 Mubashir Ahmad
Mubashir Ahmad Yasmine Hachemi
Yasmine Hachemi Kevin Paxian
Kevin Paxian Florian Mengele1,2
Florian Mengele1,2 Mascha Koenen
Mascha Koenen Jan Tuckermann
Jan Tuckermann
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 17 October 2019
Sec. Molecular Innate Immunity
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02460
This article is part of the Research TopicInnate Immunity in the Context of OsteoimmunologyView all 9 articles
Glucocorticoids (GCs) are known to have a strong impact on the immune system, metabolism, and bone homeostasis. While these functions have been long investigated separately in immunology, metabolism, or bone biology, the understanding of how GCs regulate the cellular cross-talk between innate immune cells, mesenchymal cells, and other stromal cells has been garnering attention rather recently. Here we review the recent findings of GC action in osteoporosis, inflammatory bone diseases (rheumatoid and osteoarthritis), and bone regeneration during fracture healing. We focus on studies of pre-clinical animal models that enable dissecting the role of GC actions in innate immune cells, stromal cells, and bone cells using conditional and function-selective mutant mice of the GC receptor (GR), or mice with impaired GC signaling. Importantly, GCs do not only directly affect cellular functions, but also influence the cross-talk between mesenchymal and immune cells, contributing to both beneficial and adverse effects of GCs. Given the importance of endogenous GCs as stress hormones and the wide prescription of pharmaceutical GCs, an improved understanding of GC action is decisive for tackling inflammatory bone diseases, osteoporosis, and aging.
Glucocorticoids (GCs) form one major axis of the stress response (1) and are used as immunosuppressive therapeutics in a variety of inflammatory bone diseases (2, 3). Strong impact on innate immune cells, namely macrophages, dendritic cells, and mast cells, contribute to the inhibition of inflammation. On the other hand, GCs are known to cause the most frequent secondary osteoporosis at conditions of high GC exposure. In this process myeloid cells, osteoclasts, and mesenchymal cells and their derivatives, chondrocytes, osteoblasts, and osteocytes are affected. Whereas, the cell-autonomous roles of GCs acting via the nuclear glucocorticoid receptor (GR) had been investigated intensively, the knowledge about the influence of GCs on cross-talk between innate immune cells, mesenchymal cells, and bone cells is scarce. How GCs act on cellular interactions in the osteo-immunological network is currently unraveled and is subject to this review.
Two different axes initiate the human physiological reaction to stress. While the activation of sympathetic-adrenal medulla (SAM)-axis starts a short-term stress reactions, long-term stress responses are mediated by the hypothalamus-pituitary-adrenal (HPA)-axis. Stress exposure results in the releases of corticotrophin-releasing hormone (CRH) from the hypothalamus, causing the synthesis of adrenocorticotropic hormone (ACTH) in the anterior pituitary gland, which activates the production of GCs in the adrenal cortex via induction of key enzymes of steroid synthesis (4).
Under long-term stress conditions GC release from the adrenal cortex also results in diverse physiological adaptations. Cortisol activates gluconeogenesis in the liver, decreases pancreatic insulin secretion, and promotes the release of glucagon. Furthermore, blood pressure elevates, the effect of catecholamines is potentiated, and a mild sodium/water-retention induced (5).
Since the first successful treatment of arthritis (6), GCs have been in frequent use and approximately 3% of the elderly population are being treated with GCs (7, 8), to reduce inflammatory symptoms in acute and chronic inflammatory diseases, including rheumatoid and osteoarthritis.
Adverse side effects of GCs on the human body have been observed upon extended treatment with daily prednisolone-doses of 7.5 mg and above. Besides the Cushingoid phenotype and osteoporosis, metabolic side effects as peripheral insulin resistance, type 2 diabetes and dyslipidemia are predominant (1). In addition, atrophy of skin and impact on the central nervous system can occur. To a similar extent, long-term GC treatment affects the cardiovascular system, resulting in hypertension, thrombotic stroke or myocardial infarction (9). These well-known side effects often preclude long-term treatment and cause occasional severe long lasting damage to the patient. Given the strong acute action of GCs to reduce inflammation, however, side effects are accepted to a certain extent in clinical praxis.
At the molecular level, intracellular GC-activity depends on the enzymes 11β-hydroxysteroid dehydrogenase type 1 and 2 (11β-HSD1 and 11β-HSD2). 11β-HSD1 catalyzes the conversion of cortisone into active cortisol, 11β-HSD2 mainly induces the reverse reaction by inactivating cortisol (10). A specific ratio of both isozymes is given in different tissue types, for example 11β-HSD1 being predominant in liver and adipose tissue (11). Molecular actions of GCs are initiated by binding to the mineralocorticoid receptor (MR) and the GC receptor (GR). Due to the wide expression of GR compared to MR and the inactivation of GCs by 11β-HSD2 in MR high expressing tissues, most of the GC effects are mediated by the GR as evident from knockout studies. However, the role of MR in inflammation is becoming more recognized and is reviewed elsewhere (12). The GR belongs to the nuclear receptor superfamily and acts as a ligand-induced transcription factor, resulting in transactivation or transrepression of genes (10). The GR structure is constituted by four domains: the transactivation domain AF1/2 (docking station for co-regulators and regulative enzymes), the DNA-binding-domain, the ligand-binding domain (binding locus for GCs) and the hinge-region (involved in translocation of GR) (10). When located in the cytoplasm GR, is in a state of high affinity to GCs and captured in a complex with immunophilins (FKBP51), heat-shock-proteins (Hsp90) and p23 (13). GC binding leads to an exchange of FKBP51 into FKBP52, resulting in translocation of the protein complex via interaction with the microtubules (10, 13). In case of nuclear transactivation, the GR tends to dimerize and bind to specific motives on target DNA, the GC response element (GRE). The ability of GCs to downregulate genes is mediated in part by GR-binding to negative GREs and consecutive recruitment of corepressors; all leading to deacetylation of histones and decrease of gene transcription [reviewed in (10, 14)]. A “tethering mode” whereby a GR-monomer interacts with DNA-bound inflammatory transcription factors (NF-κB, AP-1, STAT3, IRF3) instead of directly binding to DNA was observed for the repression of genes encoding pro-inflammatory mediators, such as cytokines and matrix metalloproteases (15). This way of cytokine-transrepression eventually leads to immunosuppression. Furthermore, crosstalk exists between DNA-bound GRs and NF-κB or AP-1 bound to transcription-factor binding-sites in the vicinity. However, both mechanisms—transactivation via dimerized GRs and transrepression via tethering of monomeric GR—are obligatory for complete anti-inflammatory GC actions (16). Non-genomic GR-effects can be observed under high-dose GC-application and modulated by GR-interaction with membranes or mitochondria (3).
Short term rise in physiological levels of GCs can stimulate the immune function, whereas immunosuppression resulting from chronic stress, favors infections or tumorigenesis (17). The immunomodulatory actions of GCs are amongst other functions achieved by priming of innate immunity. Under physiological stress conditions macrophage phagocytosis, natural killer-cell activity and cytokine production are increased (17). Furthermore, a wide range of stress-effects on leukocytes is observed: ranging from enhanced proliferation and distribution in the lymphatic system or better endothelial adhesion, to leukocyte margination and transmigration into the inflamed tissue (17). In contrast, chronically elevated GCs levels impair leukocyte proliferation and redistribution and cytokine and prostaglandin synthesis (17).
Accordingly Frank et al. (18) showed that GCs play an important role as an alarmin in neuroinflammatory priming. Stress induced high GC levels result in NLRP3 inflammasome priming, whereby the innate immune system (e.g., microglia) switches into activation mode (18). Frank et al. describe this paradox GC-induced neuroimmune activation under neuroinflammatory conditions to be an adaptive way of preparing against potential neuronal injuries or infections (18).
Thus, GCs via the GR suppress inflammatory reactions, but may also stimulate them, depending on pharmacological conditions. Whereas, for immune suppression several molecular mechanisms of the GR, transactivation of anti-inflammatory acting genes and repression of pro-inflammatory acting genes is required, the mode of action for immune priming remains elusive.
How the different modes of action of the GR impact osteoimmunological cross-talk by influencing bone and immune cells is discussed in this review.
Previous research focused on cell-autonomous effects of GC and GR action within bone cells toward bone homeostasis and insights were provided by the use of cell type specific mutant mouse strains compromising GC signaling.
Intriguingly, GCs at the physiological levels have anabolic effects on bone. They promote the formation of osteoblasts from mesenchymal progenitor cells and are essential for maintaining bone homeostasis (19). This is evident from patients (20), since fracture risk is increased during adrenal insufficiency (21) and was shown experimentally through the use of mice that have either impaired GC metabolism in the osteoblast lineage or a selective deletion of the GR. Overexpression of the GC inactivating enzyme 11β-HSD2 in mice in early differentiated osteoblasts (22–24), but not at late differentiation stages (25) led to a reduction of cortical and trabecular bone mass in adult mice. Furthermore, a defective mineralization in the calvaria was observed which was associated with diminished Wnt Signaling (26). A reduced trabecular bone mass was also seen in mice lacking the GR in the osteoblast lineages using the Runx2 as a driver for the cre expression in the cre-loxP system (27). Furthermore, GR deficient cells displayed strongly diminished differentiation potential in vitro. Since osteocytes are also mutant in GRRunx2Cre mice, currently it remains unclear how much the GR in osteocytes contributes to the bone mass at physiological conditions. Taken together, endogenous GC signaling via the GR promotes osteoblastogenesis. However, the GR is not essential for osteoblast generation. The embryonic lethal GR knockout mice (27) and mesenchymal specific GR knockout mice (28) displayed no absence of calcification in late stage embryos. Thus, GR is a positive modulator of osteoblastogenesis, but not a crucial factor. In contrast to the GR deletion in mesenchymal cells, deletion of GR in myeloid cells including macrophages, neutrophils, and osteoclasts, does not affect bone in adult mice in the absence of inflammation, indicating that osteo-immunological cross-talk in the absence of inflammation at physiological GC levels plays a minor role in controlling bone mass (27).
This becomes strikingly altered at conditions with high exposure of GCs as it occurs in steroid therapy. GC-induced osteoporosis is among the most common so-called secondary osteoporosis (29), when bone loss is induced as side effects by medication. Here exogenous GCs have contrasting effects to endogenous GCs on osteoblasts, which decreases their proliferation (30), differentiation, and induce apoptosis and modulate autophagy (25, 31–35). Whereas, induction of autophagy seems not to be decisive for inhibition of osteoblast and osteocyte function in vivo (36), an impaired differentiation and induction of apoptosis likely lead to decreased bone formation rate (27, 33). The molecular mechanisms of the pharmacological effects on osteoblast function are partially understood. The inhibition of proliferation and differentiation is supposed to be due to inhibition of growth factors (IGF-1, WNT proteins, BMPs), expression and inhibiting the activity of their downstream signaling pathways [reviewed in (10, 19)]. The molecular mechanisms of this inhibition involves in part the induction of inhibitory molecules such as DKK1, Sclerostin, secreted frizzled and WIF1, all antagonizing Wnt signaling (19, 37). Furthermore, negative interference of the activity of the transcription factors AP-1 and Notch had been proposed (27, 38). Recently, the involvement of miRNAs was suggested (39, 40). This was challenged by a study showing that the abrogation of dicer dependent processing of miRNAs did not inhibit decreased bone formation by GCs in osteoblast specific mutant Dicer mice (41). The induction of osteoblast and osteocyte apoptosis, another cellular phenotype associated with decreased bone formation was attributed to suppression of the pro-survival gene Bcl-XL and increase of pro-apoptotic genes BIM and BAK (42–44). Additionally the generation of reactive oxygen species by rapid activation of pro-active kinases Pyk2, and JNK were suggested (45) (Figure 1).
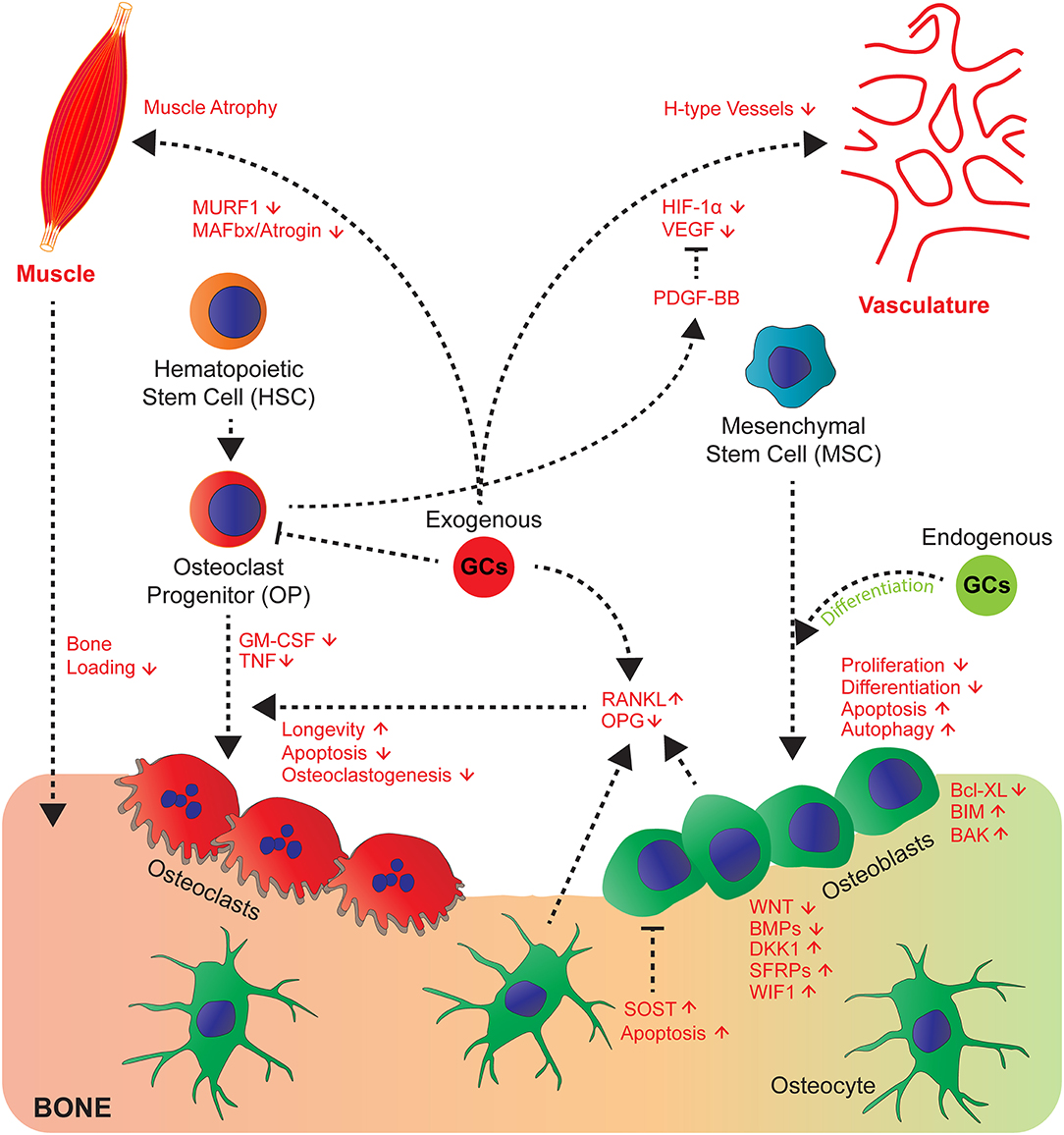
Figure 1. GCs affect cross-talk of bone cells and their communication with muscle, vasculature and myeloid cell-derived osteoclasts. GCs act directly and indirectly on bone, hematopoietic and mesenchymal cells and tissues that affect bone integrity. Endogenous GCs (green) rather favour differentiation of osteoblasts, whereas exogenous (red) rather decrease proliferation, differentiation and enhance apoptosis and autophagy of osteoblasts and osteocytes by differential regulation of signalling molecules of the Wnt and BMP pathway and pro- and anti-apoptotic molecules. Direct effects on osteoclasts are differential concerning longevity, apoptosis, osteoclastogenesis (for details see text) and indirect by altering RANKL/OPG ratio. GCs regulate cross-talk of vasculature toward bone and muscle toward bone by exerting modulatory effects on both systems (muscle atrophy) and likely impairing H-type vessels, since respective signalling molecules (VEGF and PDGF-BB are regulated by GCs).
GCs also directly act on osteoclasts stimulating initial resorption after high GC exposure (46), which then declines with prolonged GC exposures. These effects are known to be mediated through the stimulatory actions of GCs on proliferation and differentiation of osteoclast precursors as well as by prolongation of their longevity (47–49). In contrast, early progenitors are attenuated by GCs (48, 49). This latter effect might explain the decline of resorption at very long GC treatments. Nonetheless, once the osteoclasts had been formed GCs lead to enhanced longevity (46, 50), apoptosis could be suppressed, and the effects of receptor activator of nuclear factor kappa-B ligand (RANKL) potentiated. Importantly, this was abrogated in osteoclasts from GRA485T (GRdim) knock-in mice, with impaired GR dimerization (50, 51). This indicates that in contrast to GC-mediated suppression of bone formation for the increase of resorption, GR dimer dependent genetic programs are required.
Since the observation that bone formation and bone resorption are functionally coupled at the bone remodeling unit (52), cross-talk of cells in bone was considered as a hall mark of bone metabolism. This observation was supported by the discovery that osteoblasts and osteocytes are regulating bone resorption by triggering osteoclastogenesis via the induction of the pro-osteoclastogenic factor RANKL (53, 54) following exposure to M-CSF. This occurs in response to systemic hormones, such as PTH. RANKL on the other hand is counteracted by OPG. GCs seem to affect this cross-talk in part as well, since GCs induce RANKL and suppress OPG in osteoblastic cells, affecting bone resorption (27, 55–57) (Figure 1). RANKL inhibition by Denosumab in humanized mice improved some, but not all parameters of bone loss to GC effects (57). For the increase of osteoclasts in cortical bone, RANKL expression in osteocytes is decisive as shown by Piemontese et al. using mice with a conditional deletion of RANKL using RanklDmp1Cre mice (58).
Besides, the crucial soluble factors RANKL and M-CSF, TNF and TREM2 ligands play a decisive role in commitment, fusion and maturation of osteoclasts (59). Of these, TNF expression is strongly reduced by GCs at a transcriptional and post-transcriptional level. GM-CSF itself is reduced by the GR via interaction with NF-AT/AP-1 binding sites in the enhancer of the GM-CSF gene (60) (Figure 1). This is consistent with the observation that the onset of osteoclastogenesis is inhibited by GCs, which depends on cell autonomous effects (48, 49) and the down regulation of extracellular mediators. The latter was shown by coculture experiments where GCs strongly suppressed osteoclastogenesis dependent on the GR in osteoblasts despite the GR deficiency in osteoclast progenitors (27). This might play in particular a role during inflammation, where osteoclastogenesis and resorption is usually enhanced, and might be beneficially counteracted by GCs. Whether other osteoclast regulatory extracellular factors are under the control of GCs and whether this matters for osteoclastogenesis and activity is still unexplored.
Even less is understood, whether GCs affect osteoclast signals toward osteoblasts or osteocytes. This is still due to the paucity of knowledge of osteoclast-derived factors influencing osteoblasts and osteocytes. Among these identified are ephrinB2, the D2 isoforms of vacuolar (H+) ATPase (v-ATPase) V0 domain (Atp6v0d2), the complement component 3a, semaphorin 4D and microRNAs [reviewed in (61)]. It is not known whether any of these are regulated by GCs to our knowledge. Regulation of microRNAs had been shown for cell-autonomous effects in osteoblasts and osteoclasts, respectively, but whether the osteoclast-osteoblast communication or vice versa is affected is unknown. Thus, for this type of cross talk there is tremendous scope for research.
Bone is highly vascularized and previous work demonstrated that vascularization and angiogenesis is coupled with bone growth and bone homeostasis (62–64). GCs have a profound inhibitory action on vasculogenesis in bone accompanied by inhibition of HIF-1α and its target gene vascular endothelial growth factor (VEGF) (65). This is accompanied by edema formation in the femoral head in mouse bone, an area with considerable amount of vessel remodeling. In OG2-11β-HSD2 transgenic mice, overexpressing the GC inactivating enzyme 11β-HSD2 in osteocalcin expressing cells, the decrease of vasculature volume was in part prevented (62, 65). Recent studies identified the presence of a subtype of vessels, so-called H-Type vessels, positive for CD31 and endocmucin being associated with bone formation (63). These H-Type vessels were found to be reduced by GC excess, a process that could be prevented by addition of platelet-derived growth factor-BB (PDGF-BB) (66). Since PDGF-BB is in part derived from osteoclast progenitors (67), PDGF-BB could be a factor targeted by GCs.
Taken together, the precise contribution of GC signaling in cells of the vasculature vs. osteoclasts, osteoblasts and osteocytes remain to be determined, which will be of importance to decipher the effects of GC excess on bone integrity.
Since GC excess does not only influence bone strength, but also leads to muscle atrophy, this increases the risks of falls and reduces load on bone, thus accelerating bone loss and increasing fracture risk (68). GCs induce protein degradation in muscles associated with induced FoxO-dependent expression of E3 ubiquitin ligases atrophy F-Box [MAFbx/atrogin and muscle RING finger 1 (MURF1)], which is mediated in part through the GR in muscle (69–71). Surprisingly, some of these genes are also regulated in bone by excessive GC amounts (68), suggesting that some deleterious pathways might be shared between bone and muscle. The cross-talk between muscle and bone exist beyond the mechanical load. Kim et al., discovered that the muscle derived hormone Irisin binds to alphaV class of integrins in osteocytes and might stimulate resorption and increased sclerostin expression (72). Whether further soluble factors participate in this muscle bone cross-talk and whether they or Irisin signaling itself, are a target of GCs remains to be investigated. Nonetheless, both direct effects on muscle and on bone cells accelerate weakness of bone.
Interestingly, in the absence of inflammation, models of GC induced osteoporosis so far provide no clear evidence of regulation of the cross-talk between bone cells such as osteoblast/osteocytes with innate immune cells, except osteoclasts and their progenitors. This does not mean that GC mediated regulation of this cross-talk does not exist. However, this has not been addressed so far with appropriate cell conditional mouse models. This is completely different for conditions of inflammation in bone described below, where regulation of cross-talk emerges as a major theme for limiting inflammation at least in arthritis.
Innate immune cells, in particular mast cells, tissue macrophages, neutrophils and other cell types secrete inflammatory mediators (cytokines and vasodilator agents) during chronic inflammation, as it occurs e.g., during tissue damage. GCs are known to suppress the production of inflammatory mediators partially by acting on Toll-like receptor (TLR) signaling (73, 74). They also act on macrophages to inhibit the production of eicosanoids, which are lipid mediators that promote vascular dilation and permeability (75, 76). GCs also reduce the blood flow to inflammatory sites by sensitizing endothelial cells to vasoconstrictors and by inhibiting the production of vasodilators (77). In addition, GCs attenuate leukocyte extravasation by inhibiting transcription of integrins and their ligands, intercellular adhesion molecule 1 (ICAM1) as an example (78, 79). Finally, GCs inhibit the expression of many pro-inflammatory cytokines and chemokines. Mice with conditional GR ablation in macrophages or dendritic cells (DCs), produced higher levels of IL-1β, IL-6, TNF, and IL-12, and exhibited greater mortality during experimentally induced sepsis (80–82). Whereas, downregulation of chemokines, such as CC-chemokine ligand 2 (CCL2), CCL3, CCL5, restrains leukocyte migration, and deficiency of macrophage-recruiting molecule MCP-1 in mice (also known as C-C motif chemokine receptor 2 [CCR-2]), led to compromised fracture healing (83).
Interestingly, GCs reduce mast cell number, maturation and activation (84–87) and stabilize mast cells dose-dependently by inhibiting their exocytotic process. This effect is ascribed to the non-genomic actions of GCs, acting via the GR present in the plasma membrane of mast cells, and directly influencing the intracellular Ca2+ signaling pathway (88). In a mouse model of 11β-HSD1 deficiency, reduced intracellular GC action in mast cells correlated with increased activation demonstrating a clear influence of 11β-HSD1 on mast cell degranulation (89).
Despite suppressing inflammatory activity of immune cells, the concept emerges that GCs terminate inflammation by polarizing cells toward an anti-inflammatory phenotype. This has been thoroughly investigated in macrophages. Several studies demonstrated that GCs induce specific differentiation of monocytes with an anti-inflammatory phenotype and promote their survival, contributing majorly to the resolution of inflammation (90–93).
The induction of anti-inflammatory acting immune cells is decisive for resolution of inflammation during fracture healing and arthritis and is subject to GC action.
The role of GCs during fracture healing, a process that requires multiple communication steps between different cell types, is not well-understood. Fracture healing involves close interaction between bone cells and immune cells. Bone injury causes the onset of inflammation. A fracture hematoma is formed containing DAMPs and PAMPs (danger/pathogen-associated molecular patterns), erythrocytes, inflammatory cytokines and cells of the innate immunity. The inflammatory phase is followed by the repair phase where a cartilaginous callus is formed and then remodeled by osteoblast and osteoclasts (94).
Several innate immune cells are present in the early fracture hematoma such as neutrophils, macrophages and mast cells (95–98). Activated mast cells release inflammatory mediators, including histamine, KC, IL-1β, TNF, and IL-6, as well as various chemokines attracting other immune cells (99, 100). Neutrophils and macrophages migrate to the injury site in response to inflammatory mediators to phagocytose debris and pathogens (96, 97, 101, 102) (Figure 2).
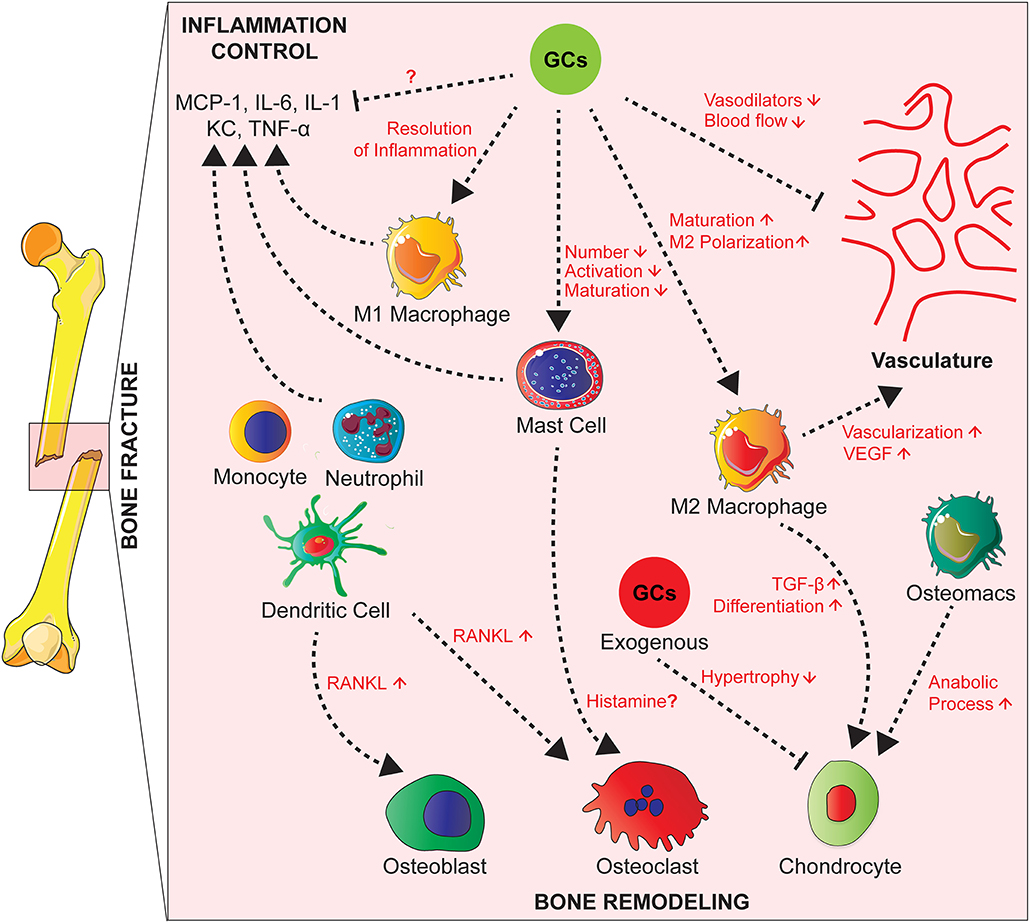
Figure 2. Effects of GCs on the cross-talk between cells of the innate immunity, bone cells, and vascularization during fracture healing. During fracture healing, cells of the innate immunity such as neutrophils, macrophages and mast cells produce pro-inflammatory cytokines and attract other phagocytes to remove debris. GCs act on these cell types to control the inflammation and resolve it partially by polarizing macrophages toward an anti-inflammatory phenotype that will in turn promote tissue repair by increasing vascularization. Presumably GCs also inhibit M1 macrophages and cytokine expression, which is not proven yet. On the other hand, GCs have counterbalancing effects by inhibiting the production of vasodilators in order to control the inflammation. Innate immune cells act on bone cells by secreting factors such as RANKL affecting then osteoblast and osteoclast activity. Also, tissue resident and infiltrating macrophages play a role in endochondral ossification by promoting chondrocyte differentiation for callus formation. Chronic GCs treatment delays chondrocyte hypertrophy and attenuates endochondral bone healing.
Depletion of neutrophils, leads to impairment of fracture healing in mice (95, 103), and a reduction of mesenchymal tissue repair in a rat model of growth plate injury (104). Macrophages persist during all phases of fracture repair (96, 97), where they are important for bone healing (105). In a mouse model of femoral fracture, Raggatt et al. showed that inflammatory macrophages were required for the initiation of the fracture repair, while both inflammatory and osteomacs, specialized resident bone macrophages, promoted anabolic processes during endochondral callus formation (106). Mast cell deficiency in mice, however, causes a reduction of the inflammatory response after fracture and disturbed callus remodeling. In the same study, in vitro investigation suggested histamine as a major mediator of mast cells action on osteoclastogenesis (98) (Figure 2).
During the repair phase, mesenchymal precursors, close to the site of the fracture, differentiate into chondrocytes and start the process of endochondral ossification. A cartilaginous soft callus is formed in order to stabilize the fracture (107). Under stable mechanical conditions the vascularization of the callus is initiated and subsequently followed by its mineralization and its conversion into bone (108). Finally, the callus is remodeled by osteoclasts and osteoblasts and the original bone architecture is restored (109).
Cells of the immune system influence the process of endochondral ossification. Tissue resident and infiltrating macrophages, in particular M2 macrophages enhance vascularization by secreting VEGF at the fracture site (97). They also release TGF-β that plays a pivotal role in chondrogenic differentiation of mesenchymal stem cells for callus formation (110). Monocytes, neutrophils, DC, and B and T lymphocytes produce RANKL and subsequently influence osteoclast and osteoblast activity (111, 112) (Figure 2).
The injury represents a stress stimulus that triggers endogenous GC release to control the inflammation. We have previously shown, in a mouse model of fracture, that mice with an induced global deletion of the GR, including bone and immune cells, had an impaired fracture healing. The presence of the GR had a protective role in our model partially by shaping the inflammatory response (113).
Few studies investigated the effects of synthetic GCs on fracture healing. It was shown that short-term treatment with GCs had minor effects on bone repair (114) while long-term treatments significantly impaired the healing process (115, 116). In a medaka fish fracture model, although both chronic and acute GC treatment affected osteoclast recruitment and osteoblast accumulation, only chronic GC treatment significantly delayed the healing (117).
The role of GCs on endochondral ossification in fracture healing hasn't been widely investigated. In a model of glucocorticoid-induced osteoporosis, endochondral ossification was impaired after fracture as chondrocyte hypertrophy was delayed (118). In a tibial metaphyseal fracture model, GR deletion in chondrocytes attenuated endochondral bone healing by momentarily increasing the cartilage content of the callus, but didn't impact negatively on the healing outcome (119). In contrast, treatment with dexamethasone had an inhibitory effect on healing in the femur shaft fracture in comparison to metaphyseal fracture, suggesting a more important role of GCs in endochondral rather than intramembranous ossification (120).
Given the distinct roles of GCs on cross-talk of immune, bone and stromal cells, and on vasculature and muscle during osteoporosis and arthritis, it is very likely that GCs shape different aspects of fracture healing positively and negatively. The exact interplay requires intensive investigations.
Osteoarthritis (OA) is the most common form of arthritis and the leading cause of pain and disability in elder people (121). The clinical picture includes not only a process of “wear and tear” but also an unbalanced remodeling of the joint associated with inflammatory processes (122). Among the main risk factors for OA are obesity, gender and age (123).
Degeneration of joints occurs as damage in articular cartilage and subchondral bone, accompanied by ectopic bone formation, so-called osteophytes. The slow turnover of extracellular matrix is dramatically enhanced in OA due to secretion of degrading proteinases and consequent loss of proteoglycans and collagen (124). This process is likely triggered by a vicious cycle of cross-talk of inflammatory cells and stromal cells, such as chondrocytes and synovial cells.
The role of endogenous GCs in this process is obscure, a recent study of Tu et al., however, showed that overexpression of the GC inactivating enzyme 11β-HSD2 in osteoblasts in transgenic mice attenuates OA in a model of destabilization of the medial meniscus (DMM) in older mice (125). This indicates that in bone cells GCs might trigger the inflammatory and erosive process (Figure 3).
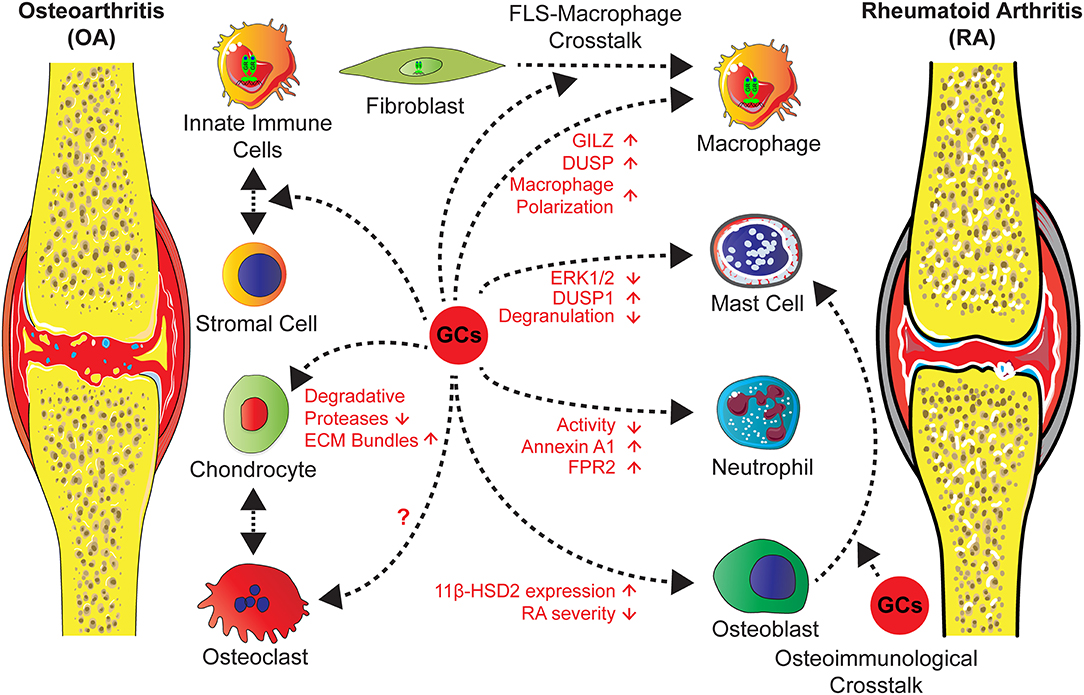
Figure 3. GCs administration in the treatment of OA and RA exert direct effects on different cell types and influence stromal-immune cell crosstalk. Actions of GCs on neutrophils and mast cells lead to an attenuated inflammation and an induction of anti-inflammatory mediators. GCs operate on macrophages either directly, causing increased levels of GILZ and decreased inflammation, or indirectly via FLS through a cross-talk between both, leading to a shift of macrophage polarization toward an anti-inflammatory phenotype and an increased efferocytosis activity. Further effects on stromal cells, in particular chondrocytes are a reduction of degradative protease levels and an increase of ECM molecules. Concerning cross-talk between osteoclasts and chondrocytes in OA or in RA the influence of GCs are unknown. Strikingly, in RA osteoblasts GC effects might lead to inflammatory and erosive processes, since the overexpression of the GC inactivating enzyme 11β-HSD2 in osteoblasts, results in an attenuated disease severity by a non defined cross-talk.
Administered GCs are accepted as short term, but not as long term agents for intra-articular injections of knee OA with few side effects [reviewed in (10)]. In the literature, the mechanisms are attributed to effects on stromal cells, by increasing the expression of ECM molecules and reduction of degradative proteases. This had been investigated in OA models, cartilage explant cultures and in cartilage cell lines [reviewed in (10, 126)] (Figure 3).
Macrophages are part of the inflammatory infiltrate in OA (127). Interestingly, a depletion of synovial macrophages led to the augmentation of OA in a model of destabilization of the medial meniscus (DMM) combined with high fat diet (128). The absence of macrophages caused intriguingly an increased numbers of T cells indicated a hyperinflammation. This indicates that anti-inflammatory polarized macrophages are essential to prevent aberrant progression of OA.
The precise contribution of GR in immune cells for GC effects on OA has not been addressed mechanistically so far. Furthermore, the suppression of VEGF by GCs (129), strongly suggests that effects on the vasculature, could be beneficial to facilitate repair processes during acute GC exposure. Long term effects on the vasculature could also be non-favorably and trigger further OA progression.
Overall, the GC action in OA is not completely understood and requires further elucidation given that GCs are frequently used for treatment, and that obesity, stress and age are known risk factors for the development of this pathology.
Beside the effects of GCs on joint erosion, not much is known about their protective effects against ectopically grown bone, so-called osteophytes. In both, inflammatory and osteoarthritis, osteophytes can be observed (130, 131) and result in pain and loss of function of joints (132). Osteophytes arise from periosteal mesenchymal stem cells (MSCs) that undergo chondrogenic differentiation, mature and produce a cartilaginous scaffold that is replaced by bone in the end-stage of osteophyte formation (133), a process closely related to endochondral ossification (130, 134–137). GCs are shown to suppress osteophyte formation (138–140), as well as endochondral ossification (141), however, it is uncertain whether the same mechanisms are involved. Interestingly, besides MSCs and chondrocytes, cross-talk with components of the innate immunity are shown to play an important role in the initiation of osteophytes (142–144). Osteophytes often develop in close proximity to synovial lining (144) and synovial inflammation is considered a key contributor to osteophyte formation (145). In this regard, it was shown that a single low-dose of avidin-conjugated dexamethasone (Dex) suppress synovial infiltration and osteophyte formation in post-traumatic OA (138). Especially, synovial macrophages, as part of the synovial infiltrate, are considered key players in osteophyte formation as their depletion significantly suppress osteophytes in two different mouse models of OA (142, 144) and GC-mediated inhibition of synovial macrophages might be beneficial to prevent osteophyte formation. Interestingly, inhibition of TNF does not result in the reduction of osteophytes in patients with psoriatic arthritis or mouse models of inflammatory arthritis (146, 147). Thus, GC-mediated suppression of pro-inflammatory cytokines alone might not be sufficient to suppress osteophyte formation. On the other hand, damage-associated molecules derived from degrading cartilage (142) can also activate synovial macrophages and depending on dosage and duration, GC treatment can protect against this cartilage degradation in OA (140, 148) and inflammatory arthritis (149). In this regard, experimental reduction of cartilage degradation reduces formation of osteophytes in mouse models of OA (150, 151).
The most prominent pathways involved in synovial macrophage activation and osteophyte growth are transforming growth factor β (TGFβ) and bone morphogenetic proteins (BMP-2/-4) (142, 144, 152, 153). TGFβ and BMPs initiate chondrogenic differentiation from periosteal MSCs and co-cultures of MSCs and macrophages enhanced spheroid formation after TGFβ treatment when compared to MSCs alone (144). Interestingly, macrophage-specific delivery of liposomal packed prednisolone results in down regulation of TGFβ in inflammatory arthritis (154) and Dex treatment was shown to suppress BMP-signaling and induce BMP-antagonists at least in osteoblast cell lines (155, 156). In addition, blockage of the hedgehog-signaling pathway also resulted in the suppression of TGFβ and BMPs and completely prevented osteophyte formation without affecting synovial inflammation (157). Thereby, GC-mediated control of TGFβ and BMPs might counteract osteophyte formation. Surprisingly, intra-articular injections of triamcinonone acetonide (TA, another GC) were associated with a higher macrophage activity, using folate-based radiotracers, but also resulted in a significant reduction of osteophytes (158). In vitro results of Siebelt et al. (158) suggested that the induction of CD163, folate receptor-β and interleukin-10 by TA might play a role in osteophyte suppression (158), however, this needs to be validated in vivo. Besides TGFβ and BMPs, dickkopf-1 (Dkk1), a master regulator of bone remodeling is strongly regulated by GCs (159) and is involved in osteophyte formation (160). Inhibition of Dkk1 results in osteophyte formation in an inflammatory mouse model that does not initially develop osteophytes (160). In addition, patients with spondylarthritis (SpA) arthritis that do develop osteophytes, show lower levels of Dkk1 (161), whereas rheumatoid arthritis (RA) patients that do not develop osteophytes have higher levels of Dkk1 (162). GCs, however, strongly induce Dkk1 expression and thereby inhibiting osteoblast differentiation and bone formation (163, 164), which might be beneficial to suppress osteophyte growth. Accordingly, overexpression of Dkk1 in the osteoblast-lineage significantly reduces osteophyte size in OA (165).
In contrast to exogenous GCs, disruption of endogenous GC signaling in the osteoblast-lineage reduces osteophyte formation in an age-related OA mouse model suggesting an osteophyte-promoting role of endogenous GC (125). Further experimental work is needed to discriminate the endogenous vs. the exogenous effects of GCs on osteophyte formation and to validate potential pathways involved in GC-mediated suppression of osteophytes to better understand the crosstalk of bone and immune cells involved in this process.
Rheumatoid arthritis (RA) is a chronic, autoimmune disease with a worldwide prevalence of 0.5–1% (166). It affects all types of patients with the highest occurrence in elderly women and a female to male ratio of 3:1 (167). RA is associated with several contributing factors, such as genetics, smoking, obesity and the environment (168). A hallmark of RA is synovial inflammation and the destruction of cartilage and bone, which makes RA a bona-fide disease of osteo-immunological interactions. The etiology is still to a certain extend unclear, but involves rheumatoid factor and anti-citrullinated peptide antibodies (ACPAs), which are at least predictive for the development of RA. The expression of pro-inflammatory mediators, like TNF and IL-6 activating the innate immune system concomitant with aberrant T- and B-Cell regulation finally leads to the development of autoantibodies (169). In the joints, osteoclasts activated by citrullinated autoantibodies, lead to bone damage. This further results in cytokine release by local cells and activation of synovial fibroblasts and macrophages (170, 171), exaggerating the inflammatory and destructive response.
Since the discovery of their anti-inflammatory action 70 years ago, GCs are still one of the most frequently used medications to treat the acute inflammatory response in RA.
Our knowledge of the mechanisms of action of GCs rely on different animal models that comply with certain aspects of the inflammatory phase in arthritis, such as collagen-induced arthritis (involving aspects of T-cells, mast cells and macrophage functions), antigen-induced arthritis (strictly T cell dependent), serum transfer-induced arthritis (T-cell independent) and TNFα transgenic mice (involving multiple cell types).
In the serum transfer-induced arthritis (STIA) and TNF-transgenic model of arthritis, it could be shown that a deficiency of 11β-HSD1 leads to an increase of inflammation, suggesting attenuation of endogenous GC action (172, 173). However, in another model of collagen-induced arthritis (CIA), 11β-HSD1 deletion caused an attenuation of inflammation indicating a pro-inflammatory role of GC activation in this model.
A clear anti-inflammatory role for the GR could be demonstrated in these models (Figure 3). For this, the capacity of the GR for dimerization seems to be required for suppression of inflammation in all arthritis models tested so far. GRA458T (GRdim) knock-in mice with attenuated GR dimerization (51), but intact monomer activity, were found refractory in arthritis models of antigen-induced arthritis, glucose-6 phosphate isomerase (G6PI)-induced arthritis and STIA (174, 175). Thus, GR dimerization-induced gene regulation seems to be a general mechanism and is in accordance with animal models with disturbed GR dimer-dependent target genes of the GR such as mitogen-activated protein kinase phosphatase 1 (MKP1), Glucocorticoid-induced leucine zipper (GILZ), and Annexin A1 (Figure 3).
GILZ interacts with several crucial signaling pathways, such as NF-κB signaling and T-cell activation (176). GILZ is constantly produced in macrophages and is stimulated by GCs and IL-10, thereby mediating the deactivation of macrophages and thus a decrease of macrophage infiltration (177). This regulation affects the balance between intensified immune reactions and immune tolerance. In mice with CIA and in human patients with RA, it could be shown that GILZ was upregulated in the synovium after the administration of GCs. Furthermore, in cultured RA synovial fibroblasts, an overexpression of GILZ inhibited the release of IL-6 and IL-8 (178).
DUSP1/MKP1 is induced by the GR dimer (179), and an important mediator of anti-inflammatory actions of the GR (81, 180, 181). It inhibits MAP Kinase signaling and DUSP-1 knockout mice have an earlier onset and higher score in CIA (182).
Annexin A1 is associated with the adaptive and the innate immunity. The anti-inflammatory effects of GCs are partly regulated by the release of Annexin A1 and the activation of its receptor formyl peptide receptor 2 (FPR2, also known as ALXR) in neutrophils and macrophages (183). Annexin A1 deficient animals render resistant to GCs in STIA (184), indicating a pivotal role for inhibition of inflammation.
Depending on the model used different cell type specific requirements for GC signaling and the GR were suggested to attenuate arthritis. For the T-cell dependent antigen-induced arthritis indeed the GR in T cells is absolutely essential for GC-mediated immune suppression in part by suppressing the generation of IL-17 producing T-cells (174). In contrast in the STIA model the deletion of GR in T-cells does not attenuate the response toward GCs (175). Strikingly, in both models GR deletion in macrophages in GRLysMCre mice hardly affected the efficiency of suppression of inflammation (174, 175). This is surprising, since there is multiple evidence for macrophages to respond to GCs during inflammation in general and the requirement of the GR in models of systemic inflammation, contact allergy and acute lung injury (80, 81, 185). In addition in STIA the presence of alternative activating macrophages is decisive for resolution of inflammation (186).
Despite other immune cells, such as type 2 innate lymphoid (ILC2) cells or others, have not been exploited yet for their functional relevance of anti-inflammatory efficacy, a new theme is emerging demonstrating the role of GR in non-immune cells.
Genetic inhibition of GC signaling in osteoblasts by overexpression of 11β-HSD2 lead surprisingly to an attenuated STIA (187). The mechanism is not clear yet, but maybe in accordance to the global 11β-HSD1 deletion in CIA.
In contrast, deletion of GR in chondrocytes in GR Col2a1CreERT2 mice leads to an accelerated inflammation in both CIA and STIA model (188). This was accompanied by an increased CXCR2 expression in the joint suggesting that GR controls chondrocyte-immune cell cross-talk on the level of CXL2/5 CXCR2 chemokine axis involved in leukocyte recruitment.
A recent study showed that GC actions in stromal cells are decisive and GR expression in immune cells alone is not sufficient to suppress inflammation in STIA (175) (Figure 3). Experiments in bone marrow chimeric mice lacking the GR in the hematopoietic compartment showed no differences in the onset or progression of STIA, nor the responsiveness to GC treatment compared to chimeric mice with a functional GR in immune cells. Furthermore, a reverse approach with chimeric mice lacking the GR globally except for the hematopoietic system revealed that GR expression in stromal cells is essential for the anti-inflammatory actions of GCs. More precisely, the study showed that for these anti-inflammatory actions, the homodimer form of the GR in stromal cells is critical. Interestingly, deficiency of GR dimerization in these cells had no effect on the suppression of inflammatory cytokines upon GC treatment. This indicates that their decrease alone is not sufficient to suppress inflammation. Additionally, GR dimers in stromal cells induce non-classical, anti-inflammatory macrophages while the levels of classical macrophages are not altered. Several anti-inflammatory markers, associated with enhanced phagocytosis and efferocytosis activity, are increased only in wildtype (wt) but not in GR dimer deficient stromal cells. This suggests an insufficient clearance of apoptotic cells after GC treatment, which leads to a persisting inflammatory condition. Finally, the study suggests that the induction of anti-inflammatory macrophages may be indirectly guided by actions of stromal cells, in particular fibroblast-like synoviocytes (FLS), since cocultures of macrophages and FLS showed an elevated efferocytosis competence when compared to cocultures of macrophages and GR dimer-deficient FLS. In addition to that, the levels of macrophage associated chemokines macrophage inflammatory protein−1α and−1β (Mip-1α / Mip-1β) are decreased in wt but not in GR dimer-deficient FLS after GC treatment. Taken together, this indicates a GC-mediated, GR dimer-dependent cross-talk between FLS and macrophages that induces an increase in the anti-inflammatory macrophage population and thereby a suppression of inflammation and STIA itself (175) (Figure 3).
Overall GCs and the GR have complex actions in bone diseases. The power of conditional mouse genetics demonstrated that GC signaling and GR action in distinct cell types of the immune system, stromal cells and bone cells have different contributions to the overall effects of GCs. Moreover, going away from this simplistic approach of interpreting cell type specific—cell autonomous effects, the field is now moving toward understanding the impact of GCs on interactions of distinct cell types or even organs.
Other issues that remain unexplored are the interplay of GC triggered immune cells in the normal pathology of postmenopausal and age-related osteoporosis. This is striking since the immune cells from the bone marrow need the bone as a niche, therefore strong interactions of immune and bone cells occur as a normal physiological process.
Given that GCs are part of the neuroendocrine regulatory network that also control inflammation and healthy bone homeostasis, a more holistic view will be needed. With the technologies of high content analysis, single cell sequencing and systemic approaches in combination with organoid models and carefully interpreted animal models, our understanding will substantially increase about the influence of these versatile hormones on the immune-metabolic crosstalk.
MA, YH, FM, KP, MK, and JT wrote individual sections of the article. MA, YH, and KP generated the figures.
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, Collaborative research Centre 1149, C02/InSt 40/492-2, Tu220/13-1, Tu 220/14-1).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. (2007) 87:873–904. doi: 10.1152/physrev.00041.2006
2. Mazziotti G, Giustina A, Canalis E, Bilezikian JP. Treatment of glucocorticoid-induced osteoporsis. Ther Adv Musculoskelet Dis. (2009) 1:27–34. doi: 10.1177/1759720X09343222
3. Buttgereit F, Burmester GR, Straub RH, Seibel MJ, Zhou H. Exogenous and endogenous glucocorticoids in rheumatic diseases. Arthritis Rheum. (2011) 63:1–9. doi: 10.1002/art.30070
4. Cruz-Topete D, Cidlowski JA. One hormone, two actions: anti- and pro-inflammatory effects of glucocorticoids. Neuroimmunomodulation. (2015) 22:20–32. doi: 10.1159/000362724
5. Becker DE. Basic and clinical pharmacology of glucocorticosteroids. Anaesthesia Prog. (2013) 60:25–32. doi: 10.2344/0003-3006-60.1.25
6. Hench PS, Kendall EC, Slocumb CH, Polley HF. The effect of a hormone of the adrenal cortex (17-hydroxy-11-dehydrocorticosterone: compound E) and of pituitary adrenocortical hormone in arthritis: preliminary report. Ann Rheum Dis. (1949) 8:97–104. doi: 10.1136/ard.8.2.97
7. Van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Use of oral corticosteroids and risk of fractures. J Bone Miner Res. (2000) 15:993–1000. doi: 10.1359/jbmr.2000.15.6.993
8. Overman RA, Yeh J-Y, Deal CL. Prevalence of oral glucocorticoid usage in the United States: a general population perspective. Arthritis Care Res. (2013) 65:294–8. doi: 10.1002/acr.21796
9. Wilson JC, Sarsour K, Gale S, Pethö-Schramm A, Jick SS, Meier CR. Incidence and risk of glucocorticoid-associated adverse effects in patients with rheumatoid arthritis. Arthritis Care Res. (2018) 71:498–511. doi: 10.1002/acr.23611
10. Hartmann K, Koenen M, Schauer S, Wittig-Blaich S, Ahmad M, Baschant U, et al. Molecular actions of glucocorticoids in cartilage and bone during health, disease, and steroid therapy. Physiol Rev. (2016) 96:409–47. doi: 10.1152/physrev.00011.2015
11. Hughes KA, Webster SP, Walker BR. 11-Beta-hydroxysteroid dehydrogenase type 1 (11beta-HSD1) inhibitors in type 2 diabetes mellitus and obesity. Expert Opin Investig Drugs. (2008) 17:481–96. doi: 10.1517/13543784.17.4.481
12. Muñoz-Durango N, Vecchiola A, Gonzalez-Gomez LM, Simon F, Riedel CA, Fardella CE, et al. Modulation of immunity and inflammation by the mineralocorticoid receptor and aldosterone. Biomed Res Int. (2015) 2015:1–14. doi: 10.1155/2015/652738
13. Heitzer MD, Wolf IM, Sanchez ER, Witchel SF, DeFranco DB. Glucocorticoid receptor physiology. Rev Endocr Metab Disord. (2007) 8:321–30. doi: 10.1007/s11154-007-9059-8
14. Weikum ER, Knuesel MT, Ortlund EA, Yamamoto KR. Glucocorticoid receptor control of transcription: precision and plasticity via allostery. Nat Rev Mol Cell Biol. (2017) 18:159–74. doi: 10.1038/nrm.2016.152
15. Beck IME, Vanden Berghe W, Vermeulen L, Yamamoto KR, Haegeman G, De Bosscher K. Crosstalk in inflammation: the interplay of glucocorticoid receptor-based mechanisms and kinases and phosphatases. Endocr Rev. (2009) 30:830–82. doi: 10.1210/er.2009-0013
16. Hübner S, Dejager L, Libert C, Tuckermann JP. The glucocorticoid receptor in inflammatory processes: transrepression is not enough. Biol Chem. (2015) 396:93–9. doi: 10.1515/hsz-2015-0106
17. Dhabhar FS, Mcewen BS. Enhancing versus suppressive effects of stress hormones on skin immune function. Proc Natl Acad Sci USA. (1999) 96:1059–64. doi: 10.1073/pnas.96.3.1059
18. Frank GF, Weber MD, Watkins LR, Maier SF. Stress sounds the alarmin: the role of the danger-associated molecular pattern HMGB1 in stress-induced neuroinflammatory priming. Brain Behav Immun. (2015) 48:1–7. doi: 10.1016/j.bbi.2015.03.010
19. Frenkel B, White W, Tuckermann J. Glucocorticoid-induced osteoporosis. Adv Exp Med Biol. (2015) 872:179–215. doi: 10.1007/978-1-4939-2895-8_8
20. Caplan A, Fett N, Rosenbach M, Werth VP, Micheletti RG. Prevention and management of glucocorticoid-induced side effects: a comprehensive review. J Am Acad Dermatol. (2017) 76:11–16. doi: 10.1016/j.jaad.2016.02.1239
21. Björnsdottir S, Sääf M, Bensing S, Kämpe O, Michaëlsson K, Ludvigsson JF. Risk of hip fracture in Addison's disease: a population-based cohort study. J Intern Med. (2011) 270:187–95. doi: 10.1111/j.1365-2796.2011.02352.x
22. Sher LB, Woitge HW, Adams DJ, Gronowicz GA, Krozowski Z, Harrison JR, et al. Transgenic expression of 11beta-hydroxysteroid dehydrogenase type 2 in osteoblasts reveals an anabolic role for endogenous glucocorticoids in bone. Endocrinology. (2004) 145:922–9. doi: 10.1210/en.2003-0655
23. Kalak R, Zhou H, Street J, Day RE, Modzelewski JRK, Spies CM, et al. Endogenous glucocorticoid signalling in osteoblasts is necessary to maintain normal bone structure in mice. Bone. (2009) 45:61–7. doi: 10.1016/j.bone.2009.03.673
24. Yang M, Trettel LB, Adams DJ, Harrison JR, Canalis E, Kream BE. Col3.6-HSD2 transgenic mice: a glucocorticoid loss-of-function model spanning early and late osteoblast differentiation. Bone. (2010) 47:573–82. doi: 10.1016/j.bone.2010.06.002
25. O'Brien CA, Jia D, Plotkin LI, Bellido T, Powers CC, Stewart SA, et al. Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology. (2004) 145:1835–41. doi: 10.1210/en.2003-0990
26. Zhou H, Mak W, Kalak R, Street J, Fong-Yee C, Zheng Y, et al. Glucocorticoid-dependent Wnt signaling by mature osteoblasts is a key regulator of cranial skeletal development in mice. Development. (2009) 136:427–36. doi: 10.1242/dev.027706
27. Rauch A, Seitz S, Baschant U, Schilling AF, Illing A, Stride B, et al. Glucocorticoids suppress bone formation by attenuating osteoblast differentiation via the monomeric glucocorticoid receptor. Cell Metab. (2010) 11:517–31. doi: 10.1016/j.cmet.2010.05.005
28. Li A, Hardy R, Stoner S, Tuckermann J, Seibel M, Zhou H. Deletion of mesenchymal glucocorticoid receptor attenuates embryonic lung development and abdominal wall closure. PLoS ONE. (2013) 8:e63578. doi: 10.1371/journal.pone.0063578
29. Weinstein RS. Clinical practice. Glucocorticoid-induced bone disease. N Engl J Med. (2011) 365:62–70. doi: 10.1056/NEJMcp1012926
30. Reid IR. Glucocorticoid effects on bone. J Clin Endocrinol Metab. (1998) 83:1860–2. doi: 10.1210/jc.83.6.1860
31. Weinstein RS. Glucocorticoid-induced osteonecrosis. Endocrine. (2012) 41:183–90. doi: 10.1007/s12020-011-9580-0
32. Plotkin LI, Manolagas SC, Bellido T. Glucocorticoids induce osteocyte apoptosis by blocking focal adhesion kinase-mediated survival. Evidence for inside-out signaling leading to anoikis. J Biol Chem. (2007) 282:24120–30. doi: 10.1074/jbc.M611435200
33. Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J Clin Invest. (1998) 102:274–82. doi: 10.1172/JCI2799
34. Weinstein RS, Nicholas RW, Manolagas SC. Apoptosis of osteocytes in glucocorticoid-induced osteonecrosis of the hip. J Clin Endocrinol Metab. (2000) 85:2907–12. doi: 10.1210/jc.85.8.2907
35. Wang L, Heckmann BL, Yang X, Long H. Osteoblast autophagy in glucocorticoid-induced osteoporosis. J Cell Physiol. (2019) 234:3207–15. doi: 10.1002/jcp.27335
36. Piemontese M, Onal M, Xiong J, Wang Y, Almeida M, Thostenson JD, et al. Suppression of autophagy in osteocytes does not modify the adverse effects of glucocorticoids on cortical bone. Bone. (2015) 75:18–26. doi: 10.1016/j.bone.2015.02.005
37. Morimoto E, Li M, Khalid AB, Krum SA, Chimge N-O, Frenkel B. Glucocorticoids Hijack Runx2 to stimulate Wif1 for suppression of osteoblast growth and differentiation. J Cell Physiol. (2017) 232:145–53. doi: 10.1002/jcp.25399
38. Pereira RMR, Delany AM, Durant D, Canalis E. Cortisol regulates the expression of Notch in osteoblasts. J Cell Biochem. (2002) 85:252–8. doi: 10.1002/jcb.10125
39. Xu D, Gao Y, Hu N, Wu L, Chen Q. miR-365 ameliorates dexamethasone-induced suppression of osteogenesis in MC3T3-E1 cells by targeting HDAC4. IJMS. (2017) 18:977. doi: 10.3390/ijms18050977
40. Li T, Li H, Li T, Fan J, Zhao RC, Weng X. MicroRNA expression profile of dexamethasone-induced human bone marrow-derived mesenchymal stem cells during osteogenic differentiation: microrna expression profile. J Cell Biochem. (2014) 115:1683–91. doi: 10.1002/jcb.24831
41. Liu P, Baumgart M, Groth M, Wittmann J, Jäck H-M, Platzer M, et al. Dicer ablation in osteoblasts by Runx2 driven cre-loxP recombination affects bone integrity, but not glucocorticoid-induced suppression of bone formation. Sci Rep. (2016) 6:32112. doi: 10.1038/srep32112
42. Espina B, Liang M, Russell RG, Hulley PA. Regulation of bim in glucocorticoid-mediated osteoblast apoptosis. J Cell Physiol. (2008) 215:488–96. doi: 10.1002/jcp.21335
43. Chang JK, Li CJ, Liao HJ, Wang CK, Wang GJ, Ho ML. Anti-inflammatory drugs suppress proliferation and induce apoptosis through altering expressions of cell cycle regulators and pro-apoptotic factors in cultured human osteoblasts. Toxicology. (2009) 258:148–56. doi: 10.1016/j.tox.2009.01.016
44. Li H, Qian W, Weng X, Wu Z, Li H, Zhuang Q, et al. Glucocorticoid receptor and sequential P53 activation by dexamethasone mediates apoptosis and cell cycle arrest of osteoblastic MC3T3-E1 cells. PLoS ONE. (2012) 7:e37030. doi: 10.1371/journal.pone.0037030
45. Almeida M, Han L, Ambrogini E, Weinstein RS, Manolagas SC. Glucocorticoids and tumor necrosis factor α increase oxidative stress and suppress Wnt protein signaling in osteoblasts. J Biol Chem. (2011) 286:44326–35. doi: 10.1074/jbc.M111.283481
46. Sivagurunathan S, Muir MM, Brennan TC, Seale JP, Mason RS. Influence of glucocorticoids on human osteoclast generation and activity. J Bone Miner Res. (2005) 20:390–8. doi: 10.1359/JBMR.041233
47. Hirayama T, Sabokbar A, Athanasou N. Effect of corticosteroids on human osteoclast formation and activity. J Endocrinol. (2002) 175:155–63. doi: 10.1677/joe.0.1750155
48. Kim H-J, Zhao H, Kitaura H, Bhattacharyya S, Brewer JA, Muglia LJ, et al. Glucocorticoids suppress bone formation via the osteoclast. J Clin Invest. (2006) 116:2152–60. doi: 10.1172/JCI28084
49. Jia D, O'Brien CA, Stewart SA, Manolagas SC, Weinstein RS. Glucocorticoids act directly on osteoclasts to increase their life span and reduce bone density. Endocrinology. (2006) 147:5592–9. doi: 10.1210/en.2006-0459
50. Conaway HH, Henning P, Lie A, Tuckermann J, Lerner UH. Activation of dimeric glucocorticoid receptors in osteoclast progenitors potentiates RANKL induced mature osteoclast bone resorbing activity. Bone. (2016) 93:43–54. doi: 10.1016/j.bone.2016.08.024
51. Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. (1998) 93:531–41. doi: 10.1016/S0092-8674(00)81183-6
52. Parfitt AM. The coupling of bone formation to bone resorption: a critical analysis of the concept and of its relevance to the pathogenesis of osteoporosis. Metab Bone Dis Relat Res. (1982) 4:1–6. doi: 10.1016/0221-8747(82)90002-9
53. Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. (1999) 397:315–23. doi: 10.1038/16852
54. Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki SI, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA. (1998) 95:3597–602. doi: 10.1073/pnas.95.7.3597
55. Hofbauer LC, Gori F, Riggs BL, Lacey DL, Dunstan CR, Spelsberg TC, et al. Stimulation of osteoprotegerin ligand and inhibition of osteoprotegerin production by glucocorticoids in human osteoblastic lineage cells: potential paracrine mechanisms of glucocorticoid-induced osteoporosis. Endocrinology. (1999) 140:4382–9. doi: 10.1210/endo.140.10.7034
56. Rauner M, Goettsch C, Stein N, Thiele S, Bornhaeuser M, De Bosscher K, et al. Dissociation of osteogenic and immunological effects by the selective glucocorticoid receptor agonist, compound A, in human bone marrow stromal cells. Endocrinology. (2011) 152:103–12. doi: 10.1210/en.2010-0456
57. Hofbauer LC, Zeitz U, Schoppet M, Skalicky M, Schüler C, Stolina M, et al. Prevention of glucocorticoid-induced bone loss in mice by inhibition of RANKL. Arthritis Rheum. (2009) 60:1427–37. doi: 10.1002/art.24445
58. Piemontese M, Xiong J, Fujiwara Y, Thostenson JD, O'Brien CA. Cortical bone loss caused by glucocorticoid excess requires RANKL production by osteocytes and is associated with reduced OPG expression in mice. Am J Physiol Endocrinol Metab. (2016) 311:E587–93. doi: 10.1152/ajpendo.00219.2016
59. Pereira M, Petretto E, Gordon S, Bassett JHD, Williams GR, Behmoaras J. Common signalling pathways in macrophage and osteoclast multinucleation. J Cell Sci. (2018) 131:cs216267. doi: 10.1242/jcs.216267
60. Smith PJ, Cousins DJ, Jee Y-K, Staynov DZ, Lee TH, Lavender P. Suppression of granulocyte-macrophage colony-stimulating factor expression by glucocorticoids involves inhibition of enhancer function by the glucocorticoid receptor binding to composite NF-AT/activator protein-1 elements. J Immunol. (2001) 167:2502–10. doi: 10.4049/jimmunol.167.5.2502
61. Chen X, Wang Z, Duan N, Zhu G, Schwarz EM, Xie C. Osteoblast–osteoclast interactions. Connect Tissue Res. (2018) 59:99–107. doi: 10.1080/03008207.2017.1290085
62. Weinstein RS, Wan C, Liu Q, Wang Y, Almeida M, O'Brien CA, et al. Endogenous glucocorticoids decrease skeletal angiogenesis, vascularity, hydration, and strength in 21-month-old mice. Aging Cell. (2010) 9:147–61. doi: 10.1111/j.1474-9726.2009.00545.x
63. Kusumbe AP, Ramasamy SK, Adams RH. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature. (2014) 507:323–8. doi: 10.1038/nature13145
64. Ramasamy SK, Kusumbe AP, Itkin T, Gur-Cohen S, Lapidot T, Adams RH. Regulation of hematopoiesis and osteogenesis by blood vessel-derived signals. Annu Rev Cell Dev Biol. (2016) 32:649–75. doi: 10.1146/annurev-cellbio-111315-124936
65. Weinstein RS, Hogan EA, Borrelli MJ, Liachenko S, O'Brien CA, Manolagas SC. The pathophysiological sequence of glucocorticoid-induced osteonecrosis of the femoral head in male mice. Endocrinology. (2017) 158:3817–31. doi: 10.1210/en.2017-00662
66. Yang P, Lv S, Wang Y, Peng Y, Ye Z, Xia Z, et al. Preservation of type H vessels and osteoblasts by enhanced preosteoclast platelet-derived growth factor type BB attenuates glucocorticoid-induced osteoporosis in growing mice. Bone. (2018) 114:1–13. doi: 10.1016/j.bone.2018.05.025
67. Xie H, Cui Z, Wang L, Xia Z, Hu Y, Xian L, et al. PDGF-BB secreted by preosteoclasts induces angiogenesis during coupling with osteogenesis. Nat Med. (2014) 20:1270–8. doi: 10.1038/nm.3668
68. Sato AY, Peacock M, Bellido T. Glucocorticoid excess in bone and muscle. Clinic Rev Bone Miner Metab. (2018) 16:33–47. doi: 10.1007/s12018-018-9242-3
69. Sandri M, Lin J, Handschin C, Yang W, Arany ZP, Lecker SH, et al. PGC-1 protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci USA. (2006) 103:16260–5. doi: 10.1073/pnas.0607795103
70. Bodine SC. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. (2001) 294:1704–8. doi: 10.1126/science.1065874
71. Watson ML, Baehr LM, Reichardt HM, Tuckermann JP, Bodine SC, Furlow JD. A cell-autonomous role for the glucocorticoid receptor in skeletal muscle atrophy induced by systemic glucocorticoid exposure. Am J Physiol Endocrinol Metab. (2012) 302:E1210–20. doi: 10.1152/ajpendo.00512.2011
72. Kim H, Wrann CD, Jedrychowski M, Vidoni S, Kitase Y, Nagano K, et al. Irisin mediates effects on bone and fat via αV integrin receptors. Cell. (2018) 175:1756–68.e17. doi: 10.1016/j.cell.2018.10.025
73. Miyata M, Lee J-Y, Susuki-Miyata S, Wang WY, Xu H, Kai H, et al. Glucocorticoids suppress inflammation via the upregulation of negative regulator IRAK-M. Nat Commun. (2015) 6:6062. doi: 10.1038/ncomms7062
74. Oppong E, Flink N, Cato ACB. Molecular mechanisms of glucocorticoid action in mast cells. Mol Cell Endocrinol. (2013) 380:119–26. doi: 10.1016/j.mce.2013.05.014
75. Kim SW, Rhee HJ, Ko J, Kim YJ, Kim HG, Yang JM, et al. Inhibition of cytosolic phospholipase A2 by annexin I. Specific interaction model and mapping of the interaction site. J Biol Chem. (2001) 276:15712–19. doi: 10.1074/jbc.M009905200
76. Yang N, Zhang W, Shi X-M. Glucocorticoid-induced leucine zipper (GILZ) mediates glucocorticoid action and inhibits inflammatory cytokine-induced COX-2 expression. J Cell Biochem. (2008) 103:1760–71. doi: 10.1002/jcb.21562
77. Perretti M, Ahluwalia A. The microcirculation and inflammation : site of action for glucocorticoids. Microcirculation. (2000) 7:147–61. doi: 10.1080/mic.7.3.147.161
78. Cronstein BN, Kimmel SC, Levin RI, Martiniuk F, Weissmann G. A mechanism for the antiinflammatory effects of corticosteroids: the glucocorticoid receptor regulates leukocyte adhesion to endothelial cells and expression of endothelial-leukocyte adhesion molecule 1 and intercellular adhesion molecule 1. Proc Natl Acad Sci USA. (1992) 89:9991–5. doi: 10.1073/pnas.89.21.9991
79. Atsuta J, Plitt J, Bochner BS, Schleimer RP. Inhibition of VCAM-1 expression in human bronchial epithelial cells by glucocorticoids. Am J Respir Cell Mol Biol. (1999) 20:643–50. doi: 10.1165/ajrcmb.20.4.3265
80. Kleiman A, Hübner S, Rodriguez Parkitna JM, Neumann A, Hofer S, Weigand MA, et al. Glucocorticoid receptor dimerization is required for survival in septic shock via suppression of interleukin-1 in macrophages. FASEB J. (2012) 26:722–9. doi: 10.1096/fj.11-192112
81. Bhattacharyya S, Brown DE, Brewer JA, Vogt SK, Muglia LJ. Macrophage glucocorticoid receptors regulate Toll-like receptor 4–mediated inflammatory responses by selective inhibition of p38 MAP kinase. Blood. (2007) 109:4313–19. doi: 10.1182/blood-2006-10-048215
82. Li CC, Munitic I, Mittelstadt PR, Castro E, Ashwell JD. Suppression of dendritic cell-derived IL-12 by endogenous glucocorticoids is protective in LPS-induced sepsis. PLoS Biol. (2015) 13:e1002269. doi: 10.1371/journal.pbio.1002269
83. Xing Z, Lu C, Hu D, Yu YY, Wang X, Colnot C, et al. Multiple roles for CCR2 during fracture healing. Dis Models Mech. (2010) 3:451–8. doi: 10.1242/dmm.003186
84. Heiman AS, Crews FT. Hydrocortisone selectively inhibits IgE-dependent arachidonic acid release from rat peritoneal mast cells. Prostaglandins. (1984) 27:335–43. doi: 10.1016/0090-6980(84)90084-4
85. Rider LG, Hirasawa N, Santini F, Beaven MA. Activation of the mitogen-activated protein kinase cascade is suppressed by low concentrations of dexamethasone in mast cells. J Immunol. (1996) 157:2374–80.
86. Eklund KK, Humphries DE, Xia Z, Ghildyal N, Friend DS, Gross V, et al. Glucocorticoids inhibit the cytokine-induced proliferation of mast cells, the high affinity IgE receptor-mediated expression of TNF-alpha, and the IL-10-induced expression of chymases. J Immunol. (1997) 158:4373–80.
87. Finotto S, Mekori YA, Metcalfe DD. Glucocorticoids decrease tissue mast cell number by reducing the production of the c-kit ligand, stem cell factor, by resident cells: in vitro and in vivo evidence in murine systems. J Clin Investig. (1997) 99:1721–8. doi: 10.1172/JCI119336
88. Mori T, Abe N, Saito K, Toyama H, Endo Y, Ejima Y, et al. Hydrocortisone and dexamethasone dose-dependently stabilize mast cells derived from rat peritoneum. Pharmacol Rep. (2016) 68:1358–65. doi: 10.1016/j.pharep.2016.09.005
89. Coutinho AE, Brown JK, Yang F, Brownstein DG, Gray M, Seckl JR, et al. Mast cells express 11beta-hydroxysteroid dehydrogenase type 1: a role in restraining mast cell degranulation. PloS ONE. (2013) 8:e54640. doi: 10.1371/journal.pone.0054640
90. Ehrchen J, Steinmüller L, Barczyk K, Tenbrock K, Nacken W, Eisenacher M, et al. Glucocorticoids induce differentiation of a specifically activated, anti-inflammatory subtype of human monocytes. Blood. (2007) 109:1265–74. doi: 10.1182/blood-2006-02-001115
91. Yona S, Gordon S. Inflammation: glucocorticoids turn the monocyte switch. Immunol Cell Biol. (2007) 85:81–2. doi: 10.1038/sj.icb.7100034
92. Varga G, Ehrchen J, Tsianakas A, Tenbrock K, Rattenholl A, Seeliger S, et al. Glucocorticoids induce an activated, anti-inflammatory monocyte subset in mice that resembles myeloid-derived suppressor cells. J Leukoc Biol. (2008) 84:644–50. doi: 10.1189/jlb.1107768
93. Barczyk K, Ehrchen J, Tenbrock K, Ahlmann M, Kneidl J, Viemann D, et al. Glucocorticoids promote survival of anti-inflammatory macrophages via stimulation of adenosine receptor A3. Blood. (2010) 116:446–55. doi: 10.1182/blood-2009-10-247106
94. Hachemi Y, Rapp AE, Hachemi Y, Rapp AE, Picke A, Weidinger G, et al. Molecular mechanisms of glucocorticoids on skeleton and bone regeneration after fracture. J Mol Endocrinol. (2018) 61:R75–90. doi: 10.1530/JME-18-0024
95. Kovtun A, Bergdolt S, Wiegner R, Radermacher P, Ignatius A. The crucial role of neutrophil granulocytes in bone fracture healing. Eur Cell Mater. (2016) 32:152–62. doi: 10.22203/eCM.v032a10
96. Sinder BP, Pettit AR, McCauley LK. Macrophages: their emerging roles in bone. J Bone Mineral Res. (2015) 30:2140–9. doi: 10.1002/jbmr.2735
97. Wu AC, Raggatt LJ, Alexander KA, Pettit AR. Unraveling macrophage contributions to bone repair. Bonekey Rep. (2013) 2:373. doi: 10.1038/bonekey.2013.107
98. Kroner J, Kovtun A, Kemmler J, Messmann JJ, Strauß G, Seitz S, et al. Mast cells are critical regulators of bone fracture-induced inflammation and osteoclast formation and activity. J Bone Miner Res. (2017) 32:2431–44. doi: 10.1002/jbmr.3234
99. Ramirez-Garcialuna JL, Chan D, Samberg R, Abou-Rjeili M, Wong TH, Li A, et al. Defective bone repair in mast cell-deficient Cpa3 Cre/+ mice. PLoS ONE. (2017) 12:e0174396. doi: 10.1371/journal.pone.0174396
100. De Filippo K, Dudeck A, Hasenberg M, Nye E, van Rooijen N, Hartmann K, et al. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood. (2013) 121:4930–7. doi: 10.1182/blood-2013-02-486217
101. Furze RC, Rankin SM. Neutrophil mobilization and clearance in the bone marrow. Immunology. (2008) 125:281–8. doi: 10.1111/j.1365-2567.2008.02950.x
102. Sadik CD, Kim ND, Luster AD. Neutrophils cascading their way to inflammation. Trends Immunol. (2011) 32:452–60. doi: 10.1016/j.it.2011.06.008
103. Chan JK, Glass GE, Ersek A, Freidin A, Williams GA, Gowers K, et al. Low-dose TNF augments fracture healing in normal and osteoporotic bone by up-regulating the innate immune response. EMBO Mol Med. (2015) 7:547–61. doi: 10.15252/emmm.201404487
104. Chung R, Cool JC, Scherer MA, Foster BK, Xian CJ. Roles of neutrophil-mediated inflammatory response in the bony repair of injured growth plate cartilage in young rats. J Leukoc Biol. (2006) 80:1272–80. doi: 10.1189/jlb.0606365
105. Alexander KA, Chang MK, Maylin ER, Kohler T, Müller R, Wu AC, et al. Osteal macrophages promote in vivo intramembranous bone healing in a mouse tibial injury model. J Bone Mineral Res. (2011) 26:1517–32. doi: 10.1002/jbmr.354
106. Raggatt LJ, Wullschleger ME, Alexander KA, Wu ACK, Millard SM, Kaur S, et al. Fracture healing via periosteal callus formation requires macrophages for both initiation and progression of early endochondral ossification. Am J Pathol. (2014) 184:3192–204. doi: 10.1016/j.ajpath.2014.08.017
107. Shapiro F. Bone development and its relation to fracture repair. The role of mesenchymal osteoblasts and surface osteoblasts. Eur Cells Mater. (2008) 15:53–76. doi: 10.22203/eCM.v015a05
108. Einhorn TA, Gerstenfeld LC. Fracture healing: mechanisms and interventions. Nat Rev Rheumatol. (2015) 11:45–54. doi: 10.1038/nrrheum.2014.164
109. Schindeler A, McDonald MM, Bokko P, Little DG. Bone remodeling during fracture repair: the cellular picture. Semin Cell Dev Biol. (2008) 19:459–66. doi: 10.1016/j.semcdb.2008.07.004
110. Schlundt C, El Khassawna T, Serra A, Dienelt A, Wendler S, Schell H, et al. Macrophages in bone fracture healing: their essential role in endochondral ossification. Bone. (2018) 106:78–89. doi: 10.1016/j.bone.2015.10.019
111. Caetano-Lopes J, Canhao H, Fonseca JE. Osteoimmunology–the hidden immune regulation of bone. Autoimmun Rev. (2009) 8:250–5. doi: 10.1016/j.autrev.2008.07.038
112. Cao X. RANKL-RANK signaling regulates osteoblast differentiation and bone formation. Bone Res. (2018) 6:1–2. doi: 10.1038/s41413-018-0040-9
113. Rapp AE, Hachemi Y, Kemmler J, Koenen M, Tuckermann J, Ignatius A. Induced global deletion of glucocorticoid receptor impairs fracture healing. FASEB J. (2018) 32:2235–45. doi: 10.1096/fj.201700459RR
114. Aslan M, Simşek G, Yildirim Ü. Effects of short-term treatment with systemic prednisone on bone healing: an experimental study in rats. Dental Traumatol. (2005) 21:222–5. doi: 10.1111/j.1600-9657.2005.00300.x
115. Waters RV, Gamradt SC, Asnis P, Vickery BH, Avnur Z, Hill E, et al. Systemic corticosteroids inhibit bone healing in a rabbit ulnar osteotomy model. Acta Orthopaedica Scand. (2000) 71:316–21. doi: 10.1080/000164700317411951
116. Liu Y, Akhter MP, Gao X, Wang X, Wang X, Zhao G, et al. Glucocorticoid-induced delayed fracture healing and impaired bone biomechanical properties in mice. CIA. (2018) 13:1465–74. doi: 10.2147/CIA.S167431
117. Azetsu Y, Chatani M, Dodo Y, Karakawa A, Sakai N, Negishi-Koga T, et al. Treatment with synthetic glucocorticoid impairs bone metabolism, as revealed by in vivo imaging of osteoblasts and osteoclasts in medaka fish. Biomed Pharmacother. (2019) 118:109101. doi: 10.1016/j.biopha.2019.109101
118. Zhang H, Shi X, Wang L, Li X, Zheng C, Gao B, et al. Intramembranous ossification and endochondral ossification are impaired differently between glucocorticoid-induced osteoporosis and estrogen deficiency-induced osteoporosis. Sci Rep. (2018) 8:1–13. doi: 10.1038/s41598-018-22095-1
119. Tu J, Henneicke H, Zhang Y, Stoner S, Cheng TL, Schindeler A, et al. Disruption of glucocorticoid signaling in chondrocytes delays metaphyseal fracture healing but does not affect normal cartilage and bone development. Bone. (2014) 69:12–22. doi: 10.1016/j.bone.2014.08.016
120. Sandberg OH, Aspenberg P. Glucocorticoids inhibit shaft fracture healing but not metaphyseal bone regeneration under stable mechanical conditions. Bone Joint Res. (2015) 4:170–5. doi: 10.1302/2046-3758.410.2000414
121. Cheng Y, Hootman J, Murphy L. Morbidity and mortality weekly report. prevalence of doctor-diagnosed arthritis and arthritis-attributable activity limitation: United States, 2007-2009. MMWR Morb Mortal Wkly Rep. (2010) 59:2007–9.
122. Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheumat. (2012) 64:1697–707. doi: 10.1002/art.34453
123. Blagojevic M, Jinks C, Jeffery A, Jordan KP. Risk factors for onset of osteoarthritis of the knee in older adults: a systematic review and meta-analysis. Osteoarthritis Cartil. (2010) 18:24–33. doi: 10.1016/j.joca.2009.08.010
124. Goldring MB, Marcu KB. Cartilage homeostasis in health and rheumatic diseases. Arthritis Res Ther. (2009) 11:224. doi: 10.1186/ar2592
125. Tu J, Zhang P, Ji Z, Henneicke H, Li J, Kim S, et al. Disruption of glucocorticoid signalling in osteoblasts attenuates age-related surgically induced osteoarthritis. Osteoarthr Cartil. (2019) 27:1518–25. doi: 10.1016/j.joca.2019.04.019
126. Savvidou O, Milonaki M, Goumenos S, Flevas D, Papagelopoulos P, Moutsatsou P. Glucocorticoid signaling and osteoarthritis. Mol Cell Endocrinol. (2019) 480:153–66. doi: 10.1016/j.mce.2018.11.001
127. Revell PA, Mayston V, Lalor P, Mapp P. The synovial membrane in osteoarthritis: a histological study including the characterisation of the cellular infiltrate present in inflammatory osteoarthritis using monoclonal antibodies. Ann Rheum Dis. (1988) 47:300–7. doi: 10.1136/ard.47.4.300
128. Wu C-L, McNeill J, Goon K, Little D, Kimmerling K, Huebner J, et al. Conditional macrophage depletion increases inflammation and does not inhibit the development of osteoarthritis in obese macrophage fas-induced apoptosis-transgenic mice: conditional macrophage depletion in mafia-transgenic mice. Arthritis Rheumatol. (2017) 69:1772–83. doi: 10.1002/art.40161
129. Smink JJ, Buchholz IM, Hamers N, van Tilburg CM, Christis C, Sakkers RJB, et al. Short-term glucocorticoid treatment of piglets causes changes in growth plate morphology and angiogenesis. Osteoarthritis Cartil. (2003) 11:864–71. doi: 10.1016/S1063-4584(03)00187-0
130. Schett G, Landewé R, van der Heijde D. Tumour necrosis factor blockers and structural remodelling in ankylosing spondylitis: what is reality and what is fiction? Ann Rheum Dis. (2007) 66:709–11. doi: 10.1136/ard.2007.071159
131. Thambyah A, Broom N. On new bone formation in the pre-osteoarthritic joint. Osteoarthr Cartil. (2009) 17:456–63. doi: 10.1016/j.joca.2008.09.005
132. Dieppe PA, Lohmander LS. Pathogenesis and management of pain in osteoarthritis. Lancet. (2005) 365:965–73. doi: 10.1016/S0140-6736(05)71086-2
133. Sandell LJ, Aigner T. Articular cartilage and changes in arthritis: cell biology of osteoarthritis. Arthritis Res Ther. (2001) 3:107. doi: 10.1186/ar148
134. Dodds RA, Merry K, Littlewood A, Gowen M. Expression of mRNA for IL1 beta, IL6 and TGF beta 1 in developing human bone and cartilage. J Histochem Cytochem. (1994) 42:733–44. doi: 10.1177/42.6.8189035
135. van der Kraan PM, van den Berg WB. Osteophytes: relevance and biology. Osteoarthritis Cartil. (2007) 15:237–44. doi: 10.1016/j.joca.2006.11.006
136. Mahjoub M, Berenbaum F, Houard X. Why subchondral bone in osteoarthritis? The importance of the cartilage bone interface in osteoarthritis. Osteoporos Int. (2012) 23:841–6. doi: 10.1007/s00198-012-2161-0
137. Xiao Z-F, Su G-Y, Hou Y, Chen S-D, Lin D-K. Cartilage degradation in osteoarthritis: a process of osteochondral remodeling resembles the endochondral ossification in growth plate? Med Hypotheses. (2018) 121:183–7. doi: 10.1016/j.mehy.2018.08.023
138. Bajpayee AG, De la Vega RE, Scheu M, Varady NH, Yannatos IA, Brown LA, et al. Sustained intra-cartilage delivery of low dose dexamethasone using a cationic carrier for treatment of post traumatic osteoarthritis. Eur Cell Mater. (2017) 34:341–64. doi: 10.22203/eCM.v034a21
139. Geannette C, Sahr M, Mayman D, Miller TT. Ultrasound diagnosis of osteophytic impingement of the popliteus tendon after total knee replacement. J Ultrasound Med. (2018) 37:2279–83. doi: 10.1002/jum.14563
140. Pelletier JP, Martel-Pelletier J. Protective effects of corticosteroids on cartilage lesions and osteophyte formation in the Pond-Nuki dog model of osteoarthritis. Arthritis Rheum. (1989) 32:181–93. doi: 10.1002/anr.1780320211
141. Lui JC, Baron J. Effects of glucocorticoids on the growth plate. In: Ghizzoni L, Cappa M, Chrousos GP, Loche S, Maghnie MS, Karger AG, editors. Endocrine Development. Basel: Karger (2010). p. 187–93.
142. Blom AB, van Lent PLEM, Holthuysen AEM, van der Kraan PM, Roth J, van Rooijen N, et al. Synovial lining macrophages mediate osteophyte formation during experimental osteoarthritis. Osteoarthr Cartil. (2004) 12:627–35. doi: 10.1016/j.joca.2004.03.003
143. Ganova P, Gyurkovska V, Belenska-Todorova L, Ivanovska N. Functional complement activity is decisive for the development of chronic synovitis, osteophyte formation and processes of cell senescence in zymosan-induced arthritis. Immunol Lett. (2017) 190:213–20. doi: 10.1016/j.imlet.2017.08.023
144. van Lent PLEM, Blom AB, van der Kraan P, Holthuysen AEM, Vitters E, van Rooijen N, et al. Crucial role of synovial lining macrophages in the promotion of transforming growth factor beta-mediated osteophyte formation. Arthritis Rheum. (2004) 50:103–11. doi: 10.1002/art.11422
145. van den Bosch MH, Blom AB, Schelbergen RFP, Vogl T, Roth JP, Slöetjes AW, et al. Induction of canonical Wnt signaling by the alarmins S100A8/A9 in murine knee joints: implications for osteoarthritis. Arthritis Rheumatol. (2016) 68:152–63. doi: 10.1002/art.39420
146. Schett G, Stolina M, Dwyer D, Zack D, Uderhardt S, Krönke G, et al. Tumor necrosis factor α and RANKL blockade cannot halt bony spur formation in experimental inflammatory arthritis. Arthritis Rheumat. (2009) 60:2644–54. doi: 10.1002/art.24767
147. Finzel S, Kraus S, Schmidt S, Hueber A, Rech J, Engelke K, et al. Bone anabolic changes progress in psoriatic arthritis patients despite treatment with methotrexate or tumour necrosis factor inhibitors. Ann Rheum Dis. (2013) 72:1176–81. doi: 10.1136/annrheumdis-2012-201580
148. Huebner KD, Shrive NG, Frank CB. Dexamethasone inhibits inflammation and cartilage damage in a new model of post-traumatic osteoarthritis. J Orthop Res. (2014) 32:566–72. doi: 10.1002/jor.22568
149. Baschant U, Lane NE, Tuckermann J. The multiple facets of glucocorticoid action in rheumatoid arthritis. Nat Rev Rheumatol. (2012) 8:645–55. doi: 10.1038/nrrheum.2012.166
150. Botter SM, Glasson SS, Hopkins B, Clockaerts S, Weinans H, van Leeuwen JPTM, et al. ADAMTS5–/– mice have less subchondral bone changes after induction of osteoarthritis through surgical instability: implications for a link between cartilage and subchondral bone changes. Osteoarthritis Cartil. (2009) 17:636–45. doi: 10.1016/j.joca.2008.09.018
151. Kamekura S, Kawasaki Y, Hoshi K, Shimoaka T, Chikuda H, Maruyama Z, et al. Contribution of runt-related transcription factor 2 to the pathogenesis of osteoarthritis in mice after induction of knee joint instability. Arthritis Rheumat. (2006) 54:2462–70. doi: 10.1002/art.22041
152. van Beuningen HM, van der Kraan PM, Arntz OJ, van den Berg WB. Transforming growth factor-beta 1 stimulates articular chondrocyte proteoglycan synthesis and induces osteophyte formation in the murine knee joint. Lab Invest. (1994) 71:279–90.
153. Scharstuhl A, Vitters EL, van der Kraan PM, van den Berg WB. Reduction of osteophyte formation and synovial thickening by adenoviral overexpression of transforming growth factor beta/bone morphogenetic protein inhibitors during experimental osteoarthritis. Arthritis Rheum. (2003) 48:3442–51. doi: 10.1002/art.11328
154. Hofkens W, Schelbergen R, Storm G, van den Berg WB, van Lent PL. Liposomal targeting of prednisolone phosphate to synovial lining macrophages during experimental arthritis inhibits M1 activation but does not favor M2 differentiation. PLoS ONE. (2013) 8:e54016. doi: 10.1371/journal.pone.0054016
155. Hayashi K, Yamaguchi T, Yano S, Kanazawa I, Yamauchi M, Yamamoto M, et al. BMP/Wnt antagonists are upregulated by dexamethasone in osteoblasts and reversed by alendronate and PTH: potential therapeutic targets for glucocorticoid-induced osteoporosis. Biochem Biophys Res Commun. (2009) 379:261–6. doi: 10.1016/j.bbrc.2008.12.035
156. Luppen CA, Smith E, Spevak L, Boskey AL, Frenkel B. Bone morphogenetic protein-2 restores mineralization in glucocorticoid-inhibited MC3T3-E1 osteoblast cultures. J Bone Miner Res. (2003) 18:1186–97. doi: 10.1359/jbmr.2003.18.7.1186
157. Ruiz-Heiland G, Horn A, Zerr P, Hofstetter W, Baum W, Stock M, et al. Blockade of the hedgehog pathway inhibits osteophyte formation in arthritis. Ann Rheum Dis. (2012) 71:400–7. doi: 10.1136/ard.2010.148262
158. Siebelt M, Korthagen N, Wei W, Groen H, Bastiaansen-Jenniskens Y, Müller C, et al. Triamcinolone acetonide activates an anti-inflammatory and folate receptor-positive macrophage that prevents osteophytosis in vivo. Arthritis Res Ther. (2015) 17:352. doi: 10.1186/s13075-015-0865-1
159. Ohnaka K, Taniguchi H, Kawate H, Nawata H, Takayanagi R. Glucocorticoid enhances the expression of dickkopf-1 in human osteoblasts: novel mechanism of glucocorticoid-induced osteoporosis. Biochem Biophys Res Commun. (2004) 318:259–64. doi: 10.1016/j.bbrc.2004.04.025
160. Diarra D, Stolina M, Polzer K, Zwerina J, Ominsky MS, Dwyer D, et al. Dickkopf-1 is a master regulator of joint remodeling. Nat Med. (2007) 13:156–63. doi: 10.1038/nm1538
161. Fassio A, Idolazzi L, Viapiana O, Benini C, Vantaggiato E, Bertoldo F, et al. In psoriatic arthritis Dkk-1 and PTH are lower than in rheumatoid arthritis and healthy controls. Clin Rheumatol. (2017) 36:2377–81. doi: 10.1007/s10067-017-3734-2
162. Ma Y, Zhang X, Wang M, Xia Q, Yang J, Wu M, et al. The serum level of Dickkopf-1 in patients with rheumatoid arthritis: a systematic review and meta-analysis. Int Immunopharmacol. (2018) 59:227–32. doi: 10.1016/j.intimp.2018.04.019
163. Brunetti G, Faienza MF, Piacente L, Ventura A, Oranger A, Carbone C, et al. High dickkopf-1 levels in sera and leukocytes from children with 21-hydroxylase deficiency on chronic glucocorticoid treatment. Am J Physiol Endocrinol Metab. (2013) 304:E546–54. doi: 10.1152/ajpendo.00535.2012
164. Wang F-S, Ko J-Y, Yeh D-W, Ke H-C, Wu H-L. Modulation of Dickkopf-1 attenuates glucocorticoid induction of osteoblast apoptosis, adipocytic differentiation, and bone mass loss. Endocrinology. (2008) 149:1793–801. doi: 10.1210/en.2007-0910
165. Funck-Brentano T, Bouaziz W, Marty C, Geoffroy V, Hay E, Cohen-Solal M. Dkk-1-mediated inhibition of Wnt signaling in bone ameliorates osteoarthritis in mice. Arthritis Rheumatol. (2014) 66:3028–39. doi: 10.1002/art.38799
166. Tobón GJ, Youinou P, Saraux A. Autoimmunity reviews the environment, geo-epidemiology, and autoimmune disease : rheumatoid arthritis. Autoimmun Rev. (2009) 9:A288–92. doi: 10.1016/j.autrev.2009.11.019
167. Alamanos Y, Drosos AA. Epidemiology of adult rheumatoid arthritis. Autoimmun Rev. (2005) 4:130–6. doi: 10.1016/j.autrev.2004.09.002
168. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. (2011) 365:2205–19. doi: 10.1056/NEJMra1004965
169. McInnes IB, Schett G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet. (2017) 389:2328–37. doi: 10.1016/S0140-6736(17)31472-1
170. Sokolove J, Zhao X, Chandra PE, Robinson WH. Immune complexes containing citrullinated fibrinogen costimulate macrophages via toll-like receptor 4 and Fcγ receptor. Arthritis Rheumat. (2011) 63:53–62. doi: 10.1002/art.30081
171. Seibl R, Birchler T, Loeliger S, Hossle JP, Gay RE, Saurenmann T, et al. Expression and regulation of Toll-like receptor 2 in rheumatoid arthritis synovium. Am J Pathol. (2003) 162:1221–7. doi: 10.1016/S0002-9440(10)63918-1
172. Coutinho AE, Gray M, Brownstein DG, Salter DM, Sawatzky DA, Clay S, et al. 11B-Hydroxysteroid dehydrogenase type 1, but not type 2, deficiency worsens acute inflammation and experimental arthritis in mice. Endocrinology. (2012) 153:234–40. doi: 10.1210/en.2011-1398
173. Hardy RS, Fenton C, Croft AP, Naylor AJ, Begum R, Desanti G, et al. 11 Beta-hydroxysteroid dehydrogenase type 1 regulates synovitis, joint destruction, and systemic bone loss in chronic polyarthritis. J Autoimmun. (2018) 92:104–13. doi: 10.1016/j.jaut.2018.05.010
174. Baschant U, Frappart L, Rauchhaus U, Bruns L, Reichardt HM, Kamradt T, et al. Glucocorticoid therapy of antigen-induced arthritis depends on the dimerized glucocorticoid receptor in T cells. Proc Natl Acad Sci USA. (2011) 108:19317–22. doi: 10.1073/pnas.1105857108
175. Koenen M, Culemann S, Vettorazzi S, Caratti G, Frappart L, Baum W, et al. Glucocorticoid receptor in stromal cells is essential for glucocorticoid-mediated suppression of inflammation in arthritis. Ann Rheum Dis. (2018) 77:1610–18. doi: 10.1136/annrheumdis-2017-212762
176. Ayroldi E, Migliorati G, Bruscoli S, Marchetti C, Zollo O, Cannarile L, et al. Modulation of T-cell activation by the glucocorticoid-induced leucine zipper factor via inhibition of nuclear factor kappaB. Blood. (2001) 98:743–53. doi: 10.1182/blood.V98.3.743
177. Berrebi D, Bruscoli S, Cohen N, Foussat A, Migliorati G, Bouchet-Delbos L, et al. Synthesis of glucocorticoid-induced leucine zipper. (GILZ) by macrophages: an anti-inflammatory and immunosuppressive mechanism shared by glucocorticoids and IL-10. Blood. (2003) 101:729–38. doi: 10.1182/blood-2002-02-0538
178. Beaulieu E, Ngo D, Santos L, Yang YH, Smith M, Jorgensen C, et al. Glucocorticoid-induced leucine zipper is an endogenous antiinflammatory mediator in arthritis. Arthritis Rheumat. (2010) 62:2651–61. doi: 10.1002/art.27566
179. Kassel O, Sancono A, Krätzschmar J, Kreft B, Stassen M, Cato AC. Glucocorticoids inhibit MAP kinase via increased expression and decreased degradation of MKP-1. EMBO J. (2001) 20:7108–16. doi: 10.1093/emboj/20.24.7108
180. Abraham SM, Lawrence T, Kleiman A, Warden P, Medghalchi M, Tuckermann J, et al. Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J Exp Med. (2006) 203:1883–9. doi: 10.1084/jem.20060336
181. Vandevyver S, Dejager L, Van Bogaert T, Kleyman A, Liu Y, Tuckermann J, et al. Glucocorticoid receptor dimerization induces MKP1 to protect against TNF-induced inflammation. J Clin Invest. (2012) 122:2130–40. doi: 10.1172/JCI60006
182. Vattakuzhi Y, Abraham SM, Freidin A, Clark AR, Horwood NJ. Dual-specificity phosphatase 1-null mice exhibit spontaneous osteolytic disease and enhanced inflammatory osteolysis in experimental arthritis. Arthritis Rheum. (2012) 64:2201–10. doi: 10.1002/art.34403
183. Perretti M, D'Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol. (2009) 9:62–70. doi: 10.1038/nri2470
184. Patel HB, Kornerup KN, Sampaio AL, D'Acquisto F, Seed MP, Girol AP, et al. The impact of endogenous annexin A1 on glucocorticoid control of inflammatory arthritis. Ann Rheum Dis. (2012) 71:1872–80. doi: 10.1136/annrheumdis-2011-201180
185. Tuckermann JP, Kleiman A, Moriggl R, Spanbroek R, Neumann A, Illing A, et al. Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact allergy. J Clin Invest. (2007) 117:1381–90. doi: 10.1172/JCI28034
186. Misharin AV, Cuda CM, Saber R, Turner JD, Gierut AK, Haines GK, et al. Nonclassical Ly6C− monocytes drive the development of inflammatory arthritis in mice. Cell Rep. (2014) 9:591–604. doi: 10.1016/j.celrep.2014.09.032
187. Buttgereit F, Zhou H, Kalak R, Gaber T, Spies CM, Huscher D, et al. Transgenic disruption of glucocorticoid signaling in mature osteoblasts and osteocytes attenuates K/BxN mouse serum-induced arthritis in vivo. Arthritis Rheum. (2009) 60:1998–2007. doi: 10.1002/art.24619
Keywords: glucocorticoids, glucocorticoid receptor, osteoporosis, arthritis, inflammation, fracture healing, conditional knockout mice
Citation: Ahmad M, Hachemi Y, Paxian K, Mengele F, Koenen M and Tuckermann J (2019) A Jack of All Trades: Impact of Glucocorticoids on Cellular Cross-Talk in Osteoimmunology. Front. Immunol. 10:2460. doi: 10.3389/fimmu.2019.02460
Received: 30 July 2019; Accepted: 02 October 2019;
Published: 17 October 2019.
Edited by:
Cristina Sobacchi, Italian National Research Council (CNR), ItalyReviewed by:
Masahiro Chatani, Showa University, JapanCopyright © 2019 Ahmad, Hachemi, Paxian, Mengele, Koenen and Tuckermann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan Tuckermann, amFuLnR1Y2tlcm1hbm5AdW5pLXVsbS5kZQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.