
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 14 November 2019
Sec. Cancer Immunity and Immunotherapy
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02683
This article is part of the Research Topic The Natural Killer Cell Interactome in the Tumor Microenvironment: Basic Concepts and Clinical Application View all 11 articles
 Michael C. Burger1,2,3,4
Michael C. Burger1,2,3,4 Congcong Zhang3,4,5
Congcong Zhang3,4,5 Patrick N. Harter2,3,4,6
Patrick N. Harter2,3,4,6 Annette Romanski7
Annette Romanski7 Florian Strassheimer1,2Christian Senft2,3,8
Florian Strassheimer1,2Christian Senft2,3,8 Torsten Tonn4,9,10,11
Torsten Tonn4,9,10,11 Joachim P. Steinbach1,2,3,4Winfried S. Wels2,3,4,5*
Joachim P. Steinbach1,2,3,4Winfried S. Wels2,3,4,5*Glioblastoma (GB) is the most common and aggressive primary brain tumor in adults and currently incurable. Despite multimodal treatment regimens, median survival in unselected patient cohorts is <1 year, and recurrence remains almost inevitable. Escape from immune surveillance is thought to contribute to the development and progression of GB. While GB tumors are frequently infiltrated by natural killer (NK) cells, these are actively suppressed by the GB cells and the GB tumor microenvironment. Nevertheless, ex vivo activation with cytokines can restore cytolytic activity of NK cells against GB, indicating that NK cells have potential for adoptive immunotherapy of GB if potent cytotoxicity can be maintained in vivo. NK cells contribute to cancer immune surveillance not only by their direct natural cytotoxicity which is triggered rapidly upon stimulation through germline-encoded cell surface receptors, but also by modulating T-cell mediated antitumor immune responses through maintaining the quality of dendritic cells and enhancing the presentation of tumor antigens. Furthermore, similar to T cells, specific recognition and elimination of cancer cells by NK cells can be markedly enhanced through expression of chimeric antigen receptors (CARs), which provides an opportunity to generate NK-cell therapeutics of defined specificity for cancer immunotherapy. Here, we discuss effects of the GB tumor microenvironment on NK-cell functionality, summarize early treatment attempts with ex vivo activated NK cells, and describe relevant CAR target antigens validated with CAR-T cells. We then outline preclinical approaches that employ CAR-NK cells for GB immunotherapy, and give an overview on the ongoing clinical development of ErbB2 (HER2)-specific CAR-NK cells currently applied in a phase I clinical trial in glioblastoma patients.
Glioblastoma (GB) is the most frequent malignant primary brain tumor in adults, without any curative treatment options available at present. Patients diagnosed with GB have a dismal prognosis, and typically succumb to the disease within 3 months if untreated. Present standard of care for most patients includes surgical resection followed by radio- and chemotherapy. Despite this aggressive treatment, median survival of glioblastoma patients is only about 15 months, and recurrence remains almost inevitable (1, 2). Still <5% of patients diagnosed with GB survive more than 5 years (3). In contrast to other cancers such as adenocarcinomas of the lung or melanoma, primary brain tumors like GB and low grade gliomas (LGG) are known as rather immunologically “cold” tumors, typically with low numbers of tumor-infiltrating lymphocytes (TILs) (4), and the mere amount of TILs is not associated with patient survival (5). Nevertheless, the composition of the immunologic tumor microenvironment undergoes changes upon radiotherapy, chemotherapy, or even after anti-angiogenic therapy (6). Hence, intensifying efforts to overcome the general resistance of glioblastoma to immunotherapy appears highly warranted (7–9).
Recent clinical trials with chimeric antigen receptor (CAR)-engineered T cells demonstrated the feasibility and safety of this approach for the treatment of recurrent glioblastoma, with signs of clinical activity and transient responses observed in some of the patients (10–12). Efforts are underway to further refine these strategies, which includes evaluation of NK cells as alternative CAR-engineered effectors. The critical role of NK cells in cancer immune surveillance is increasingly being recognized (13, 14). NK cells do not only contribute to antitumor immunity by directly eliminating malignant cells, but also by regulating tumor-specific adaptive immune responses through crosstalk with dendritic cells (DCs) (15). NK cells thereby regulate DC maturation, and so determine the effectiveness of subsequent DC-mediated T-cell activation (16). Conversely, DCs enhance the direct antitumor activity of NK cells (17), which is also relevant for glioblastoma as shown in a recent phase II clinical trial with a dendritic cell vaccine (18). However, in cancer patients NK cells are often functionally compromised due to the immunosuppressive activity of the tumor. NK cells do not carry a T-cell receptor restricted to a particular peptide epitope presented by major histocompatibility complex (MHC) molecules, but recognize stress ligands on cancer cells via germ-line encoded activating receptors, which are counter-balanced by inhibitory receptors that are triggered by self-MHC class I. Hence, for adoptive cancer immunotherapy HLA-mismatched NK cells from healthy donors are preferred, which do not recognize tumor cells as “self,” thereby bypassing inhibitory signals. Since NK cells do not carry a high risk of inducing graft-vs.-host-disease (GvHD), this approach is generally considered to be safe (19–21). The better understanding of NK-cell biology, together with the development of strategies to enhance NK-cell activity by blocking inhibitory receptor pathways or redirect NK cells to tumors using bispecific antibodies or genetic modification with CARs, have paved the way for many therapeutic approaches that are now actively pursued in a clinical setting. These extend from the treatment of different types of leukemia to various solid tumors (22–24). Such refined approaches also hold enormous potential to improve immunotherapy of glioblastoma (25), assigning to NK cells a dual role as targeted killers and modulators of innate and adaptive immunity in the GB tumor microenvironment.
Studies that evaluated infiltration of malignant gliomas by NK cells have reported different findings, ranging from the presence of insignificant NK cell numbers to large NK cell infiltrates in up to 89% of glioblastomas (26, 27). Another study described less infiltration of high-risk gliomas with NK cells and M1-like macrophages than lower grade gliomas (28). Therefore, differences in the subtype and stage of the disease, treatment history and permeability of the blood-brain barrier (BBB) appear to affect the likeliness of infiltration by NK cells. The presence of NK cells and their repertoire of activating killer cell immunoglobulin-like receptors (KIRs) can have an impact on disease progression. This was shown by Dominguez-Valentin et al. who analyzed 108 glioblastoma patients and 454 healthy individuals for HLA-A, -B, -C, NK-cell KIRs, and CMV-specific antibodies, and correlated the results with clinical parameters. The KIR allele KIR2DS4*00101 was thereby identified as an independent prognostic parameter of prolonged survival (29). Notably, all patients carrying KIR2DS4*00101 were CMV seropositive, and showed an increase in NK-cell subpopulations that expressed the cytotoxicity receptors CD16, NKG2D, and CD94/NKG2C. Healthy controls had a reduced risk to develop glioblastoma if they harbored two KIR2DS4*00101 alleles. Likewise, Gras Navarro et al. identified a subpopulation in donor-derived NK cells expressing KIR2DS2, which had a functional advantage in killing GB cells (30). Compared to KIR2DS2-negative NK cells, KIR2DS2-positive NK cells showed higher cytotoxicity, at least in part also mediated through expression of NKG2D ligands by the tumor cells. NKG2D-mediated control of glioblastoma by NK cells has also been demonstrated in experimental GB models (31).
Escape from immune surveillance is thought to contribute to the development and progression of GB (32, 33). GB tumors which are infiltrated by NK cells actively suppress NK-cell function via expression of factors such as transforming growth factor (TGF)-β, which is a main contributor to the immunosuppressive GB microenvironment (34, 35). TGF-β impairs NK cells by downregulating activating NK receptors such as NKG2D and NKp30 (36), or by repressing the mTOR pathway (37). This may be circumvented by making NK cells resistant to TGF-β, as recently achieved by expression of a dominant-negative TGF-β receptor in cord blood NK cells (38). Nevertheless, most GB cells also express high levels of MHC class I molecules, which inhibit autologous NK cells via inhibitory KIRs. Thus, blockade of such KIRs may favor a less tumor-promoting microenvironment and enhanced NK-cell mediated killing (25). GB cells can also affect NK-cell activity through expression of inhibitory molecules like regeneration and tolerance factor (RTF) and lectin-like transcript (LLT)-1 (33, 39), or indirectly through myeloid cells (40). Resident microglial cells in the brain are induced by glioma-derived TGF-β to acquire an immunosuppressive phenotype (41). Although the concept of M1 and M2 polarization of microglia in GB has been challenged as both subtypes are present in GB and prognostic relevance remains under debate (42), it is generally accepted that glioma-associated macrophages can reinforce an immunosuppressive tumor microenvironment (43, 44). Nevertheless, while actively suppressed in glioblastoma tumors, even patient-derived autologous NK cells can recover their cytolytic activity upon ex vivo culture with IL-2 or IL-15, in particular directed to GB stem-like cells (45, 46).
Combinations of donor-derived NK cells with an antibody recognizing a glioblastoma-restricted surface antigen or a histone deacetylase inhibitor (HDACi) that induced upregulation of NKG2D ligand expression by GB cells have been shown in preclinical models to overcome immunosuppressive effects of brain tumors (47, 48). Also pre-treatment with the proteasome inhibitor bortezomib sensitized GB cells toward NKG2D- or TRAIL-mediated NK-cell lysis and enhanced survival in animal models (49). Available clinical data, however, are so far still restricted to earlier approaches based on ex vivo activated autologous immune cells. Ishikawa et al. performed a phase I clinical trial with autologous NK cells combined with systemic low-dose interferon (IFN)-β in nine patients with recurrent malignant glioma, including three patients with glioblastoma (50). NK cells were expanded from peripheral blood mononuclear cells (PBMCs) using irradiated feeder cells and IL-2. Repeated doses of NK cells were either only applied intravenously, or both, intravenously and directly into the tumor cavity through an Ommaya reservoir. The NK cell therapy proved to be safe and partially effective, with two patients experiencing a partial response (PR), two patients a mixed response (MR) and three patients stable disease (SD) during different courses of treatment (50).
The majority of early studies evaluating adoptive cell therapy, however, were performed with autologous lymphokine-activated killer (LAK) cells in combination with IL-2 injected into the resection cavity of recurrent or progressive malignant gliomas, with the cells usually applied through a reservoir. LAK cells are a mixture of T and NK cells derived by ex vivo culture of peripheral blood lymphocytes in IL-2-containing medium (51). Thereby the main lytic activity of LAK cells is mediated by CD3−CD56+ NK cells, while the contribution of CD3+CD56− T cells is rather limited (52). The studies with LAK cells in glioma patients reported disease stabilization and partial or even complete responses in some of the patients, without encountering dose-limiting toxicities (53–64). Despite activation with IL-2, LAK cells can still be inhibited by immunosuppressive molecules secreted or presented by GB cells (65), which may explain why, despite the observed clinical activity in some cases, responses after therapy with autologous LAK cells were not durable. These reports are nevertheless encouraging and important for ongoing and future studies with enhanced NK-cell products such as CAR-NK cells, since they document feasibility and overall safety and tolerability of repeated intracavitary or intralesional injection of large numbers of lymphocytes together with IL-2, in some cases reaching up to 1010 cells per dose (55).
Chimeric antigen receptors were initially developed as a means for T cells to bypass MHC restriction of the T-cell receptor (TCR) and instead acquire TCR-independent, predetermined specificity for a defined cell surface antigen expressed by the target cell of interest (66–68). Since the first description of the basic CAR design encompassing a single chain fragment variable (scFv) antibody for target recognition linked to CD3ζ or FcεRIγ chains for signaling (first-generation CARs) (66), this approach has continuously been refined to enhance effector cell activity, and improve engraftment and persistence in the host upon adoptive transfer. Accordingly, receptors currently employed for CAR-T cell products approved for the treatment of malignancies of B-cell origin or undergoing clinical testing for various hematologic or solid tumor indications include in addition to CD3ζ one or more costimulatory protein domains, typically derived from CD28 and CD137 (4-1BB) (referred to as second- or third-generation CARs) (69, 70).
Also NK cells can be genetically engineered to express chimeric antigen receptors, thereby acquiring in addition to their natural cytotoxicity built-in ADCC-like activity, similar to FcγRIIIa (CD16) activation by target-specific IgG molecules (Figure 1). This was first demonstrated for a CAR-like CD4-CD3ζ fusion receptor in human NK3.3 cells (72), and a scFv-CD3ζ-based first-generation CAR in NK-92 cells (73). Subsequently, peripheral blood NK cells from healthy donors, cord blood derived NK cells, and NK cells generated from induced pluripotent stem cells (iPSCs) have successfully been used for the generation of CAR-NK cells in preclinical studies (74–76). While conventional T-cell CARs with CD3ζ and CD28 and/or CD137 domains are functional in NK cells (74–80), several groups have reported improved activity if one or more signaling domains derived from CD244 (2B4), NKG2D, DAP10 or DAP12 were included in the receptors (75, 81, 82). It is presently unknown whether a particular CAR design would thereby be preferable for all CAR-NK cells irrespective of the nature of the cell binding domain, the target antigen, and the source and differentiation state of the effector cells. So far no CAR-NK cell product has received marketing authorization, but several early phase clinical trials in different cancer indications are ongoing, mainly based on genetically engineered NK-92 and cord blood NK cells (15, 23, 83).
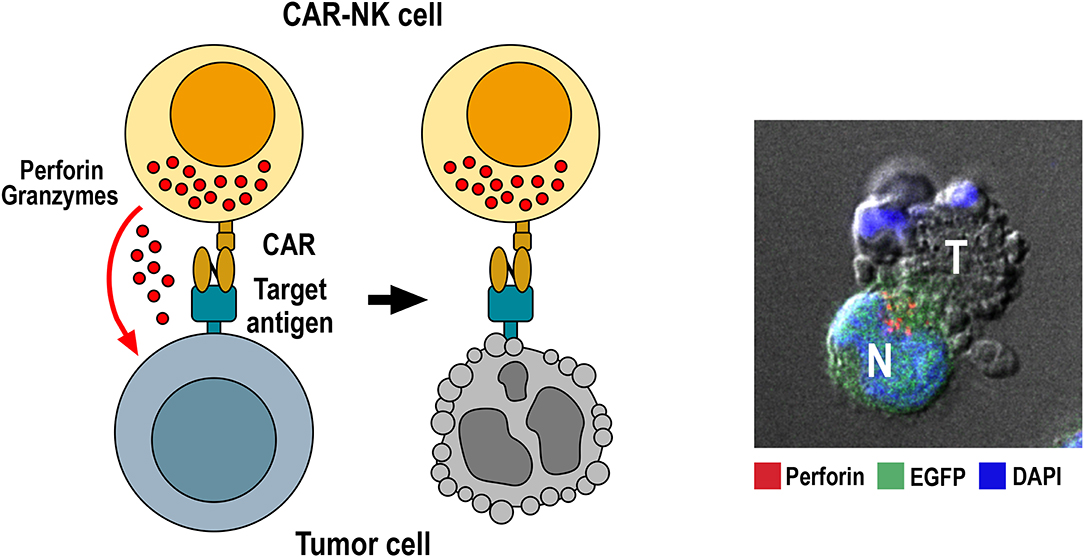
Figure 1. Killing of tumor cells by CAR-NK cells. Specific binding of the chimeric antigen receptor to its target antigen on the tumor-cell surface triggers CAR activation. This results in re-orientation of cytotoxic granules toward the immunological synapse formed between NK and target cell, followed by release of perforin and granzymes from cytotoxic granules into the synaptic cleft. Perforin and granzymes taken up by the target cell then trigger apoptotic cell death indicated by membrane blebbing and disintegration of the nucleus. The confocal microscopy image to the right shows conjugate formation between human GB cells co-expressing EGFR and EGFRvIII with NK-92/225.28.z CAR-NK cells that recognize both target antigens (71). Tumor (T) and EGFP-positive CAR-NK cells (N, green) were co-incubated for 1 h, fixed, permeabilized and stained for perforin (red) to identify cytotoxic granules. Cell nuclei were labeled with DAPI (blue).
Most studies on CAR-engineered effector cells in glioblastoma have so far focused on autologous CAR-T cells. These approaches have been reviewed extensively in recent years (84–88). Nevertheless, due to their relevance for NK-cell based therapies, some of their key findings are summarized below, concentrating in particular on tumor-associated antigens that have already been targeted in clinical trials.
Interleukin-13 receptor α 2 (IL-13Rα2) has first been described as a therapeutic target for GB two decades ago (89). It has been found overexpressed in >50% of high-grade gliomas, but is undetectable in normal brain tissue. IL-13Rα2 expression alone can induce invasiveness of GB cells without affecting cell growth, but together with the epidermal growth factor receptor variant EGFRvIII promotes GB cell proliferation (90). Initial CAR-T cell approaches targeting IL-13Rα2 employed IL-13 muteins for cell recognition in first-generation CARs (termed zetakines) to reduce binding to IL-13Rα1, which is not restricted to GB but broadly expressed in other tissues (91, 92). Later work demonstrated improved activity of such CAR-T cells in vitro and in orthotopic GB xenograft models if costimulatory protein domains were included in the CARs (93–96). To achieve exclusive binding to the target antigen, also scFv-based IL-13Rα2-specific CARs were developed, which eliminated remaining cross-reactivity to IL-13Rα1 (97–100). Safety and feasibility of local treatment of glioblastoma patients by repeated application of up to 108 CD8+ T cells expressing a first-generation IL-13 zetakine into the resection cavity through a catheter and reservoir was demonstrated in a pilot trial at the City of Hope National Medical Center with three subjects, with signs of transient clinical activity observed in two patients. For one patient, tumor material before and after therapy was available, which indicated reduced IL-13Rα2 expression in the tumor after treatment (101). More recently, the same group reported on a patient with recurrent multifocal glioblastoma from an ongoing phase I clinical trial with autologous CAR-T cells carrying an improved second-generation CAR with an IL-13 mutein and CD137 and CD3ζ signaling domains (NCT02208362, clinicaltrials.gov) (10). The patient was treated with repeated infusions of up to 107 CAR-T cells per dose into the resection cavity and the ventricular system. This resulted in the regression of intracranial and spinal tumors, with the clinical response continuing for 7.5 months after initiation of treatment. No dose-limiting toxicities were observed.
Epidermal growth factor receptor (EGFR) is a well-established therapeutic target in glioblastoma (102). EGFR gene amplification and EGFR protein overexpression is present in 40 to 60% of GB tumors (103), while the receptor is not or only minimally expressed in normal brain tissue (104). Glioblastoma cells with EGFR gene amplification often co-express a constitutively active EGFR mutant form (EGFRvIII) (105, 106), which drives tumorigenicity and mediates radio- and chemoresistance (107, 108). EGFR gene amplification and expression of EGFRvIII are both correlated with poor prognosis and shorter survival of GB patients (109). EGFRvIII harbors an in-frame deletion of exons 2–7 of the wildtype EGFR gene. This generates a tumor-specific neo-epitope at the N-terminus of the receptor which can be targeted by specific immunotherapy. Consequently, several groups investigated EGFRvIII-targeted CAR-T cells that employed second- or third-generation CARs with scFv antibody domains specifically recognizing the EGFRvIII neo-epitope. These CAR-T cells demonstrated selective cytotoxicity against EGFRvIII-positive GB cells in vitro and potent activity against subcutaneous or orthotopic human GB xenografts in mice upon intravenous or intratumoral injection (110–115). In a phase I clinical trial at the University of Pennsylvania with 10 patients suffering from recurrent glioblastoma, intravenous application of a single dose of up to 5 × 108 autologous EGFRvIII-specific CAR-T cells proved to be safe without evidence of off-tumor toxicity or cytokine release syndrome (12). One patient showed stable disease for over 18 months of follow-up. Transient expansion of CAR-T cells in the peripheral blood was found in all patients, and presence of CAR-T cells in GB tissue in five of seven patients where post-treatment tissue was available. While the level of EGFR gene amplification did not change, EGFRvIII expression had declined or was undetectable in the majority of such patients, which may have been due to the selective pressure exerted by the CAR-T cells (12). In a dose-escalation trial at the National Cancer Institute, 18 patients with recurrent EGFRvIII-positive glioblastoma were treated by intravenous infusion of up to ≥1010 autologous T cells carrying an EGFRvIII-specific third-generation CAR together with IL-2 after conditioning by lymphodepleting chemotherapy (116). One patient developed fatal and another patient serious respiratory symptoms shortly after cell infusion at the highest dose levels, which was attributed to congestion of pulmonary vasculature by activated T cells. No objective responses were observed, but one patient was still free from disease progression 6 months after therapy (116).
Like EGFR, ErbB2 (HER2) is a member of the family of EGFR-related receptor tyrosine kinases. Activating mutations of ErbB2 appear less common than in the case of EGFR (117), but overexpression of the receptor is frequently found in breast carcinomas and many other epithelial cancers. Due to its ability to form signaling-competent heterodimers with all other members of the EGFR family, overexpression of ErbB2 strongly contributes to malignant transformation (118). While absent in the adult central nervous system (119), ErbB2 protein expression was found in up to 80% of GB tumors and was correlated with impaired survival (120–124). In a recent study with 56 primary GB tumors, immunohistochemical analysis revealed high ErbB2 expression in 21.5%, more moderate expression in another 19.6%, and no or low expression in 58.9% of the samples. Thereby, in the majority of cases ErbB2 expression in relapsed tumors was comparable to or higher than that of primary tumors from the same patients (125). In a preclinical study Ahmed et al. evaluated patient-derived CAR-T cells transduced with a retroviral vector encoding an ErbB2-specific CAR with CD28 and CD3ζ signaling domains. These CAR-T cells exhibited remarkable antitumor activity against ErbB2-positive autologous GB cells in vitro including CD133-positive stem-like cells derived from primary GB tumors, and in orthotopic GB xenograft models in mice (126). In a subsequent phase I dose-escalation trial at Baylor College of Medicine, the same group treated 17 patients with progressive ErbB2-positive glioblastoma by systemic infusion of one or more doses of up to 108/m2 autologous ErbB2-specific CAR-T cells. The infusions were well-tolerated, without encountering dose-limiting toxicities. Of 16 evaluable patients, one had a partial response lasting 9 months, seven had stable disease, and eight progressed after CAR-T cell infusion. At the time of publication, three patients with stable disease were alive without evidence of progression 24 and 29 months after treatment (11).
Other antigens that were proposed as targets for CAR-T cell approaches in glioblastoma are EphA2, CSPG4, CD133, and CD70. The receptor tyrosine kinase erythropoietin-producing hepatocellular carcinoma A2 (EphA2) is overexpressed in GB and contributes to its malignancy (127, 128). CAR-T cells carrying EphA2-specific second- or third-generation CARs displayed potent activity against glioma-initiating cells growing as neurospheres and orthotopic GB xenografts in murine models (129, 130). Chondroitin sulfate proteoglycan 4 (CSPG4) is highly expressed in different solid tumors including glioblastoma, where CSPG4 expression is associated with more aggressive disease. Of 46 tumor samples tested in a recent study, 67% were positive for CSPG4 (131). CAR-T cells with CSPG4-specific second- or third-generation CARs, among other cancer entities, also lysed CSPG4-positive glioblastoma cells, including GB stem-like cells (131–133). The CAR-T cells also effectively controlled the growth of orthotopic GB xenografts upon intracranial injection in mice, without selection of antigen-loss tumor cell variants. Interestingly, TNF-α released by tumor-associated microglia further induced CSPG4 expression in the GB cells (131). CD133 (prominin-1) is a stem cell marker, also expressed by cancer stem cells of many tumor entities including glioblastoma (134). Despite concerns of on-target/off-tumor toxicity against hematopoietic stem cells, the feasibility of systemic application of CD133-specific CAR-T cells with manageable toxicity was recently shown in a phase I clinical trial in patients with hepatocellular carcinoma and different epithelial cancers (135). In preclinical models, CAR-T cells expressing CD133-specific third-generation CARs with CD28, CD137, and CD3ζ signaling domains displayed specific cytotoxicity against patient-derived GB stem cells in vitro and in orthotopic GB mouse models (136, 137). CD70 is a member of the tumor necrosis factor (TNF) family and the ligand for CD27. While CD70 expression is typically restricted to highly activated T- and B-lymphocytes and a subset of mature DCs, certain hematologic malignancies and solid tumors, including gliomas, can constitutively overexpress CD70 (138, 139). Thereby, CD70 expression in GB cells directly facilitates immune evasion by selectively inducing CD8+ T-cell death (140). T cells expressing CD70-specific first- and second-generation CARs which utilized the CD70-binding domain of CD27 for target-cell recognition specifically lysed CD70-positive primary GB cells in vitro, and inhibited growth or induced complete tumor regression in xenograft and syngeneic GB models in mice (141). To reduce the risk of immune escape due to inhomogeneous target antigen expression, also CAR-T cell products were developed which simultaneously target two or three GB antigens. Strategies validated in GB xenograft models include the combination of individual CAR-T cells targeting ErbB2 or IL-13Rα2, or co-expression of ErbB2- and IL-13Rα2-specific CARs, or receptors targeting ErbB2, IL-13Rα2 and EphA2 in the same T cells (142, 143). Alternatively, an ErbB2-specific scFv antibody and an IL-13 mutein were directly combined in a single bispecific tandem CAR (TanCAR) (144).
Since autologous CAR-T cells are made from the cancer patients' own peripheral blood lymphocytes, yield, transduction efficiency, T-cell subtype distribution, and activation state can vary, affecting overall product composition and quality. In contrast, NK cells can safely be administered to an HLA-mismatched recipient. Hence, donor-derived peripheral blood and cord blood NK cells provide readily available resources not only for donor lymphocyte infusions of unmodified effector cells, but also for the generation of genetically engineered NK cells that may be provided as cost-effective off-the-shelf products (21, 76, 145). Furthermore, NK cells carry natural cytotoxicity receptors (NCRs) and the C-type lectin-like receptor NKG2D which are triggered rapidly by engagement of ligands selectively expressed by stressed and transformed cells (146). This natural cytotoxicity of NK cells can complement CAR-mediated cell killing, and may allow CAR-NK cells to also attack tumors with heterogeneous expression of the CAR target antigen (15). Nevertheless, because of the limited life span and expansion potential of primary NK cells, ex vivo culture for the generation of the required cell numbers can be demanding (147). Consequently, also human NK cell lines that continuously expand in the presence of IL-2 are being evaluated as an alternative source for the generation of well-defined clinical grade NK-cell products (148–150).
So far most of such efforts have focused on the NK-92 cell line, which was initially isolated from a non-Hodgkin lymphoma patient (151). NK-92 cells display features of activated primary NK cells and express many activating NK-cell receptors such as NKp30, NKp46, and NKG2D as well as high levels of granzymes A and B, but lack inhibitory NK-cell receptors except for KIR2DL4, Ig-like transcript 2 (ILT-2) and NKG2A/CD94 (151–155). General safety of repeated infusions of irradiated NK-92 cells at doses up to 1010 cells/m2 has been established in phase I clinical trials in patients with advanced cancers, with durable responses observed in some of the treated subjects (156–159). NK-92 cells were also instrumental to demonstrate that it is feasible to generate tumor-targeted CAR-NK cells for cancer immunotherapy. Since the initial description of this concept (73), the number of preclinical studies evaluating CAR-engineered NK-92 cells has steadily increased, demonstrating markedly enhanced antitumor activity of the cells if targeted to surface molecules expressed by different hematologic malignancies and solid tumors (15, 83), including tumor-associated antigens such as EGFR, EGFRvIII and ErbB2 that are relevant for the development of immunotherapies for glioblastoma (71, 125, 160) (outlined in the following section). In a recent phase I clinical trial with CD33-specific CAR NK-92 cells for the treatment of acute myeloid leukemia (AML), no dose-limiting toxicities were encountered upon repeated intravenous infusions of up to 5 × 109 irradiated cells per dose (161), suggesting that the safety profile of these CAR NK-92 cells is similar to that of unmodified NK-92. Several other early phase clinical trials with CAR-engineered NK-92 cells are presently ongoing in Europe, China and the US (15, 83), including the CAR2BRAIN phase I clinical study which investigates a clonal ErbB2-specific CAR NK-92 product in glioblastoma patients and is described in more detail in a subsequent section (125, 162).
The therapeutic utility of CAR-engineered NK cells for the treatment of glioblastoma has so far mainly been investigated in preclinical studies with effector cells targeting EGFRvIII, EGFR or ErbB2 (Table 1). Müller et al. generated a CAR based on a scFv fragment of EGFRvIII-specific antibody MR1-1 (166), which was fused to an intracellular DNAX-activating protein 12 (DAP12) domain for signaling (82). DAP12 contains an immunoreceptor tyrosine-based activation motif (ITAM), and like CD3ζ which harbors three ITAMs, it transmits signals from activating NK-cell receptors (167). The CAR was expressed in the established human NK cell line YTS, with the resulting effector cells showing enhanced lysis of GB cells transfected with an EGFRvIII construct or endogenously expressing the target antigen. Intravenous injection into mice carrying subcutaneous EGFRvIII-positive GB xenografts resulted in inhibition of tumor growth and extended survival, which was further enhanced by co-expressing the chemokine receptor CXCR4 in the CAR-NK cells for improved tumor homing (82). Enhanced cytotoxicity against EGFRvIII-expressing GB cells was also found in in vitro cell culture assays with established human KHYG-1 NK cells expressing an EGFRvIII-specific CAR (163). In a study with CAR-engineered NK-92 cells, a second-generation chimeric antigen receptor was used that employed EGFRvIII-specific antibody MR1-1 as a cell binding domain and CD28 and CD3ζ domains for signaling (71). Upon contact formation, the respective CAR-NK cells re-oriented their cytotoxic granules toward EGFRvIII-positive but not EGFRvIII-negative GB cells, resulting in rapid and selective tumor cell killing. High and specific cytotoxicity of these CAR-NK cells was also demonstrated with targets growing in a tissue-like environment using patient-derived primary colon cancer organoids transduced with an EGFRvIII-encoding vector as a model (168). Repeated stereotactic injection of the EGFRvIII-specific NK-92 cells into orthotopic EGFRvIII-positive GB xenografts in immunodeficient NSG mice delayed tumor growth and markedly improved symptom-free survival (71). However, treatment of mixed tumors which similar to the clinical situation consisted of EGFR and EGFRvIII double-positive, and EGFR-expressing but EGFRvIII-negative GB cells, resulted in a less pronounced survival benefit and selective outgrowth of EGFRvIII-negative tumors still expressing the wildtype receptor. This is reminiscent of EGFRvIII-negative tumor recurrence observed in some GB patients after treatment with EGFRvIII-specific CAR-T cells (12).
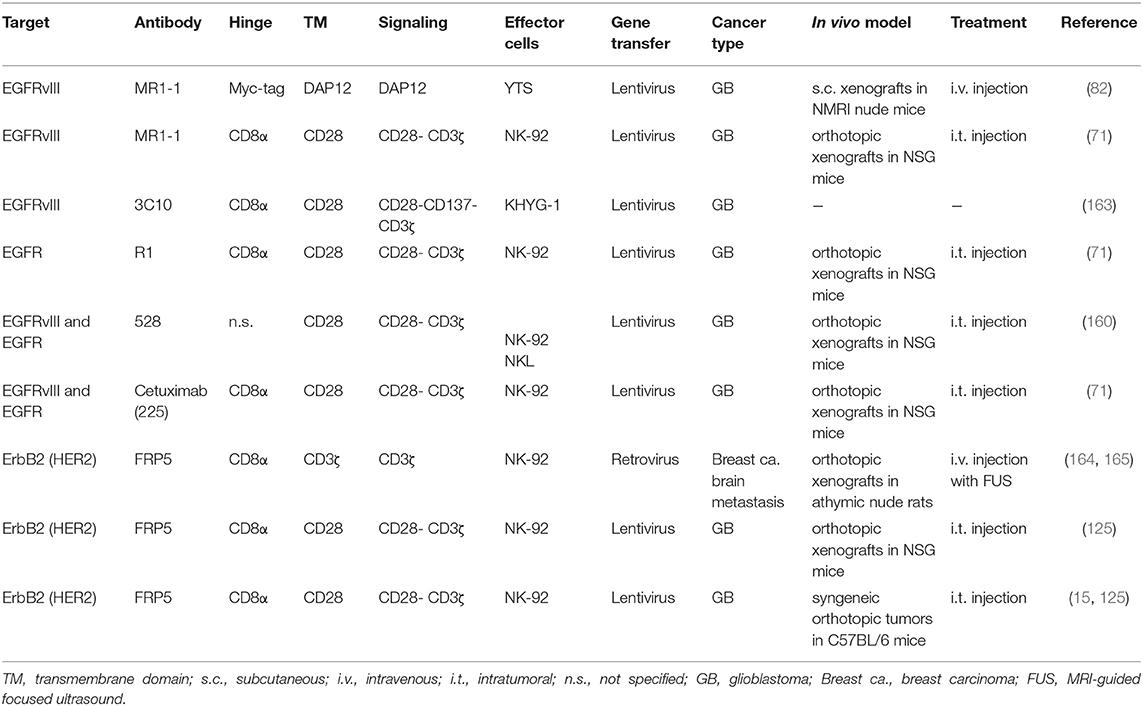
Table 1. Preclinical studies with CAR-NK cells in brain cancer models.
To circumvent the problem of heterogeneous EGFRvIII expression in GB tumors, two groups generated CAR-NK cells which simultaneously target wildtype EGFR and the receptor variant. Han et al. employed as a cell binding domain of a second-generation CAR with CD28 and CD3ζ signaling domains a scFv fragment of EGFR-specific antibody 528, which recognizes an epitope conserved in EGFRvIII (160). Established human NK-92 and NKL cells expressing this CAR displayed enhanced cytotoxicity and IFN-γ secretion when cocultured with GB cell lines or patient-derived GB stem cells expressing EGFR or EGFRvIII. Treatment of NSG mice carrying orthotopic GB xenografts expressing wildtype EGFR or EGFRvIII by repeated intracranial injection of CAR-engineered NK-92 cells inhibited tumor growth more strongly than treatment with NK-92 control cells and improved overall survival of the animals. In an independent study, Genßler et al. used a scFv fragment derived from the clinically applied antibody cetuximab as a cell binding domain for a dual-specific second-generation CAR with CD28 and CD3ζ signaling domains (71). Like antibody 528, chimeric antibody cetuximab and its murine parent 225 recognize an epitope common to EGFR and EGFRvIII (169, 170). The MR1-1-based CAR outlined above which is specific for the EGFRvIII neo-epitope and a similar CAR based on antibody R1 that recognizes an N-terminal epitope only present in wildtype EGFR were included in this study for comparison. In vitro analysis of NK-92 cells carrying these CARs revealed high and specific cytotoxicity of EGFR-targeted effectors against established and primary human GB cells, which was dependent on EGFR expression and CAR signaling. Cytotoxicity of EGFRvIII-targeted NK-92 was restricted to EGFRvIII-positive GB cells, while dual-specific NK cells were active against tumor cells positive for EGFR and/or EGFRvIII (Figure 1). Importantly, in NSG mice carrying orthotopic GB xenografts xenografts either expressing EGFR, EGFRvIII or both receptors, repeated local treatment with the dual-specific NK cells was superior to treatment with either one or a mixture of the corresponding monospecific CAR-NK cells. This led to a marked extension of survival without inducing the rapid immune escape of antigen-loss variants that was observed upon therapy with monospecific CAR-NK cells (71). In later work, Jiang et al. followed a similar strategy to co-target EGFR and EGFRvIII with CAR-T cells recognizing a shared epitope of the receptors (171). Co-targeting of EGFR and EGFRvIII has also been achieved by expressing a secreted EGFR-specific T-cell engager in EGFRvIII CAR-T cells (172). Nevertheless, due to the more limited life span and expansion potential of NK cells when compared to T cells, utilizing CAR-NK cells may be a safer approach for targeting a surface antigen such as wildtype EGFR which is highly expressed also by vital normal tissues.
In addition to EGFRvIII and EGFR, the receptor tyrosine kinase ErbB2 has been targeted with CAR-engineered NK-92 cells. The respective NK-92/5.28.z cells represent a molecularly and functionally well-defined single cell clone isolated upon transduction of NK-92 cells under GMP-compliant conditions with a lentiviral vector encoding a second-generation CAR very similar to the one used by Ahmed et al. for CAR-T cells (11), based on ErbB2-specific antibody FRP5 and a composite CD28-CD3ζ signaling domain (78, 173). In preclinical studies, these cells displayed high and selective cytotoxicity against ErbB2-positive target cells of different solid tumor origins including established GB cell lines, and primary GB stem cell cultures (78, 125). Importantly, specific cytotoxicity of NK-92/5.28.z against GB cells was retained under hypoxic conditions and in the presence of high concentrations of immunosuppressive TGF-β, indicating that the cells remain functional in an environment similar to that of a GB tumor. Systemic application in NSG mice resulted in selective enrichment of NK-92/5.28.z cells in orthotopic breast carcinoma xenografts and a reduction of pulmonary tumor nodules in an experimental renal cell carcinoma metastasis model (78). In orthotopic GB xenograft models, repeated stereotactic injection of the cells into the tumor area effectively inhibited tumor progression, and led to a marked extension of survival (125). Furthermore, in an immunocompetent GB mouse model, NK-92/5.28.z cells displayed strong immunomodulatory activity and enhanced endogenous antitumor immunity upon intratumoral injection, resulting in tumor rejection in the majority of mice carrying syngeneic intracranial GL261/ErbB2 glioblastomas. In contrast, unmodified parental NK-92 cells were unable to inhibit tumor progression (15, 125). Without further treatment, those mice that were cured from their initial tumors also rejected a rechallenge with the GB cells injected into the other brain hemisphere more than 120 days after initial therapy, indicating induction of a long-lasting protective immune response induced in the animals by treatment with NK-92/5.28.z. IgG antibodies in the sera of these mice were broadly directed against the GB cells and not limited to the CAR target antigen. Protective immunity induced by initial treatment with NK-92/5.28.z cells was also dependent on T-cell memory, since depletion of CD4+ and CD8+ T cells before rechallenge prevented tumor rejection in some of the animals (15). Because of these promising preclinical data, the ErbB2-specific NK-92/5.28.z CAR-NK cells were chosen for further development toward clinical application in glioblastoma patients (described in the following sections).
Also NK-92 cells engineered to express a CAR specific for the tumor-associated antigen and GB target CD133 have been investigated, but preclinical data are so far limited to in vitro assays with CD133-positive established and primary ovarian carcinoma cells, which were eliminated by the CAR-NK cells when combined with cisplatin (174). In addition, the disialoganglioside GD2 is considered a potential target for glioblastoma therapy (175, 176). While GD2-specific CAR-NK cells have so far not been tested in GB models, both, primary donor-derived NK cells and NK-92 cells expressing GD2-specific first- or second-generation CARs demonstrated selective antitumor activity in preclinical in vitro and in vivo models of neuroblastoma, melanoma, breast carcinoma and Ewing sarcoma (81, 177–179).
As outlined above, the use of an NK cell line such as NK-92 offers certain advantages over primary NK cells which can overcome hurdles like variable transduction efficiency of viral vectors and limited expansion potential, that can complicate clinical translation of donor-derived NK cells (15). On the downside, due to their origin from a non-Hodgkin lymphoma, there may be a risk of secondary lymphoma formation in treated patients, which is currently addressed by including γ-irradiation of NK-92 before transfusion into a recipient (156–159, 161). Preclinical titration experiments demonstrated that an irradiation dose of 10 Gy is sufficient to inhibit proliferation of unmodified NK-92 cells as well as CAR-engineered NK-92/5.28.z, without affecting their in vitro cytotoxicity for at least 48 h or reducing in vivo antitumor activity in murine models (78, 125, 148, 180). This has of course implications for the manufacturing strategy. Autologous CAR-T cell products can be frozen after transduction and will expand in vivo upon injection of relatively small amounts of the cells. In contrast, NK cells in general and especially NK cell lines need to be expanded to a therapeutic dose before infusion. Irradiation will prevent any in vivo expansion, which mandates repeated therapeutic dosages for continued control of cancer growth. Hence, it would be ideal to produce cryopreserved cell doses in advance that can be thawed before transfusion. However, NK cells represent large granular lymphocytes which are more sensitive to cryopreservation than T cells, usually displaying impaired viability and long lag phases after thawing (181). While efforts are made to improve cryopreservation of NK cells, and some NK-92 cell products under commercial development are provided in cryopreserved form (154, 182, 183), we decided to use fresh NK-92/5.28.z cells in exponential growth phase for application in glioblastoma patients.
Adapting the strategy developed for unmodified NK-92 cells (148, 157), first a qualified Master Cell Bank (MCB) of CAR-engineered NK-92/5.28.z cells was generated, providing a reliable source for subsequent production of patient doses for clinical trials (180). To bridge the lag phase of cell proliferation after thawing of vials from the MCB and to have a therapeutic dose of NK-92/5.28.z cells readily available for a patient within <1 week, a process was established which relies on a maintenance culture of the CAR NK-92 cells. From this maintenance culture therapeutic dosages can be expanded in batch culture within 5 to 6 days upon seeding the cells at an initial density of 5 × 104 cells/mL in 2 L of medium in gas permeable cell culture bags. After approximately 3.5 doublings (doubling time of NK-92/5.28.z: 32–36 h), the culture yields a total cell number of 1 × 109, which is sufficient to supply several cell dosages comprising 1 × 108 cells, which is the highest dose level currently planned for GB patients (180). Quality control performed on the maintenance culture involves monthly in-process controls for sterility, mycoplasma, endotoxin, absence of replication-competent lentivirus (p24 antigen ELISA), viability, identity, and phenotype (CD56+, CD16−, CAR+), as well as potency of the NK-92/5.28z cells (lysis of ErbB2-positive target cells) (180). Release criteria of the irradiated finished cell product comprise the same tests, but with the results of sterility testing available only after injection due to the required time period. Special care was taken to choose an excipient used as injection solution for intracranial injection of CAR-NK cells that would not elicit adverse reactions due to calcium or electrolyte imbalances. Accordingly, the NK-92/5.28.z cells are resuspended in human serum derived from a single blood donor. Using this approach, a shelf life of up to 22 h at 4 ± 2°C was established, which allows preparation and release of the investigational medicinal product 1 day before scheduled injection into a patient. The irradiated cell product is transferred to the clinical site in a bag comprising cells at a density of 5 × 107 cells/mL. The neurosurgeon then transfers the required amount of this cell suspension into a syringe at the site of operation to dilute the CAR-NK cells depending on the dose level and to perform the intracranial injections. Our experience shows that this approach is feasible and provides a ready-to-use dosage of CAR-NK cells to the application site without the need for local processing (thawing, washing, cell counting), which reduces the risk for secondary contaminations. Nevertheless, the limited shelf life demands an orchestrated and well-organized process involving all partners.
The phase I clinical trial CAR2BRAIN (NCT03383978, clinicaltrials.gov) is an investigator-initiated, prospective, open-label study carried out at the Center for Neurology and Neurosurgery at the University Hospital Frankfurt, Frankfurt am Main, Germany in patients with recurrent or refractory ErbB2-positive glioblastoma with a scheduled relapse surgery. Initially conducted as a single-center trial, it is now planned to include several other participating study centers in Germany. Main objective of the trial is to evaluate safety and tolerability of ErbB2-specific NK-92/5.28.z CAR-NK cells (78, 180) and to determine the maximum tolerated dose (MTD) or maximum feasible dose (MFD). In the dose-escalation cohort of the CAR2BRAIN study, NK-92/5.28.z cells are injected into the wall of the resection cavity during relapse surgery (Figure 2). The CAR-NK cells are subjected to γ-irradiation at a dose of 10 Gy prior to injection as part of the manufacturing process (see above). The dose levels explored are 1 × 107, 3 × 107, and 1 × 108 NK-92/5.28.z cells used as a single dose application in a total injection volume of 2 mL, distributed by the neurosurgeon with multiple injections. Patients are enrolled in cohorts of three to six patients per dose level in a 3 + 3 dose escalation scheme. Up to now we concluded the first two dose levels, with evaluation of the third and last dose level ongoing. So far, no dose-limiting toxicities were encountered.
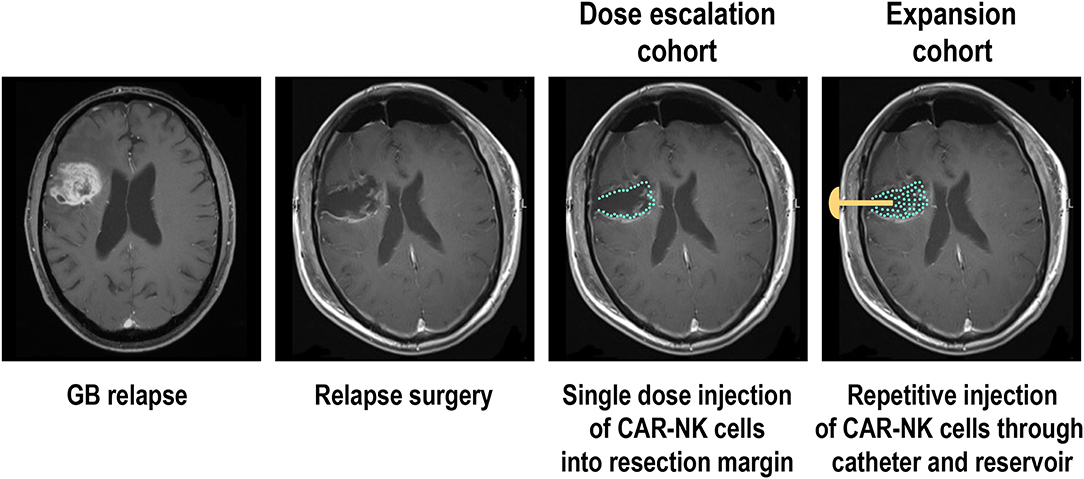
Figure 2. Phase I clinical trial with ErbB2-specific CAR NK-92 cells in glioblastoma patients. Patients with recurrent ErbB2-positive glioblastoma in the dose escalation cohort of the CAR2BRAIN phase I clinical trial (NCT03383978, clinicaltrials.gov) receive a single treatment with irradiated ErbB2-specific NK-92/5.28.z cells injected into the resection margin during relapse surgery. In the subsequent expansion cohort, patients are scheduled to receive in addition up to 12 weekly treatments applied into the resection cavity through an implanted catheter and reservoir. MRI images are for illustration only and do not show an actual patient treated in the study.
Direct injection of the CAR-NK cells into the wall of the resection cavity was chosen to allow for high effector cell density in the tumor and the infiltration zone. Unlike ErbB2-specific autologous CAR-T cells that were applied systemically in GB patients in a recent phase I dose-escalation trial (11), irradiated NK-92/5.28.z CAR-NK cells cannot engraft permanently or expand in vivo, considerably limiting the number of viable cells that may reach the brain tissue from the circulation. Systemically applied NK-92/5.28.z cells homed to orthotopically implanted breast carcinoma xenografts in mice and were effective against experimental metastasis irrespective of γ-irradiation (78). However, intravenously injected CAR NK-92 cells had no therapeutic effect against intracranial tumors in preclinical studies, suggesting that at least in rodent models they cannot cross the blood-brain barrier in sufficient numbers without applying measures such as focused ultrasound (164, 165). This may be different in GB patients where the BBB can be severely disturbed in large parts of the tumor (184), and apparently allows systemically applied CAR-T cells access to the cancer cells (11, 12). Nevertheless, encouraging data on the successful treatment of a glioblastoma patient from a phase I clinical trial by local injection of IL-13Rα2-specific CAR-T cells suggest that intracranial delivery of effector cells may indeed be the preferred route of administration (10).
While in the case of B-cell malignancies a single dose of CAR-T cells is often sufficient to achieve durable responses (69, 70), repeated application can be more beneficial for the therapy of solid tumors, and will likely be mandatory to induce a lasting response if treatment consists of irradiated effector cells such as NK-92 (15, 150). Accordingly, after concluding dose escalation in the CAR2BRAIN trial, it is planned to include additional patients in an expansion cohort, scheduled for multiple injections of the CAR-NK cells at the recommended safe dose established during dose escalation (Figure 2). In these patients, intraoperative injection of NK-92/5.28.z cells is followed by implantation of a Rickham reservoir, with the catheter of the reservoir placed in the resection cavity. Beginning 1 week after surgery and intra-operative treatment with CAR-NK cells, patients will receive up to 12 additional injections of NK-92/5.28.z into the resection cavity, applied through the reservoir and catheter. With these treatments spaced 1 week apart, resident GB cells can be exposed to NK-92/5.28.z for a prolonged time period while no potentially problematic dose accumulation of the irradiated effector cells is expected. In addition to the analysis of peripheral blood scheduled at different time points—if possible—also cerebrospinal fluid (CSF) will be collected through the catheter before each treatment for analysis of soluble factors and cells contained therein, expected to provide insights on possible effects of the CAR-NK cells on endogenous immune cells over the course of therapy. Patients will also be tested for potential immune responses to the allogeneic NK-92 cells and the CAR construct. In different phase I clinical trials with parental NK-92 cells, an anti-HLA antibody response to NK-92 was reported in one out of two, one out of seven, and six out of twelve patients evaluated (156, 157, 159). It is presently unknown to what extent such responses can affect the activity of NK-92 cells during repeated dosing.
NK-cell based adoptive immunotherapy of cancer is a rapidly expanding field. Recent advances such as the identification and effective blocking of distinct NK-cell immune checkpoints, and tumor-specific redirection of the exquisite lytic capacity of these innate lymphocytes using bispecific killer-cell engagers or chimeric antigen receptors can be expected to benefit patients suffering from many hematologic malignancies and solid tumors. Thereby glioblastoma, due to its location in the brain, its aggressiveness and highly immunosuppressive tumor microenvironment represents a particular challenge. Following similar approaches with CAR-T cells, CAR-NK cells have now entered clinical development for the treatment of malignant glioma. In addition to ongoing work with CAR-engineered NK-92 cells, other allogeneic off-the-shelf therapeutics based on donor-derived peripheral blood or cord blood NK cells, or NK cells differentiated from iPSCs may be tested for their effectiveness against brain tumors in the near future. However, overcoming inhibitory mechanisms in GB, and addressing immune escape due to inhomogeneous expression of CAR target antigens remain significant obstacles. While currently limited to a few cases, analysis of glioblastoma tissues from patients before and after treatment with CAR-T cells suggests that high effector cell activity can result in rapid selection of antigen-loss variants (12, 101). A similar problem may be encountered with CAR-NK cells. Nevertheless, NK cells naturally exhibit broad cytotoxicity triggered by interaction of their activating receptors with stress ligands expressed by tumor cells, which is retained by CAR-NK cells and may help to eliminate glioblastoma cells with low or absent expression of the CAR target antigen. Stem-like GB cells in particular appear sensitive to this natural cytotoxicity of allogeneic NK cells (185). Also preclinical data with NK-92 cells point in this direction, where ErbB2-specific CAR NK-92 cells lysed ErbB2-positive stem-like GB cells growing as neurospheres quite rapidly, but after prolonged exposure, the target cells were also eliminated by parental NK-92 cells lacking the CAR (125). The shorter life span and limited in vivo expansion of NK cells, while providing increased safety when compared to CAR-T cells, will likely restrict their long-term effectiveness and require repeated treatment. Ectopic expression of pro-inflammatory cytokines such as IL-15 together with the CAR may not only increase persistence of the CAR-NK cells themselves (76, 77), but also support T-cell infiltration and activation (186). Induction of endogenous immunological memory by CAR-NK cell treatment has been identified as the basis for cures in immunocompetent animal models of glioblastoma (15, 125). In GB patients, avoidance of prophylactic dexamethasone administration (187), and rational combination of adoptive NK-cell transfer with radiotherapy, immune checkpoint inhibitors, inhibitors of angiogenesis or modulators of the myeloid cell compartment may all be effective strategies to fully realize the cytotoxic potential of CAR-NK cells against glioblastoma (6, 49, 188, 189), and further enhance their ability to modulate innate and adaptive immune cells in the tumor microenvironment.
All authors made substantial contributions to the conception, design and writing of this review article, critically evaluated the cited literature, revised the manuscript, and approved the final version of this study. All authors agreed to be accountable for the content of this work.
Research leading to own results summarized herein was funded in part by grants from German Federal Ministry of Education and Research (BMBF) (Ci3; FKZ 131A009), LOEWE Center for Cell and Gene Therapy Frankfurt (CGT) (HMWK II L 5-518/17.004 2016), LOEWE Center Frankfurt Cancer Institute (FCI) (HMWK III L 5-519/03/03.001-0015), German Cancer Consortium (DKTK), Alfons und Gertrud Kassel-Stiftung, and institutional funds of the Georg-Speyer-Haus and the Institute for Neurooncology. The Georg-Speyer-Haus is funded jointly by the German Federal Ministry of Health and the Ministry of Higher Education, Research and the Arts of the State of Hessen (HMWK).
CZ, TT, and WW are named as inventors on patents and patent applications in the field of cancer immunotherapy owned by their respective institutions.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Lisa Sevenich and Pranav Oberoi for critical reading of the manuscript and helpful suggestions.
1. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. (2005) 352:987–96. doi: 10.1056/NEJMoa043330
2. Bähr O, Herrlinger U, Weller M, Steinbach JP. Very late relapses in glioblastoma long-term survivors. J Neurol. (2009) 256:1756–8. doi: 10.1007/s00415-009-5167-6
3. Thakkar JP, Dolecek TA, Horbinski C, Ostrom QT, Lightner DD, Barnholtz-Sloan JS, et al. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol Biomarkers Prev. (2014) 23:1985–96. doi: 10.1158/1055-9965.EPI-14-0275
4. Chakravarthy A, Furness A, Joshi K, Ghorani E, Ford K, Ward MJ, et al. Pan-cancer deconvolution of tumour composition using DNA methylation. Nat Commun. (2018) 9:3220. doi: 10.1038/s41467-018-05570-1
5. Rutledge WC, Kong J, Gao J, Gutman DA, Cooper LA, Appin C, et al. Tumor-infiltrating lymphocytes in glioblastoma are associated with specific genomic alterations and related to transcriptional class. Clin Cancer Res. (2013) 19:4951–60. doi: 10.1158/1078-0432.CCR-13-0551
6. Scholz A, Harter PN, Cremer S, Yalcin BH, Gurnik S, Yamaji M, et al. Endothelial cell-derived angiopoietin-2 is a therapeutic target in treatment-naive and bevacizumab-resistant glioblastoma. EMBO Mol Med. (2016) 8:39–57. doi: 10.15252/emmm.201505505
7. Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol. (2018) 15:422–42. doi: 10.1038/s41571-018-0003-5
8. Jackson CM, Choi J, Lim M. Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat Immunol. (2019) 20:1100–9. doi: 10.1038/s41590-019-0433-y
9. Tomaszewski W, Sanchez-Perez L, Gajewski TF, Sampson JH. Brain tumor microenvironment and host state: Implications for immunotherapy. Clin Cancer Res. (2019) 25:4202–10. doi: 10.1158/1078-0432.CCR-18-1627
10. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
11. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: A phase 1 dose-escalation trial. JAMA Oncol. (2017) 3:1094–101. doi: 10.1001/jamaoncol.2017.0184
12. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. (2017) 9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984
13. Lopez-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of metastasis by NK cells. Cancer Cell. (2017) 32:135–54. doi: 10.1016/j.ccell.2017.06.009
14. Souza-Fonseca-Guimaraes F, Cursons J, Huntington ND. The emergence of natural killer cells as a major target in cancer immunotherapy. Trends Immunol. (2019) 40:142–58. doi: 10.1016/j.it.2018.12.003
15. Zhang C, Oberoi P, Oelsner S, Waldmann A, Lindner A, Tonn T, et al. Chimeric antigen receptor-engineered NK-92 cells: An off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front Immunol. (2017) 8:533. doi: 10.3389/fimmu.2017.00533
16. Walzer T, Dalod M, Robbins SH, Zitvogel L, Vivier E. Natural-killer cells and dendritic cells: “l'union fait la force.” Blood. (2005) 106:2252–8. doi: 10.1182/blood-2005-03-1154
17. Fernandez NC, Lozier A, Flament C, Ricciardi-Castagnoli P, Bellet D, Suter M, et al. Dendritic cells directly trigger NK cell functions: cross-talk relevant in innate anti-tumor immune responses in vivo. Nat Med. (1999) 5:405–11. doi: 10.1038/7403
18. Pellegatta S, Eoli M, Cuccarini V, Anghileri E, Pollo B, Pessina S, et al. Survival gain in glioblastoma patients treated with dendritic cell immunotherapy is associated with increased NK but not CD8(+) T cell activation in the presence of adjuvant temozolomide. Oncoimmunology. (2018) 7:e1412901. doi: 10.1080/2162402X.2017.1412901
19. Davis ZB, Felices M, Verneris MR, Miller JS. Natural killer cell adoptive transfer therapy: Exploiting the first line of defense against cancer. Cancer J. (2015) 21:486–91. doi: 10.1097/PPO.0000000000000156
20. Rezvani K, Rouce RH. The application of natural killer cell immunotherapy for the treatment of cancer. Front Immunol. (2015) 6:578. doi: 10.3389/fimmu.2015.00578
21. Veluchamy JP, Kok N, van der Vliet HJ, Verheul HMW, de Gruijl TD, Spanholtz J. The rise of allogeneic natural killer cells as a platform for cancer immunotherapy: Recent innovations and future developments. Front Immunol. (2017) 8:631. doi: 10.3389/fimmu.2017.00631
22. Chiossone L, Dumas PY, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol. (2018) 18:671–88. doi: 10.1038/s41577-018-0061-z
23. Mehta RS, Rezvani K. Chimeric antigen receptor expressing natural killer cells for the immunotherapy of cancer. Front Immunol. (2018) 9:283. doi: 10.3389/fimmu.2018.00283
24. Miller JS, Lanier LL. Natural killer cells in cancer immunotherapy. Annu Rev Cancer Biol. (2019) 3:77–103. doi: 10.1146/annurev-cancerbio-030518-055653
25. Golan I, Rodriguez de la Fuente L, Costoya JA. NK cell-based glioblastoma immunotherapy. Cancers. (2018) 10:E522. doi: 10.3390/cancers10120522
26. Stevens A, Kloter I, Roggendorf W. Inflammatory infiltrates and natural killer cell presence in human brain tumors. Cancer. (1988) 61:738–43. doi: 10.1002/1097-0142(19880215)61:4<738::AID-CNCR2820610417>3.0.CO;2-E
27. Yang I, Han SJ, Sughrue ME, Tihan T, Parsa AT. Immune cell infiltrate differences in pilocytic astrocytoma and glioblastoma: evidence of distinct immunological microenvironments that reflect tumor biology. J Neurosurg. (2011) 115:505–11. doi: 10.3171/2011.4.JNS101172
28. Zhu C, Zou C, Guan G, Guo Q, Yan Z, Liu T, et al. Development and validation of an interferon signature predicting prognosis and treatment response for glioblastoma. Oncoimmunology. (2019) 8:e1621677. doi: 10.1080/2162402X.2019.1621677
29. Dominguez-Valentin M, Gras Navarro A, Rahman AM, Kumar S, Retiere C, Ulvestad E, et al. Identification of a natural killer cell receptor allele that prolongs survival of cytomegalovirus-positive glioblastoma patients. Cancer Res. (2016) 76:5326–36. doi: 10.1158/0008-5472.CAN-16-1162
30. Gras Navarro A, Kmiecik J, Leiss L, Zelkowski M, Engelsen A, Bruserud O, et al. NK cells with KIR2DS2 immunogenotype have a functional activation advantage to efficiently kill glioblastoma and prolong animal survival. J Immunol. (2014) 193:6192–206. doi: 10.4049/jimmunol.1400859
31. Friese MA, Platten M, Lutz SZ, Naumann U, Aulwurm S, Bischof F, et al. MICA/NKG2D-mediated immunogene therapy of experimental gliomas. Cancer Res. (2003) 63:8996–9006.
32. Dix AR, Brooks WH, Roszman TL, Morford LA. Immune defects observed in patients with primary malignant brain tumors. J Neuroimmunol. (1999) 100:216–32. doi: 10.1016/S0165-5728(99)00203-9
33. Roth P, Mittelbronn M, Wick W, Meyermann R, Tatagiba M, Weller M. Malignant glioma cells counteract antitumor immune responses through expression of lectin-like transcript-1. Cancer Res. (2007) 67:3540–4. doi: 10.1158/0008-5472.CAN-06-4783
34. Larsen SK, Gao Y, Basse PH. NK cells in the tumor microenvironment. Crit Rev Oncog. (2014) 19:91–105. doi: 10.1615/CritRevOncog.2014011142
35. Han J, Alvarez-Breckenridge CA, Wang QE, Yu J. TGF-β signaling and its targeting for glioma treatment. Am J Cancer Res. (2015) 5:945–55.
36. Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, et al. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci USA. (2003) 100:4120–5. doi: 10.1073/pnas.0730640100
37. Viel S, Marcais A, Guimaraes FS, Loftus R, Rabilloud J, Grau M, et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci Signal. (2016) 9:ra19. doi: 10.1126/scisignal.aad1884
38. Yvon ES, Burga R, Powell A, Cruz CR, Fernandes R, Barese C, et al. Cord blood natural killer cells expressing a dominant negative TGF-beta receptor: implications for adoptive immunotherapy for glioblastoma. Cytotherapy. (2017) 19:408–18. doi: 10.1016/j.jcyt.2016.12.005
39. Roth P, Aulwurm S, Gekel I, Beier D, Sperry RG, Mittelbronn M, et al. Regeneration and tolerance factor: a novel mediator of glioblastoma-associated immunosuppression. Cancer Res. (2006) 66:3852–8. doi: 10.1158/0008-5472.CAN-05-3062
40. Crane CA, Han SJ, Barry JJ, Ahn BJ, Lanier LL, Parsa AT. TGF-beta downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro Oncol. (2010) 12:7–13. doi: 10.1093/neuonc/nop009
41. Walentynowicz KA, Ochocka N, Pasierbinska M, Wojnicki K, Stepniak K, Mieczkowski J, et al. In search for reliable markers of glioma-induced polarization of microglia. Front Immunol. (2018) 9:1329. doi: 10.3389/fimmu.2018.01329
42. Zeiner PS, Preusse C, Golebiewska A, Zinke J, Iriondo A, Muller A, et al. Distribution and prognostic impact of microglia/macrophage subpopulations in gliomas. Brain Pathol. (2019) 29:513–29. doi: 10.1111/bpa.12690
43. Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, et al. Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep. (2016) 17:2445–59. doi: 10.1016/j.celrep.2016.10.052
44. Roesch S, Rapp C, Dettling S, Herold-Mende C. When immune cells turn bad - tumor-associated microglia/macrophages in glioma. Int J Mol Sci. (2018) 19:E436. doi: 10.3390/ijms19020436
45. Castriconi R, Daga A, Dondero A, Zona G, Poliani PL, Melotti A, et al. NK cells recognize and kill human glioblastoma cells with stem cell-like properties. J Immunol. (2009) 182:3530–9. doi: 10.4049/jimmunol.0802845
46. Avril T, Vauleon E, Hamlat A, Saikali S, Etcheverry A, Delmas C, et al. Human glioblastoma stem-like cells are more sensitive to allogeneic NK and T cell-mediated killing compared with serum-cultured glioblastoma cells. Brain Pathol. (2012) 22:159–74. doi: 10.1111/j.1750-3639.2011.00515.x
47. Horing E, Podlech O, Silkenstedt B, Rota IA, Adamopoulou E, Naumann U. The histone deacetylase inhibitor trichostatin a promotes apoptosis and antitumor immunity in glioblastoma cells. Anticancer Res. (2013) 33:1351–60.
48. Kmiecik J, Gras Navarro A, Poli A, Planaguma JP, Zimmer J, Chekenya M. Combining NK cells and mAb9.2.27 to combat NG2-dependent and anti-inflammatory signals in glioblastoma. Oncoimmunology. (2014) 3:e27185. doi: 10.4161/onci.27185
49. Gras Navarro A, Espedal H, Joseph JV, Trachsel-Moncho L, Bahador M, Tore Gjertsen B, et al. Pretreatment of glioblastoma with bortezomib potentiates natural killer cell cytotoxicity through TRAIL/DR5 mediated apoptosis and prolongs animal survival. Cancers. (2019) 11:E996. doi: 10.3390/cancers11070996
50. Ishikawa E, Tsuboi K, Saijo K, Harada H, Takano S, Nose T, et al. Autologous natural killer cell therapy for human recurrent malignant glioma. Anticancer Res. (2004) 24:1861–71.
51. Grimm EA, Robb RJ, Roth JA, Neckers LM, Lachman LB, Wilson DJ, et al. Lymphokine-activated killer cell phenomenon. III. Evidence that IL-2 is sufficient for direct activation of peripheral blood lymphocytes into lymphokine-activated killer cells. J. Exp. Med. (1983) 158:1356–61. doi: 10.1084/jem.158.4.1356
52. Phillips JH, Lanier LL. Dissection of the lymphokine-activated killer phenomenon. Relative contribution of peripheral blood natural killer cells and T lymphocytes to cytolysis. J Exp Med. (1986) 164:814–25. doi: 10.1084/jem.164.3.814
53. Merchant RE, Grant AJ, Merchant LH, Young HF. Adoptive immunotherapy for recurrent glioblastoma multiforme using lymphokine activated killer cells and recombinant interleukin-2. Cancer. (1988) 62:665–71. doi: 10.1002/1097-0142(19880815)62:4<665::AID-CNCR2820620403>3.0.CO;2-O
54. Yoshida S, Tanaka R, Takai N, Ono K. Local administration of autologous lymphokine-activated killer cells and recombinant interleukin 2 to patients with malignant brain tumors. Cancer Res. (1988) 48:5011–6.
55. Barba D, Saris SC, Holder C, Rosenberg SA, Oldfield EH. Intratumoral LAK cell and interleukin-2 therapy of human gliomas. J Neurosurg. (1989) 70:175–82. doi: 10.3171/jns.1989.70.2.0175
56. Ingram M, Buckwalter JG, Jacques DB, Freshwater DB, Abts RM, Techy GB, et al. Immunotherapy for recurrent malignant glioma: an interim report on survival. Neurol Res. (1990) 12:265–73. doi: 10.1080/01616412.1990.11739955
57. Jeffes EW 3rd, Beamer YB, Jacques S, Coss JS, Nep RL, Beckman M, et al. Therapy of recurrent high-grade gliomas with surgery, autologous mitogen-activated IL-2-stimulated (MAK) killer lymphocytes, and rIL-2: II. Correlation of survival with MAK cell tumor necrosis factor production in vitro. Lymphokine Cytokine Res. (1991) 10:89–94.
58. Lillehei KO, Mitchell DH, Johnson SD, McCleary EL, Kruse CA. Long-term follow-up of patients with recurrent malignant gliomas treated with adjuvant adoptive immunotherapy. Neurosurgery. (1991) 28:16–23. doi: 10.1227/00006123-199101000-00003
59. Jeffes EW 3rd, Beamer YB, Jacques S, Silberman RS, Vayuvegula B, Gupta S, et al. Therapy of recurrent high grade gliomas with surgery, and autologous mitogen activated IL-2 stimulated killer (MAK) lymphocytes: I. Enhancement of MAK lytic activity and cytokine production by PHA and clinical use of PHA. J Neurooncol. (1993) 15:141–55. doi: 10.1007/BF01053935
60. Boiardi A, Silvani A, Ruffini PA, Rivoltini L, Parmiani G, Broggi G, et al. Loco-regional immunotherapy with recombinant interleukin-2 and adherent lymphokine-activated killer cells (A-LAK) in recurrent glioblastoma patients. Cancer Immunol Immunother. (1994) 39:193–7. doi: 10.1007/BF01533386
61. Hayes RL, Koslow M, Hiesiger EM, Hymes KB, Hochster HS, Moore EJ, et al. Improved long term survival after intracavitary interleukin-2 and lymphokine-activated killer cells for adults with recurrent malignant glioma. Cancer. (1995) 76:840–52. doi: 10.1002/1097-0142(19950901)76:5<840::AID-CNCR2820760519>3.0.CO;2-R
62. Sankhla SK, Nadkarni JS, Bhagwati SN. Adoptive immunotherapy using lymphokine-activated killer (LAK) cells and interleukin-2 for recurrent malignant primary brain tumors. J Neurooncol. (1996) 27:133–40. doi: 10.1007/BF00177476
63. Dillman RO, Duma CM, Schiltz PM, DePriest C, Ellis RA, Okamoto K, et al. Intracavitary placement of autologous lymphokine-activated killer (LAK) cells after resection of recurrent glioblastoma. J Immunother. (2004) 27:398–404. doi: 10.1097/00002371-200409000-00009
64. Dillman RO, Duma CM, Ellis RA, Cornforth AN, Schiltz PM, Sharp SL, et al. Intralesional lymphokine-activated killer cells as adjuvant therapy for primary glioblastoma. J Immunother. (2009) 32:914–9. doi: 10.1097/CJI.0b013e3181b2910f
65. Komatsu F, Kajiwara M. CD18/CD54(+CD102), CD2/CD58 pathway-independent killing of lymphokine-activated killer (LAK) cells against glioblastoma cell lines T98G and U373MG. Oncol Res. (2000) 12:17–24. doi: 10.3727/000000001108747408
66. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA. (1993) 90:720–4. doi: 10.1073/pnas.90.2.720
67. Brocker T, Peter A, Traunecker A, Karjalainen K. New simplified molecular design for functional T cell receptor. Eur J Immunol. (1993) 23:1435–9. doi: 10.1002/eji.1830230705
68. Moritz D, Wels W, Mattern J, Groner B. Cytotoxic T lymphocytes with a grafted recognition specificity for ERBB2-expressing tumor cells. Proc Natl Acad Sci USA. (1994) 91:4318–22. doi: 10.1073/pnas.91.10.4318
69. Hartmann J, Schussler-Lenz M, Bondanza A, Buchholz CJ. Clinical development of CAR T cells-challenges and opportunities in translating innovative treatment concepts. EMBO Mol Med. (2017) 9:1183–97. doi: 10.15252/emmm.201607485
70. June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. (2018) 379:64–73. doi: 10.1056/NEJMra1706169
71. Genßler S, Burger MC, Zhang C, Oelsner S, Mildenberger I, Wagner M, et al. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. Oncoimmunology. (2016) 5:e1119354. doi: 10.1080/2162402X.2015.1119354
72. Tran AC, Zhang D, Byrn R, Roberts MR. Chimeric zeta-receptors direct human natural killer (NK) effector function to permit killing of NK-resistant tumor cells and HIV-infected T lymphocytes. J Immunol. (1995) 155:1000–9.
73. Uherek C, Tonn T, Uherek B, Becker S, Schnierle B, Klingemann HG, et al. Retargeting of natural killer-cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumor cell destruction. Blood. (2002) 100:1265–73. doi: 10.1182/blood.V100.4.1265.h81602001265_1265_1273
74. Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. (2005) 106:376–83. doi: 10.1182/blood-2004-12-4797
75. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. (2018) 23:181–92.e185. doi: 10.1016/j.stem.2018.06.002
76. Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. (2018) 32:520–31. doi: 10.1038/leu.2017.226
77. Sahm C, Schönfeld K, Wels WS. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol Immunother. (2012) 61:1451–61. doi: 10.1007/s00262-012-1212-x
78. Schönfeld K, Sahm C, Zhang C, Naundorf S, Brendel C, Odendahl M, et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol Ther. (2015) 23:330–8. doi: 10.1038/mt.2014.219
79. Oelsner S, Friede ME, Zhang C, Wagner J, Badura S, Bader P, et al. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy. (2017) 19:235–49. doi: 10.1016/j.jcyt.2016.10.009
80. Oelsner S, Waldmann A, Billmeier A, Roder J, Lindner A, Ullrich E, et al. Genetically engineered CAR NK cells display selective cytotoxicity against FLT3-positive B-ALL and inhibit in vivo leukemia growth. Int J Cancer. (2019) 145:1935–45. doi: 10.1002/ijc.32269
81. Altvater B, Landmeier S, Pscherer S, Temme J, Schweer K, Kailayangiri S, et al. 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clin Cancer Res. (2009) 15:4857–66. doi: 10.1158/1078-0432.CCR-08-2810
82. Muller N, Michen S, Tietze S, Topfer K, Schulte A, Lamszus K, et al. Engineering NK cells modified with an EGFRvIII-specific chimeric antigen receptor to overexpress CXCR4 improves immunotherapy of CXCL12/SDF-1α-secreting glioblastoma. J Immunother. (2015) 38:197–210. doi: 10.1097/CJI.0000000000000082
83. Zhang J, Zheng H, Diao Y. Natural killer cells and current applications of chimeric antigen receptor-modified NK-92 cells in tumor immunotherapy. Int J Mol Sci. (2019) 20:E317. doi: 10.3390/ijms20020317
84. Bielamowicz K, Khawja S, Ahmed N. Adoptive cell therapies for glioblastoma. Front Oncol. (2013) 3:275. doi: 10.3389/fonc.2013.00275
85. Prinzing BL, Gottschalk SM, Krenciute G. CAR T-cell therapy for glioblastoma: ready for the next round of clinical testing? Expert Rev Anticancer Ther. (2018) 18:451–61. doi: 10.1080/14737140.2018.1451749
86. Sharma P, Debinski W. Receptor-targeted glial brain tumor therapies. Int J Mol Sci. (2018) 19:E3326. doi: 10.3390/ijms19113326
87. Akhavan D, Alizadeh D, Wang D, Weist MR, Shepphird JK, Brown CE. CAR T cells for brain tumors: lessons learned and road ahead. Immunol Rev. (2019) 290:60–84. doi: 10.1111/imr.12773
88. Choi BD, Maus MV, June CH, Sampson JH. Immunotherapy for glioblastoma: adoptive T-cell strategies. Clin Cancer Res. (2019) 25:2042–8. doi: 10.1158/1078-0432.CCR-18-1625
89. Debinski W, Gibo DM, Hulet SW, Connor JR, Gillespie GY. Receptor for interleukin 13 is a marker and therapeutic target for human high-grade gliomas. Clin Cancer Res. (1999) 5:985–90.
90. Newman JP, Wang GY, Arima K, Guan SP, Waters MR, Cavenee WK, et al. Interleukin-13 receptor alpha 2 cooperates with EGFRvIII signaling to promote glioblastoma multiforme. Nat Commun. (2017) 8:1913. doi: 10.1038/s41467-017-01392-9
91. Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. (2004) 64:9160–6. doi: 10.1158/0008-5472.CAN-04-0454
92. Brown CE, Starr R, Aguilar B, Shami AF, Martinez C, D'Apuzzo M, et al. Stem-like tumor-initiating cells isolated from IL13Ralpha2 expressing gliomas are targeted and killed by IL13-zetakine-redirected T Cells. Clin Cancer Res. (2012) 18:2199–209. doi: 10.1158/1078-0432.CCR-11-1669
93. Kong S, Sengupta S, Tyler B, Bais AJ, Ma Q, Doucette S, et al. Suppression of human glioma xenografts with second-generation IL13R-specific chimeric antigen receptor-modified T cells. Clin Cancer Res. (2012) 18:5949–60. doi: 10.1158/1078-0432.CCR-12-0319
94. Krebs S, Chow KK, Yi Z, Rodriguez-Cruz T, Hegde M, Gerken C, et al. T cells redirected to interleukin-13Ralpha2 with interleukin-13 mutein–chimeric antigen receptors have anti-glioma activity but also recognize interleukin-13Ralpha1. Cytotherapy. (2014) 16:1121–31. doi: 10.1016/j.jcyt.2014.02.012
95. Brown CE, Aguilar B, Starr R, Yang X, Chang WC, Weng L, et al. Optimization of IL13Rα2-targeted chimeric antigen receptor T cells for improved anti-tumor efficacy against glioblastoma. Mol Ther. (2018) 26:31–44. doi: 10.1016/j.ymthe.2017.10.002
96. Wang D, Aguilar B, Starr R, Alizadeh D, Brito A, Sarkissian A, et al. Glioblastoma-targeted CD4+ CAR T cells mediate superior antitumor activity. JCI Insight. (2018) 3:99048. doi: 10.1172/jci.insight.99048
97. Krenciute G, Krebs S, Torres D, Wu MF, Liu H, Dotti G, et al. Characterization and functional analysis of scFv-based chimeric antigen receptors to redirect T cells to IL13Rα2-positive glioma. Mol Ther. (2016) 24:354–63. doi: 10.1038/mt.2015.199
98. Krenciute G, Prinzing BL, Yi Z, Wu MF, Liu H, Dotti G, et al. Transgenic expression of IL15 improves antiglioma activity of IL13Rα2-CAR T cells but results in antigen loss variants. Cancer Immunol Res. (2017) 5:571–81. doi: 10.1158/2326-6066.CIR-16-0376
99. Pituch KC, Miska J, Krenciute G, Panek WK, Li G, Rodriguez-Cruz T, et al. Adoptive transfer of IL13Rα2-specific chimeric antigen receptor T cells creates a pro-inflammatory environment in glioblastoma. Mol Ther. (2018) 26:986–95. doi: 10.1016/j.ymthe.2018.02.001
100. Yin Y, Boesteanu AC, Binder ZA, Xu C, Reid RA, Rodriguez JL, et al. Checkpoint blockade reverses anergy in IL-13Rα2 humanized scFv-based CAR T cells to treat murine and canine gliomas. Mol Ther Oncolytics. (2018) 11:20–38. doi: 10.1016/j.omto.2018.08.002
101. Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. (2015) 21:4062–72. doi: 10.1158/1078-0432.CCR-15-0428
102. Ronellenfitsch MW, Luger AL, Steinbach JP. EGFR and mTOR as therapeutic targets in glioblastoma. Oncotarget. (2019) 10:4721–3. doi: 10.18632/oncotarget.27094
103. Bigner SH, Humphrey PA, Wong AJ, Vogelstein B, Mark J, Friedman HS, et al. Characterization of the epidermal growth factor receptor in human glioma cell lines and xenografts. Cancer Res. (1990) 50:8017–22.
104. Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol. (1995) 19:183–232. doi: 10.1016/1040-8428(94)00144-I
105. Sugawa N, Ekstrand AJ, James CD, Collins VP. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc Natl Acad Sci USA. (1990) 87:8602–6. doi: 10.1073/pnas.87.21.8602
106. Ekstrand AJ, Sugawa N, James CD, Collins VP. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc Natl Acad Sci USA. (1992) 89:4309–13. doi: 10.1073/pnas.89.10.4309
107. Nishikawa R, Ji XD, Harmon RC, Lazar CS, Gill GN, Cavenee WK, et al. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci USA. (1994) 91:7727–31. doi: 10.1073/pnas.91.16.7727
108. Nagane M, Levitzki A, Gazit A, Cavenee WK, Huang HJ. Drug resistance of human glioblastoma cells conferred by a tumor-specific mutant epidermal growth factor receptor through modulation of Bcl-XL and caspase-3-like proteases. Proc Natl Acad Sci USA. (1998) 95:5724–9. doi: 10.1073/pnas.95.10.5724
109. An Z, Aksoy O, Zheng T, Fan QW, Weiss WA. Epidermal growth factor receptor and EGFRvIII in glioblastoma: signaling pathways and targeted therapies. Oncogene. (2018) 37:1561–75. doi: 10.1038/s41388-017-0045-7
110. Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther. (2012) 23:1043–53. doi: 10.1089/hum.2012.041
111. Ohno M, Ohkuri T, Kosaka A, Tanahashi K, June CH, Natsume A, et al. Expression of miR-17-92 enhances anti-tumor activity of T-cells transduced with the anti-EGFRvIII chimeric antigen receptor in mice bearing human GBM xenografts. J Immunother Cancer. (2013) 1:21. doi: 10.1186/2051-1426-1-21
112. Shen CJ, Yang YX, Han EQ, Cao N, Wang YF, Wang Y, et al. Chimeric antigen receptor containing ICOS signaling domain mediates specific and efficient antitumor effect of T cells against EGFRvIII expressing glioma. J Hematol Oncol. (2013) 6:33. doi: 10.1186/1756-8722-6-33
113. Choi BD, Suryadevara CM, Gedeon PC, Herndon JE 2nd, Sanchez-Perez L, Bigner DD, et al. Intracerebral delivery of a third generation EGFRvIII-specific chimeric antigen receptor is efficacious against human glioma. J Clin Neurosci. (2014) 21:189–90. doi: 10.1016/j.jocn.2013.03.012
114. Sampson JH, Choi BD, Sanchez-Perez L, Suryadevara CM, Snyder DJ, Flores CT, et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin Cancer Res. (2014) 20:972–84. doi: 10.1158/1078-0432.CCR-13-0709
115. Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med. (2015) 7:275ra222. doi: 10.1126/scitranslmed.aaa4963
116. Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, et al. Pilot trial of adoptive transfer of chimeric antigen receptor-transduced T cells targeting EGFRvIII in patients with glioblastoma. J Immunother. (2019) 42:126–35. doi: 10.1097/CJI.0000000000000260
117. Subramanian J, Katta A, Masood A, Vudem DR, Kancha RK. Emergence of ERBB2 mutation as a biomarker and an actionable target in solid cancers. Oncologist. (2019) 24. doi: 10.1634/theoncologist.2018-0845. [Epub ahead of print].
118. Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. (2009) 21:177–84. doi: 10.1016/j.ceb.2008.12.010
119. Press MF, Cordon-Cardo C, Slamon DJ. Expression of the HER-2/neu proto-oncogene in normal human adult and fetal tissues. Oncogene. (1990) 5:953–62.
120. Schlegel J, Stumm G, Brandle K, Merdes A, Mechtersheimer G, Hynes NE, et al. Amplification and differential expression of members of the erbB-gene family in human glioblastoma. J Neurooncol. (1994) 22:201–7. doi: 10.1007/BF01052920
121. Koka V, Potti A, Forseen SE, Pervez H, Fraiman GN, Koch M, et al. Role of Her-2/neu overexpression and clinical determinants of early mortality in glioblastoma multiforme. Am J Clin Oncol. (2003) 26:332–5. doi: 10.1097/01.COC.0000020922.66984.E7
122. Andersson U, Guo D, Malmer B, Bergenheim AT, Brannstrom T, Hedman H, et al. Epidermal growth factor receptor family (EGFR, ErbB2-4) in gliomas and meningiomas. Acta Neuropathol. (2004) 108:135–42. doi: 10.1007/s00401-004-0875-6
123. Liu G, Ying H, Zeng G, Wheeler CJ, Black KL, Yu JS. HER-2, gp100, and MAGE-1 are expressed in human glioblastoma and recognized by cytotoxic T cells. Cancer Res. (2004) 64:4980–6. doi: 10.1158/0008-5472.CAN-03-3504
124. Zhang JG, Eguchi J, Kruse CA, Gomez GG, Fakhrai H, Schroter S, et al. Antigenic profiling of glioma cells to generate allogeneic vaccines or dendritic cell-based therapeutics. Clin Cancer Res. (2007) 13(2 Pt 1):566–75. doi: 10.1158/1078-0432.CCR-06-1576
125. Zhang C, Burger MC, Jennewein L, Genßler S, Schönfeld K, Zeiner P, et al. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. J Natl Cancer Inst. (2016) 108:dvj375. doi: 10.1093/jnci/djv375
126. Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res. (2010) 16:474–85. doi: 10.1158/1078-0432.CCR-09-1322
127. Wykosky J, Gibo DM, Stanton C, Debinski W. EphA2 as a novel molecular marker and target in glioblastoma multiforme. Mol Cancer Res. (2005) 3:541–51. doi: 10.1158/1541-7786.MCR-05-0056
128. Wang LF, Fokas E, Bieker M, Rose F, Rexin P, Zhu Y, et al. Increased expression of EphA2 correlates with adverse outcome in primary and recurrent glioblastoma multiforme patients. Oncol Rep. (2008) 19:151–6. doi: 10.3892/or.19.1.151
129. Chow KK, Naik S, Kakarla S, Brawley VS, Shaffer DR, Yi Z, et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol Ther. (2013) 21:629–37. doi: 10.1038/mt.2012.210
130. Yi Z, Prinzing BL, Cao F, Gottschalk S, Krenciute G. Optimizing EphA2-CAR T cells for the adoptive immunotherapy of glioma. Mol Ther Methods Clin Dev. (2018) 9:70–80. doi: 10.1016/j.omtm.2018.01.009
131. Pellegatta S, Savoldo B, Di Ianni N, Corbetta C, Chen Y, Patane M, et al. Constitutive and TNFalpha-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: Implications for CAR-T cell therapy. Sci Transl Med. (2018) 10:aao2731. doi: 10.1126/scitranslmed.aao2731
132. Beard RE, Zheng Z, Lagisetty KH, Burns WR, Tran E, Hewitt SM, et al. Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J Immunother Cancer. (2014) 2:25. doi: 10.1186/2051-1426-2-25
133. Geldres C, Savoldo B, Hoyos V, Caruana I, Zhang M, Yvon E, et al. T lymphocytes redirected against the chondroitin sulfate proteoglycan-4 control the growth of multiple solid tumors both in vitro and in vivo. Clin Cancer Res. (2014) 20:962–71. doi: 10.1158/1078-0432.CCR-13-2218
134. Sanai N, Alvarez-Buylla A, Berger MS. Neural stem cells and the origin of gliomas. N Engl J Med. (2005) 353:811–22. doi: 10.1056/NEJMra043666
135. Wang Y, Chen M, Wu Z, Tong C, Dai H, Guo Y, et al. CD133-directed CAR T cells for advanced metastasis malignancies: a phase I trial. Oncoimmunology. (2018) 7:e1440169. doi: 10.1080/2162402X.2018.1440169
136. Zhu X, Prasad S, Gaedicke S, Hettich M, Firat E, Niedermann G. Patient-derived glioblastoma stem cells are killed by CD133-specific CAR T cells but induce the T cell aging marker CD57. Oncotarget. (2015) 6:171–84. doi: 10.18632/oncotarget.2767
137. Hu B, Zou Y, Zhang L, Tang J, Niedermann G, Firat E, et al. Nucleofection with plasmid DNA for CRISPR/Cas9-mediated inactivation of programmed cell death protein 1 in CD133-specific CAR T cells. Hum Gene Ther. (2019) 30:446–58. doi: 10.1089/hum.2017.234
138. Held-Feindt J, Mentlein R. CD70/CD27 ligand, a member of the TNF family, is expressed in human brain tumors. Int J Cancer. (2002) 98:352–6. doi: 10.1002/ijc.10207
139. Wischhusen J, Jung G, Radovanovic I, Beier C, Steinbach JP, Rimner A, et al. Identification of CD70-mediated apoptosis of immune effector cells as a novel immune escape pathway of human glioblastoma. Cancer Res. (2002) 62:2592–9.
140. Chahlavi A, Rayman P, Richmond AL, Biswas K, Zhang R, Vogelbaum M, et al. Glioblastomas induce T-lymphocyte death by two distinct pathways involving gangliosides and CD70. Cancer Res. (2005) 65:5428–38. doi: 10.1158/0008-5472.CAN-04-4395
141. Jin L, Ge H, Long Y, Yang C, Chang YE, Mu L, et al. CD70, a novel target of CAR T-cell therapy for gliomas. Neuro Oncol. (2018) 20:55–65. doi: 10.1093/neuonc/nox116
142. Hegde M, Corder A, Chow KK, Mukherjee M, Ashoori A, Kew Y, et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther. (2013) 21:2087–101. doi: 10.1038/mt.2013.185
143. Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. (2018) 20:506–18. doi: 10.1093/neuonc/nox182
144. Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J Clin Invest. (2016) 126:3036–52. doi: 10.1172/JCI83416
145. Shimasaki N, Coustan-Smith E, Kamiya T, Campana D. Expanded and armed natural killer cells for cancer treatment. Cytotherapy. (2016) 18:1422–34. doi: 10.1016/j.jcyt.2016.06.013
146. Koch J, Steinle A, Watzl C, Mandelboim O. Activating natural cytotoxicity receptors of natural killer cells in cancer and infection. Trends Immunol. (2013) 34:182–91. doi: 10.1016/j.it.2013.01.003
147. Granzin M, Wagner J, Kohl U, Cerwenka A, Huppert V, Ullrich E. Shaping of natural killer cell antitumor activity by ex vivo cultivation. Front Immunol. (2017) 8:458. doi: 10.3389/fimmu.2017.00458
148. Tonn T, Becker S, Esser R, Schwabe D, Seifried E. Cellular immunotherapy of malignancies using the clonal natural killer cell line NK-92. J Hematother Stem Cell Res. (2001) 10:535–44. doi: 10.1089/15258160152509145
149. Glienke W, Esser R, Priesner C, Suerth JD, Schambach A, Wels WS, et al. Advantages and applications of CAR-expressing natural killer cells. Front Pharmacol. (2015) 6:21. doi: 10.3389/fphar.2015.00021
150. Suck G, Odendahl M, Nowakowska P, Seidl C, Wels WS, Klingemann HG, et al. NK-92: an 'off-the-shelf therapeutic' for adoptive natural killer cell-based cancer immunotherapy. Cancer Immunol Immunother. (2015) 65:485–92. doi: 10.1007/s00262-015-1761-x
151. Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia. (1994) 8:652–8.
152. Maki G, Klingemann HG, Martinson JA, Tam YK. Factors regulating the cytotoxic activity of the human natural killer cell line, NK-92. J Hematother Stem Cell Res. (2001) 10:369–83. doi: 10.1089/152581601750288975
153. Kirwan SE, Burshtyn DN. Killer cell Ig-like receptor-dependent signaling by Ig-like transcript 2 (ILT2/CD85j/LILRB1/LIR-1). J Immunol. (2005) 175:5006–15. doi: 10.4049/jimmunol.175.8.5006
154. Jochems C, Hodge JW, Fantini M, Fujii R, Morillon YM 2nd, Greiner JW, et al. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget. (2016) 7:86359–73. doi: 10.18632/oncotarget.13411
155. Kloess S, Oberschmidt O, Dahlke J, Vu XK, Neudoerfl C, Kloos A, et al. Preclinical assessment of suitable natural killer cell sources for chimeric antigen receptor natural killer-based “off-the-shelf” acute myeloid leukemia immunotherapies. Hum Gene Ther. (2019) 30:381–401. doi: 10.1089/hum.2018.247
156. Arai S, Meagher R, Swearingen M, Myint H, Rich E, Martinson J, et al. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy. (2008) 10:625–32. doi: 10.1080/14653240802301872
157. Tonn T, Schwabe D, Klingemann HG, Becker S, Esser R, Koehl U, et al. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy. (2013) 15:1563–70. doi: 10.1016/j.jcyt.2013.06.017
158. Boyiadzis M, Agha M, Redner RL, Sehgal A, Im A, Hou JZ, et al. Phase 1 clinical trial of adoptive immunotherapy using “off-the-shelf” activated natural killer cells in patients with refractory and relapsed acute myeloid leukemia. Cytotherapy. (2017) 19:1225–32. doi: 10.1016/j.jcyt.2017.07.008
159. Williams BA, Law AD, Routy B, denHollander N, Gupta V, Wang XH, et al. A phase I trial of NK-92 cells for refractory hematological malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget. (2017) 8:89256–68. doi: 10.18632/oncotarget.19204
160. Han J, Chu J, Keung Chan W, Zhang J, Wang Y, Cohen JB, et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci Rep. (2015) 5:11483. doi: 10.1038/srep11483
161. Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, et al. First-in-man clinical trial of CAR NK-92 cells: Safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res. (2018) 8:1083–9.
162. Burger MC, Mildenberger I, Zhang C, Ihrig K, Wagner M, Mittelbronn M, et al. The CAR2BRAIN study: A monocentric phase I trial with ErbB2-specific NK-92/5.28.z cells in recurrent glioblastoma. Neuro Oncol. (2016) 18:24–5. doi: 10.1093/neuonc/now188.083
163. Murakami T, Nakazawa T, Natsume A, Nishimura F, Nakamura M, Matsuda R, et al. Novel human NK cell line carrying CAR targeting EGFRvIII induces antitumor effects in glioblastoma cells. Anticancer Res. (2018) 38:5049–56. doi: 10.21873/anticanres.12824
164. Alkins R, Burgess A, Ganguly M, Francia G, Kerbel R, Wels WS, et al. Focused ultrasound delivers targeted immune cells to metastatic brain tumors. Cancer Res. (2013) 73:1892–9. doi: 10.1158/0008-5472.CAN-12-2609
165. Alkins R, Burgess A, Kerbel R, Wels WS, Hynynen K. Early treatment of HER2-amplified brain tumors with targeted NK-92 cells and focused ultrasound improves survival. Neuro Oncol. (2016) 18:4–81. doi: 10.1093/neuonc/nov318
166. Beers R, Chowdhury P, Bigner D, Pastan I. Immunotoxins with increased activity against epidermal growth factor receptor vIII-expressing cells produced by antibody phage display. Clin Cancer Res. (2000) 6:2835–43.
167. Lanier LL. DAP10- and DAP12-associated receptors in innate immunity. Immunol Rev. (2009) 227:150–60. doi: 10.1111/j.1600-065X.2008.00720.x
168. Schnalzger TE, de Groot MH, Zhang C, Mosa MH, Michels B E, Roder J, et al. 3D model for CAR-mediated cytotoxicity using patient-derived colorectal cancer organoids. EMBO J. (2019) 38:e100928. doi: 10.15252/embj.2018100928
169. Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. (2005) 7:301–11. doi: 10.1016/j.ccr.2005.03.003
170. Hartmann C, Müller N, Blaukat A, Koch J, Benhar I, Wels WS. Peptide mimotopes recognized by antibodies cetuximab and matuzumab induce a functionally equivalent anti-EGFR immune response. Oncogene. (2010) 29:4517–27. doi: 10.1038/onc.2010.195
171. Jiang H, Gao H, Kong J, Song B, Wang P, Shi B, et al. Selective targeting of glioblastoma with EGFRvIII/EGFR bitargeted chimeric antigen receptor T cell. Cancer Immunol Res. (2018) 6:1314–26. doi: 10.1158/2326-6066.CIR-18-0044
172. Choi BD, Yu X, Castano AP, Bouffard AA, Schmidts A, Larson RC, et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol. (2019) 37:1049–58. doi: 10.1038/s41587-019-0192-1
173. Wels W, Harwerth IM, Zwickl M, Hardman N, Groner B, Hynes NE. Construction, bacterial expression and characterization of a bifunctional single-chain antibody-phosphatase fusion protein targeted to the human erbB-2 receptor. Biotechnology. (1992) 10:1128–32. doi: 10.1038/nbt1092-1128
174. Klapdor R, Wang S, Hacker U, Buning H, Morgan M, Dork T, et al. Improved killing of ovarian cancer stem cells by combining a novel chimeric antigen receptor-based immunotherapy and chemotherapy. Hum Gene Ther. (2017) 28:886–96. doi: 10.1089/hum.2017.168
175. Longee DC, Wikstrand CJ, Mansson JE, He X, Fuller GN, Bigner SH, et al. Disialoganglioside GD2 in human neuroectodermal tumor cell lines and gliomas. Acta Neuropathol. (1991) 82:45–54. doi: 10.1007/BF00310922
176. Golinelli G, Grisendi G, Prapa M, Bestagno M, Spano C, Rossignoli F, et al. Targeting GD2-positive glioblastoma by chimeric antigen receptor empowered mesenchymal progenitors. Cancer Gene Ther. (2018) 16:569–81. doi: 10.1038/s41417-018-0062-x
177. Esser R, Müller T, Stefes D, Kloess S, Seidel D, Gillies SD, et al. NK cells engineered to express a GD2-specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J Cell Mol Med. (2012) 16:569–81. doi: 10.1111/j.1582-4934.2011.01343.x
178. Seidel D, Shibina A, Siebert N, Wels WS, Reynolds CP, Huebener N, et al. Disialoganglioside-specific human natural killer cells are effective against drug-resistant neuroblastoma. Cancer Immunol Immunother. (2015) 64:621–34. doi: 10.1007/s00262-015-1669-5
179. Kailayangiri S, Altvater B, Spurny C, Jamitzky S, Schelhaas S, Jacobs AH, et al. Targeting Ewing sarcoma with activated and GD2-specific chimeric antigen receptor-engineered human NK cells induces upregulation of immune-inhibitory HLA-G. Oncoimmunology. (2017) 6:e1250050. doi: 10.1080/2162402X.2016.1250050
180. Nowakowska P, Romanski A, Miller N, Odendahl M, Bonig H, Zhang C, et al. Clinical grade manufacturing of genetically modified, CAR-expressing NK-92 cells for the treatment of ErbB2-positive malignancies. Cancer Immunol Immunother. (2018) 67:25–38. doi: 10.1007/s00262-017-2055-2
181. Childs RW, Berg M. Bringing natural killer cells to the clinic: ex vivo manipulation. Hematology Am Soc Hematol Educ Progr. (2013) 2013:234–46. doi: 10.1182/asheducation-2013.1.234
182. El Assal R, Abou-Elkacem L, Tocchio A, Pasley S, Matosevic S, Kaplan DL, et al. Bioinspired preservation of natural killer cells for cancer immunotherapy. Adv Sci. (2019) 6:1802045. doi: 10.1002/advs.201802045
183. Li R, Johnson R, Yu G, McKenna DH, Hubel A. Preservation of cell-based immunotherapies for clinical trials. Cytotherapy. (2019) 21:943–57. doi: 10.1016/j.jcyt.2019.07.004
184. Liebner S, Dijkhuizen RM, Reiss Y, Plate KH, Agalliu D, Constantin G. Functional morphology of the blood-brain barrier in health and disease. Acta Neuropathol. (2018) 135:311–36. doi: 10.1007/s00401-018-1815-1
185. Haspels HN, Rahman MA, Joseph JV, Gras Navarro A, Chekenya M. Glioblastoma stem-like cells are more susceptible than differentiated cells to natural killer cell lysis mediated through killer immunoglobulin-like receptors-human leukocyte antigen ligand mismatch and activation receptor-ligand Interactions. Front Immunol. (2018) 9:1345. doi: 10.3389/fimmu.2018.01345
186. Mathios D, Park CK, Marcus WD, Alter S, Rhode PR, Jeng EK, et al. Therapeutic administration of IL-15 superagonist complex ALT-803 leads to long-term survival and durable antitumor immune response in a murine glioblastoma model. Int J Cancer. (2016) 138:187–94. doi: 10.1002/ijc.29686
187. Dubinski D, Won SY, Gessler F, Quick-Weller J, Behmanesh B, Bernatz S, et al. Dexamethasone-induced leukocytosis is associated with poor survival in newly diagnosed glioblastoma. J Neurooncol. (2018) 137:503–10. doi: 10.1007/s11060-018-2761-4
188. Dar TB, Henson RM, Shiao SL. Targeting innate immunity to enhance the efficacy of radiation therapy. Front Immunol. (2018) 9:3077. doi: 10.3389/fimmu.2018.03077
Keywords: natural killer cells, NK-92, chimeric antigen receptor, adoptive cancer immunotherapy, glioblastoma
Citation: Burger MC, Zhang C, Harter PN, Romanski A, Strassheimer F, Senft C, Tonn T, Steinbach JP and Wels WS (2019) CAR-Engineered NK Cells for the Treatment of Glioblastoma: Turning Innate Effectors Into Precision Tools for Cancer Immunotherapy. Front. Immunol. 10:2683. doi: 10.3389/fimmu.2019.02683
Received: 30 August 2019; Accepted: 31 October 2019;
Published: 14 November 2019.
Edited by:
Julian Pardo, Fundacion Agencia Aragonesa para la Investigacion y el Desarrollo, SpainReviewed by:
Kerry S. Campbell, Fox Chase Cancer Center, United StatesCopyright © 2019 Burger, Zhang, Harter, Romanski, Strassheimer, Senft, Tonn, Steinbach and Wels. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Winfried S. Wels, d2Vsc0Bnc2gudW5pLWZyYW5rZnVydC5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.