 Iñigo Terrén
Iñigo Terrén Ane Orrantia
Ane Orrantia Joana Vitallé
Joana Vitallé Olatz Zenarruzabeitia
Olatz Zenarruzabeitia Francisco Borrego
Francisco Borrego- 1Immunopathology Group, Biocruces Bizkaia Health Research Institute, Barakaldo, Spain
- 2Ikerbasque, Basque Foundation for Science, Bilbao, Spain
Natural Killer (NK) cells are characterized by their potential to kill tumor cells by different means without previous sensitization and have, therefore, become a valuable tool in cancer immunotherapy. However, their efficacy against solid tumors is still poor and further studies are required to improve it. One of the major restrictions for NK cell activity is the immunosuppressive tumor microenvironment (TME). There, tumor and other immune cells create the appropriate conditions for tumor proliferation while, among others, preventing NK cell activation. Furthermore, NK cell metabolism is impaired in the TME, presumably due to nutrient and oxygen deprivation, and the higher concentration of tumor-derived metabolic end products, such as lactate. This metabolic restriction of NK cells limits their effector functions, and it could represent a potential target to focus on to improve the efficacy of NK cell-based therapies against solid tumors. In this review, we discuss the potential effect of TME into NK cell metabolism and its influence in NK cell effector functions.
Introduction
Natural Killer (NK) cells are a promising tool in cancer immunotherapy. Their activation is driven by a balance between activating and inhibitory signals, so they are able to exert antitumor responses without prior sensitization. NK cells have demonstrated their potential in the treatment of several malignancies. However, the efficacy of these cells to treat solid tumors is still unsatisfactory (1–3). One of the main reasons for this limitation is the immunosuppressive effect of the tumor microenvironment (TME). In the TME, several tumor and tumor-associated cells produce and secrete factors that directly or indirectly prevent NK cell activation, including interleukin (IL)-6, IL-10, transforming growth factor-β (TGF-β), prostaglandin E2 (PGE2), and idoleamine 2,3-dioxygenase (IDO) (4, 5). Through these cytokines and factors, tumors are able to downmodulate NK cell activating receptors, such as NKp30, NKp44, or NKG2D (3, 4, 6–8), and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) (9). Furthermore, in the TME, NK cells receive signals from inhibitory receptors such as CD94/NKG2A, which bind to HLA-E exposed on the surfaces of several solid tumors including lung, pancreas, stomach, colon, head and neck, and liver tumor tissues (10). These immunosuppressive mechanisms mainly alter the balance between activating and inhibitory signals of NK cells, a step that is decisive for NK cell activation. Nonetheless, it should be also considered the effect of the TME in NK cell metabolism, which is essential to display full effector functions (11).
It is now accepted that the metabolic profile of NK cells is different under certain pathologies, such as obesity (12, 13) or viral infection (14). Also, it can be modified by several processes, including education (15, 16), maturation (17), or cytokine stimulation (18–28). The latter is especially relevant because of the potential use of cytokine-stimulated and/or expanded NK cells for adoptive cell therapy in cancer treatment (29–32). In the tumor context, multiple factors converge to modulate NK cell metabolism. For instance, it is known that TGF-β downregulates the expression of activating receptors and effector functions (6, 33–37), but it also limits metabolic changes that accompany cell activation (18, 34). Recently, it has been demonstrated that TGF-β decreases IL-2-induced mitochondrial metabolism, including oxidative phosphorylation (OXPHOS) and maximal respiration in human NK cells (18). Another example of the impact of TME in NK cell metabolism comes from a recent report from patients with colorectal liver metastasis. The authors found that tumor-infiltrating liver-resident NK cells showed signs of mitochondrial stress, including decreased mitochondrial mass and increased reactive oxygen species (ROS) production (38). This reduced mitochondrial metabolism may be an important limitation for NK cell functionality in the TME. In this review, we will discuss how TME could shape NK cell metabolism and thus impair their antitumor activity (Figure 1).
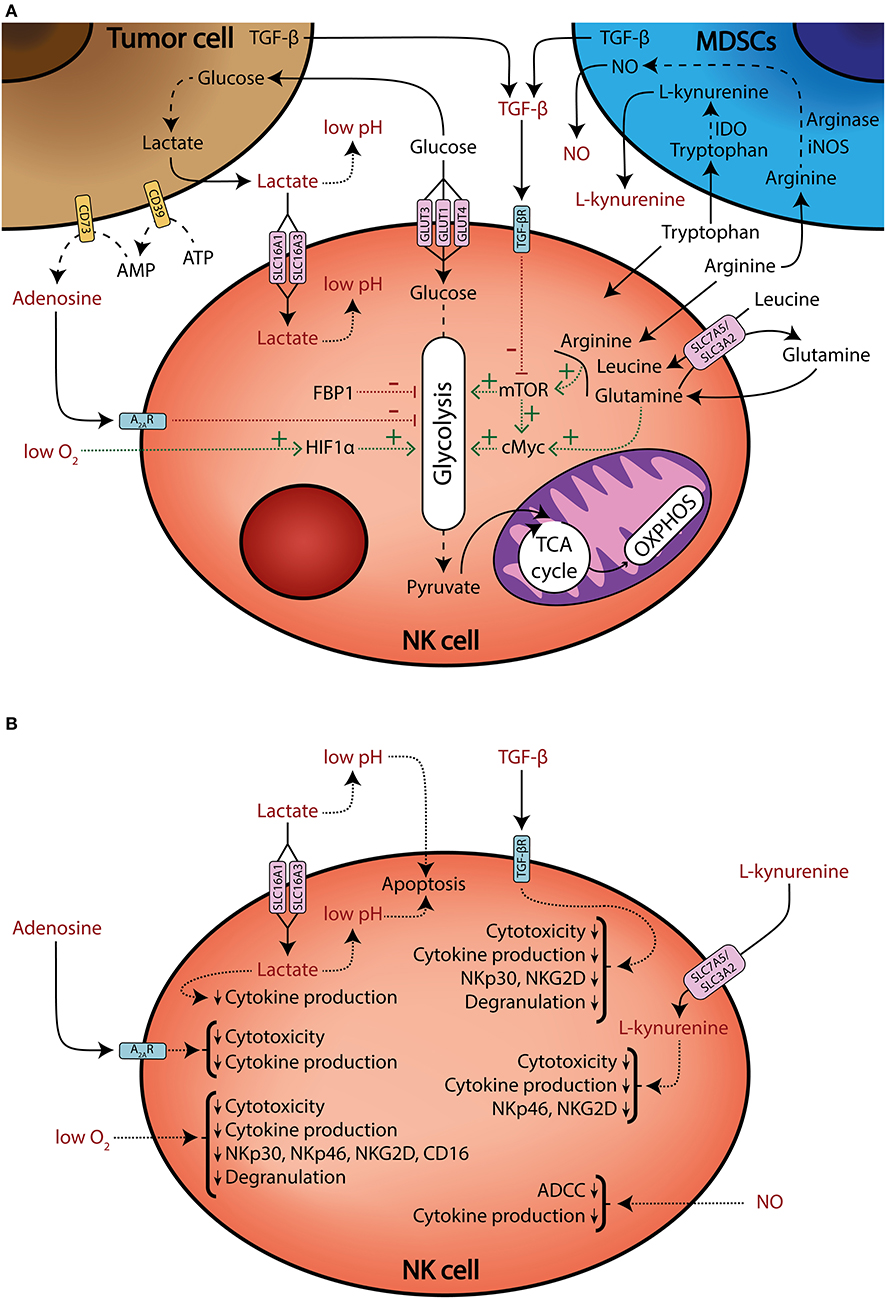
Figure 1. Tumor microenvironment shapes NK cell metabolism and effector functions. (A) Schematic representation of multiple factors that modulate NK cell metabolism (black), and factors that modulate metabolism and/or negatively affect effector functions (red). NK cells compete for nutrients against tumor and myeloid-derived suppressor cells (MDSCs). Tumor cells consume large amounts of glucose and produce lactate, which is transported into NK cells through the SLC16A1 and SLC16A3 transporters, impairing ATP production. Tumor cells also generate extracellular adenosine through the ectonucleotidases CD39 and CD73. Extracellular adenosine inhibits NK cell oxidative phosphorylation (OXPHOS) and glycolytic capacity. Additionally, tumor cells increase amino acid consumption and MDSCs upregulate arginase, IDO, and iNOS enzymes, thus generating an amino acid depleted environment and releasing the immunosuppressive metabolites NO and L-kynurenine. Some amino acids and their transport are necessary to sustain mTOR and cMyc signaling, which promote glycolysis. Moreover, mTOR signaling can be also impaired by TGF-β secreted by tumor cells and MDSCs. The glycolytic pathway can be also modulated by the FBP1 enzyme, which is found to be upregulated in the tumor-infiltrating NK cells of some cancers. Finally, high oxygen consumption by tumor cells and disorganized vascularization can generate hypoxic regions. Hypoxia impairs NK cell effector functions, but also sustains HIF1α, which promotes glycolytic metabolism. Solid lines: release or uptake of different nutrients, metabolites, and other factors. Dashed lines: metabolic processing of substrates. Dotted lines: factors that promote or sustain (green lines), or inhibit (red lines), specific pathways. (B) A schematic representation of several metabolites and other factors present in the TME that limit NK cell effector functions.
Tumor-Derived Metabolites
Besides immunosuppressive cytokines, in the TME there is also an accumulation of tumor-derived metabolites, such as adenosine and lactate that limit antitumor responses. Extracellular adenosine concentration is increased in the hypoxic conditions of tumors, and it plays an important role in immune modulation (39). Hypoxia promotes the release of ATP and AMP, and the ectonucleotidases CD39 and CD73 catalyze the conversion of extracellular ATP to AMP, and AMP to adenosine (40). The stimulation of NK cells through the adenosine A2A receptor (A2AR), the predominant subtype of adenosine receptor expressed in these cells, has been found to suppress their effector functions (40–43). Following this, it has been reported that adenosine inhibits cytotoxic activity of mouse lymphokine-activated killer (LAK) cells (44). A more recent article has shown that adenosine impairs metabolic activity of IL-12/15-stimulated human NK cells by inhibiting their OXPHOS and glycolytic capacity (45). Interestingly, it was reported that there is an elevated interferon γ (IFNγ) production in the presence of adenosine, along with a decrease in NK cell cytotoxicity, suggesting that the metabolic requirements for these two NK cell effector functions are not the same. Indeed, IL-15-stimulated NK cells showed reduced killing of A549 cancer cells in the presence of adenosine (45). A possible explanation for the elevated IFNγ production may be the downregulation of the GAPDH gene observed in IL-12/15-stimulated NK cells exposed to adenosine (45). It has been shown that GAPDH can bind to IFNγ mRNA and prevent its translation (46). However, this transcript-arresting mechanism has not been defined in NK cells yet, and it has to be considered that other mechanisms involved in the regulation of IFNγ production may explain these results.
On the other hand, lactate and low pH have been found to decrease cytotoxic activity of NK cells (47). Exposure of NK cells to lactic acid blocked their IFNγ production following PMA/Ionomycin stimulation (48). A more comprehensive analysis revealed that lactic acid inhibits the upregulation of nuclear factor of activated T cells (NFAT), which is involved in IFNγ transcription (48). Additionally, Brand et al. have also shown that lactic acid uptake by murine NK cells leads to intracellular acidification and to an impaired energy metabolism (measured as intracellular ATP levels) (48). Similar results were obtained in liver-resident NK cells treated with lactic acid, in which intracellular pH and ATP decreased, promoting apoptosis (38). The accumulation of lactate in the TME is mainly due to the metabolic reprogramming of tumors, characterized by primarily using glucose for glycolytic metabolism rather than metabolizing it via OXPHOS. This accelerated glycolysis of cancer cells, induced by multiple factors such as hypoxia and oncogenes (49), may represent a considerable obstacle for NK cell activity, since it is not only causing lactate accumulation but also reducing glucose availability in the TME. Considering that NK cells strongly rely on glucose metabolism to exert their effector functions, as we will discuss in the next section, limiting their key fuel may seriously dampen their antitumor activity. However, not only tumor cells but also many immune cells undergo metabolic reprogramming upon activation, a process that may be especially relevant in the context of the TME and have a significant impact in the tumor progression (50).
Glucose Restriction
Lymphocytes require glucose to survive and its consumption is increased following activation, to support energetic and biosynthetic demands (51). Glucose can be utilized by NK cells for ATP and NADPH generation through different metabolic pathways, or as a carbon source for other biomolecules such as amino acids and fatty acids (19). It has been reported that NK cells express GLUT1, GLUT3, and GLUT4 (15, 21, 22, 52, 53), three glucose transporters from the GLUT family. Additionally, RNA expression of GLUT8 and H+/myo-inositol co-transporter (HMIT or GLUT13) has been also measured in human NK cells (16). However, most studies have been focused on GLUT1, so the expression and regulation of the rest of glucose transporters of the GLUT family are unknown. Upon cytokine-stimulation, NK cells increase GLUT1 expression (21, 22), which is consistent with the augmented glucose uptake and glycolysis that accompanies cell activation (17, 21, 23). Several groups have studied the correlation between the glycolytic pathway and the functionality of activated NK cells, and have shown its relevance in the production of IFNγ and granzyme B, cytotoxicity and proliferative capacity (21, 23–25, 54). These findings are in accordance with those obtained in other lymphocytes. It has been demonstrated that glucose deprivation dampens T cell antitumor activity (46, 55, 56), and that metabolic competition in the TME can regulate cancer progression by impairing antigen-specific responses of tumor-infiltrating T cells (57). Therefore, it is reasonable to hypothesize that in the TME, tumor-driven glucose restriction may reduce glycolysis of NK cells and thus impair their antitumor functions. Cong et al. have addressed this issue by investigating NK cells in a murine model of lung cancer. They have found lower glycolytic rates in NK cells from the lung cancer microenvironment, which also presented attenuated cytotoxicity and cytokine production. Furthermore, Cong et al. have described the increased expression of fructose-1,6-bisphosphatase (FBP1), an enzyme that inhibits glycolysis, in NK cells of the lung cancer microenvironment. More importantly, they have demonstrated that NK cell effector functions can be restored during tumor promotion by inhibiting FBP1 (58). These findings represent a good example of how metabolism can be modulated to improve NK cell antitumor responses.
On another front, Assmann et al. thoroughly analyzed the metabolic reprogramming of cytokine-stimulated NK cells, and described the relevance of citrate-malate shuttle and its regulation by sterol regulatory element-binding proteins (SREBP). They found that SREBP activity is crucial to maintain elevated glycolytic rates and effector functions, including cytotoxicity and IFNγ and granzyme B production (19). Remarkably, some SREBP inhibitors may be increased in the TME, such as 27-hydroxycholesterol, which is found to be elevated in patients with breast, gastric and colorectal cancers (11, 59–62). Considering this fact and that citrate-malate shuttle relies on glucose metabolism, it would be interesting to study the modulation of SREBP in tumor-infiltrating NK cells and to test whether this metabolic pathway configuration is conserved in the TME, where glucose concentration is diminished. Also, it would be of utmost interest to understand the effect of TME in the modulation of other mediators linked to both NK cell metabolism and function, such as AMP-activated protein kinase (AMPK), glycogen synthase kinase 3 β (GSK-3β), diacylglycerol kinases (DGK), regulatory factor X 7 (Rfx7), or inositol-requiring enzyme 1 α (IRE1α) and its substrate X-box-binding protein 1 (XBP1) (63–69). Following this line, it would be also worthwhile to further investigate the role of the mechanistic (or mammalian) target of rapamycin (mTOR), a central metabolic regulator that promotes, among others, the glycolytic pathway. Several authors have pointed the relevance of mTOR for NK cell activation and metabolic reprogramming, and the negative effect of mTOR inhibition in terms of NK cell functionality (12, 17, 21, 23, 26, 70–72). mTOR is sensitive to nutrient availability (73) and can be repressed by TGF-β, which inhibits NK cell metabolism and functionality (18, 33, 34). It is therefore presumable that in the nutrient-deprived TME, where there is also a higher production of TGF-β, mTOR may be inhibited, thereby limiting NK cell effector functions.
Amino Acid Depletion
In addition to glucose, amino acids are also an important fuel for many cellular processes. Tumors show increased amino acid consumption (74) and synergize with tumor-associated cells to create a nutrient-depleted microenvironment. It has been reported that low arginine concentration impairs the proliferation and IFNγ production of the NK-92 cell line and primary human NK cells (75, 76). In addition, mTOR signaling has been found to be inhibited in leucine-depleted media (27, 28). As previously mentioned, mTOR plays a key role in the modulation of the glycolytic pathway, so the impairment of its signaling cascade may lead to diminished effector functions. Similarly, arginine and glutamine levels also affect mTOR signaling (77). Additionally, mTOR has been found to sustain the initial expression of cMyc, a transcription factor that supports the metabolic reprogramming (including the elevated glycolytic rates) required for the functional responses of IL-2/12-stimulated mouse NK cells (27). Moreover, glutamine and amino acid transport through the SLC7A5 transporter are also required for a sustained expression of cMyc (27).
Our current knowledge indicates that amino acid availability may be necessary for NK cell functionality, although the main role of some amino acids could be to maintain the signaling of other metabolic regulators, such as mTOR or cMyc, rather than to be used as a fuel. Indeed, Loftus et al. have reported that glutaminolysis can be inhibited without reducing NK cell functional responses (27). In this line, a previous report indicated that receptor-induced IFNγ production of murine NK cells was not impaired by limiting concentrations of glutamine (24). These findings suggest that targeting amino acid metabolism may enhance NK cell-based therapies, by impairing tumor fuel supply without reducing NK cells' functionality. However, it is necessary to thoroughly explore metabolic requirements of NK cells to validate this hypothesis and to design better therapeutic strategies.
Nonetheless, it should be also considered that amino acid consumption by tumor and tumor-associated cells leads to the accumulation of immunosuppressive catabolites in the TME. Myeloid-derived suppressor cells (MDSCs) upregulate arginase and inducible nitric oxide synthase (iNOS). Both enzymes use arginine as substrate, and the latter catabolizes its conversion to nitric oxide (NO) (6, 78). It has been found that NO impairs NK cell antibody-dependent cellular cytotoxicity, and that the inhibition of iNOS in a mouse model of breast cancer can rescue this function (79). Furthermore, tumor and tumor-associated dendritic cells and fibroblasts show increased expression of IDO, which catabolizes the conversion of tryptophan to L-kynurenine (80, 81). L-kynurenine can be transported through the SLC7A5 transporter (82), and has been found to inhibit NK cell proliferation (81, 83). Also, L-kynurenine inhibits IL-2-induced upregulation of NKp46 and NKG2D receptors and cytokine production of human NK cells in vitro (84).
Hypoxia
Most solid tumors show high oxygen consumption and disorganized vascularization, which leads to the generation of regions permanently or transiently subjected to hypoxia (85). Cells adapt to this hypoxic conditions through the hypoxia-inducible family of transcription factors (HIFs) that modulate a wide range of genes (86). In particular, most dysregulated genes of NK cells under hypoxia are related to metabolic and biosynthetic processes (87), which is consistent with the results obtained in other cells in which HIF-1α promotes or sustains glycolytic metabolism (88–91). In human NK cells, IL-15-priming synergistically acts with short term hypoxia to induce the upregulation of genes involved in the glycolytic pathway (20). Given the relevance of glycolysis for NK cell effector functions, these findings could suggest that these cells may partially conserve their functionality in hypoxic environments. Indeed, available data argue that hypoxia limits, but does not completely block, NK cell responses (92, 93). However, it is still unclear whether HIF-1α and its effect on glycolytic activity play a significant role on NK cells effector functions. It has been reported that Hif1a−/− NK cells have normal metabolic and effector functions in response to IL-2/12 stimulation (27). It would be interesting to study the relationship between HIF-1α, metabolism and effector responses of NK cells under hypoxia. Noteworthy, under hypoxic conditions, there is also a modulation of genes related to immunomodulatory functions and a downregulation of activating receptors, such as NKp30, NKp46, or NKG2D, that contributes to the diminished effector functions (87, 94). Also, hypoxia promotes tumor immune evasion through other mechanisms, such as degrading NK cell-derived granzyme B by autophagy (95). Interestingly, it has been reported that IL-2-priming can increase the expression of the above mentioned activating receptors and improve NK cell cytotoxicity, thereby overcoming the inhibitory effects of hypoxia (93, 96). Considering that there are several antitumor therapeutic strategies based on NK cell stimulation with different cytokines, including IL-2, IL-12, IL-15, IL-18, and IL-21 (29, 31, 32, 97), it would be of great interest to further analyze the effector functions of these cytokine-primed and expanded NK cells under hypoxic conditions.
Other Factors Influencing NK Cell Metabolism
As before mentioned, NK cell metabolism can be modulated by multiple factors, and some of them may have a relevant impact on the anti-cancer therapeutic efficacy. A recent report revealed that obesity impaired mTORC1 activation and limits NK cell antitumor responses (12). The same article has shown that NK cells from obese individuals have a reduced metabolic response following cytokine stimulation (12). Thus, therapies based in the administration of interleukins may show reduced efficacy in these patients. In contrast, a previous report indicated that NK cells from obese children displayed higher levels of glycolysis and mTORC1 activation both at basal levels and upon cytokine stimulation (13). These data suggest that NK cell metabolism may be differentially regulated in adults and children. It would be also interesting to explore NK cell metabolism in other pathologies, such as viral infection, and test whether the metabolic reprogramming is maintained after a prolonged period. Chronic infections have been related to the exhaustion of effector lymphocytes, and exhausted T cells show a different metabolic pattern (98, 99). It has been described that NK cells can also become exhausted during tumor progression or chronic infections (100, 101). Therefore, it would be of great interest to study metabolic requirements of exhausted NK cells, and furthermore, test whether metabolic reprogramming induced by viral infections and tumors could drive NK cell exhaustion.
Concluding Remarks
NK cell effector functions are supported by their metabolism. For instance, IFNγ production could be metabolically regulated at different levels, during transcription, translation, or post-translational processing (54). In the restrictive TME, NK cell metabolism and antitumor responses are impaired (38, 58). Similarly, it has been found that tumor cells compete with T cells for glucose, and this metabolic competition in the TME has been described as a driver of cancer progression (57). In accordance, others have demonstrated that highly glycolytic tumors impair T cell antitumor responses through multiple mechanisms (102). Hence, considering that glucose is a key fuel for NK cells, it is presumable that the metabolic competition of the TME will dampen their effector functions. In addition to glucose, reduced amino acid availability and the accumulation of tumor-derived metabolites may also have a substantial impact on NK cell functionality. Thus, targeting tumor metabolism could be a good option to improve the efficacy of NK cell-based therapies. Alternatively, or additionally, NK cell metabolism may be modified to compete more efficiently for nutrients in the TME, or to be less susceptible to the hypoxia-driven inhibition. For instance, stimulation with different cytokine combinations can upregulate the expression of several nutrient transporters, glycolysis and OXPHOS (11, 33, 103), which can be a good option to enhance NK cell metabolic competitiveness, fitness and plasticity, and thus improve their effector functions in the TME. NK cell stimulation with IL-2, IL-15, or IL-18 has been found to increase the expression of amino acid transporters (28, 72). IL-15-priming sustains murine NK cells production of IFNγ in glutamine-free media, and during the inhibition of fatty acid oxidation with etomoxir (24). Similarly, pretreatment with ALT-803, an IL-15 superagonist, confers some metabolic resistance to the inhibition of glycolysis or mTORC1 with 2-deoxy-D-glucose or rapamycin, respectively (25). However, a continuous exposure to IL-15 can lead to NK cell exhaustion, which is accompanied by a reduction in mitochondrial activity (104). It is therefore necessary to continue exploring NK cell metabolism to understand how it could be modified to resist the metabolically restrictive TME and preserve the effector functions. Undoubtedly, immunometabolism is a fascinating field of research that has been demonstrated to be relevant for NK cell effector functions. Further studies will reveal more precisely how TME shapes NK cell metabolism, which represents an attractive target to focus on to improve NK cell-based immunotherapies.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the following grants: Health Department, Basque Government (2018222038), Basque Foundation for Research and Innovation-EiTB Maratoia (BIO14/TP/003), and AECC-Spanish Association Against Cancer (PROYE16074BORR). IT was recipient of a fellowship from the Jesús de Gangoiti Barrera Foundation (FJGB18/002) and a predoctoral contract funded by the Department of Education, Basque Government (PRE_2018_1_0032). JV was recipient of a predoctoral contract funded by the Department of Education, Basque Government (PRE_2018_2_0211). OZ was recipient of a postdoctoral contract funded by Instituto de Salud Carlos III-Contratos Sara Borrell 2017 (CD17/00128) and the European Social Fund (ESF)-The ESF invests in your future. FB was an Ikerbasque Research Professor, Ikerbasque, Basque Foundation for Science.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Di Vito C, Mikulak J, Zaghi E, Pesce S, Marcenaro E, Mavilio D. NK cells to cure cancer. Semin Immunol. (2019) 41:101272. doi: 10.1016/j.smim.2019.03.004
2. Habif G, Crinier A, André P, Vivier E, Narni-Mancinelli E. Targeting natural killer cells in solid tumors. Cell Mol Immunol. (2019) 16:415–22. doi: 10.1038/s41423-019-0224-2
3. Nayyar G, Chu Y, Cairo MS. Overcoming resistance to natural killer cell based immunotherapies for solid tumors. Front Oncol. (2019) 9:51. doi: 10.3389/fonc.2019.00051
4. Konjević GM, Vuletić AM, Mirjačić Martinović KM, Larsen AK, Jurišić VB. The role of cytokines in the regulation of NK cells in the tumor environment. Cytokine. (2019) 117:30–40. doi: 10.1016/j.cyto.2019.02.001
5. Stojanovic A, Correia MP, Cerwenka A. Shaping of NK cell responses by the tumor microenvironment. Cancer Microenviron. (2013) 6:135–46. doi: 10.1007/s12307-012-0125-8
6. Vitale M, Cantoni C, Pietra G, Mingari MC, Moretta L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur J Immunol. (2014) 44:1582–92. doi: 10.1002/eji.201344272
7. Baginska J, Viry E, Paggetti J, Medves S, Berchem G, Moussay E, et al. The critical role of the tumor microenvironment in shaping natural killer cell-mediated anti-tumor immunity. Front Immunol. (2013) 4:490. doi: 10.3389/fimmu.2013.00490
8. Zenarruzabeitia O, Vitallé J, Astigarraga I, Borrego F. Natural killer cells to the attack: combination therapy against neuroblastoma. Clin Cancer Res. (2017) 23:615–17. doi: 10.1158/1078-0432.CCR-16-2478
9. Park A, Lee Y, Kim MS, Kang YJ, Park Y-J, Jung H, et al. Prostaglandin E2 secreted by thyroid cancer cells contributes to immune escape through the suppression of Natural Killer (NK) cell cytotoxicity and NK cell differentiation. Front Immunol. (2018) 9:1859. doi: 10.3389/fimmu.2018.01859
10. André P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell. (2018) 175:1731–43.e13. doi: 10.1016/j.cell.2018.10.014
11. O'Brien KL, Finlay DK. Immunometabolism and natural killer cell responses. Nat Rev Immunol. (2019) 19:282–90. doi: 10.1038/s41577-019-0139-2
12. Michelet X, Dyck L, Hogan A, Loftus RM, Duquette D, Wei K, et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat Immunol. (2018) 19:1330–40. doi: 10.1038/s41590-018-0251-7
13. Tobin LM, Mavinkurve M, Carolan E, Kinlen D, O'Brien EC, Little MA, et al. NK cells in childhood obesity are activated, metabolically stressed, and functionally deficient. JCI Insight. (2017) 2:94939. doi: 10.1172/jci.insight.94939
14. Cichocki F, Wu C-Y, Zhang B, Felices M, Tesi B, Tuininga K, et al. ARID5B regulates metabolic programming in human adaptive NK cells. J Exp Med. (2018) 215:2379–95. doi: 10.1084/jem.20172168
15. Pfeifer C, Highton AJ, Peine S, Sauter J, Schmidt AH, Bunders MJ, et al. Natural killer cell education is associated with a distinct glycolytic profile. Front Immunol. (2018) 9:3020. doi: 10.3389/fimmu.2018.03020
16. Schafer JR, Salzillo TC, Chakravarti N, Kararoudi MN, Trikha P, Foltz JA, et al. Education-dependent activation of glycolysis promotes the cytolytic potency of licensed human natural killer cells. J Allergy Clin Immunol. (2019) 143:346–58.e6. doi: 10.1016/j.jaci.2018.06.047
17. Marçais A, Cherfils-Vicini J, Viant C, Degouve S, Viel S, Fenis A, et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat Immunol. (2014) 15:749–57. doi: 10.1038/ni.2936
18. Zaiatz-Bittencourt V, Finlay DK, Gardiner CM. Canonical TGF-β signaling pathway represses human NK cell metabolism. J Immunol. (2018) 200:3934–41. doi: 10.4049/jimmunol.1701461
19. Assmann N, O'Brien KL, Donnelly RP, Dyck L, Zaiatz-Bittencourt V, Loftus RM, et al. Srebp-controlled glucose metabolism is essential for NK cell functional responses. Nat Immunol. (2017) 18:1197–206. doi: 10.1038/ni.3838
20. Velásquez SY, Killian D, Schulte J, Sticht C, Thiel M, Lindner HA. Short term hypoxia synergizes with interleukin 15 priming in driving glycolytic gene transcription and supports human natural killer cell activities. J Biol Chem. (2016) 291:12960–77. doi: 10.1074/jbc.M116.721753
21. Keating SE, Zaiatz-Bittencourt V, Loftus RM, Keane C, Brennan K, Finlay DK, et al. Metabolic reprogramming supports IFN-γ production by CD56 bright NK cells. J Immunol. (2016) 196:2552–60. doi: 10.4049/jimmunol.1501783
22. Salzberger W, Martrus G, Bachmann K, Goebels H, Heß L, Koch M, et al. Tissue-resident NK cells differ in their expression profile of the nutrient transporters Glut1, CD98 and CD71. PLoS ONE. (2018) 13:e0201170. doi: 10.1371/journal.pone.0201170
23. Donnelly RP, Loftus RM, Keating SE, Liou KT, Biron CA, Gardiner CM, et al. mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J Immunol. (2014) 193:4477–84. doi: 10.4049/jimmunol.1401558
24. Keppel MP, Saucier N, Mah AY, Vogel TP, Cooper MA. Activation-specific metabolic requirements for NK cell IFN-γ production. J Immunol. (2015) 194:1954–62. doi: 10.4049/jimmunol.1402099
25. Mah AY, Rashidi A, Keppel MP, Saucier N, Moore EK, Alinger JB, et al. Glycolytic requirement for NK cell cytotoxicity and cytomegalovirus control. JCI Insight. (2017) 2:1–17. doi: 10.1172/jci.insight.95128
26. Mao Y, van Hoef V, Zhang X, Wennerberg E, Lorent J, Witt K, et al. IL-15 activates mTOR and primes stress-activated gene expression leading to prolonged antitumor capacity of NK cells. Blood. (2016) 128:1475–89. doi: 10.1182/blood-2016-02-698027
27. Loftus RM, Assmann N, Kedia-Mehta N, O'Brien KL, Garcia A, Gillespie C, et al. Amino acid-dependent cMyc expression is essential for NK cell metabolic and functional responses in mice. Nat Commun. (2018) 9:2341. doi: 10.1038/s41467-018-04719-2
28. Almutairi SM, Ali AK, He W, Yang D, Ghorbani P, Wang L, et al. Interleukin-18 up-regulates amino acid transporters and facilitates amino acid–induced mTORC1 activation in natural killer cells. J Biol Chem. (2019) 294:4644–55. doi: 10.1074/jbc.RA118.005892
29. Fang F, Xiao W, Tian Z. NK cell-based immunotherapy for cancer. Semin Immunol. (2017) 31:37–54. doi: 10.1016/j.smim.2017.07.009
30. Becker PSA, Suck G, Nowakowska P, Ullrich E, Seifried E, Bader P, et al. Selection and expansion of natural killer cells for NK cell-based immunotherapy. Cancer Immunol Immunother. (2016) 65:477–84. doi: 10.1007/s00262-016-1792-y
31. Granzin M, Wagner J, Köhl U, Cerwenka A, Huppert V, Ullrich E. Shaping of natural killer cell antitumor activity by ex vivo cultivation. Front Immunol. (2017) 8:458. doi: 10.3389/fimmu.2017.00458
32. Terrén I, Mikelez I, Odriozola I, Gredilla A, González J, Orrantia A, et al. Implication of interleukin (IL)-12/15/18 and ruxolitinib in the phenotype, proliferation and polyfunctionality of human cytokine-preactivated NK cells. Front Immunol. (2018) 9:737. doi: 10.3389/fimmu.2018.00737
33. Viel S, Besson L, Marotel M, Walzer T, Marçais A. Regulation of mTOR, metabolic fitness, and effector functions by cytokines in natural killer cells. Cancers. (2017) 9:132. doi: 10.3390/cancers9100132
34. Viel S, Marçais A, Guimaraes FS, Loftus R, Rabilloud J, Grau M, et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci Signal. (2016) 9:ra19. doi: 10.1126/scisignal.aad1884
35. Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, et al. Transforming growth factor 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci USA. (2003) 100:4120–5. doi: 10.1073/pnas.0730640100
36. Lee J-C, Lee K-M, Kim D-W, Heo DS. Elevated TGF-β1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J Immunol. (2004) 172:7335–40. doi: 10.4049/jimmunol.172.12.7335
37. Ghiringhelli F, Ménard C, Terme M, Flament C, Taieb J, Chaput N, et al. CD4 + CD25 + regulatory T cells inhibit natural killer cell functions in a transforming growth factor–β-dependent manner. J Exp Med. (2005) 202:1075–85. doi: 10.1084/jem.20051511
38. Harmon C, Robinson MW, Hand F, Almuaili D, Mentor K, Houlihan DD, et al. Lactate-mediated acidification of tumor microenvironment induces apoptosis of liver-resident NK cells in colorectal liver metastasis. Cancer Immunol Res. (2019) 7:335–46. doi: 10.1158/2326-6066.CIR-18-0481
39. Ohta A. A metabolic immune checkpoint: adenosine in tumor microenvironment. Front Immunol. (2016) 7:109. doi: 10.3389/fimmu.2016.00109
40. Chambers AM, Lupo KB, Matosevic S. Tumor microenvironment-induced immunometabolic reprogramming of natural killer cells. Front Immunol. (2018) 9:2517. doi: 10.3389/fimmu.2018.02517
41. Lokshin A, Raskovalova T, Huang X, Zacharia LC, Jackson EK, Gorelik E. Adenosine-mediated inhibition of the cytotoxic activity and cytokine production by activated natural killer cells. Cancer Res. (2006) 66:7758–65. doi: 10.1158/0008-5472.CAN-06-0478
42. Young A, Ngiow SF, Gao Y, Patch A-M, Barkauskas DS, Messaoudene M, et al. A2AR adenosine signaling suppresses natural killer cell maturation in the tumor microenvironment. Cancer Res. (2018) 78:1003–16. doi: 10.1158/0008-5472.CAN-17-2826
43. Sek K, Mølck C, Stewart G, Kats L, Darcy P, Beavis P. Targeting adenosine receptor signaling in cancer immunotherapy. Int J Mol Sci. (2018) 19:E3837. doi: 10.3390/ijms19123837
44. Raskovalova T, Huang X, Sitkovsky M, Zacharia LC, Jackson EK, Gorelik E. Gs protein-coupled adenosine receptor signaling and lytic function of activated NK cells. J Immunol. (2005) 175:4383–91. doi: 10.4049/jimmunol.175.7.4383
45. Chambers AM, Wang J, Lupo KB, Yu H, Atallah Lanman NM, Matosevic S. Adenosinergic signaling alters natural killer cell functional responses. Front Immunol. (2018) 9:2533. doi: 10.3389/fimmu.2018.02533
46. Chang C-H, Curtis JD, Maggi LB, Faubert B, Villarino AV, O'Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. (2013) 153:1239–51. doi: 10.1016/j.cell.2013.05.016
47. Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J Immunol. (2013) 191:1486–95. doi: 10.4049/jimmunol.1202702
48. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-Associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. (2016) 24:657–71. doi: 10.1016/j.cmet.2016.08.011
49. Renner K, Singer K, Koehl GE, Geissler EK, Peter K, Siska PJ, et al. Metabolic hallmarks of tumor and immune cells in the tumor microenvironment. Front Immunol. (2017) 8:248. doi: 10.3389/fimmu.2017.00248
50. Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity. (2015) 43:435–49. doi: 10.1016/j.immuni.2015.09.001
51. MacIver NJ, Jacobs SR, Wieman HL, Wofford JA, Coloff JL, Rathmell JC. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J Leukoc Biol. (2008) 84:949–57. doi: 10.1189/jlb.0108024
52. Maratou E, Dimitriadis G, Kollias A, Boutati E, Lambadiari V, Mitrou P, et al. Glucose transporter expression on the plasma membrane of resting and activated white blood cells. Eur J Clin Invest. (2007) 37:282–90. doi: 10.1111/j.1365-2362.2007.01786.x
53. Piatkiewicz P, Bernat-Karpinska M, Miłek T, Rabijewski M, Rosiak E. NK cell count and glucotransporter 4 (GLUT4) expression in subjects with type 2 diabetes and colon cancer. Diabetol Metab Syndr. (2016) 8:38. doi: 10.1186/s13098-016-0152-6
54. Mah AY, Cooper MA. Metabolic regulation of natural killer cell IFN-γ production. Crit Rev Immunol. (2016) 36:131–47. doi: 10.1615/CritRevImmunol.2016017387
55. Ho P, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell. (2015) 162:1217–28. doi: 10.1016/j.cell.2015.08.012
56. Cham CM, Driessens G, O'Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur J Immunol. (2008) 38:2438–50. doi: 10.1002/eji.200838289
57. Chang C, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
58. Cong J, Wang X, Zheng X, Wang D, Fu B, Sun R, et al. Dysfunction of natural killer cells by FBP1-induced inhibition of glycolysis during lung cancer progression. Cell Metab. (2018) 28:243–55.e5. doi: 10.1016/j.cmet.2018.06.021
59. Li D, Long W, Huang R, Chen Y, Xia M. 27-Hydroxycholesterol inhibits sterol regulatory element-binding protein 1 activation and hepatic lipid accumulation in mice. Obesity. (2018) 26:713–22. doi: 10.1002/oby.22130
60. Wu Q, Ishikawa T, Sirianni R, Tang H, McDonald JG, Yuhanna IS, et al. 27-Hydroxycholesterol promotes cell-autonomous, ER-positive breast cancer growth. Cell Rep. (2013) 5:637–45. doi: 10.1016/j.celrep.2013.10.006
61. Guo F, Hong W, Yang M, Xu D, Bai Q, Li X, et al. Upregulation of 24(R/S),25-epoxycholesterol and 27-hydroxycholesterol suppresses the proliferation and migration of gastric cancer cells. Biochem Biophys Res Commun. (2018) 504:892–8. doi: 10.1016/j.bbrc.2018.09.058
62. Rossin D, Dias IHK, Solej M, Milic I, Pitt AR, Iaia N, et al. Increased production of 27-hydroxycholesterol in human colorectal cancer advanced stage: possible contribution to cancer cell survival and infiltration. Free Radic Biol Med. (2019) 136:35–44. doi: 10.1016/j.freeradbiomed.2019.03.020
63. Kobayashi T, Mattarollo SR. Natural killer cell metabolism. Mol Immunol. (2017). doi: 10.1016/j.molimm.2017.11.021. [Epub ahead of print].
64. Müller-Durovic B, Lanna A, Polaco Covre L, Mills RS, Henson SM, Akbar AN. Killer cell lectin-like receptor G1 inhibits NK cell function through activation of adenosine 5′-monophosphate–activated protein kinase. J Immunol. (2016) 197:2891–9. doi: 10.4049/jimmunol.1600590
65. Parameswaran R, Ramakrishnan P, Moreton SA, Xia Z, Hou Y, Lee DA, et al. Repression of GSK3 restores NK cell cytotoxicity in AML patients. Nat Commun. (2016) 7:11154. doi: 10.1038/ncomms11154
66. Fengjiao J, Zhaozhen W, Xiao H, Jiahui Z, Zihe G, Xiao H, et al. The PI3K/Akt/GSK-3β/ROS/eIF2B pathway promotes breast cancer growth and metastasis via suppression of NK cell cytotoxicity and tumor cell susceptibility. Cancer Biol Med. (2019) 16:38. doi: 10.20892/j.issn.2095-3941.2018.0253
67. Yang E, Singh BK, Paustian AMS, Kambayashi T. Diacylglycerol kinase ζ is a target to enhance NK cell function. J Immunol. (2016) 197:934–41. doi: 10.4049/jimmunol.1600581
68. Castro W, Chelbi ST, Niogret C, Ramon-Barros C, Welten SPM, Osterheld K, et al. The transcription factor Rfx7 limits metabolism of NK cells and promotes their maintenance and immunity. Nat Immunol. (2018) 19:809–20. doi: 10.1038/s41590-018-0144-9
69. Dong H, Adams NM, Xu Y, Cao J, Allan DSJ, Carlyle JR, et al. The IRE1 endoplasmic reticulum stress sensor activates natural killer cell immunity in part by regulating c-Myc. Nat Immunol. (2019) 20:865–78. doi: 10.1038/s41590-019-0388-z
70. Wang F, Meng M, Mo B, Yang Y, Ji Y, Huang P, et al. Crosstalks between mTORC1 and mTORC2 variagate cytokine signaling to control NK maturation and effector function. Nat Commun. (2018) 9:4874. doi: 10.1038/s41467-018-07277-9
71. Nandagopal N, Ali AK, Komal AK, Lee S-H. The critical role of IL-15-PI3K-mTOR pathway in natural killer cell effector functions. Front Immunol. (2014) 5:187. doi: 10.3389/fimmu.2014.00187
72. Jensen H, Potempa M, Gotthardt D, Lanier LL. Cutting edge: IL-2–induced expression of the amino acid transporters SLC1A5 and CD98 is a prerequisite for NKG2D-mediated activation of human NK cells. J Immunol. (2017) 199:1967–72. doi: 10.4049/jimmunol.1700497
73. Walls J, Sinclair L, Finlay D. Nutrient sensing, signal transduction and immune responses. Semin Immunol. (2016) 28:396–407. doi: 10.1016/j.smim.2016.09.001
74. Lukey MJ, Katt WP, Cerione RA. Targeting amino acid metabolism for cancer therapy. Drug Discov Today. (2017) 22:796–804. doi: 10.1016/j.drudis.2016.12.003
75. Lamas B, Vergnaud-Gauduchon J, Goncalves-Mendes N, Perche O, Rossary A, Vasson M-P, et al. Altered functions of natural killer cells in response to L-Arginine availability. Cell Immunol. (2012) 280:182–90. doi: 10.1016/j.cellimm.2012.11.018
76. Oberlies J, Watzl C, Giese T, Luckner C, Kropf P, Muller I, et al. Regulation of NK cell function by human granulocyte arginase. J Immunol. (2009) 182:5259–67. doi: 10.4049/jimmunol.0803523
77. Kedia-Mehta N, Finlay DK. Competition for nutrients and its role in controlling immune responses. Nat Commun. (2019) 10:2123. doi: 10.1038/s41467-019-10015-4
78. Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. (2017) 5:3–8. doi: 10.1158/2326-6066.CIR-16-0297
79. Stiff A, Trikha P, Mundy-Bosse B, McMichael E, Mace TA, Benner B, et al. Nitric oxide production by myeloid-derived suppressor cells plays a role in impairing Fc receptor–mediated natural killer cell function. Clin Cancer Res. (2018) 24:1891–904. doi: 10.1158/1078-0432.CCR-17-0691
80. Li T, Yang Y, Hua X, Wang G, Liu W, Jia C, et al. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. (2012) 318:154–61. doi: 10.1016/j.canlet.2011.12.020
81. Liu X, Shin N, Koblish HK, Yang G, Wang Q, Wang K, et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood. (2010) 115:3520–30. doi: 10.1182/blood-2009-09-246124
82. Sinclair LV, Neyens D, Ramsay G, Taylor PM, Cantrell DA. Single cell analysis of kynurenine and system L amino acid transport in T cells. Nat Commun. (2018) 9:1981. doi: 10.1038/s41467-018-04366-7
83. Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med. (2002) 196:459–68. doi: 10.1084/jem.20020121
84. Chiesa MD, Carlomagno S, Frumento G, Balsamo M, Cantoni C, Conte R, et al. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood. (2006) 108:4118–25. doi: 10.1182/blood-2006-03-006700
85. Petrova V, Annicchiarico-Petruzzelli M, Melino G, Amelio I. The hypoxic tumour microenvironment. Oncogenesis. (2018) 7:10. doi: 10.1038/s41389-017-0011-9
86. Hasmim M, Messai Y, Ziani L, Thiery J, Bouhris J-H, Noman MZ, et al. Critical role of tumor microenvironment in shaping NK cell functions: implication of hypoxic stress. Front Immunol. (2015) 6:482. doi: 10.3389/fimmu.2015.00482
87. Parodi M, Raggi F, Cangelosi D, Manzini C, Balsamo M, Blengio F, et al. Hypoxia modifies the transcriptome of human NK cells, modulates their immunoregulatory profile, and influences NK cell subset migration. Front Immunol. (2018) 9:2358. doi: 10.3389/fimmu.2018.02358
88. Gnanaprakasam JNR, Sherman JW, Wang R. MYC and HIF in shaping immune response and immune metabolism. Cytokine Growth Factor Rev. (2017) 35:63–70. doi: 10.1016/j.cytogfr.2017.03.004
89. Burrows N, Maxwell PH. Hypoxia and B cells. Exp Cell Res. (2017) 356:197–203. doi: 10.1016/j.yexcr.2017.03.019
90. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1α-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH 17 and T reg cells. J Exp Med. (2011) 208:1367–76. doi: 10.1084/jem.20110278
91. Pearce EL, Poffenberger MC, Chang C-H, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. (2013) 342:1242454. doi: 10.1126/science.1242454
92. Fink T, Ebbesen P, Koppelhus U, Zachar V. Natural killer cell-mediated basal and interferon-enhanced cytotoxicity against liver cancer cells is significantly impaired under in vivo oxygen conditions. Scand J Immunol. (2003) 58:607–12. doi: 10.1111/j.1365-3083.2003.01347.x
93. Sarkar S, Germeraad WT, Rouschop KM, Steeghs EM, van Gelder M, Bos GM, et al. Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PLoS ONE. (2013) 8:e64835. doi: 10.1371/journal.pone.0064835
94. Balsamo M, Manzini C, Pietra G, Raggi F, Blengio F, Mingari MC, et al. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur J Immunol. (2013) 43:2756–64. doi: 10.1002/eji.201343448
95. Baginska J, Viry E, Berchem G, Poli A, Noman MZ, van Moer K, et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci USA. (2013) 110:17450–5. doi: 10.1073/pnas.1304790110
96. Huenecke S, Zimmermann SY, Kloess S, Esser R, Brinkmann A, Tramsen L, et al. IL-2–driven regulation of NK cell receptors with regard to the distribution of CD16+ and CD16– subpopulations and in vivo influence after haploidentical NK cell infusion. J Immunother. (2010) 33:200–10. doi: 10.1097/CJI.0b013e3181bb46f7
97. Ewen EM, Pahl JHW, Miller M, Watzl C, Cerwenka A. KIR downregulation by IL-12/15/18 unleashes human NK cells from KIR/HLA-I inhibition and enhances killing of tumor cells. Eur J Immunol. (2018) 48:355–65. doi: 10.1002/eji.201747128
98. Wherry EJ, Ha S-J, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. (2007) 27:670–84. doi: 10.1016/j.immuni.2007.11.006
99. Schurich A, Pallett LJ, Jajbhay D, Wijngaarden J, Otano I, Gill US, et al. Distinct metabolic requirements of exhausted and functional virus-specific CD8 T cells in the same host. Cell Rep. (2016) 16:1243–52. doi: 10.1016/j.celrep.2016.06.078
101. Zhang C, Wang X, Li S, Twelkmeyer T, Wang W, Zhang S, et al. NKG2A is a NK cell exhaustion checkpoint for HCV persistence. Nat Commun. (2019) 10:1507. doi: 10.1038/s41467-019-09212-y
102. Cascone T, McKenzie JA, Mbofung RM, Punt S, Wang Z, Xu C, et al. Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab. (2018) 27:977–87.e4. doi: 10.1016/j.cmet.2018.02.024
103. Gardiner CM. NK cell metabolism. J Leukoc Biol. (2019) 105:1235–42. doi: 10.1002/JLB.MR0718-260R
Keywords: NK cell, metabolism, glucose, glycolysis, amino acid, hypoxia, tumor microenvironment, TME
Citation: Terrén I, Orrantia A, Vitallé J, Zenarruzabeitia O and Borrego F (2019) NK Cell Metabolism and Tumor Microenvironment. Front. Immunol. 10:2278. doi: 10.3389/fimmu.2019.02278
Received: 05 July 2019; Accepted: 09 September 2019;
Published: 24 September 2019.
Edited by:
Julian Pardo, Fundacion Agencia Aragonesa para la Investigacion y el Desarrollo, SpainReviewed by:
Ana Stojanovic, University of Heidelberg, GermanyUlrike Koehl, Hannover Medical School, Germany
Copyright © 2019 Terrén, Orrantia, Vitallé, Zenarruzabeitia and Borrego. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francisco Borrego, ZnJhbmNpc2NvLmJvcnJlZ29yYWJhc2NvQG9zYWtpZGV0emEuZXVz