 Yifan Zhan
Yifan Zhan Andrew M. Lew
Andrew M. Lew Michael Chopin
Michael Chopin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 15 November 2019
Sec. Antigen Presenting Cell Biology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02679
This article is part of the Research TopicMonocyte Heterogeneity and FunctionView all 19 articles
Granulocyte Macrophage-Colony Stimulating Factor (GM-CSF) is a myelopoietic growth factor that has pleiotropic effects not only in promoting the differentiation of immature precursors into polymorphonuclear neutrophils (PMNs), monocytes/macrophages (MØs) and dendritic cells (DCs), but also in controlling the function of fully mature myeloid cells. This broad spectrum of GM-CSF action may elicit paradoxical outcomes—both immunostimulation and immunosuppression—in infection, inflammation, and cancer. The complexity of GM-CSF action remains to be fully unraveled. Several aspects of GM-CSF action could contribute to its diverse biological consequences. Firstly, GM-CSF as a single cytokine affects development of most myeloid cells from progenitors to mature immune cells. Secondly, GM-CSF activates JAK2/STAT5 and also activate multiple signaling modules and transcriptional factors that direct different biological processes. Thirdly, GM-CSF can be produced by different cell types including tumor cells in response to different environmental cues; thus, GM-CSF quantity can vary greatly under different pathophysiological settings. Finally, GM-CSF signaling is also fine-tuned by other less defined feedback mechanisms. In this review, we will discuss the role of GM-CSF in orchestrating the differentiation, survival, and proliferation during the generation of multiple lineages of myeloid cells (PMNs, MØs, and DCs). We will also discuss the role of GM-CSF in regulating the function of DCs and the functional polarization of MØs. We highlight how the dose of GM-CSF and corresponding signal strength acts as a rheostat to fine-tune cell fate, and thus the way GM-CSF may best be targeted for immuno-intervention in infection, inflammation and cancer.
The Granulocyte Macrophage-Colony Stimulating Factor (GM-CSF) is a small glycoprotein that is able to stimulate generation of polymorphonuclear neutrophils (PMNs) as well as mononuclear monocytes, macrophages (MØs) and dendritic cells (DC) (1–3). When added to mouse bone marrow precursors in vitro, GM-CSF acts in two phases: an early differentiating phase of PMNs and CD11b+ mononuclear cells from progenitors, and a late phase of MØs and monocyte-derived DCs (moDC) from CD11b+ mononuclear cells. For several decades it has been known that the outcome of such cultures is greatly influenced by a number of factors, including cell density, the presence of stromal cells, co-stimulatory signals, the serum quality and the concentration of GM-CSF (1). Despite this, the molecular mechanisms underpinning the heterogeneity of the myeloid cells produced in these GM-CSF induced cultures are still ill defined. For example, while cytokines such as IL-4, IL-13, TNF-α, TLR ligands or even GM-CSF concentration could alter dramatically the ratio of generated myeloid cells (1, 4–6), the nature of this bias under different conditions has not been fully resolved at a molecular level. It has also not been fully resolved whether differentiation fate under these conditions is the result of either plasticity between MØ and moDC, or the selective expansion of a committed precursor under favorable conditions of culture. While GM-CSF is extensively used in supporting myelopoesis in vitro, the role of GM-CSF in vivo remains obscure. GM-CSF deficiency has little impact on myeloid cells except for the impairment of alveolar MØs in vivo (7–10). Nevertheless, in transgenic mice harboring high levels of GM-CSF (GM-CSF-Tg), myelopoiesis is substantially increased (11, 12).
While the importance of GM-CSF for myelopoiesis in vivo remains a matter of debate, there is cogent evidence that GM-CSF is an important mediator in inflammatory conditions such as during infection and tumor immunity (13–16). These studies suggest a role for GM-CSF in regulating biological functions of fully mature cells. Studies on GM-CSF have mainly focused on its pro-inflammatory role. Nevertheless, GM-CSF has also been linked to immuno-suppression, particularly in tumor setting. Thus, exposure of myeloid cells to GM-CSF can lead to sharp opposite extremes, and these contrasting effects of GM-CSF on myeloid cells remains hitherto unexplained.
The GM-CSF receptor (GM-CSFR) is composed of a ligand-specific alpha chain and a beta chain common with IL-3 and IL-5. Despite sharing this signaling beta chain, IL-3 or IL-5 engagement leads to distinct signaling events and myeloid cell outcomes (17). For example, IL-3 is mostly associated with differentiation of mast cells/basophils, while IL-5 is associated with differentiation of eosinophils (17). GM-CSFR is found on most myeloid cells including their precursors. Upon engagement, GM-CSFR elicits JAK2 phosphorylation, which triggers multiple intracellular signaling pathways, including STAT5, PI3K, and MAPK (15, 18). Of note, GM-CSF can selectively turn on signaling modules in a dose-dependent fashion, and can therefore differentially impact cell survival, proliferation, and differentiation at different doses (15, 18–20). GM-CSF has been shown to activate and/or upregulate many transcriptional factors such as the STAT proteins, PU.1 and interferon regulatory factors (IRFs) (18). Such factors have been implicated in the differentiation and function fate determination of myeloid cells, but it is not clear how induction and function of these transcription factors are linked to GM-CSF signaling strength.
Apart from GM-CSF abundance, GM-CSF signaling strength can be influenced by multiple factors, including post-translational modification. For example, glycosylated GM-CSF has less immunogenicity and greater in vivo pharmacokinetic availability than its non-glycosylated form Gribben et al. (21). Nevertheless, glycosylation of GM-CSF is not required for its biologic activity in vitro (22). In contrast, the GM-CSF receptor α subunit requires N-glycosylation for binding and signaling (23, 24). Thus, it has been speculated that glycosylation of the α subunit may modulate cellular responsiveness to GM-CSF (24). In addition, GM-CSF receptor signaling can also be regulated by the suppressors of cytokine signaling proteins (SOCS family members). However, the consequences of SOCS signaling in controlling GM-CSFR signaling strength and therefore myeloid cell differentiation and/or function have been little explored.
In this review, we will highlight the dynamic changes in GM-CSF quantity in different pathological situations and dose-dependent differences in the biological response to GM-CSF, ranging from immunostimulating to immunosuppressive. We dissect the differential impact of GM-CSF on the main types of myeloid cells. As the upstream events of GM-CSF signaling and the inflammatory biological outcomes have been reviewed elsewhere (17, 20), we will highlight the potential role for negative feedback regulators on GM-CSF signal strength and downstream transcriptional factors that influence myeloid differentiation trajectory and function (Figure 1). Furthermore, we will discuss the contribution of PI3K and downstream NFκB pathways upon GM-CSF engagement in controlling not only myeloid cell survival (19) but also macrophage polarization via the differential involvement of Akt1 and Akt2 subunits (25). Finally, we also discuss the role of GM-CSF in regulating end-cell function, particularly functional polarization of MØs. In the process, we aim to shed some light on the paradoxical role of GM-CSF in immune regulation and facilitate the agonistic and antagonistic targeting of GM-CSF as an immuno-intervention in infection, inflammation, and cancer. As this review covers mouse and human studies, we have indicated the species when human GM-CSF is discussed.
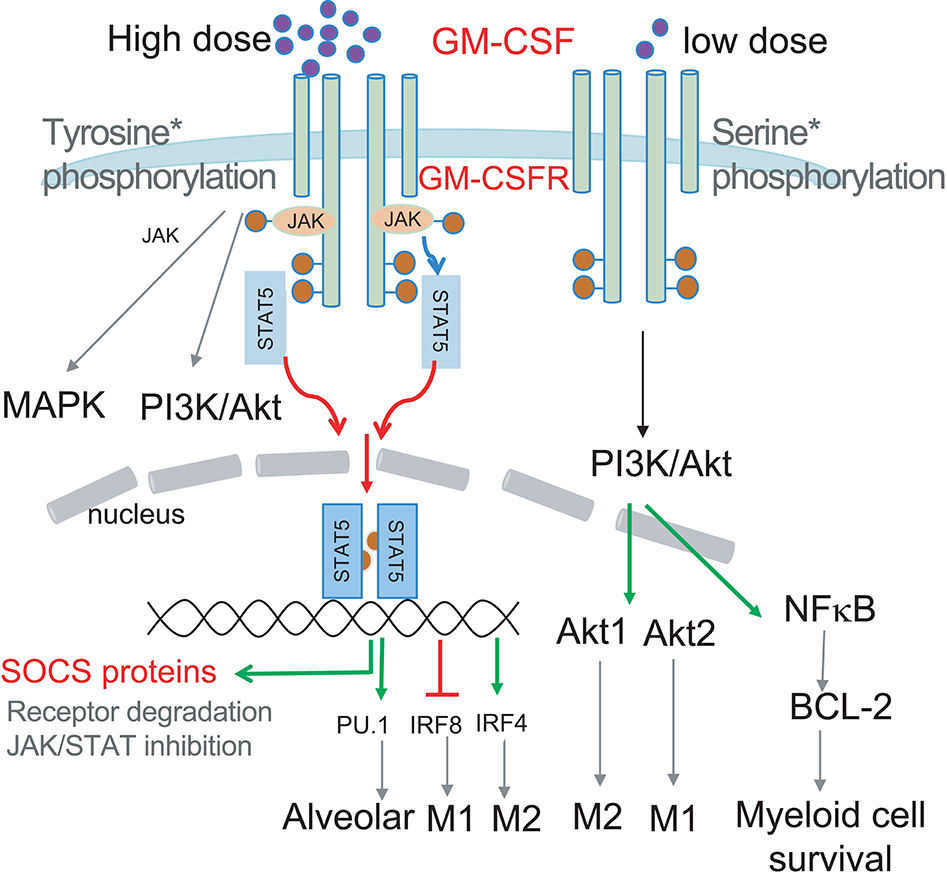
Figure 1. Schematic illustrating how GM-CSF dose selects the signal modules to be activated. Low dose GM-CSF signaling triggers Ser phosphorylation of the GM-CSFR beta chain, PI3K/Akt activation and subsequent BCL-2 enhanced survival. High dose signaling triggers Tyr phosphorylation of the GM-CSFR beta chain resulting in JAK/STAT5 activation, leading to upregulation of transcription factors PU.1 and IRF4, and downregulation of IRF8 to impact differentially on myeloid cell differentiation and function. JAK2/STAT5 activation by GM-CSF could also induce transcription of SOCS proteins to negatively regulate signaling to form a signaling regulatory loop. PI3K activation can also contribute to MØ polarization via preferential activation of Akt1 and Akt2.
The amount of GM-CSF is likely to be a key factor in determining its biological activity (19, 26). Thus, we will briefly describe the main sources of GM-CSF. A diverse set of hematopoietic and non-hematopoietic cells have been shown to secrete GM-CSF. They include T cells (27–30), human natural killer cells (31), mast cells (32), monocytes/MØs (33), human endothelial cells (34), and human fibroblasts (35). The relative contribution of each individual subset to the overall GM-CSF produced under steady-state or inflammatory conditions has not been determined. In the lung, production of GM-CSF by endothelial cells in the steady state was instrumental in the differentiation of alveolar MØs from fetal monocytes (10, 36). Under inflammatory conditions, such as collagen induced arthritis and experimental autoimmune encephalomyelitis (EAE), the production of GM-CSF by T cells has been reported to promote disease progression (28–30), although there is contention about the role of GM-CSF in EAE pathology (37). On the other hand, GM-CSF derived from radio-resistant wild-type cells in GM-CSF−/− bone marrow reconstituted irradiation chimera was sufficient to confer resistance to infection with Mycobacterium tuberculosis (38). GM-CSF is often used in the range of 10–20 ng/ml for in vitro myeloid cell differentiation (2–4, 39). It raises the question—what levels of GM-CSF can be reached in vivo? In physiological situations, concentrations of around 20 pg/mL of GM-CSF could be detected in human serum (40). Under pathological conditions, human GM-CSF was found to be significantly elevated in the serum and tissues in inflammatory diseases such as rheumatoid arthritis and colitis (41–43). GM-CSF increase was also observed in mice following LPS administration (44) and during bacterial infection (45). Notably, GM-CSF quantity can reach and persist at >10 ng per lung of mice infected with M. tuberculosis (38). When human GM-CSF was used for myeloid recovery after chemotherapy and bone marrow transplantation, patients were given with >32 μg/kg body per day for 14 days (46).
It has long been appreciated that tumor cells can produce a variety of cytokines and chemokines (47). The Broad Institute cancer cell line encyclopedia database (https://portals.broadinstitute.org/ccle) shows that a broad spectrum of solid tumor cell lines express human GM-CSF mRNA. For example, tumor cells from the kidney, pancreas and gastrointestinal tract displayed prominent GM-CSF transcription. Concordantly, an early study showed that about a third of the 75 human tumor lines tested secreted GM-CSF; this comprised a large proportion of lines from renal, prostate and colon cancers and a modest proportion of breast, cervical, ovarian and melanoma cancers (47). Indeed, 105 W-RCC renal cancer cells produced a remarkable 39 ng/mL after 16 h in culture (47). A mouse renal tumor line RenCa also produced about 0.5 ng GM-CSF/106 cells/24 h (48). In another study, a panel of mouse pancreatic ductal adenocarcinoma (PDA) tumor cell lines all produced GM-CSF (60–500 pg/mL) while benign pancreatic ductal cells did not (49). These results indicate that GM-CSF production by human and mouse tumor cells may not be uncommon.
Several reviews have described that GM-CSF has a profound immune regulatory role in health and disease (13–17). Here we briefly discuss the role of GM-CSF in tumor, autoimmunity/inflammation, and infection, with the aim to contrast the opposite roles of GM-CSF in immune regulation.
The use of murine tumor cells genetically modified to secrete cytokines has established GM-CSF as a strong immune adjuvant for vaccination to promote anti-tumor immunity (50). In a vaccination setting, Zarei et al. showed that tumor derived GM-CSF was sufficient to recruit DCs to the vaccination site in murine tumor models, thereby promoting a strong anti-tumor response and protecting from further tumor challenge (48). Hence, clinical trials using human GM-CSF as an immune adjuvant in cancer patients have been conducted with some promising outcomes (51–53). However, the use of human GM-CSF at high doses may lead to advert events such as immunosuppression (54). In mouse models, tumor derived GM-CSF has also been shown to promote the development of myeloid derived suppressor cells (49). Consequently, neutralization of GM-CSF has also been shown to reduce suppressive cells and limit tumor growth (49). Furthermore, tumor derived GM-CSF can also act in an autocrine manner to sustain tumor growth (55). Thus, GM-CSF secretion within the cancerous tissue may have very contrasting effects on either promoting anti-tumor immunity, suppressing anti-tumor immunity or promoting tumor growth directly. It is likely that the temporal and spatial abundance of GM-CSF, together with the machinery controlling GM-CSF signal strength including receptor expression and regulatory circuitry would dictate the cellular and biological outcome of tumor derived GM-CSF.
Evidence that GM-CSF is pro-inflammatory in several autoimmune diseases comes from various studies: (1) treatment with human GM-CSF to correct neutropenia results in flare-ups of rheumatoid arthritis (56, 57); (2) human GM-CSF was present in lesions of rheumatoid arthritis (41); and the cerebrospinal fluid of MS patients (58); (3) GM-CSF deficiency confers resistance to experimental collagen induced arthritis (59) and EAE (60) in mouse models. In line with the above studies, anti-GM-CSF mAb treatment was found to be effective at ameliorating the ensuing disease in mouse models, partly by reducing myeloid cell infiltration (61, 62). In clinical trials, anti-human GM-CSF mAb namilumab and MOR103 demonstrated evidence of efficacy in active rheumatoid arthritis (63, 64). Similarly, human trials of anti-GM-CSF receptor α mAb Mavrilimumab on rheumatoid arthritis had also been shown to reduce disease activity (65, 66).
However, GM-CSF is not always detrimental in autoimmune settings and has also been shown to be beneficial via the suppression of undesired immune responses (67). The supporting evidence includes: (1) treatment with human GM-CSF ameliorates Crohn's disease (68); (2) GM-CSF prevents diabetes development in NOD mice by promoting immature tolerogenic DCs and controlling the number of regulatory T cells (69); (3) GM-CSF deficiency in mouse results in the development of lupus-like disorder (70) while combined deficiency of GM-CSF and IL-3 results in the development of autoimmune diabetes (71). The cellular and molecular basis for these beneficial effects of GM-CSF is not clear. As discussed in a recent review (67), there are at least two potential mechanisms for GM-CSF to suppress autoimmunity. Firstly, GM-CSF can induce DCs and macrophages to activate antigen-specific Tregs and suppresses experimental autoimmune disease in autoimmune thyroiditis (72). GM-CSF-autoantigen conjugates had been found to be particularly effective to expand Tregs in an EAE model (73). GM-CSF can even directly expand in vitro induced Tregs to suppress disease development in a cell transfer model of type 1 diabetes (74). Secondly, GM-CSF can induce the production of monocytes with suppressive functions that dampen disease induction and severity in an IRF1 dependent fashion (75). Beyond autoimmunity, MØs can also be detrimental or beneficial to graft tolerance in organ transplantation (76). In such a context, it is interesting to note that GM-CSF mediates graft-vs. -host disease but not graft-vs. -leukemia responses, suggesting an intervention opportunity targeting GM-CSF in allogenic hematopoietic cell transplantation (77).
Studies in mice deficient in GM-CSF and GM-CSFR have highlighted the critical role for GM-CSF and its receptor in maintaining alveolar MØs in the lung (7, 10, 36, 78). Many studies have established that GM-CSF has a non-redundant role in promoting anti-pathogen immunity. Deficiency in GM-CSF reduced emergency myelopoiesis and reduced Listeria and M. tuberculosis protection in mice (79, 80). Concordantly, GM-CSF treatment enhanced protective immunity against infection with M. tuberculosis and Salmonella typhimurium (81, 82). GM-CSF also promoted resistance against various parasite infections including blood-stage malaria (83), trypanosomiasis (84), and leishmaniasis (85). Interestingly, the combined blockade of GM-CSF and IL-3 prevented the development of cerebral malaria (86). Notably, infection in human and mouse models can also lead to immunosuppression (87–91). Unfortunately, although these studies indicated an association with the generation of immunosuppressive myeloid cells, full understanding on how GM-CSF shapes immunosuppression remains elusive.
The exposure of bone marrow progenitors to GM-CSF leads to the production of two functionally distinct myeloid cells: PMNs and MØs. What determines the deviation to PMN vs. MØ pathway? In early studies using in vitro agarose cultures, high GM-CSF concentrations favored PMNs differentiation, whereas low concentrations favored MØ differentiation (26, 92); this effect was termed “differentiation downgrading.” Interestingly, a recent article has provided a mathematical interpretation for this observation, enabling the reproduction of the concentration dependent pattern of GM-CSF induced differentiation based on induction of key transcriptional factors controlling lineage commitment (93). However, when GM-CSF signaling strength that is represented by both GM-CSF quantity and receptor density is high over time, monopoiesis is favored over granulopoiesis (93). In line with this predictive model, our recent data showed that high dose GM-CSF favored monopoiesis over granulopoiesis in vitro (5). Similarly, GM-CSF transgenic mice had preferential expansion of MØs in multiple organs (5, 11, 12). Consistent with the findings above, van Nieuwenhuijze et al. described increased MØs compared to PMNs in transgenic mice expressing high level of GM-CSF (12). Conceivably, GM-CSF signal strength is not only reflected by the ratio GM-CSFR:GM-CSF but also by intracellular mechanism controlling GM-CSF signaling. We contend that all these factors ultimately play a critical role in determining myeloid cell differentiation.
Human PMNs rapidly lose viability in culture (94). Human GM-CSF but not G-CSF, IL-6, and IL-8 prevented apoptosis of PMNs, prolonging in vitro survival (94). Of note, despite sharing the βc receptor with GM-CSF, IL-3 did not improve cell survival, likely due to low expression of IL-3 receptor on mature PMNs (94, 95). We observed that the addition of small quantities of GM-CSF in vitro (80 pg/mL) can lead to substantially increased survival of murine blood PMNs (5). Interestingly, a detailed analysis of the signaling pathway induced by such low levels of GM-CSF have shown that it was sufficient to activate Ser585 of the GM-CSFR, thereby promoting downstream signaling events, in particular the PI3K-Akt pathway, that led to increased cell survival (19, 96). As pro-survival members of the BCL-2 family including BCL-2, BCL-xL, A1, MCL-1, and BCL-w have a key role in maintaining the viability of most immune cells (97), the precise contribution of individual molecules to PMN survival, specifically GM-CSF enhanced PMN survival, is unclear. Human GM-CSF has been shown to increase expression of BCL-2 but not BCL-xL in one study (96) while it increased BCL-xL transcription in another study (98). Functionally, antagonism of BCL-2 or BCL-xL has had some effects on mouse and human neutrophil count in vivo (99, 100). Similarly, A1, identified as a GM-CSF induced molecule (101), showed a pro-survival role for PMN in some studies (5, 102) but not in the most definitive study where all the functional A1 genes were ablated (103). In addition, human GM-CSF could promote granulocyte survival by maintaining MCL-1 stability (104). It is somewhat puzzling that human GM-CSF can also induce the expression of the pro-apoptotic BCL-2 family member Bim in human and mouse PMNs via a PI3K dependent fashion (105). Compared to PMNs, monocytes/MØs had better spontaneous survival in culture, and survival enhancement by GM-CSF was less remarkable than the effect observed on PMNs (5). The loss of either MCL-1 or A1 has a limited effect on murine monocyte/ MØ survival (103, 106). Overall, GM-CSF has a prominent role in promoting survival of myeloid cells. However, the molecular events responsible for the differential survival properties observed for PMNs and monocytes/MØs, with or without GM-CSF remain ill explained. Furthermore, there is little known about the role of GM-CSF in regulating multiple non-BCL-2 regulated cell death pathways including death-receptor regulated apoptosis, necroptosis and autophagy.
GM-CSF is routinely used to generate large numbers of dendritic cells from mouse bone marrow or human monocyte cultures (2, 3, 107). Yet recently, CD11c+ mononuclear cells generated in the former culture were found to contain two main populations: CD11c+MHCIIintCD11bhi CD115hiFlt3− MØs and a MHCIIhiCD11bint cell fraction enriched for Ftl3+ DCs (4). MØs and DCs within CD11c+ mononuclear cells not only differ in their gene signature but also function (4). MØs have a high capacity for producing proinflammatory cytokines while DCs have a high capacity for presenting antigens (4). In addition, recent evidence highlighted that the inflammasome activity of such cultures was due to MØs, not DCs (108).
Ontogeny analyses elegantly showed that macrophage-dendritic precursors, common monocyte progenitors, common dendritic cell progenitors, and Ly6Chigh monocytes can all become MØs or DCs, with different expansion and differentiation rates (4). Of note, Flt3+CD11c− MHCII+ PU.1hi cells within the Ly6C+ monocyte subset have been identified as precursors of GM-CSF dependent moDCs (109). Notwithstanding, there are still many unanswered questions regarding the conditions determining the differentiation fate of MØs and DCs.
GM-CSF signal strength is the net result of GM-CSF quantity, GM-CSFR expression level and positive/negative regulatory circuitry controlling GM-CSF signaling. Most in vitro studies use a range of 5–20 ng/mL GM-CSF to drive DC differentiation, with variation in cell density and culture duration. It had been shown that low dose of GM-CSF promotes the development of immature DCs featuring tolerogenic function (110). Using the recent definition of MØs and DCs within CD11c+ cells generated in GM-CSF culture (4), we and others noticed that an intermediate dose of GM-CSF favored moDC differentiation while higher doses of GM-CSF favored macrophage differentiation (5, 111). As alluded to earlier, the GM-CSFR could work as a binary switch: low doses of GM-CSF led to Ser phosphorylation, whereas high doses led to Tyr phosphorylation and STAT5 activation (19). However, it remains unclear on how this binary switch contributes to DC and MØ differentiation.
In addition to the interpretation of the abundance of the ligand, the GM-CSF induced signaling cascade can be regulated by negative regulators of cytokine signaling. One such example is the degradation of GM-CSFR through SOCS1 mediated by ubiquitination (112). Yet, the consequences of SOCS1-mediated GM-CSFR downregulation has not been examined in the context of DC differentiation. In response to GM-CSF, myeloid cells are induced to express another member of the SOCS family, CISH (113–115). CISH knockdown by shRNA was shown to impede GM-CSF-induced DC development and DC function (115). However, as authors demonstrated that CISH knockdown suppressed precursor cell proliferation, it is still unclear if CISH knockdown can directly impact on the differentiation of MØs and DCs.
Taken together, we speculate that GM-CSF induced signaling strength dictates cellular outcome, with moderate GM-CSF signaling strength enabling DC differentiation while strong GM-CSF signaling strength favors MØ differentiation.
Even at the monocyte stage when cell proliferation is very limited (4), human and mouse GM-CSF, particularly with IL-4, can differentiate human and mouse monocytes into DCs (4, 107, 109). It raises the question of whether IL-4 alters the differentiation fate for cells destined to become MØs in its absence, implying a certain degree of fate plasticity within that compartment. Consistent with the idea of a certain degree of plasticity, IL-4, through the activation of the transcription factor STAT6, has recently been shown to induce demethylation of genes favoring DC differentiation and enforced STAT6 activation in the absence of IL-4 also favors DC differentiation (116). Interestingly, the transcription factor PU.1 has been shown to be required for the induction of STAT6-mediated transcription (117) and to promote DC generation from monocytes while inhibiting MØ production (109). Thus, PU.1 and STAT6 could abet terminal DC development. However, individual STAT proteins seldom act in isolation such that functional balance between multiple STAT proteins is important to determine cell differentiation (118). Interestingly, the effects of IL-4 on GM-CSF induced DC differentiation was shown to be dependent on the dose of both IL-4 and GM-CSF (119), suggesting that differentiation trajectories are dependent on the signal strength of both cytokines. Of note, IL-4 not only altered the differentiation trajectory under GM-CSF but also increased APC function of generated dendritic cells (120). IL-4 induced the expression of IRF4 that was not only critically required for DC differentiation, but also for their antigen cross-presentation capacity and the expression of costimulatory molecules (120).
An IL-4 related Th2 cytokine IL-13 has also been shown to enhance GM-CSF stimulated DC differentiation from mouse bone marrow cells (119) and human monocytes (121, 122), although the potency and action of IL-4 and IL-13 may differ. Furthermore, TNF-α and LPS added at a late stage of bone marrow cell culture with GM-CSF have also been shown to promote DC differentiation/maturation (2, 3). At least for TNF, multiple STAT proteins including STAT6 can be activated upon stimulation. Overall, there is considerable plasticity for GM-CSF induced differentiation of mononuclear cells, subject to the conditions that activate signaling modules favoring either DC or MØ differentiation.
Despite the strong potency of GM-CSF to induce DC differentiation in vitro, GM-CSF and its receptor are redundant for the differentiation of moDCs in vivo, at least during acute infection and inflammation (9, 123, 124). It could be that infection and inflammation induce high levels of many cytokines including M-CSF and TNF-α that could influence moDC differentiation and therefore mask the role of GM-CSF. In situations where GM-CSF concentration increase is more selective (e.g., GM-CSF overexpression or engraftment of a GM-CSF-producing tumor) (109, 125), GM-CSF seems to have a positive role in inducing moDC differentiation. In an EAE model with Th17 transfer, GM-CSFR−/− moDC infiltrates in CNS tissue were significantly reduced in a competitive scenario (126). Our view is that GM-CSF is sufficient but not essential for production of moDCs in vivo. Its importance on moDCs in vivo may instead be more critical for their effector function (see below).
Many decades of work have established that the dendritic cell network is heterogenous and consists of many subsets with different phenotypic and functional features (127–129). DCs, excluding moDCs, have recently been categorized into three groups: cDC1s (for both CD8+ and CD103+ DCs), cDC2s (for CD11b+ and CD172α+), and pDCs (130). Despite the differentiation of these cells being largely independent of GM-CSF, GM-CSF has pleiotropic impacts on all these DC subsets. In Flt3L-supplemented cultures of bone marrow cells, inclusion of low dose GM-CSF (0.3 ng/mL) increased the production of cDC1s, cDC2s, and pDCs, while neutralization of endogenous GM-CSF reduced all DC generation (131). Similar findings have also been derived in vivo, particularly in mice with combined loss of GM-CSF and Flt3L (132). Enhancement of overall DC differentiation by GM-CSF is likely due to the positive effect of GM-CSF on progenitor commitment to myeloid lineages and expansion of such progenitors. However, at high doses of GM-CSF, development of cDC1s and pDCs under Flt3L stimulation was severely hampered (133, 134). At least for pDCs, it was shown that strong GM-CSF signaling leads to strong STAT5 activation and suppression of IRF8 transcription, which is critical for pDC differentiation (134). cDC1s include both lymphoid CD8+ DCs and tissue CD103+CD207+ migratory DCs (130). Even though CD8+ DCs were reduced in GM-CSF transgenic mice, the number of CD103+ DCs was increased in GM-CSF transgenic mice (135), indicating subtle differences in the two types of cDC1s differentiated at different locations. Apart from the impacts on differentiation and DC cell survival discussed above, GM-CSF has also been shown to increase the cross-presentation properties of cDC1s both in vitro and in vivo (131, 136). Functional enhancement of cDC1s by GM-CSF is also associated with an increase in CD103 expression (131, 136). However, expression of CD103 per se is not sufficient for acquisition of cross-presentation capacity as TGF-β increased CD103 expression but not cross-presentation of cDC1s (131). Together, GM-CSF has a broad impact not only on the processes driving DC differentiation but also affects DC effector function at the mature state. Once again, the nature and the extent of these GM-CSF induced changes may be greatly affected by the relative abundance of GM-CSF, the state of maturity and the microenvironment encountered by the cells.
Despite GM-CSF seeming to be redundant in the development of moDCs in vivo (9, 123, 124), GM-CSF is still required for function of monocytes/MØs in the induction and progression of EAE (123, 124). Here we will discuss the different aspects of impact on MØ function by GM-CSF with the caveats of certain degrees of ambiguity surrounding the definition of monocytes, moDCs and MØs in vivo, and the difficulty delineating the impact of GM-CSF on cell survival and function per se in some studies.
Both GM-CSF and M-CSF can generate MØs in bone marrow cultures. However, after LPS stimulation, the two factors elicit different functions. Human GM-CSF facilitates the differentiation of CD14+ monocytes into IL-23 producing M1 like MØs while M-CSF promotes differentiation of M2 like MØs (137). In murine systems, GM-CSF differentiated bone marrow derived MØs (GM-BMMØs) also produce more IL-12, IL-23, TNF-α, and IL-6 than M-CSF differentiated MØs (BMMØs) (138). Moreover, GM-BMMØs preferentially activated NFκB while BMMØs preferentially activated the IRF3-STAT1 axis (138, 139). From the cytokine pattern elicited, it was proposed that GM-BMMØs is “M1-like” (IL-12hi, IL-23hi, IL-10lo) while BMMØs is “M2-like” (IL-12lo, IL-23lo, IL-10hi) (138). An adoptive transfer study supported this proposal in that GM-BMMØs but not BMMØs induced a Th1 response via IL-12 production and transferred resistance to parasite infection (140). In EAE, GM-CSF responsiveness in CCR2+Ly6Chi monocytes/moDCs was critical for disease pathogenesis, whereas GM-CSF responsiveness in cDCs or PMNs was deemed unimportant (123, 124). Moreover, GM-CSF responsiveness in CCR2+ cells was required for IL-1β production (124), likely from MØs but not DCs (108). Overall, these studies highlight the importance of GM-CSF in priming MØs for production of proinflammatory cytokines under TLR and NLR stimulation and provides an explanation for the adjuvant effect of GM-CSF in cancer, inflammation, and infection, even when numbers of myeloid cells are not affected.
An early study showed that GM-CSF enhanced APC function by increasing IL-1β production and MHC expression (141). We and others had demonstrated that GM-CSF was required for acquisition of cross-presentation capacity by cDC1s (131, 136). Bone marrow precursors cultured with GM-CSF generated CD11c+ cells with modest levels of CD86 and MHC II, particularly in low density cultures, whereas late addition of IL-4 dramatically increased expression of CD86 and MHC II (6). Of note, in vivo treatment with human GM-CSF needed co-administration of IL-4 to enhance APC function (142). These observations suggest that GM-CSF by itself has a limited capacity to up-regulate costimulatory molecules. Consequently, CD11c+ cells derived from GM-CSF cultures alone have a weak capacity to induce T cell proliferation compared with those derived from IL-4 supplemented cultures (6). To complicate the issue, moDCs could also suppress the APC function of cDCs (125). Overall, although GM-CSF promotes APC survival and differentiation fate, it may have limited direct effect on APC function.
In the steady state, a deficiency in GM-CSF or its receptor GM-CSFR led to defective terminal differentiation of alveolar MØs, resulting in impaired surfactant catabolism and pulmonary alveolar proteinosis in both human and mice (8, 143). GM-CSF activated PU.1 to drive this differentiation pathway (144); local delivery of GM-CSF restored PU.1 and corrected the disease (144–146). In GM-CSF transgenic mice, MØs showed increased phagocytic activity and increased production of oxygen degradation products (11, 147). In vitro, GM-CSF primed GM-BMMØs for TLR-stimulated increased nitric oxide and lipid mediator LTB4 production but a reduction in PGE2 (148). In the absence of GM-CSF, MØs had reduced capacity for up-taking apoptotic cells (70).
Although GM-CSF has been viewed predominantly as a pro-inflammatory cytokine and promotes differentiation of M1-like MØs that produce proinflammatory cytokines (137, 138, 149), GM-CSF has also been associated with development of M2-like MØs (47, 49). What then determined the M1-like MØ vs. M2-like MØ fate under GM-CSF stimulation? Evidence from tumor settings indicated that GM-CSF abundance was a key factor in determining cell fate. Production of high levels of GM-CSF by tumor cells led to increased M2 like MØ accumulation within the cancerous tissues, thereby inhibiting T cell response in mouse models of melanoma and pancreatic cancer (47, 49). Conversely, GM-CSF blockade reduced the development of M2 like MØs (49). It remains unclear how GM-CSF drives M2 like MØ differentiation. A study showed that GM-CSF could activate JAK2/STAT5 which in turn suppressed IRF8 transcription (150). Functionally, IRF8 could suppress M2 like MØ differentiation since IRF8 deficiency promoted M2-like MØs differentiation in tumors, while overexpression of IRF8 reduced M2-like MØ accumulation (150). Other transcription factors influenced by GM-CSF signaling in M2-like MØ activity include C/EBPbeta (151) and RORC1 (152). Interestingly, IL-3, a cytokine sharing the signaling receptor with GM-CSF, also promoted prostaglandin E2-producing M2 like MØs in vitro (153).
Apart from difference in cytokine production, mouse M2 MØs express high levels of characteristic markers such as Arginase 1 (Arg1), Chitinase-like 3 (Chil3, YM1), and transglutaminase 2 (Tgm2) (149, 154). These molecules had been demonstrated to mediate immunosuppression, tumor metastasis and tumor growth (155, 156). While excess GM-CSF has been associated with development of M2 like MØs (47, 49), IL-4 is also known for its potent role in shaping M2 MØ differentiation and confers many functional characteristics of M2 MØs (149). However, when IL-4 was dosed in combination with GM-CSF, M2 MØs could also differentiate into fully functional APCs (47). The coordinate action of GM-CSF and IL-4 in promoting myeloid cell fate decisions remains puzzling. We reasoned that GM-CSF and IL-4 likely instruct distinct signaling modules leading to M2 MØ differentiation. As alluded to above, GM-CSF activated STAT5 which in turn suppressed IRF8, the transcription factor suppressing M2 MØ differentiation (150). On the other hand, IL-4 promoted M2 MØ differentiation via STAT6 activation and IRF4 induction in M-CSF differentiated MØs (157, 158). To complicate the issue, GM-CSF can also induce IRF4 expression in MØs (159). IRF4 also played an important role in deciding DC vs. MØ fate, as a recent study showed that IRF4 deficiency favored MØ differentiation over DC differentiation of monocytes in the presence of IL-4 and GM-CSF (120). Overall, the signaling events emanated from GM-CSF and IL-4, leading to the differentiation of functionally distinctive DCs, M1-like MØs and M2-like MØs, have not been fully defined. In addition to IL-4, another Th2 cytokine IL-13 has been shown to suppress the production of proinflammatory cytokines (160, 161). It seems that both IL-4 and IL-13 acted in a similar fashion via STAT6 activation to modulate MØ function (162).
GM-CSF can activate PI3K and NFκB pathways promoting myeloid cell survival (19) and contributing to lung inflammation (163). However, activation of PI3K pathway can also polarize MØs (25, 75, 164). Of note, the downstream signaling molecules associated with PI3K activity, Akt1, and Akt2, have been shown to have contrasting effects in controlling MØ polarization; while the latter promotes M2 MØs, Akt1 was shown to induce M1 MØ polarization (25). An unanswered question is whether and how GM-CSF, and in particular its signaling strength, promotes differential activation of Akt1 vs. Akt2.
Finally, GM-CSF can also mediate immunosuppression indirectly via promoting Treg induction (165). GM-CSF induces the expression of milk fat globule EGF8 (MFG-E8) that promotes uptake of apoptotic cells by MØs, inducing their production of TGFβ and thereby controlling Treg development (165). Interestingly, TLR stimulation or uptake of necrotic cells was shown to downregulate MFG-E8 expression and to reduce the impact of GM-CSF on MFG-E8 expression, thus preserving the pro-inflammatory action of GM-CSF in tumor immunity (165), suggesting a pathway countering GM-CSF mediated immunosuppression.
Beyond tumors, the influence of GM-CSF on M2 like MØs extends to several inflammatory situations such as autoimmunity (67), infection (166), and transplantation (167). In general, research into the impact of GM-CSF has so far mainly focused on its property to expand myeloid cells. It still remains unclear how GM-CSF steers macrophage function to M1 MØs vs. M2 MØs. While M2 MØs may be detrimental in the context of tumor immunity, they may also be beneficial in damping autoimmunity, transplant rejection and infection-associated immunopathology and therefore it is of importance to be better define GM-CSF and its signaling components as this may avail therapeutic targets during M2 MØs development and function.
GM-CSF is produced by many cells and its receptor is broadly expressed by hematopoietic cells. Engagement of GM-CSFR activates multiple signal pathways in a dose dependent manner to impact on multiple cellular processes including survival, proliferation, differentiation and function of multiple myeloid cells. Due to its promiscuous properties, GM-CSF roles in controlling pro-inflammatory or anti-inflammatory processes in healthy or diseased individuals are often complex and paradoxical. We opine that GM-CSF signaling strength likely determines biological outcome (Figure 2). At the cellular level, it drives differentiation of different cell subsets by activating different signaling modules. At the functional level, it programs antigen presentation capacity, proinflammatory function and suppressive function. Ultimately, these cellular changes will impact immunity and immunopathology in different disease settings.
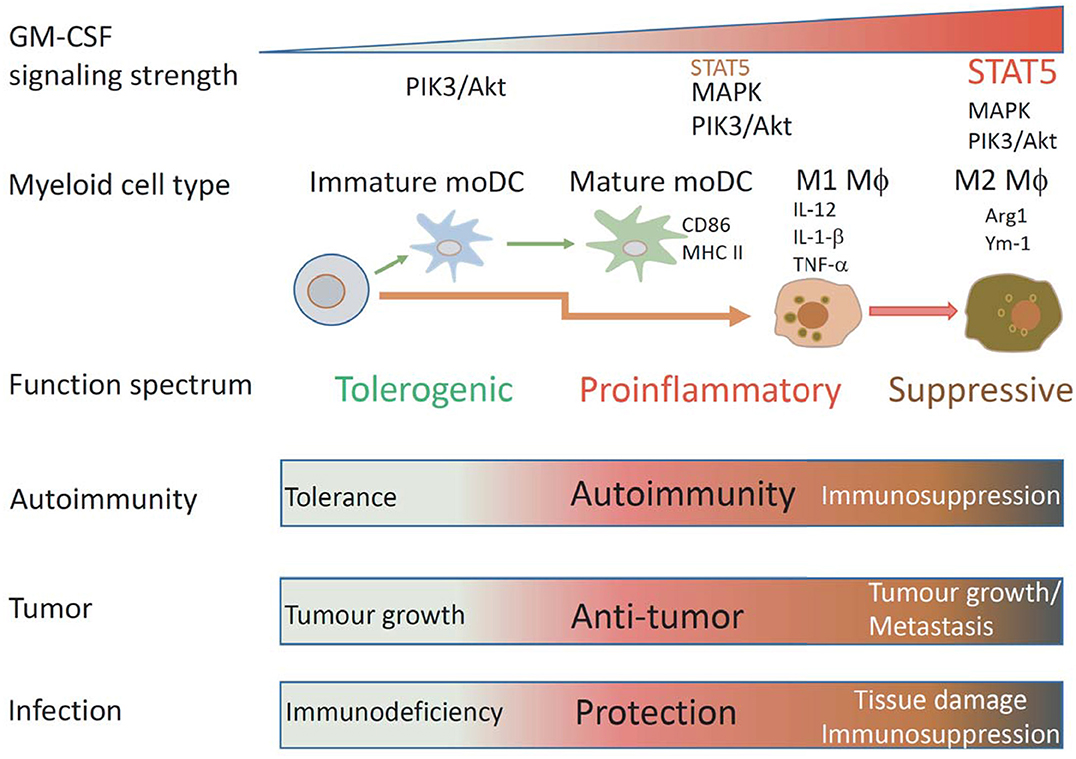
Figure 2. Schematic illustrating how GM-CSF signaling strength affects mononuclear myeloid cell differentiation and function. Under different GM-CSF signaling strength, different types of mononuclear myeloid cells with different functional properties are differentiated. Low GM-CSF signaling strength favors development of immature DCs, intermediated signaling strength favors development of MHCIIhiCD86hi mature DCs, high signaling strength favors development of proinflammatory M1 MØs, ultra-high signaling strength favors development of suppressive M2 MØs. According to these properties, these cells could have particular impacts on immunity to autoantigens, tumors, and infection.
In the tumor setting, relatively low to moderate doses of GM-CSF favored the immune adjuvant activity, while high doses of endogenous tumor-derived or exogenous GM-CSF could expand M2 like suppressor cells (54). GM-CSF also directly or indirectly expanded Tregs (67). For the latter, blocking GM-CSF could improve anti-tumor immunity (49). As GM-CSF mediated graft-vs.-host disease but not graft-vs.-leukemia response (77), blocking GM-CSF and receptor signaling could be also beneficial. Beyond ligand abundance, downstream signaling responsible for different cell fates should also be explored as intervention targets. Individual IRF members and Akt subunits have differential impacts on DCs, M1, and M2 MØs. SOCS family members naturally act as negative regulators as a brake on cytokine signaling. Their action can be potentially targeted to modify monocytic cell differentiation and function. Furthermore, directly targeting suppressive function of M2 MØs may also be considered. Both Arginase 1 and Chil3 are critical for arginine metabolism while arginine availability is key to an optimal T cell immune response (168). Arginase 1 inhibitor L-Norvaline and iNOS2 inhibitor L-NMMA had been found to enhance T cell proliferation (125, 169). It would be interesting to test whether selective targeting of these effector molecules of M2 MØs could enhance the beneficial anti-tumor effect of GM-CSF. In addition, IL-4 and IL-13 can dramatically change the differentiation trajectory of immune cells and their function. Therefore, their potential should also be considered when immune intervention strategies are explored.
In the autoimmune setting, anti-human GM-CSF mAb (63, 64) and anti-human GM-CSF receptor α mAb (65, 66) have also been shown to ameliorate rheumatoid arthritis in clinical trials, reinforcing the work of several decades that GM-CSF is a key proinflammatory cytokine. Yet, it remains unknown whether the tolerogenic roles of GM-CSF including expansion of Tregs (74) and induction of suppressive MØs (75) could also be harnessed. In addition, immunosuppression also occurs in chronic infections in which high levels of GM-CSF can persist (38). Perhaps, antagonism of GM-CSF in such settings could also be beneficial.
In summary, GM-CSF has pleiotropic effects on myeloid cell differentiation and function. The complexity of GM-CSF action provides a challenge but also an opportunity for tailored immune intervention. To fully capitalize on the agonistic and antagonistic effects of GM-CSF as in cancer, inflammation and infection, the differential impact of GM-CSF signaling strength on different target cells should be considered.
YZ, AL, and MC wrote the paper.
This work was supported by National Health and Medical Research Council of Australia (NHMRC) grants (1037321, 1105209, 1143976, 1150425, and 1080321), NHMRC Independent Research Institutes Infrastructure Support Scheme grant (361646), and Victorian State Government Operational Infrastructure Support grant.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We acknowledge the Wurundjeri people of the Kulin nation as the traditional owners and custodians of the land on which most of the work was performed. We thank Michael Zhan for his careful proofreading the manuscript.
1. Metcalf D. The molecular biology and functions of the granulocyte-macrophage colony-stimulating factors. Blood. (1986) 67:257–67. doi: 10.1182/blood.V67.2.257.257
2. Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. (1992) 176:1693–702. doi: 10.1084/jem.176.6.1693
3. Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, et al. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. (1999) 223:77–92. doi: 10.1016/S0022-1759(98)00204-X
4. Helft J, Bottcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, et al. GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c(+)MHCII(+) macrophages and dendritic cells. Immunity. (2015) 42:1197–211. doi: 10.1016/j.immuni.2015.05.018
5. Sun L, Rautela J, Delconte RB, Souza-Fonseca-Guimaraes F, Carrington EM, Schenk RL, et al. GM-CSF quantity has a selective effect on granulocytic vs. monocytic myeloid development and function. Front Immunol. (2018) 9:1922. doi: 10.3389/fimmu.2018.01922
6. Jin D, Sprent J. GM-CSF culture revisited: preparation of bulk populations of highly pure dendritic cells from mouse bone marrow. J Immunol. (2018) 201:3129–39. doi: 10.4049/jimmunol.1800031
7. Dranoff G, Crawford AD, Sadelain M, Ream B, Rashid A, Bronson RT, et al. Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science. (1994) 264:713–6. doi: 10.1126/science.8171324
8. Stanley E, Lieschke GJ, Grail D, Metcalf D, Hodgson G, Gall JA, et al. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci USA. (1994) 91:5592–6. doi: 10.1073/pnas.91.12.5592
9. Greter M, Helft J, Chow A, Hashimoto D, Mortha A, Agudo-Cantero J, et al. GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity. (2012) 36:1031–46. doi: 10.1016/j.immuni.2012.03.027
10. Schneider C, Nobs SP, Kurrer M, Rehrauer H, Thiele C, Kopf M. Induction of the nuclear receptor PPAR-gamma by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol. (2014) 15:1026–37. doi: 10.1038/ni.3005
11. Lang RA, Metcalf D, Cuthbertson RA, Lyons I, Stanley E, Kelso A, et al. Transgenic mice expressing a hemopoietic growth factor gene (GM-CSF) develop accumulations of macrophages, blindness, and a fatal syndrome of tissue damage. Cell. (1987) 51:675–86. doi: 10.1016/0092-8674(87)90136-X
12. van Nieuwenhuijze AE, Coghill E, Gray D, Prato S, Metcalf D, Alexander WS, et al. Transgenic expression of GM-CSF in T cells causes disseminated histiocytosis. Am J Pathol. (2014) 184:184–99. doi: 10.1016/j.ajpath.2013.09.014
13. Jinushi M, Hodi FS, Dranoff G. Enhancing the clinical activity of granulocyte-macrophage colony-stimulating factor-secreting tumor cell vaccines. Immunol Rev. (2008) 222:287–98. doi: 10.1111/j.1600-065X.2008.00618.x
14. Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. (2008) 8:533–44. doi: 10.1038/nri2356
15. Becher B, Tugues S, Greter M. GM-CSF: from growth factor to central mediator of tissue inflammation. Immunity. (2016) 45:963–73. doi: 10.1016/j.immuni.2016.10.026
16. Wicks IP, Roberts AW. Targeting GM-CSF in inflammatory diseases. Nat Rev Rheumatol. (2016) 12:37–48. doi: 10.1038/nrrheum.2015.161
17. Dougan M, Dranoff G, Dougan SK. GM-CSF, IL-3, and IL-5 family of cytokines: regulators of inflammation. Immunity. (2019) 50:796–811. doi: 10.1016/j.immuni.2019.03.022
18. van de Laar L, Coffer PJ, Woltman AM. Regulation of dendritic cell development by GM-CSF: molecular control and implications for immune homeostasis and therapy. Blood. (2012) 119:3383–93. doi: 10.1182/blood-2011-11-370130
19. Guthridge MA, Powell JA, Barry EF, Stomski FC, McClure BJ, Ramshaw H, et al. Growth factor pleiotropy is controlled by a receptor Tyr/Ser motif that acts as a binary switch. EMBO J. (2006) 25:479–89. doi: 10.1038/sj.emboj.7600948
20. Hercus TR, Thomas D, Guthridge MA, Ekert PG, King-Scott J, Parker MW, et al. The granulocyte-macrophage colony-stimulating factor receptor: linking its structure to cell signaling and its role in disease. Blood. (2009) 114:1289–98. doi: 10.1182/blood-2008-12-164004
21. Gribben JG, Devereux S, Thomas NS, Keim M, Jones HM, Goldstone AH, et al. Development of antibodies to unprotected glycosylation sites on recombinant human GM-CSF. Lancet. (1990) 335:434–7. doi: 10.1016/0140-6736(90)90665-R
22. Kim J, Park H, Park BT, Hwang HS, Kim JI, Kim DK, et al. O-glycans and O-glycosylation sites of recombinant human GM-CSF derived from suspension-cultured rice cells, and their structural role. Biochem Biophys Res Commun. (2016) 479:266–71. doi: 10.1016/j.bbrc.2016.09.057
23. Ding DX, Vera JC, Heaney ML, Golde DW. N-glycosylation of the human granulocyte-macrophage colony-stimulating factor receptor alpha subunit is essential for ligand binding and signal transduction. J Biol Chem. (1995) 270:24580–4. doi: 10.1074/jbc.270.41.24580
24. Niu L, Heaney ML, Vera JC, Golde DW. High-affinity binding to the GM-CSF receptor requires intact N-glycosylation sites in the extracellular domain of the beta subunit. Blood. (2000) 95:3357–62. doi: 10.1182/blood.V95.11.3357
25. Arranz A, Doxaki C, Vergadi E, Martinez de la Torre Y, Vaporidi K, Lagoudaki ED, et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc Natl Acad Sci USA. (2012) 109:9517–22. doi: 10.1073/pnas.1119038109
26. Burgess AW, Camakaris J, Metcalf D. Purification and properties of colony-stimulating factor from mouse lung-conditioned medium. J Biol Chem. (1977) 252:1998–2003.
27. Kelso A, Owens T. Production of two hemopoietic growth factors is differentially regulated in single T lymphocytes activated with an anti-T cell receptor antibody. J Immunol. (1988) 140:1159–67.
28. Campbell IK, van Nieuwenhuijze A, Segura E, O'Donnell K, Coghill E, Hommel M, et al. Differentiation of inflammatory dendritic cells is mediated by NF-κB1-dependent GM-CSF production in CD4 T cells. J Immunol. (2011) 186:5468–77. doi: 10.4049/jimmunol.1002923
29. Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. (2011) 12:560–7. doi: 10.1038/ni.2027
30. El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. (2011) 12:568–75. doi: 10.1038/ni.2031
31. Zhang AL, Colmenero P, Purath U, Teixeira de Matos C, Hueber W, Klareskog L, et al. Natural killer cells trigger differentiation of monocytes into dendritic cells. Blood. (2007) 110:2484–93. doi: 10.1182/blood-2007-02-076364
32. Wodnar-Filipowicz A, Heusser CH, Moroni C. Production of the haemopoietic growth factors GM-CSF and interleukin-3 by mast cells in response to IgE receptor-mediated activation. Nature. (1989) 339:150–2. doi: 10.1038/339150a0
33. Lenhoff S, Sallerfors B, Olofsson T. IL-10 as an autocrine regulator of CSF secretion by monocytes: disparate effects on GM-CSF and G-CSF secretion. Exp Hematol. (1998) 26:299–304.
34. Broudy VC, Kaushansky K, Harlan JM, Adamson JW. Interleukin 1 stimulates human endothelial cells to produce granulocyte-macrophage colony-stimulating factor and granulocyte colony-stimulating factor. J Immunol. (1987) 139:464–8.
35. Zucali JR, Dinarello CA, Oblon DJ, Gross MA, Anderson L, Weiner RS. Interleukin 1 stimulates fibroblasts to produce granulocyte-macrophage colony-stimulating activity and prostaglandin E2. J Clin Invest. (1986) 77:1857–63. doi: 10.1172/JCI112512
36. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. (2013) 210:1977–92. doi: 10.1084/jem.20131199
37. Pierson ER, Goverman JM. GM-CSF is not essential for experimental autoimmune encephalomyelitis but promotes brain-targeted disease. JCI Insight. (2017) 2:e92362. doi: 10.1172/jci.insight.92362
38. Rothchild AC, Stowell B, Goyal G, Nunes-Alves C, Yang Q, Papavinasasundaram K, et al. Role of granulocyte-macrophage colony-stimulating factor production by T cells during Mycobacterium tuberculosis infection. MBio. (2017) 8:e01514-17. doi: 10.1128/mBio.01514-17
39. Xu Y, Zhan Y, Lew AM, Naik SH, Kershaw MH. Differential development of murine dendritic cells by GM-CSF versus Flt3 ligand has implications for inflammation and trafficking. J Immunol. (2007) 179:7577–84. doi: 10.4049/jimmunol.179.11.7577
40. Demirci U, Coskun U, Sancak B, Ozturk B, Bahar B, Benekli M, et al. Serum granulocyte macrophage-colony stimulating factor: a tumor marker in colorectal carcinoma? Asian Pac J Cancer Prev. (2009) 10:1021–4.
41. Xu WD, Firestein GS, Taetle R, Kaushansky K, Zvaifler NJ. Cytokines in chronic inflammatory arthritis. II Granulocyte-macrophage colony-stimulating factor in rheumatoid synovial effusions. J Clin Invest. (1989) 83:876–82. doi: 10.1172/JCI113971
42. Fiehn C, Wermann M, Pezzutto A, Hufner M, Heilig B. [Plasma GM-CSF concentrations in rheumatoid arthritis, systemic lupus erythematosus and spondyloarthropathy]. Z Rheumatol. (1992) 51:121–6.
43. Noguchi M, Hiwatashi N, Liu ZX, Toyota T. Increased secretion of granulocyte-macrophage colony-stimulating factor in mucosal lesions of inflammatory bowel disease. Digestion. (2001) 63(Suppl. 1):32–6. doi: 10.1159/000051908
44. Benmerzoug S, Marinho FV, Rose S, Mackowiak C, Gosset D, Sedda D, et al. GM-CSF targeted immunomodulation affects host response to M. tuberculosis infection. Sci Rep. (2018) 8:8652. doi: 10.1038/s41598-018-26984-3
45. Cheers C, Haigh AM, Kelso A, Metcalf D, Stanley ER, Young AM. Production of colony-stimulating factors (CSFs) during infection: separate determinations of macrophage-, granulocyte-, granulocyte-macrophage-, and multi-CSFs. Infect Immun. (1988) 56:247–51.
46. Brandt SJ, Peters WP, Atwater SK, Kurtzberg J, Borowitz MJ, Jones RB, et al. Effect of recombinant human granulocyte-macrophage colony-stimulating factor on hematopoietic reconstitution after high-dose chemotherapy and autologous bone marrow transplantation. N Engl J Med. (1988) 318:869–76. doi: 10.1056/NEJM198804073181401
47. Bronte V, Chappell DB, Apolloni E, Cabrelle A, Wang M, Hwu P, et al. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J Immunol. (1999) 162:5728–37.
48. Zarei S, Schwenter F, Luy P, Aurrand-Lions M, Morel P, Kopf M, et al. Role of GM-CSF signaling in cell-based tumor immunization. Blood. (2009) 113:6658–68. doi: 10.1182/blood-2008-06-161075
49. Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. (2012) 21:822–35. doi: 10.1016/j.ccr.2012.04.025
50. Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K, et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci USA. (1993) 90:3539–43. doi: 10.1073/pnas.90.8.3539
51. Soiffer R, Lynch T, Mihm M, Jung K, Rhuda C, Schmollinger JC, et al. Vaccination with irradiated autologous melanoma cells engineered to secrete human granulocyte-macrophage colony-stimulating factor generates potent antitumor immunity in patients with metastatic melanoma. Proc Natl Acad Sci USA. (1998) 95:13141–6. doi: 10.1073/pnas.95.22.13141
52. Simons JW, Mikhak B, Chang JF, DeMarzo AM, Carducci MA, Lim M, et al. Induction of immunity to prostate cancer antigens: results of a clinical trial of vaccination with irradiated autologous prostate tumor cells engineered to secrete granulocyte-macrophage colony-stimulating factor using ex vivo gene transfer. Cancer Res. (1999) 59:5160–8.
53. Salgia R, Lynch T, Skarin A, Lucca J, Lynch C, Jung K, et al. Vaccination with irradiated autologous tumor cells engineered to secrete granulocyte-macrophage colony-stimulating factor augments antitumor immunity in some patients with metastatic non-small-cell lung carcinoma. J Clin Oncol. (2003) 21:624–30. doi: 10.1200/JCO.2003.03.091
54. Parmiani G, Castelli C, Pilla L, Santinami M, Colombo MP, Rivoltini L. Opposite immune functions of GM-CSF administered as vaccine adjuvant in cancer patients. Ann Oncol. (2007) 18:226–32. doi: 10.1093/annonc/mdl158
55. Kast RE, Hill QA, Wion D, Mellstedt H, Focosi D, Karpel-Massler G, et al. Glioblastoma-synthesized G-CSF and GM-CSF contribute to growth and immunosuppression: potential therapeutic benefit from dapsone, fenofibrate, and ribavirin. Tumour Biol. (2017) 39:1010428317699797. doi: 10.1177/1010428317699797
56. Hazenberg BP, Van Leeuwen MA, Van Rijswijk MH, Stern AC, Vellenga E. Correction of granulocytopenia in Felty's syndrome by granulocyte-macrophage colony-stimulating factor. Simultaneous induction of interleukin-6 release and flare-up of the arthritis. Blood. (1989) 74:2769–70. doi: 10.1182/blood.V74.8.2769.2769
57. de Vries EG, Willemse PH, Biesma B, Stern AC, Limburg PC, Vellenga E. Flare-up of rheumatoid arthritis during GM-CSF treatment after chemotherapy. Lancet. (1991) 338:517–8. doi: 10.1016/0140-6736(91)90594-F
58. Carrieri PB, Provitera V, De Rosa T, Tartaglia G, Gorga F, Perrella O. Profile of cerebrospinal fluid and serum cytokines in patients with relapsing-remitting multiple sclerosis: a correlation with clinical activity. Immunopharmacol Immunotoxicol. (1998) 20:373–82. doi: 10.3109/08923979809034820
59. Campbell IK, Rich MJ, Bischof RJ, Dunn AR, Grail D, Hamilton JA. Protection from collagen-induced arthritis in granulocyte-macrophage colony-stimulating factor-deficient mice. J Immunol. (1998) 161:3639–44.
60. McQualter JL, Darwiche R, Ewing C, Onuki M, Kay TW, Hamilton JA, et al. Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. J Exp Med. (2001) 194:873–82. doi: 10.1084/jem.194.7.873
61. Cook AD, Braine EL, Campbell IK, Rich MJ, Hamilton JA. Blockade of collagen-induced arthritis post-onset by antibody to granulocyte-macrophage colony-stimulating factor (GM-CSF): requirement for GM-CSF in the effector phase of disease. Arthritis Res. (2001) 3:293–8. doi: 10.1186/ar318
62. Cook AD, Turner AL, Braine EL, Pobjoy J, Lenzo JC, Hamilton JA. Regulation of systemic and local myeloid cell subpopulations by bone marrow cell-derived granulocyte-macrophage colony-stimulating factor in experimental inflammatory arthritis. Arthritis Rheum. (2011) 63:2340–51. doi: 10.1002/art.30354
63. Huizinga TW, Batalov A, Stoilov R, Lloyd E, Wagner T, Saurigny D, et al. Phase 1b randomized, double-blind study of namilumab, an anti-granulocyte macrophage colony-stimulating factor monoclonal antibody, in mild-to-moderate rheumatoid arthritis. Arthritis Res Ther. (2017) 19:53. doi: 10.1186/s13075-017-1267-3
64. Behrens F, Tak PP, Ostergaard M, Stoilov R, Wiland P, Huizinga TW, et al. MOR103, a human monoclonal antibody to granulocyte-macrophage colony-stimulating factor, in the treatment of patients with moderate rheumatoid arthritis: results of a phase Ib/IIa randomised, double-blind, placebo-controlled, dose-escalation trial. Ann Rheum Dis. (2015) 74:1058–64. doi: 10.1136/annrheumdis-2013-204816
65. Burmester GR, McInnes IB, Kremer JM, Miranda P, Vencovsky J, Godwood A, et al. Mavrilimumab, a fully human granulocyte-macrophage colony-stimulating factor receptor alpha monoclonal antibody: long-term safety and efficacy in patients with rheumatoid arthritis. Arthritis Rheumatol. (2018) 70:679–89. doi: 10.1002/art.40420
66. Weinblatt ME, McInnes IB, Kremer JM, Miranda P, Vencovsky J, Guo X, et al. A randomized phase IIb study of mavrilimumab and golimumab in rheumatoid arthritis. Arthritis Rheumatol. (2018) 70:49–59. doi: 10.1002/art.40323
67. Bhattacharya P, Thiruppathi M, Elshabrawy HA, Alharshawi K, Kumar P, Prabhakar BS. GM-CSF: an immune modulatory cytokine that can suppress autoimmunity. Cytokine. (2015) 75:261–71. doi: 10.1016/j.cyto.2015.05.030
68. Korzenik JR, Dieckgraefe BK, Valentine JF, Hausman DF, Gilbert MJ, Sargramostim in Crohn's Disease Study Group. Sargramostim for active Crohn's disease. N Engl J Med. (2005) 352:2193–201. doi: 10.1056/NEJMoa041109
69. Gaudreau S, Guindi C, Menard M, Besin G, Dupuis G, Amrani A. Granulocyte-macrophage colony-stimulating factor prevents diabetes development in NOD mice by inducing tolerogenic dendritic cells that sustain the suppressive function of CD4+CD25+ regulatory T cells. J Immunol. (2007) 179:3638–47. doi: 10.4049/jimmunol.179.6.3638
70. Enzler T, Gillessen S, Manis JP, Ferguson D, Fleming J, Alt FW, et al. Deficiencies of GM-CSF and interferon gamma link inflammation and cancer. J Exp Med. (2003) 197:1213–9. doi: 10.1084/jem.20021258
71. Enzler T, Gillessen S, Dougan M, Allison JP, Neuberg D, Oble DA, et al. Functional deficiencies of granulocyte-macrophage colony stimulating factor and interleukin-3 contribute to insulitis and destruction of beta cells. Blood. (2007) 110:954–61. doi: 10.1182/blood-2006-08-043786
72. Vasu C, Dogan RN, Holterman MJ, Prabhakar BS. Selective induction of dendritic cells using granulocyte macrophage-colony stimulating factor, but not fms-like tyrosine kinase receptor 3-ligand, activates thyroglobulin-specific CD4+/CD25+ T cells and suppresses experimental autoimmune thyroiditis. J Immunol. (2003) 170:5511–22. doi: 10.4049/jimmunol.170.11.5511
73. Moorman CD, Curtis AD II, Bastian AG, Elliott SE, Mannie MD. A GMCSF-neuroantigen tolerogenic vaccine elicits systemic lymphocytosis of CD4(+) CD25(high) FOXP3(+) regulatory T cells in myelin-specific TCR transgenic mice contingent upon low-efficiency T cell antigen receptor recognition. Front Immunol. (2018) 9:3119. doi: 10.3389/fimmu.2018.03119
74. Kared H, Leforban B, Montandon R, Renand A, Layseca Espinosa E, Chatenoud L, et al. Role of GM-CSF in tolerance induction by mobilized hematopoietic progenitors. Blood. (2008) 112:2575–8. doi: 10.1182/blood-2008-02-140681
75. Ribechini E, Hutchinson JA, Hergovits S, Heuer M, Lucas J, Schleicher U, et al. Novel GM-CSF signals via IFN-gammaR/IRF-1 and AKT/mTOR license monocytes for suppressor function. Blood Adv. (2017) 1:947–60. doi: 10.1182/bloodadvances.2017006858
76. Salehi S, Reed EF. The divergent roles of macrophages in solid organ transplantation. Curr Opin Organ Transplant. (2015) 20:446–53. doi: 10.1097/MOT.0000000000000209
77. Tugues S, Amorim A, Spath S, Martin-Blondel G, Schreiner B, De Feo D, et al. Graft-versus-host disease, but not graft-versus-leukemia immunity, is mediated by GM-CSF-licensed myeloid cells. Sci Transl Med. (2018) 10:eaat8410. doi: 10.1126/scitranslmed.aat8410
78. Nishinakamura R, Miyajima A, Mee PJ, Tybulewicz VL, Murray R. Hematopoiesis in mice lacking the entire granulocyte-macrophage colony-stimulating factor/interleukin-3/interleukin-5 functions. Blood. (1996) 88:2458–64. doi: 10.1182/blood.V88.7.2458.bloodjournal8872458
79. Zhan Y, Lieschke GJ, Grail D, Dunn AR, Cheers C. Essential roles for granulocyte-macrophage colony-stimulating factor (GM-CSF) and G-CSF in the sustained hematopoietic response of Listeria monocytogenes-infected mice. Blood. (1998) 91:863–9. doi: 10.1182/blood.V91.3.863
80. Gonzalez-Juarrero M, Hattle JM, Izzo A, Junqueira-Kipnis AP, Shim TS, Trapnell BC, et al. Disruption of granulocyte macrophage-colony stimulating factor production in the lungs severely affects the ability of mice to control Mycobacterium tuberculosis infection. J Leukoc Biol. (2005) 77:914–22. doi: 10.1189/jlb.1204723
81. Morrissey PJ, Charrier K. GM-CSF administration augments the survival of ity-resistant A/J mice, but not ity-susceptible C57BL/6 mice, to a lethal challenge with Salmonella typhimurium. J Immunol. (1990) 144:557–61.
82. Nambiar JK, Ryan AA, Kong CU, Britton WJ, Triccas JA. Modulation of pulmonary DC function by vaccine-encoded GM-CSF enhances protective immunity against Mycobacterium tuberculosis infection. Eur J Immunol. (2010) 40:153–61. doi: 10.1002/eji.200939665
83. Riopel J, Tam M, Mohan K, Marino MW, Stevenson MM. Granulocyte-macrophage colony-stimulating factor-deficient mice have impaired resistance to blood-stage malaria. Infect Immun. (2001) 69:129–36. doi: 10.1128/IAI.69.1.129-136.2001
84. Olivares Fontt E, Heirman C, Thielemans K, Vray B. Granulocyte-macrophage colony-stimulating factor: involvement in control of Trypanosoma cruzi infection in mice. Infect Immun. (1996) 64:3429–34.
85. Murray HW, Cervia JS, Hariprashad J, Taylor AP, Stoeckle MY, Hockman H. Effect of granulocyte-macrophage colony-stimulating factor in experimental visceral leishmaniasis. J Clin Invest. (1995) 95:1183–92. doi: 10.1172/JCI117767
86. Grau GE, Kindler V, Piguet PF, Lambert PH, Vassalli P. Prevention of experimental cerebral malaria by anticytokine antibodies. Interleukin 3 and granulocyte macrophage colony-stimulating factor are intermediates in increased tumor necrosis factor production and macrophage accumulation. J Exp Med. (1988) 168:1499–504. doi: 10.1084/jem.168.4.1499
87. du Plessis N, Loebenberg L, Kriel M, von Groote-Bidlingmaier F, Ribechini E, Loxton AG, et al. Increased frequency of myeloid-derived suppressor cells during active tuberculosis and after recent Mycobacterium tuberculosis infection suppresses T-cell function. Am J Respir Crit Care Med. (2013) 188:724–32. doi: 10.1164/rccm.201302-0249OC
88. Mencacci A, Montagnoli C, Bacci A, Cenci E, Pitzurra L, Spreca A, et al. CD80+Gr-1+ myeloid cells inhibit development of antifungal Th1 immunity in mice with candidiasis. J Immunol. (2002) 169:3180–90. doi: 10.4049/jimmunol.169.6.3180
89. Sponaas AM, Freitas do Rosario AP, Voisine C, Mastelic B, Thompson J, Koernig S, et al. Migrating monocytes recruited to the spleen play an important role in control of blood stage malaria. Blood. (2009) 114:5522–31. doi: 10.1182/blood-2009-04-217489
90. Goni O, Alcaide P, Fresno M. Immunosuppression during acute Trypanosoma cruzi infection: involvement of Ly6G (Gr1(+))CD11b(+)immature myeloid suppressor cells. Int Immunol. (2002) 14:1125–34. doi: 10.1093/intimm/dxf076
91. Bowen JL, Olson JK. Innate immune CD11b+Gr-1+ cells, suppressor cells, affect the immune response during Theiler's virus-induced demyelinating disease. J Immunol. (2009) 183:6971–80. doi: 10.4049/jimmunol.0902193
92. Metcalf D. Clonal analysis of proliferation and differentiation of paired daughter cells: action of granulocyte-macrophage colony-stimulating factor on granulocyte-macrophage precursors. Proc Natl Acad Sci USA. (1980) 77:5327–30. doi: 10.1073/pnas.77.9.5327
93. Weston BR, Li L, Tyson JJ. Mathematical analysis of cytokine-induced differentiation of granulocyte-monocyte progenitor cells. Front Immunol. (2018) 9:2048. doi: 10.3389/fimmu.2018.02048
94. Brach MA, deVos S, Gruss HJ, Herrmann F. Prolongation of survival of human polymorphonuclear neutrophils by granulocyte-macrophage colony-stimulating factor is caused by inhibition of programmed cell death. Blood. (1992) 80:2920–4. doi: 10.1182/blood.V80.11.2920.2920
95. Smith WB, Guida L, Sun Q, Korpelainen EI, van den Heuvel C, Gillis D, et al. Neutrophils activated by granulocyte-macrophage colony-stimulating factor express receptors for interleukin-3 which mediate class II expression. Blood. (1995) 86:3938–44. doi: 10.1182/blood.V86.10.3938.bloodjournal86103938
96. Guthridge MA, Barry EF, Felquer FA, McClure BJ, Stomski FC, Ramshaw H, et al. The phosphoserine-585-dependent pathway of the GM-CSF/IL-3/IL-5 receptors mediates hematopoietic cell survival through activation of NF-kappaB and induction of bcl-2. Blood. (2004) 103:820–7. doi: 10.1182/blood-2003-06-1999
97. Carrington EM, Tarlinton DM, Gray DH, Huntington ND, Zhan Y, Lew AM. The life and death of immune cell types: the role of BCL-2 anti-apoptotic molecules. Immunol Cell Biol. (2017) 95:870–7. doi: 10.1038/icb.2017.72
98. Cowburn AS, Summers C, Dunmore BJ, Farahi N, Hayhoe RP, Print CG, et al. Granulocyte/macrophage colony-stimulating factor causes a paradoxical increase in the BH3-only pro-apoptotic protein Bim in human neutrophils. Am J Respir Cell Mol Biol. (2011) 44:879–87. doi: 10.1165/rcmb.2010-0101OC
99. Carrington EM, Vikstrom IB, Light A, Sutherland RM, Londrigan SL, Mason KD, et al. BH3 mimetics antagonizing restricted prosurvival Bcl-2 proteins represent another class of selective immune modulatory drugs. Proc Natl Acad Sci USA. (2010) 107:10967–71. doi: 10.1073/pnas.1005256107
100. Leverson JD, Phillips DC, Mitten MJ, Boghaert ER, Diaz D, Tahir SK, et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci Transl Med. (2015) 7:279ra40. doi: 10.1126/scitranslmed.aaa4642
101. Lin EY, Orlofsky A, Berger MS, Prystowsky MB. Characterization of A1, a novel hemopoietic-specific early-response gene with sequence similarity to bcl-2. J Immunol. (1993) 151:1979–88.
102. Vier J, Groth M, Sochalska M, Kirschnek S. The anti-apoptotic Bcl-2 family protein A1/Bfl-1 regulates neutrophil survival and homeostasis and is controlled via PI3K and JAK/STAT signaling. Cell Death Dis. (2016) 7:e2103. doi: 10.1038/cddis.2016.23
103. Schenk RL, Tuzlak S, Carrington EM, Zhan Y, Heinzel S, Teh CE, et al. Characterisation of mice lacking all functional isoforms of the pro-survival BCL-2 family member A1 reveals minor defects in the haematopoietic compartment. Cell Death Differ. (2017) 24:534–45. doi: 10.1038/cdd.2016.156
104. Derouet M, Thomas L, Cross A, Moots RJ, Edwards SW. Granulocyte macrophage colony-stimulating factor signaling and proteasome inhibition delay neutrophil apoptosis by increasing the stability of Mcl-1. J Biol Chem. (2004) 279:26915–21. doi: 10.1074/jbc.M313875200
105. Andina N, Conus S, Schneider EM, Fey MF, Simon HU. Induction of Bim limits cytokine-mediated prolonged survival of neutrophils. Cell Death Differ. (2009) 16:1248–55. doi: 10.1038/cdd.2009.50
106. Dzhagalov I, St John A, He YW. The antiapoptotic protein Mcl-1 is essential for the survival of neutrophils but not macrophages. Blood. (2007) 109:1620–6. doi: 10.1182/blood-2006-03-013771
107. Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med. (1994) 179:1109–18. doi: 10.1084/jem.179.4.1109
108. Erlich Z, Shlomovitz I, Edry-Botzer L, Cohen H, Frank D, Wang H, et al. Macrophages, rather than DCs, are responsible for inflammasome activity in the GM-CSF BMDC model. Nat Immunol. (2019) 20:397–406. doi: 10.1038/s41590-019-0313-5
109. Menezes S, Melandri D, Anselmi G, Perchet T, Loschko J, Dubrot J, et al. The heterogeneity of Ly6C(hi) monocytes controls their differentiation into iNOS(+) macrophages or monocyte-derived dendritic cells. Immunity. (2016) 45:1205–18. doi: 10.1016/j.immuni.2016.12.001
110. Lutz MB, Suri RM, Niimi M, Ogilvie AL, Kukutsch NA, Rossner S, et al. Immature dendritic cells generated with low doses of GM-CSF in the absence of IL-4 are maturation resistant and prolong allograft survival in vivo. Eur J Immunol. (2000) 30, 1813–22. doi: 10.1002/1521-4141(200007)30:7<1813::AID-IMMU1813>3.0.CO;2-8
111. Na YR, Jung D, Gu GJ, Seok SH. GM-CSF grown bone marrow derived cells are composed of phenotypically different dendritic cells and macrophages. Mol Cells. (2016) 39:734–41. doi: 10.14348/molcells.2016.0160
112. Bunda S, Kommaraju K, Heir P, Ohh M. SOCS-1 mediates ubiquitylation and degradation of GM-CSF receptor. PLoS ONE. (2013) 8:e76370. doi: 10.1371/journal.pone.0076370
113. Feldman GM, Rosenthal LA, Liu X, Hayes MP, Wynshaw-Boris A, Leonard WJ, et al. STAT5A-deficient mice demonstrate a defect in granulocyte-macrophage colony-stimulating factor-induced proliferation and gene expression. Blood. (1997) 90:1768–76. doi: 10.1182/blood.V90.5.1768
114. Lehtonen A, Matikainen S, Miettinen M, Julkunen I. Granulocyte-macrophage colony-stimulating factor (GM-CSF)-induced STAT5 activation and target-gene expression during human monocyte/macrophage differentiation. J Leukoc Biol. (2002) 71:511–9. doi: 10.1189/jlb.71.3.511
115. Miah MA, Yoon CH, Kim J, Jang J, Seong YR, Bae YS. CISH is induced during DC development and regulates DC-mediated CTL activation. Eur J Immunol. (2012) 42:58–68. doi: 10.1002/eji.201141846
116. Vento-Tormo R, Company C, Rodriguez-Ubreva J, de la Rica L, Urquiza JM, Javierre BM, et al. IL-4 orchestrates STAT6-mediated DNA demethylation leading to dendritic cell differentiation. Genome Biol. (2016) 17:4. doi: 10.1186/s13059-015-0863-2
117. Pesu M, Aittomaki S, Valineva T, Silvennoinen O. PU.1 is required for transcriptional activation of the Stat6 response element in the Igepsilon promoter. Eur J Immunol. (2003) 33:1727–35. doi: 10.1002/eji.200323680
118. Yang XP, Ghoreschi K, Steward-Tharp SM, Rodriguez-Canales J, Zhu J, Grainger JR, et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol. (2011) 12:247–54. doi: 10.1038/ni.1995
119. Lutz MB, Schnare M, Menges M, Rossner S, Rollinghoff M, Schuler G, et al. Differential functions of IL-4 receptor types I and II for dendritic cell maturation and IL-12 production and their dependency on GM-CSF. J Immunol. (2002) 169:3574–80. doi: 10.4049/jimmunol.169.7.3574
120. Briseno CG, Haldar M, Kretzer NM, Wu X, Theisen DJ, Kc W, et al. Distinct transcriptional programs control cross-priming in classical and monocyte-derived dendritic cells. Cell Rep. (2016) 15:2462–74. doi: 10.1016/j.celrep.2016.05.025
121. Piemonti L, Bernasconi S, Luini W, Trobonjaca Z, Minty A, Allavena P, et al. IL-13 supports differentiation of dendritic cells from circulating precursors in concert with GM-CSF. Eur Cytokine Netw. (1995) 6:245–52.
122. Ahn JS, Agrawal B. IL-4 is more effective than IL-13 for in vitro differentiation of dendritic cells from peripheral blood mononuclear cells. Int Immunol. (2005) 17:1337–46. doi: 10.1093/intimm/dxh312
123. Ko HJ, Brady JL, Ryg-Cornejo V, Hansen DS, Vremec D, Shortman K, et al. GM-CSF-responsive monocyte-derived dendritic cells are pivotal in Th17 pathogenesis. J Immunol. (2014) 192:2202–9. doi: 10.4049/jimmunol.1302040
124. Croxford AL, Lanzinger M, Hartmann FJ, Schreiner B, Mair F, Pelczar P, et al. The cytokine GM-CSF drives the inflammatory signature of CCR2+ monocytes and licenses autoimmunity. Immunity. (2015) 43:502–14. doi: 10.1016/j.immuni.2015.08.010
125. Chow KV, Lew AM, Sutherland RM, Zhan Y. Monocyte-derived dendritic cells promote Th polarization, whereas conventional dendritic cells promote th proliferation. J Immunol. (2016) 196:624–36. doi: 10.4049/jimmunol.1501202
126. Duncker PC, Stoolman JS, Huber AK, Segal BM. GM-CSF promotes chronic disability in experimental autoimmune encephalomyelitis by altering the composition of central nervous system-infiltrating cells, but is dispensable for disease induction. J Immunol. (2018) 200:966–73. doi: 10.4049/jimmunol.1701484
127. Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat Rev Immunol. (2002) 2:151–61. doi: 10.1038/nri746
128. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. (2013) 31:563–604. doi: 10.1146/annurev-immunol-020711-074950
129. Mildner A, Jung S. Development and function of dendritic cell subsets. Immunity. (2014) 40:642–56. doi: 10.1016/j.immuni.2014.04.016
130. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol. (2014) 14:571–8. doi: 10.1038/nri3712
131. Sathe P, Pooley J, Vremec D, Mintern J, Jin JO, Wu L, et al. The acquisition of antigen cross-presentation function by newly formed dendritic cells. J Immunol. (2011) 186:5184–92. doi: 10.4049/jimmunol.1002683
132. Kingston D, Schmid MA, Onai N, Obata-Onai A, Baumjohann D, Manz MG. The concerted action of GM-CSF and Flt3-ligand on in vivo dendritic cell homeostasis. Blood. (2009) 114:835–43. doi: 10.1182/blood-2009-02-206318
133. Zhan Y, Vega-Ramos J, Carrington EM, Villadangos JA, Lew AM, Xu Y. The inflammatory cytokine, GM-CSF, alters the developmental outcome of murine dendritic cells. Eur J Immunol. (2012) 42:2889–900. doi: 10.1002/eji.201242477
134. Esashi E, Wang YH, Perng O, Qin XF, Liu YJ, Watowich SS. The signal transducer STAT5 inhibits plasmacytoid dendritic cell development by suppressing transcription factor IRF8. Immunity. (2008) 28:509–20. doi: 10.1016/j.immuni.2008.02.013
135. Jiao Z, Bedoui S, Brady JL, Walter A, Chopin M, Carrington EM, et al. The closely related CD103+ dendritic cells (DCs) and lymphoid-resident CD8+ DCs differ in their inflammatory functions. PLoS ONE. (2014) 9:e91126. doi: 10.1371/journal.pone.0091126
136. Zhan Y, Carrington EM, van Nieuwenhuijze A, Bedoui S, Seah S, Xu Y, et al. GM-CSF increases cross-presentation and CD103 expression by mouse CD8 spleen dendritic cells. Eur J Immunol. (2011) 41:2585–95. doi: 10.1002/eji.201141540
137. Verreck FA, de Boer T, Langenberg DM, Hoeve MA, Kramer M, Vaisberg E, et al. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc Natl Acad Sci USA. (2004) 101:4560–5. doi: 10.1073/pnas.0400983101
138. Fleetwood AJ, Lawrence T, Hamilton JA, Cook AD. Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J Immunol. (2007) 178:5245–52. doi: 10.4049/jimmunol.178.8.5245
139. Fleetwood AJ, Dinh H, Cook AD, Hertzog PJ, Hamilton JA. GM-CSF- and M-CSF-dependent macrophage phenotypes display differential dependence on type I interferon signaling. J Leukoc Biol. (2009) 86:411–21. doi: 10.1189/jlb.1108702
140. Doherty TM, Coffman RL. Ability of macrophage subsets to transfer resistance to murine leishmaniasis is dependent on IL-12 production. Eur J Immunol. (1999) 29:522–9. doi: 10.1002/(SICI)1521-4141(199902)29:02<522::AID-IMMU522>3.0.CO;2-U
141. Morrissey PJ, Bressler L, Park LS, Alpert A, Gillis S. Granulocyte-macrophage colony-stimulating factor augments the primary antibody response by enhancing the function of antigen-presenting cells. J Immunol. (1987) 139:1113–9.
142. Roth MD, Gitlitz BJ, Kiertscher SM, Park AN, Mendenhall M, Moldawer N, et al. Granulocyte macrophage colony-stimulating factor and interleukin 4 enhance the number and antigen-presenting activity of circulating CD14+ and CD83+ cells in cancer patients. Cancer Res. (2000) 60:1934–41.
143. Campo I. The influence of genetics on therapeutic developments in pulmonary alveolar proteinosis. Curr Opin Pulm Med. (2019) 25:294–9. doi: 10.1097/MCP.0000000000000576
144. Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity. (2001) 15:557–67. doi: 10.1016/S1074-7613(01)00218-7
145. Reed JA, Ikegami M, Cianciolo ER, Lu W, Cho PS, Hull W, et al. Aerosolized GM-CSF ameliorates pulmonary alveolar proteinosis in GM-CSF-deficient mice. Am J Physiol. (1999) 276:L556–63. doi: 10.1152/ajplung.1999.276.4.L556
146. Ohashi K, Sato A, Takada T, Arai T, Nei T, Kasahara Y, et al. Direct evidence that GM-CSF inhalation improves lung clearance in pulmonary alveolar proteinosis. Respir Med. (2012) 106:284–93. doi: 10.1016/j.rmed.2011.10.019
147. Tran HT, Metcalf D, Cheers C. Anti-bacterial activity of peritoneal cells from transgenic mice producing high levels of GM-CSF. Immunology. (1990) 71:377–82.
148. Sorgi CA, Rose S, Court N, Carlos D, Paula-Silva FW, Assis PA, et al. GM-CSF priming drives bone marrow-derived macrophages to a pro-inflammatory pattern and downmodulates PGE2 in response to TLR2 ligands. PLoS ONE. (2012) 7:e40523. doi: 10.1371/journal.pone.0040523
149. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. (2014) 6:13. doi: 10.12703/P6-13
150. Waight JD, Netherby C, Hensen ML, Miller A, Hu Q, Liu S, et al. Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. J Clin Invest. (2013) 123:4464–78. doi: 10.1172/JCI68189
151. Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity. (2010) 32:790–802. doi: 10.1016/j.immuni.2010.05.010
152. Strauss L, Sangaletti S, Consonni FM, Szebeni G, Morlacchi S, Totaro MG, et al. RORC1 regulates tumor-promoting “emergency” granulo-monocytopoiesis. Cancer Cell. (2015) 28:253–69. doi: 10.1016/j.ccell.2015.07.006
153. Kuroda E, Noguchi J, Doi T, Uematsu S, Akira S, Yamashita U. IL-3 is an important differentiation factor for the development of prostaglandin E2-producing macrophages between C57BL/6 and BALB/c mice. Eur J Immunol. (2007) 37:2185–95. doi: 10.1002/eji.200737041
154. Rath M, Muller I, Kropf P, Closs EI, Munder M. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front Immunol. (2014) 5:532. doi: 10.3389/fimmu.2014.00532
155. Martinez FO, Helming L, Milde R, Varin A, Melgert BN, Draijer C, et al. Genetic programs expressed in resting and IL-4 alternatively activated mouse and human macrophages: similarities and differences. Blood. (2013) 121:e57–69. doi: 10.1182/blood-2012-06-436212
156. Chen Y, Zhang S, Wang Q, Zhang X. Tumor-recruited M2 macrophages promote gastric and breast cancer metastasis via M2 macrophage-secreted CHI3L1 protein. J Hematol Oncol. (2017) 10:36. doi: 10.1186/s13045-017-0408-0
157. El Chartouni C, Schwarzfischer L, Rehli M. Interleukin-4 induced interferon regulatory factor (Irf) 4 participates in the regulation of alternative macrophage priming. Immunobiology. (2010) 215:821–5. doi: 10.1016/j.imbio.2010.05.031
158. Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol. (2010) 11:936–44. doi: 10.1038/ni.1920
159. Lacey DC, Achuthan A, Fleetwood AJ, Dinh H, Roiniotis J, Scholz GM, et al. Defining GM-CSF- and macrophage-CSF-dependent masHcrophage responses by in vitro models. J Immunol. (2012) 188:5752–65. doi: 10.4049/jimmunol.1103426
160. Doherty TM, Kastelein R, Menon S, Andrade S, Coffman RL. Modulation of murine macrophage function by IL-13. J Immunol. (1993) 151:7151–60.
161. Doyle AG, Herbein G, Montaner LJ, Minty AJ, Caput D, Ferrara P, et al. Interleukin-13 alters the activation state of murine macrophages in vitro: comparison with interleukin-4 and interferon-gamma. Eur J Immunol. (1994) 24:1441–5. doi: 10.1002/eji.1830240630
162. Van Dyken SJ, Locksley RM. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Annu Rev Immunol. (2013) 31:317–43. doi: 10.1146/annurev-immunol-032712-095906
163. Bozinovski S, Jones JE, Vlahos R, Hamilton JA, Anderson GP. Granulocyte/macrophage-colony-stimulating factor (GM-CSF) regulates lung innate immunity to lipopolysaccharide through Akt/Erk activation of NFkappa B and AP-1 in vivo. J Biol Chem. (2002) 277:42808–14. doi: 10.1074/jbc.M207840200
164. Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol. (2017) 198:1006–14. doi: 10.4049/jimmunol.1601515
165. Jinushi M, Nakazaki Y, Dougan M, Carrasco DR, Mihm M, Dranoff G. MFG-E8-mediated uptake of apoptotic cells by APCs links the pro- and antiinflammatory activities of GM-CSF. J Clin Invest. (2007) 117:1902–13. doi: 10.1172/JCI30966
166. Dorhoi A, Du Plessis N. Monocytic myeloid-derived suppressor cells in chronic infections. Front Immunol. (2017) 8:1895. doi: 10.3389/fimmu.2017.01895
167. Dilek N, van Rompaey N, Le Moine A, Vanhove B. Myeloid-derived suppressor cells in transplantation. Curr Opin Organ Transplant. (2010) 15:765–8. doi: 10.1097/MOT.0b013e3283401742
168. Rodriguez PC, Quiceno DG, Ochoa AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood. (2007) 109:1568–73. doi: 10.1182/blood-2006-06-031856
Keywords: GM-CSF, macrophages, dendritic cells, differentiation, function
Citation: Zhan Y, Lew AM and Chopin M (2019) The Pleiotropic Effects of the GM-CSF Rheostat on Myeloid Cell Differentiation and Function: More Than a Numbers Game. Front. Immunol. 10:2679. doi: 10.3389/fimmu.2019.02679
Received: 20 April 2019; Accepted: 30 October 2019;
Published: 15 November 2019.
Edited by:
Pierre Guermonprez, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Venetia Bigley, Newcastle University, United KingdomCopyright © 2019 Zhan, Lew and Chopin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yifan Zhan, emhhbkB3ZWhpLmVkdS5hdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.