocytes thus desensitizing them for NKG2DL-mediated tumor recognition (89). Exosomes derived from a panel of tumor cell lines and from patients with malignant pleural mesothelioma were also shown to contain TGF-β on exosomes and down-regulated NKG2D on the surface of CTLs and NK cells. Neutralizing TGF-β or MICA of exosomes indicated that TGF-β, and not MICA, is the main factor driving the observed NKG2D downregulation (94). Microvesicles derived from sera of AML patients were also shown to contain high levels of TGF-β and decreased NKG2D expression as well as NK cell cytotoxicity in a TGF-β dependent manner (117).
Mouse models suggest that metastasis formation is dependent on the tumor-protective function of platelets, but the cross-talk between tumor-coating platelets and NK cells in the blood is not yet fully understood (118, 119). Platelet-derived TGF-β may promote the immune escape of circulating disseminated tumor cells as activated platelets release factors reducing the activation and IFN-γ production of NK cells and the expression of a set of activating NK cell receptors including NKG2D. This effect is at least partially mediated by platelet-derived TGF-β (120). Platelet-derived TGF-β was shown to induce an invasive phenotype of tumor cells promoting metastasis in mouse models of colon and breast carcinoma. Abrogation of either TGF-β signaling in tumor cells or TGF-β expression by platelets suppressed metastasis formation and epithelial-mesenchymal transition (121). Accordingly, it was proposed that platelet-derived TGF-β in the circulation provides a “pulse” to tumor cells enabling them to acquire a more invasive mesenchymal-like phenotype (121). Platelets were also shown to secrete TGF-β-rich exosomes upon storage, e.g., before transfusions, that induce downregulation of NKG2D, NKp30, and DNAM-1 and modulate NK cell functions (122).
The molecular mechanism underlying the TGF-β-mediated down-modulation of NKG2D surface expression are not yet fully elucidated. Several studies reported that TGF-β treatment only results in a moderate reduction of NKG2D transcripts (64, 103) demonstrating that TGF-β mainly acts through post-transcriptional mechanisms on NKG2D expression. A more recent study provided conclusive evidence that induction of mature miR-1245 by TGF-β controls NKG2D expression in NK cells (123) (Figure 2). TGF-β augments processing of the pri-miR-1245 in NK cells and strongly increases the levels of mature miR-1245 in NK cells which acts on a target site in the 3'-UTR of NKG2D transcripts. Overexpression or silencing of miRNA-1245 markedly reduced or enhanced surface NKG2D on NK cells, respectively (123). Of note, IL-15 suppressed the maturation of miRNA-1245 which is detectable in tumor-derived exosomes in hematopoietic malignancies (123). Expression of miRNA-1245 is up-regulated by c-myc which directly binds to the miRNA-1245 promoter (124) indicating that exosomes of c-myc-driven tumors may harbor miRNA-1245 and thereby target NKG2D expression. However, TGF-β-mediated reduction of surface NKG2D levels is not completely abolished in miR-1245 knock-out cells arguing for further mechanisms (123). Accordingly, other studies reported that TGF-β treatment substantially decreases DAP10 expression both at mRNA and protein levels (64, 125). Since NKG2D cell surface expression strictly depends on complex formation with DAP10 (55, 57), the TGF-β-mediated down-regulation of DAP10 indirectly complements the direct suppression of NKG2D expression by miR-1245 (64, 123).
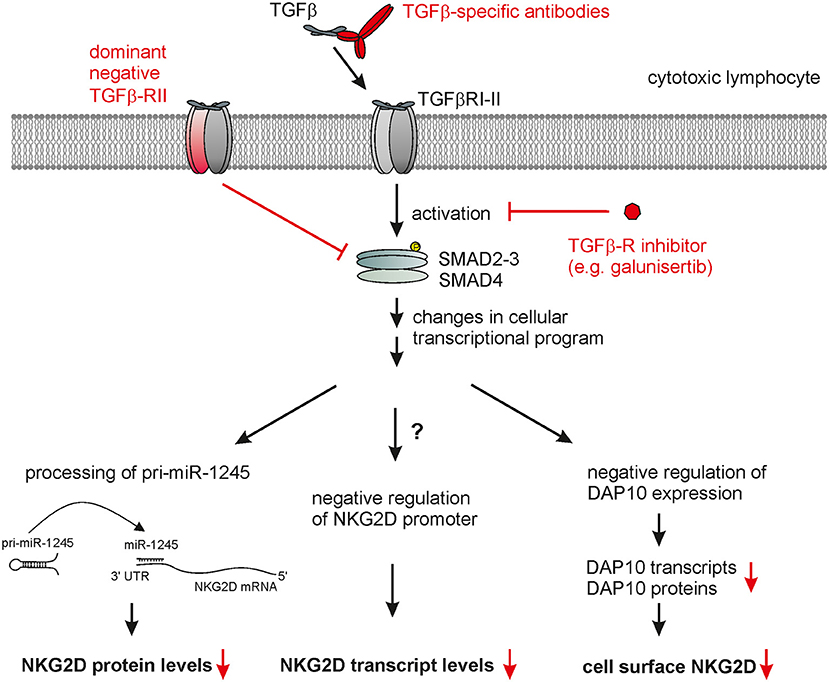
Figure 2. Therapeutic targeting of TGF-β-mediated NKG2D down-regulation by cytotoxic lymphocytes. TGF-β bound to a tetrameric complex of TGF-β-RI and TGF-β-RII homodimers causes phosphorylation of SMAD proteins, which, together with further contextual transcriptional regulators, alter the cellular transcriptional profile. This ultimately also leads to markedly reduced cell surface NKG2D expression by cytotoxic lymphocytes which appears to result from several direct and indirect effects: (i) decrease of NKG2D transcripts, (ii) maturation of miR-1245 interacting with the 3'-UTR of NKG2D transcripts thereby repressing NKG2D expression, and (iii) decreased levels of DAP10 transcripts and proteins with DAP10 being essentially required for NKG2D surface expression. Therapeutic strategies interfering with TGF-β signaling (marked in red) to rescue NKG2D expression include: (i) neutralization of TGF-β receptor through TGF-β specific antibodies or soluble TGF-β-RII, (ii) inhibition of TGF-β-RI-II activation through small molecules such as galunisertib, and (iii) engineering therapeutic lymphocytes prior to adoptive transfer with dominant negative TGF-β-RII chains.
Multiple miRNA have also been shown to down-regulate expression of human NKG2DL by human tumor cells thereby impairing NKG2D-mediated tumor recognition (97, 98, 126, 127). However, for most of these miRNA their tumor-associated regulation is not clear. In contrast, expression of the oncomiR-183, up-regulated by TGF-β in lung cancer, was shown to down-regulate MICA and MICB glycoprotein expression in lung tumor cell lines through a binding site in the 3'-UTR of MICA/B transcripts. Accordingly, shRNA-mediated knock-down of either TGF-β or miR-183 resulted in an enhanced MICA/B expression and cytolysis by CD8 T cells (128). TGF-β-induced miR-183 was also reported to impair expression and function of several activating NK receptors such as NKp44 through down-regulation of the adaptor protein DAP12 (129), and hence, targets tumor recognition by NK cells at various receptors.
The crucial role of TGF-β in tumor progression and tumor immune escape renders this cytokine an important target for therapeutic intervention in cancer. Accordingly, multiple cancer therapies targeting the TGF-β pathway are currently being evaluated in clinical trials. Therapies targeting the TGF-β pathway have, amongst others, the potential to boost tumor elimination by cytotoxic lymphocytes through harnessing the NKG2D-mediated tumor recognition and boosting cytolysis by NK cells and cytotoxic T lymphocytes. For example, galunisertib (LY2157299), a small molecule inhibiting TGF-βRI kinase activity (Figure 2), prevented in vitro the TGF-β-mediated down-regulation of surface NKG2D (as well as of NKp30, DNAM-1, TRAIL) by activated NK cells and preserved their cytotoxic activity toward various tumor cell lines (130, 131). Accordingly, administration of galunisertib markedly enhanced the anti-tumor effect of adoptively transferred activated human NK cells in NSG mice bearing human tumors (130, 131). Significant therapeutic effects in phase II clinical trials were reported with galunisertib given either in combination with gemcitabine in pancreatic cancer (132) or as a monotherapy in hepatocellular carcinoma (133). Importantly, no adverse side effects and no cardiac toxicity were reported by several clinical trials (134). Encouraging pre-clinical studies show that a combined cancer treatment using galunisertib together with checkpoint blockade antibodies strongly potentiated cancer immunity (43, 135).
Suppressive effects of TGF-β may also be overcome by targeted delivery of cytokines IL-2, IL-15, and IL-18 into the tumor. While TGF-β was shown to have a dominant effect over IL-2 or IL-15 alone with regard to NKG2D modulation on the surface of NK cells (64, 105), a combination of IL-2 and IL-18 protected NK-92MI cells from TGF-β-mediated NKG2D down-regulation and the associated impairment of NK cell function (136). An IL-15 superagonist/IL-15Rα fusion complex (ALT-803) rescued NK cytolysis of tumor cell lines from TGF-β1-mediated immunosuppression in vitro and diminished TGF-β1-mediated down-regulation of surface NKG2D (137). IRX-2, a poorly defined mixture of cytokines derived from the culture supernatants of activated lymphocytes, was tested in clinical trials for treatment of head and neck squamous cell cancer, and increased NKG2D surface expression and NKG2D-dependent NK cytotoxicity, even in the presence of TGF-β (109).
TGF-β-neutralizing macromolecules such as TGF-β-specific mAb or soluble forms of TGF-βRII are currently evaluated in several phase I and II clinical trials in treatment of patients with various solid tumors (4). A recent report on a phase I/II clinical trial for treatment of chemo-refractory metastatic breast cancer with the TGF-β-neutralizing mAb fresolimumab during radiotherapy did not observe an objective or abscopal response in tumor patients treated with fresolimumab (138, 139). Exploratory analyses of circulating T cells from these patients indicated that this treatment regimen with fresolimumab was not sufficient to reverse the impaired T cell function observed in these cancer patients (139). In addition, various chimeric molecules consisting of soluble TGF-βRII receptors, acting as TGF-β traps, linked to checkpoint blockade antibodies currently are tested in pre-clinical studies and clinical trials. Several preclinical studies have already shown substantially enhanced anti-tumor responses as compared to a monotherapy with anti-CTLA4 or anti-PD-L1 mAb in various mouse solid tumor models (140, 141). For example, administration of a bifunctional fusion protein, termed M7824, with an anti-PD-L1 mAb coupled to the extracellular domain of TGFβ-RII, provided an efficient tumor control in preclinical models of colorectal and breast tumors. M7824 administration resulted in a shift of tumor-infiltrating immune cell populations toward an increase of cytotoxic CD8 T cells and NKG2D+NKp46+NK cells which mediated tumor immunity (141). M7824 has already given to a small cohort of heavily pretreated patients with advanced solid tumors showing early signs of efficacy and a manageable safety profile (142), and is currently undergoing further clinical trials in patients with advanced solid tumors (e.g., NCT02517398, NCT02699515).
An elegant approach to shield adoptively transferred cytotoxic lymphocytes from the suppressive effects of TGF-β in cancer immunotherapy, such as NKG2D silencing, is the transduction of T cells or NK cells with a dominant negative form of TGF-βRII prior to adoptive transfer (143, 144). Transduction of cord blood NK cells with a dominant negative TGF-βRII efficiently blocked TGF-β signal transduction and supported the maintenance of the cell surface expression of activating receptors and NK cell cytotoxicity in the presence of TGF-β (144). Treatment of a small cohort of chemorefractory Hodgkin lymphoma patients with TGF-βRII-transduced autologous EBV-derived tumor antigen-specific CD8 T cells showed complete remission in four out of seven patients (145) suggesting that this type of engineered cytotoxic lymphocytes is safe and efficacious.
Another elegant strategy attempts to convert immunosuppressive signals of soluble TGF-β into stimulatory signals using the chimeric antigen receptor (CAR) concept. A recent report created a chimeric receptor consisting of a TGF-β-binding scFv fused to the transmembrane segment of CD28 and the cytoplasmic signaling domains of both CD28 and CD3ζ (146). T cells ectopically expressing such a CAR were activated by TGF-β-induced CAR dimerization that led an activation of both NFAT and NFκB with a subsequent stimulation of Th1 cytokine responses and an enhanced T cell expansion (146). It will be of great interest to address the in vivo performance of such anti-TGF-β CAR T cells utilizing TGF-β as an activating growth factor in mouse models of solid tumors.
TGF-β broadly and potently suppresses the effector functions of NK cells and cytotoxic T lymphocytes with the TGF-β-mediated impairment of the NKG2D axis representing an important facet of this phenomenon in cancer immunity. Down-regulation of both NKG2D, on cytotoxic lymphocytes, and NKG2DL surface expression, on tumor cells, facilitates the immune escape of tumor cells from induced-self recognition and elimination by cytotoxic lymphocytes. Hence, targeting TGF-β appears to represent a key intervention for an efficient boosting of tumor immunity and should be considered in future cancer treatment modalities. However, the intracellular mechanisms mediating the suppression of the NKG2D axis through TGF-β are not yet fully elucidated and further research is needed to define the underlying molecular and cellular pathways to allow for the development of more tailored and efficacious therapeutic options.
ML and AS wrote the manuscript.
ML and AS were funded by institutional grants of the University of Frankfurt.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. (2006) 24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737
2. Nandan D, Reiner NE. TGF-beta attenuates the class II transactivator and reveals an accessory pathway of IFN-gamma action. J Immunol. (1997) 158:1095–101.
3. Delvig AA, Lee JJ, Chrzanowska-Lightowlers ZM, Robinson JH. TGF-β1 and IFN-γ cross-regulate antigen presentation to CD4 T cells by macrophages. J Leukoc Biol. (2002) 72:163–6. doi: 10.1189/jlb.72.1.163
4. Batlle E, Massague J. Transforming growth factor-beta signaling in immunity and cancer. Immunity. (2019) 50:924–40. doi: 10.1016/j.immuni.2019.03.024
5. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. (2003) 198:1875–86. doi: 10.1084/jem.20030152
6. Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, Neurath MF. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25- T cells through Foxp3 induction and down-regulation of Smad7. J Immunol. (2004) 172:5149–53. doi: 10.4049/jimmunol.172.9.5149
7. Papaspyridonos M, Matei I, Huang Y, do Rosario Andre M, Brazier-Mitouart H, Waite JC, et al. Id1 suppresses anti-tumour immune responses and promotes tumour progression by impairing myeloid cell maturation. Nat Commun. (2015) 6:6840. doi: 10.1038/ncomms7840
8. Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, et al. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci USA. (1993) 90:770–4. doi: 10.1073/pnas.90.2.770
9. Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. (1992) 359:693–9. doi: 10.1038/359693a0
10. Waldhauer I, Steinle A. NK cells and cancer immunosurveillance. Oncogene. (2008) 27:5932–43. doi: 10.1038/onc.2008.267
11. Gorelik L, Flavell RA. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat Med. (2001) 7:1118–22. doi: 10.1038/nm1001-1118
12. Donkor MK, Sarkar A, Savage PA, Franklin RA, Johnson LK, Jungbluth AA, et al. T cell surveillance of oncogene-induced prostate cancer is impeded by T cell-derived TGF-beta1 cytokine. Immunity. (2011) 35:123–34. doi: 10.1016/j.immuni.2011.04.019
13. Ahmadzadeh M, Rosenberg SA. TGF-beta 1 attenuates the acquisition and expression of effector function by tumor antigen-specific human memory CD8 T cells. J Immunol. (2005) 174:5215–23. doi: 10.4049/jimmunol.174.9.5215
14. Ullrich E, Koch J, Cerwenka A, Steinle A. New prospects on the NKG2D/NKG2DL system for oncology. Oncoimmunology. (2013) 2:e26097. doi: 10.4161/onci.26097
15. Lazarova M, Steinle A. The NKG2D axis: an emerging target in cancer immunotherapy. Expert Opin Ther Targets. (2019) 23:281–94. doi: 10.1080/14728222.2019.1580693
16. David CJ, Massague J. Contextual determinants of TGFbeta action in development, immunity and cancer. Nat Rev Mol Cell Biol. (2018) 19:419–35. doi: 10.1038/s41580-018-0007-0
17. Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol. (2013) 31:413–41. doi: 10.1146/annurev-immunol-032712-095951
18. Wensveen FM, Jelencic V, Polic B. NKG2D: a master regulator of immune cell responsiveness. Front Immunol. (2018) 9:441. doi: 10.3389/fimmu.2018.00441
19. Gleizes PE, Munger JS, Nunes I, Harpel JG, Mazzieri R, Noguera I, et al. TGF-beta latency: biological significance and mechanisms of activation. Stem Cells. (1997) 15:190–7. doi: 10.1002/stem.150190
20. Munger JS, Harpel JG, Gleizes PE, Mazzieri R, Nunes I, Rifkin DB. Latent transforming growth factor-beta: structural features and mechanisms of activation. Kidney Int. (1997) 51:1376–82. doi: 10.1038/ki.1997.188
21. Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, et al. Latent TGF-beta structure and activation. Nature. (2011) 474:343–9. doi: 10.1038/nature10152
22. Tran DQ, Andersson J, Wang R, Ramsey H, Unutmaz D, Shevach EM. GARP (LRRC32) is essential for the surface expression of latent TGF-beta on platelets and activated FOXP3+ regulatory T cells. Proc Natl Acad Sci USA. (2009) 106:13445–50. doi: 10.1073/pnas.0901944106
23. Qin Y, Garrison BS, Ma W, Wang R, Jiang A, Li J, et al. A milieu molecule for TGF-beta required for microglia function in the nervous system. Cell. (2018) 174:156–71 e16. doi: 10.1016/j.cell.2018.05.027
24. Viel S, Marcais A, Guimaraes FS, Loftus R, Rabilloud J, Grau M, et al. TGF-beta inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci Signal. (2016) 9:ra19. doi: 10.1126/scisignal.aad1884
25. Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. (2006) 25:455–71. doi: 10.1016/j.immuni.2006.07.011
26. Brabletz T, Pfeuffer I, Schorr E, Siebelt F, Wirth T, Serfling E. Transforming growth factor beta and cyclosporin A inhibit the inducible activity of the interleukin-2 gene in T cells through a noncanonical octamer-binding site. Mol Cell Biol. (1993) 13:1155–62. doi: 10.1128/MCB.13.2.1155
27. Tinoco R, Alcalde V, Yang Y, Sauer K, Zuniga EI. Cell-intrinsic transforming growth factor-beta signaling mediates virus-specific CD8+ T cell deletion and viral persistence in vivo. Immunity. (2009) 31:145–57. doi: 10.1016/j.immuni.2009.06.015
28. Sanjabi S, Mosaheb MM, Flavell RA. Opposing effects of TGF-beta and IL-15 cytokines control the number of short-lived effector CD8+ T cells. Immunity. (2009) 31:131–44. doi: 10.1016/j.immuni.2009.04.020
29. Wolfraim LA, Walz TM, James Z, Fernandez T, Letterio JJ. p21Cip1 and p27Kip1 act in synergy to alter the sensitivity of naive T cells to TGF-beta-mediated G1 arrest through modulation of IL-2 responsiveness. J Immunol. (2004) 173:3093–102. doi: 10.4049/jimmunol.173.5.3093
30. Thomas DA, Massague J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. (2005) 8:369–80. doi: 10.1016/j.ccr.2005.10.012
31. Sad S, Mosmann TR. Single IL-2-secreting precursor CD4 T cell can develop into either Th1 or Th2 cytokine secretion phenotype. J Immunol. (1994) 153:3514–22.
32. Gorelik L, Constant S, Flavell RA. Mechanism of transforming growth factor beta-induced inhibition of T helper type 1 differentiation. J Exp Med. (2002) 195:1499–505. doi: 10.1084/jem.20012076
33. Lin JT, Martin SL, Xia L, Gorham JD. TGF-beta 1 uses distinct mechanisms to inhibit IFN-gamma expression in CD4+ T cells at priming and at recall: differential involvement of Stat4 and T-bet. J Immunol. (2005) 174:5950–8. doi: 10.4049/jimmunol.174.10.5950
34. Kuwahara M, Yamashita M, Shinoda K, Tofukuji S, Onodera A, Shinnakasu R, et al. The transcription factor Sox4 is a downstream target of signaling by the cytokine TGF-beta and suppresses T(H)2 differentiation. Nat Immunol. (2012) 13:778–86. doi: 10.1038/ni.2362
35. Heath VL, Murphy EE, Crain C, Tomlinson MG, O'Garra A. TGF-beta1 down-regulates Th2 development and results in decreased IL-4-induced STAT6 activation and GATA-3 expression. Eur J Immunol. (2000) 30:2639–49. doi: 10.1002/1521-4141(200009)30:9<2639::AID-IMMU2639>3.0.CO;2-7
36. Rook AH, Kehrl JH, Wakefield LM, Roberts AB, Sporn MB, Burlington DB, et al. Effects of transforming growth factor beta on the functions of natural killer cells: depressed cytolytic activity and blunting of interferon responsiveness. J Immunol. (1986) 136:3916–20.
37. Bellone G, Aste-Amezaga M, Trinchieri G, Rodeck U. Regulation of NK cell functions by TGF-beta 1. J Immunol. (1995) 155:1066–73.
38. Pierson BA, Gupta K, Hu WS, Miller JS. Human natural killer cell expansion is regulated by thrombospondin-mediated activation of transforming growth factor-beta 1 and independent accessory cell-derived contact and soluble factors. Blood. (1996) 87:180–9. doi: 10.1182/blood.V87.1.180.180
39. Malygin AM, Meri S, Timonen T. Regulation of natural killer cell activity by transforming growth factor-beta and prostaglandin E2. Scand J Immunol. (1993) 37:71–6. doi: 10.1111/j.1365-3083.1993.tb01667.x
40. Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat Immunol. (2017) 18:1004–15. doi: 10.1038/ni.3800
41. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. (2018) 554:538–43. doi: 10.1038/nature25492
42. Newsted D, Banerjee S, Watt K, Nersesian S, Truesdell P, Blazer LL, et al. Blockade of TGF-beta signaling with novel synthetic antibodies limits immune exclusion and improves chemotherapy response in metastatic ovarian cancer models. Oncoimmunology. (2019) 8:e1539613. doi: 10.1080/2162402X.2018.1539613
43. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. (2018) 554:544–8. doi: 10.1038/nature25501
44. Gorelik L, Flavell RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. (2000) 12:171–81. doi: 10.1016/S1074-7613(00)80170-3
45. Letterio JJ, Geiser AG, Kulkarni AB, Dang H, Kong L, Nakabayashi T, et al. Autoimmunity associated with TGF-beta1-deficiency in mice is dependent on MHC class II antigen expression. J Clin Invest. (1996) 98:2109–19. doi: 10.1172/JCI119017
46. Kobayashi S, Yoshida K, Ward JM, Letterio JJ, Longenecker G, Yaswen L, et al. Beta 2-microglobulin-deficient background ameliorates lethal phenotype of the TGF-beta 1 null mouse. J Immunol. (1999) 163:4013–9.
47. Hannon GJ, Beach D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature. (1994) 371:257–61. doi: 10.1038/371257a0
48. Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, et al. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. (1994) 78:59–66. doi: 10.1016/0092-8674(94)90572-X
49. Reynisdottir I, Polyak K, Iavarone A, Massague J. Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-beta. Genes Dev. (1995) 9:1831–45. doi: 10.1101/gad.9.15.1831
50. Pietenpol JA, Holt JT, Stein RW, Moses HL. Transforming growth factor beta 1 suppression of c-myc gene transcription: role in inhibition of keratinocyte proliferation. Proc Natl Acad Sci USA. (1990) 87:3758–62. doi: 10.1073/pnas.87.10.3758
51. Guasch G, Schober M, Pasolli HA, Conn EB, Polak L, Fuchs E. Loss of TGFbeta signaling destabilizes homeostasis and promotes squamous cell carcinomas in stratified epithelia. Cancer Cell. (2007) 12:313–27. doi: 10.1016/j.ccr.2007.08.020
52. Bardeesy N, Cheng KH, Berger JH, Chu GC, Pahler J, Olson P, et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. (2006) 20:3130–46. doi: 10.1101/gad.1478706
53. Cruz-Guilloty F, Pipkin ME, Djuretic IM, Levanon D, Lotem J, Lichtenheld MG, et al. Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J Exp Med. (2009) 206:51–9. doi: 10.1084/jem.20081242
54. Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. (1999) 285:727–9. doi: 10.1126/science.285.5428.727
55. Wu J, Song Y, Bakker AB, Bauer S, Spies T, Lanier LL, et al. An activating immunoreceptor complex formed by NKG2D and DAP10. Science. (1999) 285:730–2. doi: 10.1126/science.285.5428.730
56. Li PW, Morris DL, Willcox BE, Steinle A, Spies T, Strong RK. Complex structure of the activating immunoreceptor NKG2D and its MHC class I-like ligand MICA. Nat Immunol. (2001) 2:443–51. doi: 10.1038/87757
57. Garrity D, Call ME, Feng JW, Wucherpfennig KW. The activating NKG2D receptor assembles in the membrane with two signaling dimers into a hexameric structure. Proc Natl Acad Sci USA. (2005) 102:7641–6. doi: 10.1073/pnas.0502439102
58. Gumperz JE, Miyake S, Yamamura T, Brenner MB. Functionally distinct subsets of CD1d-restricted natural killer T cells revealed by CD1d tetramer staining. J Exp Med. (2002) 195:625–36. doi: 10.1084/jem.20011786
59. Andre P, Castriconi R, Espeli M, Anfossi N, Juarez T, Hue S, et al. Comparative analysis of human NK cell activation induced by NKG2D and natural cytotoxicity receptors. Eur J Immunol. (2004) 34:961–71. doi: 10.1002/eji.200324705
60. Bryceson YT, March ME, Ljunggren HG, Long EO. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood. (2006) 107:159–66. doi: 10.1182/blood-2005-04-1351
61. Groh V, Bahram S, Bauer S, Herman A, Beauchamp M, Spies T. Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc Natl Acad Sci USA. (1996) 93:12445–50. doi: 10.1073/pnas.93.22.12445
62. Maasho K, Opoku-Anane J, Marusina AI, Coligan JE, Borrego F. NKG2D is a costimulatory receptor for human naive CD8+ T cells. J Immunol. (2005) 174:4480–4. doi: 10.4049/jimmunol.174.8.4480
63. Roberts AI, Lee L, Schwarz E, Groh V, Spies T, Ebert EC, et al. NKG2D receptors induced by IL-15 costimulate CD28-negative effector CTL in the tissue microenvironment. J Immunol. (2001) 167:5527–30. doi: 10.4049/jimmunol.167.10.5527
64. Park YP, Choi SC, Kiesler P, Gil-Krzewska A, Borrego F, Weck J, et al. Complex regulation of human NKG2D-DAP10 cell surface expression: opposing roles of the gammac cytokines and TGF-beta1. Blood. (2011) 118:3019–27. doi: 10.1182/blood-2011-04-346825
65. Steinle A, Li P, Morris DL, Groh V, Lanier LL, Strong RK, et al. Interactions of human NKG2D with its ligands MICA, MICB, and homologs of the mouse RAE-1 protein family. Immunogenetics. (2001) 53:279–87. doi: 10.1007/s002510100325
66. Lanier LL. NKG2D receptor and its ligands in host defense. Cancer Immunol Res. (2015) 3:575–82. doi: 10.1158/2326-6066.CIR-15-0098
67. Cosman D, Mullberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, et al. ULBPs, novel MHC class I-related molecules bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity. (2001) 14:123–33. doi: 10.1016/S1074-7613(01)00095-4
68. Ashiru O, Lopez-Cobo S, Fernandez-Messina L, Pontes-Quero S, Pandolfi R, Reyburn HT, et al. A GPI anchor explains the unique biological features of the common NKG2D-ligand allele MICA*008. Biochem J. (2013) 454:295–302. doi: 10.1042/BJ20130194
69. Carapito R, Bahram S. Genetics, genomics, and evolutionary biology of NKG2D ligands. Immunol Rev. (2015) 267:88–116. doi: 10.1111/imr.12328
70. Wittenbrink M, Spreu J, Steinle A. Differential NKG2D binding to highly related human NKG2D ligands ULBP2 and RAET1G is determined by a single amino acid in the alpha2 domain. Eur J Immunol. (2009) 39:1642–51. doi: 10.1002/eji.200839074
71. Zoller T, Wittenbrink M, Hoffmeister M, Steinle A. Cutting an NKG2D ligand short: cellular processing of the peculiar human NKG2D ligand ULBP4. Front Immunol. (2018) 9:620. doi: 10.3389/fimmu.2018.00620
72. Schrambach S, Ardizzone M, Leymarie V, Sibilia J, Bahram S. In vivo expression pattern of MICA and MICB and its relevance to auto-immunity and cancer. PLoS ONE. (2007) 2:e518. doi: 10.1371/journal.pone.0000518
73. Cerboni C, Zingoni A, Cippitelli M, Piccoli M, Frati L, Santoni A. Antigen-activated human T lymphocytes express cell-surface NKG2D ligands via an ATM/ATR-dependent mechanism and become susceptible to autologous NK- cell lysis. Blood. (2007) 110:606–15. doi: 10.1182/blood-2006-10-052720
74. Molinero LL, Fuertes MB, Rabinovich GA, Fainboim L, Zwirner NW. Activation-induced expression of MICA on T lymphocytes involves engagement of CD3 and CD28. J Leukoc Biol. (2002) 71:791–7. doi: 10.1189/jlb.71.5.791
75. Trembath AP, Markiewicz MA. More than decoration: roles for natural killer group 2 member d ligand expression by immune cells. Front Immunol. (2018) 9:231. doi: 10.3389/fimmu.2018.00231
76. Cerboni C, Neri F, Casartelli N, Zingoni A, Cosman D, Rossi P, et al. Human immunodeficiency virus 1 Nef protein downmodulates the ligands of the activating receptor NKG2D and inhibits natural killer cell-mediated cytotoxicity. J Gen Virol. (2007) 88:242–50. doi: 10.1099/vir.0.82125-0
77. Groh V, Rhinehart R, Secrist H, Bauer S, Grabstein KH, Spies T. Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB. Proc Natl Acad Sci USA. (1999) 96:6879–84. doi: 10.1073/pnas.96.12.6879
78. Groh V, Rhinehart R, Randolph-Habecker J, Topp MS, Riddell SR, Spies T. Costimulation of CD8alphabeta T cells by NKG2D via engagement by MIC induced on virus-infected cells. Nat Immunol. (2001) 2:255–60. doi: 10.1038/85321
79. Welte SA, Sinzger C, Lutz SZ, Singh-Jasuja H, Sampaio KL, Eknigk U, et al. Selective intracellular retention of virally induced NKG2D ligands by the human cytomegalovirus UL16 glycoprotein. Eur J Immunol. (2003) 33:194–203. doi: 10.1002/immu.200390022
80. Pende D, Rivera P, Marcenaro S, Chang CC, Biassoni R, Conte R, et al. Major histocompatibility complex class I-related chain A and UL16-binding protein expression on tumor cell lines of different histotypes: analysis of tumor susceptibility to NKG2D-dependent natural killer cell cytotoxicity. Cancer Res. (2002) 62:6178–86.
81. Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity. (2008) 28:571–80. doi: 10.1016/j.immuni.2008.02.016
82. Wiemann K, Mittrucker HW, Feger U, Welte SA, Yokoyama WM, et al. Systemic NKG2D down-regulation impairs NK and CD8 T cell responses in vivo. J Immunol. (2005) 175:720–9. doi: 10.4049/jimmunol.175.2.720
83. Cerwenka A, Baron JL, Lanier LL. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc Natl Acad Sci USA. (2001) 98:11521–6. doi: 10.1073/pnas.201238598
84. Diefenbach A, Raulet DH. Strategies for target cell recognition by natural killer cells. Immunol Rev. (2001) 181:170–84. doi: 10.1034/j.1600-065X.2001.1810114.x
85. Friese MA, Platten M, Lutz SZ, Naumann U, Aulwurm S, Bischof F, et al. MICA/NKG2D-mediated immunogene therapy of experimental gliomas. Cancer Res. (2003) 63:8996–9006.
86. Salih HR, Rammensee HG, Steinle A. Cutting edge: down-regulation of MICA on human tumors by proteolytic shedding. J Immunol. (2002) 169:4098–102. doi: 10.4049/jimmunol.169.8.4098
87. Waldhauer I, Steinle A. Proteolytic release of soluble UL16-binding protein 2 from tumor cells. Cancer Res. (2006) 66:2520–6. doi: 10.1158/0008-5472.CAN-05-2520
88. Waldhauer I, Goehlsdorf D, Gieseke F, Weinschenk T, Wittenbrink M, Ludwig A, et al. Tumor-associated MICA is shed by ADAM proteases. Cancer Res. (2008) 68:6368–76. doi: 10.1158/0008-5472.CAN-07-6768
89. Ashiru O, Boutet P, Fernandez-Messina L, Aguera-Gonzalez S, Skepper JN, Vales-Gomez M, et al. Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA*008 that is shed by tumor cells in exosomes. Cancer Res. (2010) 70:481–9. doi: 10.1158/0008-5472.CAN-09-1688
90. Fernandez-Messina L, Ashiru O, Boutet P, Aguera-Gonzalez S, Skepper JN, Reyburn HT, et al. Differential mechanisms of shedding of the glycosylphosphatidylinositol (GPI)-anchored NKG2D ligands. J Biol Chem. (2010) 285:8543–51. doi: 10.1074/jbc.M109.045906
91. Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. (2002) 419:734–8. doi: 10.1038/nature01112
92. Salih HR, Holdenrieder S, Steinle A. Soluble NKG2D ligands: prevalence, release, and functional impact. Front Biosci. (2008) 13:3448–56. doi: 10.2741/2939
93. Crane CA, Han SJ, Barry JJ, Ahn BJ, Lanier LL, Parsa AT. TGF-beta downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro Oncol. (2010) 12:7–13. doi: 10.1093/neuonc/nop009
94. Clayton A, Mitchell JP, Court J, Linnane S, Mason MD, Tabi Z. Human tumor-derived exosomes down-modulate NKG2D expression. J Immunol. (2008) 180:7249–58. doi: 10.4049/jimmunol.180.11.7249
95. Jelencic V, Sestan M, Kavazovic I, Lenartic M, Marinovic S, Holmes TD, et al. NK cell receptor NKG2D sets activation threshold for the NCR1 receptor early in NK cell development. Nat Immunol. (2018) 19:1083–92. doi: 10.1038/s41590-018-0209-9
96. Coudert JD, Zimmer J, Tomasello E, Cebecauer M, Colonna M, Vivier E, et al. Altered NKG2D function in NK cells induced by chronic exposure to NKG2D ligand-expressing tumor cells. Blood. (2005) 106:1711–17. doi: 10.1182/blood-2005-03-0918
97. Heinemann A, Zhao F, Pechlivanis S, Eberle J, Steinle A, Diederichs S, et al. Tumor suppressive microRNAs miR-34a/c control cancer cell expression of ULBP2, a stress-induced ligand of the natural killer cell receptor NKG2D. Cancer Res. (2012) 72:460–71. doi: 10.1158/0008-5472.CAN-11-1977
98. Codo P, Weller M, Meister G, Szabo E, Steinle A, Wolter M, et al. MicroRNA-mediated down-regulation of NKG2D ligands contributes to glioma immune escape. Oncotarget. (2014) 5:7651–62. doi: 10.18632/oncotarget.2287
99. Baragano Raneros A, Martin-Palanco V, Fernandez AF, Rodriguez RM, Fraga MF, Lopez-Larrea C, et al. Methylation of NKG2D ligands contributes to immune system evasion in acute myeloid leukemia. Genes Immun. (2015) 16:71–82. doi: 10.1038/gene.2014.58
100. Paczulla AM, Rothfelder K, Raffel S, Konantz M, Steinbacher J, Wang H, et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature. (2019) 572:254–9. doi: 10.1038/s41586-019-1410-1
101. Derynck R, Jarrett JA, Chen EY, Eaton DH, Bell JR, Assoian RK, et al. Human transforming growth factor-beta complementary DNA sequence and expression in normal and transformed cells. Nature. (1985) 316:701–5. doi: 10.1038/316701a0
102. Kehrl JH, Wakefield LM, Roberts AB, Jakowlew S, Alvarez-Mon M, Derynck R, et al. Production of transforming growth factor beta by human T lymphocytes and its potential role in the regulation of T cell growth. J Exp Med. (1986) 163:1037–50. doi: 10.1084/jem.163.5.1037
103. Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, et al. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci USA. (2003) 100:4120–5. doi: 10.1073/pnas.0730640100
104. Friese MA, Wischhusen J, Wick W, Weiler M, Eisele G, Steinle A, et al. RNA interference targeting transforming growth factor-beta enhances NKG2D-mediated antiglioma immune response, inhibits glioma cell migration and invasiveness, and abrogates tumorigenicity in vivo. Cancer Res. (2004) 64:7596–603. doi: 10.1158/0008-5472.CAN-04-1627
105. Lee JC, Lee KM, Kim DW, Heo DS. Elevated TGF-beta1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J Immunol. (2004) 172:7335–40. doi: 10.4049/jimmunol.172.12.7335
106. Zocchi MR, Catellani S, Canevali P, Tavella S, Garuti A, Villaggio B, et al. High ERp5/ADAM10 expression in lymph node microenvironment and impaired NKG2D ligands recognition in Hodgkin lymphomas. Blood. (2012) 119:1479–89. doi: 10.1182/blood-2011-07-370841
107. Han B, Mao FY, Zhao YL, Lv YP, Teng YS, Duan M, et al. Altered NKp30, NKp46, NKG2D, and DNAM-1 expression on circulating NK cells is associated with tumor progression in human gastric cancer. J Immunol Res. (2018) 2018:6248590. doi: 10.1155/2018/6248590
108. Dasgupta S, Bhattacharya-Chatterjee M, O'Malley BW Jr, Chatterjee SK. Inhibition of NK cell activity through TGF-beta 1 by down-regulation of NKG2D in a murine model of head and neck cancer. J Immunol. (2005) 175:5541–50. doi: 10.4049/jimmunol.175.8.5541
109. Schilling B, Halstead ES, Schuler P, Harasymczuk M, Egan JE, Whiteside TL. IRX-2, a novel immunotherapeutic, enhances and protects NK-cell functions in cancer patients. Cancer Immunol Immunother. (2012) 61:1395–405. doi: 10.1007/s00262-011-1197-x
110. Sene D, Levasseur F, Abel M, Lambert M, Camous X, Hernandez C, et al. Hepatitis C virus (HCV) evades NKG2D-dependent NK cell responses through NS5A-mediated imbalance of inflammatory cytokines. PLoS Pathog. (2010) 6:e1001184. doi: 10.1371/journal.ppat.1001184
111. Sun C, Fu B, Gao Y, Liao X, Sun R, Tian Z, et al. TGF-beta1 down-regulation of NKG2D/DAP10 and 2B4/SAP expression on human NK cells contributes to HBV persistence. PLoS Pathog. (2012) 8:e1002594. doi: 10.1371/journal.ppat.1002594
112. Eisele G, Wischhusen J, Mittelbronn M, Meyermann R, Waldhauer I, Steinle A, et al. TGF-beta and metalloproteinases differentially suppress NKG2D ligand surface expression on malignant glioma cells. Brain. (2006) 129:2416–25. doi: 10.1093/brain/awl205
113. Lopez-Soto A, Huergo-Zapico L, Galvan JA, Rodrigo L, de Herreros AG, Astudillo A, et al. Epithelial-mesenchymal transition induces an antitumor immune response mediated by NKG2D receptor. J Immunol. (2013) 190:4408–19. doi: 10.4049/jimmunol.1202950
114. Ghiringhelli F, Menard C, Terme M, Flament C, Taieb J, Chaput N, et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J Exp Med. (2005) 202:1075–85. doi: 10.1084/jem.20051511
115. Smyth MJ, Teng MW, Swann J, Kyparissoudis K, Godfrey DI, Hayakawa Y. CD4+CD25+ T regulatory cells suppress NK cell-mediated immunotherapy of cancer. J Immunol. (2006) 176:1582–7. doi: 10.4049/jimmunol.176.3.1582
116. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol. (2009) 182:240–9. doi: 10.4049/jimmunol.182.1.240
117. Szczepanski MJ, Szajnik M, Welsh A, Whiteside TL, Boyiadzis M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta1. Haematologica. (2011) 96:1302–9. doi: 10.3324/haematol.2010.039743
118. Nieswandt B, Hafner M, Echtenacher B, Mannel DN. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res. (1999) 59:1295–300.
119. Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW, et al. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood. (2005) 105:178–85. doi: 10.1182/blood-2004-06-2272
120. Kopp HG, Placke T, Salih HR. Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res. (2009) 69:7775–83. doi: 10.1158/0008-5472.CAN-09-2123
121. Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. (2011) 20:576–90. doi: 10.1016/j.ccr.2011.09.009
122. Sadallah S, Schmied L, Eken C, Charoudeh HN, Amicarella F, Schifferli JA. Platelet-derived ectosomes reduce NK cell function. J Immunol. (2016) 197:1663–71. doi: 10.4049/jimmunol.1502658
123. Espinoza JL, Takami A, Yoshioka K, Nakata K, Sato T, Kasahara Y, et al. Human microRNA-1245 down-regulates the NKG2D receptor in natural killer cells and impairs NKG2D-mediated functions. Haematologica. (2012) 97:1295–303. doi: 10.3324/haematol.2011.058529
124. Song L, Dai T, Xie Y, Wang C, Lin C, Wu Z, et al. Up-regulation of miR-1245 by c-myc targets BRCA2 and impairs DNA repair. J Mol Cell Biol. (2012) 4:108–17. doi: 10.1093/jmcb/mjr046
125. Lee JC, Lee KM, Ahn YO, Suh B, Heo DS. A possible mechanism of impaired NK cytotoxicity in cancer patients: down-regulation of DAP10 by TGF-beta1. Tumori. (2011) 97:350–7. doi: 10.1177/030089161109700316
126. Stern-Ginossar N, Gur C, Biton M, Horwitz E, Elboim M, Stanietsky N, et al. Human microRNAs regulate stress-induced immune responses mediated by the receptor NKG2D. Nat Immunol. (2008) 9:1065–73. doi: 10.1038/ni.1642
127. Tsukerman P, Stern-Ginossar N, Gur C, Glasner A, Nachmani D, Bauman Y, et al. MiR-10b downregulates the stress-induced cell surface molecule MICB, a critical ligand for cancer cell recognition by natural killer cells. Cancer Res. (2012) 72:5463–72. doi: 10.1158/0008-5472.CAN-11-2671
128. Trinh TL, Kandell WM, Donatelli SS, Tu N, Tejera MM, Gilvary DL, et al. Immune evasion by TGFbeta-induced miR-183 repression of MICA/B expression in human lung tumor cells. Oncoimmunology. (2019) 8:e1557372. doi: 10.1080/2162402X.2018.1557372
129. Donatelli SS, Zhou JM, Gilvary DL, Eksioglu EA, Chen X, Cress WD, et al. TGF-beta-inducible microRNA-183 silences tumor-associated natural killer cells. Proc Natl Acad Sci USA. (2014) 111:4203–8. doi: 10.1073/pnas.1319269111
130. Tran HC, Wan Z, Sheard MA, Sun J, Jackson JR, Malvar J, et al. TGFbetaR1 blockade with galunisertib (LY2157299) enhances anti-neuroblastoma activity of the anti-GD2 antibody dinutuximab (ch14.18) with natural killer cells. Clin Cancer Res. (2017) 23:804–13. doi: 10.1158/1078-0432.CCR-16-1743
131. Otegbeye F, Ojo E, Moreton S, Mackowski N, Lee DA, de Lima M, et al. Inhibiting TGF-beta signaling preserves the function of highly activated, in vitro expanded natural killer cells in AML and colon cancer models. PLoS ONE. (2018) 13:e0191358. doi: 10.1371/journal.pone.0191358
132. Melisi D, Garcia-Carbonero R, Macarulla T, Pezet D, Deplanque G, Fuchs M, et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br J Cancer. (2018) 119:1208–14. doi: 10.1038/s41416-018-0246-z
133. Faivre SJ, Santoro A, Kelley RK, Merle P, Gane E, Douillard JY, et al. A phase 2 study of a novel transforming growth factor-beta (TGF-β 1) receptor I kinase inhibitor, LY2157299 monohydrate (LY), in patients with advanced hepatocellular carcinoma (HCC). J Clin Oncol. (2014) 32:3_suppl. doi: 10.1200/jco.2014.32.3_suppl.lba173
134. Kovacs RJ, Maldonado G, Azaro A, Fernandez MS, Romero FL, Sepulveda-Sanchez JM, et al. Cardiac safety of TGF-beta receptor I kinase inhibitor LY2157299 monohydrate in cancer patients in a first-in-human dose study. Cardiovasc Toxicol. (2015) 15:309–23. doi: 10.1007/s12012-014-9297-4
135. Holmgaard RB, Schaer DA, Li Y, Castaneda SP, Murphy MY, Xu X, et al. Targeting the TGFbeta pathway with galunisertib, a TGFbetaRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J Immunother Cancer. (2018) 6:47. doi: 10.1186/s40425-018-0356-4
136. Song H, Hur DY, Kim KE, Park H, Kim T, Kim CW, et al. IL-2/IL-18 prevent the down-modulation of NKG2D by TGF-beta in NK cells via the c-Jun N-terminal kinase (JNK) pathway. Cell Immunol. (2006) 242:39–45. doi: 10.1016/j.cellimm.2006.09.002
137. Fujii R, Jochems C, Tritsch SR, Wong HC, Schlom J, Hodge JW. An IL-15 superagonist/IL-15Ralpha fusion complex protects and rescues NK cell-cytotoxic function from TGF-beta1-mediated immunosuppression. Cancer Immunol Immunother. (2018) 67:675–89. doi: 10.1007/s00262-018-2121-4
138. Formenti SC, Lee P, Adams S, Goldberg JD, Li X, Xie MW, et al. Focal irradiation and systemic TGFbeta blockade in metastatic breast cancer. Clin Cancer Res. (2018) 24:2493–504. doi: 10.1158/1078-0432.CCR-17-3322
139. Formenti SC, Hawtin RE, Dixit N, Evensen E, Lee P, Goldberg JD, et al. Baseline T cell dysfunction by single cell network profiling in metastatic breast cancer patients. J Immunother Cancer. (2019) 7:177. doi: 10.1186/s40425-019-0633-x
140. Ravi R, Noonan KA, Pham V, Bedi R, Zhavoronkov A, Ozerov IV, et al. Bifunctional immune checkpoint-targeted antibody-ligand traps that simultaneously disable TGFbeta enhance the efficacy of cancer immunotherapy. Nat Commun. (2018) 9:741. doi: 10.1038/s41467-017-02696-6
141. Lan Y, Zhang D, Xu C, Hance KW, Marelli B, Qi J, et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-beta. Sci Transl Med. (2018) 10:eaan5488. doi: 10.1126/scitranslmed.aan5488
142. Strauss J, Heery CR, Schlom J, Madan RA, Cao L, Kang Z, et al. Phase I trial of M7824 (MSB0011359C), a bifunctional fusion protein targeting PD-L1 and TGFbeta, in advanced solid tumors. Clin Cancer Res. (2018) 24:1287–95. doi: 10.1158/1078-0432.CCR-17-2653
143. Bollard CM, Rossig C, Calonge MJ, Huls MH, Wagner HJ, Massague J, et al. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood. (2002) 99:3179–87. doi: 10.1182/blood.V99.9.3179
144. Yvon ES, Burga R, Powell A, Cruz CR, Fernandes R, Barese C, et al. Cord blood natural killer cells expressing a dominant negative TGF-beta receptor: Implications for adoptive immunotherapy for glioblastoma. Cytotherapy. (2017) 19:408–18. doi: 10.1016/j.jcyt.2016.12.005
145. Bollard CM, Tripic T, Cruz CR, Dotti G, Gottschalk S, Torrano V, et al. Tumor-specific T-cells engineered to overcome tumor immune evasion induce clinical responses in patients with relapsed hodgkin lymphoma. J Clin Oncol. (2018) 36:1128–39. doi: 10.1200/JCO.2017.74.3179
Keywords: NK cell, NKG2D, TGF-β1, tumor immune evasion, immunotherapy
Citation: Lazarova M and Steinle A (2019) Impairment of NKG2D-Mediated Tumor Immunity by TGF-β. Front. Immunol. 10:2689. doi: 10.3389/fimmu.2019.02689
Received: 29 July 2019; Accepted: 31 October 2019;
Published: 15 November 2019.
Edited by:
Roberta Castriconi, University of Genoa, ItalyReviewed by:
Jörg Wischhusen, Julius Maximilian University of Würzburg, GermanyCopyright © 2019 Lazarova and Steinle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander Steinle, YWxleGFuZGVyLnN0ZWlubGVAa2d1LmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.