 Abhirup Jayasimhan
Abhirup Jayasimhan Eliana Mariño
Eliana Mariño
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 29 October 2019
Sec. Mucosal Immunity
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02429
This article is part of the Research TopicThe Role of Physical and Biological Gut Barriers in Modulating Crosstalk between the Microbiota and the Immune SystemView all 16 articles
Microbial metabolites have a profound effect on the development of type 1 diabetes (T1D). The cross-talk between the gut microbiota, the nervous system, and immune system is necessary to establish and maintain immune and gut tolerance. As quoted by Hippocrates, “All disease begins in the gut.” Although this has been recognized for 2,000 years, the connection between the gut and autoimmune T1D is not yet well-understood. Here, we outline new advances supported by our research and others that have contributed to elucidate the impact of microbial metabolites on the physiology of the pancreas and the gut through their remarkable effect on the immune and nervous system. Among many of the mechanisms involved in the gut–beta-cell–immune cross-talk, glial fibrillary acidic protein (GFAP)-expressing cells are critical players in the development of invasive insulitis. Besides, this review reveals a novel mechanism for microbial metabolites by stimulating IL-22, an essential cytokine for gut homeostasis and beta-cell survival. The close connections between the gut and the pancreas are highlighted through our review as microbial metabolites recirculate through the whole body and intimately react with the nervous system, which controls essential disorders associated with diabetes. As such, we discuss the mechanisms of action of microbial metabolites or short-chain fatty acids (SCFAs), IL-22, and GFAP on beta-cells, gut epithelial cells, neurons, and glial cells via metabolite sensing receptors or through epigenetic effects. The fine-tuned gut–neuro–immune network may be profoundly affected by SCFA deficiency related to dysbiosis and diet alterations at very early stages of the initiation of the disease. Thus, dampening the initial immune response or preventing the perpetuation of the immune response by maintaining the integrity of the gut is among the alternative approaches to prevent T1D.
Type 1 diabetes (T1D) is a chronic autoimmune disease in which T cells destroy the insulin-producing beta-cells of the pancreas (1–3). The beta-cell's attack happens when T cells recognize autoantigens such as glutamic acid decarboxylase (GAD), islet cell autoantigen 69 (ICA69), insulinoma-associated antigen 2 (IA2), islet-specific glucose-6-phosphatase catalytic subunit–related protein (IGRP), and proinsulin, which are widely accepted as the initiating autoantigens in T1D (4, 5). Antigenic targets for T cell priming are not solely expressed in beta-cells, but also in multiple tissues distal to islets, and they can be found in food like insulin or produced by bacteria like GAD (6–8). This all leads to many questions. How antigen expression in other distal tissues to beta-cells control the invasive infiltration of immune cells into the pancreas? Is the gut an important compartment as a source of antigens that trigger T1D? Is the gut microbiota influencing T cell priming against beta-cells? The microenvironment regulates beta-cell function and maturity, in particular close interaction with endocrine cells, neuronal, immune, and vascular cells (9, 10). Pancreatic ducts are physiologically neighboring to the beta-cells, and their primary function is to deliver enzymes or pancreatic juices provided from the exocrine pancreas into the duodenum to help digestion. As such, the pancreatic beta-cells can be influenced by the gut, which is intimately connected not only through the pancreatic ducts but also by lymph ducts (11). Beyond the pancreas, there is hardly any tissue in the body that has not been somehow in contact with gut microbial SCFAs. From food fermentation, bacteria in the large bowel produce many metabolites that are used by the epithelial cells in the gut. The remaining amount is transported to the liver where they are metabolized and then released to systemic circulation. As such, SCFAs have a broad spectrum of remarkable beneficial properties that affect many systems, in particular under inflammatory conditions, regulating metabolic, and immune responses (12–14).
One example is the nervous system, which is also critical for the pancreas to function (15). Both the endocrine and the exocrine part of the pancreas are innervated by the sympathetic and the parasympathetic nervous system, as such pancreatic sensory neurons have been shown to play a critical role in controlling islet inflammation (16). Similarly, the enteric nervous system (ENS) via the enteric glial cells (EGCs) is vital to maintain gut and immune homeostasis (17, 18), given that diabetic animals and patients presented gastrointestinal motility disorders (19). In this review, we will discuss the gut–neuro–immune axis in T1D and its effect on beta-cell priming. In particular, we will focus on the role of GFAP-positive cell types as critical players in T1D and on the impact of the gut microbiota, SCFAs, and their mechanisms of action through interleukin 22 (IL-22).
Beta-cells are involved in late T cell priming, suggesting that they are not required during the induction of T1D (20, 21). So, a critical consideration is—what might be driving the initiation of T1D? It has been shown in the pancreas that GFAP-expressing peri-islet Schwann cells (pSC or glial cells) can attract and recruit autoreactive cells, which precedes the attack on beta-cells. Two studies support the finding that immune responses to autoantigens expressed in pSCs precede the immune response to beta-cells (6, 7). In particular, they showed that early T cell attack on GFAP-expressing pSCs progressively results in the release of glial cell antigens, GFAP, and insulin (6, 7). GFAP epitopes for autoreactive T and B cells have now been identified in non-obese diabetic (NOD) mice and humans with T1D. Serum GFAP antibodies are now used as a predictive marker for the development of T1D, and it has been shown that administration of GFAP as a vaccine delayed the progression of T1D by regulating T cell differentiation (22, 23). GFAP-expressing glial cells of the peripheral nervous system require TRPV1 expression for their proper maturation, and studies have shown that depleting TRPV1-expressing cells reduced the development of insulitis in NOD mice (16, 24). It is fascinating that a cytoskeletal protein widely expressed in pancreatic ductal cells and also in pancreatic glial cells of the central and peripheral nervous system may work as an early autoantigen in T1D.
Exploring further this idea, Slattery's group has recently shown that ablation of autoantigen presentation in GFAP-expressing cell types reduced the development of invasive insulitis in NOD mice (25). We can speculate that reduction, but not total elimination of invasive infiltration, may be due to the absence of presentation of autoantigens other than insulin by GFAP-expressing cells, suggesting that Ag-derived ductal cell is one of the critical requirements in orchestrating the initiation of autoimmune responses to beta-cell antigens.
After many years of efforts and studies focusing on the destruction of the beta-cells in the pancreas, there is still no cure or method of prevention for T1D. So, it makes us wonder whether we have been losing the battle only because we are not looking beyond the walls of the pancreas. T1D can be viewed as an orchestrated autoimmune response originated in the gut. This is evident from the observation that in many autoimmune diseases including T1D, the integrity of the epithelial barrier is compromised, leading to a phenomenon termed as “leaky gut” (26, 27). Pathogens, microbial products, and food-derived antigens find the leaky gut as a route to encounter the resident immune cells. For example, Gram-negative bacteria produce lipopolysaccharides (LPS), an identified endotoxin that can induce immune responses via the toll-like receptor 4 (TLR-4) expressed on monocytes (28). Given the gut connects to the pancreas through pancreatic lymph nodes (PLNs) and mesenteric lymph nodes (MLNs), bacterial and food products can hyperactivate resident T and B autoreactive cells in the gut or the gut-associated MLNs (29). Alternatively, it has been shown that gut microbial products can reach PLNs and locally modify the presentation of pancreatic self-antigens (30). Therefore, excess of food, chemicals, and microbial antigens can skew the intestinal immune system toward a perpetually pro-inflammatory state that may trigger T1D. Newly-diagnosed children with T1D present autoantibodies to GAD, a pancreatic beta-cell autoantigen that is also produced by many bacterial species (31). For instance, T1D patients present antibodies against a heat shock protein from the Mycobacterium avium subspecies paratuberculosis, MAP Hsp65, which has a high degree of homology with human GAD65, suggesting that cross-reactivity between MAP Hsp65 and GAD65 potentially could be a mechanism of triggering TID (32). Strong homology has been found between the islet-antigen IGRP and several gut- and oral-derived microbial peptides. These peptide sequences encode for magnesium transporter (Mgt), for hypothetical protein IEM_00289 and NAD synthetase, respectively, which activate NY8.3 CD8+ T cells with comparable potency to IGRP native peptide (33). Thus, molecular mimicry between excess of gut microbial antigens and islet cell autoantigens may be a mechanism by which gut dysbiosis leads to T1D development.
T1D is a multifactorial condition; diet and environment play an inevitable role in disease modulation (1, 13). Human and murine studies have demonstrated that defects in the induction of central and peripheral tolerance checkpoints (34) also correlate with an altered gut microbiota (35–39), which are notable contributors to T1D pathology. Building on previously extensive reviews on the gut microbiota topic, we have firmly discussed that an altered microbiota and SCFA deficiency are primary causal factors triggering T1D (12–14, 40). The gut microbiota through the production of dietary SCFAs plays a significant role in host defense by modulating the immune system and metabolism. Studies conducted by our group have shown that the combination of a diet rich in acetate and butyrate protected 90% of the NOD mice against T1D, yielding exceptionally high levels of the corresponding SCFAs to the feces (35). In this study, SCFA-induced T1D protection happened via changes in gut/immune regulation-expanding regulatory T (Treg) cells and reducing pathogenic B cells, CD4+, and CD8+ T cells. Diet rich in SCFA acetate and butyrate not only reduced the levels of serum LPS and pro-inflammatory interleukin 21 (IL-21) but also increased the level of serum IL-22, an important cytokine that maintains a healthy commensal microbiota, gut epithelial integrity, and mucosal immunity and ameliorates metabolic disease (41–44). Alternatively, SCFAs can also reduce islet-specific immune responses by increasing the production of antimicrobial peptides (AMPs) in the beta-cells (45). As it has been shown, C-type lectin regenerating islet-derived protein IIIγ (REGIIIγ) and defensins disrupt surface membranes of bacteria, thus enabling a broad regulation of commensal and pathogenic bacteria in the gut (46–48). Diana's group showed that insulin-secreting beta-cells produced the cathelicidin-related antimicrobial peptide (CRAMP), which was defective in NOD mice. Intraperitoneal administration of SCFA butyrate stimulates the production of CRAMP on pancreatic beta-cells via G protein-coupled receptors (GPCRs), which also correlated with the conversion of inflammatory immune cells to a regulatory phenotype (45). Likewise, another study has shown that microbial SCFAs contribute to the increasing concentrations of serum IL-22 (35) required for beta-cell regeneration by up-regulating the expression of Regenerating Reg1 and Reg2 genes in the islets (49).
There are pieces of evidence of compromised gut integrity, dysbiosis, and associated inflammation of the gastrointestinal tract (GI) in NOD mice and patients with T1D (50–55), similar to what has been shown in other inflammatory or autoimmune gut diseases (i.e., infection, celiac disease, IBD). The gut microbiota and the ENS play a critical role in diabetic gastrointestinal motility disorders, as individuals with diabetes suffer from symptoms such as nausea, heartburn, vomiting, diarrhea, abdominal pain, and constipation (56, 57). For example, it is known that slow motility of the GI leads to alterations of the gut microbiota that favors pathogenic bacterial overgrowth and subsequently diarrhea (58, 59). As such, the abundance and diversity of bacteria needed to maintain the integrity of the gut were significantly lower in children with T1D compared to healthy controls (60). On the other hand, animal studies have suggested that accelerated colonic transit time, relative to constipation, could be caused by autonomic neuropathy and diabetes-induced denervation of sympathetic nerve terminals (56, 61). Diet and/or deficiency of dietary SCFAs can also modulate the intestinal motility and survival of enteric neurons by miRNAs, which are involved in energy homeostasis, lipid metabolism, and proliferation and development of GI smooth muscles. miRNAs have been vastly studied in organ damage caused by diabetes, and one study has shown in mice that high-fat diets delay the GI transit, partly by inducing apoptosis in enteric neuronal cells, and this effect was shown to be mediated by Mir375 associated with reduced levels of Pdk (62). There is still too much to understand about the intrinsic mechanisms underlying the connection between the gut microbiota and the ENS and how this affects the course of T1D. Particularly high-fiber or specialized acylated starch diets that boost the microbial production of SCFAs are effective in the control of gut infections and diarrhea, as it has been shown to promote commensal acetate-producing bacteria (63).
Activation of IL-22 through microbial SCFAs contribute to the maintenance of gut homeostasis by the close connection between the intestinal-resident innate lymphoid cell 3 (ILC3) and EGCs. IL-22 is expressed by ILC3, which lies close to EGCs (64), but its role in T1D is still elusive (14). ILCs sustain appropriate immune responses to commensals and pathogens at mucosal barriers by potentiating adaptive immunity and regulating tissue inflammation (65, 66). Likewise, EGCs have critical roles in maintaining gut homeostasis, as they can sense the pathogenic bacteria through toll-like receptors (TLRs). EGCs surround neurons and also connect with blood vessels and lymphatics (67), which allowed EGC-derived signaling molecules to modulate mucosal immunity. As such, EGCs sense environmental stimuli and extend their stellate projections into the ILC3 aggregates within the crypto-patches of the intestinal lamina propria and release neurotrophic factors that stimulate IL-22 secretion from ILC3s (68). The notion that gut microbiota affects the development and maturation of EGCs was shown in germ-free (GF) mice, which present a defective influx of EGCs into the intestinal mucosa (69). This occurs via expression of the neuroregulatory receptor (RET), as ablation of RET in ILC3 leads to reduced IL-22 production and compromised epithelial protection in colon inflammation mouse models (69).
Aligned with this idea, does the early autoreactivity to GFAP observed during insulitis originate in the gut? This is possible to the connections between the pancreas, the ENS, and the gut. The fine-tuned neuro–beta-cell cross-talk is more likely to be broken by the pathological changes occurring in a perturbed gut. Alterations of the gut microbiota, referred to as dysbiosis, decrease epithelial permeability, causing inflammation, and associated tissue damage that exposes numerous self-antigens harbored in the gut and associated enteric neuronal tissues. Gut microbial products can also sense enteric neurons and EGCs partly by pattern recognition receptors, such as TLRs. Indeed, pathogenic and commensal SCFA-producing bacteria up-regulate differentially toll-like receptor 2 (TLR2) expression on human EGCs (70). Expression of TLR2 on enteric neurons and EGCs controls nNOS+ neurons and acetylcholine-esterase-stained fibers in the myenteric ganglia. For example, Escherichia coli promoted expression of MHC II molecules on EGCs and significantly induced S100B protein overexpression and nitric oxide (NO) release from EGC, which was counteracted by pre-treatment with TLR and S100B inhibitors (70). As such, the myenteric plexus of TLR2Ko mice presented reduced expression of glial markers, GFAP, and S100B. Overexpression of GFAP has been observed to correlate with inflammatory responses in the gut (71). S100B is considered as a neurotrophin, due to its either tropic or toxic effects depending on the concentration in the extracellular milieu. Excess amount of S100B acts on RAGE (receptor for advanced glycation end-products), leading to the phosphorylation of mitogen-activated protein kinases (MAPK) and subsequent activation of the nuclear factor κB (NF-κB) and the associated release of NO. Excess NO causes damage to the tissue, resulting in inflammation and reduced integrity of the guts (72, 73). The protective role of EGCs in the maintenance of the gut epithelial integrity has been demonstrated in mice lacking GFAP-positive (+) glia that presented fatal hemorrhagic jejuno-ileitis (74).
During chronic tissue inflammation, significantly increased expression of GFAP on glial cells after stimulation with LPS and pro-inflammatory cytokines has been shown (75), similar to what has been seen in Crohn's disease (CD) and necrotizing enterocolitis (NEC). On the other hand, the presence of MHC class II expression on activated EGCs suggests that these cell types can present antigens (76, 77) derived from multiples sources, including microbes and host. EGCs, with the help of their stellate projections, sample microbial antigens crossing the epithelial barrier and activate diabetogenic T cells. This is given under dysbiosis, predominant in T1D and many autoimmune diseases, and the release of microbial antigens such as LPS may break the tolerance of EGCs leading to overexpression of glial cell markers GFAP and S100B. Thus, GFAP-expressing glial cells may have a protective role in maintaining the integrity of the gut, but under uncontrolled inflammatory conditions, it may lead to autoreactivity. As such, glial cell-derived protein GFAP is now an identified autoantigen in T1D and autoantibodies to GFAP has been detected in NOD mice and humans with T1D (23), thus showing the relevance of the microbiota–EGC pathways in T1D.
One study has shown that SCFA butyrate can induce increasing excitatory choline acetyltransferase (ChAT) neurons through the butyrate transporter monocarboxylate transporter (MCT), which is expressed by enteric neurons (78). However, it is still unknown what factors control neuronal MCT2 expression. Further studies will be necessary to determine how SCFAs regulate MCT2 expression and control the activity of intestinal neural circuits. SCFAs exert their function through two mechanisms, via metabolite sensing GPCRs or inhibition of histone deacetylase (HDAC) activity (13, 35, 79, 80). There are three receptors for SCFA acetate, butyrate, and propionate, namely GPR43 (FFA2), GPR41 (FFA3), and GPR109a. GPR43 is activated by SCFAs with varying potency—acetate > propionate > butyrate. GPR43 is expressed on gut epithelial cells and certain immune cells (81). GPR109a is expressed on a variety of immune cells, as well as adipocytes, hepatocytes, gut and retinal epithelium, vascular endothelium, and neuronal tissue (82). GPR109a is primarily activated by both niacin and butyrate ligands. While niacin levels are not high enough to activate the receptor under normal physiological conditions, levels of butyrate, obtained from the gut environment, and its oxidized form, β-hydroxybutyrate, are sufficient to stimulate a response (82). Similarly, GPR41 has been reported to be expressed on EGCs and enteric neurons (83, 84). GPR41 also binds the three major SCFAs, but with differing affinities (85).
Similar to the effects exerted through the GPCRs, SCFAs can influence the function and development of immune cells directly through epigenetic regulation of gene expression such as inhibition of HDACs (13, 86). HDACs allow the conversion of repressive chromatin structures, which takes place on lysine residues on N-terminal tails of histones 3 and 4, to increase gene transcription. HDACs are a group of 18 known enzymes that remove acetyl groups from the histones tails that bind DNA (87). Although little is known about the effects of SCFAs on EGCs through epigenetic modifications, it has been shown that butyrate treatment increases acetylation of the H3K9 in primary enteric neurons and the EGC in vitro (84).
SCFAs can also modulate gut motility by the production of serotonin by epithelial enterochromaffin cells (ECs) (88, 89). For instance, GF mice present gut dysmotility that was reversed by inoculation with SCFA-producing bacteria. Tested in human-derived EC cell lines, SCFAs increased serotonin (5-hydroxytryptamine [5-HT]) by up-regulating THER expression of tryptophan hydroxylase 1 (Tph1) (89) and by the serotonin-selective reuptake transporter (SERT), which is expressed by intestinal epithelial cells (90). Another critical role of SCFAs on the ENS is evidenced by the conversion of primary bile acids synthesized de novo into secondary bile acids in the liver (91). Aside from their role in dietary fat absorption, secondary bile acids can activate several GPCRs and nuclear hormone receptors, including the G-protein-coupled bile acid receptor 1 (TGR5) and farnesoid X receptor (FXR), highly expressed in enteric neurons and enteroendocrine L cells that improved intestinal inflammation and glucose tolerance in HFD-fed mice (92). TGR5 also affect peristalsis that is mediated partly by serotonin 5-HT (93), implicating its potential for the treatment of constipation and diarrhea. Altogether, this suggests the relevance of the gut–neuro–immune axis in T1D (Figure 1).
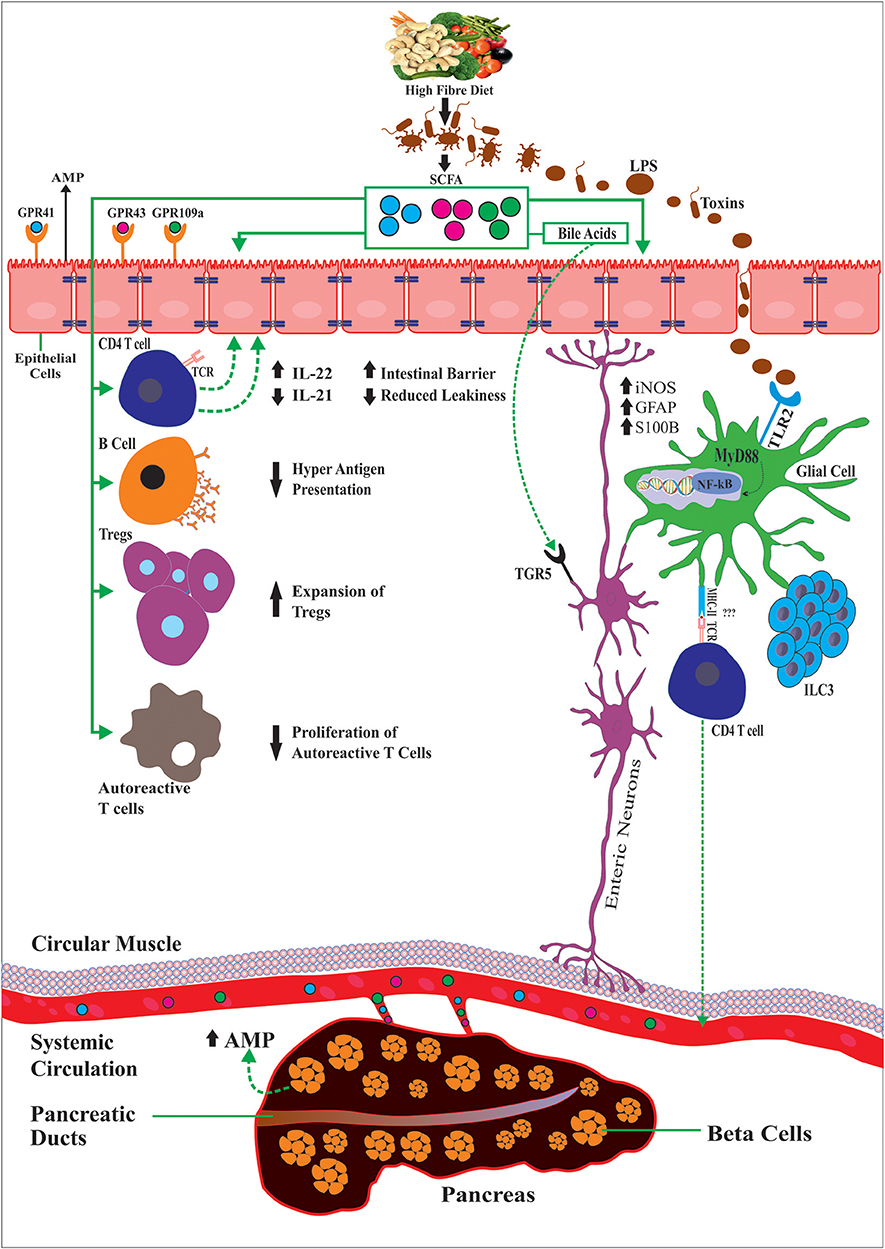
Figure 1. Diet and gut microbiota through the production of dietary SCFAs exert anti-inflammatory effects by controlling the activity of multiple immune cell types, outside or locally in the intestinal mucosa, the enteric glial cells and neurons but also glial cells in the pancreatic islets and the beta-cells. As such SCFAs promote IL-22 production CD4+ T cells or by supporting ILC3 cells, the major producers of IL-22. Also SCFAs can reduce production of pro-inflammatory cytokines IL-21, LPS, induce beta cell regeneration via AMPS, regulate GFAP in the gut and beta-cells, modulate the expansion of regulatory T and reduction of autoreactive CD8 T cells and reducing B cell hyperactive antigen presentation capacity. Activation of GPRCs (GPR41 and GPR43) on enteroendocrine cells of the intestinal epithelium and TLR signaling (e.g., TLR2 and TLR4) maintains subsets of enteric neurons resulting changes in gut motility, conversion of primary bile acids into secondary bile acids, which activate TGRS expressed by enteroendocrine cells and enteric neurons among many others.
Among the described effects that SCFAs have on modulating the immune system, beta-cell biology, and gut homeostasis, we have uncovered a novel role for SCFAs by modulating the ENS in the gut, central for the control and prevention of T1D. Overall, an immune response to antigens presented not only by GFAP-expressing pSCs in the pancreas but also by GFAP-expressing EGCs in the gut is a novel finding involved in the initiation of the autoimmune process. Could it trigger antigen-experienced autoreactive cells to move up the gut and reach the ductal and beta-cells, and break the GFAP-expressing neuronal mantle of the islets? This is an unexplored field and requires further research. Given the close location and connection between the gut and the pancreas and their intrinsic dependence from the nervous system, this fine-tuned immuno–neuro-islet cross-talk may be profoundly affected by perturbed gut homeostasis at very early stages of the initiation of the T1D. Dampening the initial immune response or preventing the perpetuation of the islet-specific immune response by maintaining the integrity of the gut is among the possible therapeutic approaches to reprogram T1D (12, 14). Thus, any hope for a cure may lie in methods that can halt immune-mediated beta-cell damage by maintaining or improving gut–immune tolerance.
EM developed the conceptual idea, wrote and edited the manuscript. AJ wrote and edited the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Jayasimhan A, Mansour Kristy P, Slattery Robyn M. Advances in our understanding of the pathophysiology of type 1 diabetes: lessons from the NOD mouse. Clin Sci. (2014) 126:1–18. doi: 10.1042/CS20120627
2. Paschou SA, Papadopoulou-Marketou N, Chrousos GP, Kanaka-Gantenbein C. On type 1 diabetes mellitus pathogenesis. Endocrine Connect. (2017) 7:R38–46. doi: 10.1530/EC-17-0347
3. Katsarou A, Gudbjörnsdottir S, Rawshani A, Dabelea D, Bonifacio E, Anderson BJ, et al. Type 1 diabetes mellitus. Nat Rev Dis Primers. (2017) 3:17016. doi: 10.1038/nrdp.2017.16
4. Moriyama H, Abiru N, Paronen J, Sikora K, Liu E, Miao D, et al. Evidence for a primary islet autoantigen (preproinsulin 1) for insulitis and diabetes in the nonobese diabetic mouse. Proc Natl Acad Sci USA. (2003) 100:10376–81. doi: 10.1073/pnas.1834450100
5. Arvan P, Pietropaolo M, Ostrov D, Rhodes CJ. Islet autoantigens: structure, function, localization, and regulation. Cold Spring Harbor Perspect Med. (2012) 2: a007658. doi: 10.1101/cshperspect.a007658
6. Winer S, Tsui H, Lau A, Song A, Li X, Cheung RK, et al. Autoimmune islet destruction in spontaneous type 1 diabetes is not β-cell exclusive. Nat Med. (2003) 9:198. doi: 10.1038/nm818
7. Tsui H, Chan Y, Tang L, Winer S, Cheung RK, Paltser G, et al. Targeting of pancreatic glia in type 1 diabetes. Diabetes. (2008) 57:918–28. doi: 10.2337/db07-0226
8. Mallone R, Brezar V, Boitard C. T cell recognition of autoantigens in human type 1 diabetes: clinical perspectives. Clin Dev Immunol. (2011) 2011:513210. doi: 10.1155/2011/513210
9. Eberhard D, Lammert E. The pancreatic beta-cell in the islet and organ community. Curr Opin Genet Dev. (2009) 19:469–75. doi: 10.1016/j.gde.2009.07.003
10. Brissova M, Aamodt K, Brahmachary P, Prasad N, Hong JY, Dai C, et al. Islet microenvironment, modulated by vascular endothelial growth factor-A signaling, promotes beta cell regeneration. Cell Metab. (2014) 19:498–511. doi: 10.1016/j.cmet.2014.02.001
11. Bernier-Latmani J, Petrova TV. Intestinal lymphatic vasculature: structure, mechanisms and functions. Nat Rev Gastroenterol Hepatol. (2017) 14:510–26. doi: 10.1038/nrgastro.2017.79
12. McLeod K, Richards JL, Yap YA, Mariño E. Dietary short chain fatty acids: how the gut microbiota fight against autoimmune and inflammatory diseases. In: Watson RR, Preedy VR, editors. Bioactive Food as Dietary Interventions for Arthritis and Related Inflammatory Diseases. 2nd ed. London: Elsevier (2019). p. 139–52. doi: 10.1016/B978-0-12-813820-5.00007-6
13. Richards JL, Yap YA, McLeod KH, Mackay CR, Mariño E. Diet and gut Microbiota: An Alternative way to control inflamatory and autoimmune diseases. Clin Transl Immunol. (2016) 2:e82. doi: 10.1038/cti.2016.29
14. Yap YA, Marino E. An insight into the intestinal web of mucosal immunity, microbiota, and diet in inflammation. Front Immunol. (2018) 9:2617. doi: 10.3389/fimmu.2018.02617
15. Begg DP, Woods SC. Interactions between the central nervous system and pancreatic islet secretions: a historical perspective. Adv Physiol Educ. (2013) 37:53–60. doi: 10.1152/advan.00167.2012
16. Razavi R, Chan Y, Afifiyan FN, Liu XJ, Wan X, Yantha J, et al. TRPV1+ sensory neurons control beta cell stress and islet inflammation in autoimmune diabetes. Cell. (2006) 127:1123–35. doi: 10.1016/j.cell.2006.10.038
17. Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science. (2011) 334:255–8. doi: 10.1126/science.1209791
18. Yoo BB, Mazmanian SK. The enteric network: interactions between the immune and nervous systems of the gut. Immunity. (2017) 46:910–26. doi: 10.1016/j.immuni.2017.05.011
19. Yarandi SS, Srinivasan S. Diabetic gastrointestinal motility disorders and the role of enteric nervous system: current status and future directions. Neurogastroenterol Motil. (2014) 26:611–24. doi: 10.1111/nmo.12330
20. de Jersey J, Snelgrove SL, Palmer SE, Teteris SA, Mullbacher A, Miller JF, et al. Beta cells cannot directly prime diabetogenic CD8 T cells in nonobese diabetic mice. Proc Natl Acad Sci USA. (2007) 104:1295–300. doi: 10.1073/pnas.0610057104
21. Hamilton-Williams EE, Palmer SE, Charlton B, Slattery RM. Beta cell MHC class I is a late requirement for diabetes. Proc Natl Acad Sci USA. (2003) 100:6688–93. doi: 10.1073/pnas.1131954100
22. Pang Z, Higuchi M, Koriyama H, Yoshida S, Kurinami H, Shimamura M, et al. Evaluating the potential of the GFAP-KLH immune-tolerizing vaccine for type 1 diabetes in mice. FEBS Lett. (2017) 591:129–36. doi: 10.1002/1873-3468.12511
23. Pang Z, Kushiyama A, Sun J, Kikuchi T, Yamazaki H, Iwamoto Y, et al. Glial fibrillary acidic protein (GFAP) is a novel biomarker for the prediction of autoimmune diabetes. FASEB J. (2017) 31:4053–63. doi: 10.1096/fj.201700110R
24. Bour-Jordan H, Bluestone JA. Sensory neurons link the nervous system and autoimmune diabetes. Cell. (2006) 127:1097–9. doi: 10.1016/j.cell.2006.11.030
25. Jayasimhan A, Ellis DP, Ziegler AI, Slattery RM. Pancreatic ductal cell antigens are important in the development of invasive insulitis in non-obese diabetic mice. J Neuroimmunol. (2019) 327:1–9. doi: 10.1016/j.jneuroim.2019.01.001
26. Vaarala O. Leaking gut in type 1 diabetes. Curr Opin Gastroenterol. (2008) 24:701–6. doi: 10.1097/MOG.0b013e32830e6d98
27. Vaarala O. Is the origin of type 1 diabetes in the gut? Immunol Cell Biol. (2012) 90:271–6. doi: 10.1038/icb.2011.115
28. Sabroe I, Read RC, Whyte MKB, Dockrell DH, Vogel SN, Dower SK. Toll-like receptors in health and disease: complex questions remain. J Immunol. (2003) 171:1630. doi: 10.4049/jimmunol.171.4.1630
29. Jaakkola I, Jalkanen S, Hanninen A. Diabetogenic T cells are primed both in pancreatic and gut-associated lymph nodes in NOD mice. Eur J Immunol. (2003) 33:3255–64. doi: 10.1002/eji.200324405
30. Turley SJ, Lee JW, Dutton-Swain N, Mathis D, Benoist C. Endocrine self and gut non-self intersect in the pancreatic lymph nodes. Proc Natl Acad Sci USA. (2005) 102:17729–33. doi: 10.1073/pnas.0509006102
31. Mulder SJ. Bacteria of food and human intestine are the most possible sources of the gad-trigger of type 1 diabetes. Med Hyp. (2005) 65:308–11. doi: 10.1016/j.mehy.2005.02.027
32. Bodin J, Stene LC, Nygaard UC. Can exposure to environmental chemicals increase the risk of diabetes type 1 development? Biomed Res Int. (2015) 2015:208947. doi: 10.1155/2015/208947
33. Tai N, Peng J, Liu F, Gulden E, Hu Y, Zhang X, et al. Microbial antigen mimics activate diabetogenic CD8 T cells in NOD mice. J Exp Med. (2016) 213:2129–46. doi: 10.1084/jem.20160526
34. Herold KC, Vignali DA, Cooke A, Bluestone JA. Type 1 diabetes: translating mechanistic observations into effective clinical outcomes. Nat Rev Immunol. (2013) 13:243–56. doi: 10.1038/nri3422
35. Marino E, Richards JL, McLeod KH, Stanley D, Yap YA, Knight J, et al. Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat Immunol. (2017) 18:552–62. doi: 10.1038/ni.3713
36. Vatanen T, Franzosa EA, Schwager R, Tripathi S, Arthur TD, Vehik K, et al. The human gut microbiome in early-onset type 1 diabetes from the TEDDY study. Nature. (2018) 562:589–94. doi: 10.1038/s41586-018-0620-2
37. Vatanen T, Kostic AD, d'Hennezel E, Siljander H, Franzosa EA, Yassour M, et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell. (2016) 165:842–53. doi: 10.1016/j.cell.2016.04.007
38. Gavin PG, Mullaney JA, Loo D, Cao KL, Gottlieb PA, Hill MM, et al. Intestinal metaproteomics reveals host-microbiota interactions in subjects at risk for type 1 diabetes. Diabetes Care. (2018) 41:2178–86. doi: 10.2337/dc18-0777
39. Mullaney JA, Stephens JE, Costello ME, Fong C, Geeling BE, Gavin PG, et al. Type 1 diabetes susceptibility alleles are associated with distinct alterations in the gut microbiota. Microbiome. (2018) 6:35. doi: 10.1186/s40168-018-0438-z
40. Marino E. The gut microbiota and immune-regulation: the fate of health and disease. Clin Transl Immunol. (2016) 5:e107. doi: 10.1038/cti.2016.61
41. Hasnain SZ, Borg DJ, Harcourt BE, Tong H, Sheng YH, Ng CP, et al. Glycemic control in diabetes is restored by therapeutic manipulation of cytokines that regulate beta cell stress. Nat Med. (2014) 20:1417–26. doi: 10.1038/nm.3705
42. Wang X, Ota N, Manzanillo P, Kates L, Zavala-Solorio J, Eidenschenk C, et al. Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature. (2014) 514:237–41. doi: 10.1038/nature13564
43. Dudakov JA, Hanash AM, van den Brink MR. Interleukin-22: immunobiology and pathology. Annu Rev Immunol. (2015) 33:747–85. doi: 10.1146/annurev-immunol-032414-112123
44. Sabat R, Ouyang W, Wolk K. Therapeutic opportunities of the IL-22-IL-22R1 system. Nat Rev Drug Discov. (2014) 13:21–38. doi: 10.1038/nrd4176
45. Sun J, Furio L, Mecheri R, van der Does AM, Lundeberg E, Saveanu L, et al. Pancreatic beta-cells limit autoimmune diabetes via an immunoregulatory antimicrobial peptide expressed under the influence of the gut microbiota. Immunity. (2015) 43:304–17. doi: 10.1016/j.immuni.2015.07.013
46. Gallo RL, Hooper LV. Epithelial antimicrobial defence of the skin and intestine. Nat Rev Immunol. (2012) 12:503–16. doi: 10.1038/nri3228
47. Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol. (2011) 9:356–68. doi: 10.1038/nrmicro2546
48. Mukherjee S, Hooper LV. Antimicrobial defense of the intestine. Immunity. (2015) 42:28–39. doi: 10.1016/j.immuni.2014.12.028
49. Hill T, Krougly O, Nikoopour E, Bellemore S, Lee-Chan E, Fouser LA, et al. The involvement of interleukin-22 in the expression of pancreatic beta cell regenerative Reg genes. Cell Regen. (2013) 2:2. doi: 10.1186/2045-9769-2-2
50. Alam C, Valkonen S, Palagani V, Jalava J, Eerola E, Hänninen A. Inflammatory tendencies and overproduction of IL-17 in the colon of young NOD mice are counteracted with diet change. Diabetes. (2010) 59:2237–46. doi: 10.2337/db10-0147
51. Bolla AM, Pellegrini S, Sordi V, Bonfanti R, Bosi E, Piemonti L, et al. Duodenal mucosa of patients with type 1 diabetes shows distinctive inflammatory profile and microbiota. J Clin Endocrinol Metab. (2017) 102:1468–77. doi: 10.1210/jc.2016-3222
52. Bosi E, Molteni L, Radaelli MG, Folini L, Fermo I, Bazzigaluppi E, et al. Increased intestinal permeability precedes clinical onset of type 1 diabetes. Diabetologia. (2006) 49:2824–7. doi: 10.1007/s00125-006-0465-3
53. Lee AS, Gibson DL, Zhang Y, Sham HP, Vallance BA, Dutz JP. Gut barrier disruption by an enteric bacterial pathogen accelerates insulitis in NOD mice. Diabetologia. (2010) 53:741–8. doi: 10.1007/s00125-009-1626-y
54. Leeds JS, Hopper AD, Hadjivassiliou M, Tesfaye S, Sanders DS. Inflammatory bowel disease is more common in type 1 diabetes mellitus. Gut. (2011) 60(Suppl 1):A208. doi: 10.1136/gut.2011.239301.438
55. Halling ML, Kjeldsen J, Knudsen T, Nielsen J, Hansen LK. Patients with inflammatory bowel disease have increased risk of autoimmune and inflammatory diseases. World J Gastroenterol. (2017) 23:6137–46. doi: 10.3748/wjg.v23.i33.6137
56. Du YT, Rayner CK, Jones KL, Talley NJ, Horowitz M. Gastrointestinal symptoms in diabetes: prevalence, assessment, pathogenesis, and management. Diabetes care. (2018) 41:627–37. doi: 10.2337/dc17-1536
57. Maleki D, Locke GR 3rd, Camilleri M, Zinsmeister AR, Yawn BP, Leibson C, et al. Gastrointestinal tract symptoms among persons with diabetes mellitus in the community. Arch Intern Med. (2000) 160:2808–16. doi: 10.1001/archinte.160.18.2808
58. Nguyen NQ, Debreceni TL, Bambrick JE, Bellon M, Wishart J, Standfield S, et al. Rapid gastric and intestinal transit is a major determinant of changes in blood glucose, intestinal hormones, glucose absorption and postprandial symptoms after gastric bypass. Obesity. (2014) 22:2003–9. doi: 10.1002/oby.20791
59. Sellin JH, Hart R. Glucose malabsorption associated with rapid intestinal transit. Am J Gastroenterol. (1992) 87:584–9.
60. Murri M, Leiva I, Gomez-Zumaquero JM, Tinahones FJ, Cardona F, Soriguer F, et al. Gut microbiota in children with type 1 diabetes differs from that in healthy children: a case-control study. BMC Med. (2013) 11:46. doi: 10.1186/1741-7015-11-46
61. Rosa-e-Silva L, Troncon LE, Oliveira RB, Foss MC, Braga FJ, Gallo Junior L. Rapid distal small bowel transit associated with sympathetic denervation in type I diabetes mellitus. Gut. (1996) 39:748–56. doi: 10.1136/gut.39.5.748
62. Nezami BG, Mwangi SM, Lee JE, Jeppsson S, Anitha M, Yarandi SS, et al. MicroRNA 375 mediates palmitate-induced enteric neuronal damage and high-fat diet-induced delayed intestinal transit in mice. Gastroenterology. (2014) 146:473–83.e3. doi: 10.1053/j.gastro.2013.10.053
63. Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K, et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature. (2011) 469:543–7. doi: 10.1038/nature09646
64. Ibiza S, Garcia-Cassani B, Ribeiro H, Carvalho T, Almeida L, Marques R, et al. Glial-cell-derived neuroregulators control type 3 innate lymphoid cells and gut defence. Nature. (2016) 535:440–3. doi: 10.1038/nature18644
65. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell. (2018) 174:1054–66. doi: 10.1016/j.cell.2018.07.017
66. Wang S, Xia P, Chen Y, Qu Y, Xiong Z, Ye B, et al. Regulatory innate lymphoid cells control innate intestinal inflammation. Cell. (2017) 171:201–16.e18. doi: 10.1016/j.cell.2017.07.027
67. Sternini C, Anselmi L, Rozengurt E. Enteroendocrine cells: a site of ‘taste' in gastrointestinal chemosensing. Curr Opin Endocrinol Diabetes Obes. (2008) 15:73–8. doi: 10.1097/MED.0b013e3282f43a73
68. Ouyang W, O'Garra A. IL-10 Family cytokines IL-10 and IL-22: from basic science to clinical translation. Immunity. (2019) 50:871–91. doi: 10.1016/j.immuni.2019.03.020
69. Kabouridis PS, Lasrado R, McCallum S, Chng SH, Snippert HJ, Clevers H, et al. Microbiota controls the homeostasis of glial cells in the gut lamina propria. Neuron. (2015) 85:289–95. doi: 10.1016/j.neuron.2014.12.037
70. Turco F, Sarnelli G, Cirillo C, Palumbo I, De Giorgi F, D'Alessandro A, et al. Enteroglial-derived S100B protein integrates bacteria-induced toll-like receptor signalling in human enteric glial cells. Gut. (2014) 63:105–15. doi: 10.1136/gutjnl-2012-302090
72. Schulzke JD, Ploeger S, Amasheh M, Fromm A, Zeissig S, Troeger H, et al. Epithelial tight junctions in intestinal inflammation. Ann N Y Acad Sci. (2009) 1165:294–300. doi: 10.1111/j.1749-6632.2009.04062.x
74. Bush TG, Savidge TC, Freeman TC, Cox HJ, Campbell EA, Mucke L, et al. Fulminant jejuno-ileitis following ablation of enteric glia in adult transgenic mice. Cell. (1998) 93:189–201. doi: 10.1016/S0092-8674(00)81571-8
75. von Boyen GB, Steinkamp M, Reinshagen M, Schafer KH, Adler G, Kirsch J. Proinflammatory cytokines increase glial fibrillary acidic protein expression in enteric glia. Gut. (2004) 53:222–8. doi: 10.1136/gut.2003.012625
76. Taniguchi Y, Ono K, Yoshida S, Tanaka R. Antigen-presenting capability of glial cells under glioma-harboring conditions and the effect of glioma-derived factors on antigen presentation. J Neuroimmunol. (2000) 111:177–85. doi: 10.1016/S0165-5728(00)00361-1
77. Schetters STT, Gomez-Nicola D, Garcia-Vallejo JJ, Van Kooyk Y. Neuroinflammation: microglia and T cells get ready to tango. Front Immunol. (2017) 8:1905. doi: 10.3389/fimmu.2017.01905
78. Soret R, Chevalier J, De Coppet P, Poupeau G, Derkinderen P, Segain JP, et al. Short-chain fatty acids regulate the enteric neurons and control gastrointestinal motility in rats. Gastroenterology. (2010) 138:1772–82. doi: 10.1053/j.gastro.2010.01.053
79. Macia L, Tan J, Vieira AT, Leach K, Stanley D, Luong S, et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat Commun. (2015) 6:6734. doi: 10.1038/ncomms7734
80. Thorburn AN, McKenzie CI, Shen S, Stanley D, Macia L, Mason LJ, et al. Evidence that asthma is a developmental origin disease influenced by maternal diet and bacterial metabolites. Nat Commun. (2015) 6:7320. doi: 10.1038/ncomms8320
81. Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. (2009) 461:1282–6. doi: 10.1038/nature08530
82. Elangovan S, Pathania R, Ramachandran S, Ananth S, Padia RN, Lan L, et al. The niacin/butyrate receptor GPR109A suppresses mammary tumorigenesis by inhibiting cell survival. Cancer Res. (2014) 74:1166–78. doi: 10.1158/0008-5472.CAN-13-1451
83. Nohr MK, Pedersen MH, Gille A, Egerod KL, Engelstoft MS, Husted AS, et al. GPR41/FFAR3 and GPR43/FFAR2 as cosensors for short-chain fatty acids in enteroendocrine cells vs FFAR3 in enteric neurons and FFAR2 in enteric leukocytes. Endocrinology. (2013) 154:3552–64. doi: 10.1210/en.2013-1142
84. Cossais F, Durand T, Chevalier J, Boudaud M, Kermarrec L, Aubert P, et al. Postnatal development of the myenteric glial network and its modulation by butyrate. Am J Physiol Gastrointestinal Liver Physiol. (2016) 310:G941–51. doi: 10.1152/ajpgi.00232.2015
85. Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, et al. The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem. (2003) 278:11312–9. doi: 10.1074/jbc.M211609200
86. Thorburn AN, Macia L, Mackay CR. Diet, metabolites, and “western-lifestyle” inflammatory diseases. Immunity. (2014) 40:833–42. doi: 10.1016/j.immuni.2014.05.014
87. Mathias RA, Guise AJ, Cristea IM. Post-translational modifications regulate class IIa histone deacetylase (HDAC) function in health and disease. Mol Cell Proteomics. (2015) 14:456–70. doi: 10.1074/mcp.O114.046565
88. Yano JM, Yu K, Donaldson GP, Shastri GG, Ann P, Ma L, et al. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell. (2015) 161:264–76. doi: 10.1016/j.cell.2015.02.047
89. Reigstad CS, Salmonson CE, Rainey JF III, Szurszewski JH, Linden DR, Sonnenburg JL, et al. Gut microbes promote colonic serotonin production through an effect of short-chain fatty acids on enterochromaffin cells. FASEB J. (2015) 29:1395–403. doi: 10.1096/fj.14-259598
90. Bian X, Patel B, Dai X, Galligan JJ, Swain G. High mucosal serotonin availability in neonatal guinea pig ileum is associated with low serotonin transporter expression. Gastroenterology. (2007) 132:2438–47. doi: 10.1053/j.gastro.2007.03.103
91. Poole DP, Godfrey C, Cattaruzza F, Cottrell GS, Kirkland JG, Pelayo JC, et al. Expression and function of the bile acid receptor GpBAR1 (TGR5) in the murine enteric nervous system. Neurogastroenterol Motil. (2010) 22:814–25, e227–8. doi: 10.1111/j.1365-2982.2010.01487.x
92. Chavez-Talavera O, Tailleux A, Lefebvre P, Staels B. Bile acid control of metabolism and inflammation in obesity, type 2 diabetes, dyslipidemia, and nonalcoholic fatty liver disease. Gastroenterology. (2017) 152:1679–94.e3. doi: 10.1053/j.gastro.2017.01.055
Keywords: SCFA (short chain fatty acids), GFAP—glial fibrillary acidic protein, gut microbiota, glial cell, interleukin 22 (IL-22), ILC3s, beta cells, diabetes
Citation: Jayasimhan A and Mariño E (2019) Dietary SCFAs, IL-22, and GFAP: The Three Musketeers in the Gut–Neuro–Immune Network in Type 1 Diabetes. Front. Immunol. 10:2429. doi: 10.3389/fimmu.2019.02429
Received: 10 July 2019; Accepted: 30 September 2019;
Published: 29 October 2019.
Edited by:
Julien Diana, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Jasenka Zubcevic, University of Florida, United StatesCopyright © 2019 Jayasimhan and Mariño. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eliana Mariño, ZWxpYW5hLm1hcmlub0Btb25hc2guZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.