 Gloria B. Kim
Gloria B. Kim Kristen Hege
Kristen Hege James L. Riley
James L. Riley
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 27 September 2019
Sec. Viral Immunology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02310
This article is part of the Research TopicHIV and Cancer Immunotherapy: Similar Challenges and Converging ApproachesView all 14 articles
Re-directing T cells via chimeric antigen receptors (CARs) was first tested in HIV-infected individuals with limited success, but these pioneering studies laid the groundwork for the clinically successful CD19 CARs that were recently FDA approved. Now there is great interest in revisiting the concept of using CAR-expressing T cells as part of a strategy to cure HIV. Many lessons have been learned on how to best engineer T cells to cure cancer, but not all of these lessons apply when developing CARs to treat and cure HIV. This mini review will focus on how early CAR T cell studies in HIV paved the way for cancer CAR T cell therapy and how progress in cancer CAR therapy has and will continue to be instructive for the development of HIV CAR T cell therapy. Additionally, the unique challenges that must be overcome to develop a successful HIV CAR T cell therapy will be highlighted.
From a T cell perspective, controlling HIV replication and cancer growth share many of the same challenges: antigen escape, antigen persistence resulting in T cell exhaustion, and active mechanisms employed by both HIV and tumors to avoid T cell recognition and elimination. Thus, the use of CARs to redirect T cells toward both HIV and cancer as a means to bolster T cell control of these maladies was an attractive concept, which led to the preclinical studies using both HIV and cancer models. In the 1990s when antiretroviral therapy (ART) was in its infancy and not yet able to provide durable control of HIV replication, the rationale to treat HIV infection with CAR T cell therapy advanced more rapidly, and in this setting, the first CAR T cell trials were performed. These studies tested the ability of T cells expressing a major histocompatibility complex (MHC)-unrestricted chimeric receptor consisting of CD4, as the natural ligand of the HIV Envelope (Env) glycoprotein, and the CD3 zeta (ζ) chain (1) to suppress viral replication in HIV–infected individuals (2–4). While clinical success was not achieved with these early efforts in the just-emerging CAR T cell field, these efforts were not a “failure,” but in fact, successfully laid fundamental groundwork that enabled success using CAR T cells to treat CD19-expressing tumors. Several key observations and discoveries foundational to the overall field of CAR T cell therapy were made during the clinical investigation of CD4-ζ CAR T cells. For one, the field gained an appreciation that a combination of CAR-modified CD4 and CD8 T cells, rather than purified CD8 T cells alone, resulted in a marked improvement in CAR T cell persistence (3). This was ultimately confirmed by demonstration of >10 years of durable CD4-ζ CAR T cell detection in treated subjects (5). Additionally, these early studies demonstrated that rapid and reproducible CAR T cell manufacturing could be achieved both from uninfected and viremic HIV-infected subjects following 10-day culture incorporating T cell co-stimulation with anti-CD3 and anti-CD28 immuno-magnetic beads. This manufacturing process resulted in improved functional properties of CD4-ζ CAR T cells as well as stable and durable in vivo persistence (3–5). Moreover, evidence in randomized trials suggested modest anti-viral activity in HIV-infected subjects through demonstration of trends in reduction of blood- and gut-associated HIV reservoirs, and a reduction in transient viral rebound in plasma (or “blips”) in aviremic subjects (2, 4). Finally, these studies demonstrated a lack of immunogenicity of the fully human CD4-ζ construct and an absence of depletion of MHC class II expressing cells, suggesting that CD4-MHC class II interaction was not sufficient to trigger CAR activity. Of note, these early trials with CD4-ζ CAR T cells were performed with the first generation CAR constructs using gamma-retroviral vectors and including only the CD3-ζ cytoplasmic domain without the benefit of co-stimulatory molecules, such as CD28 or 4-1BB, included in successful modern CAR T cell trials. Additionally, these early HIV-specific CAR T cells were not protected from HIV infection, a risk that is further exacerbate by using CD4 as a retargeting domain. Recently, a CD4-based CAR that was re-engineered (see details below) to incorporate lessons learned from successful cancer targeting CARs (6), was shown to confer greater antiviral activity than widely-investigated broadly neutralizing antibody (BNAb) based CARs. This CAR coupled with agents to protect the CAR from HIV infection (7–10) has recently entered the clinic (NCT03617198) to determine whether these changes augment HIV CAR T cell activity and provide some durable control of HIV replication and/or reduce the latent reservoir. The evolution of CAR design is summarized in Table 1.

Table 1. Evolution of CARs used in HIV and cancer cell and gene therapy.
Persistence of antigen at high levels drives exhaustion of T cells, which limits the functional properties of T cells and is characterized by high expression of immune checkpoint (IC) molecules, such as programmed death-1 (PD-1), and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), ultimately hindering clearance of tumors and chronic infections (13–16). An advantage of CAR T cell therapy is that new, fully functional T cells can be redirected toward HIV or tumor antigens. Once re-infused, however, these CAR T cells are susceptible to becoming exhausted if they are unable to clear the targeted antigen in a timely manner. Thus, the reversal or prevention of T cell exhaustion may represent a mechanism whereby dysregulated immunity is prevented, allowing CAR T cells to have a longer therapeutic window to control either HIV replication or tumor cell growth.
Antibodies targeting ICs (e.g., PD-1, PD-L1 or programmed death-ligand 1, and CTLA-4) have shown clinical responses in multiple tumor types, including melanoma, renal cell carcinoma, non-small cell lung cancer (17), and bladder cancer (18). So far, there are six U.S. FDA-approved immune checkpoint inhibitors (ipilimumab, nivolumab, pembrolizumab, avelumab, atezolizumab, and durvalumab) and their objective response rates have ranged from 27% in melanoma patients, to 30% in non-small cell lung cancer patients, and 63% in Kaposi sarcoma patients (19). However, there have been significant immune-related toxicities, including onset of type 1 diabetes, colitis, and dermatological issues (20) that may represent an acceptable risk/benefit to advanced cancer patients, but may be unacceptable to HIV-infected individuals whose viral load is well-controlled by ART. Several clinical trials are currently underway to explore the effect of anti-PD-1 based therapies in HIV-infected individuals who also have tumors known to be responsive to PD-1 blockade (NCT03367754, NCT02408861) (19) and one trial is treating non-tumor bearing HIV-infected individuals (NCT03787095). It will be interesting to see if and, if so to what extent, anti- PD-1 therapies can re-invigorate the HIV-1 specific immune response and whether side effects of this anti-PD-1 therapy in this otherwise healthy population confer an overall benefit/risk sufficient to permit wider exploration in HIV Cure studies.
Furthermore, some studies show that PD-1 also contributes to the establishment and maintenance of HIV latency, so checkpoint blockade may be a promising approach to reverse latency (21). In order for the remaining hidden pool of virus to become recognized by HIV–specific T cells, it must be reactivated first and this could be accomplished by various latency reversing agents (LRAs) (e.g., histone deacetylase inhibitors (HDACis) and protein kinase C class drugs) (22). IC blockades could also function to reverse HIV latency through limiting inhibitory signals sent from IC molecules into cells harboring latent HIV. CTLA-4 blockade results in significant increases in plasma viremia and T cell activation (23). Thus, the combination of IC blockade coupled with HIV CAR T cell therapy may be an effective “shock and kill” (24) strategy.
If systemic checkpoint inhibitor approaches prove too toxic for routine use in HIV-infected individuals, specific targeting of checkpoint genes within HIV-specific CAR T cells via clustered regularly interspaced short palindromic repeats (CRISPR) or small hairpin RNA (shRNA) technologies may prove an effective and safe way to make HIV-specific CAR T cells exhaustion resistant because only the HIV CAR T cells will have their checkpoint genes disabled (25, 26). Here, cancer-based therapies are paving the way for HIV-specific therapies. A clinical trial using a CRISPR-based approach to disable PD-1 is currently underway (NCT03399448) to determine if this improves the anti-tumor efficacy of engineered New York esophageal squamous cell carcinoma 1 (NY-ESO-1), a cancer-testis antigen expressed in a wide range of tumor types -targeted T cells. If successful, this trial could establish sufficient safety and feasibility to warrant coupling HIV CARs with PD-1 CRISPRs. Other immune checkpoint inhibitors, such as those targeting T-cell immunoglobulin and mucin-domain containing-3 (Tim-3), lymphocyte-activation gene 3 (LAG-3), and T-cell immunoreceptor with Ig and ITIM domains (TIGIT), may also help enhance anti-HIV CAR T cell therapy by overcoming T cell exhaustion, possibly with a more acceptable safety profile (27–30).
Antigen escape and efforts to limit T cell recognition of targeted cells are major hurdles for effective T cell-based HIV and cancer control (13). Most common mechanisms of antigen escape in cancers are (1) the immune selection of cancer cells, which lack or mutate immunogenic tumor antigens or lose expression of the antigens targeted by CAR T cells, (2) the acquisition of defects or deficiencies in antigen presentation [e.g., loss of major histocompatibility (MHC) expression], or (3) deficits of antigen processing machinery (31–33). Multiple compelling studies suggest that aberrant signal transducer and activator of transcription 3 (STAT3) signaling plays a key role in facilitating tumor escape from immune detection by impairing antigen presentation and reducing production of immunostimulatory molecules (34). Thus, STAT3 inhibition in concert with other immunostimulatory agents, such as toll-like receptor (TLR) 3, TLR7, and TLR8 agonists like stimulator of interferon genes (STING) or retinoic acid inducible gene (RIG)-I, could provide promising combination immunotherapeutic strategies. Additionally, a variety of CD19 mutations and alternative splicing have been observed with development of acquired resistance of acute lymphocytic leukemia (ALL) to CD19 targeted CAR T cells (35). In this regard, CARs targeting distinct motifs on the tumor surface may be an effective strategy to prevent resistance through tumor escape. For example, Ruella et al. demonstrated that the combination of CD123-targeted and CD19-targeted CAR T cells prevented relapses caused by antigen loss in preclinical models (36). Another study used bispecific CARs that targeted both CD19 and CD20 in order to minimize antigen escape from CD19-negative leukemia. Those bispecific CAR T cells were able to eradicate heterogeneous populations of leukemic cells in NSG mice (37).
In the case of HIV, the virus has evolved features to escape from immune monitoring with quick selection for cytotoxic T lymphocytes (CTL) escape mutations prior to antiretroviral therapy (ART) due to an error prone reverse transcriptase (10). Additionally, the HIV-1 negative regulatory factor (Nef) protein modulates expression of MHC class I, CD28, and other proteins involved in immune recognition to evade CTLs (38–40). As a result, recent efforts have focused on introducing a potent engineered immune response designed to overcome HIV's escape mechanisms instead of solely relying on the endogenous immune response to control HIV replication in the absence of ART (41–43). One advantage of CARs to target HIV is that HIV Env expression on the cell surface is not affected by Nef; thus, CAR T cells may recognize HIV-infected cells better than natural HIV-specific T cells. HIV can rapidly escape from a single BNAb (44–46), and will likely escape from a CAR that uses a BNAb as its targeting domain, though those targeting the CD4 binding site seem to be more resistant to escape (47). However, like in cancer, bi- or multi-specific HIV CARs have been constructed and have demonstrated superior efficacy against several HIV-1 primary isolates in vitro, warranting further in vivo investigation (8, 10, 48, 49). Moreover, it is not clear whether use of BNAb is advantageous as a means to redirect T cells to HIV as BNAb binding relative to non-BNAb binding promotes Env internalization (50). Thus, in both HIV and cancer, loss of target recognition by CAR T cells via antigen escape is an issue, but through simultaneous targeting of multiple antigens or the targeting of biologically important functions such as HIV binding to CD4, this issue seems to be solvable.
Immune privileged sites are anatomical regions (CNS, testes, and eyes) in which the immune response is purposely attenuated, usually to protect sensitive tissue from immune-related, off-target damage. These immune sanctuaries are often used by HIV and some tumors to hide from the immune attack. To overcome these issues, recent preclinical studies have shown the antitumor efficacy and safety of intracranial administration of EGFRvIII, HER2, and IL13Rα2 redirected CAR T/NK cells. Brown et al. described a patient who received multiple infusions of IL13Rα2-CAR T cells over 220 days via infusions to the resected tumor cavity and the ventricular system (51, 52).
Immune privilege coupled with HIV latency is an even more daunting problem for T cell-based therapies targeting HIV. Recent data have highlighted the fact that the >99% of all HIV-infected CD4+ T cells are found outside the vasculature within secondary lymphoid organs (SLOs), gut, brain, lung, and other tissues (53). Immunologic clearance of these infected cells is thought to largely involve cytotoxic CD8+ T cells, specifically CD8+ T cells with a fully differentiated “CTL” phenotype (CCR7-CD62L-CD27-CD45RA+) (54–59). CTLs, however, do not bear the markers (CCR7 and CD62L) necessary to enter lymphoid tissue (60–63). Betts and colleagues recently demonstrated that peripheral blood CTLs are rarely found in HIV-infected lymph nodes, and instead lymph nodes are populated by HIV-specific CD8+ T cells with very limited cytotoxic function (64, 65). In addition, it has been demonstrated that intestinal mucosal tissue is similarly populated with CD8+ T cells that have limited cytotoxic function (66). HIV-infected CD4+ T follicular helper cells (TFH cells) in B cell follicles of lymphoid tissue are a major compartment for persistent virus replication during combination ART (cART) (67–69). Even though virus-specific CTLs have been detected in lymph nodes, they are largely absent from the B cell follicles because they lack expression of CXC-chemokine receptor 5 (CXCR5), which is responsible for the trafficking of cells into the B cell zone along a CXC-chemokine ligand 13 (CXCL13) concentration gradient (70, 71). Therefore, the lack of CXCR5 expression on virus-specific CTLs is one mechanism that promotes the persistence of infected CD4+ TFH cells within an immune-privileged site (72). On the other hand, increasing evidence suggests the existence of tissue-resident macrophages as HIV-1 reservoirs (73, 74). Allers et al. found that macrophages were significantly enriched in the gut of untreated HIV patients (75). This also corresponds with a decrease in blood monocytes and increased expression of gut homing receptors (e.g., chemokine receptor CCR9 and integrin α4β7) on those monocytes, suggesting that blood monocytes may be a major source of macrophages that infiltrate gut mucosa. It has been reported that α4β7 is able to bind HIV-1 Env protein gp120 and is 3-fold larger than CD4 receptor, allowing it to capture HIV efficiently (76). Lastly, it is unclear whether engineered T cells will be able to transverse the blood brain barrier in HIV-infected individuals in order to target the HIV reservoir hiding in the CNS (77).
Taken together, there are at least three major issues facing HIV CAR T cells: (1) Will the latent reservoir of HIV-infected cells express sufficient levels of the target antigen (e.g., HIV Env) to drive CAR T cell recognition after a latency reversal agent is used? (2) Will the HIV CAR T cell be able to traffic to the site where the HIV-infected cell is hiding? and (3) if it is expressing antigen and the HIV CAR T cell is able to recognize the infected cell, will the CAR T cell have the necessary machinery (perforin and granzyme) that may be lost as part of the T cell exhaustion program to kill the HIV-infected cell and eliminate the latent reservoir?
Unless employed to prevent tumor relapse or treat minimal residual disease, cancer-specific CAR T cells are generally infused when there is abundant target antigen available. CAR T cells that quickly recognize their target have an engraftment advantage (78, 79). Moreover, CAR T cell recognition and killing of target cells can result in massive expansion of CAR T cells. In one celebrated case, a single CAR T cell whose vector integrated into and disrupted the function of the Tet methylcytosine dioxygenase 2 (Tet2) gene preferentially expanded to >90% of all of the CAR T cells within the body and this clone was able to maintain durable control of the targeted leukemia (80), indicating that CAR T cells have massive expansion potential. Thus, for individuals with established tumors, it may be possible to infuse a small number of well-engineered T cells and let the body serve as the bioreactor to generate enough T cells to eradicate the targeted tumor. However, CAR T cells that enter a body without significant target antigen may massively contract with a small subset becoming memory T cells, similar to what happens in a natural T cell response once antigen is cleared. Initial studies (NCT03617198) propose to infuse HIV CAR T cells in individuals whose ART has fully suppressed viral replication. It is unclear how well these adoptively transferred T cells will engraft in the absence of high levels of target antigen; however, it is reassuring that first generation CAR T cells targeting CD4-ζ demonstrated brisk expansion and prolonged persistence following infusion into aviremic patients effectively managed with ART therapy (2). For approaches that attempt to block viral rebound once ART is removed, there needs to be a sufficient quantity of T cells present that are widely distributed throughout the body to recognize the vast majority of cells expressing HIV Env as soon as they emerge. Thus, strategies such as infusion of very high numbers of T cells or vaccination approaches that maintain high levels of HIV-specific CAR T cells in the presence of minimal antigen may be required for HIV-specific CAR T cells to be used as part an HIV cure strategy (68).
The search for a CAR target that uniquely recognizes a tumor has proven very challenging. Currently, targets fall into two categories: (1) those with acceptable on-target/off-tumor toxicity, i.e., loss of “expendable” tissue such as B cells in the case of CART-19 therapies or (2) targets highly expressed on tumors and weakly expressed on a limited set of healthy cells, which may allow the CAR T cells to preferentially kill tumor with minimal effects on healthy cells. On-target/off-tumor recognition of CAR T cells has been observed in a variety of organ systems, including gastrointestinal, hematologic, and pulmonary (81). A fatal example of on target/off tumor CAR T cell recognition was observed with the cancer-associated antigen HER-2/neu. Rapid respiratory failure, multi-organ dysfunction, and subsequent death was attributed to reactivity against pulmonary tissue expression of HER-2/neu (82). Fortunately, for HIV CAR T cell therapy, HIV is non-self and thus highly specific agents can be developed that are unlikely to cross-react with human tissue. However, while HIV can be uniquely targeted, there are some challenges: (1) only the HIV Env protein is expressed on the cell surface after latency reversal, making it the only target suitable for CAR T cell therapy, and thus limiting some combinatorial approaches that may improve the efficacy and/or safety; (2) extensive sequence diversity within Env making it challenging to find antibody-based targeting agents that can bind all strains of HIV. Consequently, the natural HIV ligand, CD4, is attractive for use in a CAR construct, because HIV escape from binding to CD4, would likely result in a virus with greatly reduced fitness; (3) HIV Env expression levels are not fixed as in most cancer targets. Rather, the number of HIV Env targets on the cell surface increases over time as HIV replicates within the cell. However, the best chance for HIV CAR T cells to control HIV replication is to recognize and kill HIV-infected targets as soon as possible after infection when there is minimal HIV Env on the cell surface in order to limit the spread of the virus. Thus, CAR constructs that can redirect T cells to recognize minute levels of HIV Env on the cell surface will likely be very successful to limit HIV spread. This race between the CAR T cell to recognize HIV and HIV's effort to infect new cells has no clear parallel to cancer CAR T cells. It will therefore be interesting to see how this difference impacts the ability of HIV CAR T cells to control HIV replication in HIV-infected individuals.
Additionally, whereas tumors create hostile environments for T cells to function (83), HIV actively infects and kills T cells. While CD4 is a necessary binding receptor for most HIV strains, CD8 T cells can temporally express CD4 after T cell activation permits making both CD4 and CD8 HIV-specific CAR T cells susceptible to infection (6, 84, 85). For these reasons, HIV-specific CAR T cells will need to be protected from HIV infection. A variety of strategies exist including chemokine co-receptor disruption and fusion inhibitors that provide robust protection of T cells from infection (41). The only challenge in these strategies is the additional engineering that is required during the T cell manufacturing process.
Current cancer treatments such as chemotherapy, surgery, and/or radiation, have significant side effects and in most cases low rates of cure in advanced disease settings. CAR T therapies are currently being explored in patients with advanced/refractory malignancies and are FDA approved in chemotherapy refractory leukemia and lymphoma. Clinical success and FDA approval for Sipuleucel-T (Provenge), a dendritic cell-based therapeutic vaccine, was based on ~4 month increase in survival time for prostate cancer patients. In contrast, ART is nearly universally successful in compliant individuals with access to healthcare, and those individuals whose virus remains undetected due to ART have lifespans approaching those of non-HIV infected individuals (86). Thus, both commercial and clinical success for cancer therapies is measured by increasing mean survival time whereas for HIV, only a cure, whether functional or sterilizing (87, 88), is considered a success. Given that only two people have been cured of HIV infection (89, 90), having a lifetime cure as the only measure of success is quite a high bar. This is why analytical treatment interruptions (ATIs) are crucial to advance the HIV CAR T cell field. Here, individuals involved in an IRB approved clinical trial voluntary stop taking ART after receiving an experimental agent and the time to viral rebound is measured. Most individuals rebound within 2–4 weeks; therefore, individuals who are part of an interventional study that is able to limit the virus from replicating significantly longer provide evidence that the experimental therapy is having some effect. As the field matures and many approaches are studied, one can then analyze ATI data to propose combination trials to determine whether further delays in viral rebound occur. This combinatorial, iterative approach is likely the best chance we have to develop an effective and safe HIV Cure regimen. To date, carefully monitored ATIs have not resulted in ART escape or increased the viral reservoir (91–93), suggesting that there are no long term adverse outcomes for individuals participating in clinical trials that have ATIs (94).
The development of cancer and HIV CAR T cell therapy has a long, intertwined, and symbiotic relationship (95), and this relationship is highlighted in Table 2. Exactly how did success with cancer CAR T cell therapy inform the design and implementation of HIV CAR T cell therapy? The initial CD4-ζ CAR was housed in a murine gammaretroviral vector, contained the CD4 transmembrane domain, lacked costimulatory domains, and was driven by the phosphoglycerate kinase (PGK) promoter (1, 112). In a side-by-side, step-by-step study, Leibman et al. compared this first generation HIV CAR with the vector design of CARs that achieved FDA approval for CD19-expressing tumors (6). Surprisingly, the choice of vector delivery made a huge difference in CAR expression and this translated into greater control of HIV replication. Substituting the EF-1a promoter resulted in both more stable and higher CD4 CAR expression. Replacing the CD4 transmembrane domain with the CD8 hinge region resulted in slightly less expression, but rendered the HIV CAR T cells less susceptible to infection and improved the overall efficacy of these T cells. Lastly, endowing the CD4 CAR with 4-1BB costimulation promoted both the survival and expansion in vivo as previously observed in tumor models (6, 113).
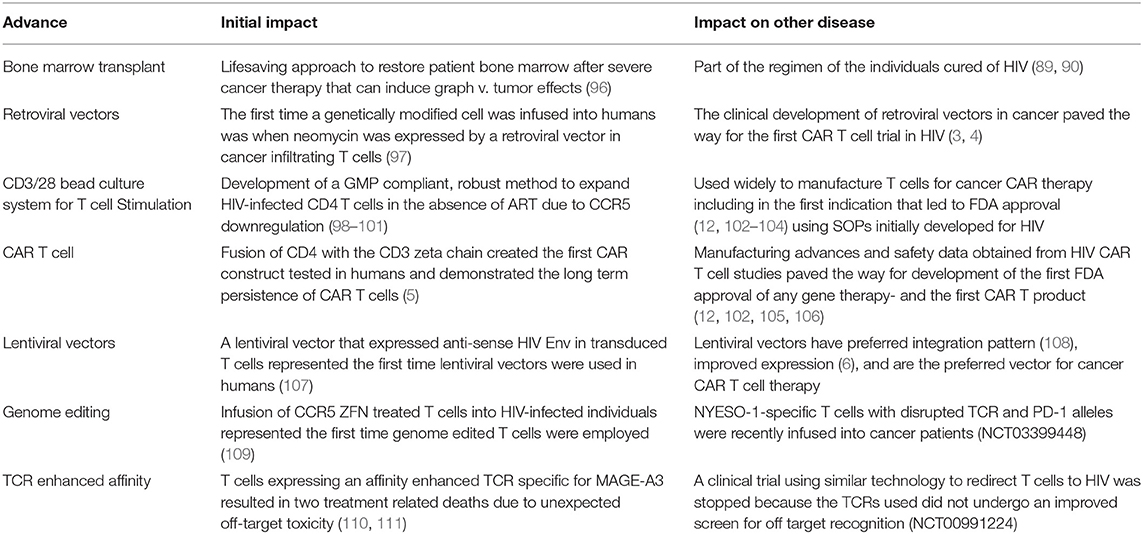
Table 2. Synergy between HIV and cancer cell and gene therapy.
In a convergence of fields, much attention is now focused on where a CAR vector integrates. Pioneering studies by the Bushman lab demonstrated that HIV (and HIV-based vectors) prefers to integrate in coding regions, whereas murine gammaretroviruses target promoter regions (108, 114). More recently, the site of HIV integration has been shown to play a role in whether T cells will become part of the latent reservoir (115), suggesting that the site of integration can impact a T cell's long term persistence and ability to homeostatically expand. Using approaches to study how HIV integrates, Fraietta et al. uncovered how a CD19 CAR vector fortuitously integrated into the TET2 locus, and this integration resulted in a central memory-like T cell phenotype with an incredible ability to expand and function (116). As genome engineering becomes more effective, safer and less expensive (117), one can imagine that it will be possible to specifically insert a CAR vector into a precise spot in the genome to provide a functional advantage or survival benefit to either HIV or cancer CAR T cells.
As mentioned in the beginning, the field of T cell manufacturing was in its infancy when the first HIV CAR T cell therapy trials were performed. The field has matured considerably, but there is much more to learn in order to improve how T cells are produced for use in adoptive T cell applications. Cancer CAR T therapy has seen a strong correlation in how well T cells expand ex vivo with their in vivo function and persistence (118). Additionally, it has been demonstrated that changes in T cell manufacturing such as expanding T cells in the absence of human serum (119) improves the in vivo efficacy of CAR T cells. Here, developers of cancer CAR and HIV CAR can support each other as many of the developments in T cell manufacturing are likely to benefit both fields. One possible difference is that for HIV CAR T therapy large quantities of HIV CAR T cells may be required to have enough effectors on hand to prevent viral rebound after ART removal since there is minimal antigen present to induce in vivo CAR T cell expansion. In contrast, for cancer CAR T cell therapy, infusion of less CAR T cells may be safer, less expensive and just as effective so the manufacturing for these two therapies are reasonably similar now but they may diverge considerably once we learn more about what is required to obtain therapeutic responses. Lastly, HIV-infected individuals are currently excluded from receiving CAR T therapy in part because the commercial manufacturers have not developed a process by which HIV-infected T cells can be GMP manufactured. Perhaps one of the last gifts HIV CAR therapy can give to cancer CAR therapy is to share the best practices by which HIV CAR T cells are manufactured using T cells from HIV-infected individuals so that HIV-infected individuals can benefit from this life saving, cancer CAR T cell technology.
GK, KH, and JR co-wrote and edited the manuscript.
Support for these studies was provided by the NIH (T32CA009140, U19AI117950, and UM1AI126620).
KH is an employee of Celgene and holds equity in Celgene, Mersana Therapeutics, and Arcus Biosciences. None of these companies have active HIV therapeutics programs. JR holds equity in Tmunity Therapeutics.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank members of the Riley Lab and Julie Jadlowsky for their thoughtful comments and support; Carl June, Richard Carroll, and Bruce Levine for their long term partnership and support; and members of the Center of Cellular Immunotherapies at the University of Pennsylvania for providing an exciting environment to study CAR T cell therapy.
1. Roberts MR, Qin L, Zhang D, Smith DH, Tran AC, Dull TJ, et al. Targeting of human immunodeficiency virus-infected cells by CD8+ T lymphocytes armed with universal T-cell receptors. Blood. (1994) 84:2878–89.
2. Deeks SG, Wagner B, Anton PA, Mitsuyasu RT, Scadden DT, Huang C, et al. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol Ther. (2002) 5:788–97. doi: 10.1006/mthe.2002.0611
3. Walker RE, Bechtel CM, Natarajan V, Baseler M, Hege KM, Metcalf JA, et al. Long-term in vivo survival of receptor-modified syngeneic T cells in patients with human immunodeficiency virus infection. Blood. (2000) 96:467–74.
4. Mitsuyasu RT, Anton PA, Deeks SG, Scadden DT, Connick E, Downs MT, et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood. (2000) 96:785–93.
5. Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, Hege KM, et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med. (2012) 4:132ra53. doi: 10.1126/scitranslmed.3003761
6. Leibman RS, Richardson MW, Ellebrecht CT, Maldini CR, Glover JA, Secreto AJ, et al. Supraphysiologic control over HIV-1 replication mediated by CD8 T cells expressing a re-engineered CD4-based chimeric antigen receptor. PLoS Pathog. (2017) 13:e1006613. doi: 10.1371/journal.ppat.1006613
7. Zhen A, Kamata M, Rezek V, Rick J, Levin B, Kasparian S, et al. HIV-specific immunity derived from chimeric antigen receptor-engineered stem cells. Mol Ther. (2015) 23:1358–67. doi: 10.1038/mt.2015.102
8. Liu L, Patel B, Ghanem MH, Bundoc V, Zheng Z, Morgan RA, et al. Novel CD4-based bispecific chimeric antigen receptor designed for enhanced anti-HIV potency and absence of HIV entry receptor activity. J Virol. (2015) 89:6685–94. doi: 10.1128/JVI.00474-15
9. MacLean AG, Walker E, Sahu GK, Skowron G, Marx P, von Laer D, et al. A novel real-time CTL assay to measure designer T-cell function against HIV Env(+) cells. J Med Primatol. (2014) 43:341–8. doi: 10.1111/jmp.12137
10. Hale M, Mesojednik T, Romano Ibarra GS, Sahni J, Bernard A, Sommer K, et al. Engineering HIV-resistant, anti-HIV chimeric antigen receptor T cells. Mol Ther. (2017) 25:570–9. doi: 10.1016/j.ymthe.2016.12.023
11. Yang OO, Tran AC, Kalams SA, Johnson RP, Roberts MR, Walker BD. Lysis of HIV-1-infected cells and inhibition of viral replication by universal receptor T cells. Proc Natl Acad Sci USA. (1997) 94:11478–83. doi: 10.1073/pnas.94.21.11478
12. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. (2013) 368:1509–18. doi: 10.1056/NEJMoa1215134
13. Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J Clin Invest. (2011) 121:2350–60. doi: 10.1172/JCI46102
14. Zajac A, Blattman J, Murali-Krishna K, Sourdive D, Suresh M, Altman J, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. (1998) 188:2205–13. doi: 10.1084/jem.188.12.2205
15. Gallimore A, Glithero A, Godkin A, Tissot AC, Pluckthun A, Elliott T, et al. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I peptide complexes. J Exp Med. (1998) 187:1383–93. doi: 10.1084/jem.187.9.1383
16. Virgin HW, Wherry EJ, Ahmed R, Redefining chronic viral infection. Cell. (2009) 138:30–50. doi: 10.1016/j.cell.2009.06.036
17. Brahmer JR, Tykodi SS, Chow LQM, Hwu W-J, Topalian SL, Hwu P, et al. Activity of anti–PD-L1 antibody in patients with advanced cancer. New Engl J Med. (2012) 366:2455–65. doi: 10.1056/NEJMoa1200694
18. Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. (2014) 515:558–62. doi: 10.1038/nature13904
19. Cook MR, Kim C. Safety and efficacy of immune checkpoint inhibitor therapy in patients with HIV infection and advanced-stage cancer: a systematic review. JAMA Oncol. (2019) 5:1049–54. doi: 10.1001/jamaoncol.2018.6737
20. Riley JL. Combination checkpoint blockade–taking melanoma immunotherapy to the next level. N Engl J Med. (2013) 369:187–9. doi: 10.1056/NEJMe1305484
21. Evans VA, van der Sluis RM, Solomon A, Dantanarayana A, McNeil C, Garsia R, et al. Programmed cell death-1 contributes to the establishment and maintenance of HIV-1 latency. Aids. (2018) 32:1491–7. doi: 10.1097/QAD.0000000000001849
22. Rasmussen TA, Tolstrup M, Brinkmann CR, Olesen R, Erikstrup C, Solomon A, et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. Lancet HIV. (2014) 1:e13–21. doi: 10.1016/S2352-3018(14)70014-1
23. Cecchinato V, Tryniszewska E, Ma ZM, Vaccari M, Boasso A, Tsai WP, et al. Immune activation driven by CTLA-4 blockade augments viral replication at mucosal sites in simian immunodeficiency virus infection. J Immunol. (2008) 180:5439–47. doi: 10.4049/jimmunol.180.8.5439
24. Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang HC, et al. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity. (2012) 36:491–501. doi: 10.1016/j.immuni.2012.01.014
25. Ren J, Zhang X, Liu X, Fang C, Jiang S, June CH, et al. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget. (2017) 8:17002–11. doi: 10.18632/oncotarget.15218
26. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Investig. (2016) 126:3130–44. doi: 10.1172/JCI83092
27. Chew GM, Fujita T, Webb GM, Burwitz BJ, Wu HL, Reed JS, et al. TIGIT marks exhausted T cells, correlates with disease progression, and serves as a target for immune restoration in HIV and SIV infection. PLoS Pathog. (2016) 12:e1005349. doi: 10.1371/journal.ppat.1005349
28. Fujita T, Burwitz BJ, Chew GM, Reed JS, Pathak R, Seger E, et al. Expansion of dysfunctional Tim-3-expressing effector memory CD8+ T cells during simian immunodeficiency virus infection in rhesus macaques. J Immunol. (2014) 193:5576–83. doi: 10.4049/jimmunol.1400961
29. Hoffmann M, Pantazis N, Martin GE, Hickling S, Hurst J, Meyerowitz J, et al. Exhaustion of activated CD8 T cells predicts disease progression in primary HIV-1 infection. PLoS Pathog. (2016) 12:e1005661. doi: 10.1371/journal.ppat.1005661
30. Tian X, Zhang A, Qiu C, Wang W, Yang Y, Qiu C, et al. The upregulation of LAG-3 on T cells defines a subpopulation with functional exhaustion and correlates with disease progression in HIV-infected subjects. J Immunol. (2015) 194:3873–82. doi: 10.4049/jimmunol.1402176
31. Iorgulescu JB, Braun D, Oliveira G, Keskin DB, Wu CJ. Acquired mechanisms of immune escape in cancer following immunotherapy. Genome Med. (2018) 10:87. doi: 10.1186/s13073-018-0598-2
32. Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Canc Res. (2015) 21:687–92. doi: 10.1158/1078-0432.CCR-14-1860
33. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. (2011) 331:1565–70. doi: 10.1126/science.1203486
34. Su YL, Banerjee S, White SV, Kortylewski M. STAT3 in tumor-associated myeloid cells: multitasking to disrupt immunity. Int J Mol Sci. (2018) 19:1803. doi: 10.3390/ijms19061803
35. Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. (2015) 5:1282–95. doi: 10.1158/2159-8290.CD-15-1020
36. Ruella M, Barrett DM, Kenderian SS, Shestova O, Hofmann TJ, Perazzelli J, et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Invest. (2016) 126:3814–26. doi: 10.1172/JCI87366
37. Zah E, Lin MY, Silva-Benedict A, Jensen MC, Chen YY. T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol Res. (2016) 4:639–41. doi: 10.1158/2326-6066.CIR-16-0108
38. Landi A, Iannucci V, Nuffel AV, Meuwissen P, Verhasselt B. One protein to rule them all: modulation of cell surface receptors and molecules by HIV Nef. Curr HIV Res. (2011) 9:496–504. doi: 10.2174/157016211798842116
39. Schwartz O, Marechal V, LeGall S, Lemonnier F, Heard JM. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat Med. (1996) 2:338–42. doi: 10.1038/nm0396-338
40. Arhel NJ, Kirchhoff F. Implications of Nef: host cell interactions in viral persistence and progression to AIDS. Curr Top Microbiol Immunol. (2009) 339:147–75. doi: 10.1007/978-3-642-02175-6_8
41. Leibman RS, Riley JL. Engineering T cells to functionally cure HIV-1 infection. Mol Ther. (2015) 23:1149–59. doi: 10.1038/mt.2015.70
42. Kiem HP, Jerome KR, Deeks SG, McCune JM. Hematopoietic-stem-cell-based gene therapy for HIV disease. Cell Stem Cell. (2012) 10:137–47. doi: 10.1016/j.stem.2011.12.015
43. Hoxie JA, June CH, Novel cell and gene therapies for HIV. Cold Spring Harb Perspect Med. (2012) 2:a007179. doi: 10.1101/cshperspect.a007179
44. Bar KJ, Sneller MC, Harrison LJ, Justement JS, Overton ET, Petrone ME, et al. Effect of HIV antibody VRC01 on viral rebound after treatment interruption. N Engl J Med. (2016) 375:2037–50. doi: 10.1056/NEJMoa1608243
45. Scheid JF, Horwitz JA, Bar-On Y, Kreider EF, Lu CL, Lorenzi JC, et al. HIV-1 antibody 3BNC117 suppresses viral rebound in humans during treatment interruption. Nature. (2016) 535:556–60. doi: 10.1038/nature18929
46. Caskey M, Schoofs T, Gruell H, Settler A, Karagounis T, Kreider EF, et al. Antibody 10-1074 suppresses viremia in HIV-1-infected individuals. Nat Med. (2017) 23:185–91. doi: 10.1038/nm.4268
47. Wang CY, Wong WW, Tsai HC, Chen YH, Kuo BS, Lynn S, et al. Effect of anti-CD4 antibody UB-421 on HIV-1 rebound after treatment interruption. N Engl J Med. (2019) 380:1535–45. doi: 10.1056/NEJMoa1802264
48. Ghanem MH, Bolivar-Wagers S, Dey B, Hajduczki A, Vargas-Inchaustegui DA, Danielson DT, et al. Bispecific chimeric antigen receptors targeting the CD4 binding site and high-mannose Glycans of gp120 optimized for anti-human immunodeficiency virus potency and breadth with minimal immunogenicity. Cytotherapy. (2018) 20:407–19. doi: 10.1016/j.jcyt.2017.11.001
49. Ali A, Kitchen SG, Chen ISY, Ng HL, Zack JA, Yang OO. HIV-1-specific chimeric antigen receptors based on broadly neutralizing antibodies. J Virol. (2016) 90:6999–7006. doi: 10.1128/JVI.00805-16
50. Anand SP, Grover JR, Tolbert WD, Prevost J, Richard J, Ding S, et al. Antibody-induced internalization of HIV-1 env proteins limits surface expression of the closed conformation of Env. J Virol. (2019) 93:e00293–19. doi: 10.1128/JVI.00293-19
51. Brown CE, Badie B, Barish ME, Weng LH, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13R alpha 2-redirected chimeric antigen receptor CD8(+) T cells in patients with recurrent glioblastoma. Clin Cancer Res. (2015) 21:4062–72. doi: 10.1158/1078-0432.CCR-15-0428
52. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
53. Estes JD, Kityo C, Ssali F, Swainson L, Makamdop KN, Del Prete GQ, et al. Defining total-body AIDS-virus burden with implications for curative strategies. Nat Med. (2017) 23:1271–6. doi: 10.1038/nm.4411
54. Migueles SA, Laborico AC, Shupert WL, Sabbaghian MS, Rabin R, Hallahan CW, et al. HIV-specific CD8+ T cell proliferation is coupled to perforin expression and is maintained in nonprogressors. Nat Immunol. (2002) 3:1061–8. doi: 10.1038/ni845
55. Migueles SA, Osborne CM, Royce C, Compton AA, Joshi RP, Weeks KA, et al. Lytic granule loading of CD8+ T cells is required for HIV-infected cell elimination associated with immune control. Immunity. (2008) 29:1009–21. doi: 10.1016/j.immuni.2008.10.010
56. Hersperger AR, Martin JN, Shin LY, Sheth PM, Kovacs CM, Cosma GL, et al. Increased HIV-specific CD8+ T-cell cytotoxic potential in HIV elite controllers is associated with T-bet expression. Blood. (2011) 117:3799–808. doi: 10.1182/blood-2010-12-322727
57. Hersperger AR, Pereyra F, Nason M, Demers K, Sheth P, Shin LY, et al. Perforin expression directly ex vivo by HIV-specific CD8 T-cells is a correlate of HIV elite control. PLoS Pathog. (2010) 6:e1000917. doi: 10.1371/journal.ppat.1000917
58. Chattopadhyay PK, Betts MR, Price DA, Gostick E, Horton H, Roederer M, et al. The cytolytic enzymes granyzme A, granzyme B, and perforin: expression patterns, cell distribution, and their relationship to cell maturity and bright CD57 expression. J Leukoc Biol. (2009) 85:88–97. doi: 10.1189/jlb.0208107
59. Appay V, Nixon DF, Donahoe SM, Gillespie GM, Dong T, King A, et al. HIV-specific CD8(+) T cells produce antiviral cytokines but are impaired in cytolytic function. J Exp Med. (2000) 192:63–75. doi: 10.1084/jem.192.1.63
60. Sallusto F, Langenkamp A, Geginat J, Lanzavecchia A. Functional subsets of memory T cells identified by CCR7 expression. Curr Top Microbiol Immunol. (2000) 251:167–71. doi: 10.1007/978-3-642-57276-0_21
61. Picker LJ, Treer JR, Ferguson-Darnell B, Collins PA, Bergstresser PR, Terstappen LW. Control of lymphocyte recirculation in man. II. Differential regulation of the cutaneous lymphocyte-associated antigen, a tissue-selective homing receptor for skin-homing T cells. J Immunol. (1993) 150:1122–36.
62. Picker LJ, Treer JR, Ferguson-Darnell B, Collins PA, Buck D, Terstappen LW. Control of lymphocyte recirculation in man. I. Differential regulation of the peripheral lymph node homing receptor L-selectin on T cells during the virgin to memory cell transition. J Immunol. (1993) 150:1105–21.
63. Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GM, Papagno L, et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med. (2002) 8:379–85. doi: 10.1038/nm0402-379
64. Buggert M, Nguyen S, Salgado-Montes de Oca G, Bengsch B, Darko S, Ransier A, et al. Identification and characterization of HIV-specific resident memory CD8(+) T cells in human lymphoid tissue. Sci Immunol. (2018) 3:eaar4526. doi: 10.1126/sciimmunol.aar4526
65. Reuter MA, Del Rio Estrada PM, Buggert M, Petrovas C, Ferrando-Martinez S, Nguyen S, et al. HIV-specific CD8(+) T cells exhibit reduced and differentially regulated cytolytic activity in lymphoid tissue. Cell Rep. (2017) 21:3458–70. doi: 10.1016/j.celrep.2017.11.075
66. Kiniry BE, Hunt PW, Hecht FM, Somsouk M, Deeks SG, Shacklett BL. Differential expression of CD8(+) T cell cytotoxic effector molecules in blood and gastrointestinal mucosa in HIV-1 infection. J Immunol. (2018) 200:1876–88. doi: 10.4049/jimmunol.1701532
67. Banga R, Procopio FA, Noto A, Pollakis G, Cavassini M, Ohmiti K, et al. PD-1(+) and follicular helper T cells are responsible for persistent HIV-1 transcription in treated aviremic individuals. Nat Med. (2016) 22:754–61. doi: 10.1038/nm.4113
68. Maldini CR, Ellis GI, Riley JL. CAR T cells for infection, autoimmunity and allotransplantation. Nat Rev Immunol. (2018) 18:605–16. doi: 10.1038/s41577-018-0042-2
69. Perreau M, Savoye AL, De Crignis E, Corpataux JM, Cubas R, Haddad EK, et al. Follicular helper T cells serve as the major CD4 T cell compartment for HIV-1 infection, replication, and production. J Exp Med. (2013) 210:143–56. doi: 10.1084/jem.20121932
70. Ansel KM, Ngo VN, Hyman PL, Luther SA, Forster R, Sedgwick JD, et al. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature. (2000) 406:309–14. doi: 10.1038/35018581
71. Forster R, Mattis AE, Kremmer E, Wolf E, Brem G, Lipp M. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell. (1996) 87:1037–47. doi: 10.1016/S0092-8674(00)81798-5
72. Fukazawa Y, Lum R, Okoye AA, Park H, Matsuda K, Bae JY, et al. B cell follicle sanctuary permits persistent productive simian immunodeficiency virus infection in elite controllers. Nat Med. (2015) 21:132–9. doi: 10.1038/nm.3781
73. Bradley CA. Urethral macrophages - a new HIV-1 reservoir. Nat Rev Urol. (2019) 16:205. doi: 10.1038/s41585-019-0158-6
74. Ganor Y, Real F, Sennepin A, Dutertre CA, Prevedel L, Xu L, et al. HIV-1 reservoirs in urethral macrophages of patients under suppressive antiretroviral therapy. Nat Microbiol. (2019) 4:633–44. doi: 10.1038/s41564-018-0335-z
75. Allers K, Fehr M, Conrad K, Epple HJ, Schurmann D, Geelhaar-Karsch A, et al. Macrophages accumulate in the gut mucosa of untreated HIV-infected patients. J Infect Dis. (2014) 209:739–48. doi: 10.1093/infdis/jit547
76. Cicala C, Arthos J, Fauci AS. HIV-1 envelope, integrins and co-receptor use in mucosal transmission of HIV. J Transl Med. (2011) 9(Suppl 1):S8. doi: 10.1186/1479-5876-9-S1-S2
77. Marban C, Forouzanfar F, Ait-Ammar A, Fahmi F, El Mekdad H, Daouad F, et al. Targeting the brain reservoirs: toward an HIV cure. Front Immunol. (2016) 7:397. doi: 10.3389/fimmu.2016.00397
78. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4(+): CD8(+) composition in adult B cell ALL patients. J Clin Investig. (2016) 126:2123–38. doi: 10.1172/JCI85309
79. Milone MC, Bhoj VG. The pharmacology of T cell therapies. Mol Ther Methods Clin Dev. (2018) 8:210–21. doi: 10.1016/j.omtm.2018.01.010
80. Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature. (2018) 558:307–312. doi: 10.1038/s41586-018-0178-z
81. Guedan S, Ruella M, June CH. Emerging cellular therapies for cancer. Annu Rev Immunol. (2019) 37:145–71. doi: 10.1146/annurev-immunol-042718-041407
82. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. (2010) 18:843–51. doi: 10.1038/mt.2010.24
83. Ecker C, Riley JL. Translating In Vitro T cell metabolic findings to in vivo tumor models of nutrient competition. Cell Metab. (2018) 28:190–5. doi: 10.1016/j.cmet.2018.07.009
84. Kitchen SG, Korin YD, Roth MD, Landay A, Zack JA. Costimulation of naive CD8(+) lymphocytes induces CD4 expression and allows human immunodeficiency virus type 1 infection. J Virol. (1998) 72:9054–60.
85. Yang LP, Riley JL, Carroll RG, June CH, Hoxie J, Patterson BK, et al. Productive infection of neonatal CD8+ T lymphocytes by HIV-1. J Exp Med. (1998) 187:1139–44. doi: 10.1084/jem.187.7.1139
86. Teeraananchai S, Kerr SJ, Amin J, Ruxrungtham K, Law MG. Life expectancy of HIV-positive people after starting combination antiretroviral therapy: a meta-analysis. HIV Med. (2017) 18:256–66. doi: 10.1111/hiv.12421
87. Badia R, Ballana E, Castellvi M, Garcia-Vidal E, Pujantell M, Clotet B, et al. CD32 expression is associated to T-cell activation and is not a marker of the HIV-1 reservoir. Nat Commun. (2018) 9:2739. doi: 10.1038/s41467-018-05157-w
88. Abdel-Mohsen M, Kuri-Cervantes L, Grau-Exposito J, Spivak AM, Nell RA, Tomescu C, et al. CD32 is expressed on cells with transcriptionally active HIV but does not enrich for HIV DNA in resting T cells. Sci Transl Med. (2018) 10:eaar6759. doi: 10.1126/scitranslmed.aar6759
89. Gupta RK, Abdul-Jawad S, McCoy LE, Mok HP, Peppa D, Salgado M, et al. HIV-1 remission following CCR5Δ32/Δ32 haematopoietic stem-cell transplantation. Nature. (2019) 568:244–8. doi: 10.1038/s41586-019-1027-4
90. Hutter G, Nowak D, Mossner M, Ganepola S, Mussig A, Allers K, et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med. (2009) 360:692–8. doi: 10.1056/NEJMoa0802905
91. Salantes DB, Zheng Y, Mampe F, Srivastava T, Beg S, Lai J, et al. HIV-1 latent reservoir size and diversity are stable following brief treatment interruption. J Clin Invest. (2018) 128:3102–15. doi: 10.1172/JCI120194
92. Strongin Z, Sharaf R, VanBelzen DJ, Jacobson JM, Connick E, Volberding P, et al. Effect of short-term antiretroviral therapy interruption on levels of integrated HIV DNA. J Virol. (2018) 92:e00285–18. doi: 10.1128/JVI.00285-18
93. Papasavvas E, Lada SM, Joseph J, Yin X, Liu Q, Azzoni L, et al. Analytical antiretroviral therapy interruption does not irreversibly change preinterruption levels of cellular HIV. AIDS. (2018) 32:1763–72. doi: 10.1097/QAD.0000000000001909
94. Wen Y, Bar KJ, Li JZ. Lessons learned from HIV antiretroviral treatment interruption trials. Curr Opin HIV AIDS. (2018) 13:416–21. doi: 10.1097/COH.0000000000000484
95. June CH. Toward synthetic biology with engineered T cells: a long journey just begun. Hum Gene Ther. (2014) 25:779–84. doi: 10.1089/hum.2014.2533
96. Thomas ED. The nobel lectures in immunology. The nobel prize for physiology or medicine, 1990. Bone marrow transplantation–past, present and future. Scand J Immunol. (1994) 39:339–45. doi: 10.1111/j.1365-3083.1994.tb03382.x
97. Rosenberg SA, Aebersold P, Cornetta K, Kasid A, Morgan RA, Moen R, et al. Gene transfer into humans–immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. N Engl J Med. (1990) 323:570–8. doi: 10.1056/NEJM199008303230904
98. Riley JL, Carroll RG, Levine BL, Bernstein W, St Louis DC, Weislow OS, et al. Intrinsic resistance to T cell infection with HIV type 1 induced by CD28 costimulation. J Immunol. (1997) 158:5545–53.
99. Carroll RG, Riley JL, Levine BL, Feng Y, Kaushal S, Ritchey DW, et al. Differential regulation of HIV-1 fusion cofactor expression by CD28 costimulation of CD4+ T cells. Science. (1997) 276:273–6. doi: 10.1126/science.276.5310.273
100. Levine BL, Mosca JD, Riley JL, Carroll RG, Vahey MT, Jagodzinski LL, et al. Antiviral effect and ex vivo CD4+ T cell proliferation in HIV-positive patients as a result of CD28 costimulation. Science. (1996) 272:1939–43. doi: 10.1126/science.272.5270.1939
101. Maier DA, Brennan AL, Jiang S, Binder-Scholl GK, Lee G, Plesa G, et al. Efficient clinical scale gene modification via zinc finger nuclease-targeted disruption of the HIV co-receptor CCR5. Hum Gene Ther. (2013) 24:245–58. doi: 10.1089/hum.2012.172
102. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. (2015) 7:303ra139. doi: 10.1126/scitranslmed.aac5415
103. Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. (2011) 3:95ra73. doi: 10.1126/scitranslmed.3002842
104. Porter DL, Levine BL, Bunin N, Stadtmauer EA, Luger SM, Goldstein S, et al. A phase 1 trial of donor lymphocyte infusions expanded and activated ex vivo via CD3/CD28 costimulation. Blood. (2006) 107:1325–31. doi: 10.1182/blood-2005-08-3373
105. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
106. Porter BB, Harty JT. The onset of CD8+-T-cell contraction is influenced by the peak of Listeria monocytogenes infection and antigen display. Infect Immun. (2006) 74:1528–36. doi: 10.1128/IAI.74.3.1528-1536.2006
107. Levine BL, Humeau LM, Boyer J, MacGregor RR, Rebello T, Lu X, et al. Gene transfer in humans using a conditionally replicating lentiviral vector. Proc Natl Acad Sci USA. (2006) 103:17372–7. doi: 10.1073/pnas.0608138103
108. Wang GP, Levine BL, Binder GK, Berry CC, Malani N, McGarrity G, et al. Analysis of lentiviral vector integration in HIV+ study subjects receiving autologous infusions of gene modified CD4+ T cells. Mol Ther. (2009) 17:844–50. doi: 10.1038/mt.2009.16
109. Tebas P, Stein D, Tang W, Frank I, Wang S, Lee G, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. New Engl J Med. (2014) 370:901–10. doi: 10.1056/NEJMoa1300662
110. Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med. (2013) 5:197ra103. doi: 10.1126/scitranslmed.3006034
111. Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. (2013) 122:863–71. doi: 10.1182/blood-2013-03-490565
112. Finer MH, Dull TJ, Qin L, Farson D, Roberts MR. kat: a high-efficiency retroviral transduction system for primary human T lymphocytes. Blood. (1994) 83:43–50.
113. Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. (2009) 17:1453–64. doi: 10.1038/mt.2009.83
114. Mitchell RS, Beitzel BF, Schroder AR, Shinn P, Chen H, Berry CC, et al. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. (2004) 2:E234. doi: 10.1371/journal.pbio.0020234
115. Symons J, Cameron PU, Lewin SR. HIV integration sites and implications for maintenance of the reservoir. Curr Opin HIV AIDS. (2018) 13:152–9. doi: 10.1097/COH.0000000000000438
116. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. (2018) 24:563–71. doi: 10.1038/s41591-018-0010-1
117. Simeonov DR, Marson A. CRISPR-based tools in immunity. Annu Rev Immunol. (2019) 37:571–97. doi: 10.1146/annurev-immunol-042718-041522
118. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. New Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
Keywords: T cell, lentiviral (LV) vector, immune escape and surveillance, clinical trials, immune privilege
Citation: Kim GB, Hege K and Riley JL (2019) CAR Talk: How Cancer-Specific CAR T Cells Can Instruct How to Build CAR T Cells to Cure HIV. Front. Immunol. 10:2310. doi: 10.3389/fimmu.2019.02310
Received: 20 May 2019; Accepted: 12 September 2019;
Published: 27 September 2019.
Edited by:
Kavita M. Dhodapkar, Emory University, United StatesReviewed by:
Morgane Bomsel, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceCopyright © 2019 Kim, Hege and Riley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James L. Riley, cmlsZXlqQHVwZW5uLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.