 Weiting Qin†
Weiting Qin† Xueli Zhang
Xueli Zhang Xu Wang
Xu Wang- State Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China
The recent success of PD-1 and PD-L1 blockade in cancer therapy illustrates the important role of the PD-1/PD-L1 pathway in the regulation of antitumor immune responses. However, signaling regulated by the PD-1/PD-L pathway is also associated with substantial inflammatory effects that can resemble those in autoimmune responses, chronic infection, and sepsis, consistent with the role of this pathway in balancing protective immunity and immunopathology, as well as in homeostasis and tolerance. Targeting PD-1/PD-L1 to treat cancer has shown benefits in many patients, suggesting a promising opportunity to target this pathway in autoimmune and inflammatory disorders. Here, we systematically evaluate the diverse biological functions of the PD-1/PD-L pathway in immune-mediated diseases and the relevant mechanisms that control these immune reactions.
Introduction
Immune checkpoint inhibitory receptors [such as cytotoxic T lymphocyte antigen 4 (CTLA4) and programmed cell death protein 1 (PD-1)] expressed on immune cells trigger immunosuppressive signaling pathways. CTLA4 competes with CD28 for binding to CD80 and CD86 (1). PD-1 binds to PD-L1 or PD-L2 and resists positive signals through T-cell receptors (TCRs) and CD28 (2). These immunosuppressive molecules function as brakes to regulate the adaptive immune response. To date, suppressive signals have been used in a variety of ways to maintain the balance of the immune system. Their use has also been translated to the clinic. Recent years have witnessed the wide application of anti-PD-1, anti-CTLA4, and anti-PD-L1 antibodies in various types of cancer (3). Based on this application, the PD-1/PD-L pathway has stood out as an immune checkpoint.
In general, expression of PD-L1 is observed on T, B, and antigen-presenting cells and in some non-lymphoid tissues. Ligand binding to PD-1 on the surface of T cells mediates immune inhibition. In addition, PD-L1 is detected in the cardiac endothelium, placenta, and pancreatic islets, which indicates a role of PD-L1 in immunological tolerance (4). PD-L2 is another ligand of PD-1. The function of PD-L2 is not as well-known as that of PD-L1; thus, its clinical utility is still being explored. It has been reported that PD-L2 plays an inhibitory role similar to that of PD-L1 (5). In addition, it has been shown that PD-L2 plays an opposing role to PD-L1 in T-cell function in a PD-1-independent manner (6).
Recent reviews have extensively discussed the role and application of the PD-1/PD-L1 pathway in cancer. Recent studies have shown that inhibitory pathways are also involved in immune evasion by pathogens, suggesting that PD-1/PD-L may play a vital role in infections (7). In addition, studies using animal models of autoimmune diseases have shown that the interaction between PD-1 and PD-Ls is pivotal for the regulation of peripheral tolerance and autoimmunity (8). These data suggest that beyond their role in cancer, PD-1 and PD-Ls can be good candidates for the treatment of these diseases. However, whether this targeting could succeed may need to be further explored.
Here, we systematically evaluate the diverse biological functions of PD-1/PD-L in diseases other than cancer, including autoimmunity, chronic infections, and sepsis. For each disease, we cover the current understanding of the roles that PD-1 and PD-Ls play and the relevant mechanisms that control these immune reactions.
PD-1/PD-L Axis
PD-1 (CD279), a member of the CD28/CTLA4 family, is a receptor for PD-L1 and PD-L2 (1, 2, 5). PD-L1 (CD274) and PD-L2 (CD273) are both B7 family members, sharing 60% amino acid homology in humans and 77% amino acid homology in mice (9). Moreover, there is high diversity in the structure of PD-1 between humans and mice. Binding of PD-L1 to PD-1 has been found to exhibit similar affinity in both species (humans and mice) in vitro. Recently, the structures of mouse (m) PD-1/human (h) PD-L1, mPD-1/mPD-L2, and hPD-1/hPD-L1 have been reported, which may promote protein affinity engineering (10). With the breakthrough in the structural elucidation of the PD-1/PD-L1 complex, targeting the PD-1/PD-L1 immunological checkpoint with monoclonal antibodies and small molecular drugs has seen great progress (Figure 1). To date, five antibody-based inhibitors targeting PD-1/PD-L1 have been approved by the Food and Drug Administration (FDA), including two anti-PD-1 antibodies (nivolumab and pembrolizumab) and three anti-PD-L1 antibodies (avelumab, atezolizumab, and durvalumab). Nivolumab has been approved for non-small-cell lung cancer (NSCLC), renal cell carcinoma (RCC), bladder cancer (BC), colorectal cancer (CRC) with microsatellite instability or mismatch repair deficiency (MSI-H/dMMR), hepatocellular carcinoma (HCC), classic Hodgkin lymphoma (cHL), melanoma, and head and neck squamous cell carcinoma (HNSCC). Pembrolizumab has been approved for melanoma, HNSCC, cervical cancer, cHL, NSCLC, BC, stomach and gastroesophageal cancers, and all advanced solid tumors classified as MSI-H/dMMR. Avelumab has been approved for Merkel cell carcinoma and BC. Atezolizumab has been approved for NSCLC and BC. Durvalumab has been approved for BC and NSCLC (stage III) (11). In addition, an experimental anti-PD-L1 antibody (BMS-936559) and a nanobody targeting PD-L1 (KN035) have been developed as potential new drugs (12). KN035 is being evaluated in phase I clinical trials in patients with advanced solid tumors. Furthermore, several promising small molecules, including non-peptide and peptide-based molecules, are also designed to target this immune checkpoint pathway [see (13) for detailed information on small-molecule inhibitors]. For instance, a novel small-molecule inhibitor named “22” can inhibit the interaction between PD-1 and PD-L1 via binding at the PD-1–PD-L1 interface (14).
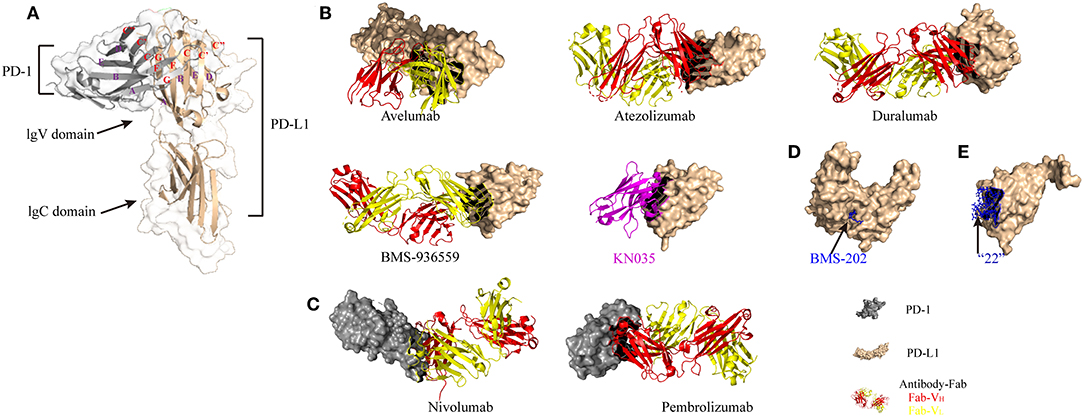
Figure 1. Structural basis of PD-1/PD-L1 checkpoint blockade by antibodies and small molecules. (A) Structure of the PD-1 and PD-L1 complex (modified from PDB ID 3BIK). (B) Overall structure of the hPD-L1 and avelumab-Fab complex, the hPD-L1 and atezolizumab-Fab complex, the hPD-L1 and durvalumab-Fab complex, the hPD-L1 and BMS-936559-Fab complex, and the hPD-L1 and KN035 nanobody complex (magenta ribbon model) (modified from PDB IDs 5GRJ, 5X8L, 5X8M, 5GG7, and 5JDS, respectively). The heavy chain (VH) and the light chain (VL) of the antibody-Fab fragments are represented by red and yellow ribbon models, respectively. (C) Overall structure of the hPD-1 and nivolumab-Fab complex and the hPD-1 and pembrolizumab-Fab complex (modified from PDB IDs 5GGR and 5GGS, respectively). (D) Crystal structure of BMS-202 (non-peptide small molecule inhibitor, blue stick model) on the surface of hPD-L1 (light yellow ribbon structure) (modified from PDB ID 5J89). (E) Structure of macrocyclic peptide inhibitor “22” in complex with hPD-L1 (light yellow ribbon model = hPD-L1; blue ball and stick model = peptide) (modified from PDB ID 5O45).
PD-1 and its ligands are transmembrane proteins and include an extracellular domain, a transmembrane region, and a cytoplasmic tail. PD-L1 is a type I transmembrane glycoprotein composed of typical IgC and IgV domains. Figure 1A shows a 1:1 receptor/ligand stoichiometry, with interactions primarily between the faces of the IgV domains GFCC' β sheet (indicated by the red letters). Notably, ligands of PD-1 with a short cytoplasmic tail may not transduce signaling through this pathway. Blockade of TCR and costimulatory signals is the primary effect of PD-1/PD-L1 signaling. Like in TCR signaling, CD28 may be the key target (15–17). Signaling through PD-1 is well-understood [see (4, 18) for detailed information on the PD-1/PD-L1 pathway], while there are fewer studies focusing on PD-L1 signaling. It has been reported that in the intracellular domain of PD-L1, there is a specific site that can be phosphorylated. This site shares 16% homology with B7-2 (CD86), which has been shown to transduce reverse signaling via cross-linking with mAbs. Furthermore, in vitro treatment with PD-L1 agonists (PD-L1 mAbs or PD-1 Ig) in Epstein-Barr virus (EBV)-transformed B cells can trigger cell apoptosis via cross-linking of PD-L1 (19). The above studies suggest that reverse signaling through PD-L1 exists, and further research should be conducted on this process.
PD-1/PD-L Axis Expression
Expression of PD-1 can be observed on immature CD4−CD8− thymocytes, a small fraction of murine thymocytes, lymph node, spleen, and bone marrow cells [for a review, see (20)]. The level of PD-1 (mRNA or protein) is rarely detectable, only becoming evident after a period of stimulation. Several factors can induce PD-1 expression. Generally, in lymphocytes, cytokines and the factors that stimulate B-cell receptor (BCR) or TCR signaling [such as Con A, phorbol 12-myristate-13-acetate (PMA)/ionomycin, and anti-IgM antibody] have the capacity to promote the expression of PD-1 (21, 22). Notably, it was reported that estrogen stimulation can also promote PD-1 expression on T lymphocytes and antigen-presenting cells (APCs) (23). Recently, it has been found that in mouse cytomegalovirus infection, endogenous glucocorticoids can induce selective and tissue-specific expression of PD-1 on NK cells (24), suggesting that NK cells are potential PD-1 blockade responders.
Currently, PD-L1 is expressed not only on hematopoietic cells [T cells, B cells, macrophages, dendritic cells (DCs), and neutrophils] but also in some non-hematopoietic tissues (heart, pancreas, placenta, vascular endothelium, muscle, liver, lung, eye, and skin tissues). PD-L1 expression in normal tissues suggests that the PD-1/PD-L1 signaling pathway may prevent tissue inflammation and contribute to homeostasis maintenance [for reviews, see (4, 25)]. However, PD-L1 expression can also be observed in cancers and plays a critical role in the avoidance of immune surveillance by cancerous cells. Unlike PD-L1 expression, PD-L2 expression is mainly restricted to APCs (26). Given the importance of the PD-1/PD-L1 axis, it is interesting to explore the regulatory mechanisms underlying its components.
PD-L1 is located on chromosome 9p24.1 and is only 42 KB from PD-L2. It has been implied that the elevated PD-L1 level is associated with amplification and translocation of this gene. In cases of NSCLC, SCLC, Hodgkin lymphoma, mediastinal large B-cell lymphoma, squamous cell carcinoma of the oral cavity, and Epstein-Barr virus-positive gastric cancer, amplification of the chromosomal portion containing PD-L1 has been reported [for a review, see (16)]. In addition, inflammatory signaling has been reported to modulate PD-L1 levels. Over the past few years, various soluble factors produced by immune cells have been described as inducers of PD-L1. Interferon IFN-γ has been considered one of the most potent inducers of PD-L1 expression. Importantly, the regulation of PD-L1 expression by IFN-γ is different between tumor and immune cells. For instance, in a mouse model of sarcoma, with IFN-γ blockade, the expression of PD-L1 was greatly decreased on tumor cells but only partially on tumor-associated macrophages (TAMs), which indicated that the expression of PD-L1 on TAMs is partially regulated by IFN-γ (27). This context-dependent effect on PD-L1 expression was also observed for transforming growth factor (TGF)-β and interleukin (IL)-12. Specifically, in vitro treatment of cultured monocytes and tubular epithelial cells with TGF-β inhibited PD-L1 expression (28). In a pancreatic islet transplantation model, TGF-β was required for sustained expression of PD-L1 on monocytes and tubular epithelial cells (29). Similarly, IL-12 promotes PD-L1 expression in endothelial cells and monocyte-derived macrophages but negatively regulates PD-L1 levels in THP-1 cell line-derived macrophages (30). In addition, other stimulators have been shown to induce PD-L1 expression [for review, see (16, 18)]. Monocytes and macrophages, as well as T cells, express elevated levels of PD-L1 when stimulated with IL-2, IL-17, and IL-15. IL-4 and granulocyte-macrophage colony-stimulating factor (GM-CSF) upregulate PD-L1 in macrophages. DCs express increased levels of PD-L1 when treated with IL-1β, IL-6, IL-10, and IL-27. Tumor necrosis factor (TNF)-α induces PD-L1 expression in dendritic cells, monocytes, endothelial cells, myelodysplastic syndrome blast cells, RCC cells, prostate cells, breast cancer cells, and colon cancer cells. Lipopolysaccharide (LPS) increases PD-L1 expression in macrophages, monocytes, DCs, neutrophils, and B cells. Poly(I:C) upregulates PD-L1 expression in lung cells.
Notably, the expression of PD-L1 can also be controlled by microRNAs, which regulate the PD-L1 gene by binding to its 3 prime untranslated region (3′UTR). The reported microRNAs include miR-155, miR-513, miR-34a, miR-142-5p, miR-93, miR-106b, miR-138-5p, miR-200, miR-152, miR-570, miR-17-5p, miR-15a, miR-193a, miR-16 [for a review, see (16)], miR-148a-3p, and miR-873 (31, 32). Consistent with this finding, deletion of the 3′UTR of the PD-L1 gene enhances PD-L1 mRNA stability in human and murine cells (33). In addition, cells that express high PD-L1 levels are observed to have loss of the 3′UTR in the PD-L1 gene, which is reported to result from gene trap vector integration (34). MicroRNAs (such as miR-20, miR-21, miR-130b, miR-197, and miR-223) can also regulate the expression of PD-L1 in an indirect way (35, 36). For example, mDCs from miR-223-KO mice display elevated PD-L1 expression mainly via regulation of the expression of STAT1, which is a crucial regulator of PD-L1 expression. Notably, the PD-L1 protein can be glycosylated, leading to inaccurate immunohistochemical readouts resulting from the inaccessibility of PD-L1 antibodies to PD-L1 polypeptide antigens. Lee et al. (37) found that deglycosylation can significantly ameliorate the above problem and provide an effective approach to guide anti-PD-1/PD-L1 therapy.
PD-1/PD-L Axis Function
Although the PD-L2 binds to PD-1 with higher affinity than does PD-L1, the primary ligand of PD-1 is PD-L1, as supported by the studies in PD-L1 knockout mice. The relevant mechanism is still unknown. As mentioned above, both PD-1 and its ligands are transmembrane proteins that include an extracellular domain, a transmembrane region, and a cytoplasmic tail. Upon binding of PD-L1, PD-1 activation leads to phosphorylation of the immunoreceptor tyrosine-based switch motif (ITSM) in the cytoplasmic tail and recruitment of Src homology region 2 domain containing phosphatase 1/2 (SHP1/2) and the newly identified slam-associated protein (SAP) (SH2D1A) (38). Then, through PD-1/PD-L1 signaling, TCR (such as ZAP 70) and costimulatory (such as PI3K/AKT) signaling cascades are inhibited (39). Moreover, as reported, like in TCR signaling, CD28 may be the key target. In detail, SHP1/2 is recruited to the ITSM, leading to dephosphorylation of the TCR and CD28 proximal signaling molecules (e.g., ZAP70, PKCθ, and other kinases) (40). Then, the inhibitory signaling contributes to the inactivation of downstream signaling pathways, such as the PI3K/AKT and Ras-MEK-ERK pathways. Additionally, SHP-2 can dephosphorylate casein kinase 2 (CK-2), leading to PTEN activation, in turn suppressing PI3K/AKT signaling (41). SHP-2 targets phospholipase Cγ and can inhibit the Ras-MEK-ERK cascade (42).
Binding of PD-L1 to PD-1 on different cells contributes to different functions. For instance, in effector T cells expressing PD-1, PD-L1 binding suppresses TCR or costimulatory signaling, resulting in T-cell apoptosis, anergy, and exhaustion (2, 43–46). In innate lymphoid cells (ILC2s), PD-L1 binding to the PD-1 receptor dampens ILC2 proliferation and function (47). However, PD-L1 binding to PD-1 on regulatory T (Treg) cells leads to their proliferation. Moreover, PD-L1 binding to PD-1 on naive T cells results in polarization of CD4+ T cells toward the T helper type 1 (TH1) subtype with effector T (Teff) cell proliferation [for review, see (16, 48)]. In follicular T helper (Tfh) cells expressing PD-1, PD-L1 binding regulates Tfh cell recruitment, restricts their localization to the germinal center (GC), and maintains the stringency of affinity selection in the GC (49). In some settings, PD-1 can protect cells from T-cell attack by shortening the interaction time between cytotoxic T lymphocytes (CTLs) and target cells (29). Therefore, PD-1/PD-L1 signaling plays a vital role in immune tolerance and T-cell exhaustion and has emerged as a key target in the treatment of cancer.
Interestingly, in some settings, the promotion of inflammation by PD-L1 and PD-L2 does not require PD-1 expression on target cells, suggesting the existence of a currently unknown proinflammatory receptor for PD-L1 and PD-L2. Wang et al. (50) found that PD-L1 signals through receptors that remain unidentified to activate T cells. B7-1 (CD80) cross-linking activates CD4+ T cells and increases the production of proinflammatory cytokines by T cells (50). Notably, another form of PD-L1, soluble PD-L1 (sPD-l1), has been identified. Via matrix metalloproteinase activity, sPD-L1 is released from the cell surface (51); therefore, the level of sPD-L1 indirectly accounts for the PD-L1 level, and it has been suggested that PD-1 interaction with PD-L1 may be blocked by sPD-L1.
The above findings illustrate the PD-1/PD-L1 pathway. However, the role of PD-L2 in regulating the immune response is still unclear. In general, on the one hand, interaction of PD-L2 with PD-1 may inhibit the activation of T cells (5). On the other hand, PD-L2, in a PD-1-independent manner, plays an antitumor role, inducing the production of TH1 cytokines and CTLs (6). Notably, it has been shown that an aggregated form of PD-L2 can suppress the interaction between PD-1 and PD-L1, which was observed on DCs (52).
Above, we reviewed the fundamental biology of the PD-1/PD-L immune checkpoint. In the following sections, the therapeutic potential of the PD-1/PD-L axis in cancers and other disorders (focusing on autoimmunity, chronic infection, and sepsis) will be thoroughly discussed.
Cancer
PD-L1 and/or PD-L2 expression either in tumor or in infiltrating immune cells has been verified in numerous tumors, indicating a role for the PD-1/PD-L1 axis as a prognostic trait and therapeutic target. Researchers have found that PD-1 expression on tumor-infiltrating lymphocytes (TILs) correlates with aggressive features (8, 53) and is linked to poor patient outcome. Furthermore, transgenic expression of PD-L1 in mouse tumor cell lines (such as melanoma, mastocytoma, myeloma/plasmocytomas) has additional escape ability from the host T cells and enhances the invasiveness in vivo (8). Till now, the regulation of PD-L1 has been well–researched, and it involves many pathways [see (54, 55) for detailed information on PD-L1-expressing regulation]. Studies show that the oncogenic signaling could regulate the expression of PD-L1 in tumor. For instance, in anaplastic lymphoma kinase (ALK)-carrying T-cell lymphoma, PD-L1 expression can be induced by ALK signal transducer, and this function is also observed in signal transducer and activator of transcription 3 (STAT3) in chemoresistant NSCLC (35). Besides, IFNs, especially IFN γ, and some other stimuli [e.g., IL-4, IL-10, vascular endothelial growth factor (VEGF), G-CSF, and LPS] are involved in PD-L1 regulation (56).
It has been demonstrated that PD-L1 protects cancer cells from direct attack by cytotoxic T cells. Binding of cancer cells expressing PD-L1 on their surface to PD-1 expressed on activated CD8+ T cells results in anergy and apoptosis of CD8+ T cells (57). It has been reported that thymocyte selection-associated high-mobility group box (TOX) expression coincides with PD-1 expression and can reinforce the phenotype and longevity of these exhausted T cells (58, 59). Interestingly, PD-L1 expressed on cancer cells can also serve as an antiapoptotic factor, leading to resistance to lysis by CD8+ T cells (60). Thus, cancer can escape from immune system attack via the PD-1/PD-L1 pathway, which suggests that blockade of this pathway may generate valuable results. The FDA approved two anti-PD-1 antibodies (pembrolizumab and nivolumab) in 2014, one anti-PD-L1 antibody (atezolizumab) in May 2016, and two PD-L1 antibodies (durvalumab and avelumab) in 2017 (61). The clinical success of these therapies has been shown in patients with melanoma, metastatic lung cancer, kidney cancer, BC, head and neck cancer, urothelial carcinoma, hepatocellular carcinoma, gastric cancer, metastatic Merkel cell carcinoma, and Hodgkin lymphoma (62). Certainly, it is unfeasible to attain stable and long-term disease remission with a single agent. In addition, a proportion of patients fails to respond to these therapies. The next wave in the treatment of cancer will focus on combination therapies (63, 64). Currently, the combination of PD-1 inhibitors with either CTLA-4 blockade or chemotherapy has been approved by the FDA and has shown improved clinical response rates compared with the single agents (65, 66).
Importantly, the interplay between the tumor and the immune system is complex, and both entities affect the outcome. In general, tumor foreignness, patient health status, other inhibitory processes within the tumor, functional exhaustion of tumor-infiltrating lymphocytes, and the sensitivity of tumor cells to tumor-specific T cells may affect the outcome of PD-1/PD-L1 blockade. Some other strategies for combination therapies with PD-1/PD-L1 blockade are being developed, including blockade of other inhibitory receptors and immunoregulatory cytokines, administration of agonists of costimulatory molecules, and treatment with homeostatic cytokines or engineered T cells [see (39) for detailed information on combination strategies]. More recently, Li et al. (67) showed that B7S1-B7S1R signaling additionally regulates CD8+ T-cell responses by cooperating with PD-1/PD-L1 checkpoint signaling, which emphasizes the importance of improving the understanding of T-cell checkpoint signaling and strategies to maximally and intelligently employ these therapeutic approaches alone or in combination.
Autoimmunity
Autoimmune diseases are defined as aberrant immune responses of an organism to its own cells and tissues. The incidence of autoimmunity is increasing worldwide, affecting ~5% of the population (68). The immunological abnormalities can precede autoimmune diseases by months to years (69). Autoimmunity can result from the escape of antigen-specific autoreactive T cells from the thymus to the periphery in the perinatal period (70).
PD-1 and PD-Ls have been demonstrated to be involved in the modulation of both central and peripheral tolerance. In particular, during thymocyte development, PD-1 plays a critical role by regulating signaling thresholds during positive selection (71). Consequently, the population of CD4+CD8+ thymocytes is increased in the absence of PD-1 or PD-L1 (72). Moreover, the PD-1 pathway serves as a negative regulator in autoreactive T and B cells to maintain tolerance (73).
In the setting of autoimmunity, PD-1 and PD-L1 have been studied in PD-1 knockout (PD-1−/−) mice, where breakdown of peripheral tolerance results in negative regulation of lymphocyte activation. PD-1 knockout can lead to different autoimmune features depending on the genetic background (44). PD-1 knockout mice in the BALB/c background, C57BL/6 background, and non-obese diabetic (NOD) background may develop dilated cardiomyopathies, lupus-like diseases, and type 1 diabetes mellitus (T1DM), respectively. Since the genetic background is a key factor in PD-1/PD-L signaling, it is important to elucidate the specific role of this axis in humans. Notably, the role of PD-L1 and PD-L2 in susceptibility regulation and autoimmune response progression may be different. For example, PD-1 and PD-L1 knockout mice showed more severe experimental autoimmune encephalomyelitis (EAE) symptoms than PD-L2 knockout or control mice (74). Furthermore, in some experimental animal models (diabetes, EAE, and autoimmune enteritis), administration of anti-PD-1/PD-L1 antibodies accelerates autoimmunity (75–77). Indeed, administration of a PD-L1 Ig recombinant fusion protein, PD-1-specific toxins [αPD-1-ABD-PE: anti-PD-1 single-chain variable fragment (αPD-1), an albumin-binding domain (ABD) and Pseudomonas exotoxin (PE)], or adenovirus expressing Fc-PD-L1 can reduce the severity of inflammatory diseases [rheumatoid arthritis (RA), colitis, lupus-like nephritis, EAE, systemic lupus erythematosus (SLE) and psoriasis] (Table 1) (78–80, 94, 95). In addition, upregulation of PD-1/PD-L1 expression can decrease disease onset and severity (autoimmune diabetes and EAE) (96). Furthermore, cells (such as umbilical cord mesenchymal stem cells and tonsil/bone mesenchymal stem cells) expressing PD-L1, which has the potential to inhibit lymphocyte proliferation, exhibited reduced immunogenicity and improved immunosuppressive capacity, which have been reported to lead to effective immunotherapy for diseases such as psoriasis (97).
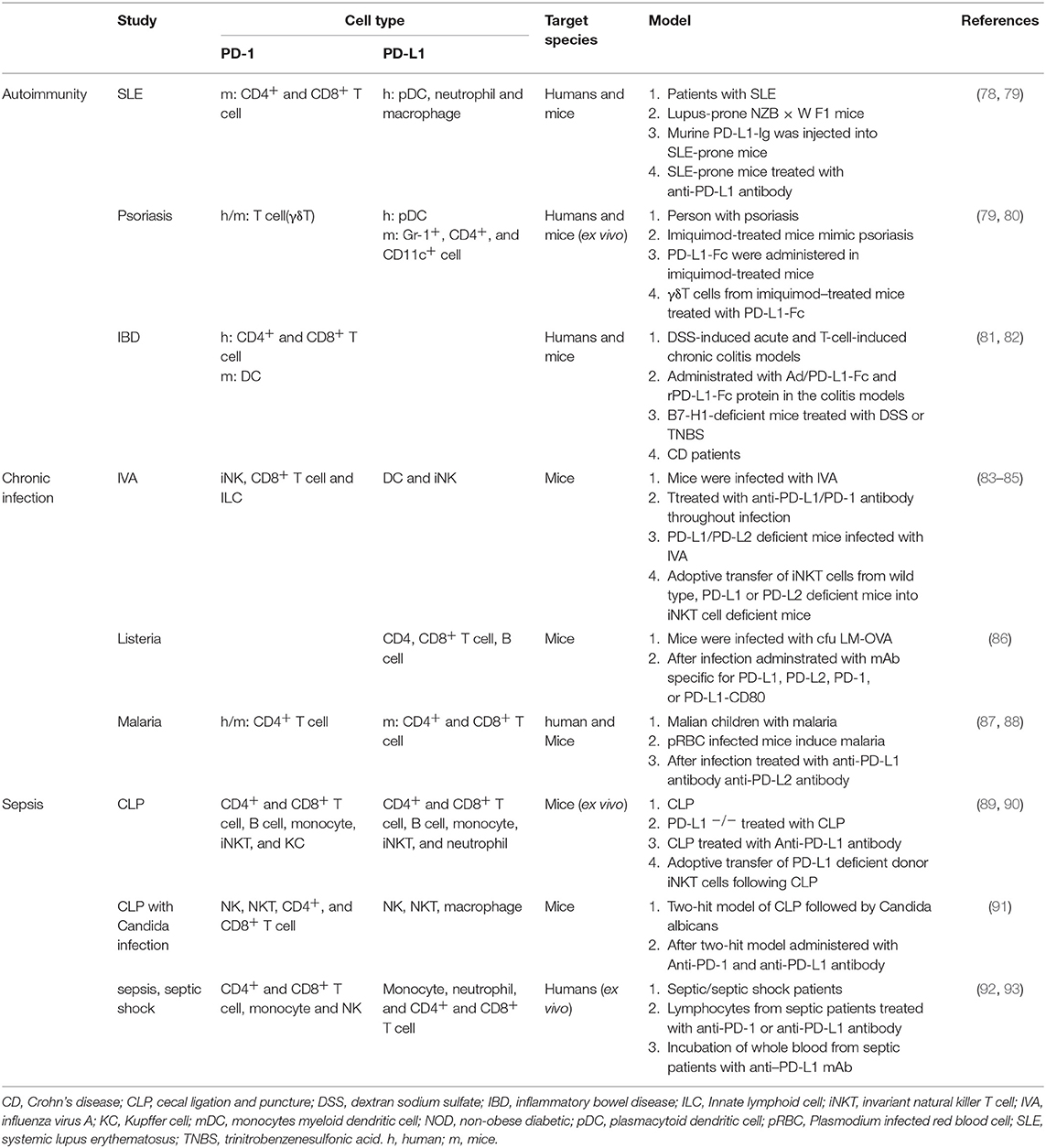
Table 1. Summary of studies reporting PD-1/PD-L1 in autoimmunity and inflammatory diseases.
The PD-1/PD-L1 pathway protects normal host tissues mainly via two aspects: promoting Treg development and function and directly inhibiting self-reactive T cells. The interaction between PD-1 and PD-L1 reduces antigen-specific T-cell activation, proliferation, and effector function (98). PD-1−/− mice demonstrated enhanced occurrence and severity of collagen-induced arthritis, which was linked to elevated T-cell proliferation and cytokine secretion (IFN-γ and IL-17) (99). Experiments in a murine model of SLE have shown that Tregs suppress autoreactive B cells via the interaction of PD-1 with PD-L1 (100). In addition, in actively induced and adoptively transferred experimental uveitis, treatment of mice with interphotoreceptor retinoid binding protein (IRBP)-specific T cells preincubated with PD-L1hi retinal pigment epithelial (RPE) cells, which acquired the Treg phenotype, delayed the induction of uveitis (75). Moreover, PD-1 can regulate chemokine receptor expression on T cells. Endothelial cell expression of PD-L1 may inhibit the migration of T cells to non-lymphoid tissues (101). Further studies should be performed to assess the effects of PD-1/PD-L1 signaling on T-cell migration. Additionally, the PD-1/PD-L1 pathway can regulate the progression of diseases via the regulation of other immune cells (DCs and NKT cells). In different murine colitis models [dextran sulfate sodium (DSS)-induced and T cell-induced colitis], administration of PD-L1-Fc reduced the Th17 cell frequency, DC function, and disease activity (Table 1) (81, 82). Consistent with this finding, PD-L1-Fc can also mitigate the pathogenesis of immune thrombocytopenia (ITP) by increasing T-cell apoptosis and by suppressing T-cell activation and proliferation and cytokine (IFN-γ, IL-2) production (102).
The widespread expression of PD-1/PD-L1 on both hematopoietic and non-hematopoietic cells indicates its pivotal role in tissue tolerance maintenance. Generally, in tissues expressing high levels of PD-1 on autoreactive T cells, PD-L1 binding regulates autoreactive T cells. Islet β-cells express high levels of PD-L1 and can suppress autoreactive T cells. Recently, in a Japanese cohort of autoimmune diabetic patients (type 1A), CD4+ T cells expressed lower levels of PD-1 than those of patients with type 1 (fulminant type 1) or type 2 diabetes or healthy control subjects, which may indicate that CD4+ T cells with decreased PD-1 expression result in Type 1A autoimmune diabetes via T-cell activation (103). Consistent with this finding, low levels of PD-1 expression led to active disease in SLE patients. Interestingly, in multiple sclerosis (MS) lesions, the PD-L1 level is increased, while CD8+ T cells in such lesions without PD-1 expression are insensitive to PD-L1 interaction (104). In autoimmune thyroid diseases (AITDs), the PD-1/PD-L1 axis is activated but cannot suppress disease progression probably due to the limited PD-L1 expression in vivo (105). Notably, in individuals with RA, high levels of soluble PD-1 (sPD-1) can inhibit the PD-1/PD-L1 pathway, which appears to affect Treg maintenance (106). Studies have reported that the serum level of sPD-1 is correlated with the 28-joint disease activity score. In line with this finding, in celiac disease (CD) patients, excessive sPD-1 can inhibit the PD-1/PD-L1 pathway and cause aberrant T-cell proliferation (107). Additionally, recent studies have reported that a single-nucleotide polymorphism (SNP) in PDCD1 is associated with the development of autoimmune diseases (SLE, type 1 diabetes, progressive MS, Vogt-Koyanagi-Harada disease, Graves' disease, and RA), suggesting that PDCD1 may be involved in the maintenance of tolerance (20, 108). For instance, the SNP rs4143815 in PD-L1 is associated with T1DM and could be a new biomarker for predicting T1DM susceptibility (109, 110).
Chronic Infection
Chronic infection is characterized by persistently high levels of antigen exposure, causing T cells to progressively lose effector functions and progress to exhaustion, similar to the state in cancer. Given its success in the treatment of cancer, anti-PD-1/PD-L1 therapy has potential in the treatment of severe chronic infections.
High PD-1 expression on CD8+ T cells was first reported in acute lymphocyte choriomeningitis virus (LCMV) infection. In addition, in infection with HIV, simian immunodeficiency virus, hepatitis B virus (HBV), and hepatitis C virus (HCV), PD-1 was observed to be expressed on virus-specific T cells (111–114). Studies have shown that several transcription factor binding sites regulate PD-1 expression on exhausted T cells. In the context of viral infection, binding of the transcription factor IFN regulatory factor 9 (IRF9) to the Pdcd1 promoter induces Pdcd1 transcription, which is also observed for the transcription factor forkhead box protein O 1 (FoxO1) (115, 116). Furthermore, during chronic infection, high expression of PD-1 can be induced by demethylation of the Pdcd1 promoter, which is observed in exhausted CD8+ T cells (117). Additionally, Alfei et al. (58) found that a new TOX molecule is correlated with PD-1 expression and can maintain the population of exhausted T cells. Metabolic processes play a vital role in the execution of the T-cell exhaustion program. Intriguingly, a recent report showed that PD-1 regulates several metabolic changes in the development of T-cell exhaustion (118). In chronic infection, PD-1 on T cells interacts with its ligands, which are found to be increased in monocytes, macrophages, and neutrophils, shutting down the T-cell response and leading to immunosuppression. CD4+ T cells expressing high PD-1 during HBV infection appeared to exhibit loss of helper T-cell function, and after treatment with PD-L1 blockade, the function of CD4+ T cells was partially restored (119). Combined blockade of the costimulatory molecule OX40 (CD134) and PD-L1 also improved the function of HBV-specific CD4+ T cells (120). Recently, Zhou (121) found that during LCMV and adenovirus infection, liver-resident NK (LrNK) cells expressing PD-L1 can control hepatic T-cell antiviral activity. In addition, blockade of PD-L1 can abolish the suppressive effect of LrNK cells on T cells (121). Notably, PD-L1-expressing neutrophils should attract attention for their importance in contributing to T-cell exhaustion because they are much more numerous than other immune cells. Moreover, recent studies have shown a link between PD-1/PD-L1 expression and clinical outcomes. In acute infection and untreated chronic infection, upregulation of PD-1 expression was linked to decreased effectiveness of CD4+ T-cell antiretroviral therapy (ART) (7, 122). PD-1 expression on CD8+ T cells after ART has been associated with impaired CD4+ T-cell immune reconstitution (123), microvascular diseases (124), and a reduced time to viral rebound after ART cessation (125). During the acute inflammatory phase of chronic HBV, elevated expression of PD-1/PD-L1 is required to offset the increasing positive costimulatory signals to prevent severe damage from an overvigorous immune response (7). Intriguingly, in Listeria-infected mice, PD-L1 binds to unknown receptors (not PD-1 or CD80) and is characterized as a costimulatory factor for antigen-specific CD8+ T cells (Table 1) (86).
PD-1/PD-L1 inhibitors can reinvigorate exhausted T cells. During LCMV infection, PD-1/PD-L1 pathway blockade can restore virus-specific CD8+ T-cell functions, promoting proliferation, cytokine production, and killing capacity. Additionally, PD-1/PD-L1 pathway blockade can reduce the viral load (111). Consistent with this finding, studies of HIV (a non-human primate model of simian immunodeficiency virus infection) and HBV/HCV (human infection with HBV/HCV and mouse and woodchuck models of HBV infection) infection showed similar results (112–114). In addition, studies have reported that PD-1 is required for maintaining T-cell exhaustion, but PD-1 inhibitors can enhance exhausted T-cell function. Therefore, the timing of PD-1/PD-L1 pathway modulation is critical (126). In addition to bolstering T-cell responses, treatment with anti-PD-1 antibody improved B-cell responses in a non-human primate model of HIV infection (127). In line with this finding, the combination of Tim-3 and PD-1 inhibitors reinvigorated exhausted CD8+ T cells and decreased the viral load in a mouse model of chronic infection (128). Intriguingly, in some cases, PD-1 blockade first restored virus-specific CD8+ T-cell function, but CD8+ T cells subsequently became exhausted again, displaying resistance to PD-1 immunotherapy (129). Studies have shown that during chronic infection, T cells that are sensitive to PD-1 inhibitors are the “progenitor-like” subset, and the less sensitive subset is the “terminally differentiated” subset (130–132). Progenitor-like T cells can transform into terminally differentiated T cells via proliferation, which may explain the above intriguing phenomenon (130, 131, 133).
Furthermore, PD-1/PD-L1 blockade may yield conflicting results in some infectious diseases. In influenza infection, in vivo blockade of PD-L1 was shown to reduce virus titers and increase the number of CD8+ T cells (Table 1) (83–85). In M. tuberculosis (Mtb) infection, PD-1 and PD-L1 expressions were enhanced in TB patients and Mtb-treated mice (134). PD-1 inhibitors can restore the effector functions (such as cytokine production and cytolytic activity) of NK cells and T cells (135), and PD-L1 blockade can increase the cytotoxicity of Mtb-specific CD8+ T cells to CD14+ cells from human tuberculous pleural effusion samples (136). However, the survival rate of PD-1-deficient mice challenged with Mtb infection is low (137, 138). Notably, high levels of Treg cells and mesenchymal stem cell infiltration may suppress specific T cells, leading to high mortality (Table 1) (138). In parallel, in a model of murine cerebral malaria, blocking PD-L1 induced inflammation and hemorrhage in the brain compared to its effects in untreated controls (Table 1) (87). However, PD-1 and LAG-3 blockade proved beneficial in murine malaria, with parasitemia reduction and improved CD4+ and CD8+ T-cell responses (88). In addition to the functions of PD-1/PD-L1, recent studies of malaria revealed a novel regulatory function of PD-L2. Expression of the PD-L2 protein on DCs that also express PD-L1 improved immune responses by inhibiting the PD-L1–PD-1 interaction. Moreover, administration of PD-L2 fused with the Fc region of immunoglobulin (PD-L2-Fc) to mice infected with malaria exerted a protective effect in this lethal infection (52).
Considering the success of PD-1/PD-L1 blockade in tumors, it is possible to promote enhanced immunity to chronic infection. Evaluation of the impact of PD-1/PD-L1 blockade in patients with chronic infection has been proposed. An anti-PD-L1 antibody (BMS-936559) underwent a phase II dose-escalation study in HIV infection with ART, the only trial in the setting of HIV infection without malignancy. Recently, this trial was ceased because of potential retinal toxicity, which was reported in a simultaneous macaque study (139). To date, anti-PD1 and anti-CTLA-4 antibodies both alone (NCT02595866) and in combination (NCT02408861) are in current clinical trials to evaluate their effects on HIV-associated malignancies (140). In addition, the anti-PD-1 antibody nivolumab, with and without an HBV vaccine, has been shown to suppress chronic HBV infection. A pilot study recently reported that in most virally suppressed HBV patients with negative HBeAg, treatment with nivolumab and the HBV vaccine (GS-4774) appeared to lead to HBsAg decline (7, 141).
Sepsis
Sepsis, associated with high mortality in intensive care units, is characterized by severe immunosuppression after the first proinflammatory hours (142). Postmortem examination of deceased septic patients highlights key immunological defects that impair host immunity (143). Sepsis has immunosuppressive mechanisms similar to those of cancer. Given the potency of CTLA-4- and PD-1-specific antibodies in improving host immunity and increasing survival in cancer patients, these agents are expected to open a promising avenue to the development of novel medicines for sepsis (144).
It has been reported that in different animal models of sepsis, including bacterial, fungal, and burn sepsis, treatment with PD-1 or PD-L1 antibodies shows survival benefits (Table 1) (89, 91, 145, 146). Further studies found that PD-1- or PD-L1-deficient animals undergoing septic challenge also showed improved survival rates compared with WT animals (147). Importantly, in patients with sepsis and in septic animal models, the widespread expression of PD-1 and PD-L1 [on immune effector cells (monocytes, T lymphocytes, neutrophils, and NK cells), endothelial cells, and bronchial epithelial cells] suggests the potential ability of PD-1 and PD-L1 to be indicators of immunosuppression and therapeutic agents. Hotchkiss et al. found that in septic patients, PD-1 expression on T cells was elevated, which was linked to poor proliferation of T cells as well as a high risk of secondary nosocomial infections and mortality (91). In addition, in septic shock patients, increased expression of PD-L1 in monocytes is correlated with 28-day mortality (Table 1) (92), and the abundance of PD-L1-expressing neutrophils is a marker of disease severity and predicts prognosis in sepsis (Table 1) (90, 148).
The beneficial effects of anti-PD1 and anti-PD-L1 therapy have been confirmed, but the mechanism underlying these effects is unclear. Given the complexity of the cellular targets and activities of PD-1 and PD-L1, different mechanisms may be involved. Generally, PD-1 is expressed on innate immune cells and binds to PD-L1. For example, binding of PD-L1 to PD-1 expressed on DCs leads to an increase in the apoptosis of DCs and a decrease in the levels of proinflammatory cytokines (149). Peripheral blood monocytes from patients with sepsis showed increased levels of PD-L1, and binding with PD-1 reduced cell survival and function, while treatment with anti-PD-1 antibody restored the production of the key cytokines IFN-γ and IL-2 by monocytes (Table 1) (93). However, recent reports indicate that PD-1 and PD-L1 function very differently and possibly separately. For example, macrophages from septic animals retain increased PD-1 expression in the absence of PD-L1 (147). In addition, Huang et al. (150) demonstrated that during sepsis, peritoneal macrophages upregulate PD-1 expression, and the ability of macrophages to phagocytose and clear bacteria is PD-1-dependent; PD-L1 has a minor role in this process. Notably, the site at which PD-1 and PD-L are expressed may affect their functions. Studies show that in animal models of sepsis, high expression of PD-L1 in the liver can protect the liver from damage (151). A study in a mouse model of hemorrhagic shock/CLP-induced indirect acute lung injury (iALI) found that PD-L1 can modulate the ability of Tregs to suppress iALI, but this ability was abolished when wild-type (WT) Tregs were adoptively transferred to PD-L1−/− recipient mice (152). Another post-mortem study showed detectable PD-L1 expression in the lungs of septic patients. Septic patients are often susceptible to secondary lung infections, which may contribute to this increase in PD-L1 expression, thereby suppressing T-cell function (143, 153).
Furthermore, immunotherapeutic approaches that use anti-PD-1/PD-L1 antibodies warrant further investigation. To date, some anti-PD-1 and anti-PD-L1 antibodies, including nivolumab (anti-PD-1) and BMS-936559 (anti-PD-L1), are in current clinical trials to evaluate their effects on severe sepsis/septic shock (154).
Others
The immunoregulatory role of the PD-1/PD-L pathway in other disorders, including ischemia–reperfusion injury (IRI), stroke, Alzheimer's disease (AD), and pain, has been reported.
Studies in mouse models with blockade of PD-L1 and/or PD-L2 or knockout of PD-Ls confirmed that PD-L1 and PD-L2 play a protective role in kidney IRI (155). Furthermore, when recipient mice were treated with inhibitors of either PD-L1 or PD-L2 before adoptive transfer of Tregs, the protective effect of Treg transfer was abolished, suggesting that the therapeutic mechanisms of adoptive Treg transfer are attributed to ligand inhibition. This phenomenon was also observed in a model of acute liver IRI, in which blocking PD-L1 increased liver damage and treatment with a PD-L1 Ig fusion protein reduced liver inflammation (156).
The PD-1/PD-L pathway has also been studied in post-stroke inflammation. Interestingly, in ischemic brain injury, peripheral B cells inhibit the activation of effector T cells, resident microglia, and infiltrated macrophages via the PD-1/PD-L1 pathway, thereby reducing inflammation (157). In an experimental model of stroke, 4 days after transient middle cerebral artery occlusion, PD-L1 and PD-L2 expression was increased on B cells in the central nervous system (CNS), blood, and spleen (158). In addition, in the acute phase after shock, the expression of PD-L1 is enhanced on Tregs, suppressing the release of matrix metalloproteinase-9 (MMP-9) from neutrophils, thereby alleviating damage to the blood–brain barrier (159). In contrast, knockout of PD-L1 or PD-L2 in mice exhibits an adverse effect on stroke outcomes and elevates inflammation in the post-stroke period. Notably, PD-L1 also plays a role in enhancing the severity of stoke (160). The current study demonstrated that the enhanced post-stroke inflammation and brain injury are due to the decreased number of CD8+CD12+ suppressor T cells in the CNS, which are PD-L1-dependent. The studies mentioned above show the different aspects of the PD-1/PD-L pathway, which reflects the dualistic nature of the immune system.
Patients with mild cognitive impairment or a diagnosis of AD express lower levels of PD-1 on CD4+ T cells and lower levels of PD-L1 on CD14+ macrophages/monocytes (161). With the disruption of the PD-1/PD-L1 pathway, the release of IL-10 is decreased (162). It is known that IL-10 can ameliorate pathology in animal models of AD, indicating that the remission of AD is, at least in part, due to the PD-1/PD-L1 pathway (162). However, the molecular and cellular mechanisms of the PD-1/PD-L1 interaction in AD and the influence of this interaction on immune cells in the CNS need to be further studied.
PD-L1 can also serve as an endogenous pain inhibitor and a neuromodulator. It can also be expressed in the spinal cord, dorsal root ganglia (DRG), nerves, and skin (163). In naive mice, PD-L1 can exert an analgesic effect by interacting with PD-1 and, conversely, blockade of either PD-1 or PD-L1 elicits mechanical allodynia. Moreover, PD-L1, as a unique neuromodulator, can suppress spinal cord synaptic transmission in the pain circuit. Mechanistically, PD-1 is expressed in nociceptive neurons in the DRG, and ligand binding to PD-1 leads to SHP-1 phosphorylation and triggers hyperpolarization of TREK2 K+ channels. Nociceptive neurons have been inhibited by PD-L1, and we anticipate a future painkiller based on PD-L1.
Resistance to and Toxicity of PD-1/PD-L1 Inhibitors
PD-1/PD-L1 inhibitors are a major advance, but they are not without drawbacks. As PD-1 and PD-L1 play a crucial role in peripheral immune tolerance, blockade of this immune checkpoint could lead to novel immune-related adverse events (irAEs). The most commonly observed irAEs include colitis, hepatitis, myositis, pneumonitis, endocrinopathies, kidney injury, and skin toxicities (164). Serous diarrhea and colitis often occur under combination therapy with ipilimumab and nivolumab (15%) but are less frequently observed in patients receiving anti-PD-1 monotherapy (1–4%) (165–167). Hepatitis occurrence varies with the immune checkpoint inhibitor (ICI) agent. Hepatitis occurred in up to 7% of individuals treated with anti-PD-1 monotherapy and in up to 33% of individuals treated with anti-PD-1 in combination with anti-CTLA-4 (168, 169). Usually, this hepatitis presents as asymptomatic elevated transaminase levels with or without elevation of the bilirubin concentration (167). Cardiac irAEs due to PD-1 axis inhibitors occur in <1% of treated individuals. However, high mortality rates have drawn attention to these events. Based on pharmacovigilance studies, compared with nivolumab monotherapy, treatment with a combination of ipilimumab and nivolumab resulted in an increased incidence of myocarditis (0.27 vs. 0.06%) and increased severity of myocarditis (0.17 vs. 0.01%) (170). With anti-PD-1/PD-L1 monotherapy, the incidence of pneumonitis is ~5%, and in those with combination immunotherapy, the incidence can be up to 10%. The onset of pneumonitis is often earlier and more severe in patients with NSCLC than in patients with other malignancies treated with PD-1 inhibitors (164, 165). Endocrinopathy irAEs due to PD-1 axis inhibitors include hypophysitis, thyroiditis, hypothyroidism, hyperthyroidism, and T1DM (171). Unlike the majority of irAEs, endocrinopathies are usually irreversible (165). Under anti-PD-1 monotherapy, patients often experience worsening renal function (13–22%) (172). Patients treated with ICI combination therapy were more susceptible to acute kidney injury (AKI) than those who received ipilimumab, nivolumab, or pembrolizumab monotherapy (173). Under treatment with anti-PD-1 agents, 30–40% of patients experience skin toxicities (174). The most common skin toxicities are lichenoid reactions, vitiligo, pruritus, and eczema (175). Intriguingly, vitiligo was observed only in patients with metastatic melanoma and not in patients with any other tumors (176). In addition to the above irAEs, some rare toxicities mediated by checkpoint inhibitors are also observed, such as neurological, ocular, and hematological toxicities (172). In clinical trials, there is no consensus definition to guide reporting of irAEs because irAEs are a novel presentation of toxicity from ICI treatment. Therefore, it is challenging for the physician to give an accurate report of the occurrence of irAEs. It has been reported that irAEs may be linked to dysregulated immune cell subpopulations. For instance, PD-1 blockade in viral infections or cancer increases the risk of cardiovascular complications, which is at least partially due to the effect of PD-1 on proatherogenic T cells (11). Patients (mainly those with melanoma and NSCLC) treated with PD-1/PD-L1 blockade were diagnosed with checkpoint inhibitor pneumonitis showing increased bronchoalveolar lavage (BAL) lymphocytosis, especially involving central memory T cells, and decreased expression of CTLA-4 and PD-1 in BAL Tregs (177). Moreover, the extent of autoantibodies could also lead to immune-related adverse events. In a report of thyroid disorders, these events occurred when patients with antithyroid antibodies received anti-PD-1 therapy, suggesting that anti-PD-1 treatment may regulate preexisting antithyroid antibodies (178). Similar T-cell clones were found in normal and tumor tissue, suggesting that T-cell cross-reactivity may result in injury to normal tissue. This phenomenon has been observed in patients with melanoma treated with an ICI, which led to vitiligo (179). In addition to the adverse effects of PD-1/PD-L1 inhibitors, resistance to PD-1/PD-L1 inhibitors is observed in some cancer patients, which is reported to be partially associated with an abnormal gut microbiome composition. Routy et al. (180) used fecal microbiota transplantation (avatar mice) to determine the relationship between the anticancer efficacy of PD-1 blockade and the dominance of commensal species and found that the intestinal microbiota influences the outcome of PD-1 blockade in mice and patients. Additionally, Chen et al. (181) found that tumor cells expressing CD38, which regulates the adenosine production pathway, escape from checkpoint inhibitor-mediated immune attack. In patients with advanced melanoma, resistance to PD-1 blockade therapy was reported to be linked to genetic aberrations in the cyclin D–cyclin-dependent kinase 4 (CDK4) pathway (182). Moreover, anti-PD-1 antibody binding to Fc receptors expressed on tumor-associated macrophages can partially be removed from tumors, which may reduce the effect of PD-1 antibodies (183). Furthermore, with high levels of exosomal PD-L1, T cells may no longer be reinvigorated via anti-PD-1 treatment, shedding light on the possible cause of resistance to PD-1 inhibitor therapy (184). Notably, in some tumors, such as melanoma, PD-L1 can mask pain; conversely, suppression of PD-L1 or PD-1 induced pain in tumor-bearing mice. In patients diagnosed with cancer, the pain level should be assessed before, during, and after immunotherapy (163). Recent reports have shown that in certain cancer patients, treatment with anti-PD-1 mAb can promote rapid disease progression, which is called hyperprogressive disease (HPD). Kamada et al. (185) found that in patients and mice with advanced gastric cancer, anti-PD-1 mAb increased the proliferation of FoxP3highCD45RA−CD4+PD-1+T cells [effector Treg (eTreg) cells], which could suppress antitumor immunity. In addition, depletion of these Treg cells could be beneficial for preventing HPD in the setting of PD-1 blockade therapy (185). Therefore, the use of PD-1 and PD-L1 inhibitors should be considered when diagnosing and treating diseases.
Concluding Remarks
The PD-1/PD-L signaling pathway plays a pivotal role in peripheral immune tolerance, which prevents inappropriate immune responses under physiological conditions. However, in infection or cancer, the alternative role of this pathway leading to immune suppression could cause serious issues. Thus, the functions of PD-1/PD-L in different diseases should be considered. The discrepant roles of PD-1/PD-L pathway observed in various diseases may be attributed to the different phases of disease, the dual effects of the PD-1/PD-L axis (Figure 2), the specific cells expressing PD-1/PD-Ls, and the unknown receptors to which PD-L1 binds. Moreover, the influence of the gut microbiome and the potential side effects should always be considered. As the mechanisms by which PD-1/PD-L1 blockade therapy functions and the mechanisms of resistance and toxicities are elucidated, the development of more precise treatments for different diseases is anticipated. Furthermore, the implementation of these treatments may inform researchers' and clinicians' choice of the appropriate combinatorial approaches to produce more rapid results.
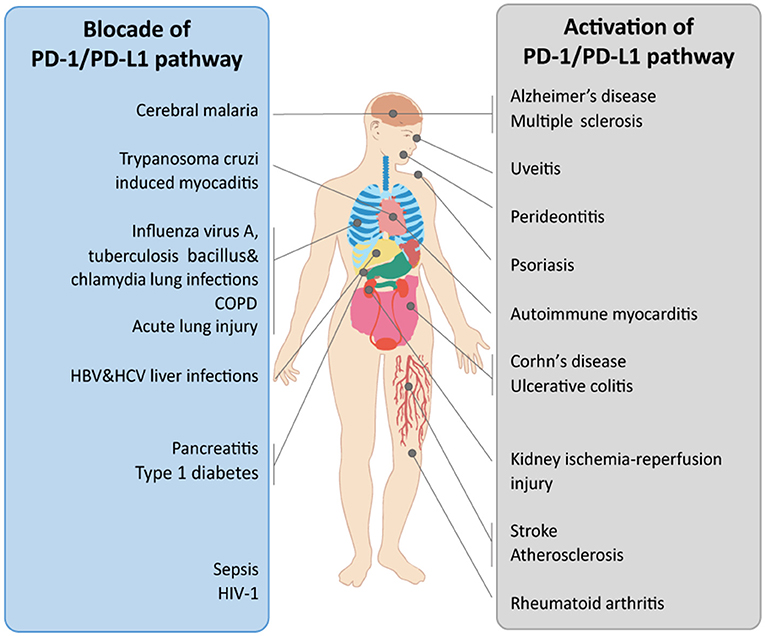
Figure 2. The therapeutic potential of the PD-1/PD-L1 pathway in organ-related diseases. The PD-1/PD-L1 pathway is implicated in multiple diseases. In different diseases, the PD-1/PD-L1 pathway shows diverse functions. The therapeutic potential of the PD-1/PD-L1 pathway when treated via blockade or activation in diverse organ-related diseases is shown. COPD, chronic obstructive pulmonary disease.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by grants from the National High Technology Research and Development Program of China (2014AA020609), National Natural Science Foundation of China (81301815, 81472678, 81201624, 81802890, and 81701945), State Key Laboratory for Oncogenes and Related Genes (YSJJ-2014-01), Natural Science Foundation of Shanghai (18ZR1436900), China Postdoctoral Science Foundation (2018M640403), and additional grants from the Shanghai Key Medical Specialty Program (ZK2012A22).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. (2005) 23:515–48. doi: 10.1146/annurev.immunol.23.021704.115611
2. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. (2000) 192:1027–34. doi: 10.1084/jem.192.7.1027
3. Teng F, Meng X, Kong L, Yu J. Progress and challenges of predictive biomarkers of anti PD-1/PD-L1 immunotherapy: a systematic review. Cancer Lett. (2018) 414:166–173. doi: 10.1016/j.canlet.2017.11.014
4. Chinai JM, Janakiram M, Chen F, Chen W, Kaplan M, Zang X. New immunotherapies targeting the PD-1 pathway. Trends Pharmacol Sci. (2015) 36:587–95. doi: 10.1016/j.tips.2015.06.005
5. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. (2001) 2:261–8. doi: 10.1038/85330
6. Shin T, Yoshimura K, Shin T, Crafton EB, Tsuchiya H, Housseau F, et al. In vivo costimulatory role of B7-DC in tuning T helper cell 1 and cytotoxic T lymphocyte responses. J Exp Med. (2005) 201:1531–41. doi: 10.1084/jem.20050072
7. Wykes MN, Lewin SR. Immune checkpoint blockade in infectious diseases. Nat Rev Immunol. (2018) 18:91–104. doi: 10.1038/nri.2017.112
8. Kuol N, Stojanovska L, Nurgali K, Apostolopoulos V. PD-1/PD-L1 in disease. Immunotherapy. (2018) 10:149–60. doi: 10.2217/imt-2017-0120
9. Lin DY, Tanaka Y, Iwasaki M, Gittis AG, Su HP, Mikami B, et al. The PD-1/PD-L1 complex resembles the antigen-binding Fv domains of antibodies and T cell receptors. Proc Natl Acad Sci USA. (2008) 105:3011–6. doi: 10.1073/pnas.0712278105
10. Pascolutti R, Sun X, Kao J, Maute RL, Ring AM, Bowman GR, et al. Structure and dynamics of PD-L1 and an ultra-high-affinity PD-1 receptor mutant. Structure. (2016) 24:1719–28. doi: 10.1016/j.str.2016.06.026
11. Postow MA, Sidlow R, Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med. (2018) 378:158–68. doi: 10.1056/NEJMra1703481
12. Zhang F, Wei H, Wang X, Bai Y, Wang P, Wu J, et al. Structural basis of a novel PD-L1 nanobody for immune checkpoint blockade. Cell Discov. (2017) 3:17004. doi: 10.1038/celldisc.2017.4
13. Yang J, Hu L. Immunomodulators targeting the PD-1/PD-L1 protein-protein interaction: From antibodies to small molecules. Med Res Rev. (2019) 39:265–301. doi: 10.1002/med.21530
14. Magiera-Mularz K, Skalniak L, Zak KM, Musielak B, Rudzinska-Szostak E, Berlicki L, et al. Bioactive macrocyclic inhibitors of the PD-1/PD-L1 immune checkpoint. Angew Chem Int Ed Engl. (2017) 56:13732–5. doi: 10.1002/anie.201707707
15. Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci USA. (2001) 98:13866–71. doi: 10.1073/pnas.231486598
16. Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity. (2018) 48:434–52. doi: 10.1016/j.immuni.2018.03.014
17. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. (2017) 355:1428–33. doi: 10.1126/science.aaf1292
18. Dermani FK, Samadi P, Rahmani G, Kohlan AK, Najafi R. PD-1/PD-L1 immune checkpoint: Potential target for cancer therapy. J Cell Physiol. (2019) 234:1313–25. doi: 10.1002/jcp.27172
19. Kim YS, Park GB, Lee HK, Song H, Choi IH, Lee WJ, et al. Cross-linking of B7-H1 on EBV-transformed B cells induces apoptosis through reactive oxygen species production, JNK signaling activation, and fasL expression. J Immunol. (2008) 181:6158–69. doi: 10.4049/jimmunol.181.9.6158
20. Gianchecchi E, Delfino DV, Fierabracci A. Recent insights into the role of the PD-1/PD-L1 pathway in immunological tolerance and autoimmunity. Autoimmun Rev. (2013) 12:1091–100. doi: 10.1016/j.autrev.2013.05.003
21. Kinter AL, Godbout EJ, McNally JP, Sereti I, Roby GA, O'Shea MA, et al. The common gamma-chain cytokines IL-2, IL-7, IL-15, and IL-21 induce the expression of programmed death-1 and its ligands. J Immunol. (2008) 181:6738–46. doi: 10.4049/jimmunol.181.10.6738
22. Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. (1996) 8:765–72. doi: 10.1093/intimm/8.5.765
23. Polanczyk MJ, Hopke C, Vandenbark AA, Offner H. Estrogen-mediated immunomodulation involves reduced activation of effector T cells, potentiation of Treg cells, and enhanced expression of the PD-1 costimulatory pathway. J Neurosci Res. (2006) 84:370–8. doi: 10.1002/jnr.20881
24. Quatrini L, Wieduwild E, Escaliere B, Filtjens J, Chasson L, Laprie C, et al. Endogenous glucocorticoids control host resistance to viral infection through the tissue-specific regulation of PD-1 expression on NK cells. Nat Immunol. (2018) 19:954–62. doi: 10.1038/s41590-018-0185-0
25. Dai S, Jia R, Zhang X, Fang Q, Huang L. The PD-1/PD-Ls pathway and autoimmune diseases. Cell Immunol. (2014) 290:72–9. doi: 10.1016/j.cellimm.2014.05.006
26. Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y, et al. Expression of programmed death 1 ligands by murine T cells and APJ Immunol C. (2002) 169:5538–45. doi: 10.4049/jimmunol.169.10.5538
27. Noguchi T, Ward JP, Gubin MM, Arthur CD, Lee SH, Hundal J, et al. Temporally distinct PD-L1 expression by tumor and host cells contributes to immune escape. Cancer Immunol Res. (2017) 5:106–17. doi: 10.1158/2326-6066.CIR-16-0391
28. Ou JN, Wiedeman AE, Stevens AM. TNF-alpha and TGF-beta counter-regulate PD-L1 expression on monocytes in systemic lupus erythematosus. Sci Rep. (2012) 2:295. doi: 10.1038/srep00295
29. Baas M, Besancon A, Goncalves T, Valette F, Yagita H, Sawitzki B, et al. TGFbeta-dependent expression of PD-1 and PD-L1 controls CD8(+) T cell anergy in transplant tolerance. Elife. (2016) 5:e08133. doi: 10.7554/eLife.08133
30. Xiong HY, Ma TT, Wu BT, Lin Y, Tu ZG. IL-12 regulates B7-H1 expression in ovarian cancer-associated macrophages by effects on NF-kappaB signalling. Asian Pac J Cancer Prev. (2014) 15:5767–72. doi: 10.7314/APJCP.2014.15.14.5767
31. Ashizawa M, Okayama H, Ishigame T, Thar Min AK, Saito K, Ujiie D, et al. miRNA-148a-3p Regulates Immunosuppression in DNA Mismatch repair-deficient colorectal cancer by targeting PD-L1. Mol Cancer Res. (2019) 17:1403–13. doi: 10.1158/1541-7786.MCR-18-0831
32. Gao L, Guo Q, Li X, Yang X, Ni H, Wang T, et al. MiR-873/PD-L1 axis regulates the stemness of breast cancer cells. EBioMedicine. (2019) 41:395–407. doi: 10.1016/j.ebiom.2019.02.034
33. Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, et al. Aberrant PD-L1 expression through 3'-UTR disruption in multiple cancers. Nature. (2016) 534:402–6. doi: 10.1038/nature18294
34. Mezzadra R, Sun C, Jae LT, Gomez-Eerland R, de Vries E, Wu W, et al. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature. (2017) 549:106–10. doi: 10.1038/nature23669
35. Fujita Y, Yagishita S, Hagiwara K, Yoshioka Y, Kosaka N, Takeshita F, et al. The clinical relevance of the miR-197/CKS1B/STAT3-mediated PD-L1 network in chemoresistant non-small-cell lung cancer. Mol Ther. (2015) 23:717–27. doi: 10.1038/mt.2015.10
36. Zhu J, Chen L, Zou L, Yang P, Wu R, Mao Y, et al. MiR-20b,−21, and−130b inhibit PTEN expression resulting in B7-H1 over-expression in advanced colorectal cancer. Hum Immunol. (2014) 75:348–53. doi: 10.1016/j.humimm.2014.01.006
37. Lee HH, Wang YN, Xia W, Chen CH, Rau KM, Ye L, et al. Removal of N-linked glycosylation enhances PD-L1 Detection and Predicts Anti-PD-1/PD-L1 therapeutic efficacy. Cancer Cell. (2019) 36:168–78.e4 doi: 10.1016/j.ccell.2019.06.008
38. Peled M, Tocheva AS, Sandigursky S, Nayak S, Philips EA, Nichols KE, et al. Affinity purification mass spectrometry analysis of PD-1 uncovers SAP as a new checkpoint inhibitor. Proc Natl Acad Sci USA. (2018) 115:E468–77. doi: 10.1073/pnas.1710437115
39. Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol. (2018) 18:153–67. doi: 10.1038/nri.2017.108
40. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. (2012) 209:1201–17. doi: 10.1084/jem.20112741
41. Patsoukis N, Li L, Sari D, Petkova V, Boussiotis VA. PD-1 increases PTEN phosphatase activity while decreasing PTEN protein stability by inhibiting casein kinase 2. Mol Cell Biol. (2013) 33:3091–8. doi: 10.1128/MCB.00319-13
42. Bivona TG, Perez De Castro I, Ahearn IM, Grana TM, Chiu VK, Lockyer PJ, et al. Phospholipase Cgamma activates Ras on the Golgi apparatus by means of RasGRP1. Nature. (2003) 424:694–8. doi: 10.1038/nature01806
43. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. (2007) 27:111–22. doi: 10.1016/j.immuni.2007.05.016
44. Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. (2006) 203:883–95. doi: 10.1084/jem.20051776
45. Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, et al. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci USA. (2004) 101:10691–6. doi: 10.1073/pnas.0307252101
46. Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. (1999) 5:1365–9. doi: 10.1038/70932
47. Mallett G, Laurence A, Amarnath S. Programmed cell death-1 receptor (PD-1)-mediated regulation of innate lymphoid cells. Int J Mol Sci. (2019) 20:E2836. doi: 10.3390/ijms20112836
48. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331
49. Shi J, Hou S, Fang Q, Liu X, Liu X, Qi H. PD-1 Controls follicular t helper cell positioning and function. Immunity. (2018) 49:264–74. doi: 10.1016/j.immuni.2018.06.012
50. Wang S, Bajorath J, Flies DB, Dong H, Honjo T, Chen L. Molecular modeling and functional mapping of B7-H1 and B7-DC uncouple costimulatory function from PD-1 interaction. J Exp Med. (2003) 197:1083–91. doi: 10.1084/jem.20021752
51. Chen Y, Wang Q, Shi B, Xu P, Hu Z, Bai L, et al. Development of a sandwich ELISA for evaluating soluble PD-L1 (CD274) in human sera of different ages as well as supernatants of PD-L1+ cell lines. Cytokine. (2011) 56:231–8. doi: 10.1016/j.cyto.2011.06.004
52. Karunarathne DS, Horne-Debets JM, Huang JX, Faleiro R, Leow CY, Amante F, et al. Programmed Death-1 Ligand 2-Mediated Regulation of the PD-L1 to PD-1 Axis Is Essential for Establishing CD4(+) T Cell Immunity. Immunity. (2016) 45:333–45. doi: 10.1016/j.immuni.2016.07.017
53. Ghebeh H, Tulbah A, Mohammed S, Elkum N, Bin Amer SM, Al-Tweigeri T, et al. Expression of B7-H1 in breast cancer patients is strongly associated with high proliferative Ki-67-expressing tumor cells. Int J Cancer. (2007) 121:751–8. doi: 10.1002/ijc.22703
54. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. (2015) 27:450–61. doi: 10.1016/j.ccell.2015.03.001
55. Guan J, Lim KS, Mekhail T, Chang CC. Programmed Death Ligand-1 (PD-L1) Expression in the Programmed Death Receptor-1 (PD-1)/PD-L1 blockade: a key player against various cancers. Arch Pathol Lab Med. (2017) 141:851–61. doi: 10.5858/arpa.2016-0361-RA
56. He J, Hu Y, Hu M, Li B. Development of PD-1/PD-L1 Pathway in tumor immune microenvironment and treatment for non-small cell lung cancer. Sci Rep. (2015) 5:13110. doi: 10.1038/srep13110
57. Mandai M PD-1/PD-L1 blockage in cancer treatment-from basic research to clinical application. Int J Clin Oncol. (2016) 21:447. doi: 10.1007/s10147-016-0969-x
58. Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli P, et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature. (2019) 571:265–9. doi: 10.1038/s41586-019-1326-9
59. Seo H, Chen J, Gonzalez-Avalos E, Samaniego-Castruita D, Das A, Wang YH, et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc Natl Acad Sci USA. (2019) 116:12410–5. doi: 10.1073/pnas.1905675116
60. Azuma T, Yao S, Zhu G, Flies AS, Flies SJ, Chen L. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood. (2008) 111:3635–43. doi: 10.1182/blood-2007-11-123141
61. Kythreotou A, Siddique A, Mauri FA, Bower M, Pinato DJ. Pd-L1. J Clin Pathol. (2018) 71:189–94. doi: 10.1136/jclinpath-2017-204853
62. Page DB, Postow MA, Callahan MK, Allison JP, Wolchok JD. Immune modulation in cancer with antibodies. Annu Rev Med. (2014) 65:185–202. doi: 10.1146/annurev-med-092012-112807
63. Hellmann MD, Friedman CF, Wolchok JD. Combinatorial Cancer Immunotherapies. Adv Immunol. (2016) 130:251–77. doi: 10.1016/bs.ai.2015.12.005
64. Sharma P, Allison JP. The future of immune checkpoint therapy. Science. (2015) 348:56–61. doi: 10.1126/science.aaa8172
65. Larkin J, Hodi FS, Wolchok JD. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. (2015) 373:1270–1. doi: 10.1056/NEJMc1509660
66. Langer CJ, Gadgeel SM, Borghaei H, Papadimitrakopoulou VA, Patnaik A, Powell SF, et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol. (2016) 17:1497–508. doi: 10.1016/S1470-2045(16)30498-3
67. Li J, Lee Y, Li Y, Jiang Y, Lu H, Zang W, et al. Co-inhibitory molecule B7 superfamily member 1 expressed by tumor-infiltrating myeloid cells induces dysfunction of anti-tumor CD8(+) T cells. Immunity. (2018) 48:773–86. doi: 10.1016/j.immuni.2018.03.018
68. Tobon GJ, Pers JO, Canas CA, Rojas-Villarraga A, Youinou P, Anaya JM. Are autoimmune diseases predictable? Autoimmun Rev. (2012) 11:259–66. doi: 10.1016/j.autrev.2011.10.004
69. Shapira Y, Agmon-Levin N, Shoenfeld Y. Defining and analyzing geoepidemiology and human autoimmunity. J Autoimmun. (2010) 34:J168–77. doi: 10.1016/j.jaut.2009.11.018
70. Fierabracci A, Milillo A, Locatelli F, Fruci D. The putative role of endoplasmic reticulum aminopeptidases in autoimmunity: insights from genomic-wide association studies. Autoimmun Rev. (2012) 12:281–8. doi: 10.1016/j.autrev.2012.04.007
71. Nishimura H, Honjo T, Minato N. Facilitation of beta selection and modification of positive selection in the thymus of PD-1-deficient mice. J Exp Med. (2000) 191:891–8. doi: 10.1084/jem.191.5.891
72. Keir ME, Freeman GJ, Sharpe AH. PD-1 regulates self-reactive CD8+ T cell responses to antigen in lymph nodes and tissues. J Immunol. (2007) 179:5064–70. doi: 10.4049/jimmunol.179.8.5064
73. Blank C, Brown I, Marks R, Nishimura H, Honjo T, Gajewski TF. Absence of programmed death receptor 1 alters thymic development and enhances generation of CD4/CD8 double-negative TCR-transgenic T cells. J Immunol. (2003) 171:4574–81. doi: 10.4049/jimmunol.171.9.4574
74. Carter LL, Leach MW, Azoitei ML, Cui J, Pelker JW, Jussif J, et al. PD-1/PD-L1, but not PD-1/PD-L2, interactions regulate the severity of experimental autoimmune encephalomyelitis. J Neuroimmunol. (2007) 182:124–34. doi: 10.1016/j.jneuroim.2006.10.006
75. Ke Y, Sun D, Jiang G, Kaplan HJ, Shao H. PD-L1(hi) retinal pigment epithelium (RPE) cells elicited by inflammatory cytokines induce regulatory activity in uveitogenic T cells. J Leukoc Biol. (2010) 88:1241–9. doi: 10.1189/jlb.0610332
76. Pauken KE, Jenkins MK, Azuma M, Fife BT. PD-1, but not PD-L1, expressed by islet-reactive CD4+ T cells suppresses infiltration of the pancreas during type 1 diabetes. Diabetes. (2013) 62:2859–69. doi: 10.2337/db12-1475
77. Liu Y, Carlsson R, Comabella M, Wang J, Kosicki M, Carrion B, et al. FoxA1 directs the lineage and immunosuppressive properties of a novel regulatory T cell population in EAE and MS. Nat Med. (2014) 20:272–82. doi: 10.1038/nm.3485
78. Luo Q, Huang Z, Ye J, Deng Y, Fang L, Li X, et al. PD-L1-expressing neutrophils as a novel indicator to assess disease activity and severity of systemic lupus erythematosus. Arthritis Res Ther. (2016) 18:47. doi: 10.1186/s13075-016-0942-0
79. Alculumbre SG, Saint-Andre V, Di Domizio J, Vargas P, Sirven P, Bost P, et al. Diversification of human plasmacytoid predendritic cells in response to a single stimulus. Nat Immunol. (2018) 19:63–75. doi: 10.1038/s41590-017-0012-z
80. Kim JH, Choi YJ, Lee BH, Song MY, Ban CY, Kim J, et al. Programmed cell death ligand 1 alleviates psoriatic inflammation by suppressing IL-17A production from programmed cell death 1-high T cells. J Allergy Clin Immunol. (2016) 137:1466–76 e3. doi: 10.1016/j.jaci.2015.11.021
81. Song MY, Hong CP, Park SJ, Kim JH, Yang BG, Park Y, et al. Protective effects of Fc-fused PD-L1 on two different animal models of colitis. Gut. (2015) 64:260–71. doi: 10.1136/gutjnl-2014-307311
82. Scandiuzzi L, Ghosh K, Hofmeyer KA, Abadi YM, Lazar-Molnar E, Lin EY, et al. Tissue-expressed B7-H1 critically controls intestinal inflammation. Cell Rep. (2014) 6:625–32. doi: 10.1016/j.celrep.2014.01.020
83. Yu Y, Tsang JC, Wang C, Clare S, Wang J, Chen X, et al. Single-cell RNA-seq identifies a PD-1(hi) ILC progenitor and defines its development pathway. Nature. (2016) 539:102–6. doi: 10.1038/nature20105
84. Maazi H, Singh AK, Speak AO, Lombardi V, Lam J, Khoo B, et al. Lack of PD-L1 expression by iNKT cells improves the course of influenza A infection. PLoS ONE. (2013) 8:e59599. doi: 10.1371/journal.pone.0059599
85. Rutigliano JA, Sharma S, Morris MY, Oguin TH III, McClaren JL, Doherty PC, et al. Highly pathological influenza A virus infection is associated with augmented expression of PD-1 by functionally compromised virus-specific CD8+ T cells. J Virol. (2014) 88:1636–51. doi: 10.1128/JVI.02851-13
86. Xu D, Fu HH, Obar JJ, Park JJ, Tamada K, Yagita H, et al. A potential new pathway for PD-L1 costimulation of the CD8-T cell response to Listeria monocytogenes infection. PLoS ONE. (2013) 8:e56539. doi: 10.1371/journal.pone.0056539
87. Hafalla JC, Claser C, Couper KN, Grau GE, Renia L, de Souza JB, et al. The CTLA-4 and PD-1/PD-L1 inhibitory pathways independently regulate host resistance to Plasmodium-induced acute immune pathology. PLoS Pathog. (2012) 8:e1002504. doi: 10.1371/journal.ppat.1002504
88. Butler NS, Moebius J, Pewe LL, Traore B, Doumbo OK, Tygrett LT, et al. Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection. Nat Immunol. (2011) 13:188–95. doi: 10.1038/ni.2180
89. Zhang Y, Zhou Y, Lou J, Li J, Bo L, Zhu K, et al. PD-L1 blockade improves survival in experimental sepsis by inhibiting lymphocyte apoptosis and reversing monocyte dysfunction. Crit Care. (2010) 14:R220. doi: 10.1186/cc9354
90. Wang JF, Li JB, Zhao YJ, Yi WJ, Bian JJ, Wan XJ, et al. Up-regulation of programmed cell death 1 ligand 1 on neutrophils may be involved in sepsis-induced immunosuppression: an animal study and a prospective case-control study. Anesthesiology. (2015) 122:852–63. doi: 10.1097/ALN.0000000000000525
91. Chang KC, Burnham CA, Compton SM, Rasche DP, Mazuski RJ, McDonough JS, et al. Blockade of the negative co-stimulatory molecules PD-1 and CTLA-4 improves survival in primary and secondary fungal sepsis. Crit Care. (2013) 17:R85. doi: 10.1186/cc12711
92. Shao R, Fang Y, Yu H, Zhao L, Jiang Z, Li CS. Monocyte programmed death ligand-1 expression after 3-4 days of sepsis is associated with risk stratification and mortality in septic patients: a prospective cohort study. Crit Care. (2016) 20:124. doi: 10.1186/s13054-016-1301-x
93. Patera AC, Drewry AM, Chang K, Beiter ER, Osborne D, Hotchkiss RS. Frontline science: defects in immune function in patients with sepsis are associated with PD-1 or PD-L1 expression and can be restored by antibodies targeting PD-1 or PD-L1. J Leukoc Biol. (2016) 100:1239–54. doi: 10.1189/jlb.4HI0616-255R
94. van der Vlist M, Kuball J, Radstake TR, Meyaard L. Immune checkpoints and rheumatic diseases: what can cancer immunotherapy teach us? Nat Rev Rheumatol. (2016) 12:593–604. doi: 10.1038/nrrheum.2016.131
95. Wang Z, Huang CA. A PD-1-specific toxin suppresses autoimmunity. Nat Biomed Eng. (2019) 3:253–4. doi: 10.1038/s41551-019-0384-5
96. Chamoto K, Al-Habsi M, Honjo T. Role of PD-1 in immunity and diseases. Curr Top Microbiol Immunol. (2017) 410:75–97. doi: 10.1007/82_2017_67
97. Kim JY, Park M, Kim YH, Ryu KH, Lee KH, Cho KA, et al. Tonsil-derived mesenchymal stem cells (T-MSCs) prevent Th17-mediated autoimmune response via regulation of the programmed death-1/programmed death ligand-1 (PD-1/PD-L1) pathway. J Tissue Eng Regen Med. (2018) 12:e1022–33. doi: 10.1002/term.2423
98. Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. (2010) 236:219–42. doi: 10.1111/j.1600-065X.2010.00923.x
99. Raptopoulou AP, Bertsias G, Makrygiannakis D, Verginis P, Kritikos I, Tzardi M, et al. The programmed death 1/programmed death ligand 1 inhibitory pathway is up-regulated in rheumatoid synovium and regulates peripheral T cell responses in human and murine arthritis. Arthritis Rheum. (2010) 62:1870–80. doi: 10.1002/art.27500
100. Wong M, La Cava A, Hahn BH. Blockade of programmed death-1 in young (New Zealand Black x New Zealand White)F1 mice promotes the suppressive capacity of CD4+ regulatory T cells protecting from lupus-like disease. J Immunol. (2013) 190:5402–10. doi: 10.4049/jimmunol.1202382
101. Frebel H, Nindl V, Schuepbach RA, Braunschweiler T, Richter K, Vogel J, et al. Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. J Exp Med. (2012) 209:2485–99. doi: 10.1084/jem.20121015
102. Nie M, Liu Y, Li XX, Min YN, Yang DD, Li Q, et al. PD-1/PD-L Pathway Potentially Involved in ITP Immunopathogenesis. Thromb Haemost. (2019) 119:758–65. doi: 10.1055/s-0039-1679909
103. Fujisawa R, Haseda F, Tsutsumi C, Hiromine Y, Noso S, Kawabata Y, et al. Low programmed cell death-1 (PD-1) expression in peripheral CD4(+) T cells in Japanese patients with autoimmune type 1 diabetes. Clin Exp Immunol. (2015) 180:452–7. doi: 10.1111/cei.12603
104. Trabattoni D, Saresella M, Pacei M, Marventano I, Mendozzi L, Rovaris M, et al. Costimulatory pathways in multiple sclerosis: distinctive expression of PD-1 and PD-L1 in patients with different patterns of disease. J Immunol. (2009) 183:4984–93. doi: 10.4049/jimmunol.0901038
105. Alvarez-Sierra D, Marin-Sanchez A, Ruiz-Blazquez P, de Jesus Gil C, Iglesias-Felip C, Gonzalez O, et al. Analysis of the PD-1/PD-L1 axis in human autoimmune thyroid disease: insights into pathogenesis and clues to immunotherapy associated thyroid autoimmunity. J Autoimmun. (2019) 103:102285. doi: 10.1016/j.jaut.2019.05.013
106. Liu C, Jiang J, Gao L, Wang X, Hu X, Wu M, et al. Soluble PD-1 aggravates progression of collagen-induced arthritis through Th1 and Th17 pathways. Arthritis Res Ther. (2015) 17:340. doi: 10.1186/s13075-015-0859-z
107. Ponce de Leon C, Angel Lopez-Casado M, Lorite P, Palomeque T, Isabel Torres M. Dysregulation of the PD-1/PD-L1 pathway contributes to the pathogenesis of celiac disease. Cell Mol Immunol. (2019) 16:777–9. doi: 10.1038/s41423-019-0256-7
108. Bertsias GK, Nakou M, Choulaki C, Raptopoulou A, Papadimitraki E, Goulielmos G, et al. Genetic, immunologic, and immunohistochemical analysis of the programmed death 1/programmed death ligand 1 pathway in human systemic lupus erythematosus. Arthritis Rheum. (2009) 60:207–18. doi: 10.1002/art.24227
109. Qian C, Guo H, Chen X, Shi A, Li S, Wang X, et al. Association of PD-1 and PD-L1 genetic polymorphyisms with type 1 diabetes susceptibility. J Diabetes Res. (2018) 2018:1614683. doi: 10.1155/2018/1614683
110. Pizarro C, Garcia-Diaz DF, Codner E, Salas-Perez F, Carrasco E, Perez-Bravo F. PD-L1 gene polymorphisms and low serum level of PD-L1 protein are associated to type 1 diabetes in Chile. Diabetes Metab Res Rev. (2014) 30:761–6. doi: 10.1002/dmrr.2552
111. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. (2006) 439:682–7. doi: 10.1038/nature04444
112. Fuller MJ, Callendret B, Zhu B, Freeman GJ, Hasselschwert DL, Satterfield W, et al. Immunotherapy of chronic hepatitis C virus infection with antibodies against programmed cell death-1 (PD-1). Proc Natl Acad Sci USA. (2013) 110:15001–6. doi: 10.1073/pnas.1312772110
113. Gardiner D, Lalezari J, Lawitz E, DiMicco M, Ghalib R, Reddy KR, et al. A randomized, double-blind, placebo-controlled assessment of BMS-936558, a fully human monoclonal antibody to programmed death-1 (PD-1), in patients with chronic hepatitis C virus infection. PLoS ONE. (2013) 8:e63818. doi: 10.1371/journal.pone.0063818
114. Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. (2009) 458:206–10. doi: 10.1038/nature07662
115. Terawaki S, Chikuma S, Shibayama S, Hayashi T, Yoshida T, Okazaki T, et al. IFN-alpha directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J Immunol. (2011) 186:2772–9. doi: 10.4049/jimmunol.1003208
116. Staron MM, Gray SM, Marshall HD, Parish IA, Chen JH, Perry CJ, et al. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8(+) T cells during chronic infection. Immunity. (2014) 41:802–14. doi: 10.1016/j.immuni.2014.10.013
117. Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, West EE, et al. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8(+) T cells. Immunity. (2011) 35:400–12. doi: 10.1016/j.immuni.2011.06.015
118. McKinney EF, Smith KG C Metabolic exhaustion in infection cancer and autoimmunity. Nat Immunol. (2018) 19:213–21. doi: 10.1038/s41590-018-0045-y
119. Dong Y, Li X, Zhang L, Zhu Q, Chen C, Bao J, et al. CD4(+) T cell exhaustion revealed by high PD-1 and LAG-3 expression and the loss of helper T cell function in chronic hepatitis B. BMC Immunol. (2019) 20:27. doi: 10.1186/s12865-019-0309-9
120. Jacobi FJ, Wild K, Smits M, Zoldan K, Csernalabics B, Flecken T, et al. OX40 stimulation and PD-L1 blockade synergistically augment HBV-specific CD4 T cells in patients with HBeAg-negative infection. J Hepatol. (2019) 70:1103–13. doi: 10.1016/j.jhep.2019.02.016
121. Zhou J, Peng H, Li K, Qu K, Wang B, Wu Y, et al. Liver-resident NK cells control antiviral activity of hepatic T cells via the PD-1-PD-L1 axis. Immunity. (2019) 50:403–17. doi: 10.1016/j.immuni.2018.12.024
122. Hoffmann M, Pantazis N, Martin GE, Hickling S, Hurst J, Meyerowitz J, et al. Exhaustion of activated CD8 T cells predicts disease progression in primary HIV-1 infection. PLoS Pathog. (2016) 12:e1005661. doi: 10.1371/journal.ppat.1005661
123. Shive CL, Clagett B, McCausland MR, Mudd JC, Funderburg NT, Freeman ML, et al. Inflammation perturbs the IL-7 axis, promoting senescence and exhaustion that broadly characterize immune failure in treated HIV infection. J Acquir Immune Defic Syndr. (2016) 71:483–92. doi: 10.1097/QAI.0000000000000913
124. Sinha A, Ma Y, Scherzer R, Hur S, Li D, Ganz P, et al. Role of T-cell dysfunction, inflammation, and coagulation in microvascular disease in HIV. J Am Heart Assoc. (2016) 5:e004243. doi: 10.1161/JAHA.116.004243
125. Hurst J, Hoffmann M, Pace M, Williams JP, Thornhill J, Hamlyn E, et al. Immunological biomarkers predict HIV-1 viral rebound after treatment interruption. Nat Commun. (2015) 6:8495. doi: 10.1038/ncomms9495
126. Odorizzi PM, Pauken KE, Paley MA, Sharpe A, Wherry EJ. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J Exp Med. (2015) 212:1125–37. doi: 10.1084/jem.20142237
127. Titanji K, Velu V, Chennareddi L, Vijay-Kumar M, Gewirtz AT, Freeman GJ, et al. Acute depletion of activated memory B cells involves the PD-1 pathway in rapidly progressing SIV-infected macaques. J Clin Invest. (2010) 120:3878–90. doi: 10.1172/JCI43271
128. Jin HT, Ahmed R, Okazaki T. Role of PD-1 in regulating T-cell immunity. Curr Top Microbiol Immunol. (2011) 350:17–37. doi: 10.1007/82_2010_116
129. Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science. (2016) 354:1160–5. doi: 10.1126/science.aaf2807
130. Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. (2016) 537:417–21. doi: 10.1038/nature19330
131. He R, Hou S, Liu C, Zhang A, Bai Q, Han M, et al. Follicular CXCR5- expressing CD8(+) T cells curtail chronic viral infection. Nature. (2016) 537:412–28. doi: 10.1038/nature19317
132. Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science. (2012) 338:1220–5. doi: 10.1126/science.1229620
133. Blackburn SD, Shin H, Freeman GJ, Wherry EJ. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc Natl Acad Sci USA. (2008) 105:15016–21. doi: 10.1073/pnas.0801497105
134. Cao S, Li J, Lu J, Zhong R, Zhong H. Mycobacterium tuberculosis antigens repress Th1 immune response suppression and promotes lung cancer metastasis through PD-1/PDl-1 signaling pathway. Cell Death Dis. (2019) 10:44. doi: 10.1038/s41419-018-1237-y
135. Rao M, Valentini D, Dodoo E, Zumla A, Maeurer M. Anti-PD-1/PD-L1 therapy for infectious diseases: learning from the cancer paradigm. Int J Infect Dis. (2017) 56:221–8. doi: 10.1016/j.ijid.2017.01.028
136. Suarez GV, Melucci Ganzarain CDC, Vecchione MB, Trifone CA, Marin Franco JL, Genoula M, et al. PD-1/PD-L1 Pathway Modulates Macrophage Susceptibility to Mycobacterium tuberculosis specific CD8(+) T cell induced death. Sci Rep. (2019) 9:187. doi: 10.1038/s41598-018-36403-2
137. Barber DL, Mayer-Barber KD, Feng CG, Sharpe AH, Sher A. CD4 T cells promote rather than control tuberculosis in the absence of PD-1-mediated inhibition. J Immunol. (2011) 186:1598–607. doi: 10.4049/jimmunol.1003304
138. Tousif S, Singh Y, Prasad DV, Sharma P, Van Kaer L, Das G. T cells from Programmed Death-1 deficient mice respond poorly to Mycobacterium tuberculosis infection. PLoS ONE. (2011) 6:e19864. doi: 10.1371/journal.pone.0019864
139. Gay CL, Bosch RJ, Ritz J, Hataye JM, Aga E, Tressler RL, et al. Clinical trial of the Anti-PD-L1 antibody BMS-936559 in HIV-1 infected participants on suppressive antiretroviral therapy. J Infect Dis. (2017) 215:1725–33. doi: 10.1093/infdis/jix191
140. Rasmussen TA, Anderson JL, Wightman F, Lewin SR. Cancer therapies in HIV cure research. Curr Opin HIV AIDS. (2017) 12:96–104. doi: 10.1097/COH.0000000000000328
141. Gane E, Verdon DJ, Brooks AE, Gaggar A, Nguyen AH, Subramanian GM, et al. Anti-PD-1 blockade with nivolumab with and without therapeutic vaccination for virally suppressed chronic hepatitis B: a pilot study. J Hepatol. (2019). doi: 10.1016/j.jhep.2019.06.028. [Epub ahead of print].
142. Delano MJ, Ward PA. Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest. (2016) 126:23–31. doi: 10.1172/JCI82224
143. Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA. (2011) 306:2594–605. doi: 10.1001/jama.2011.1829
144. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. (2012) 366:2443–54. doi: 10.1056/NEJMoa1200690
145. Brahmamdam P, Inoue S, Unsinger J, Chang KC, McDunn JE, Hotchkiss RS. Delayed administration of anti-PD-1 antibody reverses immune dysfunction and improves survival during sepsis. J Leukoc Biol. (2010) 88:233–40. doi: 10.1189/jlb.0110037
146. Patil NK, Guo Y, Luan L, Sherwood ER. Targeting immune cell checkpoints during sepsis. Int J Mol Sci. (2017) 18:2413. doi: 10.3390/ijms18112413
147. Huang X, Chen Y, Chung CS, Yuan Z, Monaghan SF, Wang F, et al. Identification of B7-H1 as a novel mediator of the innate immune/proinflammatory response as well as a possible myeloid cell prognostic biomarker in sepsis. J Immunol. (2014) 192:1091–9. doi: 10.4049/jimmunol.1302252
148. Patel JM, Sapey E, Parekh D, Scott A, Dosanjh D, Gao F, et al. Sepsis induces a dysregulated neutrophil phenotype that is associated with increased mortality. Mediators Inflamm. (2018) 2018:4065362. doi: 10.1155/2018/4065362
149. Yao S, Wang S, Zhu Y, Luo L, Zhu G, Flies S, et al. PD-1 on dendritic cells impedes innate immunity against bacterial infection. Blood. (2009) 113:5811–8. doi: 10.1182/blood-2009-02-203141
150. Huang X, Venet F, Wang YL, Lepape A, Yuan Z, Chen Y, et al. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc Natl Acad Sci USA. (2009) 106:6303–8. doi: 10.1073/pnas.0809422106
151. Zhu W, Bao R, Fan X, Tao T, Zhu J, Wang J, et al. PD-L1 blockade attenuated sepsis-induced liver injury in a mouse cecal ligation and puncture model. Mediators Inflamm. (2013) 2013:361501. doi: 10.1155/2013/361501
152. Tang L, Bai J, Chung CS, Lomas-Neira J, Chen Y, Huang X, et al. Active players in resolution of shock/sepsis induced indirect lung injury: immunomodulatory effects of Tregs and PD-1. J Leukoc Biol. (2014) 96:809–20. doi: 10.1189/jlb.4MA1213-647RR
153. Boomer JS, Green JM, Hotchkiss RS. The changing immune system in sepsis: is individualized immuno-modulatory therapy the answer? Virulence. (2014) 5:45–56. doi: 10.4161/viru.26516
154. Riva A, Chokshi S. Immune checkpoint receptors: homeostatic regulators of immunity. Hepatol Int. (2018) 12:223–36. doi: 10.1007/s12072-018-9867-9
155. Jaworska K, Ratajczak J, Huang L, Whalen K, Yang M, Stevens BK, et al. Both PD-1 ligands protect the kidney from ischemia reperfusion injury. J Immunol. (2015) 194:325–33. doi: 10.4049/jimmunol.1400497
156. Ji H, Shen X, Gao F, Ke B, Freitas MC, Uchida Y, et al. Programmed death-1/B7-H1 negative costimulation protects mouse liver against ischemia and reperfusion injury. Hepatology. (2010) 52:1380–9. doi: 10.1002/hep.23843
157. Zhao S, Li F, Leak RK, Chen J, Hu X. Regulation of neuroinflammation through programed death-1/programed death ligand signaling in neurological disorders. Front Cell Neurosci. (2014) 8:271. doi: 10.3389/fncel.2014.00271
158. Ren X, Akiyoshi K, Vandenbark AA, Hurn PD, Offner H. Programmed death-1 pathway limits central nervous system inflammation and neurologic deficits in murine experimental stroke. Stroke. (2011) 42:2578–83. doi: 10.1161/STROKEAHA.111.613182
159. Li P, Mao L, Liu X, Gan Y, Zheng J, Thomson AW, et al. Essential role of program death 1-ligand 1 in regulatory T-cell-afforded protection against blood-brain barrier damage after stroke. Stroke. (2014) 45:857–64. doi: 10.1161/STROKEAHA.113.004100
160. Bodhankar S, Chen Y, Vandenbark AA, Murphy SJ, Offner H. PD-L1 enhances CNS inflammation and infarct volume following experimental stroke in mice in opposition to PD-1. J Neuroinflammation. (2013) 10:111. doi: 10.1186/1742-2094-10-111
161. Saresella M, Calabrese E, Marventano I, Piancone F, Gatti A, Farina E, et al. A potential role for the PD1/PD-L1 pathway in the neuroinflammation of Alzheimer's disease. Neurobiol Aging. (2012) 33:624 e11–22. doi: 10.1016/j.neurobiolaging.2011.03.004
162. Koronyo-Hamaoui M, Ko MK, Koronyo Y, Azoulay D, Seksenyan A, Kunis G, et al. Attenuation of AD-like neuropathology by harnessing peripheral immune cells: local elevation of IL-10 and MMP-9. J Neurochem. (2009) 111:1409–24. doi: 10.1111/j.1471-4159.2009.06402.x
163. Chen G, Kim YH, Li H, Luo H, Liu DL, Zhang ZJ, et al. PD-L1 inhibits acute and chronic pain by suppressing nociceptive neuron activity via PD-1. Nat Neurosci. (2017) 20:917–26. doi: 10.1038/nn.4571
164. Wang DY, Johnson DB, Davis EJ. Toxicities associated with PD-1/PD-L1 blockade. Cancer J. (2018) 24:36–40. doi: 10.1097/PPO.0000000000000296
165. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. (2017) 377:1345–56. doi: 10.1056/NEJMoa1709684
166. Schachter J, Ribas A, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet. (2017) 390:1853–62. doi: 10.1016/S0140-6736(17)31601-X
167. Spain L, Diem S, Larkin J. Management of toxicities of immune checkpoint inhibitors. Cancer Treat Rev. (2016) 44:51–60. doi: 10.1016/j.ctrv.2016.02.001
168. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. (2013) 369:134–44. doi: 10.1056/NEJMoa1305133
169. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. (2015) 373:23–34. doi: 10.1056/NEJMoa1504030
170. Johnson DB, Balko JM, Compton ML, Chalkias S, Gorham J, Xu Y, et al. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med. (2016) 375:1749–55. doi: 10.1056/NEJMoa1609214
171. Hahn AW, Gill DM, Agarwal N, Maughan BL. PD-1 checkpoint inhibition: Toxicities and management. Urol Oncol. (2017) 35:701–7. doi: 10.1016/j.urolonc.2017.08.005
172. Heinzerling L, de Toni EN, Schett G, Hundorfean G, Zimmer L. Checkpoint inhibitors. Dtsch Arztebl Int. (2019) 116:119–26. doi: 10.3238/arztebl.2019.0119
173. Cortazar FB, Marrone KA, Troxell ML, Ralto KM, Hoenig MP, Brahmer JR, et al. Clinicopathological features of acute kidney injury associated with immune checkpoint inhibitors. Kidney Int. (2016) 90:638–47. doi: 10.1016/j.kint.2016.04.008
174. Naidoo J, Page DB, Li BT, Connell LC, Schindler K, Lacouture ME, et al. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann Oncol. (2015) 26:2375–91. doi: 10.1093/annonc/mdv383
175. Collins LK, Chapman MS, Carter JB, Samie FH. Cutaneous adverse effects of the immune checkpoint inhibitors. Curr Probl Cancer. (2017) 41:125–8. doi: 10.1016/j.currproblcancer.2016.12.001
176. Sibaud V, Meyer N, Lamant L, Vigarios E, Mazieres J, Delord JP. Dermatologic complications of anti-PD-1/PD-L1 immune checkpoint antibodies. Curr Opin Oncol. (2016) 28:254–63. doi: 10.1097/CCO.0000000000000290
177. Suresh K, Naidoo J, Zhong Q, Xiong Y, Mammen J, Villegas de Flores M, et al. The alveolar immune cell landscape is dysregulated in checkpoint inhibitor pneumonitis. J Clin Invest. (2019) 130:128654. doi: 10.1172/JCI128654
178. Osorio JC, Ni A, Chaft JE, Pollina R, Kasler MK, Stephens D, et al. Antibody-mediated thyroid dysfunction during T-cell checkpoint blockade in patients with non-small-cell lung cancer. Ann Oncol. (2017) 28:583–9. doi: 10.1093/annonc/mdw640
179. Byrne EH, Fisher DE. Immune and molecular correlates in melanoma treated with immune checkpoint blockade. Cancer. (2017) 123:2143–53. doi: 10.1002/cncr.30444
180. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. (2018) 359:91–7. doi: 10.1126/science.aan3706
181. Chen L, Diao L, Yang Y, Yi X, Rodriguez BL, Li Y, et al. CD38-mediated immunosuppression as a mechanism of tumor cell escape from PD-1/PD-L1 blockade. Cancer Discov. (2018) 8:1156–75. doi: 10.1158/2159-8290.CD-17-1033
182. Kong Y, Yu J, Yan J, Guo Q, Chi Z, Tang B, et al. Genetic aberrations in the CDK4 pathway are associated with innate resistance to PD-1 blockade in Chinese patients with non-cutaneous melanoma. Clin Cancer Res. (2019). doi: 10.1158/1078-0432.CCR-19-0475. [Epub ahead of print].
183. Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci Transl Med. (2017) 9:eaal3604. doi: 10.1126/scitranslmed.aal3604
184. Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. (2018) 560:382–6. doi: 10.1038/s41586-018-0392-8
Keywords: PD-1, PD-L1, autoimmune, chronic infection, sepsis
Citation: Qin W, Hu L, Zhang X, Jiang S, Li J, Zhang Z and Wang X (2019) The Diverse Function of PD-1/PD-L Pathway Beyond Cancer. Front. Immunol. 10:2298. doi: 10.3389/fimmu.2019.02298
Received: 10 May 2019; Accepted: 11 September 2019;
Published: 04 October 2019.
Edited by:
Philippe Saas, INSERM U1098 Interactions Hôte-Greffon-Tumeur & Ingénierie Cellulaire et Génique, FranceReviewed by:
Daniel Olive, Aix Marseille Université, FranceThierry Mp Gauthier, National Institutes of Health (NIH), United States
Copyright © 2019 Qin, Hu, Zhang, Jiang, Li, Zhang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhigang Zhang, enpoYW5nQHNoc2NpLm9yZw==; Xu Wang, anNkeHd4QDEyNi5jb20=
†These authors share co-first authorship