
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 13 September 2019
Sec. Inflammation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02204
This article is part of the Research TopicPlatelets and Immune Responses during ThromboinflammationView all 17 articles
 Li Guo1
Li Guo1 Matthew T. Rondina1,2,3,4*
Matthew T. Rondina1,2,3,4*Platelets are anucleate cells produced by megakaryocytes. In recent years, a robust body of literature supports the evolving role of platelets as key sentinel and effector cells in infectious diseases, especially critical in bridging hemostatic, inflammatory, and immune continuums. Upon intravascular pathogen invasion, platelets can directly sense viral, parasitic, and bacterial infections through pattern recognition receptors and integrin receptors or pathogen: immunoglobulin complexes through Fc and complement receptors—although our understanding of these interactions remains incomplete. Constantly scanning for areas of injury or inflammation as they circulate in the vasculature, platelets also indirectly respond to pathogen invasion through interactions with leukocytes and the endothelium. Following antigen recognition, platelets often become activated. Through a diverse repertoire of mechanisms, activated platelets can directly sequester or kill pathogens, or facilitate pathogen clearance by activating macrophages and neutrophils, promoting neutrophil extracellular traps (NETs) formation, forming platelet aggregates and microthrombi. At times, however, platelet activation may also be injurious to the host, exacerbating inflammation and promoting endothelial damage and thrombosis. There are many gaps in our understandings of the role of platelets in infectious diseases. However, with the emergence of advanced technologies, our knowledge is increasing. In the current review, we mainly discuss these evolving roles of platelets under four different infectious pathogen infections, of which are dengue, malaria, Esterichia coli (E. coli) and staphylococcus aureus S. aureus, highlighting the complex interplay of these processes with hemostatic and thrombotic pathways.
Infectious diseases remain a leading cause of morbidity and mortality worldwide. Low platelet number, termed thrombocytopenia, is common in infectious diseases (also referred to at times as sepsis). The mechanisms are often multifactorial, but increased platelet clearance and/or decreased platelet production are common. Sepsis-associated thrombocytopenia has been recognized for many years and is a predictor of adverse outcomes.
Until more recently, the involvement of platelets in the pathogenesis of acute infectious diseases has been less often studied. Part of the reason might be the traditional dogma that platelets are primary effectors of hemostasis and thrombosis, rather than participating in host immune responses against infections. With the expansion of our knowledge in the last decade or so, it is increasingly recognized that hemostasis, thrombosis, and inflammation are tightly interconnected processes and that platelets are often the cell that bridge these three processes.
Classically, hemostasis is referred to as the process of clot formation under normal physiological situations to stop bleeding upon blood vessel damage (e.g., wound). Thrombosis is defined as clot formation under pathological situations (e.g., atherosclerosis plaque rupture, deep vein thrombosis, pulmonary embolism, and stroke – among other examples). Infections, particularly when acute, are known to increase the risk of thrombosis. For example, Kaplan et al. identified a cohort of septic patients, that approximately one-third of patients developed a deep vein thrombosis and/or pulmonary embolism (1–3). Although the reasons that infection trigger thrombosis are multifactorial and incompletely understood, inflammation is thought to be a contributing factor. In particular, there is increasing recognition that inflammation may directly activate the hemostatic systems, resulting in thrombosis (4, 5).
Inflammation is generally considered a process of various immune responses that causes clinical symptoms include heat, pain, redness, and swelling. Similar to thrombosis, inflammation can also be triggered by wounds, tissue damage, and pathogen invasion. The traditional separation of “thrombosis” and inflammation,” while helping to understand the key physiological processes upon injury or infection, may hinder an understanding of the complete picture. Moreover, pursuits of novel therapeutics too focused on one aspect of the disease may not be successful (6).
In recent years, words like “thromboinflammation,” “immunothrombosis,” and “immunohemostasis” have been used to describe responses/mechanisms that are involved in both thrombosis and inflammation (7–11). To our knowledge, “thromboinflammation” was initially used by Blair et al. in 2009 to describe their discovery that the activation of platelet toll-like receptor 2 (TLR2), a receptor best known for its role in triggering inflammation, also promotes thrombosis (7). In 2013, Engelmann used the word “Immunothrombosis” to describe thrombosis “triggered by” or “involved with” innate immune responses (8). While not of our invention, we and others have used “immunohemostasis” to describe hemostatic responses that involves immune players under physiologic situations, as compared to the pathologic property of “thrombosis” (12). To clarify our discussion below, here we will define “immunohemostasis” as an integrated process that includes the classic coagulation, clot formation, as well as the immune responses for pathogen trapping upon blood vessel damage without pathological consequences. In contrast, “thromboinflammation” will refer to pathological responses within the vasculature following blood vessel injury, invasion by a variety of pathogens, or non-infectious inflammatory triggers. We hope that by this definition, the process of thromboinflammation be an umbrella that considers thrombus formation, coagulation system activation, and innate and adaptive immunity as an integrated detrimental process. Thus, thromboinflammation is associated with diseases that are historically under separated categories. Some examples include thrombotic diseases like stroke, deep vein thrombosis, and myocardial infarction, infectious diseases such as bacteremia, viremia, and parasitemia, cancer metastasis through blood vessels, and disseminated intravascular coagulation (DIC).
In the past decade, efforts in better understanding the pathogenesis of infectious diseases have led to new discoveries of the critical roles that platelets have in thromboinflammation. In some settings, platelets may be protective through limiting pathogen dissemination, directly killing pathogens, and eliciting timely and adaptive host immune responses timely. In other situations, however, platelet responses during infection may be harmful. In our current review, we will focus on the interactions between platelets and classic immune cells during infectious diseases. We use dengue, malaria, Esterichia coli (E. coli) and Staphylococcus aureus (S. aureus) infections as specific pathogen examples to illustrate the thromboinflammation as an integrated process in which platelets actively participate. Platelets are also involved in many other infectious diseases outside the scope of this review. Readers are referred to several other excellent summaries of this topic (13–19).
Platelets are the smallest cells in blood circulation with a diameter of 2–3 μm under resting conditions (20). They are anucleate cells produced by fragmentation of the megakaryocyte extrusions into the vasculature, formed mostly in the bone marrow, although other sites of platelet genesis include the lung, spleen, and liver (21). Differentiated from hematopoietic stem cells or common myeloid progenitors (22–24), megakaryocytes and platelets share many myeloid lineage features, such as the expression of a panel of pattern recognition receptors (PRRs), phagocytosis of exogenous antigens, interactions with other immune cells, and the release of chemokines and cytokines upon activation (15, 20, 25, 26). Having a life span of 7–10 days in healthy humans and about 5 days in the mouse, it is estimated that about 100 billion platelets are produced every day in humans, with about 2 billion a day in mice (20). Platelets are mighty patrollers that constantly scan over the vascular endothelium and circulating leukocytes in a “touch and go” manner (27). Upon activation, platelets rapidly undergo massive plasma membrane extension (spreading), become activated, translocate and/or express multiple receptors on their surface that further enhance their aggregation with nearby platelets or leukocytes, or directly bind to and sequester extracellular pathogens (20). Platelets also degranulate and release pre-packed (or in some cases) newly-synthesized microbicide proteins and chemokines that facilitate pathogen destruction, signal immune cells, and promote inflammation (20). The reader is also referred to a recent review by Rossaint et al. that provides more detailed information on the platelet receptors and chemokines involved in inflammation (28).
Work from our group and others demonstrates that during pathogen invasion, platelets have alterations in their transcriptome and proteome that augment host defense mechanisms (29–31), although these changes may also result in adverse outcomes in some settings. These and other features make platelets effective and dynamic sentinels against bloodborne pathogens.
Dengue virus (DENV) is a mosquito-borne, positive sense, single-stranded RNA virus of the Flaviviridae family, with five serotypes documented so far (32, 33). The annual incidence of dengue is estimated to be 390 million globally, with about 100 million individuals having clinically apparent symptoms (34). While in most individuals the disease is self-limited with a high fever as the only symptom, about 10% of patients have thrombocytopenia and hemorrhagic symptoms (termed dengue hemorrhagic fever, DHF). In severe cases of dengue infection, patients may develop dengue hemorrhagic shock (DHS) (35). Morbidity and mortality rates in these last two types of dengue infection can be rather high. Emerging and established studies highlight that platelets are implicated centrally in the pathogenesis of the disease. Figure 1 summarizes some of the responses of platelets during dengue infection.
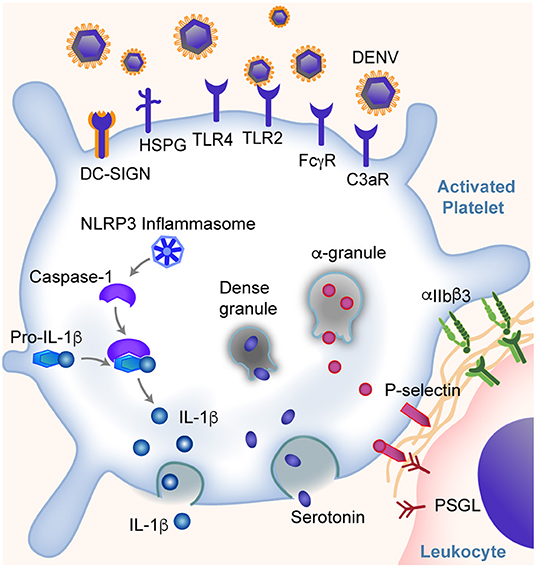
Figure 1. Platelets are key sentinel and effector cells during dengue infection. Platelets can directly bind dengue virus (DENV) and virus: IgG immune complexes through DC-SIGN and other receptors as listed. This binding leads to altered gene and protein expression in platelets and the activation of platelets. Some of these responses include the expression of IFITM3, assembly of NLRP3 inflammasomes, production and release of IL-1β, secretion of α-granule and dense granule contents, translocation of P-selectin to the platelet surface (allowing platelet-leukocyte interactions and signaling), and integrin αIIbβ3 activation. Changes in platelets further trigger the platelet aggregation and thrombosis, endothelial inflammation and vascular leakage, and monocyte activation and cytokine production, and more. These responses span the classic pathogenesis of thrombosis and inflammation, and may contribute to hemorrhagic symptoms and shock in some patients.
Dengue virus can directly bind and activate human platelets via multiple receptors. The direct interaction was suggested from observations of dengue virus RNA and dengue-like particles in platelets from patients with dengue (36). Later studies demonstrated that the direct interactions between dengue and platelets are mediated through dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) and heparin sulfate proteoglycan receptors (HSPG) on platelets (37, 38). Intriguingly, the expression of DC-SIGN on platelets appears to decrease in some patients as dengue infection resolves (39). In addition, Chao et al. showed direct platelet activation by DENV nonstructural protein 1 (NS1) through interactions with TLR4 and TLR2 (40). Recently, Sung et al. showed DENV induces platelet activation via CLEC-2 (41). Following DENV binding, platelets become activated, undergo a series of changes that further amplify the thromboinflammation, which will be discussed later.
Similar to myeloid cells, platelets also express Fc receptors that are capable of binding IgG opsonized DENV complexes. This mediates, in part, the development of thrombocytopenia and DHF/DHS in patients infected with DENV. Fc receptors have recently been shown to play a central role in antibody-dependent enhancement (ADE) during dengue infection. ADE is the clinical setting whereby individuals previously exposed to (or immunized against) one dengue serotype have an increased risk of more severe dengue (e.g., DHF/DHS) when they become infected with a different dengue serotype (42, 43), suggesting an intimate relationship between antibody mediated inflammatory responses and platelets. The work by Katzelnick et al., which included a large cohort of more than 8,000 patients with dengue, reported that ADE and severity of dengue is associated with DENV-antibody titers between 1:21 and 1:80 (43). Moreover, during the early phase of DENV infection, the glycosylation of Fc regions and the ratio between IgG subclasses (IgG1/IgG2) appears to regulate platelet counts (42). Human platelets express FcγRIIA and FcγRIIIA receptors that can engage both IgG1 and IgG2 (31, 42, 44–46). Increased afucosylation (the absence of fucose on the Fc glycan) of the CH2 domain of the Fc region of both IgG1 and IgG2, together with an increased IgG1/IgG2 ratio, leading to altered binding affinity between IgG and FcγR receptors on platelets (42). In humanized FcR transgenic mice, this results in significant thrombocytopenia (42). Following binding of IgG immune complexes to platelets, platelets degranulate and release stored serotonin; this pathway is downstream of FcγRIIA signaling (47). In some settings, increased circulating levels of serotonin can contribute to systemic shock (47, 48).
Upon DENV infection, platelets can also be activated indirectly by complement C3, serotonin, or PAF. For example, platelets express complement receptor C3R (49, 50). Platelets from patients infected with dengue have increased IgG and C3 binding that is associated with increased platelet activation and clearance. This could not be completely blocked by FcγRIIA inhibition (51, 52). Moreover, platelets could be activated by serotonin via 5HT2A receptors, which leads to increased platelet clearance and development of thrombocytopenia (53). Recently, Masri et al. showed that the serotonin is mainly synthesized in mast cells during DENV infection and subsequently internalized by platelets via 5HT2 receptors (53). Similarly, PAF receptor (PAFR) deficient mice also exhibited elevated platelet counts and decreased vascular permeability after DENV-II inoculation, as compared to wild type (WT) mice, suggesting that PAFR is also involved in the development of thrombocytopenia (54, 55). This further supports our notion that platelets have many myeloid cell features that mediate thromboinflammation.
Following activation of platelets during dengue infection, the global gene expression, proteomic, and lipodomic profilings of platelets are all altered to facilitate viral clearance (56–58). Unbiased next-generation RNA sequencing of platelets from patients and in megakaryocytes infected in vitro with dengue virus by recent work from Campbell et al. (56) suggests that megakaryocytes sense dengue infection and/or agonists generated during infection, and in response alter the repertoire of mRNAs invested into newly produced platelets (56). One of transcripts significantly upregulated in megakaryocytes and platelets during DENV infection was interferon-induced transmembrane protein 3 (IFITM3). IFITM3 protein was also increased in platelets from patients infected with DENV and cultured megakaryocytes exposed to DENV in vitro. IFITM3 in other cells restricts viral replication, therefore enhancing resistance to DENV infection (56). Interestingly, IFITM3 induction in human megakaryocytes not only reduced DENV infection of megakaryocytes but also reduced DENV infection in stem cells in the surrounding bone marrow niche. This study highlights the ability of megakaryocytes to participate effective in innate antiviral immune responses.
As mentioned above, through DC-SIGN and HSPG, TLR4, CLEC-2, and other receptors, DENV could activate platelets, cause thrombocytopenia and thromboinflammation in patients. Platelet activation is associated with the severity of thrombocytopenia and increased risk of DHF in patients with DENV (37, 51, 59, 60). A hallmark of platelet activation is the conformational change of integrin αIIbβ3, which binds to fibrinogen and von Willebrand factor (vWF), triggering platelet adhesion to vascular endothelial cells and causing thrombosis (61). Activation of platelets also triggers degranulation and the release of a number of proteins including P-selectin, PAF, soluble CD40L (CD154), and serotonin—proteins that are often dichotomized into either pro-thrombotic or pro-inflammatory, but which could fall together under the term “thromboinflammation” (54, 55, 62, 63). P-selectin on platelets engage its ligand, P-selectin glycoprotein ligand (PSGL) on leukocytes, promoting proinflammatory cytokine production such as IL-1β and IL-8 by both platelets and monocytes (40, 62, 64). These cytokines induce enhanced permeability of endothelial cells and vascular leakage, and are associated with an increased risk of DHS in patients (64). P-selectin also tether platelets to PSGL on endothelial cells, and induces vascular endothelial damage (65, 66). Moreover, serotonin not only contributes to the development of thrombocytopenia. The stored serotonin could be released from a large number of platelets following platelet activation, lead to an increased concentration of serotonin in circulation, cause vasodilation and hypotension, and development of systemic shock (47, 48). Upon activation, platelets undergo apoptosis, with decreased mitochondrial potential, assembly of inflammasomes, increased phosphatidylserine (PS) exposure and P-selectin expression on the cell surface (37, 38, 67). This process further catalyzes thromboinflammation. For example, the apoptosis of platelets not only increases the phagocytosis of platelets by monocytes, but also triggers activation of the coagulation system, generation of thrombin, and formation of thrombi (68–70). In addition, during DENV infection, the release of angiopoetin-1 by platelets is reduced. This is associated with dampened inhibition of angiopoetin-2, and increased endothelial damage in patients with DHS (71).
Dengue activated platelets are associated with increased assembly of inflammasome NLRP3, which activates caspase-1 and triggers apoptosis within platelets, and promotes platelet aggregation (67, 72). This leads to a reduced platelet lifespan and contributes to the development of thrombocytopenia in dengue patients (63, 67). The NLRP3 inflammasome assembly in platelets also leads to the synthesis of IL-1β by platelets and its subsequent secretion into plasma and packaging into microvesicles (67). Platelet activation and apoptosis is higher in patients with DHF than without DHF, and correlates with in vitro phagocytosis of platelets by macrophages through a phosphatidylserine-recognizing pathway (37, 51). Furthermore, NLRP3 and FcγRIIIA have been shown to induce dengue-triggered hemorrhage in mice synergistically (73). NLRP3 has also been correlated with increased low-density lipoproteins (LDL) and decreased high-density lipoproteins (HDL), suggesting extravascular effects to host lipid metabolism following NLRP3 activation (74). However, the role of platelets in this pathological process remains unclear.
In addition to antibody-dependent platelet activation, platelets may also engage antigen specific T cells by presenting dengue antigens through MHC class I. For example, a proteomic study by Trugilho et al. revealed proteasome subunit proteins and HLA class I antigen presentation pathway proteins as the most significantly upregulated in platelets during dengue (57). In addition, platelets express several T cell co-signaling ligands, such as CD40, CD86, ICOSL, and upon activation, are capable of cross-presenting exogenous antigens in vitro and stimulating antigen specific T cell responses (75). It seems possible that following direct binding of virus or through FcγRIIA, platelets internalize dengue virus, degrade dengue antigens in their immunoproteasomes, and present antigen peptides through HLA class I for recognition by CD8+ T cells.
Platelets are also involved in other pathological mechanisms of dengue infection. For example, platelets may increase endothelial barrier permeability due to decreased S1P levels, or promote DENV replication in monocytes through the release of platelet factor 4 (PF4, also known as CXCL4) (64, 76–78). Platelet-derived microparticles may also play a role in the pathogenesis of the disease, as they are notably increased in thrombocytopenic patients without bleeding, and decreased in thrombocytopenic patients with bleeding (67). A summary of some of these identified mechanisms can be found in Figure 1.
Malaria, also a mosquito-borne infectious disease, is caused by systemic infection with parasites of the Plasmodium group. In humans, five species of Plasmodium have been identified that cause malaria: P. falciparum, P. malariae, P. ovale, P. vivax, and P. knowlesi. Of these, P. falciparum is the most common (79). Malaria has been a major health concern worldwide for decades, with an estimated incidence of more than 200 million cases a year (79). The parasitic sporozoites are transmitted from the mosquito's saliva into human bloodstream, and subsequently infect hepatocytes where they mature into schizonts. Eventually, the infected hepatocytes rupture, releasing merozoites into the bloodstream. Plasmodium then invade erythrocytes, replicating until the cells burst. This cycle is repeated, typically causing fever each time the erythrocytes burst.
Common characteristics of malaria include headache, cyclical fevers, anemia, and thrombocytopenia. In the study by de Mast et al. healthy volunteers were infected with P. falciparum and developed thrombocytopenia at the earliest phase of blood-stage infection (80). Thrombocytopenia is so common in patients that platelet count has been proposed as an affordable and fast diagnostic test for malaria in low-income regions, with a reported sensitivity of 60% and specificity of 88% (81–83). Recently, Gardinassi et al. integrated plasma metabolomic data and whole blood transcriptomic data obtained from volunteers infected with P. vivax (84). They found that platelet activation together with changes in IFN signature modules and T cell signaling are the top three most significantly changed processes (84).
A major cause of morbidity and mortality in patients with malaria is cerebral malaria (CM) (85, 86). Increased platelet accumulation has been documented in the brain microvasculature of children who died of CM, as well as in animals with experimental cerebral malaria (ECM) (85–89). These platelets were often found aggregated with Plasmodium parasites or leukocytes, together with increased vWF staining (18, 90–92). Platelet activation and thrombosis may precede leukocyte infiltration (88). Moreover, recent proteomic studies performed on postmortem brain tissue obtained from CM patients revealed that proteins involved in platelet activation and coagulation were upregulated (93). Thus, platelets and leukocytes (as well as platelet-leukocyte aggregates) are associated with CM. Figure 2 highlights a schematic representation of some of the interactions between activated platelets and other circulating blood cells during malaria infection.
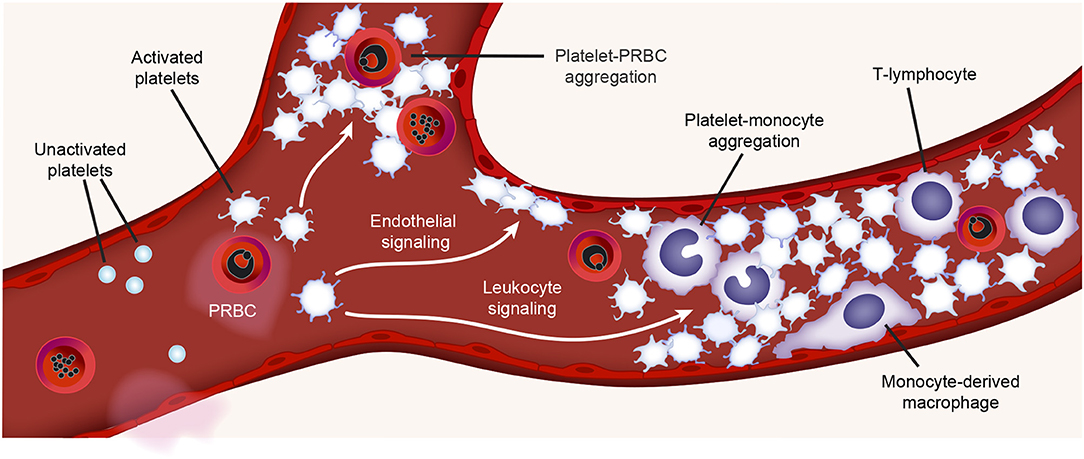
Figure 2. Platelet activities during malaria infection. During malaria infection, platelets may be found in the vasculature closely surrounding plasmodium-infected RBCs (PRBCs) or interacting with monocytes (platelet-monocyte aggregates) or lymphocytes. Toxins generated during malaria infection (e.g., hemozoin) may active platelets. Platelet interactions with monocytes trigger pro-inflammatory cytokine synthesis by monocytes. Figure is adapted from Rondina et al. (94) with permission obtained from Elsevier.
Both platelets and platelet derived microparticles (PMPs) can directly bind Plasmodium infected RBCs (PRBCs) (88, 89, 95, 96). This binding is mainly mediated by CD31 (PECAM-1) and CD36 (GPIV) on platelets and the P. falciparum erythrocyte membrane protein 1 variant on PRBCs (89, 95, 96). After binding, platelets can directly kill the intraerythrocytic Plasmodium in vitro (97, 98). In patients, platelets can directly kill all major malaria parasites in vivo, and the decreased platelet count has partially been attributed to the increased binding to PRBCs (99). In some—but not all—studies, the severity of thrombocytopenia has been associated with increased parasitic density and adverse outcomes (98, 100, 101). The significance of platelets in parasite clearance remains somewhat uncertain, as the physiological platelet to RBC ratio ranges from ~1:10 to 1:50, and platelet-bound PRBCs have been found in less than 5% of the total PRBCs in patients infected with malaria (98, 99, 102). Nevertheless, considering that about 100 billion platelets are produced daily (for comparison, about 200 billion RBCs are produced daily), and platelet-mediated parasite killing could be occur very rapidly (e.g., minutes), it remains plausible that platelets contribute to parasitic killing in humans, directly or indirectly. This remains an area of active investigation in the field.
One chemokine implicated in platelet-mediated parasite clearance is PF4. Following direct platelet-PRBC binding, platelets release PF4 which engage the Duffy antigen receptor (Df) on PRBCs. This engagement induces disruption of the parasite digestive vacuoles and terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick-end labeling of parasite nuclei (TUNEL+), without lysing PRBCs (99, 103). Following this initial discovery, subsequent studies have shown that PF4-driven antiparasitic activity is mainly mediated by the C-terminus of PF4. A synthetic PF4 derived cyclic peptide, named cPF4PD, showed promising killing of parasites in PRBCs without lysing normal RBCs (104, 105). Recent work by Wang et al. showed that the host can upregulate PF4 production in malaria by activating the transcription factor E74 like ETS transcription factor 4 (ELF4) in megakaryocytes (106).
However, the PF4 released from platelets is not entirely protective, as in some settings PF4 release may trigger inflammation. For example, in a mouse model of experimental cerebral malaria (ECM), platelet accumulation and PF4 release in the brain led to significantly increased blood brain barrier permeability and T cell infiltration. In these models, PF4 deficient mice have decreased T cell infiltration and were protected from the development of ECM (89). Further studies showed that platelet-derived PF4 activated the transcription factor KLF4 in monocytes and promoted monocytes to produce proinflammatory cytokines such as IL-6 and TNFα, which are important in the pathogenesis of ECM (88).
Platelets also have been shown to participate in thromboinflammation during malaria infection through other mechanisms. For example, platelets are the major source of IL-1β, which protects against ECM development in experimental models of malaria (107). Additionally, plasma vWF levels (including ultra-large vWF multimers) are increased in patients with malaria (18, 92, 108, 109), also associated with increased GPIb shedding from platelets and the development of thrombocytopenia (110). The readers are referred to several excellent reviews on this topic (18, 81, 111).
Bacteria are commonly classified into Gram-positive and Gram-negative species based on their cell wall structures. Gram-positive bacteria feature a thick layer of peptidoglycan, which is evident by Gram staining. Gram-negative bacteria have a thin layer of peptidoglycan covered by a layer of outer membrane and lipopolysaccharides (LPS). Infection by pathogenic bacteria can cause thromboinflammation locally or systemically, such as via endothelium barrier damage and lung edema, intestine inflammation and diarrhea, platelet activation, and thrombocytopenia, coagulation activation and disseminated intravascular coagulation (DIC). In experimental models of infection, depleting platelets prior to infection often leads to increased mortality, suggesting an important role of platelets in host survival during bacterial infections (112–116).
The Gram-negative bacterium E. coli is a major cause of urinary tract infections. Strains of E. coli, as well as LPS purified from E. coli are widely used in research and these tools have improved our understanding of many host immune responses in platelets and other cells. We and others have shown direct interactions between platelets and E. coli in vitro (117). Direct interactions between platelets and E. coli have been captured in vivo recently using super-resolution microscopy (118). These investigators captured real-time, in vivo platelet migration toward E. coli in mouse liver sinusoids within hours after infection, as well as in vitro platelet migration toward and bundling of E. coli bacteria within minutes (118).
Human platelets express toll-like receptors (TLRs) 1–10 (at the mRNA and/or protein level), and mouse platelets express TLRs 1-8 (31, 119–121). Of these, TLR4, an LPS specific receptor, was the first TLR well characterized on platelets (122). In macrophages and dendritic cells, LPS activates the TLR4/MyD88 signaling, triggers inflammation (123, 124). In platelets, LPS activates the TLR4/MyD88 and signals downstream via cGMP/PKG-dependent pathway (125). Administration of LPS to rats resulted in platelet activation and adhesion to the endothelium through P-selectin and integrin GPIb receptors and the development of thrombocytopenia in rats within 30 min (122). Subsequently, Cognasse, Andoneigui and Aslam et al. showed independently that both human and mouse platelets have functional TLR4 on their surface (113, 121, 126).
Upon LPS stimulation and TLR4 activation, a number of events happen to platelets that cause thromboinflammation (127). LPS stimulation signals to human platelets to process tissue factor (TF) pre-mRNA into mature mRNA, with subsequent increases in TF protein, which is procoagulant (117, 128). This is a unique bacteria stimulated platelet response that has been not reported in any other infectious diseases. LPS also induces some other changes in platelets as we have mentioned above. Similar like activation during DENV infection, platelets can also be activated and undergo apoptosis upon LPS stimulation (129), and triggers the secretion of thrombo-inflammatory factors, including P-selectin at low LPS concentrations and CD40L, TNF-α, and PF4 at higher LPS concentrations (127, 130, 131). From an evolutionary perspective, this might serve as an adaptive rheostat to the host, perhaps by limiting the scale of thromboinflammation when the bacterial load is low (125, 126, 128, 131). Moreover, LPS stimulates platelets to opsonize IgG immune complexes and enhances the phagocytosis of platelets by macrophages, suggesting there may be a synergistic effect of TLR4 and FcR on platelets (132). LPS also stimulates platelets to interact with neutrophils through the interactions between P-selectin, PSGL-1, and lymphocyte function-associated antigen 1 (LFA-1) (112, 114, 133). Neutrophils reciprocally trigger platelet activation and thromboxane generation during E. coli infection (114). In addition, upon LPS stimulation, platelets maintain vascular integrity and prevent leukocyte infiltration via CLEC-2 (116).
Upon Gram-negative bacterial infections, platelets are actively involved in the generation of neutrophil extracellular traps (NETs) (112). NETs are extracellular lattices of chromatin, histones, and granule enzymes extruded by neutrophils upon activation (Figure 3A), via a unique process termed “NETosis” (134). NET formation is an efficient mechanism for trapping both Gram-positive and Gram-negative bacteria. Platelets mediate key aspects of NET formation. Although neutrophils can form NETs without platelets when infected with Gam-positive or Gram-negative bacteria, platelets are able to significantly expand the surface area of NETs in vitro (112). In vivo depletion of neutrophils or platelets impairs NET formation and significantly impedes bacterial clearance (112, 135).
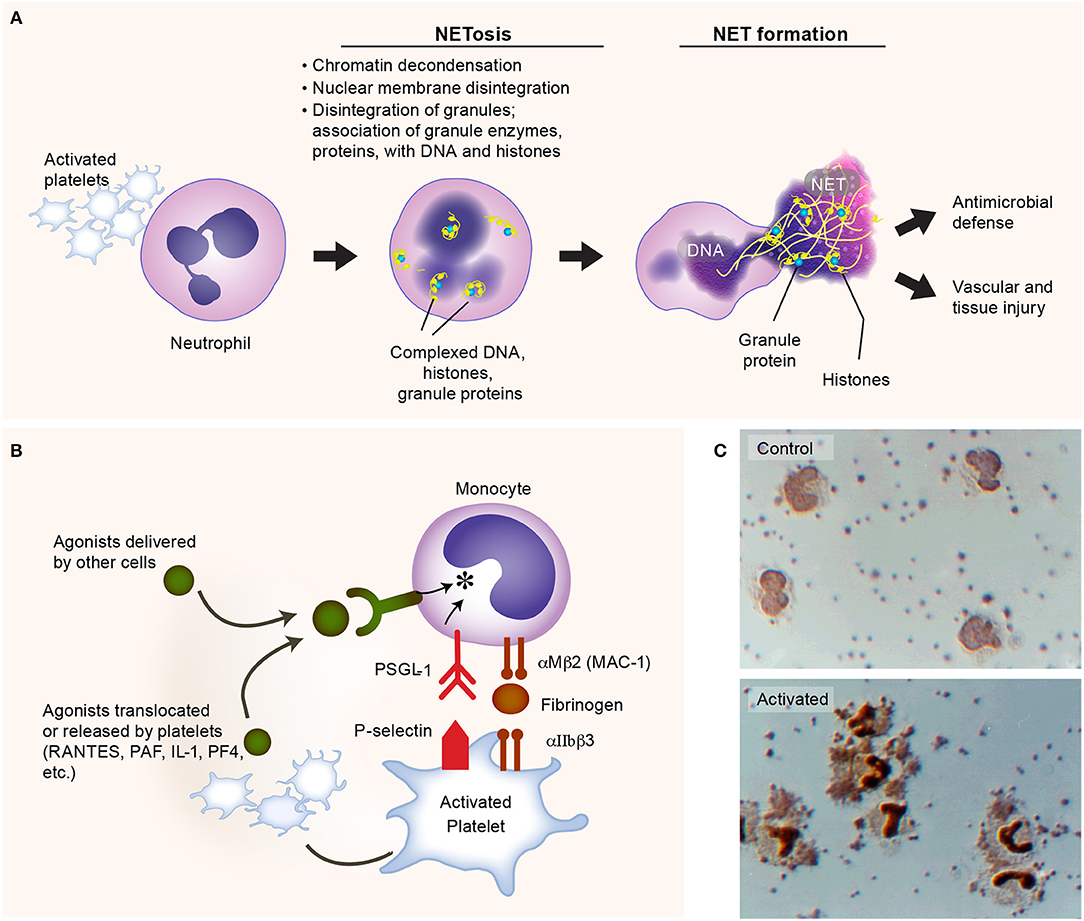
Figure 3. Activated platelets induce formation of neutrophil extracellular traps (NETs). (A) Platelets are commonly activated during Gram-negative bacteria infections, and express multiple receptors that could facilitate the process of NET formation (NETosis), such as P-selectin. The lattices of chromatin, histones, and granule enzymes (NETs) play a critical role in pathogen clearance and may also induce thromboinflammatory responses, potentially contributing to vascular and tissue injury. (B) Activated platelets interact with monocytes, inducing the synthesis of inflammatory mediators. (C) Upper panel: isolated human platelets and monocytes incubated under control conditions. Lower panel: formation of platelet-monocyte aggregates and nuclear translocation of nuclear factor kappa B (NF-κB) in monocytes when the platelets were activated with nanomolar concentrations of thrombin. Figure is adapted from Rondina et al. (94) with permission obtained from Elsevier.
Multiple surface receptors on platelets and secreted factors by platelets regulate NET formation. For instance, platelet-derived P selectin plays an important role in the early stage of NETosis, facilitating platelet-neutrophil direct interactions (114, 136). Platelet GPIbα, a well-studied receptor in hemostasis and thrombosis, has been found recently triggers the activation of neutrophils and extracellular vesicle release from neutrophils (114). In addition, another important receptor in hemostasis and thrombosis, the integrin αIIbβ3 on platelets, this also mediates NETosis, as the deficiency of integrin αIIbβ3 impairs NETs formation (135). Although NETs trap bacteria efficiently, exaggerated or misplaced NET formation may also be deleterious. Histones, serine proteases, and cathepsin G released by activated neutrophils can activate platelets, promoting coagulation, endothelial damage, and thrombosis (133, 137–139). Understanding the roles of NET formation in adaptive and maladaptive host responses remains an active area of investigation. We refer the reader to the recent review article by Zucoloto et al. for more detailed discussions on the platelet-neutrophil interactions that illustrate the intimate relationship between inflammation and thrombosis (19, 140).
Another example of the versatility of platelets and thromboinflammation as an integrated process is the synergistic signaling of FcγRIIA receptors and platelet integrin αIIbβ3 (a canonical hemostatic receptor) upon E. coli infection. In addition to activation through TLR4, platelets could also be activated by E. coli via FcγRIIA receptors, provided simultaneous integrin αIIbβ3 signaling (45). Either absence of IgG or blockage of αIIbβ3 signaling would abolish the aggregation of platelets when incubated with E. coli (45, 141). Recent work by Palankar et al. demonstrates that human platelets directly kill E. coli in mechanisms that require FcγRIIA and PF4 (142). As with malaria, PF4 is also central to effective E. coli killing. Disruption of platelet cytoskeletal functions also reduced the efficacy of E. coli killing by platelets. Moreover, the complement C3 opsonization of E. coli facilitates the formation of platelet-bacteria aggregates which is important in the induction of adaptive immune responses (143). Together, these findings suggest that platelets accumulate on bacteria, releasing antimicrobial α-granule contents that effectively kill E. coli (142). While not a central focus of this review, platelets interact with other leukocytes, including monocytes, forming stable platelet-leukocyte aggregates that promote the release of agonists from platelets and subsequent pro-inflammatory cytokine synthesis by monocytes (Figures 3B,C). Interestingly, in aging (where the risk of infection rises substantially), these interactions may be upregulated—potentially contributing to cytokine release injurious to the host (144). Whether these aging-dependent responses contribute to adverse clinical outcomes in sepsis remains an area of active investigation.
Bacteria also possess endogenous mechanisms to counteract host defenses. Some of these directly affect human platelet activities. As one example, work from our group demonstrated that pathogenic E. coli bacteria isolated from infected patients induced platelet apoptosis via calpain-mediated degradation of the cell survival protein Bcl-xL (145). This was accompanied by impaired mitochondrial membrane potential and lateral condensation of actin. Degradation of Bcl-xL was driven by alpha hemolysin, a pore-forming toxin produced by E. coli. Interestingly, clinical isolates of S. aureus (a Gram-positive bacteria discussed below) that produced alpha toxin (α toxin, also known as α hemolysin) degraded in Bcl-xL platelets (145). These findings suggest a mechanism whereby bacterial pathogens contribute to thrombocytopenia.
Gram-positive bacterial infections, especially multidrug resistant pathogens, are a major global health challenge (146). S. aureus, Listeria monocytogenes (L. monocytogenes), and Streptococcus pneumoniae (S. pneumoniae, or pneumococcus) are common pathogens causing substantial morbidity and mortality. Here we highlight the mechanisms of platelet-mediated thromboinflammation during S. aureus infections, but readers are referred to the recent review by Anderson and Feldman on platelets in pneumonia and other excellent reviews as we have mentioned above (13, 14, 147).
For decades, S. aureus, including methicillin sensitive and resistant S. aureus, i.e., MSSA and MRSA, was known to interact with, and activate platelets. In 1964, Siegal et al. showed that a toxin from staphylococcal bacteria could induce platelet morphological changes observed by electron microscopy images and inhibition of platelet rich plasma clotting (148). In the 1970s, Clawson and White showed that among several strains of bacteria able to directly bind platelets and induce platelet aggregation and adhesion, S. aureus was the most potent (149, 150). Interestingly, at low bacterial numbers, platelets bind to bacteria without forming substantial platelet-platelet aggregates, suggesting there may be favored interactions between bacteria and platelets under these conditions. At higher bacterial numbers, platelet aggregation is induced and bacteria can be found encased in platelet-platelet aggregates (Figure 4) (149, 150). More recently, Wong et al. showed that upon MRSA infection in vivo, platelets scan liver Kupffer cells and rapidly (within minutes of infection) recognize MRSA on the surface of Kupffer cells (27). This triggers aggregation within sinusoids that limits bacterial spreading (27). Additionally, platelet depleted mice have significantly increased mortality upon MRSA infection in vivo (27).
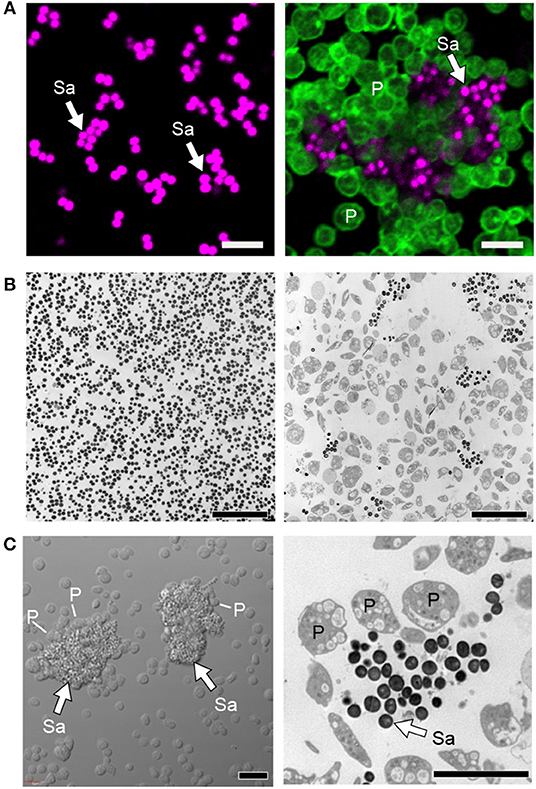
Figure 4. Platelets sequester S. aureus and promote thromboinflammation. (A) Confocal and (B) transmission electron microscopy of cultured staphylococcus aureus (Sa) incubated in the presence (right panels) or absence of platelets (P, time = 240 min). (C) Differential interference contrast (left panel) and transmission electron (right panel) microscopy of clusters of S. aureus (Sa, white arrows) surrounded by platelets (P). Scale bars = 5 μm. After sequestration of S. aureus, platelets become activated, form aggregates in vitro and microthrombosis in vivo, interact with macrophages and neutrophils, trigger NETs formation, and also shuttle bacteria to splenic dendritic cells to activate CD8+ T cell responses. Figure adapted from Kraemer et al. (151) with permission.
There is heterogeneity for clinically isolated S. aureus species from patients to bind platelets (27, 152). When S. aureus binds to platelets, it appears to be in a saturable and reversible manner, suggesting there are receptor-ligand mediated interactions between platelets and S. aureus (153). Recently, platelets are shown to be capable of spreading and enclosing bacteria in vitro (118, 151).
Platelets can directly bind S. aureus antigens through multiple receptors. For example, activated human platelets express gC1qR, which could directly bind S. aureus protein A without complement opsonization (154). In addition, platelets could be directly activated by the α toxin, the Staphylococcal superantigen-like 5 (SSL5), extracellular adherence protein (Eap), chemotaxis inhibitory protein of S. aureus (CHIPS), the formyl peptide receptor-like 1 inhibitory protein (FLIPr) and other proteins of S. aureus (151, 155–158). Alpha toxin is the major cytotoxic virulent factor of S. aureus, capable of forming heptamers and develop pores on target cell membrane, causing cell death. Incubation of α toxin with washed platelets induced morphological changes of platelets in suspension, including visibly lysed shape, smaller in size, and decreased content of intracellular granules (159). The incubation of α toxin also impaired the spreading capability of platelets on collagen and fibrinogen (159). In addition, α toxin can form complex with ADAM10 on platelets, a widely expressed zinc-dependent metalloprotease, and activate the latter (160). The activated ADAM10 then proteolyze GPVI on platelets, and impede platelet adhesion to collagen, and in vivo reduce platelets activation and accumulation at sites of infection (155, 161). These mechanisms facilitate bacteria evasion and spreading. Furthermore, in vivo, the α toxin induced platelet aggregation in the liver and kidney and associated organ failure has been shown by Surewaard et al. (162). In contrast, platelets also evolved with protective responses following α-toxin encounters. For example, α toxin induces the release of multiple microbicidal proteins from platelets, such as thrombin-induced platelet microbicidal protein-1 (tPMP-1) and human β defensin-1, which significantly suppress bacterial growth and trigger NETs formation (151, 152). Alpha toxin also triggers integrin αIIbβ3 dependent platelet aggregation and increased protein synthesis of Bcl3, which promotes clot retraction, stabilizes thrombus within the vasculature (159). In addition, α toxin can indirectly bind platelets when opsonized by complement C3b, which promotes platelet aggregates as well as platelet-macrophage and platelet-neutrophil interactions (161, 162). These platelet-monocyte interactions can induce NLRP3 inflammasome and IL-1β production in monocytes; this may happen in platelets but as of yet remains unproven (163–165). SSL5 is a member of the SSL-family proteins that has been shown evolved in S. aureus evasion. It can directly bind either GPVI or GPIbα on platelets and signal downstream, induce activation of integrin αIIbβ3 and P selectin on platelets (157). Activated αIIbβ3 further promotes platelet aggregation, platelet-leukocyte aggregates, and adhesion to endothelial cells. Increased P-selectin also binds SSL5, as well as monocytes and neutrophils (156). In addition to direct binding, platelets can rapidly (within ~1 min of infection) bind complement opsonized bacteria, including S. aureus, in vivo (166). This may play a critical role in shuttling bacteria to the splenic DCs to trigger host CD8+ T cell responses (143, 166).
Small in size and abundant in number, platelets are effective sentinels constantly roaming the vasculature, quickly sensing and responding to invading pathogens. Many of the responses by platelets to pathogen invasion bridges hemostatic, inflammatory, and immune continuums: the activation of platelets leads to the expression of activated integrin αIIbβ3 and P-selection on the plasma membrane, formation of platelet aggregates and thrombosis, adherence and damage to the endothelium, increased interactions with macrophages and neutrophils, promotion of NETs formation, release of cytokines. These bridging features contributed to the evolution of the concept of thromboinflammation. Undoubtedly, the field will continue to see new discoveries expanding the armamentarium of platelet functions during infectious diseases. Exciting technological innovations are likely to continue to facilitate many of these discoveries. Sequencing techniques, such as next-generation RNA-sequencing and ribosomal footprint profiling, have already uncovered important new insights into the rich and dynamic nature of the platelet transcriptome and proteome (31, 167–170). Moreover, efforts to integrate sequencing data with machine-learning strategies may uncover new insights (84, 170). Proteomic studies of platelet lysates and granules have provided another layer of unbiased information about numerous proteins released by platelets and/or stored in their granules. Many of these include proteins synthesized within platelets, packaged from megakaryocytes, or internalized from the extracellular environment (84, 171–173). The application of super-resolution microscopy, including single-molecule localization microscopy (SMLM) and structured illumination microscopy (SIM), will provide unparalleled opportunities to visualize platelet granules and gather information about intracellular protein localization (174–176). The incredible evolution of our understanding of anucleate platelets in functions beyond just hemostasis has been an exciting journey and further discoveries will likely continue to expand the role of platelets in infectious diseases.
LG and MR designed and wrote the paper. Both authors reviewed and critically edited the manuscript.
This work was supported by the NHLBI and NIA (HL142804, HL130541, AG059877, and AG04802 to MR). This material is the result of work supported with resources and the use of facilities at the George E. Wahlen VA Medical Center, Salt Lake City, Utah (I01CX001696 to MR). The sponsor had no role in the design or preparation of paper. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Ms. Diana Lim for her creativity and excellent figure preparation, Dr. Guy Zimmerman for his critical comments, and Ms. Toni Blair for her excellent editorial assistance.
1. Kaplan D, Casper TC, Elliott CG, Men S, Pendleton RC, Kraiss LW, et al. VTE incidence and risk factors in patients with severe sepsis and septic shock. Chest. (2015) 148:1224–30. doi: 10.1378/chest.15-0287
2. Iba T, Levy JH. Inflammation and thrombosis: roles of neutrophils, platelets and endothelial cells and their interactions in thrombus formation during sepsis. J Thromb Haemost. (2018) 16:231–41. doi: 10.1111/jth.13911
3. Levi M, Schultz M, van der Poll T. Sepsis and thrombosis. Semin Thromb Hemost. (2013) 39:559–66. doi: 10.1055/s-0033-1343894
4. Branchford BR, Carpenter SL. The role of inflammation in venous thromboembolism. Front Pediatr. (2018) 6:142. doi: 10.3389/fped.2018.00142
5. Mukhopadhyay S, Johnson TA, Duru N, Buzza MS, Pawar NR, Sarkar R, et al. Fibrinolysis and inflammation in venous thrombus resolution. Front Immunol. (2019) 10:1348. doi: 10.3389/fimmu.2019.01348
6. Marshall JC. Why have clinical trials in sepsis failed? Trends Mol Med. (2014) 20:195–203. doi: 10.1016/j.molmed.2014.01.007
7. Blair P, Rex S, Vitseva O, Beaulieu L, Tanriverdi K, Chakrabarti S, et al. Stimulation of Toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ Res. (2009) 104:346–54. doi: 10.1161/CIRCRESAHA.108.185785
8. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. (2013) 13:34–45. doi: 10.1038/nri3345
9. Kambas K, Chrysanthopoulou A, Vassilopoulos D, Apostolidou E, Skendros P, Girod A, et al. Tissue factor expression in neutrophil extracellular traps and neutrophil derived microparticles in antineutrophil cytoplasmic antibody associated vasculitis may promote thromboinflammation and the thrombophilic state associated with the disease. Ann Rheum Dis. (2014) 73:1854–63. doi: 10.1136/annrheumdis-2013-203430
10. Gould TJ, Lysov Z, Liaw PC. Extracellular DNA and histones: double-edged swords in immunothrombosis. J Thromb Haemost. (2015) 13 (Suppl. 1):S82–91. doi: 10.1111/jth.12977
11. Gaertner F, Massberg S. Blood coagulation in immunothrombosis-At the frontline of intravascular immunity. Semin Immunol. (2016) 28:561–9. doi: 10.1016/j.smim.2016.10.010
12. Rondina MT, Garraud O. Emerging evidence for platelets as immune and inflammatory effector cells. Front Immunol. (2014) 5:653. doi: 10.3389/fimmu.2014.00653
13. Yeaman MR. The role of platelets in antimicrobial host defense. In: Michelson A, Cattaneo M, frelinger A, Newman P, editors. Platelets. 4th ed. Academic Press (2019). doi: 10.1016/B978-0-12-813456-6.00029-1
14. Hamzeh-Cognasse H, Damien P, Chabert A, Pozzetto B, Cognasse F, Garraud O. Platelets and infections - complex interactions with bacteria. Front Immunol. (2015) 6:82. doi: 10.3389/fimmu.2015.00082
15. Cognasse F, Nguyen KA, Damien P, McNicol A, Pozzetto B, Hamzeh-Cognasse H, et al. The inflammatory role of platelets via their TLRs and siglec receptors. Front Immunol. (2015) 6:83. doi: 10.3389/fimmu.2015.00083
16. Hottz ED, Bozza FA, Bozza PT. Platelets in immune response to virus and immunopathology of viral infections. Front Med. (2018) 5:121. doi: 10.3389/fmed.2018.00121
17. Morrell CN. Platelets: killers of parasites or patients? Blood. (2017) 129:1571–2. doi: 10.1182/blood-2017-01-764621
18. O'Sullivan JM, Preston RJ, O'Regan N, O'Donnell JS. Emerging roles for hemostatic dysfunction in malaria pathogenesis. Blood. (2016) 127:2281–8. doi: 10.1182/blood-2015-11-636464
19. Zucoloto AZ, Jenne CN. Platelet-neutrophil interplay: insights into neutrophil extracellular trap (NET)-driven coagulation in infection. Front Cardiovasc Med. (2019) 6:85. doi: 10.3389/fcvm.2019.00085
20. Michelson AD. Platelets. Amsterdam: Elsevier (2013). Available online at: http://search.ebscohost.com/login.aspx?direct=true&scope=site&db=nlebk&AN=485886 (accessed August 15, 2019).
21. Lefrancais E, Ortiz-Munoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM, et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature. (2017) 544:105–9. doi: 10.1038/nature21706
22. Woolthuis CM, Park CY. Hematopoietic stem/progenitor cell commitment to the megakaryocyte lineage. Blood. (2016) 127:1242–8. doi: 10.1182/blood-2015-07-607945
23. Rodriguez-Fraticelli AE, Wolock SL, Weinreb CS, Panero R, Patel SH, Jankovic M, et al. Clonal analysis of lineage fate in native haematopoiesis. Nature. (2018) 553:212–6. doi: 10.1038/nature25168
24. Psaila B, Mead AJ. Single-cell approaches reveal novel cellular pathways for megakaryocyte and erythroid differentiation. Blood. (2019) 133:1427–35. doi: 10.1182/blood-2018-11-835371
25. Hamzeh-Cognasse H, Berthelot P, Tardy B, Pozzetto B, Bourlet T, Laradi S, et al. Platelet toll-like receptors are crucial sensors of infectious danger moieties. Platelets. (2018) 29:533–40. doi: 10.1080/09537104.2018.1445842
26. Cognasse F, Garraud O, Pozzetto B, Laradi S, Hamzeh-Cognasse H. How can non-nucleated platelets be so smart? J Thromb Haemost. (2016) 14:794–6. doi: 10.1111/jth.13262
27. Wong CH, Jenne CN, Petri B, Chrobok NL, Kubes P. Nucleation of platelets with blood-borne pathogens on Kupffer cells precedes other innate immunity and contributes to bacterial clearance. Nat Immunol. (2013) 14:785–92. doi: 10.1038/ni.2631
28. Rossaint J, Margraf A, Zarbock A. Role of platelets in leukocyte recruitment and resolution of inflammation. Front Immunol. (2018) 9:2712. doi: 10.3389/fimmu.2018.02712
29. Rondina MT, Weyrich AS. Regulation of the genetic code in megakaryocytes and platelets. J Thromb Haemost. (2015) 13 (Suppl. 1):S26–32. doi: 10.1111/jth.12965
30. Rowley JW, Schwertz H, Weyrich AS. Platelet mRNA: the meaning behind the message. Curr Opin Hematol. (2012) 19:385–91. doi: 10.1097/MOH.0b013e328357010e
31. Rowley JW, Oler AJ, Tolley ND, Hunter BN, Low EN, Nix DA, et al. Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood. (2011) 118:e101–11. doi: 10.1182/blood-2011-03-339705
32. Normile D. Tropical medicine. Surprising new dengue virus throws a spanner in disease control efforts. Science. (2013) 342:415. doi: 10.1126/science.342.6157.415
33. Mustafa MS, Rasotgi V, Jain S, Gupta V. Discovery of fifth serotype of dengue virus (DENV-5): A new public health dilemma in dengue control. Med J Armed Forces India. (2015) 71:67–70. doi: 10.1016/j.mjafi.2014.09.011
34. Castro MC, Wilson ME, Bloom DE. Disease and economic burdens of dengue. Lancet Infect Dis. (2017) 17:e70–8. doi: 10.1016/S1473-3099(16)30545-X
35. Pang X, Zhang R, Cheng G. Progress towards understanding the pathogenesis of dengue hemorrhagic fever. Virol Sin. (2017) 32:16–22. doi: 10.1007/s12250-016-3855-9
36. Noisakran S, Gibbons RV, Songprakhon P, Jairungsri A, Ajariyakhajorn C, Nisalak A, et al. Detection of dengue virus in platelets isolated from dengue patients. Southeast Asian J Trop Med Public Health. (2009) 40:253–62.
37. Hottz ED, Oliveira MF, Nunes PC, Nogueira RM, Valls-de-Souza R, Da Poian AT, et al. Dengue induces platelet activation, mitochondrial dysfunction and cell death through mechanisms that involve DC-SIGN and caspases. J Thromb Haemost. (2013) 11:951–62. doi: 10.1111/jth.12178
38. Simon AY, Sutherland MR, Pryzdial EL. Dengue virus binding and replication by platelets. Blood. (2015) 126:378–85. doi: 10.1182/blood-2014-09-598029
39. Tomo S, Mohan S, Ramachandrappa VS, Samadanam DM, Suresh S, Pillai AB, et al. Dynamic modulation of DC-SIGN and FcUpsilonR2A receptors expression on platelets in dengue. PLoS ONE. (2018) 13:e0206346. doi: 10.1371/journal.pone.0206346
40. Chao CH, Wu WC, Lai YC, Tsai PJ, Perng GC, Lin YS, et al. Dengue virus nonstructural protein 1 activates platelets via Toll-like receptor 4, leading to thrombocytopenia and hemorrhage. PLoS Pathog. (2019) 15:e1007625. doi: 10.1371/journal.ppat.1007625
41. Sung PS, Huang TF, Hsieh SL. Extracellular vesicles from CLEC2-activated platelets enhance dengue virus-induced lethality via CLEC5A/TLR2. Nat Commun. (2019) 10:2402. doi: 10.1038/s41467-019-10360-4
42. Wang TT, Sewatanon J, Memoli MJ, Wrammert J, Bournazos S, Bhaumik SK, et al. IgG antibodies to dengue enhanced for FcgammaRIIIA binding determine disease severity. Science. (2017) 355:395–8. doi: 10.1126/science.aai8128
43. Katzelnick LC, Gresh L, Halloran ME, Mercado JC, Kuan G, Gordon A, et al. Antibody-dependent enhancement of severe dengue disease in humans. Science. (2017) 358:929–32. doi: 10.1126/science.aan6836
44. Qiao J, Al-Tamimi M, Baker RI, Andrews RK, Gardiner EE. The platelet Fc receptor, FcgammaRIIa. Immunol Rev. (2015) 268:241–52. doi: 10.1111/imr.12370
45. Watson CN, Kerrigan SW, Cox D, Henderson IR, Watson SP, Arman M. Human platelet activation by Escherichia coli: roles for FcgammaRIIA and integrin alphaIIbbeta3. Platelets. (2016) 27:535–40. doi: 10.3109/09537104.2016.1148129
46. Simon LM, Edelstein LC, Nagalla S, Woodley AB, Chen ES, Kong X, et al. Human platelet microRNA-mRNA networks associated with age and gender revealed by integrated plateletomics. Blood. (2014) 123:e37–45. doi: 10.1182/blood-2013-12-544692
47. Cloutier N, Allaeys I, Marcoux G, Machlus KR, Mailhot B, Zufferey A, et al. Platelets release pathogenic serotonin and return to circulation after immune complex-mediated sequestration. Proc Natl Acad Sci USA. (2018) 115:E1550–9. doi: 10.1073/pnas.1720553115
48. Cui L, Lee YH, Thein TL, Fang J, Pang J, Ooi EE, et al. Serum metabolomics reveals serotonin as a predictor of severe dengue in the early phase of dengue fever. PLoS Negl Trop Dis. (2016) 10:e0004607. doi: 10.1371/journal.pntd.0004607
49. Peerschke EI, Yin W, Grigg SE, Ghebrehiwet B. Blood platelets activate the classical pathway of human complement. J Thromb Haemost. (2006) 4:2035–42. doi: 10.1111/j.1538-7836.2006.02065.x
50. Verschoor A, Kemper C, Köhl J. Complement receptors. In: eLS. John Wiley & Sons, Ltd. (2017). p. 1–17. doi: 10.1002/9780470015902.a0000512.pub3
51. Ojha A, Nandi D, Batra H, Singhal R, Annarapu GK, Bhattacharyya S, et al. Platelet activation determines the severity of thrombocytopenia in dengue infection. Sci Rep. (2017) 7:41697. doi: 10.1038/srep41697
52. Wang S, He R, Patarapotikul J, Innis BL, Anderson R. Antibody-enhanced binding of dengue-2 virus to human platelets. Virology. (1995) 213:254–7. doi: 10.1006/viro.1995.1567
53. Masri MFB, Mantri CK, Rathore APS St. John AL. Peripheral serotonin causes dengue-induced thrombocytopenia through 5HT2 receptors. Blood. (2019). doi: 10.1182/blood-2018-08-869156
54. Ishii S, Kuwaki T, Nagase T, Maki K, Tashiro F, Sunaga S, et al. Impaired anaphylactic responses with intact sensitivity to endotoxin in mice lacking a platelet-activating factor receptor. J Exp Med. (1998) 187:1779–88. doi: 10.1084/jem.187.11.1779
55. Souza DG, Fagundes CT, Sousa LP, Amaral FA, Souza RS, Souza AL, et al. Essential role of platelet-activating factor receptor in the pathogenesis of Dengue virus infection. Proc Natl Acad Sci USA. (2009) 106:14138–43. doi: 10.1073/pnas.0906467106
56. Campbell RA, Schwertz H, Hottz ED, Rowley JW, Manne BK, Washington AV, et al. Human megakaryocytes possess intrinsic anti-viral immunity through regulated induction of IFITM3. Blood. (2019) 133:2013–26. doi: 10.1182/blood-2018-09-873984
57. Trugilho MRO, Hottz ED, Brunoro GVF, Teixeira-Ferreira A, Carvalho PC, Salazar GA, et al. Platelet proteome reveals novel pathways of platelet activation and platelet-mediated immunoregulation in dengue. PLoS Pathog. (2017) 13:e1006385. doi: 10.1371/journal.ppat.1006385
58. Samadanam DM, Muthuraman KR, Mariappan V, Kadhiravan T, Parameswaran N, Balakrishna Pillai AK, et al. Altered platelet fatty acids in dengue cases by gas chromatography-mass spectrometry analysis. Intervirology. (2019) 62:57–64. doi: 10.1159/000501015
59. Lam PK, Ngoc TV, Thu Thuy TT, Hong Van NT, Nhu Thuy TT, Hoai Tam DT, et al. The value of daily platelet counts for predicting dengue shock syndrome: Results from a prospective observational study of 2301 Vietnamese children with dengue. PLoS Negl Trop Dis. (2017) 11:e0005498. doi: 10.1371/journal.pntd.0005498
60. Premaratne MK, Perera SS, Malavige GN, Jayasinghe S. Mathematical modelling of immune parameters in the evolution of severe dengue. Comput Math Methods Med. (2017) 2017:2187390. doi: 10.1155/2017/2187390
61. Estevez B, Du X. New concepts and mechanisms of platelet activation signaling. Physiology. (2017) 32:162–77. doi: 10.1152/physiol.00020.2016
62. Nunez-Avellaneda D, Mosso-Pani MA, Sanchez-Torres LE, Castro-Mussot ME, Corona-de la Pena NA, Salazar MI. Dengue virus induces the release of sCD40L and changes in levels of membranal CD42b and CD40L molecules in human platelets. Viruses. (2018) 10:E357. doi: 10.3390/v10070357
63. Ghosh K, Gangodkar S, Jain P, Shetty S, Ramjee S, Poddar P, et al. Imaging the interaction between dengue 2 virus and human blood platelets using atomic force and electron microscopy. J Electron Microsc. (2008) 57:113–8. doi: 10.1093/jmicro/dfn007
64. Hottz ED, Medeiros-de-Moraes IM, Vieira-de-Abreu A, de Assis EF, Vals-de-Souza R, Castro-Faria-Neto HC, et al. Platelet activation and apoptosis modulate monocyte inflammatory responses in dengue. J Immunol. (2014) 193:1864–72. doi: 10.4049/jimmunol.1400091
65. Michels M, Alisjahbana B, De Groot PG, Indrati AR, Fijnheer R, Puspita M, et al. Platelet function alterations in dengue are associated with plasma leakage. Thromb Haemost. (2014) 112:352–62. doi: 10.1160/TH14-01-0056
66. Krishnamurti C, Peat RA, Cutting MA, Rothwell SW. Platelet adhesion to dengue-2 virus-infected endothelial cells. Am J Trop Med Hyg. (2002) 66:435–41. doi: 10.4269/ajtmh.2002.66.435
67. Hottz ED, Lopes JF, Freitas C, Valls-de-Souza R, Oliveira MF, Bozza MT, et al. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood. (2013) 122:3405–14. doi: 10.1182/blood-2013-05-504449
68. Marcus AJ. The role of lipids in blood coagulation. Adv Lipid Res. (1966) 4:1–37. doi: 10.1016/B978-1-4831-9940-5.50008-9
69. Marcus AJ, Spaet TH. Platelet phosphatides: their separation, identification, and clotting activity. J Clin Invest. (1958) 37:1836–47. doi: 10.1172/JCI103776
70. Lentz BR. Exposure of platelet membrane phosphatidylserine regulates blood coagulation. Prog Lipid Res. (2003) 42:423–38. doi: 10.1016/S0163-7827(03)00025-0
71. Michels M, van der Ven AJ, Djamiatun K, Fijnheer R, de Groot PG, Griffioen AW, et al. Imbalance of angiopoietin-1 and angiopoetin-2 in severe dengue and relationship with thrombocytopenia, endothelial activation, and vascular stability. Am J Trop Med Hyg. (2012) 87:943–6. doi: 10.4269/ajtmh.2012.12-0020
72. Qiao J, Wu X, Luo Q, Wei G, Xu M, Wu Y, et al. NLRP3 regulates platelet integrin alphaIIbbeta3 outside-in signaling, hemostasis and arterial thrombosis. Haematologica. (2018) 103:1568–76. doi: 10.3324/haematol.2018.191700
73. Lien TS, Sun DS, Chang CM, Wu CY, Dai MS, Chan H, et al. Dengue virus and antiplatelet autoantibodies synergistically induce haemorrhage through Nlrp3-inflammasome and FcgammaRIII. Thromb Haemost. (2015) 113:1060–70. doi: 10.1160/TH14-07-0637
74. Marin-Palma D, Sirois CM, Urcuqui-Inchima S, Hernandez JC. Inflammatory status and severity of disease in dengue patients are associated with lipoprotein alterations. PLoS ONE. (2019) 14:e0214245. doi: 10.1371/journal.pone.0214245
75. Chapman LM, Aggrey AA, Field DJ, Srivastava K, Ture S, Yui K, et al. Platelets present antigen in the context of MHC class I. J Immunol. (2012) 189:916–23. doi: 10.4049/jimmunol.1200580
76. Schaphorst KL, Chiang E, Jacobs KN, Zaiman A, Natarajan V, Wigley F, et al. Role of sphingosine-1 phosphate in the enhancement of endothelial barrier integrity by platelet-released products. Am J Physiol Lung Cell Mol Physiol. (2003) 285:L258–67. doi: 10.1152/ajplung.00311.2002
77. Ojha A, Bhasym A, Mukherjee S, Annarapu GK, Bhakuni T, Akbar I, et al. Platelet factor 4 promotes rapid replication and propagation of Dengue and Japanese encephalitis viruses. EBioMedicine. (2019) 39:332–47. doi: 10.1016/j.ebiom.2018.11.049
78. Gomes L, Fernando S, Fernando RH, Wickramasinghe N, Shyamali NL, Ogg GS, et al. Sphingosine 1-phosphate in acute dengue infection. PLoS ONE. (2014) 9:e113394. doi: 10.1371/journal.pone.0113394
80. de Mast Q, Groot E, Lenting PJ, de Groot PG, McCall M, Sauerwein RW, et al. Thrombocytopenia and release of activated von Willebrand Factor during early Plasmodium falciparum malaria. J Infect Dis. (2007) 196:622–8. doi: 10.1086/519844
81. Thachil J. Platelets and infections in the resource-limited countries with a focus on malaria and viral haemorrhagic fevers. Br J Haematol. (2017) 177:960–70. doi: 10.1111/bjh.14582
82. Lathia TB, Joshi R. Can hematological parameters discriminate malaria from nonmalarious acute febrile illness in the tropics? Indian J Med Sci. (2004) 58:239–44.
83. Gupta P, Guddattu V, Saravu K. Characterization of platelet count and platelet indices and their potential role to predict severity in malaria. Pathog Glob Health. 113:86–93. doi: 10.1080/20477724.2019.1600855
84. Gardinassi LG, Arevalo-Herrera M, Herrera S, Cordy RJ, Tran V, Smith MR, et al. Integrative metabolomics and transcriptomics signatures of clinical tolerance to Plasmodium vivax reveal activation of innate cell immunity and T cell signaling. Redox Biol. (2018) 17:158–70. doi: 10.1016/j.redox.2018.04.011
85. von Zur Muhlen C, Sibson NR, Peter K, Campbell SJ, Wilainam P, Grau GE, et al. A contrast agent recognizing activated platelets reveals murine cerebral malaria pathology undetectable by conventional MRI. J Clin Invest. (2008) 118:1198–207. doi: 10.1172/JCI33314
86. Grau GE, Mackenzie CD, Carr RA, Redard M, Pizzolato G, Allasia C, et al. Platelet accumulation in brain microvessels in fatal pediatric cerebral malaria. J Infect Dis. (2003) 187:461–6. doi: 10.1086/367960
87. Grau GE, Tacchini-Cottier F, Vesin C, Milon G, Lou JN, Piguet PF, et al. TNF-induced microvascular pathology: active role for platelets and importance of the LFA-1/ICAM-1 interaction. Eur Cytokine Netw. (1993) 4:415–9.
88. Srivastava K, Field DJ, Aggrey A, Yamakuchi M, Morrell CN. Platelet factor 4 regulation of monocyte KLF4 in experimental cerebral malaria. PLoS ONE. (2010) 5:e10413. doi: 10.1371/journal.pone.0010413
89. Srivastava K, Cockburn IA, Swaim A, Thompson LE, Tripathi A, Fletcher CA, et al. Platelet factor 4 mediates inflammation in experimental cerebral malaria. Cell Host Microbe. (2008) 4:179–87. doi: 10.1016/j.chom.2008.07.003
90. Hora R, Kapoor P, Thind KK, Mishra PC. Cerebral malaria–clinical manifestations and pathogenesis. Metab Brain Dis. (2016) 31:225–37. doi: 10.1007/s11011-015-9787-5
91. O'Regan N, Gegenbauer K, O'Sullivan JM, Maleki S, Brophy TM, Dalton N, et al. A novel role for von Willebrand factor in the pathogenesis of experimental cerebral malaria. Blood. (2016) 127:1192–201. doi: 10.1182/blood-2015-07-654921
92. Larkin D, de Laat B, Jenkins PV, Bunn J, Craig AG, Terraube V, et al. Severe Plasmodium falciparum malaria is associated with circulating ultra-large von Willebrand multimers and ADAMTS13 inhibition. PLoS Pathog. (2009) 5:e1000349. doi: 10.1371/journal.ppat.1000349
93. Kumar M, Varun CN, Dey G, Ravikumar R, Mahadevan A, Shankar SK, et al. Identification of host-response in cerebral malaria patients using quantitative proteomic analysis. Proteomics Clin Appl. (2018) 12:e1600187. doi: 10.1002/prca.201600187
94. Rondina MT, Zimmerman GA. Chapter 28. The role of platelets in inflammation. In: Michelson AD, Cattaneo M, Frelinger A, Newman P, editors. Platelets. 4th ed. Elsevier (2019). p. 505–22.
95. Pain A, Ferguson DJ, Kai O, Urban BC, Lowe B, Marsh K, et al. Platelet-mediated clumping of Plasmodium falciparum-infected erythrocytes is a common adhesive phenotype and is associated with severe malaria. Proc Natl Acad Sci USA. (2001) 98:1805–10. doi: 10.1073/pnas.98.4.1805
96. Faille D, Combes V, Mitchell AJ, Fontaine A, Juhan-Vague I, Alessi MC, et al. Platelet microparticles: a new player in malaria parasite cytoadherence to human brain endothelium. FASEB J. (2009) 23:3449–58. doi: 10.1096/fj.09-135822
97. Peyron F, Polack B, Lamotte D, Kolodie L, Ambroise-Thomas P. Plasmodium falciparum growth inhibition by human platelets in vitro. Parasitology. (1989) 99 (Pt 3):317–22. doi: 10.1017/S0031182000059011
98. McMorran BJ, Marshall VM, de Graaf C, Drysdale KE, Shabbar M, Smyth GK, et al. Platelets kill intraerythrocytic malarial parasites and mediate survival to infection. Science. (2009) 323:797–800. doi: 10.1126/science.1166296
99. Kho S, Barber BE, Johar E, Andries B, Poespoprodjo JR, Kenangalem E, et al. Platelets kill circulating parasites of all major Plasmodium species in human malaria. Blood. (2018) 132:1332–44. doi: 10.1182/blood-2018-05-849307
100. Jadhav UM, Patkar VS, Kadam NN. Thrombocytopenia in malaria–correlation with type and severity of malaria. J Assoc Physicians India. (2004) 52:615–8.
101. Gerardin P, Rogier C, Ka AS, Jouvencel P, Brousse V, Imbert P. Prognostic value of thrombocytopenia in African children with falciparum malaria. Am J Trop Med Hyg. (2002) 66:686–91. doi: 10.4269/ajtmh.2002.66.686
102. Gramaglia I, Velez J, Combes V, Grau GE, Wree M, van der Heyde HC. Platelets activate a pathogenic response to blood-stage Plasmodium infection but not a protective immune response. Blood. (2017) 129:1669–79. doi: 10.1182/blood-2016-08-733519
103. McMorran BJ, Wieczorski L, Drysdale KE, Chan JA, Huang HM, Smith C, et al. Platelet factor 4 and Duffy antigen required for platelet killing of Plasmodium falciparum. Science. (2012) 338:1348–51. doi: 10.1126/science.1228892
104. Love MS, Millholland MG, Mishra S, Kulkarni S, Freeman KB, Pan W, et al. Platelet factor 4 activity against P. falciparum and its translation to nonpeptidic mimics as antimalarials. Cell Host Microbe. (2012) 12:815–23. doi: 10.1016/j.chom.2012.10.017
105. Lawrence N, Dennis ASM, Lehane AM, Ehmann A, Harvey PJ, Benfield AH, et al. Defense peptides engineered from human platelet factor 4 kill plasmodium by selective membrane disruption. Cell Chem Biol. (2018) 25:1140–50 e5. doi: 10.1016/j.chembiol.2018.06.009
106. Wang D, Zhang Z, Cui S, Zhao Y, Craft S, Fikrig E, et al. ELF4 facilitates innate host defenses against Plasmodium by activating transcription of Pf4 and Ppbp. J Biol Chem. (2019) 294:7787–96. doi: 10.1074/jbc.RA118.006321
107. Aggrey AA, Srivastava K, Ture S, Field DJ, Morrell CN. Platelet induction of the acute-phase response is protective in murine experimental cerebral malaria. J Immunol. (2013) 190:4685–91. doi: 10.4049/jimmunol.1202672
108. Hollestelle MJ, Donkor C, Mantey EA, Chakravorty SJ, Craig A, Akoto AO, et al. von Willebrand factor propeptide in malaria: evidence of acute endothelial cell activation. Br J Haematol. (2006) 133:562–9. doi: 10.1111/j.1365-2141.2006.06067.x
109. de Mast Q, Groot E, Asih PB, Syafruddin D, Oosting M, Sebastian S, et al. ADAMTS13 deficiency with elevated levels of ultra-large and active von Willebrand factor in P. falciparum and P. vivax malaria. Am J Trop Med Hyg. (2009) 80:492–8. doi: 10.4269/ajtmh.2009.80.492
110. de Mast Q, de Groot PG, van Heerde WL, Roestenberg M, van Velzen JF, Verbruggen B, et al. Thrombocytopenia in early malaria is associated with GP1b shedding in absence of systemic platelet activation and consumptive coagulopathy. Br J Haematol. (2010) 151:495–503. doi: 10.1111/j.1365-2141.2010.08399.x
111. Essien EM, Emagha UT. Blood platelet: a review of its characteristics and function in acute malaria infection. Afr J Med Med Sci. (2014) 43:287–94.
112. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. (2007) 13:463–9. doi: 10.1038/nm1565
113. Andonegui G, Kerfoot SM, McNagny K, Ebbert KV, Patel KD, Kubes P. Platelets express functional Toll-like receptor-4. Blood. (2005) 106:2417–23. doi: 10.1182/blood-2005-03-0916
114. Rossaint J, Kuhne K, Skupski J, Van Aken H, Looney MR, Hidalgo A, et al. Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat Commun. (2016) 7:13464. doi: 10.1038/ncomms13464
115. McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe. (2012) 12:324–33. doi: 10.1016/j.chom.2012.06.011
116. Rayes J, Lax S, Wichaiyo S, Watson SK, Di Y, Lombard S, et al. The podoplanin-CLEC-2 axis inhibits inflammation in sepsis. Nat Commun. (2017) 8:2239. doi: 10.1038/s41467-017-02402-6
117. Rondina MT, Schwertz H, Harris ES, Kraemer BF, Campbell RA, Mackman N, et al. The septic milieu triggers expression of spliced tissue factor mRNA in human platelets. J Thromb Haemost. (2011) 9:748–58. doi: 10.1111/j.1538-7836.2011.04208.x
118. Gaertner F, Ahmad Z, Rosenberger G, Fan S, Nicolai L, Busch B, et al. Migrating platelets are mechano-scavengers that collect and bundle bacteria. Cell. (2017) 171:1368–82 e23. doi: 10.1016/j.cell.2017.11.001
119. Koupenova M, Mick E, Mikhalev E, Benjamin EJ, Tanriverdi K, Freedman JE. Sex differences in platelet toll-like receptors and their association with cardiovascular risk factors. Arterioscler Thromb Vasc Biol. (2015) 35:1030–7. doi: 10.1161/ATVBAHA.114.304954
120. Kapur R, Semple JW. The nonhemostatic immune functions of platelets. Semin Hematol. (2016) 53 (Suppl. 1):S2–6. doi: 10.1053/j.seminhematol.2016.04.002
121. Cognasse F, Hamzeh H, Chavarin P, Acquart S, Genin C, Garraud O. Evidence of Toll-like receptor molecules on human platelets. Immunol Cell Biol. (2005) 83:196–8. doi: 10.1111/j.1440-1711.2005.01314.x
122. Katayama T, Ikeda Y, Handa M, Tamatani T, Sakamoto S, Ito M, et al. Immunoneutralization of glycoprotein Ibalpha attenuates endotoxin-induced interactions of platelets and leukocytes with rat venular endothelium in vivo. Circ Res. (2000) 86:1031–7. doi: 10.1161/01.RES.86.10.1031
123. Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. (1998) 282:2085–8. doi: 10.1126/science.282.5396.2085
124. Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. (1999) 11:115–22. doi: 10.1016/S1074-7613(00)80086-2
125. Zhang G, Han J, Welch EJ, Ye RD, Voyno-Yasenetskaya TA, Malik AB, et al. Lipopolysaccharide stimulates platelet secretion and potentiates platelet aggregation via TLR4/MyD88 and the cGMP-dependent protein kinase pathway. J Immunol. (2009) 182:7997–8004. doi: 10.4049/jimmunol.0802884
126. Aslam R, Speck ER, Kim M, Crow AR, Bang KW, Nestel FP, et al. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood. (2006) 107:637–41. doi: 10.1182/blood-2005-06-2202
127. Rondina MT, Weyrich AS, Zimmerman GA. Platelets as cellular effectors of inflammation in vascular diseases. Circ Res. (2013) 112:1506–19. doi: 10.1161/CIRCRESAHA.113.300512
128. Matus V, Valenzuela JG, Hidalgo P, Pozo LM, Panes O, Wozniak A, et al. Human platelet interaction with E. coli O111 promotes tissue-factor-dependent procoagulant activity, involving Toll like receptor 4. PLoS ONE. (2017) 12:e0185431. doi: 10.1371/journal.pone.0185431
129. Feng G, Yang X, Li Y, Wang X, Tan S, Chen F. LPS enhances platelets aggregation via TLR4, which is related to mitochondria damage caused by intracellular ROS, but not extracellular ROS. Cell Immunol. (2018) 328:86–92. doi: 10.1016/j.cellimm.2018.04.002
130. Stahl AL, Svensson M, Morgelin M, Svanborg C, Tarr PI, Mooney JC, et al. Lipopolysaccharide from enterohemorrhagic Escherichia coli binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood. (2006) 108:167–76. doi: 10.1182/blood-2005-08-3219
131. Cognasse F, Hamzeh-Cognasse H, Lafarge S, Delezay O, Pozzetto B, McNicol A, et al. Toll-like receptor 4 ligand can differentially modulate the release of cytokines by human platelets. Br J Haematol. (2008) 141:84–91. doi: 10.1111/j.1365-2141.2008.06999.x
132. Semple JW, Aslam R, Kim M, Speck ER, Freedman J. Platelet-bound lipopolysaccharide enhances Fc receptor-mediated phagocytosis of IgG-opsonized platelets. Blood. (2007) 109:4803–5. doi: 10.1182/blood-2006-12-062695
133. McDonald B, Davis RP, Kim SJ, Tse M, Esmon CT, Kolaczkowska E, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood. (2017) 129:1357–67. doi: 10.1182/blood-2016-09-741298
134. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
135. Slaba I, Wang J, Kolaczkowska E, McDonald B, Lee WY, Kubes P. Imaging the dynamic platelet-neutrophil response in sterile liver injury and repair in mice. Hepatology. (2015) 62:1593–605. doi: 10.1002/hep.28003
136. Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood. (2015) 126:242–6. doi: 10.1182/blood-2015-01-624023
137. Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, et al. Extracellular histones are major mediators of death in sepsis. Nat Med. (2009) 15:1318–21. doi: 10.1038/nm.2053
138. Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. (2010) 16:887–96. doi: 10.1038/nm.2184
139. Sakurai K, Miyashita T, Okazaki M, Yamaguchi T, Ohbatake Y, Nakanuma S, et al. Role for Neutrophil Extracellular Traps (NETs) and platelet aggregation in early sepsis-induced hepatic dysfunction. In Vivo. (2017) 31:1051–8. doi: 10.21873/invivo.11169
140. Lisman T. Platelet-neutrophil interactions as drivers of inflammatory and thrombotic disease. Cell Tissue Res. (2018) 371:567–76. doi: 10.1007/s00441-017-2727-4
141. Moriarty RD, Cox A, McCall M, Smith SG, Cox D. Escherichia coli induces platelet aggregation in an FcgammaRIIa-dependent manner. J Thromb Haemost. (2016) 14:797–806. doi: 10.1111/jth.13226
142. Palankar R, Kohler TP, Krauel K, Wesche J, Hammerschmidt S, Greinacher A. Platelets kill bacteria by bridging innate and adaptive immunity via platelet factor 4 and FcgammaRIIA. J Thromb Haemost. (2018) 16:1187–97. doi: 10.1111/jth.13955
143. Broadley SP, Plaumann A, Coletti R, Lehmann C, Wanisch A, Seidlmeier A, et al. Dual-track clearance of circulating bacteria balances rapid restoration of blood sterility with induction of adaptive immunity. Cell Host Microbe. (2016) 20:36–48. doi: 10.1016/j.chom.2016.05.023
144. Campbell RA, Franks Z, Bhatnagar A, Rowley JW, Manne BK, Supiano MA, et al. Granzyme A in human platelets regulates the synthesis of proinflammatory cytokines by monocytes in aging. J Immunol. (2018) 200:295–304. doi: 10.4049/jimmunol.1700885
145. Kraemer BF, Campbell RA, Schwertz H, Franks ZG, Vieira de Abreu A, Grundler K, et al. Bacteria differentially induce degradation of Bcl-xL, a survival protein, by human platelets. Blood. (2012) 120:5014–20. doi: 10.1182/blood-2012-04-420661
146. Woodford N, Livermore DM. Infections caused by Gram-positive bacteria: a review of the global challenge. J Infect. (2009) 59 (Suppl. 1):S4–16. doi: 10.1016/S0163-4453(09)60003-7
147. Anderson R, Feldman C. Review manuscript: Mechanisms of platelet activation by the pneumococcus and the role of platelets in community-acquired pneumonia. J Infect. (2017) 75:473–85. doi: 10.1016/j.jinf.2017.09.013
148. Siegel I, Cohen S. Action of staphylococcal toxin on human platelets. J Infect Dis. (1964) 114:488–502. doi: 10.1093/infdis/114.5.488
149. Clawson CC, White JG. Platelet interaction with bacteria. I. Reaction phases and effects of inhibitors. Am J Pathol. (1971) 65:367–80.
150. Clawson CC. Platelet interaction with bacteria. 3. Ultrastructure. Am J Pathol. (1973) 70:449–71.
151. Kraemer BF, Campbell RA, Schwertz H, Cody MJ, Franks Z, Tolley ND, et al. Novel anti-bacterial activities of beta-defensin 1 in human platelets: suppression of pathogen growth and signaling of neutrophil extracellular trap formation. PLoS Pathog. (2011) 7:e1002355. doi: 10.1371/journal.ppat.1002355
152. Yeaman MR, Norman DC, Bayer AS. Staphylococcus aureus susceptibility to thrombin-induced platelet microbicidal protein is independent of platelet adherence and aggregation in vitro. Infect Immun. (1992) 60:2368–74.
153. Yeaman MR, Sullam PM, Dazin PF, Norman DC, Bayer AS. Characterization of Staphylococcus aureus-platelet binding by quantitative flow cytometric analysis. J Infect Dis. (1992) 166:65–73. doi: 10.1093/infdis/166.1.65
154. Nguyen T, Ghebrehiwet B, Peerschke EI. Staphylococcus aureus protein A recognizes platelet gC1qR/p33: a novel mechanism for staphylococcal interactions with platelets. Infect Immun. (2000) 68:2061–8. doi: 10.1128/IAI.68.4.2061-2068.2000
155. Powers ME, Becker RE, Sailer A, Turner JR, Bubeck Wardenburg J. Synergistic action of Staphylococcus aureus alpha-toxin on platelets and myeloid lineage cells contributes to lethal sepsis. Cell Host Microbe. (2015) 17:775–87. doi: 10.1016/j.chom.2015.05.011
156. de Haas CJ, Weeterings C, Vughs MM, de Groot PG, Van Strijp JA, Lisman T. Staphylococcal superantigen-like 5 activates platelets and supports platelet adhesion under flow conditions, which involves glycoprotein Ibalpha and alpha IIb beta 3. J Thromb Haemost. (2009) 7:1867–74. doi: 10.1111/j.1538-7836.2009.03564.x
157. Hu H, Armstrong PC, Khalil E, Chen YC, Straub A, Li M, et al. GPVI and GPIbalpha mediate staphylococcal superantigen-like protein 5 (SSL5) induced platelet activation and direct toward glycans as potential inhibitors. PLoS ONE. (2011) 6:e19190. doi: 10.1371/journal.pone.0019190
158. Binsker U, Palankar R, Wesche J, Kohler TP, Prucha J, Burchhardt G, et al. Secreted immunomodulatory proteins of Staphylococcus aureus activate platelets and induce platelet aggregation. Thromb Haemost. (2018) 118:745–57. doi: 10.1055/s-0038-1637735
159. Schubert S, Schwertz H, Weyrich AS, Franks ZG, Lindemann S, Otto M, et al. Staphylococcus aureus alpha-toxin triggers the synthesis of B-cell lymphoma 3 by human platelets. Toxins. (2011) 3:120–33. doi: 10.3390/toxins3020120
160. Wilke GA, Bubeck Wardenburg J. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc Natl Acad Sci USA. (2010) 107:13473–8. doi: 10.1073/pnas.1001815107
161. Parimon T, Li Z, Bolz DD, McIndoo ER, Bayer CR, Stevens DL, et al. Staphylococcus aureus alpha-hemolysin promotes platelet-neutrophil aggregate formation. J Infect Dis. (2013) 208:761–70. doi: 10.1093/infdis/jit235
162. Surewaard BGJ, Thanabalasuriar A, Zeng Z, Tkaczyk C, Cohen TS, Bardoel BW, et al. alpha-toxin induces platelet aggregation and liver injury during Staphylococcus aureus sepsis. Cell Host Microbe. (2018) 24:271–84 e3. doi: 10.1016/j.chom.2018.06.017
163. Munoz-Planillo R, Franchi L, Miller LS, Nunez G. A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the Nlrp3 inflammasome. J Immunol. (2009) 183:3942–8. doi: 10.4049/jimmunol.0900729
164. Craven RR, Gao X, Allen IC, Gris D, Bubeck Wardenburg J, McElvania-Tekippe E, et al. Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS ONE. (2009) 4:e7446. doi: 10.1371/journal.pone.0007446
165. Kebaier C, Chamberland RR, Allen IC, Gao X, Broglie PM, Hall JD, et al. Staphylococcus aureus alpha-hemolysin mediates virulence in a murine model of severe pneumonia through activation of the NLRP3 inflammasome. J Infect Dis. (2012) 205:807–17. doi: 10.1093/infdis/jir846
166. Verschoor A, Neuenhahn M, Navarini AA, Graef P, Plaumann A, Seidlmeier A, et al. A platelet-mediated system for shuttling blood-borne bacteria to CD8alpha+ dendritic cells depends on glycoprotein GPIb and complement C3. Nat Immunol. (2011) 12:1194–201. doi: 10.1038/ni.2140
167. Mills EW, Wangen J, Green R, Ingolia NT. Dynamic regulation of a ribosome rescue pathway in erythroid cells and platelets. Cell Rep. (2016) 17:1–10. doi: 10.1016/j.celrep.2016.08.088
168. Mills EW, Green R, Ingolia NT. Slowed decay of mRNAs enhances platelet specific translation. Blood. (2017) 129:e38–8. doi: 10.1182/blood-2016-08-736108
169. Best MG, Sol N, Kooi I, Tannous J, Westerman BA, Rustenburg F, et al. RNA-Seq of Tumor-educated platelets enables blood-based pan-cancer, multiclass, and molecular pathway cancer diagnostics. Cancer Cell. (2015) 28:666–76. doi: 10.1016/j.ccell.2015.09.018
170. Best MG, Sol N, In 't Veld S, Vancura A, Muller M, Niemeijer AN, et al. Swarm intelligence-enhanced detection of non-small-cell lung cancer using tumor-educated platelets. Cancer Cell. (2017) 32:238–52 e9. doi: 10.1016/j.ccell.2017.07.004
171. Coppinger JA, Cagney G, Toomey S, Kislinger T, Belton O, McRedmond JP, et al. Characterization of the proteins released from activated platelets leads to localization of novel platelet proteins in human atherosclerotic lesions. Blood. (2004) 103:2096–104. doi: 10.1182/blood-2003-08-2804
172. Maynard DM, Heijnen HF, Horne MK, White JG, Gahl WA. Proteomic analysis of platelet alpha-granules using mass spectrometry. J Thromb Haemost. (2007) 5:1945–55. doi: 10.1111/j.1538-7836.2007.02690.x
173. Zufferey A, Schvartz D, Nolli S, Reny JL, Sanchez JC, Fontana P. Characterization of the platelet granule proteome: evidence of the presence of MHC1 in alpha-granules. J Proteomics. (2014) 101:130–40. doi: 10.1016/j.jprot.2014.02.008
174. Kamykowski J, Carlton P, Sehgal S, Storrie B. Quantitative immunofluorescence mapping reveals little functional coclustering of proteins within platelet alpha-granules. Blood. (2011) 118:1370–3. doi: 10.1182/blood-2011-01-330910
175. Aslan JE, Phillips KG, Healy LD, Itakura A, Pang J, McCarty OJ. Histone deacetylase 6-mediated deacetylation of alpha-tubulin coordinates cytoskeletal and signaling events during platelet activation. Am J Physiol Cell Physiol. (2013) 305:C1230–9. doi: 10.1152/ajpcell.00053.2013
Keywords: platelet, sepsis, inflammation, infection, dengue, malaria, bacteria, thromboinflammation
Citation: Guo L and Rondina MT (2019) The Era of Thromboinflammation: Platelets Are Dynamic Sensors and Effector Cells During Infectious Diseases. Front. Immunol. 10:2204. doi: 10.3389/fimmu.2019.02204
Received: 10 May 2019; Accepted: 30 August 2019;
Published: 13 September 2019.
Edited by:
Craig N. Jenne, University of Calgary, CanadaReviewed by:
Olivier Garraud, Université de Lyon, FranceCopyright © 2019 Guo and Rondina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew T. Rondina, bWF0dGhldy5yb25kaW5hQGhzYy51dGFoLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.