 Amy Lewis
Amy Lewis Philip M. Elks
Philip M. Elks
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 27 September 2019
Sec. Molecular Innate Immunity
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02321
Macrophage phenotypes are poorly characterized in disease systems in vivo. Appropriate macrophage activation requires complex coordination of local microenvironmental cues and cytokine signaling. If the molecular mechanisms underpinning macrophage activation were better understood, macrophages could be pharmacologically tuned during disease situations. Here, using zebrafish tnfa:GFP transgenic lines as in vivo readouts, we show that physiological hypoxia and stabilization of Hif-1α promotes macrophage tnfa expression. We demonstrate a new mechanism of Hif-1α-induced macrophage tnfa expression via a cyclooxygenase/prostaglandin E2 axis. These findings uncover a macrophage HIF/COX/TNF axis that links microenvironmental cues to macrophage phenotype, with important implications during inflammation, infection, and cancer, where hypoxia is a common microenvironmental feature and where cyclooxygenase and TNF are major mechanistic players.
Innate immune activation during homeostasis and disease are tightly regulated, in part by the coordination of microenvironmental cues and cytokine signaling. Macrophages are important innate immune cells in disease and their activation status, commonly termed polarization, is especially important during the response to injury and infection. Mammalian macrophages have been classified into two broad polarization states: M1 (or classically activated) and M2 (alternatively activated) (1, 2). M1 macrophages are highly antimicrobial and can phagocytose and efficiently kill bacteria. M2 macrophages are central players in tissue healing and restoration of homeostasis post injury/infection (3, 4). M1/M2 polarization states have largely been determined in vitro by addition of exogenous factors, e.g., cytokines, to the growth media, however the complex regulation of the diverse macrophage phenotypes observed in vivo remains poorly understood. It is only in whole organism in vivo models, where tissues remain intact, that microenvironmental cues of macrophage activation can be fully investigated.
Leukocytes, including macrophages and neutrophils, are exquisitely sensitive to tissue hypoxia, a signature of many disease microenvironments (e.g., in inflammatory diseases, infections, and cancers) due to lack of local blood supply and a high turnover of local oxygen by pathogens (5, 6). Tissue hypoxia can polarize macrophages, via stabilization of the Hypoxia Inducible Factor (HIF)-1α transcription factor (7). HIF-1α is a master transcriptional regulator of the cellular response to hypoxia (8) and has been shown to have activating effects on macrophages in vitro, increasing their pro-inflammatory cytokine profile and bactericidal capabilities, yet the molecular mechanisms behind these observations are not well-understood in vivo (6).
Eicosanoids are lipid signaling molecules and important pro-inflammatory mediators during homeostasis and disease (9, 10). For example, tissue hypoxia increases cyclooxygenase (COX)/prostaglandin E2 (PGE2) production in epithelial cells in cancer models (11–13). Recent evidence indicates that hypoxia-induced tumor necrosis factor alpha (TNF) expression in osteoblasts (bone producing cells) is mediated through cyclooxygenase enzymes by an, as yet, unknown mechanism (14). This HIF/COX/TNF axis has not, to date, been observed in macrophages, and could be a potential novel pathway to macrophage activation in diseases where hypoxia is a microenvironmental factor.
Here, we investigated the regulation of expression of the important, primarily pro-inflammatory, cytokine tnfa by Hif-1α stabilization in vivo using optically transparent zebrafish larvae. We demonstrate that genetic, pharmacological, and hypoxia-mediated Hif-1α stabilization upregulated macrophage tnfa. We utilize well-characterized, non-invasive, zebrafish models of inflammation and infection to show that Hif-1α-mediated tnfa acts via an alternative, cyclooxygenase dependent mechanism, that differs from better understood DAMP/PAMP mediated pathways that are cyclooxygenase independent (15, 16). We show that the Hif-1α-mediated tnfa acts via active PGE2. Furthermore, this new macrophage HIF/COX/TNF axis is found in primary human macrophages. These findings have important implications in inflammation, infection, and cancer biology where macrophage phenotypes are influenced by microenvironmental hypoxia, and where HIF, COX, and TNF are major mechanistic players.
Zebrafish were raised and maintained on a 14:10 h light/dark cycle at 28°C, according to standard protocols (17), in UK Home Office approved facilities at The Bateson Center aquaria at the University of Sheffield. Strains used were Nacre (wildtype), TgBAC(tnfa:GFP)pd1028 (Source: Bagnat lab, Duke University, USA), Tg(tnfa:eGFP-F)ump5Tg:Tg(mpeg1:mCherry-F)ump2Tg (Source: Lutfalla Lab, Montpellier University, France), Tg(mpeg1:mCherryCAAX)sh378 (Source: University of Sheffield), Tg(lyz:Ds-RED2)nz50 (Source: Hall Lab, University of Auckland, New Zealand), TgBAC(il-1β:eGFP)sh445 (Source: University of Sheffield), and Tg(phd3:EGFP)i144 (Source: University of Sheffield) (18–22).
Inflammation was induced in zebrafish embryos by tail transection at 2 days post-fertilization (dpf). Larvae were anesthetized and the tailfin was transected using a sterile scalpel blade (23). Larvae were imaged by confocal microscopy at 16 h post-wounding (hpw) on a Leica TCS-SPE confocal on an inverted Leica DMi8 base and imaged using 20× or 40× objective lenses.
Mycobacterium marinum (Mm) M (ATCC #BAA-535), containing a psMT3-mCherry or psMT3 mCrimson vector were used (24). Injection inoculum was prepared from an overnight liquid culture in the log-phase of growth resuspended in 2% polyvinylpyrrolidone40 (PVP40) solution (CalBiochem, CAS 9003-39-8) (25). One hundred or one hundred and fifty colony forming units (CFU) were injected into the caudal vein at 28–30 h post-fertilization (hpf) (26).
Larvae were mounted in 0.8–1% low melting point agarose (Sigma-Aldrich, A9414) and imaged on a Leica TCS-SPE confocal on an inverted Leica DMi8 base and imaged using 20× or 40× objective lenses.
For quantification purposes acquisition settings and area of imaging were kept the same across groups. Corrected total cell fluorescence was calculated for each cell using Image J by drawing around the cell and correcting for background levels of staining (27, 28).
Embryos were injected with dominant hif-1αb (ZFIN: hif1ab) variant RNA at the one cell stage as previously described (23). hif-1α variants used were dominant active (DA) that is a constitutively stable Hif-1α and dominant negative (DN) hif-1α that cannot signal due to lack of transactivation domain. Phenol red (PR) (Sigma-Aldrich, P0290) was used as a vehicle control.
Mm mCherry infected zebrafish larvae were imaged at 4 days post-fertilization (dpi) on an inverted Leica DMi8 with a 2.5× objective lens. Brightfield and fluorescent images were captured using a Hammamatsu OrcaV4 camera. Bacterial burden was assessed using dedicated pixel counting software as previously described (29).
Unless otherwise stated, embryos were treated from 4 h pre-Mm infection to 24 h post-infection (hpi) by addition to the embryo water in six well plates and dimethyl sulfoxide (DMSO, Sigma-Aldrich, D8418), was used as a negative solvent control. The pan hydroxylase inhibitor, DMOG (dimethyloxaloylglycine, Enzo Life Sciences, BML-EI347-0010), was used at a 100 μM concentration by incubation in E3 embryo media (23). The selective PHD inhibitors JNJ-402041935 (Cayman Chemicals, 14316) and FG-4592 (Selleckchem, FG-34595) were used at 100 and 5 μM, respectively (30, 31). The selective inhibitor of COX-1, SC-560 (Cayman Chemical, 70340), was used at 30 μM and the selective inhibitor of COX-2, NS-398 (Cayman Chemical, 70590), was used at 15 μM by incubation in E3 embryo media (32). The 15-LOX inhibitor PD146176 (Tocris, 2850) was microinjected (1 nl of 40 nM) at 1 h post-infection (hpi) (33). The leukotriene B4 receptor 1 (BLTR1) inhibitor, U-75302 (Abcam, ab141736), was used at 30 mM by incubation in E3 embryo media (33) together with the BLTR2 inhibitor, LY255283 (Abcam, ab144472), which was used at 1 mM (34). Exogenous prostaglandin E2 (Cayman Chemical, 14010) and 15-keto-prostaglandin E2 (Cayman Chemical, 14720) were added by incubation in E3 at a concentration of 1 μM (35). The EP4 antagonist AH23848 (Cayman Chemical, 19023) was used at 1 μM (36).
Embryos were incubated in 5% oxygen (with 5% carbon dioxide) in a hypoxia hood (Baker-Ruskinn Sci-tive UM-027) from 32 hpi for 6 h and were imaged at 2 dpf. Embryos from the same clutch kept in incubated normoxic room air were used as controls (37).
Peripheral blood mononuclear cells (PBMC) were isolated by Ficoll-Paque PLUS (GE Healthcare, GE17-1440-03) density centrifugation of whole blood from healthy donors (National Research Ethics Service reference 07/Q2305/7). PBMC were seeded in RMPI 1640 media (Gibco, 11875-093) containing 2 mmol/L L-glutamine (Lonza, BE17-605F) and 10% newborn calf serum (Gibco, 26010074). Cells were cultured in 5% CO2 at 37°C for 14 days in RPMI 1640 supplemented with 2 mmol/L L-glutamine and 10% heat-inactivated fetal bovine serum (FBS) (PAN Biotech, P30-3306) (38).
RPMI 1640 + 25 mM HEPES (Gibco, 11550496) containing 2 mmol/L L-glutamine and 10% heat-inactivated FBS and drugs were pre-equilibrated in normoxia or hypoxia (0.8% O2, 5% CO2, 70% humidity at 37°C, in a Baker-Ruskinn Sci-Tive UM-027) for 24 h prior to the experiment (39). Cells were treated in duplicate or triplicate wells with 100 μg/ml LPS (InvivoGen, tlrl-b5lps), DMSO (Sigma-Aldrich, D8418), or 15 mM COX-2 inhibitor (NS398) (Cayman Chemical, 70590) and incubated for 18 h. For normoxic controls, cells were treated identically within a class-2 tissue culture hood and transferred to a normoxic tissue-culture incubator.
Cell supernatants were collected following 18 h incubation and were assayed in triplicate using a human TNF ELISA MAX (Biolegend, 430204).
All data were analyzed (Prism 7.0, GraphPad Software) using unpaired, two-tailed t-tests for comparisons between two groups and ANOVA (with Bonferonni post-test adjustment) for other data. P-values shown are: *P < 0.05, **P < 0.01, and ***P < 0.001.
Tumor necrosis factor (TNF) is a central regulator of macrophage pro-inflammatory phenotypes during homeostasis, inflammation, and infection. Here, we use two transgenic zebrafish lines that express GFP under the control of a tnfa promoter region; TgBAC(tnfa:GFP)pd1028 and Tg(tnfa:eGFP-F)ump5Tg (18, 20). The TgBAC(tnfa:GFP)pd1028 is a BAC (bacterial artificial chromosome) transgenic line that contains 50 kb of promoter region (18). The Tg(tnfa:eGFP-F)ump5Tg line has a smaller promoter region (3.8 kb) than the BAC line (20) and has previously been demonstrated to be upregulated in macrophages after tailfin injury in a zebrafish model (20). Here, we demonstrate that injury-induced upregulation of macrophage tnfa:GFP is also found in the BAC transgenic line at 16 h post-wound (hpw), shown in double transgenic TgBAC(tnfa:GFP)pd1028; Tg(mpeg1:mCherryCAAX)sh378 larvae (Figure 1A).
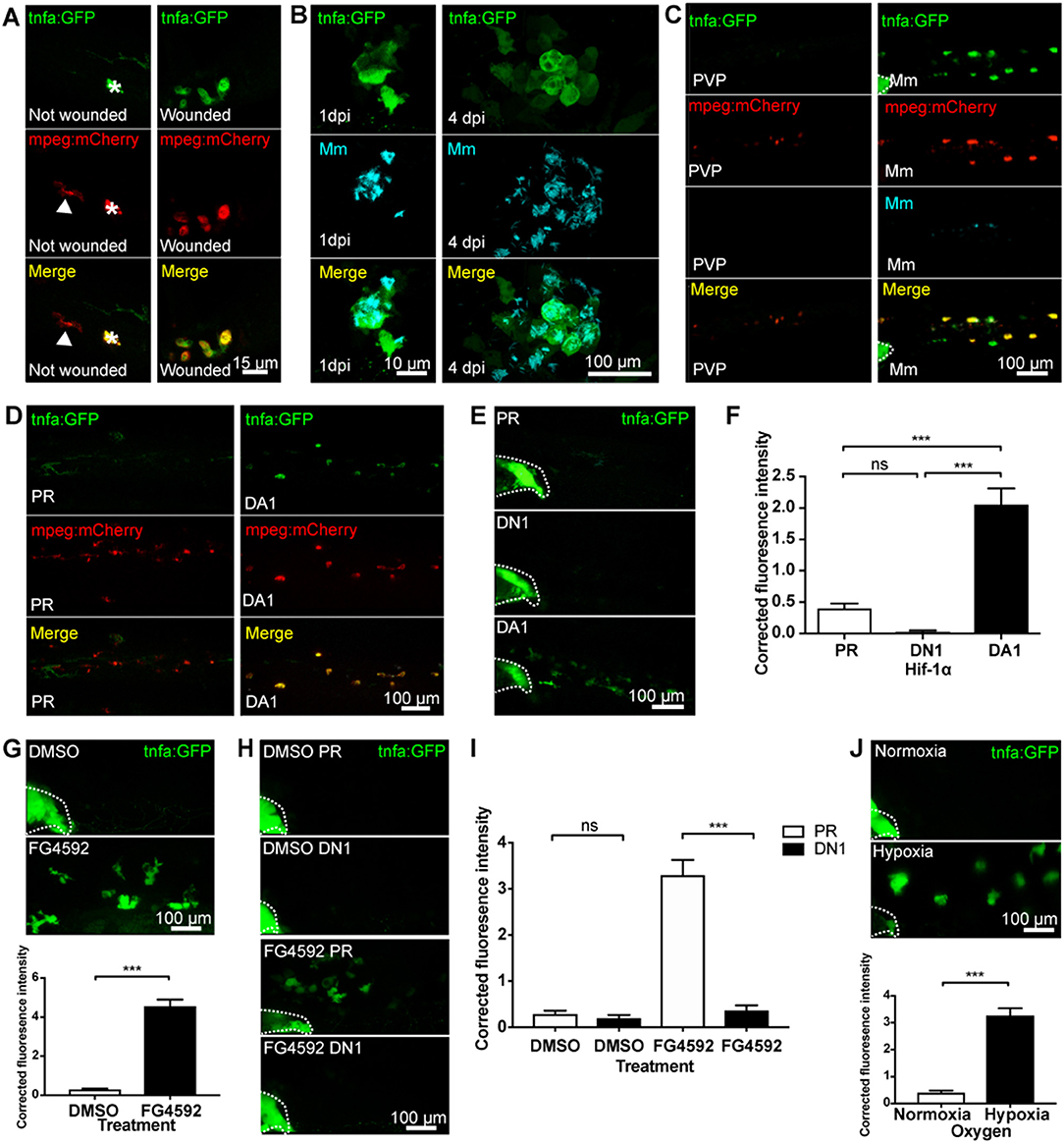
Figure 1. Macrophage tnfa:GFP is upregulated by injury, Mm infection and Hif-1α stabilization. (A) Confocal micrographs of 3 days post-fertilization larvae with or without tailfin wound (16 hpw) at the tailfin site. tnfa expression was detected using the TgBAC(tnfa:GFP)pd1028 transgenic line. Macrophages are shown in red using a Tg(mpeg1:mCherryCAAX)sh378 line. The white arrowhead indicates the presence of a tnfa:GFP negative, mpeg:mCherry positive macrophage in the tailfin of a non-wounded control. Asterisk indicates a neuromast that is fluorescent in all channels as a marker of position. Representative images of three independent experiments with 20–30 larvae per group per experiment. (B) Confocal micrographs of 1 dpi Mm mCrimson infected larvae, prior to granuloma formation (left panels), and 4 dpi, early granuloma (right panels) stages of infection using the TgBAC(tnfa:GFP)pd1028 line. Representative images of three independent experiments with 20–30 larvae per group per experiment. (C) Confocal micrographs of 1 day post-infection (dpi) with Mm mCrimson at the caudal vein region of infection in the TgBAC(tnfa:GFP)pd1028 line. Macrophages are shown in red using a Tg(mpeg1:mCherryCAAX)sh378 line. Mm in right panels with PVP control in left panels. a Dotted lines indicate the yolk extension of the larvae where there is non-specific fluorescence. Representative images of three independent experiments with 20–30 larvae per group per experiment. (D) Confocal micrographs of 2 dpf larvae in the caudal vein region in the TgBAC(tnfa:GFP)pd1028 line. Macrophages are shown in red using a Tg(mpeg1:mCherryCAAX)sh378 line. Larvae were injected at the 1 cell stage with dominant active (DA) Hif-1α or phenol red (PR) control. Representative images of three independent experiments with 20–30 larvae per group per experiment. (E) Confocal micrographs of 2 dpf zebrafish imaged around the caudal vein region in the TgBAC(tnfa:GFP)pd1028 line. Larvae were injected at the 1 cell stage with dominant negative (DN) or dominant active (DA) Hif-1α or phenol red (PR) control. (F) Corrected fluorescence intensity levels of tnfa:GFP in larvae in (E). Mean ± SEM, n = 66 cells from 12 embryos accumulated from three independent experiments and corresponds to images in (E). P-values shown are: *P < 0.05, **P < 0.01, and ***P < 0.001, one way ANOVA with Bonferonni post-test adjustment. (G) Confocal micrographs of 2 dpf TgBAC(tnfa:GFP)pd1028 larvae treated with the PHD inhibitor FG4592 or a DMSO solvent control. Graph shows corrected fluorescence intensity levels of tnfa:GFP. Mean ± SEM, n = 54 cells from nine embryos accumulated from three independent experiments. P-values shown are: *P < 0.05, **P < 0.01, and ***P < 0.001, unpaired, two-tailed t-test. (H) Confocal micrographs of 2 dpf TgBAC(tnfa:GFP)pd1028 larvae treated with the PHD inhibitor FG4592 or a DMSO solvent control. Larvae were injected at the 1 cell stage with dominant negative (DN) Hif-1α or phenol red (PR) control. (I) Corrected fluorescence intensity levels of tnfa:GFP in larvae in (H). Mean ± SEM, n = 60 cells from 10 embryos accumulated from three independent experiments. P-values shown are: *P < 0.05, **P < 0.01, and ***P < 0.001, two way ANOVA with Bonferonni post-test adjustment. (J) Confocal micrographs of 2 dpf TgBAC(tnfa:GFP)pd1028 larvae incubated in room normoxia or 5% oxygen (hypoxia) for 6 h between 32 and 38 hpf. Graph shows corrected fluorescence intensity levels of tnfa:GFP. Mean ± SEM, n = 72 cells from 12 embryos accumulated from three independent experiments. P-values shown are: *P < 0.05, **P < 0.01, and ***P < 0.001, unpaired, two-tailed t-test.
TNF has been implicated in many bacterial infections, however the cell types involved have been difficult to observe in vivo. To investigate this we used a well-characterized zebrafish infection model, the M. marinum (Mm) model of tuberculosis infection (40). We used the tnfa BAC promoter GFP line to establish the transcriptional regulation of tnfa during pre- and early- larval Mm granuloma formation stages. tnfa:GFP expression was induced in larvae following Mm infection before granuloma onset (at 1 day post-infection, dpi), and during granuloma formation (at 4 dpi) (Figure 1B). tnfa:GFP expression was predominantly found in macrophages, shown by co-localization of fluorescence with mpeg-1:mCherry expressing macrophages (Figure 1C). Macrophage expression was confirmed using the other tnfa promoter driven line, Tg(tnfa:eGFP-F)ump5Tg; Tg(mpeg1:mCherry-F)ump2Tg (Figure S1). Our data demonstrate that injury and Mm induced tnfa expression occurs in macrophages as part of an early response.
Hypoxia and Hif-1α stabilization are key features of diseased tissue microenvironments that can trigger macrophage pro-inflammatory responses. We first tested whether Hif-1α promotes tnfa in vivo, by stabilizing Hif-1α genetically in the tnfa:GFP BAC transgenic line (18, 20). Dominant active (DA) Hif-1α RNA (23) resulted in upregulation of macrophage tnfa:GFP expression (Figures 1D–F) indicating a shift of macrophage phenotype toward a pro-inflammatory response. Dominant negative (DN) Hif-1α caused no change in tnfa:GFP expression (Figures 1E,F).
To ensure that endogenous levels of Hif-1α could trigger a tnfa response, Hif-α was stabilized in embryos pharmacologically. Treatment with the well-described hypoxia mimetic DMOG (dimethyloxaloylglycine, a pan-hydroxylase inhibitor that stabilizes endogenous Hif-1α via inactivation of regulatory prolyl hydroxylase enzymes) alongside newer hydroxylase inhibitors with reportedly greater prolyl hydroxylase selectively than DMOG, JNJ-42041935, and FG4592 were tested in the Tg(phd3:GFP)i144 hypoxia reporter zebrafish. phd3 is a Hif-1α transcriptional target that is highly upregulated after hypoxia/Hif-1α stabilization (22). Treatment with all the hydroxylase inhibitors increased phd3:GFP levels compared to DMSO controls demonstrating stabilization of the Hif-α pathway (Figure S2) with FG4592 treatment leading to the highest phd3:GFP levels (23, 30, 31, 37). FG4592 (Figure 1G), DMOG (Figure S3A), and JNJ-42041935 (Figure S3B) treatment upregulated tnfa:GFP expression compared to a DMSO solvent control, phenocopying the DA Hif-1α response. tnfa:GFP induced by hydroxylase inhibition could be blocked using dominant negative (DN) Hif-1α, both in the case of FG4592 (Figures 1H,I) and DMOG (Figure S4), demonstrating that hydroxylase inhibitors are acting via Hif-1α to induce tnfa:GFP.
Finally, we subjected the tnfa:GFP line to physiological hypoxia (5% oxygen for 6 h at 32 hpf) and looked for GFP expression at 48 hpf. This level of hypoxia was sufficient to turn on phd3:GFP expression in Tg(phd3:GFP)i144 hypoxia reporter zebrafish (Figure S5) (37). Hypoxia induced tnfa:GFP expression, compared to normoxic controls at 48 hpf, to a similar level as that observed with genetic or pharmacological Hif-1α stabilization (Figure 1J).
Together, these data indicate that tnfa expression is induced in macrophages in response to hypoxia and Hif-1α, a response that is targetable by pharmacological agents and has the potential to manipulate macrophage responses during disease.
Eicosanoids are lipid signaling molecules and can be produced by macrophages as part of a pro-inflammatory response (9, 10). We tested whether cyclooxygenase inhibition affected Hif-1α-mediated macrophage tnfa:GFP expression, using Cox-1 and Cox-2 inhibitors (SC560 and NS398, respectively) (32). First, we showed that neither SC560 nor NS398 caused increased Hif-α transcriptional activity, using the Tg(phd3:GFP)i144 hypoxia reporter zebrafish (Figure S6). In the absence of Hif-1α stabilization tnfa:GFP was not altered by SC560 or NS398 compared to DMSO treated negative controls (Figures 2A,B). Strikingly, the DA Hif-1α-induced tnfa:GFP expression was reduced to basal levels by both SC560 and NS398 (Figures 2A,B). These findings were replicated using the hydroxylase inhibitors FG4592 (Figures 2C,D) and DMOG with NS398 (Figure S7), (N.B. DMOG with SC560 caused toxicity and developmental delay therefore data is not shown for this group). The production of an unrelated Hif-1α target gene phd-3 by hydroxylase inhibitors, shown in the Tg(phd3:GFP)i144 transgenic line, was not altered by SC560 or NS398 treatment, demonstrating that Hif-1α transcriptional potential is not affected by cyclooxygenase inhibition (Figure S8).
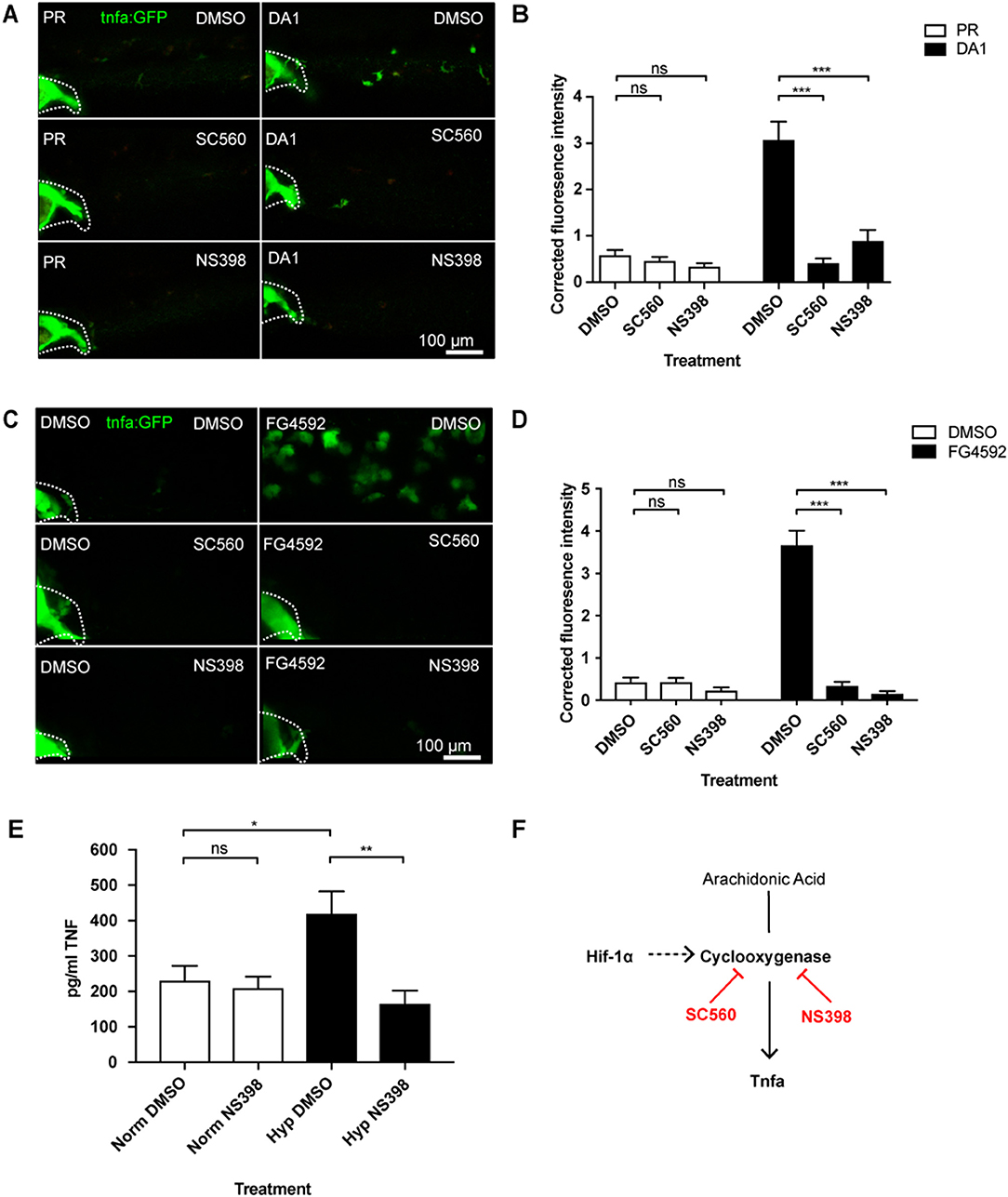
Figure 2. Hif-1α-activated tnfa is cyclooxygenase dependent. (A) Confocal micrographs of 2 dpf caudal vein region of larvae in the TgBAC(tnfa:GFP)pd1028 line. Phenol red (PR) and dominant active Hif-1α (DA1) injected larvae were treated with DMSO, SC560 (Cox-1 inhibitor), and NS398 (Cox-2 inhibitor). Dotted lines indicate the yolk extension of the larvae where there is non-specific fluorescence. (B) Corrected fluorescence intensity levels of tnfa:GFP in larvae in (A). Mean ± SEM, n = 36 cells from six embryos representative of three independent experiments. P-values shown are: *P < 0.05, **P < 0.01, and ***P < 0.001, two way ANOVA with Bonferonni post-test adjustment. (C) Confocal micrographs of 2 dpf caudal vein region of larvae in the TgBAC(tnfa:GFP)pd1028 line. DMSO and FG4592 treated larvae were co-treated with DMSO, SC560 (Cox-1 inhibitor), and NS398 (Cox-2 inhibitor). Dotted lines indicate the yolk extension of the larvae where there is non-specific fluorescence. (D) Corrected fluorescence intensity levels of tnfa:GFP in larvae in (C). Mean ± SEM, n = 48 cells from eight embryos representative of three independent experiments. P-values shown are: *P < 0.05, **P < 0.01, and ***P < 0.001, two way ANOVA with Bonferonni post-test adjustment. (E) TNF ELISA of human monocyte derived macrophages treated with LPS and incubated in normoxia or 0.8% hypoxia with or without treatment with NS398. LPS negative controls were performed but TNF produced in these groups was below detectable levels. Mean ± SEM, n = 5–8 biological repeats from 3 to 4 donors. P-values shown are: *P < 0.05, **P < 0.01, and ***P < 0.001, two way ANOVA with Bonferonni post-test adjustment. (F) Schematic of the arachidonic pathway indicating that stabilizing Hif-1α upregulates tnfa, an effect that is blocked using the Cox1/2 inhibitors SC560/NS398.
We tested whether HIF-1α stabilization induced TNF in human monocyte derived macrophages (hMDMs). We found that human monocyte derived macrophages produced higher levels of TNF protein in hypoxia (0.8% oxygen) than those in normoxia, measured by an anti-human TNF ELISA (Figure 2E). Furthermore, treatment with the COX-2 inhibitor, NS398, reduced this hypoxia-induced TNF to equivalent levels found in normoxia (Figure 2E). This was replicated when HIF-1α was stabilized in hMDMs using the hypoxia mimetic FG4592 (Figure S9). These data indicate that a HIF/COX/TNF axis is found in zebrafish and human macrophages, and could be a druggable target in disease situations (Figure 2F).
We have previously shown that DA Hif-1α induces the expression of another important pro-inflammatory cytokine, il-1β, shown by an il-1β:GFP transgenic line (Figure S10A) (37). Cox-1 inhibition by SC560 did not abrogate Hif-1α-induced il-1β:GFP, while Cox-2 inhibition with NS398 caused a small decrease that did not reach basal levels (Figures S10B,C). These data indicate that Hif-1α-induced tnfa expression is caused by a product of the cyclooxygenase arm of the arachidonic acid pathway via a novel macrophage HIF/COX/TNF axis, while Hif-1α-induced il-1β does not fully act via this pathway suggestive of complex regulation of inflammatory cytokines by Hif-1α stabilization.
We next tested whether injury- and infection-induced tnfa are Hif-1α and cyclooxygenase dependent processes. Induction of tnfa:GFP expression after injury was not blocked by dominant negative Hif-1α (Figures 3A,B), and this was also the case for Mm infection-induced tnfa:GFP expression (Figures 3C,D). Similarly, neither genetic nor pharmacological Hif-1α stabilization were additive to the tnfa:GFP expression caused by either injury (Figures 3E,F) or Mm infection (Figures 3G,H; (Figures S11A,B). These data indicate that the early tnfa response of injury and Mm infection do not require active Hif-1α. Macrophage tnfa:GFP expression induced after injury was not abrogated by cyclooxygenase inhibition using NS398 (Figures 3E,F). Similarly, cyclooxygenase inhibition using either SC560 or NS398 did not alter the expression of Mm-induced tnfa:GFP (Figures 3G,H). SC560 and NS398 mediated inhibition of Hif-1α-induced tnfa expression did not diminish the host-protective effect of DA Hif-1α (Figures S12A–C) nor DMOG treated larvae (Figures S12D–F), suggesting that the presence of Mm induced tnfa is sufficient for protection. Our data indicate that Hif-1α-induced macrophage tnfa expression via cyclooxygenase differs from DAMP- (injury) or PAMP- (Mm) induced tnfa mechanisms.
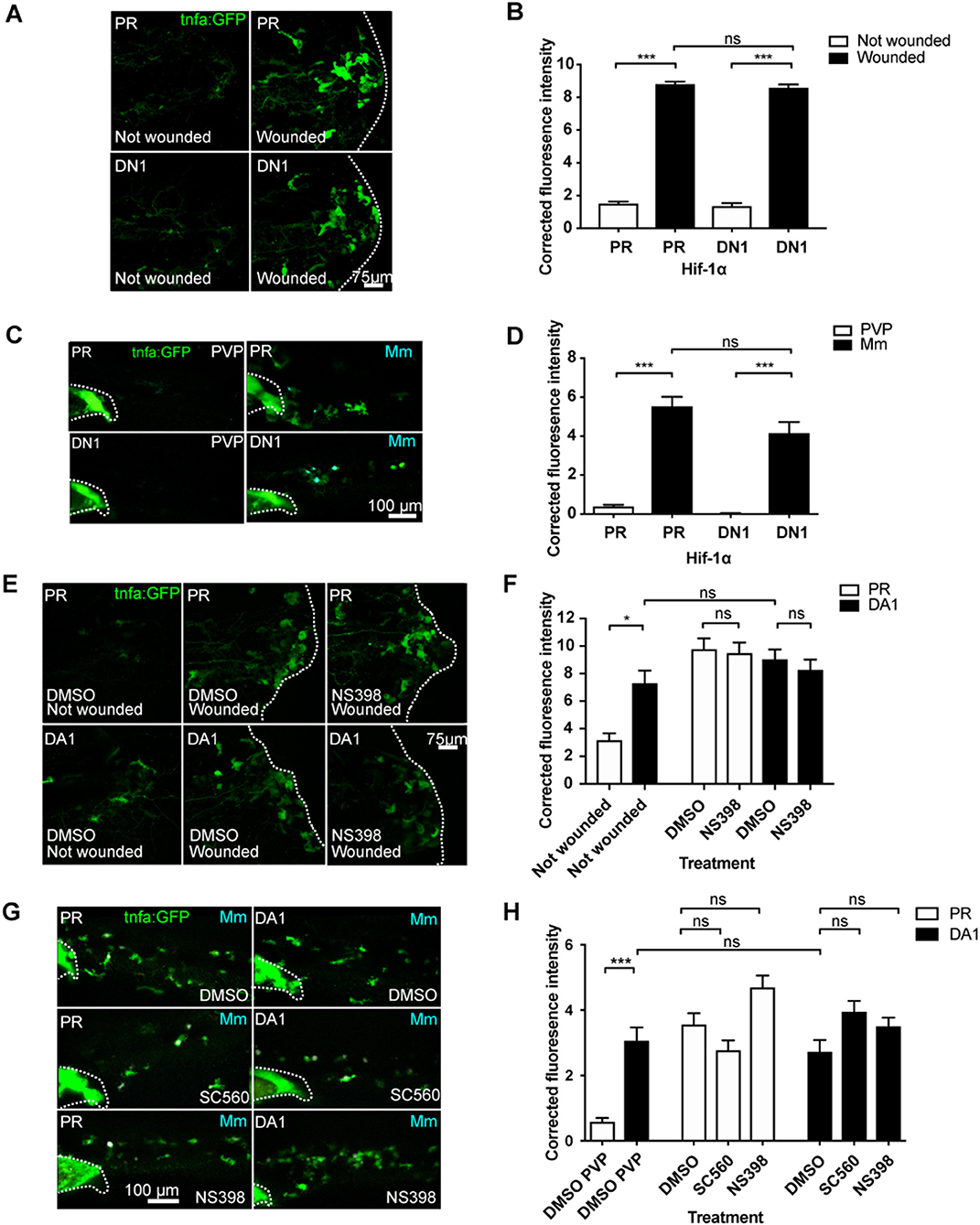
Figure 3. Injury and infection induced tnfa:GFP is cyclooxygenase independent. (A) Confocal micrographs of the TgBAC(tnfα:GFP)pd1028 line at 3 days post-fertilization larvae with or without tailfin wound (induced 16 h previously) in phenol red (PR) or dominant negative (DN) Hif-1α embryos. The dotted line indicates the border of the tailfin wound. (B) Corrected fluorescence intensity levels of tnfa:GFP in larvae in (A). Mean ± SEM, n = 60 cells from 10 embryos accumulated from three independent experiments. P-values shown are: *P < 0.05, **P < 0.01, and ***P < 0.001, two way ANOVA with Bonferonni post-test adjustment. (C) Confocal micrographs of 1 dpi caudal vein region of larvae in the TgBAC(tnfa:GFP)pd1028 line. Phenol red (PR) and dominant negative Hif-1α (DN1) injected larvae were infected with Mm Crimson or PVP controls. (D) Corrected fluorescence intensity levels of tnfa:GFP in larvae in (C). Mean ± SEM, n = 30 cells from five embryos accumulated from two independent experiments. P-values shown are: *P < 0.05, **P < 0.01, and ***P < 0.001, two way ANOVA with Bonferonni post-test adjustment. (E) Confocal micrographs of the TgBAC(tnfα:GFP)pd1028 line at 3 days post fertilization larvae with or without tailfin wound (induced 16 h previously) in phenol red (PR) or dominant active Hif-1α embryos. Embryos were treated with DMSO or NS398 (Cox-2 inhibitor). (F) Corrected fluorescence intensity levels of tnfa:GFP in larvae in (E). Mean ± SEM, n = 24 (in not wounded) or n = 36 (in wounded) cells from six embryos accumulated from two independent experiments. n.b. Not-wounded fish had fewer macrophages at the tailfin site. P-values shown are: *P < 0.05, **P < 0.01, and ***P < 0.001, two way ANOVA with Bonferonni post-test adjustment. (G) Confocal micrographs of 1 dpi caudal vein region of Mm Crimson infected larvae in the TgBAC(tnfa:GFP)pd1028 line. Phenol red (PR) and dominant active Hif-1α (DA1) injected larvae were treated with DMSO, SC560 (Cox-1 inhibitor), and NS398 (Cox-2 inhibitor) and infected with Mm Crimson. (H) Corrected fluorescence intensity levels of tnfa:GFP in larvae in (G). Mean ± SEM, n = 36 cells from six embryos representative of three independent experiments. P-values shown are: *P < 0.05, **P < 0.01, and ***P < 0.001, two way ANOVA with Bonferonni post-test adjustment.
To investigate whether the effect of the cyclooxygenase inhibitors on tnfa was specific to the prostaglandin path of arachidonic acid signaling, we targeted the lipoxin and leukotriene producing arms using the 15-Lipoxygenase inhibitor PD146176 and leukotriene B4 receptor antagonists (Figure 4A) (33, 34). The 15-Lipoxygenase inhibitor PD146176 did not block the tnfa:GFP induced by DA Hif-1α (Figures 4B,C). Furthermore, PD147176 increased tnfa:GFP expression on its own in the absence of infection, although not to the same extent as DA Hif-α (Figures 4B,C). PD146176 also did not affect tnfa:GFP expression after Mm infection (Figures 4B,C), nor did it block the protective effect of DA Hif-1α. Treatment with PD146176 was sufficient to decrease Mm burden, but not to the same extent as DA Hif-1α (Figure 4D). Leukotriene B4 inhibition, using the BLTR1/2 antagonists U75302 and LY255283, did not increase tnfa:GFP levels and also did not block DA Hif-1α-induced tnfa:GFP (Figures 4E,F). These data indicate that blocking components of the lipoxygenase dependent arms of the arachidonic acid pathway does not block the Hif-1α effect on tnfa expression and do not replicate the effects observed by blocking the cyclooxygenase dependent arm.
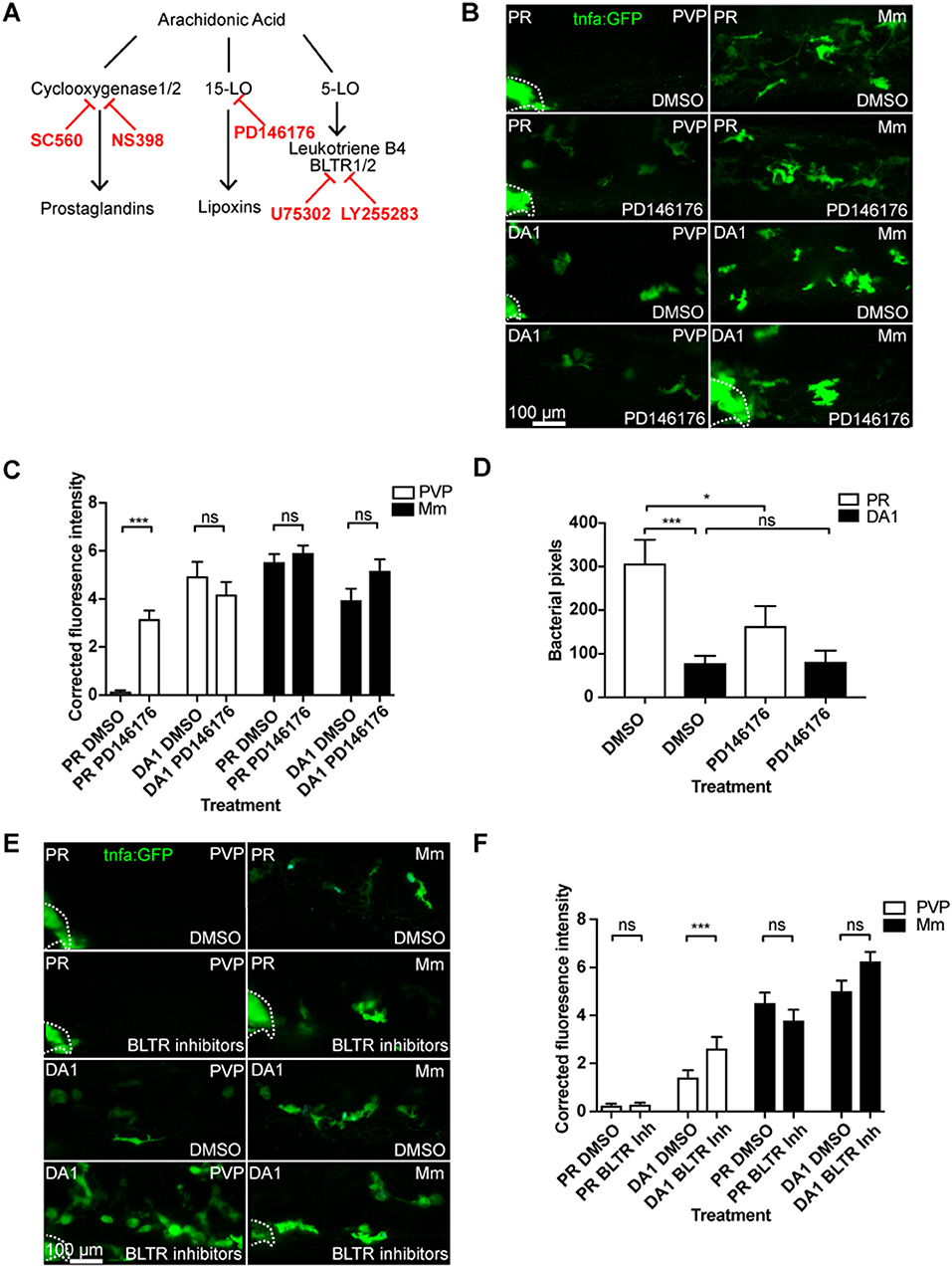
Figure 4. Blocking 15-lipoxygenase or leukotriene B4 receptors does not abrogate DA-Hif-1α-upregulation of tnfa:GFP. (A) Schematic of the arachidonic pathway indicating where the inhibitors act. (B) Confocal micrographs of 1 dpi caudal vein region of Mm mCrimson and PVP infected larvae in the TgBAC(tnfa:GFP)pd1028 line. Phenol red (PR) and dominant active Hif-1α (DA1) injected larvae were treated with DMSO or PD146176 (15-Lipoxygenase inhibitor). Dotted lines indicate the yolk extension of the larvae where there is non-specific fluorescence. (C) Corrected fluorescence intensity levels of tnfa:GFP in larvae in (B). Mean ± SEM, n = 42 cells from seven embryos accumulated from three independent experiments. P-values shown are: *P < 0.05, **P < 0.01, and ***P < 0.001, two way ANOVA with Bonferonni post-test adjustment. (D) Bacterial burden at 4 dpi after injection of DA Hif-1α (DA1) and 24 h of the 15-lipoxygenase inhibitor PD146176, using DMSO as a negative solvent control. Data shown are mean ± SEM, n = 48–50 as accumulated from three independent experiments. P-values shown are: *P < 0.05, **P < 0.01, and ***P < 0.001, two way ANOVA with Bonferonni post-test adjustment. (E) Confocal micrographs of 1 dpi caudal vein region of Mm mCrimson and PVP infected larvae in the TgBAC(tnfa:GFP)pd1028 line. Phenol red (PR) and dominant active Hif-1α (DA1) injected larvae were treated with DMSO or U75302/LY255283 (BLTR1/2 inhibitors). (F) Corrected fluorescence intensity levels of tnfa:GFP confocal z-stacks in uninfected larvae (PVP, empty bars) and infected larvae (Mm, filled bars) at 1 dpi of data shown in (E) after treatment with DMSO or U75302/LY255283 (BLTR1/2 inhibitors). Data shown are mean ± SEM, n = 30 cells from five embryos accumulated from two independent experiments. P-values shown are: *P < 0.05, **P < 0.01, and ***P < 0.001, two way ANOVA with Bonferonni post-test adjustment.
A key family of immune regulators downstream of arachidonic acid and cyclooxygenases are prostaglandins. The best characterized of these as a regulator of macrophage function is prostaglandin E2 (PGE2) (41). We tested whether PGE2 was a mediator in the HIF/COX/TNF pathway by addition of exogenous PGE2 to DA Hif-1α larvae (35). Exogenous PGE2 had no effect on tnfa:GFP expression in the absence of infection (Figures 5A,B). However, PGE2 was able to rescue the decrease in tnfa:GFP expression after inhibition of either Cox-1 (Figure S13) or Cox-2 (Figures 5A,B) in DA Hif-1α larvae. Furthermore, addition of PGE2 alone was sufficient to increase the DA Hif-1α-induced tnfa:GFP expression (Figures 4A,B). These tnfa:GFP activating effects were not observed by addition of exogenous 15-keto prostaglandin E2, an immunologically inactive degradation product of PGE2 (Figures 5C,D) (35, 42, 43). These data indicate that Hif-1α-induced tnfa:GFP requires active prostaglandin E2.
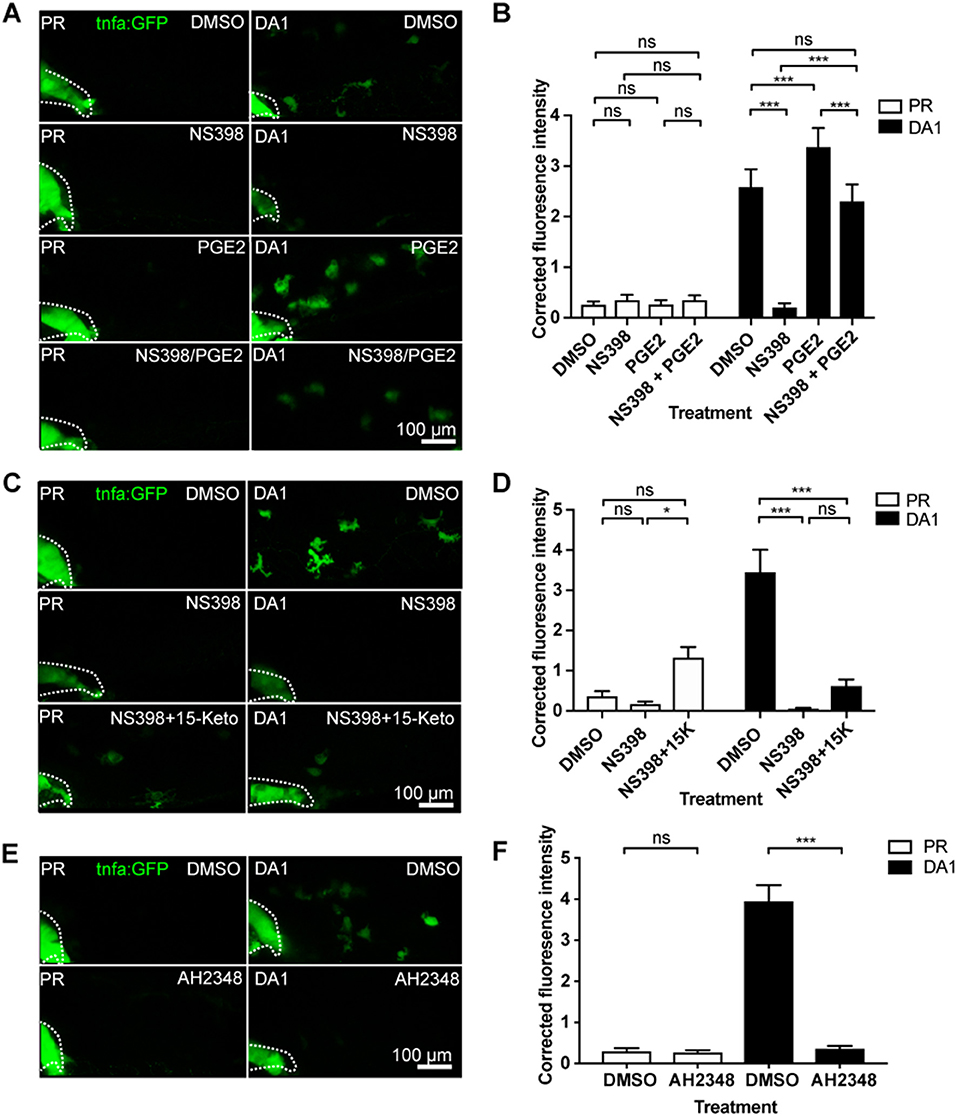
Figure 5. Hif-1α-induced tnfa:GFP requires active prostaglandin E2. (A) Confocal micrographs of 1 dpi caudal vein region in the TgBAC(tnfa:GFP)pd1028 line. Phenol red (PR) and dominant active Hif-1α (DA1) injected larvae were treated with DMSO or NS398 (Cox-2 inhibitor) in the presence or absence of endogenous prostaglandin E2 (PGE2). All larvae are PVP injected. (B) Corrected fluorescence intensity levels of tnfa:GFP in larvae in (A). Mean ± SEM, n = 54 cells from nine embryos accumulated from three independent experiments. P-values shown are: *P < 0.05, **P < 0.01, and ***P < 0.001, two way ANOVA with Bonferonni post-test adjustment. (C) Confocal micrographs of 1 dpi caudal vein region in the TgBAC(tnfa:GFP)pd1028 transgenic line. Phenol red (PR) and dominant active Hif-1α (DA1) injected larvae were treated with DMSO or NS398 (Cox-2 inhibitor) in the presence or absence of endogenous 15-keto prostaglandin E2 (15K). All larvae are PVP injected. P-values shown are: *P < 0.05, **P < 0.01, and ***P < 0.001, two way ANOVA with Bonferonni post-test adjustment. (D) Corrected fluorescence intensity levels of tnfa:GFP in larvae in (C). Mean ± SEM, n = 24 cells from four embryos accumulated from two independent experiments. (E) Confocal micrographs of 1 dpi caudal vein region in the TgBAC(tnfa:GFP)pd1028 transgenic line. Phenol red (PR) and dominant active Hif-1α (DA1) injected larvae were treated with DMSO or AH2348 (EP4 inhibitor). All larvae are PVP injected. (F) Corrected fluorescence intensity levels of tnfa:GFP in larvae in (E). Mean ± SEM, n = 72 cells from 12 embryos accumulated from three independent experiments. P-values shown are: *P < 0.05, **P < 0.01, and ***P < 0.001, two way ANOVA with Bonferonni post-test adjustment.
PGE2 signals via prostanoid receptors. In human and zebrafish there are four different receptors, EP1-4 (44). Of these, EP4 is the best characterized in immune cells and is expressed on human macrophages. When EP4 is blocked, macrophage pro-inflammatory cytokine release is decreased (45). We therefore tested a previously published EP4 antagonist AH23848 (36). We first demonstrated that AH23848 did not activate Hif-1α transcription using the Tg(phd3:GFP) hypoxia reporter line (Figure S6). The increased level of phd3:GFP induced by hydroxylase inhibitors was also not altered by AH23484 treatment, demonstrating that Hif-1α transcriptional potential is not affected by EP4 inhibition (Figure S8). Addition of AH23848 abrogated dominant active Hif-1α-induced tnfa:GFP, indicating that Hif-1α stabilization acts through PGE2 signaling via EP4 to upregulate Tnfa expression (Figures 5E,F).
Control of macrophage function during homeostasis and disease are critical for maintaining healthy tissues and must integrate changes in the local microenvironment with cytokine signaling. Understanding of the signaling pathways that link microenvironment with macrophage phenotypic outcomes may identify novel therapeutic avenues for control of macrophages during disease.
Here, we show that a disease relevant microenvironmental cue, hypoxia signaling via Hif-1α, upregulates macrophage tnfa expression in a cyclooxygenase dependent. TNF upregulation by hypoxia has been previously demonstrated in a range of mammalian cells, both directly, through HIF responsive elements (HREs) in its promotor region, and indirectly (46–49). Here, we addressed hypoxia-induced tnfa levels using promoter driven GFP expression in two zebrafish transgenic lines as a proxy for endogenous tnfa expression (18, 20). Changes in endogenous tnfa expression on a wholebody-level after Hif-1α stabilization were not observable by qPCR (data not shown), presumably because the changes in transcription at the macrophage level are too small to be detected using these methods due to macrophages representing a small proportion of the zebrafish larvae, a challenge that was previously reported in wholebody detection of another pro-inflammatory cytokine, il-1β (37). These promoter driven tnfa transgenic lines have therefore opened up opportunities to study tnfa induction in situ in the in vivo zebrafish. We observed increased tnfa expression with stabilized Hif-1α, demonstrating that hypoxia signaling alone can lead to a change of macrophage phenotype, consistent with our previous observations with il-1β expression, suggestive of a pro-inflammatory response (37). Our findings are also in line with HIF-1α stabilization being important for the production of TNF and other pro-inflammatory molecules, such as nitric oxide (NO), leading to host-protective effects against group A Streptococcus in in vivo murine models (50). Not only could tnfa activation be achieved by genetic stabilization of Hif-1α, but also using hypoxia mimetics (hydroxylase inhibitors) and physiological hypoxia, that stabilize endogenous Hif-1α. These findings indicate that the Hif-1α-Tnfa axis is targetable by pharmaceuticals and could be druggable during disease.
A similar tnfa induction was induced by infection and inflammation in our in vivo zebrafish models. It has been widely demonstrated that Tnfa is required for control of early Mm infection, with perturbation of Tnfa signaling leading to high infectious burdens (51). While Hif-1α-induced tnfa was downregulated by cyclooxygenase inhibition, it was notable that the high level of tnfa expression after inflammation or infection was not affected by cyclooxygenase inhibition. These findings are consistent with the novel HIF/COX/TNF macrophage axis being independent of infection/inflammation-induced macrophage Tnfa. Mm-induced Tnfa is widely reported as being toll-like receptor (TLR) mediated, and our data indicate that Mm-induced levels of Tnfa are sufficient for control of bacteria in the presence of stabilized Hif-1α (28, 52). In the absence of the TLR adapter protein MyD88, bacterial induction of Tnfa is lost, demonstrating the central role of TLR pathways in post-infection Tnfa induction (28, 52–54). The best characterized exogenous TLR signal, LPS via TLR4, can induce Tnfa production via activation of the central pro-inflammatory regulator NFκB (54, 55). As cyclooxygenase inhibition did not alter either inflammation or infection driven tnfa it suggests that these complex damage/infection driven mechanisms of Tnfa induction are not dependent on the HIF/COX/TNF pathway. Our data demonstrate that Hif-1α stabilization can stimulate a macrophage tnfa producing response in the absence of a TLR stimulus, which may have important implications in sterile diseased tissue and infers an alternative macrophage tuning mechanism.
Hypoxic regulation of TNF via COX has previously been demonstrated in mammalian osteoblasts, however, little is known about this interaction (14). Here, we demonstrate that Hif-1α upregulation of macrophage tnfa is likely to be via the production of PGE2 signaling via the EP4 receptor. PGE2 is an inflammatory mediator that is known to affect macrophage polarization states during infection, and synergises with cytokines to amplify pro-inflammatory responses (41, 56). The degradation metabolite 15-keto-PGE2 did not rescue the loss of tnfa expression after cyclooxygenase inhibition, consistent with previous reports that 15-keto-PGE2 is unable to bind the prostaglandin E2 EP receptors, thus demonstrating a requirement for active PGE2 (57).
Here, we demonstrate that a macrophage HIF/COX/TNF pathway is found in human monocyte derived macrophages, indicating that this pathway may have conserved roles in macrophage homeostasis and disease, and opening up possibilities for pharmaceutical intervention to promote proinflammatory macrophage phenotypes in human disease. Our findings in human MDMs indicate that the HIF/COX/TNF pathway may, at least in part, be cell-autonomous to macrophages, however, this was not tested in vivo where it is likely that contributing signals from other celltypes, for example endothelial cells, may play important roles in macrophage phenotype outcomes (58).
Although the effect of the HIF/COX/TNF axis in early Mm infection was negligible, this pathway is likely to have important roles in other disease situations. Mycobacterial infection, along with many other bacterial pathogens, stimulates early TNF production, however some pathogens remain undetected by macrophages during early disease. An example of this is in Lyme disease, caused by Lyme borreliosis, where early low TNF production is associated with worse disease outcomes (59). Multiple infectious/inflammatory disease pathologies have hypoxia as a feature of the tissue microenvironment, concurrent with the presence of macrophages. For example, Hif-1α mediated TNF activation may be pertinent in later TB infection situations where granulomas have stabilized Hif-1α due to necrotic and hypoxic centers (60, 61). Hypoxia is a key hallmark of cancers with high levels of HIF-1α widely found in those that produce large tumors where the center is hypoxic (62). These tumors can also produce cyclooxygenase/PGE2, where these important immunomodulatory signals are required for cellular adaption to the tumor microenvironment (63). Macrophages play central roles in cancer-inflammation that could be exploited utilizing pharmaceutical control of this novel HIF/COX/TNF mechanism. Further investigation of the HIF/COX/TNF axis in models where hypoxia is a key hallmark of disease pathology is required to uncover the full therapeutic relevance of this potentially important novel macrophage pathway.
In conclusion, we have identified a novel mechanism for macrophage tnfa production via Hif-1α and cyclooxygenase that is found in zebrafish and humans. Importantly, this axis links a commonly found microenvironmental cue, hypoxia, to a macrophage cytokine, TNF. We provide strong evidence to show that this response acts via the cyclooxygenase/PGE2 arm of the arachidonic pathway. Due to the central roles of these modulators in disease microenvironments we anticipate that this HIF/COX/TNF pathway may have important implications in conditions such as inflammation, infection and cancer.
The datasets generated for this study are available on request to the corresponding author.
The studies involving human participants were reviewed and approved by the South Sheffield Research Ethics Committee (07/Q2305/7), University of Sheffield, Firth Court, Western Bank, S10 2TN. The healthy participants provided their written informed consent to participate in this study. Animal work was performed following UK law: Animal (Scientific Procedures) Act 1986, under Project License P1A4A7A5E. Ethical approval was granted by the University of Sheffield Local Ethical Review Panel, University of Sheffield, Firth Court, Western Bank, Sheffield, S10 2TN.
PE and AL conceived and designed the experiments, performed the experiments, and analyzed the data. PE wrote the paper.
AL and PE are funded by a Sir Henry Dale Fellowship jointly funded by the Wellcome Trust and the Royal Society (Grant Number 105570/Z/14/Z) held by PE.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank The Bateson Aquarium Team for fish care and the IICD Technical Team for practical assistance (University of Sheffield). We thank, Michel Bagnat (Duke University) for providing their TgBAC(tnfa:GFP)pd1028 transgenic line, Georges Lutfalla (Montpellier University) for providing the Tg(tnfa:eGFP-F)ump5Tg line, Annemarie Meijer (University of Leiden) for M. marinum strains. Thanks to David Dockrell (University of Edinburgh), Paul Collini (University of Sheffield), and Jon Kilby (University of Sheffield) for help with hMDM experiments. Thanks to Alison Condliffe and Benjamin Durham (University of Sheffield) for use of, and invaluable help with, their hypoxia hood. We thank Robert Evans (The Francis Crick, London) and Catherine Loynes (University of Sheffield) for helpful discussions and advice on prostaglandin modulation. Thanks to Simon Johnston, Hannah Isles, and Stephen Renshaw (University of Sheffield) for constructive comments on the manuscript. A version of this manuscript has been released as a Pre-Print at bioRxiv (64).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.02321/full#supplementary-material
LOX, lipoxygenase; BLTR, leukotriene receptor; COX, cyclooxygenase; DAMP, damage associated molecular pattern; DMOG, dimethyloxaloylglycine; DMSO, dimethyl sulfoxide; Dpf, day post-fertilization; Dpi, day post-infection; FBS, fetal bovine serum; GFP, green fluorescent protein; HEPES, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; HIF, hypoxia inducible factor; hMDM, human monocyte derived macrophage; Hpf, hour post-fertilization; Hpi, hour post-infection; Hpw, hour post-wound; HRE, HIF responsive element; IL, Interleukin; Mm, Mycobacterium marinum; NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells; PAMP, pathogen associated molecular pattern; PGE2, Prostaglandin E2; PMBC, peripheral blood mononuclear cell; TLR, toll-like receptor; TNF, tumor necrosis factor.
1. Benoit M, Desnues B, Mege J-L. Infections macrophage polarization in bacterial macrophage polarization in bacterial infections. J Immunol Ref J Immunol. (2008) 181:3733–9. doi: 10.4049/jimmunol.181.6.3733
2. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. (2000) 164:6166–73. doi: 10.4049/jimmunol.164.12.6166
3. Wiegertjes GF, Wentzel AS, Spaink HP, Elks PM, Fink IR. Polarization of immune responses in fish: the ‘macrophages first' point of view. Mol Immunol. (2016) 69:146–56. doi: 10.1016/j.molimm.2015.09.026
4. Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. (2013) 229:176–85. doi: 10.1002/path.4133
5. Peyssonnaux C, Cejudo-Martin P, Doedens A, Zinkernagel AS, Johnson RS, Nizet V. Cutting edge: essential role of hypoxia inducible factor-1alpha in development of lipopolysaccharide-induced sepsis. J Immunol. (2007) 178:7516–9. doi: 10.4049/jimmunol.178.12.7516
6. Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat Rev Immunol. (2009) 9:609–17. doi: 10.1038/nri2607
7. Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage M1-M2 polarization balance. Front Immunol. (2014) 5:614. doi: 10.3389/fimmu.2014.00614
8. Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, et al. HIF-1α is essential for myeloid cell-mediated inflammation. Cell. (2003) 112:645–57. doi: 10.1016/S0092-8674(03)00154-5
9. Norris PC, Reichart D, Dumlao DS, Glass CK, Dennis EA. Specificity of eicosanoid production depends on the TLR-4-stimulated macrophage phenotype. J Leukoc Biol. (2011) 90:563–74. doi: 10.1189/jlb.0311153
10. Gupta S, Maurya MR, Stephens DL, Dennis EA, Subramaniam S. An integrated model of eicosanoid metabolism and signaling based on lipidomics flux analysis. Biophys J. (2009) 96:4542–51. doi: 10.1016/j.bpj.2009.03.011
11. Kaidi A, Qualtrough D, Williams AC, Paraskeva C. Direct transcriptional up-regulation of cyclooxygenase-2 by hypoxia-inducible factor (HIF)-1 promotes colorectal tumor cell survival and enhances HIF-1 transcriptional activity during hypoxia. Cancer Res. (2006) 66:6683–91. doi: 10.1158/0008-5472.CAN-06-0425
12. Lee JJ, Natsuizaka M, Ohashi S, Wong GS, Takaoka M, Michaylira CZ, et al. Hypoxia activates the cyclooxygenase-2-prostaglandin E synthase axis. Carcinogenesis. (2010) 31:427–34. doi: 10.1093/carcin/bgp326
13. Zhao L, Wu Y, Xu Z, Wang H, Zhao Z, Li Y, et al. Involvement of COX-2/PGE2signalling in hypoxia-induced angiogenic response in endothelial cells. J Cell Mol Med. (2012) 16:1840–55. doi: 10.1111/j.1582-4934.2011.01479.x
14. Xing Y, Wang R, Chen D, Mao J, Shi R, Wu Z, et al. COX2 is involved in hypoxia-induced TNF-α expression in osteoblast. Sci Rep. (2015) 5:10020. doi: 10.1038/srep10020
15. Meijer AH. Protection and pathology in TB: learning from the zebrafish model. Semin Immunopathol. (2016) 38:261–73. doi: 10.1007/s00281-015-0522-4
16. Loynes CA, Martin JS, Robertson A, Trushell DM, Ingham PW, Whyte MK, et al. Pivotal advance: pharmacological manipulation of inflammation resolution during spontaneously resolving tissue neutrophilia in the zebrafish. J Leukoc Biol. (2010) 87:203–12. doi: 10.1189/jlb.0409255
17. Nusslein-Volhard C DR. Zebrafish: A Practical Approach. 1st ed. Oxford: Oxford University Press (2002).
18. Marjoram L, Alvers A, Deerhake ME, Bagwell J, Mankiewicz J, Cocchiaro JL, et al. Epigenetic control of intestinal barrier function and inflammation in zebrafish. Proc Natl Acad Sci USA. (2015) 112:2770–5. doi: 10.1073/pnas.1424089112
19. Bojarczuk A, Miller KA, Hotham R, Lewis A, Ogryzko NV, Kamuyango AA, et al. Cryptococcus neoformans intracellular proliferation and capsule size determines early macrophage control of infection. Sci Rep. (2016) 6:21489. doi: 10.1038/srep21489
20. Nguyen-Chi M, Laplace-Builhe B, Travnickova J, Luz-Crawford P, Tejedor G, Phan QT, et al. Identification of polarized macrophage subsets in zebrafish. Elife. (2015) 4:e07288. doi: 10.7554/eLife.07288
21. Hall C, Flores M V, Storm T, Crosier K, Crosier P. The zebrafish lysozyme C promoter drives myeloid-specific expression in transgenic fish. BMC Dev Biol. (2007) 7:42. doi: 10.1186/1471-213X-7-42
22. Santhakumar K, Judson EC, Elks PM, McKee S, Elworthy S, van Rooijen E, et al. A zebrafish model to study and therapeutically manipulate hypoxia signaling in tumorigenesis. Cancer Res. (2012) 72:4017–27. doi: 10.1158/0008-5472.CAN-11-31480008-5472.CAN-11-3148
23. Elks PM, van Eeden FJ, Dixon G, Wang X, Reyes-Aldasoro CC, Ingham PW, et al. Activation of hypoxia-inducible factor-1α (Hif-1 α) delays inflammation resolution by reducing neutrophil apoptosis and reverse migration in a zebrafish inflammation model. Blood. (2011) 118:712–22. doi: 10.1182/blood-2010-12-324186
24. van der Sar AM, Spaink HP, Zakrzewska A, Bitter W, Meijer AH. Specificity of the zebrafish host transcriptome response to acute and chronic mycobacterial infection and the role of innate and adaptive immune components. Mol Immunol. (2009) 46:2317–32. doi: 10.1016/j.molimm.2009.03.024
25. Cui C, Benard EL, Kanwal Z, Stockhammer OW, van der Vaart M, Zakrzewska A, et al. Infectious disease modeling and innate immune function in zebrafish embryos. Methods Cell Biol. (2011) 105:273–308. doi: 10.1016/B978-0-12-381320-6.00012-6
26. Benard EL, van der Sar AM, Ellett F, Lieschke GJ, Spaink HP, Meijer AH. Infection of zebrafish embryos with intracellular bacterial pathogens. J Vis Exp. (2012) 15:3781. doi: 10.3791/37813781
27. Elks PM, Brizee S, van der Vaart M, Walmsley SR, van Eeden FJ, Renshaw SA, et al. PLoS Pathogens: hypoxia inducible factor signaling modulates susceptibility to mycobacterial infection via a nitric oxide dependent mechanism. PLoS Pathog. (2013) 9:e1003789. doi: 10.1371/journal.ppat.1003789
28. Elks PM, Van Der Vaart M, Van Hensbergen V, Schutz E, Redd MJ, Murayama E, et al. Mycobacteria counteract a TLR-mediated nitrosative defense mechanism in a zebrafish infection model. PLoS ONE. (2014) 9:e100928. doi: 10.1371/journal.pone.0100928
29. Stoop EJ, Schipper T, Huber SK, Nezhinsky AE, Verbeek FJ, Gurcha SS, et al. Zebrafish embryo screen for mycobacterial genes involved in the initiation of granuloma formation reveals a newly identified ESX-1 component. Dis Model Mech. (2011) 4:526–36. doi: 10.1242/dmm.006676
30. Barrett TD, Palomino HL, Brondstetter TI, Kanelakis KC, Wu X, Haug PV, et al. Pharmacological characterization of 1-(5-Chloro-6-(trifluoromethoxy)-1H-benzoimidazol-2-yl)-1H-pyrazole-4-carboxylic Acid (JNJ-42041935), a potent and selective hypoxia-inducible factor prolyl hydroxylase inhibitor. Mol Pharmacol. (2011) 79:910–20. doi: 10.1124/mol.110.070508
31. Wu K, Zhou K, Wang Y, Zhou Y, Tian N, Wu Y, et al. Stabilization of HIF-1α by FG-4592 promotes functional recovery and neural protection in experimental spinal cord injury. Brain Res. (2016) 1632:19–26. doi: 10.1016/j.brainres.2015.12.017
32. Feng Y, Renshaw S, Martin P. Live imaging of tumor initiation in zebrafish larvae reveals a trophic role for leukocyte-derived PGE2. Curr Biol. (2012) 22:1253–9. doi: 10.1016/j.cub.2012.05.010
33. Tobin DM, Vary JC Jr., Ray JP, Walsh GS, Dunstan SJ, Bang ND, et al. The lta4h locus modulates susceptibility to mycobacterial infection in zebrafish and humans. Cell. (2010) 140:717–30. doi: 10.1016/j.cell.2010.02.013S0092-8674(10)00128-5
34. Kojo K, Ito Y, Eshima K, Nishizawa N, Ohkubo H, Yokomizo T, et al. BLT 1 signalling protects the liver against acetaminophen hepatotoxicity by preventing excessive accumulation of hepatic neutrophils. Sci Rep. (2016) 6:29650. doi: 10.1038/srep29650
35. Evans RJ, Pline K, Loynes CA, Needs S, Aldrovandi M, Tiefenbach J, et al. 15-keto-prostaglandin E2 activates host peroxisome proliferator-activated receptor gamma (PPAR-γ) to promote Cryptococcus neoformans growth during infection. PLoS Pathog. (2019) 15:e1007597. doi: 10.1371/journal.ppat.1007597
36. Loynes CA, Lee JA, Robertson AL, Steel MJG, Ellett F, Feng Y, et al. PGE2 production at sites of tissue injury promotes an anti-inflammatory neutrophil phenotype and determines the outcome of inflammation resolution in vivo. Sci Adv. (2018) 4:eaar8320. doi: 10.1126/sciadv.aar8320
37. Ogryzko NV, Lewis A, Wilson HL, Meijer AH, Renshaw SA, Elks PM. Hif-1α-induced expression of Il-1β protects against mycobacterial infection in zebrafish. J Immunol. (2019) 202:494–502. doi: 10.4049/jimmunol.1801139
38. Dockrell DH, Lee M, Lynch DH, Read RC. Immune-mediated phagocytosis and killing of Streptococcus pneumoniae are associated with direct and bystander macrophage apoptosis. J Infect Dis. (2001) 184:713–22. doi: 10.1086/323084
39. Ghezzi P, Dinarello CA, Bianchi M, Rosandich ME, Repine JE, White CW. Hypoxia increases production of interleukin-1 and tumor necrosis factor by human mononuclear cells. Cytokine. (1991) 3:189–94. doi: 10.1016/1043-4666(91)90015-6
40. Tobin DM, Roca FJ, Oh SF, McFarland R, Vickery TW, Ray JP, et al. Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell. (2012) 148:434–46. doi: 10.1016/j.cell.2011.12.023
41. Harizi H. The immunobiology of prostanoid receptor signaling in connecting innate and adaptive immunity. Biomed Res Int. (2013) 2013:683405. doi: 10.1155/2013/683405
42. Chen IJ, Hee SW, Liao CH, Lin SY, Su L, Shun CT, et al. Targeting the 15-keto-PGE2-PTGR2 axis modulates systemic inflammation and survival in experimental sepsis. Free Radic Biol Med. (2018) 115:113–26. doi: 10.1016/j.freeradbiomed.2017.11.016
43. Tai HH, Ensor CM, Tong M, Zhou H, Yan F. Prostaglandin catabolizing enzymes. Prostaglandins Other Lipid Mediat. (2002) 68–69:483–93. doi: 10.1016/S0090-6980(02)00050-3
44. Kalinski P. Regulation of immune responses by prostaglandin E 2. J Immunol. (2012) 188:21–8. doi: 10.4049/jimmunol.1101029
45. Ikegami R, Sugimoto Y, Segi E, Katsuyama M, Karahashi H, Amano F, et al. The expression of prostaglandin E receptors EP 2 and EP 4 and their different regulation by lipopolysaccharide in C3H/HeN peritoneal macrophages. J Immunol. (2001) 166:4689–96. doi: 10.4049/jimmunol.166.7.4689
46. Ghosh S, Paul A, Sen E. Tumor necrosis factor alpha-induced hypoxia-inducible factor 1α-β-catenin axis regulates major histocompatibility complex class I gene activation through chromatin remodeling. Mol Cell Biol. (2013) 33:2718–31. doi: 10.1128/MCB.01254-12
47. Charbonneau M, Harper K, Grondin F, Pelmus M, McDonald PP, Dubois CM. Hypoxia-inducible factor mediates hypoxic and tumor necrosis factor alpha-induced increases in tumor necrosis factor-alpha converting enzyme/ADAM17 expression by synovial cells. J Biol Chem. (2007) 282:33714–24. doi: 10.1074/jbc.M704041200
48. Imtiyaz HZ, Simon MC. Hypoxia-inducible factors as essential regulators of inflammation. Curr Top Microbiol Immunol. (2010) 345:105–20. doi: 10.1007/82-2010-74
49. Sandau KB, Zhou J, Kietzmann T, Brüne B. Regulation of the hypoxia-inducible factor 1α by the inflammatory mediators nitric oxide and tumor necrosis factor-α in contrast to desferroxamine and phenylarsine oxide. J Biol Chem. (2001) 276:39805–11. doi: 10.1074/jbc.M107689200
50. Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, et al. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest. (2005) 115:1806–15. doi: 10.1172/JCI23865
51. Clay H, Volkman HE, Ramakrishnan L. Tumor necrosis factor signaling mediates resistance to mycobacteria by inhibiting bacterial growth and macrophage death. Immunity. (2008) 29:283–94. doi: 10.1016/j.immuni.2008.06.011S1074-7613(08)00327-0
52. Basu J, Shin D-M, Jo E-K. Mycobacterial signaling through toll-like receptors. Front Cell Infect Microbiol. (2012) 2:145. doi: 10.3389/fcimb.2012.00145
53. van der Vaart M, van Soest JJ, Spaink HP, Meijer AH. Functional analysis of a zebrafish myd88 mutant identifies key transcriptional components of the innate immune system. Dis Model Mech. (2013) 6:841–54. doi: 10.1242/dmm.010843
54. Kissner TL, Cisney ED, Ulrich RG, Fernandez S, Saikh KU. Staphylococcal enterotoxin A induction of pro-inflammatory cytokines and lethality in mice is primarily dependent on MyD88. Immunology. (2010) 130:516–26. doi: 10.1111/j.1365-2567.2010.03249.x
55. Zuckerman SH, Evans GF, Guthrie L. Transcriptional and post-transcriptional mechanisms involved in the differential expression of LPS-induced IL-1 and TNF mRNA. Immunology. (1991) 73(4): 460–5.
56. Mancini AD, Di Battista JD. The cardinal role of the phospholipase A(2)/cyclooxygenase-2/prostaglandin E synthase/prostaglandin E(2) (PCPP) axis in inflammostasis. Inflamm Res. (2011) 60:1083–92. doi: 10.1007/s00011-011-0385-7
57. Coggins KG, Latour A, Nguyen MS, Audoly L, Coffman TM, Koller BH. Metabolism of PGE2by prostaglandin dehydrogenase is essential for remodeling the ductus arteriosus. Nat Med. (2002) 8:91–2. doi: 10.1038/nm0202-91
58. He H, Xu J, Warren CM, Duan D, Li X, Wu L, et al. Endothelial cells provide an instructive niche for the differentiation and functional polarization of M2-like macrophages. Blood. (2012) 120:3152–62. doi: 10.1182/blood-2012-04-422758
59. Widhe M, Grusell M, Ekerfelt C, Vrethem M, Forsberg P, Ernerudh J. Cytokines in lyme borreliosis: lack of early tumour necrosis factor-α and transforming growth factor-β1 responses are associated with chronic neuroborreliosis. Immunology. (2002) 107:46–55. doi: 10.1046/j.1365-2567.2002.01500.x
60. Duque-Correa MA, Kuhl AA, Rodriguez PC, Zedler U, Schommer-Leitner S, Rao M, et al. Macrophage arginase-1 controls bacterial growth and pathology in hypoxic tuberculosis granulomas. Proc Natl Acad Sci USA. (2014) 111:E4024–32. doi: 10.1073/pnas.1408839111
61. Qualls JE, Murray PJ. Immunometabolism within the tuberculosis granuloma: amino acids, hypoxia, and cellular respiration. Semin Immunopathol. (2016) 38:139–52. doi: 10.1007/s00281-015-0534-0
62. Ajdukovic J. HIF-1–a big chapter in the cancer tale. Exp Oncol. (2016) 38:9–12. doi: 10.31768/2312-8852.2016.38(1):9-12
63. Greenhough A, Smartt HJM, Moore AE, Roberts HR, Williams AC, Paraskeva C, et al. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis. (2009) 30:377–86. doi: 10.1093/carcin/bgp014
Keywords: hypoxia, TNF–tumour necrosis factor, COX–cyclooxygenase, zebrafish, prostaglandin, macrophage, in vivo, HIF–hypoxia inducible factor
Citation: Lewis A and Elks PM (2019) Hypoxia Induces Macrophage tnfa Expression via Cyclooxygenase and Prostaglandin E2 in vivo. Front. Immunol. 10:2321. doi: 10.3389/fimmu.2019.02321
Received: 17 May 2019; Accepted: 13 September 2019;
Published: 27 September 2019.
Edited by:
Alexandre Corthay, Oslo University Hospital, NorwayReviewed by:
Andreas Weigert, Goethe University Frankfurt, GermanyCopyright © 2019 Lewis and Elks. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Philip M. Elks, cC5lbGtzQHNoZWZmaWVsZC5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.