
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 18 October 2019
Sec. T Cell Biology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02486
This article is part of the Research Topic Control of Regulatory T cell Stability, Plasticity and Function in Health and Disease View all 21 articles
 Guoping Deng1*
Guoping Deng1* Xiaomin Song2
Xiaomin Song2 Shigeyoshi Fujimoto3
Shigeyoshi Fujimoto3 Ciriaco A. Piccirillo4,5
Ciriaco A. Piccirillo4,5 Yasuhiro Nagai6
Yasuhiro Nagai6 Mark I. Greene6*
Mark I. Greene6*Regulatory T cells (Tregs) are engaged in maintaining immune homeostasis and preventing autoimmunity. Treg cells include thymic Treg cells and peripheral Treg cells, both of which can suppress the immune response via multiple distinct mechanisms. The differentiation, proliferation, suppressive function and survival of Treg cells are affected by distinct energy metabolic programs. Tissue-resident Treg cells hold unique features in comparison with the lymphoid organ Treg cells. Foxp3 transcription factor is a lineage master regulator for Treg cell development and suppressive activity. Accumulating evidence indicates that the activity of Foxp3 protein is modulated by various post-translational modifications (PTMs), including phosphorylation, O-GlcNAcylation, acetylation, ubiquitylation and methylation. These modifications affect multiple aspects of Foxp3 function. In this review, we define features of Treg cells and roles of Foxp3 in Treg biology, and summarize current research in PTMs of Foxp3 protein involved in modulating Treg function. This review also attempts to define Foxp3 dimer modifications relevant to mediating Foxp3 activity and Treg suppression. Understanding Foxp3 protein features and modulation mechanisms may help in the design of rational therapies for immune diseases and cancer.
CD4+CD25+ T cells have been identified as a suppressive T cell subset, which have a dominant role in mediating peripheral immune tolerance (1). The Treg population includes two main subsets according to their origins: thymic Treg cells (tTregs) develop with stimulations of self-antigens, cytokines, and costimulatory molecules in thymus; peripheral Treg cells (pTregs) are differentiated from naïve T cells by the act of cytokine TGF-β in periphery. Both tTregs and pTregs require expression of the Foxp3 transcription factor to maintain their lineage stability and effective suppression of immune responses (2, 3).
The definition of a stable Treg lineage can be evaluated by several features, including constitutive expression of Foxp3 and a variety of Treg feature markers (4). Expression of Foxp3 is a prerequisite for Treg lineage commitment and maintenance. Foxp3 expression relies on demethylation of the conserved non-coding sequence 2 (CNS2, also known as the Treg cell-specific demethylated region TSDR) in the Foxp3 locus. A deletion of CNS2 results in loss of Foxp3 expression during Treg cell expansion and destabilizes Treg cells (5–7). High-resolution quantitative proteomics and transcriptomics approaches have revealed that expression patterns of the core Treg properties, including CD25, CTLA-4, Helios, and FOXP3 gene TSDR methylation, appear relatively stable in culture in vitro (8). The role of Foxp3 in Treg function will be discussed below. Moreover, Treg cells are endowed with unique processes to rapidly respond to environmental cues, and can achieve this through distinct mechanisms of regulation of global or gene-specific mRNA translation. Unlike gene transcription, translational regulation is advantageous for environmental-sensing as it provides a rapid and energetically favorable mechanism to shape the proteome of a given cell, and to tailer cell function to the extracellular context (9). Indeed, distinct translational signatures distinguish Treg and Teff cells (10).
Treg cells are phenotypically diverse in migration, homeostasis, and function (11). Tregs are divided into CD44lowCD62Lhigh central Tregs (cTregs) and CD44highCD62Llow effector Tregs (eTregs). cTregs are quiescent, IL-2 signaling dependent and long-lived, and they function in the secondary lymphoid tissues to suppress T cell priming; in contrast, eTregs are highly activated and ICOS signaling dependent with potent suppressive function in specific non-lymphoid tissues to dampen immune responses (12). eTregs have increased mTORC1 signaling and glycolysis compared with cTregs. Consistently, inhibition of mTORC1 activity by administration of rapamycin (mTORC1 inhibitor) promotes generation of long-lived cTreg cells in vivo (13). Treg cells lacking Ndfip1, a coactivator of Nedd4-family E3 ubiquitin ligases, elevate mTORC1 signaling and glycolysis, which increases eTreg cells but impairs Treg stability in terms of Foxp3 expression and pro-inflammatory cytokine production (14).
Treg cells suppress immune response via multiple mechanisms [as reviewed in (15–17)]. Treg cells highly express CD25 (the IL-2 receptor α-chain, IL-2Rα) and may compete with effector T cells leading to consumption of cytokine IL-2 (18). Treatment with low-dose rhIL-2 selectively promotes Treg frequency and function, and ameliorates diseases in patients with systemic lupus erythematosus (SLE) (19). The constitutive expression of CD25, a direct target of Foxp3, is essential to engage a strong STAT5 signal for Treg proliferation, survival, and Foxp3 expression (20). CTLA-4 activation can down-regulate CD80 and CD86 expression on antigen-presenting cells (21). Treg cells also produce inhibitory cytokines, IL-10, TGF-β, and IL-35, to enhance immune tolerance along with cell-contact suppression (22–24). Treg cells may mediate specific suppression by depleting cognate peptide-MHC class II from dendritic cells in vivo (25). Of note, Treg cells recognize cognate antigen and require T cell receptor (TCR) signaling for optimal activation, differentiation, and function (26). Polyclonal expanded Treg cell mixed populations exhibit suppressive potency for certain autoimmune diseases (27). Engineering Treg cells with antigen-specific TCR appears to lead to antigen-specific suppression with increased potency (28).
Treg cells exploit distinct energy metabolism programs for their differentiation, proliferation, suppressive function, and survival (29, 30). Rather than glucose metabolism, Treg cells have activated AMP-activated protein kinase (AMPK) and use lipid oxidation as an energy source. AMPK stimulation by Met can decrease Glut1 and increase Treg generation (31). Further proteomic analysis showed that fresh-isolated human Treg cells are highly glycolytic, while non-proliferating Tconv cells mainly use fatty-acid oxidation (FAO) as an energy source. When cultured in vitro, Treg proliferation and suppression require both glycolysis and FAO, while Tconv cells mainly rely on glucose metabolism for proliferation and function. This finding indicates that Treg cells and Tconv cells may adopt different metabolic programs in vitro and in vivo (32). Treg cells cannot only use anabolic glycolysis to produce sufficient fundamental building blocks to fuel cell expansion, but also efficiently generate ATP energy via catabolic fatty acid oxidation (FAO) driven oxidative phosphorylation (OXPHOS) by the mitochondria to support activation and suppression function (33).
Treg cells have greater mitochondrial mass and higher ROS production than Tconv cells. Tregs are more vulnerable to OXPHOS inhibition, which underscores the unique metabolic features of Treg cell (34). Loss of subunit of the mitochondrial complex III RISP in Treg cells diminishes oxygen consumption rate (OCR), but increases in glycolytic flux. Treg cells require the activity of mitochondrial complex III to maintain Treg feature gene expression and suppression (35). Foxp3 expression increases mitochondrial respiratory capacity and promotes ATP generation through oxidative phosphorylation. Increased fatty acid metabolism can protect Treg cells from fatty acid–induced apoptosis accordingly (36). Toll-like receptor (TLR) signals activate PI(3)K-Akt-mTORC1 signaling and glycolysis to promote cell proliferation, however, Foxp3 opposes PI(3)K-Akt-mTORC1 signaling but increases oxidative phosphorylation for effector function. Thus, Foxp3 and inflammatory TLR signals control the proliferation and suppressive function of Treg cells through balancing the metabolic activity between glycolysis and oxidative phosphorylation (37). Genetic deletion of mTOR gene (Frap1) or pharmacological inhibition of mTOR activity by rapamycin promotes Treg cell maintenance (38, 39). A recent study shows that the deficiency of Foxp3 dysregulates mTORC2 signaling, which promotes aerobic glycolysis and oxidative phosphorylation. Genetic deletion of the mTORC2 adaptor gene Rictor or pharmacological treatment with mTOR inhibitors antagonizes the Teff cell-like program and restores the suppressive function of Foxp3-deficient Treg cells (40).
Treg cells adopt distinct patterns of tissue- or immune-context-specific suppression mechanisms (41). Treg function in vivo requires a timely recruitment and/or accumulation in non-lymphoid tissues where immune responses are frequently occurring. In these sites, Treg cells adapt their function (i.e., effector mechanism) based on local immune mediators (42). For instance, TNFα-induced dephosphorylation of FOXP3 at Ser-418 by protein phosphatase 1 (PP1) compromises Treg suppressive activity within the inflamed synovium, which is responsible for the pathogenicity of rheumatoid arthritis (43). Here, we attempt to review some unique features and underlying mechanisms of tissue-resident Treg cells in skin, visceral adipose tissue and tumor, which has attracted extensive research interests recently (44–46).
Skin is an immunologically active organ that protects against pathogen invasion but prevents collateral tissue damage (47). Both murine and human skin contain a large number of Tregs, which accumulate through multiple chemokines and ligands, such as CCR4, CCR6, CCL17, CCL20, and CCL22 (48, 49). Functionally, Tregs inhibit proinflammatory cytokine IFN-γ production and macrophage accumulation in wounded skin to facilitate cutaneous wound closure and healing (50). Skin-resident Treg cells localize to hair follicles and facilitate stem cell-mediated hair follicle regeneration through Notch-Jagged signaling pathway (51). Tregs contribute to the establishment of immune tolerance to commensal microbes in neonatal skin and colon (51–53).
Visceral adipose tissue (VAT)-resident Treg cells guard against abnormal inflammation of the adipose tissue in obesity, type 2 diabetes and atherosclerosis (54, 55). Progressively decreased frequency of Tregs in visceral adipose tissue leads to the adipose inflammation and insulin resistance in obese animal models, while the induction of Tregs by administration of IL-2 contained complex or anti-CD3 monoclonal antibody can ameliorate glucose tolerance and insulin sensitivity (56, 57). VAT-Treg cells show unique gene signatures implicated in leukocyte migration, extravasation, and cytokine production (56). Peroxisome-proliferator-activated receptor γ (PPARγ) acts as a crucial orchestrator for the accumulation, phenotype, and function of VAT-Treg cells; a deficiency of PPARγ in Treg cells down-regulates the specific VAT Treg gene expression signature and Treg frequency in the visceral adipose tissue particularly (58). VAT-Treg cells maintain adipose tissue homeostasis by catabolizing prostaglandin E2 (PGE2) into the metabolite 15-keto PGE2, which limits conventional T cell activation and proliferation in visceral adipose tissue (59).
Treg cells are often enriched in tumor tissue, and a high ratio of tumor-infiltrating Treg cells (TIL-Tregs) to effector T cells generally predicts poor clinical outcomes of certain types of cancers, including ovarian cancer, breast cancer, melanoma, and hepatocellular carcinoma (60). Depleting intra-tumoral Tregs by treatment with immune checkpoint inhibitor monoclonal antibody against cytotoxic T lymphocyte–associated antigen 4 (anti-CTLA-4) promotes anti-tumor immune activity (61, 62). A recent study shows that the antitumor efficacy of anti-CTLA-4 can be enhanced by the combination therapy with Toll-like receptor 1/2 (TLR1/2) ligand via a FcγRIV-dependent manner (63). The origins of TIL-Tregs are diverse, including: (1) selective recruitment to tumor sites; (2) conversion from conventional CD4+ T cells; (3) Treg cell expansion (64). TIL-Tregs exhibit Foxp3 TSDR hypomethylation, and are highly proliferated and apoptotic due to the oxidative stress within tumor lesions (65, 66). Treg cells can thrive in tissues with ischemic injury or the tumor microenvironment, characterized by low-glucose and high-lactate. Treg cells favor oxidation of L-lactate to pyruvate to increase the intracellular NAD:NADH ratio, and resist the suppressive effects of L-lactate on cell proliferation (67). In addition, Tregs are endowed with the relative advantage of a circuitry of glycolysis, fatty acid synthesis, and oxidation to prevail over conventional T cells in the hostile tumor microenvironment (68).
Overall, tissue-resident Treg cells play unique roles in distinct non-lymphoid tissues via multiple molecular mechanisms, including regulation by post-translational modification of FOXP3 protein (which will be discussed in detail in the section of “Post-translational modifications of Foxp3”). It holds promise to harness the behavior and function of tissue-resident Tregs, specifically to treat certain immune disorders, such as skin autoimmunity and regeneration, obesity, type 2 diabetes and cancer.
The Forkhead box (FOX) protein superfamily of transcriptional regulators play pleiotropic roles in cell proliferation, differentiation, survival, and apoptosis during embryonic development and homeostasis of adult tissues (69). The FOXP family include FOXP1, FOXP2, FOXP3, and FOXP4 members. Foxp1 interacts with and regulates Foxp3 chromatin binding to coordinate Treg cell suppressive function and homeostasis (70–72). In humans, the FOXP3 gene (originally named JM2 or Scurfin) is found on the X-chromosome at Xp11.23-Xq13.3 (73). Genetic analysis has demonstrated that Treg cell dysfunction caused by the FOXP3 mutation is responsible for the immune dysregulation polyendocrinopathy enteropathy X-linked syndrome [IPEX, also called X-linked autoimmunity–allergic dysregulation syndrome (XLAAD)], a recessive immune disorder occurring in newborns and children (74). In mice, conditional deletion of Foxp3 in CD4+ T cells leads to fatally lymphoproliferative autoimmunity, which includes high IgE levels, enteropathy, type 1 diabetes and failure to thrive, while ectopic expression of Foxp3 can re-program conventional CD4+ T cells as anti-inflammatory Treg cells (75).
The Foxp3 protein contains multiple structural domains. There is a proline-rich N-terminal domain that interacts with many molecular factors to regulate transcriptional activity. Another central zinc-finger and leucine-zipper domain is involved in oligomer formation, and a conserved C-terminal forkhead/winged helix domain (FKH) is responsible for DNA binding (76). Chromatin immuno-precipitation combining genome-wide analysis revealed that Foxp3 interacts with up to ~700 genes. Interestingly, Foxp3 can activate or repress its target genes to cooperatively regulate Treg cell development, function, and homeostasis (77, 78). Foxp3 protein complexes define the transcriptional network of Treg cells (79). Foxp3 forms heterogeneous super-complexes of 400–800 kDa and associates with 361 partner proteins of which ~30% are transcription related. These proteins include Foxp1, Foxp4, Stat3, IKZF1, Runx1, and GATA-3 (71, 72). These interactions highlight the master role of Foxp3 in programming Treg cell lineage.
FOXP3 mutations that occur in IPEX patients include missense point mutations, frameshift, and missplicing, causing a premature stop codon, as well as mutations in the polyadenylation site. The missense mutations can preserve a normal or reduced expression of FOXP3 in IPEX patients. Although various distinct FOXP3 mutations have been reported with IPEX patients, individuals with the same mutation can develop disease manifestations that are not similar, indicating heterogeneity of severity among IPEX cases (80). IPEX patients may present with different intestinal lesions (81), and similar genotypes come out with different symptoms and severity (82); which suggests the complex relevance of genotype and phenotype in IPEX patients, and reflects complex intracellular interactions and post-translational modifications of FOXP3 (83).
Post-translational modifications (PTMs) of proteins link cellular signals to the functional properties. The transcriptional activity of Foxp3 can be modulated by various post-translational modifications, such as phosphorylation, O-GlcNAcylation, acetylation, ubiquitination, and methylation (Figure 1).

Figure 1. Post-translational modifications of Foxp3 and their roles in Treg suppression. A schematic representation shows the Foxp3 protein structure domains and post-translational modification sites (top). The table lists the kind of modification, modified sites, modifier enzymes, and roles in Treg suppression (bottom) (Guoping Deng and Mark I. Greene).
Protein phosphorylation, occurring at serine, threonine or tyrosine resides of proteins, is a reversible and transient modification catalyzed by certain kinases and phosphatases. Protein phosphorylation is involved in protein intracellular stability, interaction, and localization. TCR engagement triggers a signaling cascade with rapid phosphorylation events, and reprograms the proteomes and bioenergetic features for T cell activation, proliferation, and differentiation (84). In humans, TCR stimulation can induce transient FOXP3 expression (85). TCR signaling enhances Foxp3 phosphorylation through incubation of anti-CD3/CD28 antibodies or pharmacological treatment with PMA (phorbol 12-myristate 13-acetate) and Ionomycin (86). A recent report found that TCR stimulation can activate TAK1-NLK signaling, leading to phosphorylation of Foxp3, which decreases its interaction with the STUB1 E3-ubiquitin ligase to modify ubiquitination and proteasome-mediated degradation rates. Using mass spectrometry, seven distinct phosphorylated residues (S19, S156, S189, S273, S278, S295, and T341) of Foxp3 have been identified upon the co-expression of NLK in cell line. Although there may be other substrates targeted by NLK kinase other than Foxp3, Treg cell-specific NLK deficiency results in auto-inflammation and renders animals susceptible to induced experimental autoimmune encephalomyelitis (EAE) (87).
Levels of the inhibitor of the cyclin-dependent kinases (CDKs) p27kip1 are up-regulated in anergic T cells. CDK2 is a direct target of p27kip1 and CDK2 promotes cytokine production by CD4+ T cells, while limiting Treg function. Treg cells lacking CDK2 had increased capacity to inhibit Tconv cell proliferation and ameliorate immunopathogenesis in colitis (88). The N-terminal domain of Foxp3 protein contains several putative CDK motifs. CDK2 kinase phosphorylates Ser-19 and Thr-175 residues of Foxp3. Mutation of CDK motifs via a substitution of Ser/Thr to Ala increases Foxp3 protein stability and transcriptional activity, which manifests that Foxp3 phosphorylation by CDK2 kinase negatively regulates Treg function (89). These findings demonstrate that CDK2 negatively regulates Treg function and peripheral tolerance by phosphorylating Foxp3.
The Foxp3 N-terminal domain is important for Foxp3 transcriptional repression and nuclear transport (90, 91). Pim-2 kinase, an oncogenic serine/threonine kinase, can phosphorylate multiple sites of Foxp3 N-terminal domain as identified by mass spectrum analyses. The deficiency of Pim-2 increases Treg suppression in in vitro assay, and Pim-2-deficient mice appear resistant to DSS-induced acute colitis (92). Pim-2 kinase can promote rapamycin-resistant survival, growth and proliferation of lymphocytes, including Treg cells (93, 94). The Pim-1 kinase phosphorylates Ser422 of the forkhead domain of human FOXP3, which attenuates FOXP3 DNA binding activity and down-regulates expression of Treg feature genes. Knockdown of Pim-1 in Tregs enhances suppressive activity (95). These findings demonstrate that phosphorylation of Foxp3 by Pim-1 and Pim-2 negatively regulates Foxp3 transcriptional activity and Treg suppressive function. Kaempferol is a natural flavonoid found in vegetables and fruits, which may reduce PIM1-mediated FOXP3 phosphorylation at S422 and enhance Treg cell suppressive capability. The anti-inflammatory effect of Kaempferol may facilitate therapies of certain autoimmune diseases (96).
Phosphorylation of the Ser-418 located in the FOXP3 C-terminal forkhead domain plays a positive role in regulating Treg suppressive function (43). TNF-α induces protein phosphatase 1 (PP1), which can dephosphorylate Ser-418 of FOXP3 and limit Treg cell activity, while increasing pathogenic Th17 and Th1 CD4 T cells within the inflamed synovium. TNF-α antagonist therapy (treatment with TNF-α-specific antibody: Infliximab) increased Treg cell function in patients with rheumatoid arthritis (43). Ser-418 phosphorylation of FOXP3 affected C-terminal cleavage of Foxp3 protein by proprotein convertase (PC) (97) and also modulated Foxp3 DNA affinity by interactions with other proximal modification, such as acetylation (98). FOXP3 can be phosphorylated at the Tyr-342 site by Lymphocyte-specific protein tyrosine kinase (LCK) in the MCF-7 cell line. Tyr-342 phosphorylation of FOXP3 down-regulates expression of SKP2, VEGF-A, and MMP9 genes (99). Taken together, Foxp3 undergoes phosphorylation modifications, and these modifications can either positively or negatively modulate Treg function according to the various disease settings.
Protein O-GlcNAcylation modification occurs at serine and threonine residues, as well as phosphorylation, which is catalyzed by O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA) oppositely. O-GlcNAcylation may counteract ubiquitination to stabilize FOXP3 protein, and loss of O-GlcNAcylation destabilizes FOXP3 protein. Deficiency of OGT in Treg cells leads to lethal autoimmune diseases in mice (100).
Acetylation occurs on histone proteins and other cellular proteins (101). Protein acetylation is catalyzed by histone acetyltransferases (HTAs) and histone/protein deacetylases (HDACs) oppositely. HATs can transfer the acetyl moiety of acetyl-coenzyme A to Nα-amino groups of methionine residue or the Nε-amino groups of lysine residues on proteins. Nα-acetylation occurs as a co-translational process of protein N-terminal methionine cleavage (102), and is a reversible process that modulates protein biological activity in response to internal or external cell stimuli (103).
Based on structural and functional similarity of their catalytic domains, HATs can be grouped into three main families: (1) the Gcn5/PCAF family including Gcn5, PCAF, and related proteins; (2) the p300/CBP family; (3) the MYST family are involved in control of transcription and cell growth and survival (104).
TIP60, p300, and CBP, orchestrate multiple aspects of Treg development, function, and lineage stability (105). TIP60 belongs to the MYST HAT family. The TIP60 histone acetylase complex also plays an important role in DNA repair and apoptosis (106). TIP60 can act as both a transcriptional co-repressor (107, 108) and a co-activator (109). TIP60 is the first identified HAT that acts as an essential subunit of the FOXP3 repression complex. The N-terminal 106–190 aa of FOXP3 associates with TIP60 and HDAC7. The FOXP3–TIP60–HDAC7 complex is required for IL-2 repression by Foxp3 in T cells (110).
p300 acetylates Foxp3 to prevent proteasome-mediated degradation and increase Foxp3 protein levels, as well as Foxp3-mediated transcriptional repression of IL-2 production (111). Mass spectrometry analysis has identified three acetylation sites in murine Foxp3 (K31, K262, and K267), upon co-expression with p300 acetyltransferase in 293T cells (112). A conditional deletion of Ep300 (which encodes p300) in Treg cells compromises Treg suppressive capability and leads to autoimmunity at around 10 weeks of age. On the other hand, deficiency of p300 impairs Treg cell infiltration into tumor, accompanied with enhanced anti-tumor immunity (113). Administration of the p300 inhibitor C646 or a peptidic p300i (Lys-CoA-Tat) abrogates Treg cell–dependent allograft survival, while increasing the anti-tumor immune responses in animal tumor models (113, 114).
CBP, a p300 paralog, is also important in regulating Treg function in certain inflammatory or lymphopenic conditions. Double-deletion of CBP and p300 in Treg cells leads to fatal autoimmunity by 3–4 weeks of age (115). Likewise, TIP60 and p300 cooperatively regulate FOXP3 activity. p300 interacts with TIP60 and facilitates autoacetylation of TIP60 at K327. This modification increases TIP60 protein stability and promotes FOXP3 acetylation. Reciprocally, TIP60 promotes p300 acetylation and HAT activity as well (116). In contrast to the modest phenotype of p300-deficient mice, a deficiency of TIP60 in Tregs results in severe and fatal autoimmune diseases at an early age. These studies indicate that TIP60 plays a more unique and dominant role than p300 (for which there are similar and redundant HAT), in terms of regulation of Treg cell development and function (116). A recent study by Bin Dhuban et al. elegantly shows that some inheritable mutations in the forkhead domain of FOXP3 can specifically disrupt FOXP3-TIP60 association, in turn, compromising human Treg cell development and function. Restoring FOXP3-TIP60 in this setting with allosteric modification of TIP60 also rescued Treg cell function (117, 118).
FOXP3 forms dynamic homo- or hetero- dimer via its zinc-finger and leucine-zipper domain. This dimer structure is characterized as a two-stranded anti-parallel a-helical coiled-coil. The crystal structure indicates that lysine residues, K250 and K252 of FOXP3, are electrostatically involved in the interface network for Foxp3 coiled-coil dimerization. Acetylation of K250 and K252 of FOXP3 by p300 results in dimer relaxation and down-regulates Foxp3 suppressive activity (119). Of note, FOXP3 acetylation and related chromatin DNA binding activity were induced by treatment of TGF-β, through residues other than K250 and K252 (86, 119). These studies suggest a work model that regulates the Treg suppression by integrating the conformation, dimerization and post-translational modification of the FOXP3 protein (Figure 2).
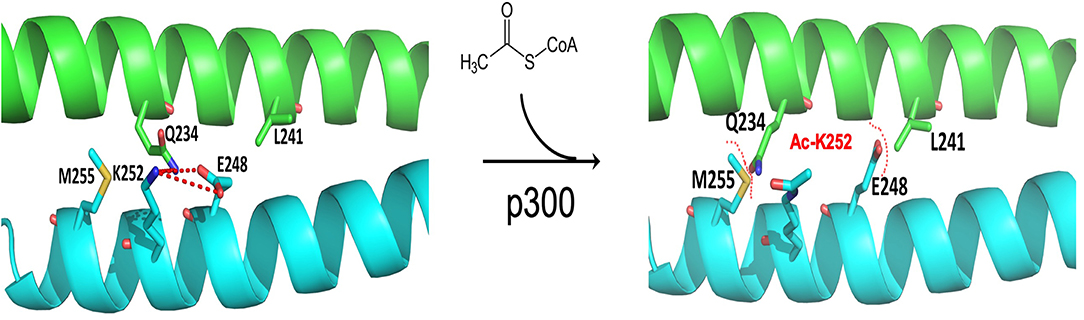
Figure 2. Structure model of acetylation at FOXP3 K252 downregulating suppression by destabilizing homodimerization. FOXP3 forms dynamic homodimer via zinc-finger and leucine-zipper domains, which featured as a two-stranded, anti-parallel a-helical coiled-coil. The inter-subunit hydrogen bond formed between Q234 and E248 stabilizes FOXP3 dimer via electrostatic interactions. The Q234-E248 hydrogen bond depends on the unique conformation of the E248 side chain held by K252 residue (left). Acetylation of K252 by p300 neutralizes the positive charges of K252 side chain and decreases its interaction with E248, and as a consequence, breaks the Q234-E248 inter-subunit hydrogen bond. E248-L241 and Q234-M255 form the steric tension to relax and destabilize the FOXP3 homodimerization (right). (Xiaomin Song, Guoping Deng, and Mark I. Greene).
Eighteen human histone deacetylation enzymes (HDACs) have been identified and grouped into four subsets based on their molecular phylogenetic analysis of primary structure, intracellular localization, and homology to yeast counterpart enzymes (120). Mechanistically, the classical (including classes I, II, and IV) HDAC enzymes depend on zinc ions (Zn2+), whereas the Sirtuins (class III) HDACs utilize nicotinamide adenine dinucleotide (NAD+) as a coenzyme (121).
Treg cells express 11 classical Zn2+-dependent HDACs, and several NAD-dependent HDACs belonging to the Sirtuin family. Although targeting HDAC enzymes in mediating multiple aspects of Treg cell function has received extensive attention (122–124), we focus on reviewing the direct effects of HDAC on Foxp3 protein acetylation and related Treg function regulation. Treatment with a pan-HDACi, trichostatin-A (TSA) boosts the production of thymic Foxp3+ Treg cells, Treg feature gene expression, Treg suppression and Foxp3 protein acetylation. Combined therapy with TSA and rapamycin (TSA-RPM) can extend cardiac or islet allograft survival in animal models (75). Moreover, HDAC inhibitors, including sodium butyrate, valproate acid, SAHA, MS-275, Bufexamac, and BML210, can increase human Treg suppression (125, 126).
HDAC inhibitor therapy has been examined for its ability to modify Treg function in vivo and effects on protection of allograft and prevention of autoimmunity (127). The class III HDAC SIRT1 also decreases Foxp3 acetylation and Treg stability. Pharmacological treatment with either the HDACs pan-inhibitor, TSA, or SIRT specific inhibitor, NAM, increases Foxp3 levels and Treg suppressor capacity in an in vitro suppression assay. Treatment with the SIRT activator, resveratrol, decreases numbers of FOXP3+ cells in human PBMC and skin samples (111). Deficiency of Sirt1 promotes Foxp3 stability in iTreg cells and restrains the generation of pathogenic T cells. Administration of the Sirt-1 inhibitor Ex-527 attenuates GVHD but preserves the therapeutic effect of graft-vs.-leukemia (128).
HDAC6, HDAC9, and Sirt1 may deacetylate Foxp3, however, these HDACs regulate Foxp3 gene expression by affecting different transcription factors. HDAC6, HDAC9, and Sirt1 have shared and individual mechanisms of action, and the combined loss of HDAC6, HDAC9, and Sirt1 activity may augment Treg function (129–131).
Protein ubiquitylation modification is catalyzed by a sequential and concerted action of three distinct classes of enzymes: the ubiquitin-activating enzyme, E1, the ubiquitin-conjugating enzyme, E2, and the ubiquitin-ligase, E3 (132). Ubiquitin consists of a 76-amino acid polypeptide that has seven lysine residues (K6, K11, K27, K29, K33, K48, and K63), each of which may participate in the formation of diverse poly-ubiquitin chains. K48-linked polyubiquitin chains represent targeting signals for proteasomal degradation, while K63 polyubiquitylation plays non-proteolytic roles, including DNA damage response, receptor endocytosis and protein trafficking (133).
Ubiquitylation signals regulate multiple aspects of immune cell function and immune response (134). Upon T cell activation, the non-degradative ubiquitylation, including K29, K33, and K63 polyubiquitylation, increases largely in CD4+ T cells, which suggests the importance of non-degradative ubiquitylation in T cell signaling (135).
As the master regulator of the Treg cell lineage, Foxp3 protein level and turnover rate determine Treg cell identity and function. Inflammatory stress destabilizes Treg cell functions (136, 137). The Foxp3 protein undergoes polyubiquitylation and proteasome-based degradation especially under certain stressful conditions. HIF-1α promotes expression of Th17 signature genes and Th17 cell development, but attenuates Treg development by enhancing Foxp3 ubiquitylation and proteasomal degradation (138). Stimulation by proinflammatory cytokines and lipopolysaccharides (LPS) promotes Foxp3 K48-linked polyubiquitination by the E3 ubiquitin ligase, Stub1, in an Hsp70-dependent manner. The absence of endogenous activity of Stub1 or Hsp70 prevents Foxp3 from proteasomal degradation (139).
Upon TCR stimulation, E3 ubiquitin ligase, Cbl-b, together with Stub1, targets Foxp3 for ubiquitylation and degradation; and a deficiency of Cbl-b can partially rescue defective development of thymic Treg cells in Cd28−/− mice (140). Cimetidine (CIM) can promote immune responses, suppress Treg cell function via activation of PI3K-AKT-mTOR signaling and increase of Stub1-mediated degradation of Foxp3, respectively (141). As mentioned above, Foxp3 phosphorylation by NLK prevents its association with the STUB1 and increases Foxp3 protein stabilization (87). These studies highlight the critical role of Stub1 ubiquitin E3 ligase in mediating Foxp3 protein level and Treg suppressive function.
Besides K48-linked ubiquitylation, Foxp3 also undergoes non-proteolytic ubiquitylation. Tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6) mediates K63-linked polyubiquitylation (142). TRAF6 plays an essential role in maintaining Treg cell function to inhibit Th2 type autoimmunity (143). A recent study found that deficiency of TRAF6 in Treg cells compromises Treg suppression in vivo. Mechanistically, TRAF6 interacts with FOXP3 and mediates K63 polyubiquitination at lysine 262 residue, which ensures proper nuclear localization of FOXP3 and facilitates FOXP3 transcriptional activity in Tregs. Deficiency of TRAF6 in Tregs enhances anti-tumor immunity (144). The E3 ubiquitin ligase ring finger protein 31 (RNF31) catalyzes FOXP3 atypical ubiquitylation, and promotes FOXP3 protein stability and Treg suppression. RNF31 expression has been correlated with intratumoral Treg cell activities in gastric cancer, indicating a potential role of RNF31 in tumor immunity (145).
Protein ubiquitylation can be reversed by the deubiquitinases (DUBs, also known as deubiquitinating enzymes) (146). The deubiquitinases consist of five families: ubiquitin C-terminal hydrolases (UCHs), ubiquitin-specific proteases (USPs), ovarian tumor proteases (OTUs), Josephins and JAB1/ MPN/MOV34 metalloenzymes (JAMMs). The UCH, USP, OTU, and Josephin families are Cys proteases, whereas the JAMM/MPN+ family members are zinc metalloproteases (147).
It has been well-documented that deubiquitinases are involved in T cell development, activation, differentiation and tolerance (148). The K63-specific deubiquitinase Cylindromatosis (CYLD) negatively regulates CARMA1, which is required for NF-kB activation and IL-2 receptor signaling. A deficiency of CYLD causes constitutive NF-kB activation and enhanced TGF-b signaling, which increases the frequency of Treg cells in peripheral lymphoid organs and promotes Treg cell differentiation in vitro (149, 150). Similarly, Treg cell number increases in a non-functional CYLD splice variant CYLDex7/8, however, the suppressive capacity is impaired and correlated with decreased expression of CD25 and CTLA4 (151).
USP7 is upregulated in Treg cells, and the ectopic expression of USP7 decreases Foxp3 polyubiquitylation and increases Foxp3 expression. Treg cells pretreated with deubiquitinase inhibitor enhance their modifying functions in adoptive transfer induced colitis (152). In addition, USP7 promotes Treg suppression by enhancing the multimerization of Tip60 and Foxp3 (153). USP21 associates with GATA3 and Foxp3 transcriptional factors. Mice depleted of Usp21 in Treg cells develop Th1-type inflammation (154, 155). USP4 stabilizes the IRF8 protein via a K48-linked deubiquitinase, which promotes the suppressive function of Treg cells (156).
Arginine methylations are mainly catalyzed by the Protein Arginine Methyltranasferase (PRMT) family. PRMT family can be divided into 3 subfamilies: the Type 1 PRMTs include PRMT1, 2, 3, 4 (also known as CARM1), and 6, which asymmetrically methylate target arginine residues; the Type 2 PRMTs include PRMT5 and 9, which symmetrically methylate target arginine residues; the Type 3 PRMT7 mono-methylates target arginine residues (157, 158).
T cell activation induces protein methylation modification (159). Employing isomethionine methyl-SILAC (iMethyl)-SILAC and mass spectrometry approaches, various transcription factors, including Foxp3, that influence T cell differentiation and lineage specificities, have been identified as being methylated (160). FOXP3 can be di-methylated at arginine(R) 51 position, although whether this methylation is asymmetry or symmetry, and which PRMT is responsible for this modification both remain unclear (129). More recent mass spectrometric analysis reveals that human FOXP3 contains several possible di-methylation sites at R27, R51, and R147 residues (161). PRMT1 and PRMT5 are reported to be involved in FOXP3 methylation (162). PRMT5 preferentially binds to FOXP3, and a conditional deletion of PRMT5 gene in Tregs leads to deadly scurfy-like autoimmune diseases. Lymph node Tregs from those animals showed significantly less suppressive function, indicating that PRMT5 is essential for Treg function. Site-mutation of R51K dramatically decreases symmetric arginine signals, as detected by symmetric arginine specific antibody sym10, suggesting that PRMT5 di-methylates FOXP3 at R51. Of note, the arginine methylation at the N-terminal position of FOXP3 regulates Treg function via a Foxp3-DNA-binding-activity independent manner. Pharmacological ablation of PRMT5 activity by DS-437 can enhance the anti-tumor efficacy of anti-erbB2/neu monoclonal antibody target therapy by reducing human Treg functions (161).
Targeting protein methylation modification provides novel insights into clinical therapeutic designing for certain diseases, including autoimmune diseases and cancer. PRMT5 forms a complex with MEP50, an essential cofactor, which then causes higher enzymatic activity and allows the binding to target proteins. PRMT5 uses S-adenosyl methionine (SAM) for transferring a methyl group to the target substrate. It is also known that PRMT5 forms a large complex with different cofactors, for changing its target proteins (158). PRMT5 is highly expressed in a variety of cancers, and several inhibitors have been developed that target PRMT5 for cancer therapy (157). PRMT5 forms a large complex and has several different cofactors; the effect of PRMT5 inhibitors may be dependent on the screening strategy, such as substrate- or SAM- competitive. SAM-competitive inhibitor, DS-437, has more efficient inhibition of FOXP3 methylation than the substrate-competitive inhibitor, EPZ015666 (161). Thus, for targeting FOXP3 methylation in Treg cells, it may rationalize to use SAM-competitive inhibitors. Likewise, Foxp3 associates with PRMT1 and is asymmetrically di-methylated at R48 and R51 positions. FOXP3 methylation and its function is compromised by administration of PRMT1/6 inhibitor MS023 (162). Based on the current findings, more research is needed to clarify the biological significance of the asymmetric methylation on FOXP3 in modulating Treg suppression.
Both lymphoid-organ Treg cells and tissue-resident Treg cells require the expression of the Foxp3 transcription factor. Foxp3 protein functions in transcriptional molecule complexes. Ensemble formation is regulated by various interactions and post-translational modifications (PTMs), including phosphorylation, O-GlcNAcylation, acetylation, ubiquitination, and methylation. Modifications influence each other to orchestrate Foxp3 activity and Treg suppression. However, the currently available, published results are still not sufficient to fully reveal roles of Foxp3 PTMs in modulating Treg suppression, and some caveats should be considered when interpreting the data. For example, to what extent does the ectopic expression of protein in the transfected cell line represent the real situation in Treg cells? Are there potential substrates other than Foxp3 responsible for the phenotypes observed in those enzyme-deficient mice? What are the possible cross-talks among different Foxp3 PTMs under certain tissue- microenvironment? What is the clinical relevance and significance of Foxp3 PTMs? Moreover, are there other types of post-translational modifications occurring on Foxp3 that have not been identified yet, such as lysine crotonylation or succinylation?
The understanding of Foxp3 protein PTMs in modulating Treg suppression may facilitate the design of rational therapies for immune disorders developed in IPEX patients. Based on the high-resolution crystal structure of Foxp3, combination therapy by targeting Foxp3 post-translational modifications with other therapies may enhance therapeutic approaches in a variety of diseases. Moreover, a full understanding of the contribution of other regulatory cells, such as Suppressor T cells, may lead to additional therapeutic alternatives (163–165).
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work is supported by Start Funding of Peking University Health Science Center (BMU2018YJ011) and 2019 Clinical Medicine Plus X- Young Scholars Project, Peking University.
SF is employed by company Seishin Medical Group, Takara Clinic.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer ZZ declared a shared affiliation, with no collaboration, with the author XS to the handling editor at the time of review.
1. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. (1995)155:1151–64.
2. Dhamne C, Chung Y, Alousi AM, Cooper LJ, Tran DQ. Peripheral and thymic foxp3(+) regulatory T cells in search of origin, distinction, and function. Front Immunol. (2013) 4:253. doi: 10.3389/fimmu.2013.00253
3. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. (2003) 4:330–6. doi: 10.1038/ni904
4. Sakaguchi S, Vignali DA, Rudensky AY, Niec RE, Waldmann H. The plasticity and stability of regulatory T cells. Nat Rev Immunol. (2013) 13:461–7. doi: 10.1038/nri3464
5. Feng Y, Arvey A, Chinen T, van der Veeken J, Gasteiger G, Rudensky AY. Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell. (2014) 158:749–63. doi: 10.1016/j.cell.2014.07.031
6. Li X, Liang Y, LeBlanc M, Benner C, Zheng Y. Function of a Foxp3 cis-element in protecting regulatory T cell identity. Cell. (2014) 158:734–48. doi: 10.1016/j.cell.2014.07.030
7. Ogawa C, Tone Y, Tsuda M, Peter C, Waldmann H, Tone M. TGF-beta-mediated Foxp3 gene expression is cooperatively regulated by Stat5, Creb, and AP-1 through CNS2. J Immunol. (2014) 192:475–83. doi: 10.4049/jimmunol.1301892
8. Cuadrado E, van den Biggelaar M, de Kivit S, Chen YY, Slot M, Doubal I, et al. Proteomic analyses of human regulatory T cells reveal adaptations in signaling pathways that protect cellular identity. Immunity. (2018) 48:1046–59.e1046. doi: 10.1016/j.immuni.2018.04.008
9. Piccirillo CA. Environmental sensing and regulation of gene expression in CD4+ T cell subsets. Curr Opin Immunol. (2013) 25:564–70. doi: 10.1016/j.coi.2013.09.006
10. Bjur E, Larsson O, Yurchenko E, Zheng L, Gandin V, Topisirovic I, et al. Distinct translational control in CD4+ T cell subsets. PLoS Genet. (2013) 9:e1003494. doi: 10.1371/journal.pgen.1003494
11. Smigiel KS, Richards E, Srivastava S, Thomas KR, Dudda JC, Klonowski KD, et al. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J Exp Med. (2014) 211:121–36. doi: 10.1084/jem.20131142
12. Campbell DJ. Control of regulatory T cell migration, function, and homeostasis. J Immunol. (2015) 195:2507–13. doi: 10.4049/jimmunol.1500801
13. Sun IH, Oh MH, Zhao L, Patel CH, Arwood ML, Xu W, et al. mTOR complex 1 signaling regulates the generation and function of central and effector Foxp3(+) regulatory T Cells. J Immunol. (2018) 201:481–92. doi: 10.4049/jimmunol.1701477
14. Layman AAK, Deng G, O'Leary CE, Tadros S, Thomas RM, Dybas JM, et al. Ndfip1 restricts mTORC1 signalling and glycolysis in regulatory T cells to prevent autoinflammatory disease. Nat Commun. (2017) 8:15677. doi: 10.1038/ncomms15677
15. Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. (2009) 30:636–45. doi: 10.1016/j.immuni.2009.04.010
16. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. (2008) 8:523–32. doi: 10.1038/nri2343
17. von Boehmer H. Mechanisms of suppression by suppressor T cells. Nat Immunol. (2005) 6:338–44. doi: 10.1038/ni1180
18. Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol. (2007) 8:1353–62. doi: 10.1038/ni1536
19. He J, Zhang X, Wei Y, Sun X, Chen Y, Deng J, et al. Low-dose interleukin-2 treatment selectively modulates CD4(+) T cell subsets in patients with systemic lupus erythematosus. Nat Med. (2016) 22:991–3. doi: 10.1038/nm.4148
20. Cheng G, Yu A, Malek TR. T-cell tolerance and the multi-functional role of IL-2R signaling in T-regulatory cells. Immunol Rev. (2011) 241:63–76. doi: 10.1111/j.1600-065X.2011.01004.x
21. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86, a molecular basis for the cell-extrinsic function of CTLA-4. Science. (2011) 332:600–3. doi: 10.1126/science.1202947
22. Asseman C, Mauze S, Leach MW, Coffman RL, Powrie F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J Exp Med. (1999) 190:995–1004. doi: 10.1084/jem.190.7.995
23. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. (2007) 450:566–9. doi: 10.1038/nature06306
24. Joetham A, Takeda K, Taube C, Miyahara N, Matsubara S, Koya T, et al. Naturally occurring lung CD4(+)CD25(+) T cell regulation of airway allergic responses depends on IL-10 induction of TGF-beta. J Immunol. (2007) 178:1433–42. doi: 10.4049/jimmunol.178.3.1433
25. Akkaya B, Oya Y, Akkaya M, Al Souz J, Holstein AH, Kamenyeva O, et al. Regulatory T cells mediate specific suppression by depleting peptide-MHC class II from dendritic cells. Nat Immunol. (2019) 20:218–31. doi: 10.1038/s41590-018-0280-2
26. Li MO, Rudensky AY. T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat Rev Immunol. (2016) 16:220–33. doi: 10.1038/nri.2016.26
27. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med. (2015) 7:315ra189. doi: 10.1126/scitranslmed.aad4134
28. Hull CM, Nickolay LE, Estorninho M, Richardson MW, Riley JL, Peakman M, et al. Generation of human islet-specific regulatory T cells by TCR gene transfer. J Autoimmun. (2017) 79:63–73. doi: 10.1016/j.jaut.2017.01.001
29. Cobbold SP. The mTOR pathway and integrating immune regulation. Immunology. (2013) 140:391–8. doi: 10.1111/imm.12162
30. Saravia J, Chapman NM, Chi H. Helper T cell differentiation. Cell Mol Immunol. (2019) 16:634–43. doi: 10.1038/s41423-019-0220-6
31. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. (2011) 186:3299–303. doi: 10.4049/jimmunol.1003613
32. Procaccini C, Carbone F, Di Silvestre D, Brambilla F, De Rosa V, Galgani M, et al. The proteomic landscape of human ex vivo regulatory and conventional T cells reveals specific metabolic requirements. Immunity. (2016) 44:406–21. doi: 10.1016/j.immuni.2016.01.028
33. Howie D, Waldmann H, Cobbold S. Nutrient sensing via mTOR in T cells maintains a tolerogenic microenvironment. Front Immunol. (2014) 5:409. doi: 10.3389/fimmu.2014.00409
34. Beier UH, Angelin A, Akimova T, Wang L, Liu Y, Xiao H, et al. Essential role of mitochondrial energy metabolism in Foxp3(+) T-regulatory cell function and allograft survival. FASEB J. (2015) 29:2315–26. doi: 10.1096/fj.14-268409
35. Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, Martinez-Reyes I, et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature. (2019) 565:495–9. doi: 10.1038/s41586-018-0846-z
36. Howie D, Ten Bokum A, Necula AS, Cobbold SP, Waldmann H. The role of lipid metabolism in T lymphocyte differentiation and survival. Front Immunol. (2017) 8:1949. doi: 10.3389/fimmu.2017.01949
37. Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, et al. Foxp3 and Toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat Immunol. (2016) 17:1459–66. doi: 10.1038/ni.3577
38. Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. (2009) 30:832–44. doi: 10.1016/j.immuni.2009.04.014
39. Procaccini C, De Rosa V, Galgani M, Abanni L, Cali G, Porcellini A, et al. An oscillatory switch in mTOR kinase activity sets regulatory T cell responsiveness. Immunity. (2010) 33:929–41. doi: 10.1016/j.immuni.2010.11.024
40. Charbonnier LM, Cui Y, Stephen-Victor E, Harb H, Lopez D, Bleesing JJ, et al. Functional reprogramming of regulatory T cells in the absence of Foxp3. Nat Immunol. (2019) 20:1208–19. doi: 10.1038/s41590-019-0442-x
41. Ulges A, Schmitt E, Becker C, Bopp T. Context- and tissue-specific regulation of immunity and tolerance by regulatory T cells. Adv Immunol. (2016) 132:1–46. doi: 10.1016/bs.ai.2016.08.002
42. Alvarez F, Istomine R, Shourian M, Pavey N, Al-Aubodah TA, Qureshi S, et al. The alarmins IL-1 and IL-33 differentially regulate the functional specialisation of Foxp3(+) regulatory T cells during mucosal inflammation. Mucosal Immunol. (2019) 12:746–60. doi: 10.1038/s41385-019-0153-5
43. Nie H, Zheng Y, Li R, Guo TB, He D, Fang L, et al. Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-alpha in rheumatoid arthritis. Nat Med. (2013) 19:322–8. doi: 10.1038/nm.3085
44. Burzyn D, Benoist C, Mathis D. Regulatory T cells in nonlymphoid tissues. Nat Immunol. (2013) 14:1007–13. doi: 10.1038/ni.2683
45. DiSpirito JR, Zemmour D, Ramanan D, Cho J, Zilionis R, Klein AM, et al. Molecular diversification of regulatory T cells in nonlymphoid tissues. Sci Immunol. (2018) 3:eaat5861. doi: 10.1126/sciimmunol.aat5861
46. Panduro M, Benoist C, Mathis D. Tissue tregs. Annu Rev Immunol. (2016) 34:609–33. doi: 10.1146/annurev-immunol-032712-095948
47. Kupper TS, Fuhlbrigge RC. Immune surveillance in the skin: mechanisms and clinical consequences. Nat Rev Immunol. (2004) 4:211–22. doi: 10.1038/nri1310
48. Iellem A, Colantonio L, D'Ambrosio D. Skin-versus gut-skewed homing receptor expression and intrinsic CCR4 expression on human peripheral blood CD4+CD25+ suppressor T cells. Eur J Immunol. (2003) 33:1488–96. doi: 10.1002/eji.200323658
49. Sather BD, Treuting P, Perdue N, Miazgowicz M, Fontenot JD, Rudensky AY, et al. Altering the distribution of Foxp3(+) regulatory T cells results in tissue-specific inflammatory disease. J Exp Med. (2007) 204:1335–47. doi: 10.1084/jem.20070081
50. Nosbaum A, Prevel N, Truong HA, Mehta P, Ettinger M, Scharschmidt TC, et al. Cutting edge: regulatory T cells facilitate cutaneous wound healing. J Immunol. (2016) 196:2010–4. doi: 10.4049/jimmunol.1502139
51. Ali N, Zirak B, Rodriguez RS, Pauli ML, Truong HA, Lai K, et al. Regulatory T cells in skin facilitate epithelial stem cell differentiation. Cell. (2017) 169:1119–29.e1111. doi: 10.1016/j.cell.2017.05.002
52. Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, et al. Peripheral education of the immune system by colonic commensal microbiota. Nature. (2011) 478:250–4. doi: 10.1038/nature10434
53. Scharschmidt TC, Vasquez KS, Truong HA, Gearty SV, Pauli ML, Nosbaum A, et al. A wave of regulatory T cells into neonatal skin mediates tolerance to commensal microbes. Immunity. (2015) 43:1011–21. doi: 10.1016/j.immuni.2015.10.016
54. Cipolletta D, Kolodin D, Benoist C, Mathis D. Tissular T(regs): a unique population of adipose-tissue-resident Foxp3+CD4+ T cells that impacts organismal metabolism. Semin Immunol. (2011) 23:431–7. doi: 10.1016/j.smim.2011.06.002
55. Cipolletta D. Adipose tissue-resident regulatory T cells: phenotypic specialization, functions and therapeutic potential. Immunology. (2014) 142:517–25. doi: 10.1111/imm.12262
56. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. (2009) 15:930–9. doi: 10.1038/nm.2002
57. Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med. (2009) 15:921–9. doi: 10.1038/nm.2001
58. Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, et al. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. (2012) 486:549–53. doi: 10.1038/nature11132
59. Schmidleithner L, Thabet Y, Schonfeld E, Kohne M, Sommer D, Abdullah Z, et al. Enzymatic activity of HPGD in Treg cells suppresses Tconv cells to maintain adipose tissue homeostasis and prevent metabolic dysfunction. Immunity. (2019) 50:1232–48.e1214. doi: 10.1016/j.immuni.2019.03.014
60. Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. (2012) 12:298–306. doi: 10.1038/nrc3245
61. Marabelle A, Kohrt H, Sagiv-Barfi I, Ajami B, Axtell RC, Zhou G, et al. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J Clin Invest. (2013) 123:2447–63. doi: 10.1172/JCI64859
62. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. (2013) 210:1695–710. doi: 10.1084/jem.20130579
63. Sharma N, Vacher J, Allison JP. TLR1/2 ligand enhances antitumor efficacy of CTLA-4 blockade by increasing intratumoral Treg depletion. Proc Natl Acad Sci USA. (2019) 116:10453–62. doi: 10.1073/pnas.1819004116
64. Deng G. Tumor-infiltrating regulatory T cells: origins and features. Am J Clin Exp Immunol. (2018) 7:81–7.
65. Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat Immunol. (2017) 18:1332–41. doi: 10.1038/ni.3868
66. Waight JD, Takai S, Marelli B, Qin G, Hance KW, Zhang D, et al. Cutting edge: epigenetic regulation of Foxp3 defines a stable population of CD4+ regulatory T cells in tumors from mice and humans. J Immunol. (2015) 194:878–82. doi: 10.4049/jimmunol.1402725
67. Angelin A, Gil-de-Gomez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. (2017) 25:1282–93.e1287. doi: 10.1016/j.cmet.2016.12.018
68. Pacella I, Procaccini C, Focaccetti C, Miacci S, Timperi E, Faicchia D, et al. Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc Natl Acad Sci USA. (2018) 115:E6546–55. doi: 10.1073/pnas.1720113115
69. Lam EW, Brosens JJ, Gomes AR, Koo CY. Forkhead box proteins: tuning forks for transcriptional harmony. Nat Rev Cancer. (2013) 13:482–95. doi: 10.1038/nrc3539
70. Konopacki C, Pritykin Y, Rubtsov Y, Leslie CS, Rudensky AY. Transcription factor Foxp1 regulates Foxp3 chromatin binding and coordinates regulatory T cell function. Nat Immunol. (2019) 20:232–42. doi: 10.1038/s41590-018-0291-z
71. Li B, Samanta A, Song X, Iacono KT, Brennan P, Chatila TA, et al. FOXP3 is a homo-oligomer and a component of a supramolecular regulatory complex disabled in the human XLAAD/IPEX autoimmune disease. Int Immunol. (2007) 19:825–35. doi: 10.1093/intimm/dxm043
72. Rudra D, deRoos P, Chaudhry A, Niec RE, Arvey A, Samstein RM, et al. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat Immunol. (2012) 13:1010–9. doi: 10.1038/ni.2402
73. Bennett CL, Yoshioka R, Kiyosawa H, Barker DF, Fain PR, Shigeoka AO, et al. X-linked syndrome of polyendocrinopathy, immune dysfunction, and diarrhea maps to Xp11.23–Xq13.3. Am J Hum Genet. (2000) 66:461–8. doi: 10.1086/302761
74. d'Hennezel E, Bin Dhuban K, Torgerson T, Piccirillo CA. The immunogenetics of immune dysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. J Med Genet. (2012) 49:291–302. doi: 10.1136/jmedgenet-2012-100759
75. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. (2003) 299:1057–61. doi: 10.1126/science.1079490
76. Lopes JE, Torgerson TR, Schubert LA, Anover SD, Ocheltree EL, Ochs HD, et al. Analysis of FOXP3 reveals multiple domains required for its function as a transcriptional repressor. J Immunol. (2006) 177:3133–42. doi: 10.4049/jimmunol.177.5.3133
77. Marson A, Kretschmer K, Frampton GM, Jacobsen ES, Polansky JK, MacIsaac KD, et al. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature. (2007) 445:931–5. doi: 10.1038/nature05478
78. Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, Rudensky AY. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. (2007) 445:936–40. doi: 10.1038/nature05563
79. Hori S. The Foxp3 interactome: a network perspective of T(reg) cells. Nat Immunol. (2012) 13:943–5. doi: 10.1038/ni.2424
80. Bacchetta R, Barzaghi F, Roncarolo MG. From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation. Ann N Y Acad Sci. (2018) 1417:5–22. doi: 10.1111/nyas.13011
81. Patey-Mariaud de Serre N, Canioni D, Ganousse S, Rieux-Laucat F, Goulet O, Ruemmele F, et al. Digestive histopathological presentation of IPEX syndrome. Mod Pathol. (2009) 22:95–102. doi: 10.1038/modpathol.2008.161
82. Gambineri E, Perroni L, Passerini L, Bianchi L, Doglioni C, Meschi F, et al. Clinical and molecular profile of a new series of patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome: inconsistent correlation between forkhead box protein 3 expression and disease severity. J Allergy Clin Immunol. (2008) 122:1105–12.e1101. doi: 10.1016/j.jaci.2008.09.027
83. d'Hennezel E, Ben-Shoshan M, Ochs HD, Torgerson TR, Russell LJ, Lejtenyi C, et al. FOXP3 forkhead domain mutation and regulatory T cells in the IPEX syndrome. N Engl J Med. (2009) 361:1710–3. doi: 10.1056/NEJMc0907093
84. Tan H, Yang K, Li Y, Shaw TI, Wang Y, Blanco DB, et al. Integrative proteomics and phosphoproteomics profiling reveals dynamic signaling networks and bioenergetics pathways underlying T cell activation. Immunity. (2017) 46:488–503. doi: 10.1016/j.immuni.2017.02.010
85. Gavin MA, Torgerson TR, Houston E, DeRoos P, Ho WY, Stray-Pedersen A, et al. Single-cell analysis of normal and FOXP3-mutant human T cells: FOXP3 expression without regulatory T cell development. Proc Natl Acad Sci USA. (2006) 103:6659–64. doi: 10.1073/pnas.0509484103
86. Samanta A, Li B, Song X, Bembas K, Zhang G, Katsumata M, et al. TGF-beta and IL-6 signals modulate chromatin binding and promoter occupancy by acetylated FOXP3. Proc Natl Acad Sci USA. (2008) 105:14023–7. doi: 10.1073/pnas.0806726105
87. Fleskens V, Minutti CM, Wu X, Wei P, Pals C, McCrae J, et al. Nemo-like kinase drives Foxp3 Stability and is critical for maintenance of immune tolerance by regulatory T cells. Cell Rep. (2019) 26:3600–12.e3606. doi: 10.1016/j.celrep.2019.02.087
88. Chunder N, Wang L, Chen C, Hancock WW, Wells AD. Cyclin-dependent kinase 2 controls peripheral immune tolerance. J Immunol. (2012) 189:5659–66. doi: 10.4049/jimmunol.1202313
89. Morawski PA, Mehra P, Chen C, Bhatti T, Wells AD. Foxp3 protein stability is regulated by cyclin-dependent kinase 2. J Biol Chem. (2013) 288:24494–502. doi: 10.1074/jbc.M113.467704
90. Deng G, Xiao Y, Zhou Z, Nagai Y, Zhang H, Li B, Greene MI. Molecular and biological role of the FOXP3 N-terminal domain in immune regulation by T regulatory/suppressor cells. Exp Mol Pathol. (2012) 93:334–8. doi: 10.1016/j.yexmp.2012.09.013
91. Hancock WW, Ozkaynak E. Three distinct domains contribute to nuclear transport of murine Foxp3. PLoS ONE. (2009) 4:e7890. doi: 10.1371/journal.pone.0007890
92. Deng G, Nagai Y, Xiao Y, Li Z, Dai S, Ohtani T, et al. (2015). Pim-2 kinase influences regulatory T cell function and stability by mediating Foxp3 protein N-terminal phosphorylation. J Biol Chem. (2009) 290:20211–20. doi: 10.1074/jbc.M115.638221
93. Basu S, Golovina T, Mikheeva T, June CH, Riley JL. Cutting edge: Foxp3-mediated induction of pim 2 allows human T regulatory cells to preferentially expand in rapamycin. J Immunol. (2008) 180:5794–8. doi: 10.4049/jimmunol.180.9.5794
94. Fox CJ, Hammerman PS, Thompson CB. The Pim kinases control rapamycin-resistant T cell survival and activation. J Exp Med. (2005) 201:259–66. doi: 10.1084/jem.20042020
95. Li Z, Lin F, Zhuo C, Deng G, Chen Z, Yin S, et al. PIM1 kinase phosphorylates the human transcription factor FOXP3 at serine 422 to negatively regulate its activity under inflammation. J Biol Chem. (2014) 289:26872–81. doi: 10.1074/jbc.M114.586651
96. Lin F, Luo X, Tsun A, Li Z, Li D, Li B. Kaempferol enhances the suppressive function of Treg cells by inhibiting FOXP3 phosphorylation. Int Immunopharmacol. (2015) 28:859–65. doi: 10.1016/j.intimp.2015.03.044
97. de Zoeten EF, Lee I, Wang L, Chen C, Ge G, Wells AD, et al. Foxp3 processing by proprotein convertases and control of regulatory T cell function. J Biol Chem. (2009) 284:5709–16. doi: 10.1074/jbc.M807322200
98. Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. (2007) 13:1299–307. doi: 10.1038/nm1652
99. Nakahira K, Morita A, Kim NS, Yanagihara I. Phosphorylation of FOXP3 by LCK downregulates MMP9 expression and represses cell invasion. PLoS ONE. (2013) 8:e77099. doi: 10.1371/journal.pone.0077099
100. Liu B, Salgado OC, Singh S, Hippen KL, Maynard JC, Burlingame AL, et al. The lineage stability and suppressive program of regulatory T cells require protein O-GlcNAcylation. Nat Commun. (2019) 10:354. doi: 10.1038/s41467-019-08300-3
101. Verdin E, Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol. (2015) 16:258–64. doi: 10.1038/nrm3931
102. Polevoda B, Sherman F. Nalpha -terminal acetylation of eukaryotic proteins. J Biol Chem. (2000) 275:36479–82. doi: 10.1074/jbc.R000023200
103. VanDrisse CM, Escalante-Semerena JC. Protein acetylation in bacteria. Annu Rev Microbiol. (2019) 73:111–32. doi: 10.1146/annurev-micro-020518-115526
104. Yang XJ. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res. (2004) 32:959–76. doi: 10.1093/nar/gkh252
105. Xiao Y, Li B, Zhou Z, Hancock WW, Zhang H, Greene MI. Histone acetyltransferase mediated regulation of FOXP3 acetylation and Treg function. Curr Opin Immunol. (2010) 22:583–91. doi: 10.1016/j.coi.2010.08.013
106. Ikura T, Ogryzko VV, Grigoriev M, Groisman R, Wang J, Horikoshi M, et al. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell. (2000) 102:463–73. doi: 10.1016/S0092-8674(00)00051-9
107. Ai W, Zheng H, Yang X, Liu Y, Wang TC. Tip60 functions as a potential corepressor of KLF4 in regulation of HDC promoter activity. Nucleic Acids Res. (2007) 35:6137–49. doi: 10.1093/nar/gkm656
108. Xiao H, Chung J, Kao HY, Yang YC. Tip60 is a co-repressor for STAT3. J Biol Chem. (2003) 278:11197–204. doi: 10.1074/jbc.M210816200
109. Brady ME, Ozanne DM, Gaughan L, Waite I, Cook S, Neal DE, et al. Tip60 is a nuclear hormone receptor coactivator. J Biol Chem. (1999) 274:17599–604. doi: 10.1074/jbc.274.25.17599
110. Li B, Samanta A, Song X, Iacono KT, Bembas K, Tao R, et al. FOXP3 interactions with histone acetyltransferase and class II histone deacetylases are required for repression. Proc Natl Acad Sci USA. (2007) 104:4571–6. doi: 10.1073/pnas.0700298104
111. van Loosdregt J, Vercoulen Y, Guichelaar T, Gent YY, Beekman JM, van Beekum O, et al. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood. (2010) 115:965–74. doi: 10.1182/blood-2009-02-207118
112. Kwon HS, Lim HW, Wu J, Schnolzer M, Verdin E, Ott M. Three novel acetylation sites in the Foxp3 transcription factor regulate the suppressive activity of regulatory T cells. J Immunol. (2012) 188:2712–21. doi: 10.4049/jimmunol.1100903
113. Liu Y, Wang L, Predina J, Han R, Beier UH, Wang LC, et al. Inhibition of p300 impairs Foxp3(+) T regulatory cell function and promotes antitumor immunity. Nat Med. (2013) 19:1173–7. doi: 10.1038/nm.3286
114. Lau OD, Kundu TK, Soccio RE, Ait-Si-Ali S, Khalil EM, Vassilev A, et al. HATs off: selective synthetic inhibitors of the histone acetyltransferases p300 and PCAF. Mol Cell. (2000) 5:589–95. doi: 10.1016/S1097-2765(00)80452-9
115. Liu Y, Wang L, Han R, Beier UH, Akimova T, Bhatti T, et al. Two histone/protein acetyltransferases, CBP and p300, are indispensable for Foxp3+ T-regulatory cell development and function. Mol Cell Biol. (2014) 34:3993–4007. doi: 10.1128/MCB.00919-14
116. Xiao Y, Nagai Y, Deng G, Ohtani T, Zhu Z, Zhou Z, et al. Dynamic interactions between TIP60 and p300 regulate FOXP3 function through a structural switch defined by a single lysine on TIP60. Cell Rep. (2014) 7:1471–80. doi: 10.1016/j.celrep.2014.04.021
117. Bin Dhuban K, d'Hennezel E, Nagai Y, Xiao Y, Shao S, Istomine R, et al. Suppression by human FOXP3(+) regulatory T cells requires FOXP3-TIP60 interactions. Sci Immunol. (2017) 2:eaai9297. doi: 10.1126/sciimmunol.aai9297
118. Deng G, Song X, Greene MI. FoxP3 in Treg cell biology: a molecular and structural perspective. Clin Exp Immunol. (2019). doi: 10.1111/cei.13357. [Epub ahead of print].
119. Song X, Li B, Xiao Y, Chen C, Wang Q, Liu Y, et al. (2012). Structural and biological features of FOXP3 dimerization relevant to regulatory T cell function. Cell Rep. (2017) 1:665–75. doi: 10.1016/j.celrep.2012.04.012
120. Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. (2004) 338:17–31. doi: 10.1016/j.jmb.2004.02.006
121. Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, et al. Chemical phylogenetics of histone deacetylases. Nat Chem Biol. (2010) 6:238–43. doi: 10.1038/nchembio.313
122. Akimova T, Beier UH, Liu Y, Wang L, Hancock WW. Histone/protein deacetylases and T-cell immune responses. Blood. (2012) 119:2443–51. doi: 10.1182/blood-2011-10-292003
123. Wang L, de Zoeten EF, Greene MI, Hancock WW. Immunomodulatory effects of deacetylase inhibitors: therapeutic targeting of FOXP3+ regulatory T cells. Nat Rev Drug Discov. (2009) 8:969–81. doi: 10.1038/nrd3031
124. Wang L, Beier UH, Akimova T, Dahiya S, Han R, Samanta A, et al. Histone/protein deacetylase inhibitor therapy for enhancement of Foxp3+ T-regulatory cell function posttransplantation. Am J Transplant. (2018) 18:1596–603. doi: 10.1111/ajt.14749
125. Akimova T, Ge G, Golovina T, Mikheeva T, Wang L, Riley JL, et al. Histone/protein deacetylase inhibitors increase suppressive functions of human FOXP3+ Tregs. Clin Immunol. (2010) 136:348–63. doi: 10.1016/j.clim.2010.04.018
126. Saouaf SJ, Li B, Zhang G, Shen Y, Furuuchi N, Hancock WW, et al. Deacetylase inhibition increases regulatory T cell function and decreases incidence and severity of collagen-induced arthritis. Exp Mol Pathol. (2009) 87:99–104. doi: 10.1016/j.yexmp.2009.06.003
127. Beier UH, Akimova T, Liu Y, Wang L, Hancock WW. Histone/protein deacetylases control Foxp3 expression and the heat shock response of T-regulatory cells. Curr Opin Immunol. (2011) 23:670–8. doi: 10.1016/j.coi.2011.07.002
128. Daenthanasanmak A, Iamsawat S, Chakraborty P, Nguyen HD, Bastian D, Liu C, et al. Targeting Sirt-1 controls GVHD by inhibiting T-cell allo-response and promoting Treg stability in mice. Blood. (2019) 133:266–79. doi: 10.1182/blood-2018-07-863233
129. Beier UH, Wang L, Han R, Akimova T, Liu Y, Hancock WW. Histone deacetylases 6 and 9 and sirtuin-1 control Foxp3+ regulatory T cell function through shared and isoform-specific mechanisms. Sci Signal. (2012) 5:ra45. doi: 10.1126/scisignal.2002873
130. de Zoeten EF, Wang L, Sai H, Dillmann WH, Hancock WW. Inhibition of HDAC9 increases T regulatory cell function and prevents colitis in mice. Gastroenterology. (2010) 138:583–94. doi: 10.1053/j.gastro.2009.10.037
131. de Zoeten EF, Wang L, Butler K, Beier UH, Akimova T, Sai H, et al. Histone deacetylase 6 and heat shock protein 90 control the functions of Foxp3(+) T-regulatory cells. Mol Cell Biol. (2011) 31:2066–78. doi: 10.1128/MCB.05155-11
132. Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. (2001) 70:503–33. doi: 10.1146/annurev.biochem.70.1.503
133. Adhikari A, Chen ZJ. Diversity of polyubiquitin chains. Dev Cell. (2009) 16:485–6. doi: 10.1016/j.devcel.2009.04.001
134. Bhoj VG, Chen ZJ. Ubiquitylation in innate and adaptive immunity. Nature. (2009) 458:430–7. doi: 10.1038/nature07959
135. Dybas JM, O'Leary CE, Ding H, Spruce LA, Seeholzer SH, Oliver PM. Integrative proteomics reveals an increase in non-degradative ubiquitylation in activated CD4(+) T cells. Nat Immunol. (2019) 20:747–55. doi: 10.1038/s41590-019-0381-6
136. Komatsu N, Mariotti-Ferrandiz ME, Wang Y, Malissen B, Waldmann H, Hori S. Heterogeneity of natural Foxp3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc Natl Acad Sci USA. (2009) 106:1903–8. doi: 10.1073/pnas.0811556106
137. Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, et al. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity. (2008) 28:687–97. doi: 10.1016/j.immuni.2008.03.016
138. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. (2011) 146:772–84. doi: 10.1016/j.cell.2011.07.033
139. Chen Z, Barbi J, Bu S, Yang HY, Li Z, Gao Y, et al. The ubiquitin ligase Stub1 negatively modulates regulatory T cell suppressive activity by promoting degradation of the transcription factor Foxp3. Immunity. (2013) 39:272–85. doi: 10.1016/j.immuni.2013.08.006
140. Zhao Y, Guo H, Qiao G, Zucker M, Langdon WY, Zhang J. E3 ubiquitin ligase Cbl-b regulates thymic-derived CD4+CD25+ regulatory T cell development by targeting Foxp3 for ubiquitination. J Immunol. (2015) 194:1639–45. doi: 10.4049/jimmunol.1402434
141. Zhang Y, Chen Z, Luo X, Wu B, Li B, Wang B. Cimetidine down-regulates stability of Foxp3 protein via Stub1 in Treg cells. Hum Vaccin Immunother. (2016) 12:2512–8. doi: 10.1080/21645515.2016.1191719
142. Fu TM, Shen C, Li Q, Zhang P, Wu H. Mechanism of ubiquitin transfer promoted by TRAF6. Proc Natl Acad Sci USA. (2018) 115:1783–8. doi: 10.1073/pnas.1721788115
143. Muto G, Kotani H, Kondo T, Morita R, Tsuruta S, Kobayashi T, et al. TRAF6 is essential for maintenance of regulatory T cells that suppress Th2 type autoimmunity. PLoS ONE. (2013) 8:e74639. doi: 10.1371/journal.pone.0074639
144. Ni X, Kou W, Gu J, Wei P, Wu X, Peng H, et al. TRAF6 directs FOXP3 localization and facilitates regulatory T-cell function through K63-linked ubiquitination. EMBO J. (2019) 38:e99766. doi: 10.15252/embj.201899766
145. Zhu F, Yi G, Liu X, Zhu F, Zhao A, Wang A, et al. Ring finger protein 31-mediated atypical ubiquitination stabilizes forkhead box P3 and thereby stimulates regulatory T-cell function. J Biol Chem. (2018) 293:20099–111. doi: 10.1074/jbc.RA118.005802
146. Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell. (2005) 123:773–86. doi: 10.1016/j.cell.2005.11.007
147. Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. (2009) 10:550–63. doi: 10.1038/nrm2731
148. Yang XD, Sun SC. Deubiquitinases as pivotal regulators of T cell functions. Front Med. (2018) 12:451–62. doi: 10.1007/s11684-018-0651-y
149. Lee AJ, Wu X, Cheng H, Zhou X, Cheng X, Sun SC. CARMA1 regulation of regulatory T cell development involves modulation of interleukin-2 receptor signaling. J Biol Chem. (2010) 285:15696–703. doi: 10.1074/jbc.M109.095190
150. Zhao Y, Thornton AM, Kinney MC, Ma CA, Spinner JJ, Fuss IJ, et al. The deubiquitinase CYLD targets Smad7 protein to regulate transforming growth factor beta (TGF-beta) signaling and the development of regulatory T cells. J Biol Chem. (2011) 286:40520–30. doi: 10.1074/jbc.M111.292961
151. Reissig S, Hovelmeyer N, Weigmann B, Nikolaev A, Kalt B, Wunderlich TF, et al. The tumor suppressor CYLD controls the function of murine regulatory T cells. J Immunol. (2012) 189:4770–6. doi: 10.4049/jimmunol.1201993
152. van Loosdregt J, Fleskens V, Fu J, Brenkman AB, Bekker CP, Pals CE, et al. Stabilization of the transcription factor Foxp3 by the deubiquitinase USP7 increases Treg-cell-suppressive capacity. Immunity. (2013) 39:259–71. doi: 10.1016/j.immuni.2013.05.018
153. Wang L, Kumar S, Dahiya S, Wang F, Wu J, Newick K, et al. Ubiquitin-specific protease-7 inhibition impairs Tip60-dependent Foxp3+ T-regulatory cell function and promotes antitumor immunity. EBio Med. (2016) 13:99–112. doi: 10.1016/j.ebiom.2016.10.018
154. Li Y, Lu Y, Wang S, Han Z, Zhu F, Ni Y, et al. USP21 prevents the generation of T-helper-1-like Treg cells. Nat Commun. (2016) 7:13559. doi: 10.1038/ncomms13559
155. Zhang J, Chen C, Hou X, Gao Y, Lin F, Yang J, et al. Identification of the E3 deubiquitinase ubiquitin-specific peptidase 21 (USP21) as a positive regulator of the transcription factor GATA3. J Biol Chem. (2013) 288:9373–82. doi: 10.1074/jbc.M112.374744
156. Lin R, Nie J, Ren J, Liang R, Li D, Wang P, et al. USP4 interacts and positively regulates IRF8 function via K48-linked deubiquitination in regulatory T cells. FEBS Lett. (2017) 591:1677–86. doi: 10.1002/1873-3468.12668
157. Jarrold J, Davies CC. PRMTs and arginine methylation: cancer's best-kept secret? Trends Mol Med. (2019). doi: 10.1016/j.molmed.2019.05.007. [Epub ahead of print].
158. Stopa N, Krebs JE, Shechter D. The PRMT5 arginine methyltransferase: many roles in development, cancer and beyond. Cell Mol Life Sci. (2015) 72:2041–59. doi: 10.1007/s00018-015-1847-9
159. Blanchet F, Cardona A, Letimier FA, Hershfield MS, Acuto O. CD28 costimulatory signal induces protein arginine methylation in T cells. J Exp Med. (2005) 202:371–7. doi: 10.1084/jem.20050176
160. Geoghegan V, Guo A, Trudgian D, Thomas B, Acuto O. Comprehensive identification of arginine methylation in primary T cells reveals regulatory roles in cell signalling. Nat Commun. (2015) 6:6758. doi: 10.1038/ncomms7758
161. Nagai Y, Ji MQ, Zhu F, Xiao Y, Tanaka Y, Kambayashi T, et al. PRMT5 associates with the FOXP3 homomer and when disabled enhances targeted p185(erbB2/neu) tumor immunotherapy. Front Immunol. (2019) 10:174. doi: 10.3389/fimmu.2019.00174
162. Kagoya Y, Saijo H, Matsunaga Y, Guo T, Saso K, Anczurowski M, et al. Arginine methylation of FOXP3 is crucial for the suppressive function of regulatory T cells. J Autoimmun. (2019) 97:10–21. doi: 10.1016/j.jaut.2018.09.011
163. Fujimoto S, Greene MI, Sehon AH. Regualtion of the immune response to tumor antigens. I. Immunosuppressor cells in tumor-bearing hosts. J Immunol. (1976) 116:791–9.
164. Fujimoto S, Greene MI, Sehon AH. Regulation of the immune response to tumor antigens. II. The nature of immunosuppressor cells in tumor-bearing hosts. J Immunol. (1976) 116:800–6.
Keywords: regulatory T cells, Foxp3, post-translational modification, phosphorylation, O-GlcNAcylation, acetylation, ubiquitylation, methylation
Citation: Deng G, Song X, Fujimoto S, Piccirillo CA, Nagai Y and Greene MI (2019) Foxp3 Post-translational Modifications and Treg Suppressive Activity. Front. Immunol. 10:2486. doi: 10.3389/fimmu.2019.02486
Received: 23 July 2019; Accepted: 04 October 2019;
Published: 18 October 2019.
Edited by:
Lucy S. K. Walker, University College London, United KingdomReviewed by:
Jochen Huehn, Helmholtz Center for Infection Research, GermanyCopyright © 2019 Deng, Song, Fujimoto, Piccirillo, Nagai and Greene. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guoping Deng, Z2RlbmdAcGt1LmVkdS5jbg==; Mark I. Greene, Z3JlZW5lQHJlby5tZWQudXBlbm4uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.