
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 09 October 2019
Sec. Inflammation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02376
This article is part of the Research Topic Role of Neutrophils in Inflammatory Diseases View all 11 articles
 Chih-Chao Chiang1,2,3†
Chih-Chao Chiang1,2,3† Wei-Jen Cheng1,4,5†
Wei-Jen Cheng1,4,5† Michal Korinek6,7,8
Michal Korinek6,7,8 Cheng-Yu Lin6
Cheng-Yu Lin6 Tsong-Long Hwang4,6,7,9,10,11*
Tsong-Long Hwang4,6,7,9,10,11*Neutrophils are the most abundant innate immune cells. The pathogenic roles of neutrophils are related to chronic inflammation and autoimmune diseases. Psoriasis is a chronic systemic inflammatory disease affecting ~2–3% of the world population. The abundant presence of neutrophils in the psoriatic skin lesions serves as a typical histopathologic hallmark of psoriasis. Recent reports indicated that oxidative stress, granular components, and neutrophil extracellular traps from psoriatic neutrophils are related to the initial and maintenance phases of psoriasis. This review provides an overview on the recent (up to 2019) advances in understanding the role of neutrophils in the pathophysiology of psoriasis, including the effects of respiratory burst, degranulation, and neutrophil extracellular trap formation on psoriatic immunity and the clinical relationships.
Neutrophils are the most abundant cells in innate immunity. The main offensive functions of neutrophils include respiratory burst accompanied by reactive oxygen species (ROS) generation, degranulation (release of granules), and the formation of neutrophil extracellular traps (NETs) (Figure 1) (1, 2). Neutrophils shape adaptive immunity because they communicate and interact with the antigen-presenting cells and lymphocytes at the sites of inflammation (3, 4). Recently great attention was brought to the role of neutrophils in the development and progression of autoimmune diseases such as psoriasis.
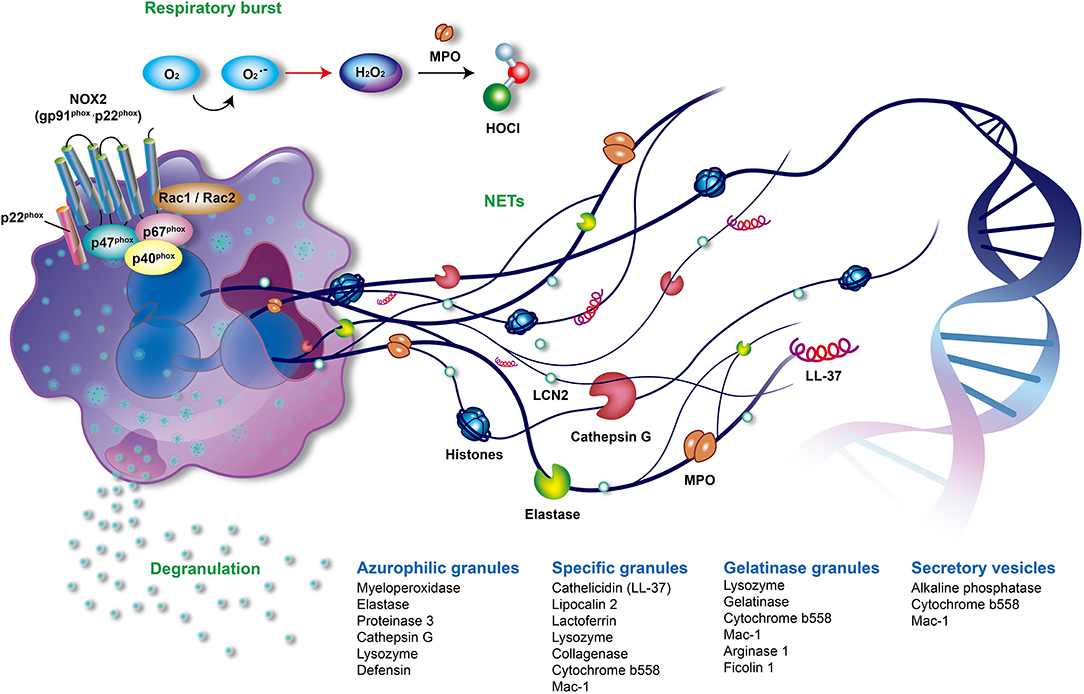
Figure 1. The offensive mechanisms of neutrophils in inflammation. In the activated neutrophils, respiratory burst, degranulation, and the formation of neutrophil extracellular traps (NETs) are the main mechanisms responsible for inflammation. During the respiratory burst, neutrophils utilize oxygen to produce reactive oxygen species (ROS) such as superoxide (), peroxide H2O2 or hypochlorous acid (HOCl) by activating NADPH oxidase (NOX2) and myeloperoxidase (MPO). As activation processes are triggered by diverse stimuli, neutrophils mobilize different granules including azurophilic granules, specific granules, gelatinase granules, and secretory vesicles. Subsequently, neutrophils degranulate and release various granular compounds of which neutrophil elastase (NE), MPO, proteinase 3, LL37, and lipocalin 2 play an important role in the pathogenesis of psoriasis. NETs are novel and pivotal components of neutrophils composed of extruded sticky and decondensed chromatin decorated with many antimicrobial compounds, such as histones, MPO, NE, and cathepsin G. The formation of NETs is involved in complicated inflammatory reactions and contributes to the pathogenesis of psoriasis. MPO, myeloperoxidase; NADPH, reduced nicotinamide adenine dinucleotide phosphate; NE, neutrophil elastase; NET, neutrophil extracellular trap; NOX2, NADPH oxidase.
Psoriatic neutrophils produce augmented respiratory burst with an overt accumulation of oxidative stress involving complicated inflammatory pathways. Proteases released in the degranulation step by neutrophils, such as myeloperoxidase (MPO), neutrophil elastase (NE), proteinase 3, and cathepsin G, participate in the generation of ROS, proteolytical activation of inflammatory mediators, and formation of autoantigens in psoriasis. The complexity and prevalence of psoriasis among the population since antiquity encouraged scientists to study the etiology of the disease and its relation to the immune system and inflammatory responses. Elegant reviews described the role of inflammation and immune system in the development of psoriasis (5, 6), the importance of biologic therapies targeting systemic inflammation (7), the significance of utilizing specific antibodies (8), the contribution of platelets to regulation of NET formation (9), the antimicrobial and pathogenetic roles of neutrophils in autoimmune, autoinflammatory, metabolic (10) and cardiovascular diseases (11). Also, recent books summarized the causes, symptoms, and treatment options of psoriasis (12–15).
However, the role of neutrophils in psoriasis was not deeply analyzed and summarized based on recent literature. Thus, in this work, we focus on summarizing recent findings on the main offensive features of neutrophils including respiratory burst, degranulation and NETs and their direct connection with development and progression of psoriasis (Figure 2). We hope that our work would provide a foundation for further studies to attenuate overstimulation of neutrophils in psoriasis and aid patients with a debilitating disease.
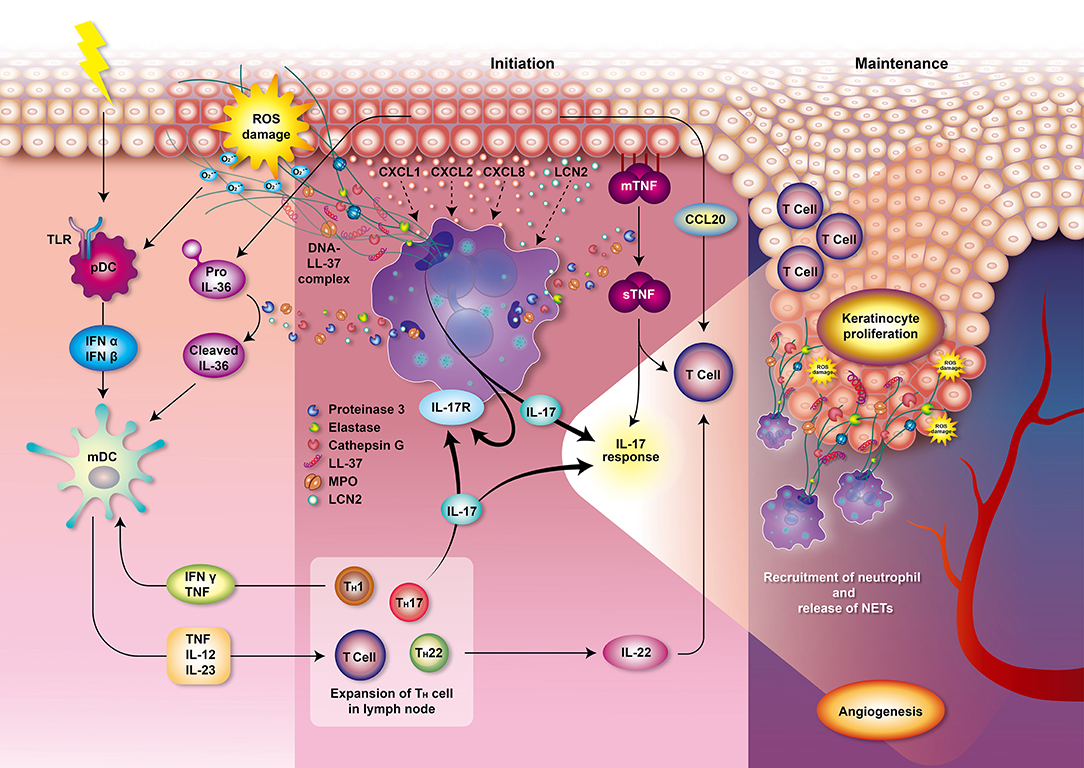
Figure 2. Role of neutrophils in psoriasis. Various and diverse endogenous and exogenous impulses such as antigens, trauma, infection, or emotional stress can trigger the complex immune reactions leading to psoriasis. The interplay of neutrophils, dendritic cells, and T cells bridges the innate immune and adaptive immune systems. T cells and keratinocytes release chemokines, such as IL-17, CXCL1, CXCL2, and CXCL8, that mediate the recruitment of neutrophils. Circulating neutrophils migrate to the psoriatic lesions and induce respiratory burst, degranulation, and formation of NETs, thereby contributing to the immunopathogenesis of psoriasis which involves T cell imbalance, keratinocyte proliferation, angiogenesis, and auto-antigen formation. Neutrophils from patients with psoriasis have enhanced NOX2 and MPO activity and augmented respiratory burst. MPO also participates in generating oxidative stress and upregulating degranulation. The accumulated oxidative stress produced by neutrophils could stimulate the antigen-presenting cells pDCs via TLR receptors 7 and 9 which stimulate antigen-specific CD8+ T cells (memory T cells in dermis and naive T cells in lymph node) to release cytokines, chemokines, and other innate immune mediators. These T cells may also migrate to epidermis and trigger local inflammation and keratinocyte proliferation via MHC I receptor of keratinocytes. Production of IFN-α and IFN-β by pDCs then stimulates mDCs to secrete pro-inflammatory mediators such as TNF, IL-12 and IL-23. Proteinase 3 released from neutrophils cleaves pro-IL-36 to activated IL-36 cytokine amplifying the response of mDCs. TNF, IL-12, and IL-23 play an important role in the initiation of the Th1, Th17, and Th22 cells immune response in lymph node, leading to a secretion of various cytokines and chemokines. IL-1 is further amplifying Th17 response while TNF and IFN-γ is creating a back loop to mDCs activation. Th17 activation then leads to the production of IL-17 activating neutrophils and keratinocytes via IL-17 receptors which generates profound IL-17 response. Keratinocytes produce TNF and CCL20, a chemotactic for T cell and DCs. Neutrophils degranulate and release MPO, NE, proteinase 3, cathepsin G and lipocalin. Proteinase 3 cleaves and converts the resting TNFα located in membrane of epithelial cells (mTNFα) to an activated state called soluble TNFα (sTNFα). Proteinase 3 also contributes to the formation of the LL-37 which serves as autoantigen. The chromatin of NETs (DNA) combined with LL-37 have a profound role in the initiation and maintenance of immune response in psoriasis. NETs further supply IL-17 and induce Th17 cells to release more IL-17, which plays a decisive role in the maintenance of psoriasis. These processes participate in the psoriasis complex inflammatory reactions and lead to the escalation of local psoriatic tissue inflammation. IL-22 contributes to the formation of characteristic psoriatic skin lesions including epidermal hyperplasia and acanthosis (thickening of skin). The activation of the following transcription factors promote TNF and IL-17 production and formation of downstream amplification loops in psoriasis: the Janus kinase (JAK)–signal transducer and activator of transcription (STAT) family, nuclear factor-κB (NF-κB) and cyclic AMP. Furthermore, the activation of endothelial cells induces vascular proliferation, angiogenesis and the expression of adhesion molecules in the endothelium to recruit additional inflammatory cells into the skin such as mast cells and macrophages contributing to the pathogenesis of psoriasis. CCL, CC-chemokine ligand; CXCL, chemokine (C-X-C motif) ligand; IL, interleukin; IFN, interfenon; MHC I, major histocompatibility complex class I, MPO, myeloperoxidase; NET, neutrophil extracellular trap; NOX, NADPH oxidase; NE, neutrophil elastase; pDCs, plasmacytoid dendritic cells; mDCs, myeloid dendritic cells; TLR, Toll-like receptor; TNF, tumor necrosis factor.
Psoriasis affects ~2–3% of the world population (>125 million people). Psoriasis is a common, chronic, immune-mediated disease that is manifested mainly as skin lesions and extracutaneous comorbidities (16, 17). It is associated with systemic inflammation, similar to that observed in obesity, malignancy, psoriatic arthritis, cardiovascular disorders, chronic obstructive pulmonary diseases, type 2 diabetes mellitus, liver and renal diseases, and inflammatory bowel diseases (18–20). Psoriasis affects men and women equally and usually starts to be manifested at the age of 20 to 30, but children and teenagers can be also affected (21). Clinical types of psoriasis include psoriasis vulgaris, guttate psoriasis, inverse psoriasis, pustular psoriasis, and erythrodermic psoriasis (22). Typical skin manifestations of psoriasis include erythematous, indurated, and scaling plaques that are painful, itchy, and have a burning sensation (16, 23). Psoriasis decreases patients' quality of life due to unpleasant symptoms and related public stigma (24, 25). The unpleasant skin appearance contributes to reduced employment levels and thus, affects the financial status of patients (26). Depression and suicidal tendencies are also increased in patients with psoriasis (27, 28). Therefore, psoriasis results in long-term physical, psychological, and economic burden at both the individual and societal levels.
Psoriasis is an immunogenetic disease that is associated with the interactions between the innate and adaptive immune systems (29). The immunology disturbance in psoriasis is related to overstimulation of neutrophils, dendritic cells, T cells, keratinocytes, fibroblasts (6, 30), mast cells (31), and melanocytes (30, 32). Munro's microabscesses filled with neutrophils, which were first described in 1898, are considered as one of the major histopathological hallmarks of psoriasis (33). Neutrophils are now thought to be regulators between the innate and adaptive immune systems (34, 35).
There is no cure for psoriasis, however the symptoms can be reduced either by avoiding triggers or by medications. Currently, the available treatments such as phototherapy, topical therapy (corticosteroids, vitamin D analogs), systemic therapy (methotrexate, apremilast, cyclosporin), and biological treatments offer a relieve for patients with different severity of psoriasis. However, the risk-benefit ratio must be well-considered on individual basis, particularly, considering the chronic course of the disease and limitations of the long-term use of certain drugs (16). Although T cell immunology-related treatments have emerged as attractive options for psoriasis, according to a systematic review of adherence and satisfaction to current treatment covering studies conducted in 2002 (36) or between beginning of 2009 and end of 2014 in European Union (37), psoriatic patients expressed only moderate satisfaction with the available treatments as evidenced by the poor adherence rates, in particular to topical treatments (36, 37). That might be the biggest motivating factor for the use of alternative treatment methods such as traditional Chinese medicine and herbs (Scutellaria baicalensis, Zingiber officinale, Indigo naturalis, Mahonia aquifolium, Aloe vera) (38), dietary supplements (fish oil, vitamin D) or other (39). Furthermore, the combination of certain alternative medicines with conventional drug therapies has been shown to improve the treatment efficacy, which points to the importance of evaluation of safety of combined treatments, education of doctors but also improvements in patient-doctor interactions (40). Indeed, modern biological therapies demonstrate improved safety and efficacy, as well as better satisfaction in patients, but belong to an expensive class of drugs, with limited availability (41). Biological therapies of psoriasis include monoclonal antibodies or inhibitors targeting tumor necrosis factor (TNF)α (infliximab, adalimumab; etanercept) (42–44), interleukin (IL)-23/IL-12 (ustekinumab) (42), IL-23 (guselkumab, tildrakizumab, risankizumab) (45, 46), or IL-17 (secukinumab, ixekizumab, brodalumab) (11, 47, 48). The available therapies indirectly affect the function and numbers of neutrophils. According to previous studies, the neutrophil-to-lymphocyte ratio (NLR) and platelet-to-lymphocyte ratio (PLR) are significantly increased in patients with psoriasis (49). While the elevated NLR and PLR values are particularly associated with psoriasis, they do not indicate the severity of the condition (50). The importance of NLR level in the progression of the disease was demonstrated by the reduction in NLR level following psoriasis treatment (51). That brings in question the effectiveness of certain treatments such as narrow-band ultraviolet B phototherapy which does not affect the increased level of NLR (52). Increased NLR and PLR levels emerged as unrecognized predictors of subclinical atherosclerosis in patients with psoriasis (53). The presence of excessive amount of neutrophils is characteristic for the generalized pustular psoriasis. Recent reports indicated that the depletion of neutrophils significantly relieves the symptoms of pustular psoriasis in patients that did not respond well to conventional treatments (54). Understanding the role of neutrophils in psoriasis attracted the attention of scientific community aiming to develop treatment protocols focusing on attenuating neutrophil overstimulation in this disease (55).
Circulating neutrophils are recruited to inflammatory sites following inflammatory signals. They are then activated to generate and release large amounts of ROS in a phenomenon known as respiratory burst. NADPH oxidase (NOX2) and MPO are two key enzymes that contribute to the respiratory burst (56, 57). NOX2 is composed of transmembrane cytochrome b558 (p22phox and gp91phox) and cytosolic subunits (p40phox, p47phox, p67phox, and Rac1/2). The assembled NOX2 complex, at the phagosomal and plasma membranes, is fully activated to generate superoxide anion (), which is the origin of various ROS produced by neutrophils. Superoxide () is rapidly converted to hydrogen peroxide (H2O2) by superoxide dismutase (SOD). MPO, a heme peroxidase enzyme which is released in a process known as degranulation, utilizes H2O2 to produce many secondary reactive products. These products include hypochlorous acid (HOCl), chloramines (R-NHCl), and hypothiocyanite (OSCN–), as well as organic radicals such as products of lipid peroxidation (Figure 1) (58, 59).
ROS production is an integral part of the antimicrobial activity of neutrophils. However, the overproduction or inadequate clearance of ROS can cause various oxidative stress-related dysfunctions. These include cell and tissue damage; peroxidation and modification of DNA, lipids, and proteins; autoimmune NET formation; and autoantibody generation (58, 60). Neutrophils obtained from patients with psoriasis were shown to possess increased MPO and NOX2 activities, and release more ROS compared with neutrophils from healthy individuals (61, 62). Keratinocytes and T cells in psoriatic lesions produce priming agents of neutrophils, which results in an augmented respiratory burst by neutrophils with the overproduction of ROS (63–66). Accumulation of oxidative radicals also contributes to the pathogenesis of psoriasis. In response to the overproduction of ROS, dendritic cells are stimulated to present antigens to the T cells which results in an imbalance of T helper cell (Th)1 and Th2 cells, stimulation of keratinocytes proliferation, and promotion of angiogenesis (Figure 2). ROS then serve as the second messenger to activate mitogen-activated protein kinase (MAPK), nuclear factor-kappa B (NF-κB), or the Janus kinase-signal transducer and activator of transcription proteins (JAK-STAT)-related inflammatory pathways (67, 68). Suppression of respiratory burst of neutrophils emerged as a plausible pathway of attenuating overly immune response associated with psoriatic symptoms.
Neutrophils possess a multi-lobed nucleus, few mitochondria, and many specific storage organelles called granules. Granules are classified into azurophilic granules, specific granules, gelatinase granules, and secretory vesicles depending on their size, reaction with peroxidase-reactive dye, staining with 3,3'-diaminobenzidine, protein content, and tendency to mobilize (Figure 1) (69, 70). Azurophilic (peroxidase-positive or primary) granules are packed with MPO, bactericidal/permeability-increasing protein, defensins, lysozyme, and serine proteases, such as neutrophil elastase (NE), proteinase 3, and cathepsin G. Lysozyme is also found in specific and gelatinase granules (55). Specific (secondary) granules contain distinctive iron-binding glycoprotein lactoferrin, neutrophil gelatinase-associated lipocalin (NGAL, also called serum lipocalin-2, LCN2), collagenase, cytochrome b558, MAC-1 (CD11/CD18), and importantly, cathelicidins such as LL-37 (71). Cytochrome b558 and MAC-1 are also present in gelatinase granules and secretory vesicles (55). Gelatinase (tertiary) granules store gelatinase, lysozyme, arginase 1, and ficolin 1 (72). The secretory vesicles contain a characteristic alkaline phosphatase (57).
As activation processes of neutrophils are triggered by diverse stimuli such as bacterial or proinflammatory lipid mediators, neutrophils mobilize different granules and release the aforementioned granular components in a process known as degranulation or exocytosis (57). Degranulation is regulated by complicated control mechanisms, such as calcium signaling and actin remodeling (72–74). Azurophilic granules discharge toxic components into phagosomes and at inflammation sites. The secretion of specific and gelatinase granules promotes migration of neutrophils and the antimicrobial activity. The main purpose of releasing secretory vesicles is to facilitate neutrophils recruitment. Therefore, the degranulation process promotes firm adhesion, migration, respiratory burst, and successive NET formation of activated neutrophils (75). However, the dysregulation of neutrophil degranulation could damage tissues as observed in various diseases, such as hypoxia-related airway injury (76), severe pneumonia and chronic obstructive pulmonary diseases (77), atherosclerosis (78, 79), acute inflammatory liver injury (80), and rheumatoid arthritis (81, 82).
In psoriasis, MPO is significantly increased in skin plaques and is positively correlated with the severity of psoriasis (83). Serum MPO is also increased in patients with psoriasis, which may be related to recruited leukocytes in psoriatic skin lesions (84). MPO, the major enzymatic content of neutrophil granules, accounts for ~5% of the dry weight of the cell and represents the most toxic enzyme expressed by neutrophils (85). MPO is involved in the respiratory burst and can bind to CD11b/CD18 integrins, thereby contributing to the upregulation and augmentation of neutrophil degranulation in psoriasis (86). Furthermore, the neutrophil granule-derived serine proteinases, such as NE, proteinase 3, and cathepsin G, can activate interleukin (IL)-36 cytokine and lead to the escalation of local psoriatic tissue inflammation (87, 88). Proteinase 3 cleaves and converts the resting TNFα located in membrane of epithelial cells (mTNFα) to an activated state called soluble TNFα (sTNFα), which participates in the psoriasis complex inflammatory reactions (Figure 2). Proteinase 3 also contributes to the formation of the LL-37, an antimicrobial peptide belonging to cathelicidin family of polypeptides (89, 90), which serves as autoantigen mediating immune response in psoriasis (33). Antimicrobial peptides, synthesized by various leukocytes and epithelial cells, act via DNA/RNA complexes binding Toll-like receptors (TLR) 7, 8, and 9 to facilitate skin inflammation (91, 92). In addition, NE proteolytically activates the epidermal growth factor receptor (EGFR) signaling pathway resulting in excessive keratinocyte proliferation in psoriasis (93). Thus, the inhibition of neutrophils degranulation process or some of the enzymes contributing to psoriasis (NE, MPO, proteinase 3) are feasible targets for alleviating psoriatic symptoms.
The process of forming neutrophil extracellular traps (NETs) was first reported in 2004 (94). NETs are composed of extruded sticky chromatin ornamented with many antimicrobial components including histones, MPO, NE, cathepsin G, high mobility group protein B1 (HMGB1) and antimicrobial peptides, such as LL-37 (Figure 1) (95). NETs can catch and destroy pathogens in order to prevent microbes from spreading (96). However, the dysregulated formation and clearance of NETs can result in many diseases. These include autoimmune diseases, such as systemic lupus erythematosus, anti-neutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis, rheumatoid arthritis, gout (97), and autoimmune hepatitis (98); cardiovascular diseases, such as atherosclerosis, thrombosis, and abdominal aortic aneurysm (99–101); respiratory disorders inclusive of asthma, chronic obstructive pulmonary disease, cystic fibrosis, tuberculosis, bacterial and viral pneumonia, and transfusion-related acute lung injury (102, 103); digestive diseases, such as inflammatory bowel diseases, primary sclerosing cholangitis, primary biliary cholangitis (98); and cancer-related organ damage, metastasis, and thrombosis (9). Recently, a role of NETs in awakening of dormant cancer cells was discovered (104).
The process of NET formation is termed NETosis, which is subdivided into lytic NETosis and non-lytic NETosis. In lytic NETosis (also called suicidal NETosis), activated neutrophils generate NETs (it takes 2–4 h) and undergo a programmed cell death, which differs from necrosis, necroptosis, and apoptosis (105). Lytic NET formation is triggered by various stimuli undergoing different pathways. For instance, phorbol myristate acetate (PMA) increases cytosol calcium, activates protein kinase C (PKC)/Raf/MEK/ERK pathway, and induces NOX2 to generate ROS. ROS then acts as the second messenger to disintegrate the nuclear membrane and stimulate MPO to translocate NE to the nucleus where it causes proteolysis of histones and decondensation of chromatin. Afterward, peptidyl arginine deiminase 4 (PAD4)-mediated hypercitrullination of histones allows decondensed chromatin, the main component of NETs, to be readily expelled from the cell nucleus. Finally, as the plasma membrane dissolves, the chromatin decorated with granular components is released as extracellular traps (106). Other pathways of lytic NETosis include stimulation by fungi (such as Aspergillus spp.) through Dectin 2 and complement receptor 3 (CR3) (107), LPS (lipopolysaccharide) under special conditions described later in detail (108) or by monosodium urate crystals via receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and RIPK3 pathway (109). All of these pathways involve NOX2, MPO, and NE activation (105). Nevertheless, there are other stimulators of NETosis acting independently of NOX2 such as ionomycin, or immune complexes (110). Ionomycin induces NETs via small conductance calcium-activated potassium channel protein 3 (SK3) and protein kinase C ζ (PKCζ), mitochondrial ROS (mitoROS), NE, and protein-arginine deiminase type 4 (PAD4) (111). Immune complexes related NETosis through FcγRIIIb are highly dependent on mitoROS (110, 112). The lytic-NETosis inducers such as PMA, ionomycin, or living bacteria were confirmed using a live imaging confocal microscopy, however, dead bacteria, LPS, glucose, or activated platelets alone failed to induce NETosis in the in vitro experiment (113). Such discrepancy might be due to variations in the experimental design of various studies.
On the other hand, non-lytic NETosis (also called vital NETosis) does not require neutrophils lysis or even the breach of the plasma membrane. Following the release of NETs, neutrophils are alive and keep their functions, such as chemotactic movement, phagocytotic ability, and respiratory burst power (98). This form of NETosis usually occurs early in infection by Gram-positive bacteria in human and mice. The process is very rapid (5–60 min to form NETs), requires both TLR 2 and complement-mediated opsonization, and is independent of NOX2 (114). Non-lytic NETosis can be induced by Staphylococcus aureus via a unique mechanism where the inner and outer nuclear membranes are separated, and the vesicles filled with nuclear DNA are extruded intact into the extracellular space where they rupture and release chromatins. Despite that this type of NETs keep a limited amount of proteolytic activity it is still able to kill S. aureus (115). Non-lytic NETosis can also be stimulated by Candida albicans via interaction with CR3 and fibronectin (116). Moreover, a special type of non-lytic NETosis, which releases mitochondrial DNA and is dependent on ROS, is stimulated by the granulocyte-macrophage colony-stimulating factor (GM-CSF) and LPS (10). Interestingly, Leishmania parasites induce both lytic and non-lytic NETosis (117). In that case, the chromatin decondensed by PAD4 is mixed with granular proteins and subsequently excreted with a minor nuclear envelope disruption and without cell membrane disorganization (10, 98). Delgado-Rizo et al. previously summarized the microbial inducers of NETs (10) but we would like to clarify the effect of LPS. Lipopolysaccharide (LPS) is an important component of the outer membrane of gram-negative bacteria known to trigger immune response (118). For a long time, it was unclear whether the direct interaction between LPS and neutrophils causes NETs release, because several reports showed LPS-induced lytic NET formation (94, 119) while other not (113, 120). Recently, it has been shown that only species- and serotype-specific LPS is able to induce NETs by direct interaction with neutrophils. It was demonstrated that LPS has to be derived from specific bacterial strain of Escherichia coli (O128:B12) and Pseudomonas aeruginosa (serotype 10) and must be present at sufficient concentration (8 pg per neutrophil). The neutrophils then undergo a lytic-NETosis independent of TLR4. However, non-lytic NETosis is triggered when sufficient amount of LPS regardless of bacterial origin interacts with TLR4 of platelets (108). The process is followed by binding of platelets to the P-selectin glycoprotein ligand-1 (PSGL-1) of neutrophils, and the release of HMGB1 by platelets (9, 95). Moreover, there is a growing evidence of crucial role of the other endogenous and immune factors in the process of NET formation, such as presence of platelets (95, 120), glucose (10), or other effectors (121). To orchestrate inflammatory response, NETs in combination with LPS were shown to induce the production of IL-1β by J774 macrophages via the caspase-1 and caspase-8 pathways (122).
In patients with psoriasis, neutrophils are pre-activated and form NETs in psoriatic skin lesions (55, 123). NETs are increased in blood samples and correlate with the severity of psoriasis (124, 125). They create an extremely immunogenic environment and participate in the initial and maintenance phases of psoriasis (126, 127). NETs stimulate epidermis to release inflammatory cytokines via TLR4 and IL-36 receptor crosstalk (123). Various exogenous and endogenous stimuli and ROS generated by neutrophils initiate immune reaction leading to psoriasis which involves T cell imbalance, keratinocyte proliferation, angiogenesis, and auto-antigen formation (Figure 2). The chromatin of NETs in psoriasis plaques is accompanied with antimicrobial peptide LL-37 released by keratinocytes to stimulate the synthesis of inflammatory mediators including IFN-α and IFN-β in plasmacytoid dendritic cells (pDCs) (16). Myeloid DCs (mDCs) are then activated to release many pro-inflammatory mediators, such as IL-6, IL-12, IL-23, and TNFα (91, 92), which play an important role in the initiation of the Th1, Th17, and Th22 cells immune response (16). Proteinase 3 released from neutrophils cleaves pro-IL-36 to activated IL-36 cytokine which is together with TNF and IFN-γ amplifying the response of mDCs. Th17 activation then leads to the production of IL-17 activating neutrophils and keratinocytes via IL-17 receptors which generates profound IL-17 response (16). Secretory leukocyte protease inhibitor (SLPI), a component of NETs with an inhibitory function on NET formation, may bind to DNA and NE in psoriatic skin lesions and activate the pDCs to produce type 1 interferons (IFN-α, IFN-β, etc.) which regulates autoimmunity in psoriasis (128–130). In addition, NETs allow the mDCs to readily sense the neutrophilic antigens and allow the T cells to be primed directly (45, 131, 132). Thus, NETs play an important role in the pathophysiology of psoriasis due to their link between the innate and adaptive immune systems. Psoriasis is accompanied with increased serum levels of TNF-α, interferon (IFN)-γ, IL-1, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12, IL-17, IL-18 (133), IL-22 (134), chemerin, resistin (135), lipocalin-2 (LCN2) (123), soluble E-selectin (sE-selectin) (136), complement 3 (137), and decreased levels of transforming growth factor-beta (TGF-β) and adiponectin (133). These cytokines may therefore serve as potential biomarkers for psoriasis and treatment response in patients. According to a cross-sectional study, psoriasis patients had increased proinflammatory macrophage type 1 (IL-1, IL-6, TNF-α), Th1 (IL-2, IL-12, IFN-γ), Th17 (IL-6, IL-17) but also anti-inflammatory Th2/T regulatory (Treg) (IL-4, IL-10) profiles which may be correlated to the severity of psoriasis (133). Among the important mediators in psoriasis, LCN2 acts as an antimicrobial protein as well as adipokine associated with obesity, insulin resistance, and atherosclerotic disease, and is also responsible for the activation of the immune system in response to inflammatory and toxic stimuli. Importantly, serum LCN2 levels are elevated in psoriatic patients (138) and correlate with the severity of itching and thus might be used as a clinical marker for itching in psoriasis (139). Not only granulocytes but also keratinocytes of epidermis secrete LCN2, which drives the chemotaxis of neutrophils and sustains NET formation, and thereby in turn maintains the psoriatic inflammation (123). The increased LCN2 blood levels were observed in patients with palmoplantar pustular psoriasis (140) as well as other chronic inflammatory skin diseases such as acne inversa (141) or atopic dermatitis (142). Wolk et al. reported a positive correlation between the LCN2 production and IL-1β levels in the epidermis, which was further enhanced by IL-17 and TNF-α, but not by IL-22. The contribution of LCN2 on skin neutrophil infiltration is apparent (141). In the clinical setting, tissue LCN2 was found to be also significantly higher in psoriasis, regardless of dyslipidemia, or metabolic disturbance in patients. But the LCN2 levels together with psoriasis area and severity index (PASI) score significantly dropped after NBUVB treatment (143). The in vivo effects of LCN2 on topical imiquimod (IMQ)-induced psoriasis-like skin in BALB/c mice were evaluated by Hau group (138). In addition to markedly exacerbated erythema and scaling in IMQ-treated murine skin, LCN2 increased the mRNA expression of interleukin (IL)-17A, IL-17F, IL-22, IL-23, CC-chemokine ligand (CCL)20, TNF-α, chemokine (C-X-C motif) ligand (CXCL)1, CXCL2, LCN2, and S100A7 while it did not affect the mRNA levels of IFN-γ, or CXCL10 in the skin. Similar effects were observed in vitro on human keratinocytes (138). The data suggest a link between NETs-related cytokines and Th-17 activation in psoriasis.
Psoriasis has been considered as a T-helper (Th)1/Th17-mediated, chronic inflammatory dermatosis with relation to metabolic syndromes (144). Apart from keratinocytes and T lymphocytes (145), neutrophils are one of the major cellular sources of IL-17 via NET formation in psoriasis (146), and also mast cells were reported to generate extracellular traps (147). NETs activation has been linked with Th17 responses in psoriasis and has drawn particular attention recently (148). In an experimental model, NETs induced the generation of CD3+CD4+IL-17+ (Th17) cells from peripheral blood mononuclear cells, which requires monocyte and cell-to-cell contact. Th17 induction was enhanced by a psoriasis risk-associated variant in the TRAF3IP2 gene encoding the D10N variant of Act1 which serves as a key mediator of IL-17 signal transduction. That provides an evidence of genetic basis for the enhanced IL-17 expression in psoriasis. IL-17-expressing T cells and neutrophils were suggested to have a cross-talk because IL-17-expressing T cells produce cytokines which promote the development, recruitment, and lifespan of neutrophils (149).
Although many immune diseases including psoriasis or atopic dermatitis have been traditionally classified as Th1/Th2 biphasic disorders, there is a growing evidence supporting a rather systemic activation of other multiple Th-cell subsets, such as Th17 cells producing IL-17 and IL-22 (Figure 2). Interestingly, in comparison with psoriasis, atopic dermatitis showed reduced genomic expression of IL-23, IL-17, IFN-γ, and other innate defense genes (hBD2, elafin, LL-37) (150). Elevated IL-17 is detected in psoriatic skin lesions and in the blood (151). IL-17 mainly stimulates keratinocytes to produce neutrophil-tropic chemokines CXCL-1, CXCL-2, CXCL-8 (IL-8), and antimicrobial peptides, such as LL-37. IL-17 serves as an autocrine-amplifying mediator that is simultaneously involved in the recruitment, activation, and survival of neutrophils (6). There are several subtypes of IL-17 family cytokines binding to IL-17 receptors, namely IL-17A, IL-17B, IL-17C, IL-17D, IL-17E (also known as IL-25) and IL-17F (145). IL-17A from neutrophils stimulates keratinocytes to express CCL20, attracting Th17 cells with CCR6 expression to release IL-17A, and finally resulting in positive feedback and the development of the psoriatic lesions (8). IL-17C is a unique cytokine that is produced by keratinocytes and that is involved in such synergistic loops that may be responsible for amplifying the inflammation in both psoriasis and atopic dermatitis. This may ultimately lead to induction of S100As and other molecules that accompany epidermal hyperplasia. Thus, antagonism of IL-17C may be beneficial for psoriasis and atopic dermatitis treatment (152). IL-17E then recruits neutrophils by activating macrophages and contributes to the infiltration of psoriatic neutrophils (153). Besides other innate immune cells, neutrophils significantly contribute to IL-17-related immune regulations in psoriasis by employing several mechanisms including the formation of NETs (45). Moreover, IL-17 released by NETs leads to endothelial dysfunction in atherosclerosis and keratinocyte proliferation in psoriasis, which may explain why patients with psoriasis also suffer from increased risk of atherosclerosis (11). Anti-IL-17 drugs, such as secukinumab, clear the neutrophils in the epidermis and can be used to effectively treat psoriasis (48). The targeted biologic therapies are of great importance with regards to an increasing number of comorbidities associated with psoriasis together with its systemic inflammation nature indicating that these diseases are sharing some common pathological mechanisms (7). In summary, NETs were demonstrated as potential upstream drug targets for the treatment of psoriasis.
Neutrophils in psoriasis are of interest, particularly, because of their crucial roles in the innate and adaptive immune system. The respiratory burst with ROS generation, degranulation, and formation of NETs are the main offensive functions of neutrophils and contribute to the immunopathogenesis of psoriasis. Recently, great attention was brought to the role of NETs in psoriasis because activated neutrophils producing NETs are abundant in psoriatic skin plaques and pustules, as well as in the serum of patients with psoriasis. Overexpression of NETs leads to the activation of other cells releasing IL-17, which stimulates the synthesis of inflammatory mediators and in turn leads to auto-amplification of neutrophils. Therefore, further development of inhibitors and biologic drugs targeting overexpressed offensive features of neutrophils, i.e., respiratory burst, degranulation, and NET formation, is of great importance. We believe that the consideration of the role of neutrophil defense mechanisms in the pathogenesis of psoriasis offered in this review highlights the need to further investigate neutrophils for possible improvements of available treatments in the future.
C-CC and W-JC wrote and revised the manuscript. MK consulted and revised the manuscript. C-YL drew the figures. T-LH initiated the concept and supervised the writing. All authors read and approved the final manuscript.
This review was supported by the grants from the Ministry of Science Technology (MOST 108-2320-B-255-003-MY3, MOST 106-2320-B-255-003-MY3, and MOST 104-2320-B-255-004-MY3), Ministry of Education (EMRPD1I0441), and Chang Gung Memorial Hospital (CMRPG5F0161 and BMRP450). The funding source had no role in this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to Assoc. Prof. Mohamed El-Shazly for valuable comments and revision of the manuscript. Special thanks to Editage English Editorial Office, Taiwan for English editing. The authors wish to thank Miss Ingrid Kuo and the Center for Big Data Analytics and Statistics (Grant CLRPG 3D0045) at Chang Gung Memorial Hospital for creating the illustrations used herein.
CCL, CC-chemokine ligand; CXCL, chemokine (C-X-C motif) ligand; IL, interleukin; IFN, interfenon; LPS, lipopolysaccharide; mDCs, myeloid dendritic cells; MPO, myeloperoxidase; NET, neutrophil extracellular trap; NOX, NADPH oxidase; NE, neutrophil elastase; NLR, neutrophil-to-lymphocyte ratio, pDCs, plasmacytoid dendritic cells; PLR, platelet-to-lymphocyte ratio; Th, T helper cell; TLR, Toll-like receptor; TNF, tumor necrosis factor.
1. Tecchio C, Cassatella MA. Neutrophil-derived chemokines on the road to immunity. Semin Immunol. (2016) 28:119–28. doi: 10.1016/j.smim.2016.04.003
2. Nauseef WM, Borregaard N. Neutrophils at work. Nat Immunol. (2014) 15:602–11. doi: 10.1038/ni.2921
3. Leliefeld PH, Koenderman L, Pillay J. How Neutrophils Shape Adaptive Immune Responses. Front Immunol. (2015) 6:471. doi: 10.3389/fimmu.2015.00471
4. Rosales C, Lowell CA, Schnoor M, Uribe-Querol E. Neutrophils: Their Role in Innate and Adaptive Immunity 2017. J Immunol Res. (2017) 2017:9748345. doi: 10.1155/2017/9748345
5. Georgescu SR, Tampa M, Caruntu C, Sarbu MI, Mitran CI, Mitran MI, et al. Advances in understanding the immunological pathways in psoriasis. Int J Mol Sci. (2019) 20:739. doi: 10.3390/ijms20030739
6. Chiricozzi A, Romanelli P, Volpe E, Borsellino G, Romanelli M. Scanning the Immunopathogenesis of Psoriasis. Int J Mol Sci. (2018) 19:E179. doi: 10.3390/ijms19010179
7. Korman NJ. Management of psoriasis as a systemic disease: What is the evidence? Br J Dermatol. (2019). doi: 10.1111/bjd.18245. [Epub ahead of print].
8. Katayama H. Development of psoriasis by continuous neutrophil infiltration into the epidermis. Exp Dermatol. (2018) 27:1084–91. doi: 10.1111/exd.13746
9. Cedervall J, Hamidi A, Olsson AK. Platelets, NETs and cancer. Thromb Res. (2018) 164(Suppl 1):S148–52. doi: 10.1016/j.thromres.2018.01.049
10. Delgado-Rizo V, Martínez-Guzmán MA, Iñiguez-Gutierrez L, García-Orozco A, Alvarado-Navarro A, Fafutis-Morris M. Neutrophil extracellular traps and its implications in inflammation: an overview. Front Immunol. (2017) 8:81. doi: 10.3389/fimmu.2017.00081
11. Sanda GE, Belur AD, Teague HL, Mehta NN. Emerging Associations Between Neutrophils, Atherosclerosis, and Psoriasis. Curr Atheroscler Rep. (2017) 19:53. doi: 10.1007/s11883-017-0692-8
12. Bagchi D, Kundu-Raychaudhuri SK, Raychaudhuri SP. Psoriasis and Psoriatic Arthritis: Pathophysiology, Therapeutic Intervention, and Complementary Medicine. Portland, OR: CRC Press/Taylor & Francis Group (2018). p. 413.
14. Warren R MA. Handbook of Psoriasis and Psoriatic Arthritis. Springer International Publishing (2016). p. 116.
15. Adebajo A, Gladman DD, Mease PJ. Psoriatic Arthritis and Psoriasis: Pathology and Clinical Aspects. Springer International Publishing (2016). p. 316.
16. Greb JE, Goldminz AM, Elder JT, Lebwohl MG, Gladman DD, Wu JJ, et al. Psoriasis. Nat Rev Dis Primers. (2016) 2:16082. doi: 10.1038/nrdp.2016.83
17. Sticherling M. Psoriasis and autoimmunity. Autoimmun Rev. (2016) 15:1167–70. doi: 10.1016/j.autrev.2016.09.004
18. Kaushik SB, Lebwohl MG. CME Part I Psoriasis: which therapy for which patient psoriasis comorbidities and preferred systemic agents. J Am Acad Dermatol. (2019) 80:27–40. doi: 10.1016/j.jaad.2018.06.057
19. Geller S, Xu H, Lebwohl M, Nardone B, Lacouture ME, Kheterpal M. Malignancy risk and recurrence with psoriasis and its treatments: a concise update. Am J Clin Dermatol. (2018) 19:363–75. doi: 10.1007/s40257-017-0337-2
20. Gelfand JM. Psoriasis, type 2 diabetes mellitus, and obesity: weighing the evidence. JAMA Dermatol. (2016) 152:753–4. doi: 10.1001/jamadermatol.2016.0670
22. Raychaudhuri SK, Maverakis E, Raychaudhuri SP. Diagnosis and classification of psoriasis. Autoimmun Rev. (2014) 13:490–5. doi: 10.1016/j.autrev.2014.01.008
23. Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. (2009) 361:496–509. doi: 10.1056/NEJMra0804595
24. Vanderpuye-Orgle J, Zhao Y, Lu J, Shrestha A, Sexton A, Seabury S, et al. Evaluating the economic burden of psoriasis in the United States. J Am Acad Dermatol. (2015) 72:961–7.e5. doi: 10.1016/j.jaad.2015.02.1099
25. Brezinski EA, Dhillon JS, Armstrong AW. Economic burden of psoriasis in the United States: a systematic review. JAMA Dermatol. (2015) 151:651–8. doi: 10.1001/jamadermatol.2014.3593
26. Hawro T, Zalewska A, Hawro M, Kaszuba A, Krolikowska M, Maurer M. Impact of psoriasis severity on family income and quality of life. J Eur Acad Dermatol Venereol. (2015) 29:438–43. doi: 10.1111/jdv.12572
27. Singh S, Taylor C, Kornmehl H, Armstrong AW. Psoriasis and suicidality: a systematic review and meta-analysis. J Am Acad Dermatol. (2017) 77:425–40.e2. doi: 10.1016/j.jaad.2017.05.019
28. Dowlatshahi EA, Wakkee M, Arends LR, Nijsten T. The prevalence and odds of depressive symptoms and clinical depression in psoriasis patients: a systematic review and meta-analysis. J Invest Dermatol. (2014) 134:1542–51. doi: 10.1038/jid.2013.508
29. Prinz JC. Autoimmune aspects of psoriasis: heritability and autoantigens. Autoimmun Rev. (2017) 16:970–9. doi: 10.1016/j.autrev.2017.07.011
30. Hwang ST, Nijsten T, Elder JT. Recent highlights in psoriasis research. J Invest Dermatol. (2017) 137:550–6. doi: 10.1016/j.jid.2016.11.007
31. Harvima IT, Nilsson G, Suttle MM, Naukkarinen A. Is there a role for mast cells in psoriasis? Arch Dermatol Res. (2008) 300:461–78. doi: 10.1007/s00403-008-0874-x
32. Prinz JC. Melanocytes: target cells of an HLA-C*06:02-restricted autoimmune response in psoriasis. J Invest Dermatol. (2017) 137:2053–8. doi: 10.1016/j.jid.2017.05.023
33. Mrowietz U. Neutrophils' sexiness is independent of trendy fashion. Exp Dermatol. (2017) 26:312–3. doi: 10.1111/exd.13102
34. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. (2011) 11:519–31. doi: 10.1038/nri3024
35. Jaillon S, Galdiero MR, Del Prete D, Cassatella MA, Garlanda C, Mantovani A. Neutrophils in innate and adaptive immunity. Semin Immunopathol. (2013) 35:377–94. doi: 10.1007/s00281-013-0374-8
36. Dubertret L, Mrowietz U, Ranki A, van de Kerkhof PC, Chimenti S, Lotti T, et al. European patient perspectives on the impact of psoriasis: the EUROPSO patient membership survey. Br J Dermatol. (2006) 155:729–36. doi: 10.1111/j.1365-2133.2006.07405.x
37. Belinchón I, Rivera R, Blanch C, Comellas M, Lizán L. Adherence, satisfaction and preferences for treatment in patients with psoriasis in the European Union: a systematic review of the literature. Patient Prefer Adherence. (2016) 10:2357–67. doi: 10.2147/PPA.S117006
38. Meng S, Lin Z, Wang Y, Wang Z, Li P, Zheng Y. Psoriasis therapy by Chinese medicine and modern agents. Chin Med. (2018) 13:16. doi: 10.1186/s13020-018-0174-0
39. Damevska K, Franca K, Lotti T, Nikolovska S, Pollozhani N. Complementary and integrative therapies for psoriasis: looking forward. Dermatol Ther. (2018) 31:e12627. doi: 10.1111/dth.12627
40. Talbott W, Duffy N. Complementary and alternative medicine for psoriasis: what the dermatologist needs to know. Am J Clin Dermatol. (2015) 16:147–65. doi: 10.1007/s40257-015-0128-6
41. Costanzo A, Malara G, Pelucchi C, Fatiga F, Barbera G, Franchi A, et al. Effectiveness end points in real-world studies on biological therapies in psoriasis: systematic review with focus on drug survival. Dermatology. (2018) 234:1–12. doi: 10.1159/000488586
42. Yamanaka K, Umezawa Y, Yamagiwa A, Saeki H, Kondo M, Gabazza EC, et al. Biologic therapy improves psoriasis by decreasing the activity of monocytes and neutrophils. J Dermatol. (2014) 41:679–85. doi: 10.1111/1346-8138.12560
43. de Groot M, Picavet DI, van Kuijk AW, Tak PP, Bos JD, de Rie MA, et al. A prospective, randomized, placebo-controlled study to identify biomarkers associated with active treatment in psoriatic arthritis: effects of adalimumab treatment on lesional and nonlesional skin. Dermatology. (2012) 225:298–303. doi: 10.1159/000343290
44. Tan JK, Aphale A, Malaviya R, Sun Y, Gottlieb AB. Mechanisms of action of etanercept in psoriasis. J Investig Dermatol Symp Proc. (2007) 12:38–45. doi: 10.1038/sj.jidsymp.5650037
45. Schon MP, Erpenbeck L. The Interleukin-23/Interleukin-17 Axis Links Adaptive and Innate Immunity in Psoriasis. Front Immunol. (2018) 9:1323. doi: 10.3389/fimmu.2018.01323
46. Chen F, Cao A, Yao S, Evans-Marin HL, Liu H, Wu W, et al. mTOR Mediates IL-23 Induction of Neutrophil IL-17 and IL-22 Production. J Immunol. (2016) 196:4390–9. doi: 10.1097/01.ccm.0000508877.60312.7c
47. Liu L, Lu J, Allan BW, Tang Y, Tetreault J, Chow CK, et al. Generation and characterization of ixekizumab, a humanized monoclonal antibody that neutralizes interleukin-17A. J Inflamm Res. (2016) 9:39–50. doi: 10.2147/JIR.S100940
48. Reich K, Papp KA, Matheson RT, Tu JH, Bissonnette R, Bourcier M, et al. Evidence that a neutrophil-keratinocyte crosstalk is an early target of IL-17A inhibition in psoriasis. Exp Dermatol. (2015) 24:529–35. doi: 10.1111/exd.12710
49. Polat M, Bugdayci G, Kaya H, Oguzman H. Evaluation of neutrophil-to-lymphocyte ratio and platelet-to-lymphocyte ratio in Turkish patients with chronic plaque psoriasis. Acta Dermatovenerol Alp Pannonica Adriat. (2017) 26:97–100. doi: 10.15570/actaapa.2017.28
50. Paliogiannis P, Satta R, Deligia G, Farina G, Bassu S, Mangoni AA, et al. Associations between the neutrophil-to-lymphocyte and the platelet-to-lymphocyte ratios and the presence and severity of psoriasis: a systematic review and meta-analysis. Clin Exp Med. (2019) 19:37–45. doi: 10.1007/s10238-018-0538-x
51. Balevi A, Olmuscelik O, Ustuner P, Ozdemir M. Is there any correlation between red cell distribution width, mean platelet volume neutrophil count, lymphocyte count, and psoriasis area severity index in patients under treatment for psoriasis? Acta Dermatovenerol Croat. (2018) 26:199–205.
52. Erek Toprak A, Ozlu E, Uzuncakmak TK, Yalcinkaya E, Sogut S, Karadag AS. Neutrophil/lymphocyte ratio, serum endocan, and nesfatin-1 levels in patients with psoriasis vulgaris undergoing phototherapy treatment. Med Sci Monit. (2016) 22:1232–7. doi: 10.12659/MSM.898240
53. Yurtdas M, Yaylali YT, Kaya Y, Ozdemir M, Ozkan I, Aladag N. Neutrophil-to-lymphocyte ratio may predict subclinical atherosclerosis in patients with psoriasis. Echocardiography. (2014) 31:1095–104. doi: 10.1111/echo.12511
54. Ikeda S, Takahashi H, Suga Y, Eto H, Etoh T, Okuma K, et al. Therapeutic depletion of myeloid lineage leukocytes in patients with generalized pustular psoriasis indicates a major role for neutrophils in the immunopathogenesis of psoriasis. J Am Acad Dermatol. (2013) 68:609–17. doi: 10.1016/j.jaad.2012.09.037
55. Schon MP, Broekaert SM, Erpenbeck L. Sexy again: the renaissance of neutrophils in psoriasis. Exp Dermatol. (2017) 26:305–11. doi: 10.1111/exd.13067
56. Belambri SA, Rolas L, Raad H, Hurtado-Nedelec M, Dang PM, El-Benna J. NADPH oxidase activation in neutrophils: role of the phosphorylation of its subunits. Eur J Clin Invest. (2018) 2:e12951. doi: 10.1111/eci.12951
57. Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol. (2012) 30:459–89. doi: 10.1146/annurev-immunol-020711-074942
58. Glennon-Alty L, Hackett AP, Chapman EA, Wright HL. Neutrophils and redox stress in the pathogenesis of autoimmune disease. Free Radic Biol Med. (2018) 125:25–35. doi: 10.1016/j.freeradbiomed.2018.03.049
59. Winterbourn CC, Kettle AJ, Hampton MB. Reactive oxygen species and neutrophil function. Annu Rev Biochem. (2016) 85:765–92. doi: 10.1146/annurev-biochem-060815-014442
60. Hoffmann MH, Griffiths HR. The dual role of ROS in autoimmune and inflammatory diseases: Evidence from preclinical models. Free Radic Biol Med. (2018) 125:62–71. doi: 10.1016/j.freeradbiomed.2018.03.016
61. Bloomfield FJ, Young MM. Enhanced release of inflammatory mediators from lithium-stimulated neutrophils in psoriasis. Br J Dermatol. (1983) 109:9–13. doi: 10.1111/j.1365-2133.1983.tb03985.x
62. Bloomfield FJ, Young MM. Enhanced chemiluminescence production by phagocytosing neutrophils in psoriasis. Inflammation. (1988) 12:153–9. doi: 10.1007/BF00916398
63. Martin G, Guerard S, Fortin MM, Rusu D, Soucy J, Poubelle PE, et al. Pathological crosstalk in vitro between T lymphocytes and lesional keratinocytes in psoriasis: necessity of direct cell-to-cell contact. Lab Invest. (2012) 92:1058–70. doi: 10.1038/labinvest.2012.69
64. Vogt KL, Summers C, Chilvers ER, Condliffe AM. Priming and de-priming of neutrophil responses in vitro and in vivo. Eur J Clin Invest. (2018) 47:e12967. doi: 10.1111/eci.12967
66. Guerard S, Allaeys I, Martin G, Pouliot R, Poubelle PE. Psoriatic keratinocytes prime neutrophils for an overproduction of superoxide anions. Arch Dermatol Res. (2013) 305:879–89. doi: 10.1007/s00403-013-1404-z
67. Lin X, Huang T. Oxidative stress in psoriasis and potential therapeutic use of antioxidants. Free Radic Res. (2016) 50:585–95. doi: 10.3109/10715762.2016.1162301
68. Lai R, Xian D, Xiong X, Yang L, Song J, Zhong J. Proanthocyanidins: novel treatment for psoriasis that reduces oxidative stress and modulates Th17 and Treg cells. Redox Rep. (2018) 23:130–5. doi: 10.1080/13510002.2018.1462027
69. Lacy P. Mechanisms of degranulation in neutrophils. Allergy Asthma Clin Immunol. (2006) 2:98–108. doi: 10.1186/1710-1492-2-3-98
70. Sheshachalam A, Srivastava N, Mitchell T, Lacy P, Eitzen G. Granule protein processing and regulated secretion in neutrophils. Front Immunol. (2014) 5:448. doi: 10.3389/fimmu.2014.00448
71. Kahlenberg JM, Kaplan MJ. Little peptide, big effects: the role of LL-37 in inflammation and autoimmune disease. J Immunol. (2013) 191:4895–901. doi: 10.4049/jimmunol.1302005
72. Cowland JB, Borregaard N. Granulopoiesis and granules of human neutrophils. Immunol Rev. (2016) 273:11–28. doi: 10.1111/imr.12440
73. Mitchell T, Lo A, Logan MR, Lacy P, Eitzen G. Primary granule exocytosis in human neutrophils is regulated by Rac-dependent actin remodeling. Am J Physiol Cell Physiol. (2008) 295:C1354–65. doi: 10.1152/ajpcell.00239.2008
74. Jog NR, Rane MJ, Lominadze G, Luerman GC, Ward RA, McLeish KR. The actin cytoskeleton regulates exocytosis of all neutrophil granule subsets. Am J Physiol Cell Physiol. (2007) 292:C1690–700. doi: 10.1152/ajpcell.00384.2006
75. Yin C, Heit B. Armed for destruction: formation, function and trafficking of neutrophil granules. Cell Tissue Res. (2018) 371:455–71. doi: 10.1007/s00441-017-2731-8
76. Hoenderdos K, Lodge KM, Hirst RA, Chen C, Palazzo SG, Emerenciana A, et al. Hypoxia upregulates neutrophil degranulation and potential for tissue injury. Thorax. (2016) 71:1030–8. doi: 10.1136/thoraxjnl-2015-207604
77. Polverino E, Rosales-Mayor E, Dale GE, Dembowsky K, Torres A. The role of neutrophil elastase inhibitors in lung diseases. Chest. (2017) 152:249–62. doi: 10.1016/j.chest.2017.03.056
78. Chistiakov DA, Bobryshev YV, Orekhov AN. Neutrophil's weapons in atherosclerosis. Exp Mol Pathol. (2015) 99:663–71. doi: 10.1016/j.yexmp.2015.11.011
79. Sivalingam Z, Larsen SB, Grove EL, Hvas AM, Kristensen SD, Magnusson NE. Neutrophil gelatinase-associated lipocalin as a risk marker in cardiovascular disease. Clin Chem Lab Med. (2017) 56:5–18. doi: 10.1515/cclm-2017-0120
80. Ramaiah SK, Jaeschke H. Role of neutrophils in the pathogenesis of acute inflammatory liver injury. Toxicol Pathol. (2007) 35:757–66. doi: 10.1080/01926230701584163
81. Wright HL, Moots RJ, Bucknall RC, Edwards SW. Neutrophil function in inflammation and inflammatory diseases. Rheumatology. (2010) 49:1618–31. doi: 10.1093/rheumatology/keq045
82. Wright HL, Moots RJ, Edwards SW. The multifactorial role of neutrophils in rheumatoid arthritis. Nat Rev Rheumatol. (2014) 10:593–601. doi: 10.1038/nrrheum.2014.80
83. Dilek N, Dilek AR, Taskin Y, Erkinuresin T, Yalcin O, Saral Y. Contribution of myeloperoxidase and inducible nitric oxide synthase to pathogenesis of psoriasis. Postepy Dermatol Alergol. (2016) 33:435–9. doi: 10.5114/ada.2016.63882
84. Cao LY, Soler DC, Debanne SM, Grozdev I, Rodriguez ME, Feig RL, et al. Psoriasis and cardiovascular risk factors: increased serum myeloperoxidase and corresponding immunocellular overexpression by Cd11b. (+) CD68. (+) macrophages in skin lesions. Am J Transl Res. (2013) 6:16–27.
85. Strzepa A, Pritchard KA, Dittel BN. Myeloperoxidase: a new player in autoimmunity. Cell Immunol. (2017) 317:1–8. doi: 10.1016/j.cellimm.2017.05.002
86. Lau D, Mollnau H, Eiserich JP, Freeman BA, Daiber A, Gehling UM, et al. Myeloperoxidase mediates neutrophil activation by association with CD11b/CD18 integrins. Proc Natl Acad Sci USA. (2005) 102:431–6. doi: 10.1073/pnas.0405193102
87. Guo J, Tu J, Hu Y, Song G, Yin Z. Cathepsin G cleaves and activates IL-36γ and promotes the inflammation of psoriasis. Drug Des Devel Ther. (2019) 13:581–8. doi: 10.2147/DDDT.S194765
88. Henry CM, Sullivan GP, Clancy DM, Afonina IS, Kulms D, Martin SJ. Neutrophil-derived proteases escalate inflammation through activation of IL-36 family cytokines. Cell Rep. (2016) 14:708–22. doi: 10.1016/j.celrep.2015.12.072
89. Xhindoli D, Pacor S, Benincasa M, Scocchi M, Gennaro R, Tossi A. The human cathelicidin LL-37–A pore-forming antibacterial peptide and host-cell modulator. Biochim Biophys Acta. (2016) 1858:546–66. doi: 10.1016/j.bbamem.2015.11.003
90. Durr UH, Sudheendra US, Ramamoorthy A. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim Biophys Acta. (2006) 1758:1408–25. doi: 10.1016/j.bbamem.2006.03.030
91. Ganguly D, Chamilos G, Lande R, Gregorio J, Meller S, Facchinetti V, et al. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med. (2009) 206:1983–94. doi: 10.1084/jem.20090480
92. Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. (2007) 449:564–9. doi: 10.1038/nature06116
93. Meyer-Hoffert U, Wingertszahn J, Wiedow O. Human leukocyte elastase induces keratinocyte proliferation by epidermal growth factor receptor activation. J Invest Dermatol. (2004) 123:338–45. doi: 10.1111/j.0022-202X.2004.23202.x
94. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
95. Kim SJ, Jenne CN. Role of platelets in neutrophil extracellular trap. (NET) production and tissue injury. Semin Immunol. (2016) 28:546–54. doi: 10.1016/j.smim.2016.10.013
96. Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, et al. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol. (2014) 15:1017–25. doi: 10.1038/ni.2987
97. Apel F, Zychlinsky A, Kenny EF. The role of neutrophil extracellular traps in rheumatic diseases. Nat Rev Rheumatol. (2018) 14:467–75. doi: 10.1038/s41584-018-0039-z
98. Honda M, Kubes P. Neutrophils and neutrophil extracellular traps in the liver and gastrointestinal system. Nat Rev Gastroenterol Hepatol. (2018) 15:206–21. doi: 10.1038/nrgastro.2017.183
99. Fernandez-Ruiz I. Inflammation: NETs are involved in AAA. Nat Rev Cardiol. (2018) 15:257. doi: 10.1038/nrcardio.2018.28
100. Doring Y, Soehnlein O, Weber C. Neutrophil extracellular traps in atherosclerosis and atherothrombosis. Circ Res. (2017) 120:736–43. doi: 10.1161/CIRCRESAHA.116.309692
101. Dabrowska D, Jablonska E, Garley M, Sawicka-Powierza J, Nowak K. The Phenomenon of Neutrophil Extracellular Traps in Vascular Diseases. Arch Immunol Ther Exp. (2018) 66:273–81. doi: 10.1007/s00005-018-0505-y
102. Porto BN, Stein RT. Neutrophil extracellular traps in pulmonary diseases: too much of a good thing? Front Immunol. (2016) 7:311. doi: 10.3389/fimmu.2016.00311
103. Liu T, Wang FP, Wang G, Mao H. Role of neutrophil extracellular traps in asthma and chronic obstructive pulmonary disease. Chin Med J. (2017) 130:730–6. doi: 10.4103/0366-6999.201608
104. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. (2018) 361:eaao4227. doi: 10.1126/science.aao4227
105. Kenny EF, Herzig A, Kruger R, Muth A, Mondal S, Thompson PR, et al. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife. (2017) 6:e24437. doi: 10.7554/eLife.24437
106. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. (2010) 191:677–91. doi: 10.1083/jcb.201006052
107. Loures FV, Rohm M, Lee CK, Santos E, Wang JP, Specht CA, et al. Recognition of Aspergillus fumigatus hyphae by human plasmacytoid dendritic cells is mediated by dectin-2 and results in formation of extracellular traps. PLoS Pathog. (2015) 11:e1004643. doi: 10.1371/journal.ppat.1004643
108. Pieterse E, Rother N, Yanginlar C, Hilbrands LB, van der Vlag J. Neutrophils discriminate between lipopolysaccharides of different bacterial sources and selectively release neutrophil extracellular traps. Front Immunol. (2016) 7:484. doi: 10.3389/fimmu.2016.00484
109. Desai J, Kumar SV, Mulay SR, Konrad L, Romoli S, Schauer C, et al. PMA and crystal-induced neutrophil extracellular trap formation involves RIPK1-RIPK3-MLKL signaling. Eur J Immunol. (2016) 46:223–9. doi: 10.1002/eji.201545605
110. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. (2018) 18:134–47. doi: 10.1038/nri.2017.105
111. Douda DN, Khan MA, Grasemann H, Palaniyar N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc Natl Acad Sci USA. (2015) 112:2817–22. doi: 10.1073/pnas.1414055112
112. Behnen M, Leschczyk C, Moller S, Batel T, Klinger M, Solbach W, et al. Immobilized immune complexes induce neutrophil extracellular trap release by human neutrophil granulocytes via FcgammaRIIIB and Mac-1. J Immunol. (2014) 193:1954–65. doi: 10.4049/jimmunol.1400478
113. Hoppenbrouwers T, Autar ASA, Sultan AR, Abraham TE, van Cappellen WA, Houtsmuller AB, et al. In vitro induction of NETosis: Comprehensive live imaging comparison and systematic review. PLoS ONE. (2017) 12:e0176472. doi: 10.1371/journal.pone.0176472
114. Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med. (2012) 18:1386–93. doi: 10.1038/nm.2847
115. Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol. (2010) 185:7413–25. doi: 10.4049/jimmunol.1000675
116. Byrd AS, O'Brien XM, Johnson CM, Lavigne LM, Reichner JS. An extracellular matrix-based mechanism of rapid neutrophil extracellular trap formation in response to Candida albicans. J Immunol. (2013) 190:4136–48. doi: 10.4049/jimmunol.1202671
117. Rochael NC, Guimaraes-Costa AB, Nascimento MT, DeSouza-Vieira TS, Oliveira MP, Garcia e Souza LF, et al. Classical ROS-dependent and early/rapid ROS-independent release of neutrophil extracellular traps triggered by Leishmania parasites. Sci Rep. (2015) 5:18302. doi: 10.1038/srep18302
118. Chandler CE, Ernst RK. Bacterial lipids: powerful modifiers of the innate immune response. F1000Res. (2017) 6:F1000. doi: 10.12688/f1000research.11388.1
119. Petretto A, Bruschi M, Pratesi F, Croia C, Candiano G, Ghiggeri G, et al. Neutrophil extracellular traps. (NET) induced by different stimuli: a comparative proteomic analysis. PLoS ONE. (2019) 14:e0218946. doi: 10.1371/journal.pone.0218946
120. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. (2007) 13:463–9. doi: 10.1038/nm1565
121. Liu S, Su X, Pan P, Zhang L, Hu Y, Tan H, et al. Neutrophil extracellular traps are indirectly triggered by lipopolysaccharide and contribute to acute lung injury. Sci Rep. (2016) 6:37252. doi: 10.1038/srep37252
122. Hu Z, Murakami T, Tamura H, Reich J, Kuwahara-Arai K, Iba T, et al. Neutrophil extracellular traps induce IL-1β production by macrophages in combination with lipopolysaccharide. Int J Mol Med. (2017) 39:549–58. doi: 10.3892/ijmm.2017.2870
123. Shao S, Fang H, Dang E, Xue K, Zhang J, Li B, et al. Neutrophil extracellular traps promote inflammatory responses in psoriasis via activating epidermal TLR4/IL-36R crosstalk. Front Immunol. (2019) 10:746. doi: 10.3389/fimmu.2019.00746
124. Hu SC, Yu HS, Yen FL, Lin CL, Chen GS, Lan CC. Neutrophil extracellular trap formation is increased in psoriasis and induces human β-defensin-2 production in epidermal keratinocytes. Sci Rep. (2016) 6:31119. doi: 10.1038/srep31119
125. Lee KH, Kronbichler A, Park DD, Park Y, Moon H, Kim H, et al. Neutrophil extracellular traps (NETs) in autoimmune diseases: a comprehensive review. Autoimmun Rev. (2017) 16:1160–73. doi: 10.1016/j.autrev.2017.09.012
126. Lowes MA, Suárez-Fariñas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol. (2014) 32:227–55. doi: 10.1146/annurev-immunol-032713-120225
127. Pinegin B, Vorobjeva N, Pinegin V. Neutrophil extracellular traps and their role in the development of chronic inflammation and autoimmunity. Autoimmun Rev. (2015) 14:633–40. doi: 10.1016/j.autrev.2015.03.002
128. Skrzeczynska-Moncznik J, Wlodarczyk A, Zabieglo K, Kapinska-Mrowiecka M, Marewicz E, Dubin A, et al. Secretory leukocyte proteinase inhibitor-competent DNA deposits are potent stimulators of plasmacytoid dendritic cells: implication for psoriasis. J Immunol. (2012) 189:1611–7. doi: 10.4049/jimmunol.1103293
129. Zabieglo K, Majewski P, Majchrzak-Gorecka M, Wlodarczyk A, Grygier B, Zegar A, et al. The inhibitory effect of secretory leukocyte protease inhibitor. (SLPI) on formation of neutrophil extracellular traps. J Leukoc Biol. (2015) 98:99–106. doi: 10.1189/jlb.4AB1114-543R
130. Majewski P, Majchrzak-Gorecka M, Grygier B, Skrzeczynska-Moncznik J, Osiecka O, Cichy J. Inhibitors of serine proteases in regulating the production and function of neutrophil extracellular traps. Front Immunol. (2016) 7:261. doi: 10.3389/fimmu.2016.00261
131. Tillack K, Breiden P, Martin R, Sospedra M. T lymphocyte priming by neutrophil extracellular traps links innate and adaptive immune responses. J Immunol. (2012) 188:3150–9. doi: 10.4049/jimmunol.1103414
132. Sangaletti S, Tripodo C, Chiodoni C, Guarnotta C, Cappetti B, Casalini P, et al. Neutrophil extracellular traps mediate transfer of cytoplasmic neutrophil antigens to myeloid dendritic cells toward ANCA induction and associated autoimmunity. Blood. (2012) 120:3007–18. doi: 10.1182/blood-2012-03-416156
133. Cataldi C, Mari NL, Lozovoy MAB, Martins LMM, Reiche EMV, Maes M, et al. Proinflammatory and anti-inflammatory cytokine profiles in psoriasis: use as laboratory biomarkers and disease predictors. Inflamm Res. (2019) 68:557–67. doi: 10.1007/s00011-019-01238-8
134. Murakami M, Hagforsen E, Morhenn V, Ishida-Yamamoto A, Iizuka H. Patients with palmoplantar pustulosis have increased IL-17 and IL-22 levels both in the lesion and serum. Exp Dermatol. (2011) 20:845–7. doi: 10.1111/j.1600-0625.2011.01325.x
135. Gisondi P, Lora V, Bonauguri C, Russo A, Lippi G, Girolomoni G. Serum chemerin is increased in patients with chronic plaque psoriasis and normalizes following treatment with infliximab. Br J Dermatol. (2013) 168:749–55. doi: 10.1111/bjd.12118
136. Shveta, Agarwal K, Ranchander, Agarwal S. Serum sE-selectin levels in newly diagnosed psoriasis patients and correlation with psoriasis area and severity index score. J Clin Diagnost Res. (2017) 11:EC09–12. doi: 10.7860/JCDR/2017/29957.10831
137. Torres T, Bettencourt N, Mendonca D, Vasconcelos C, Silva BM, Selores M. Complement C3 as a marker of cardiometabolic risk in psoriasis. Arch Dermatol Res. (2014) 306:653–60. doi: 10.1007/s00403-014-1467-5
138. Hau CS, Kanda N, Tada Y, Shibata S, Uozaki H, Fukusato T, et al. Lipocalin-2 exacerbates psoriasiform skin inflammation by augmenting T-helper 17 response. J Dermatol. (2016) 43:785–94. doi: 10.1111/1346-8138.13227
139. Aizawa N, Ishiuji Y, Tominaga M, Sakata S, Takahashi N, Yanaba K, et al. Relationship between the degrees of itch and serum lipocalin-2 levels in patients with psoriasis. J Immunol Res. (2019) 2019:8171373. doi: 10.1155/2019/8171373
140. Wolk K, Frambach Y, Jacobi A, Wilsmann-Theis D, Phillipp S, Witte-Handel E, et al. Increased levels of lipocalin 2 in palmoplantar pustular psoriasis. J Dermatol Sci. (2018) 90:68–74. doi: 10.1016/j.jdermsci.2017.12.018
141. Wolk K, Wenzel J, Tsaousi A, Witte-Handel E, Babel N, Zelenak C, et al. Lipocalin-2 is expressed by activated granulocytes and keratinocytes in affected skin and reflects disease activity in acne inversa/hidradenitis suppurativa. Br J Dermatol. (2017) 177:1385–93. doi: 10.1111/bjd.15424
142. Esaki H, Brunner PM, Renert-Yuval Y, Czarnowicki T, Huynh T, Tran G, et al. Early-onset pediatric atopic dermatitis is TH2 but also TH17 polarized in skin. J Allergy Clin Immunol. (2016) 138:1639–51. doi: 10.1016/j.jid.2016.02.064
143. Abdel Hay R, Samir N, Safwat M, Rashed L, Soliman M. Tissue lipocalin-2 in psoriasis: is it a marker of metabolic disturbance or a possible marker of therapeutic efficacy after narrow band ultraviolet B? J Dermatolog Treat. (2019) 17:1–5. doi: 10.1080/09546634.2019.1605141
144. Cai Y, Fleming C, Yan J. New insights of T cells in the pathogenesis of psoriasis. Cell Mol Immunol. (2012) 9:302–9. doi: 10.1038/cmi.2012.15
145. Brembilla NC, Senra L, Boehncke WH. The IL-17 family of cytokines in psoriasis: IL-17A and beyond. Front Immunol. (2018) 9:1682. doi: 10.3389/fimmu.2018.01682
146. Blauvelt A, Chiricozzi A. The immunologic role of IL-17 in psoriasis and psoriatic arthritis pathogenesis. Clin Rev Allergy Immunol. (2018) 55:379–90. doi: 10.1007/s12016-018-8702-3
147. Lin AM. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Immunol. (2011) 187:490–500. doi: 10.4049/jimmunol.1100123
148. Di Domizio J, Gilliet M. Psoriasis caught in the NET. J Invest Dermatol. (2019) 139:1426–9. doi: 10.1016/j.jid.2019.04.020
149. Lambert S, Hambro CA, Johnston A, Stuart PE, Tsoi LC, Nair RP, et al. Neutrophil extracellular traps induce human Th17 cells: effect of psoriasis-associated TRAF3IP2 genotype. J Invest Dermatol. (2019) 139:1245–53. doi: 10.1016/j.jid.2018.11.021
150. Guttman-Yassky E, Lowes MA, Fuentes-Duculan J, Zaba LC, Cardinale I, Nograles KE, et al. Low expression of the IL-23/Th17 pathway in atopic dermatitis compared to psoriasis. J Immunol. (2008) 181:7420–7. doi: 10.4049/jimmunol.181.10.7420
151. Chen K, Kolls JK. Interluekin-17A (IL17A). Gene. (2017) 614:8–14. doi: 10.1016/j.gene.2017.01.016
152. Guttman-Yassky E, Krueger JG. IL-17C: a unique epithelial cytokine with potential for targeting across the spectrum of atopic dermatitis and psoriasis. J Invest Dermatol. (2018) 138:1467–9. doi: 10.1016/j.jid.2018.02.037
Keywords: neutrophils, psoriasis, immunity, respiratory burst, degranulation, neutrophil extracellular traps
Citation: Chiang C-C, Cheng W-J, Korinek M, Lin C-Y and Hwang T-L (2019) Neutrophils in Psoriasis. Front. Immunol. 10:2376. doi: 10.3389/fimmu.2019.02376
Received: 18 April 2019; Accepted: 23 September 2019;
Published: 09 October 2019.
Edited by:
Onur Boyman, University of Zurich, SwitzerlandReviewed by:
Nicolas Gaudenzio, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceCopyright © 2019 Chiang, Cheng, Korinek, Lin and Hwang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tsong-Long Hwang, aHRsQG1haWwuY2d1LmVkdS50dw==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.