 Doan C. Nguyen
Doan C. Nguyen Chester J. Joyner1,2
Chester J. Joyner1,2 Iñaki Sanz
Iñaki Sanz F. Eun-Hyung Lee
F. Eun-Hyung Lee
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 11 September 2019
Sec. B Cell Biology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02138
This article is part of the Research Topic Advances in Plasma Cells in Health and Disease View all 10 articles
Antibody secreting cells (ASCs) are terminally differentiated cells of the humoral immune response and must adapt morphologically, transcriptionally, and metabolically to maintain high-rates of antibody (Ab) secretion. ASCs differentiate from activated B cells in lymph nodes and transiently circulate in the blood. Most of the circulating ASCs undergo apoptosis, but a small fraction of early ASCs migrate to the bone marrow (BM) and eventually mature into long-lived plasma cells (LLPCs). LLPC survival is controlled both intrinsically and extrinsically. Their differentiation and maintenance programs are governed by many intrinsic mechanisms involving anti-apoptosis, autophagy, and metabolism. The extrinsic factors involved in LLPC generation include BM stromal cells, cytokines, and chemokines, such as APRIL, IL-6, and CXCL12. In humans, the BM CD19−CD38hiCD138+ ASC subset is the main repository of LLPCs, and our recent development of an in vitro BM mimic provides essential tools to study environmental cues that support LLPC survival and the critical molecular mechanisms of maturation from early minted blood ASCs to LLPCs. In this review, we summarize the evidence of LLPC generation and maintenance and provide novel paradigms of LLPC maturation.
A key aspect of the adaptive immune response is the rapid production of high-affinity antibodies (Abs). This antibody production is the function of an antibody secreting cell (ASC), which arises from naïve (or memory) B cells as they encounter antigen, activate, proliferate, and differentiate. In draining lymph nodes, the B cell to ASC differentiation can occur either in the germinal center (GC) reactions (which has implications for ASC longevity; see below) or outside GC, as a part of an extrafollicular response (1–3) (Figure 1A). Differentiated ASCs subsequently egress out of lymph nodes and circulate in the blood (Figure 1B). Despite their critical function, ASCs are rare and comprise of no more than ~0.01–1% of the total cellularity in the circulation and lymphoid tissues.
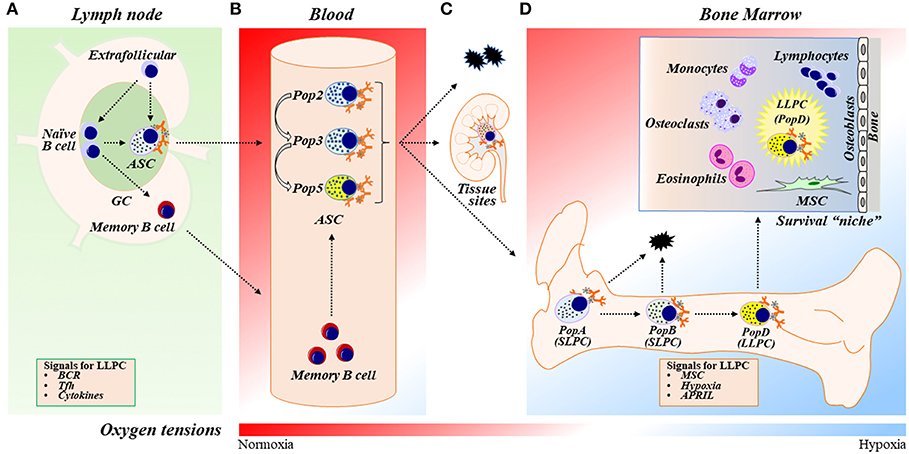
Figure 1. ASC differentiation, maturation, and LLPC generation and maintenance. (A) In response to vaccination or infection, naïve B cells proliferate and differentiate into ASCs within germinal center (GC) reactions in secondary lymphoid organs (SLOs) such as lymph nodes (LNs) or outside of GCs in extrafollicular responses. ASCs then egress out of SLOs and circulate in the blood transiently. Naïve B cells can also differentiate into memory B cells, circulate systemically, and further differentiate into ASCs upon activation. The two-step model of LLPC generation consists of the GC reaction and the BM maturation process [see also (D)]. In the GC reaction, B cells may receive signals such as BCR stimulation, Tfh (or other Th cell) interactions, and cytokines (such as IL-21 and IFNγ) in the GC that are important during ASC differentiation to ultimately become a LLPC. (B) Human blood ASCs are heterogeneous and can be classified into distinct populations (Pops), including Pop2, Pop3, and Pop5, based on their surface protein expression (see also Table 1). (C) Most blood ASCs apoptose. However, a fraction migrates to inflammatory or other tissue “niches” (e.g., mucosa and bone marrow) and become tissue-specific ASCs. (D) A small number of blood ASCs emigrate to the BM, a physiologically hypoxic environment. Human BM ASCs can be identified based on surface protein expression as PopA (SLPCs), PopB (SLPCs), and PopD (LLPCs), respectively, in the BM (Table 1). LLPCs acquire longevity and are maintained within the dedicated BM survival “niche.” ASC survival factors include BM MSCs (and/or their secreted factors), cytokines (i.e., APRIL), and hypoxic conditions. BM, bone marrow; GC, germinal center; ASC, antibody-secreting cells; SLPC, short-lived plasma cells; LLPC, long-lived plasma cells; BCR, B-cell receptor; Tfh, T follicular helper cells; MSC, mesenchymal stromal/stem cells.
In the blood and secondary lymph organs (SLOs), the ASC presence is transient (days to a few weeks) during primary and secondary immune responses (4–7). After vaccination or during infection, ASCs appear in circulation for days and quickly disappear from the blood while serum Ab titers remain (6, 8–10). The bimodal characteristics of serum Ab decay after infection or long-lived vaccines suggests that the large initial burst of ASCs is responsible for the peak of serum Ab titers and the secondary decay is due to ASCs with longer half-lives (11).
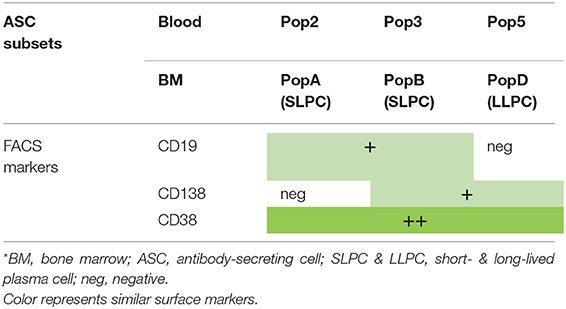
Table 1. Phenotype of blood and BM ASC subsets*.
In the circulation, the majority of circulating ASCs undergo apoptosis; yet a specialized fraction further matures into long-lived plasma cells (LLPCs) as they migrate to BM or other tissues (12) (Figures 1C,D). LLPCs are quiescent, terminally-differentiated, non-dividing cells that survive after the antigen vanishes and is responsible for protection (4, 13–15). It is thought that LLPCs constitutively produce specific Abs for years or even decades after an initial infection or immunization (12), with evidence that they can persist independently of memory B cells (11, 15–17). Hence, generating LLPCs is the main objective of an ideal vaccine.
Many studies show that ASCs derived from GC reactions, rather than extrafollicular responses, have the potential for LLPC formation and survival (12, 18–21). After B cell receptor (BCR) cross-linking and costimulation, a number of cytokines (particularly IL-21 and IFNγ), through help from T follicular helper cells (Tfh), and affinity maturation have significant intrinsic influence on an ASC generated through GC responses to become a LLPC (20–28). Most recently, IFNγ through B cell intrinsic T-bet expression is required for LLPC generation (29). Thus, the GC appears to be important for the formation of LLPCs, yet the overall molecular mechanisms and programs that govern these processes are largely unexplored.
Some investigators show that LLPCs can be generated independently of B cell maturation (i.e., in the absence of GC formation) (30–32). However, these studies showed persistence of ASCs for only 3–4 months and it is unclear if they are indeed long-lived. Nonetheless, whether a memory B cell generated initially through a GC response and re-stimulated extrafollicularly has similar intrinsic LLPC potential is not entirely known. Thus, it is still debated if some ASCs derived extrafollicularly from memory vs. naïve B cell origins may have long-lived survival potential.
In this review, we focus on the molecular and cellular processes involved in the adaptation of ASCs after exiting the GC into the medullary cords and efferent lymphatics and upon entering the blood, and eventually the BM niche (Figure 1D). Throughout the review, we will refer to “differentiation” as the process in which a B cell becomes an ASC (through GC or non-GC reactions), and “maturation” as the process in which an early minted ASC develops or transforms into a LLPC. The term “maintenance” will be used for survival of the ASCs. On this basis, we will discuss the role of BM “microniches” in both the maturation and the maintenance of BM LLPCs.
The prime directive of ASCs is to synthesize and secrete Abs that provide protection against microorganisms. ASCs produce large amounts of Abs, typically at a rate of between ~10 and 1,000 (and can be up to ~5,000–10,000) Ab molecules per cell per second, which corresponds to ~0.2–22 pg/cell/day (33–38). To perform this heroic feat, ASCs undergo fundamental changes in morphology and homeostasis during terminal differentiation, which include altering cellular structure, surface protein expression profile, metabolism, and other cellular and molecular programs. In this section, we discuss these changes by contrasting the recent findings of human blood and BM ASCs.
To accommodate the production of copious amounts of proteins, ASCs expand their cytoplasm, endoplasmic reticulum (ER), and Golgi apparatus during differentiation and maturation, enabling increased capacity for Ab synthesis and secretion (39) (Figure 2). Early ASCs lose their proliferative capacity and reduce the nucleus size by condensing their chromatin. The decrease in nuclear area and the repositioning of the nucleus closer to the cell membrane enable ASCs to accommodate additional cellular machinery necessary for protein production. Overall, these morphological changes are orchestrated to result in assembling an Ab factory.
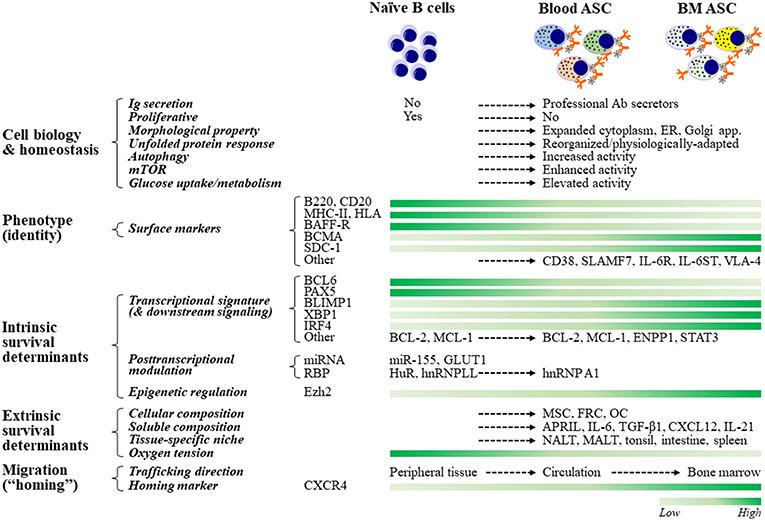
Figure 2. ASC cell biology, fate determination, and resultant phenotypes. ASCs adapt their cellular biology, machinery, and morphology to maintain homeostasis while producing copious amounts of antibodies. For example, ASCs must upregulate the unfolded protein response (UPR) to inhibit apoptotic pathways that would be engaged due to increased Ig synthesis. Other pathways that act in concert with the UPR to support ASC homeostasis are autophagy, mTOR, and glucose metabolism. Although no phenotype-based “universal identity” of blood and bone marrow ASCs presently exists, ASCs are typically identified by increased expression of CD38, CD138, and BCMA, and decreased expression of B220, MHCII, and BCR. The commitment to the ASC fate of naïve B cells is regulated transcriptionally, post-transcriptionally, and epigenetically. Maintenance of B cell programs is mainly controlled by Bcl6 and Pax5, and promotion of B cell differentiation and ASC commitment are essentially governed by Blimp1 (encoded by Prdm1), Xbp1, and Irf4. The role of post-transcriptional (through certain miRNAs and RBPs) and epigenetic (such as via Ezh2) regulation is increasingly being recognized. The lifespan of ASCs ranges from a few days to several decades and is thought to be chiefly determined extrinsically, which is largely dependent on their residency. Accordingly, LLPCs reside in a specialized, dedicated BM compartment (“survival niche”) that is physiologically hypoxic and is formed and maintained by both cellular (such as MSCs, FRCs, and OCs) and soluble (such as APRIL, IL-6, TGF-β1, and CXCL12) components. Other tissue-specific niches capable of maintaining ASCs have also been described. ASC migration (or “homing”) from peripheral tissues or SLOs toward survival niches is primarily directed by binding of CXCR4 to its ligand, MSC-secreted chemokine CXCL12 (SDF1α). Certain miRNAs and RBPs include miR-155, which is involved in cytokine production, and HuR and hnRNPLL, which modulate IgH mRNA turnover and translation. ASC, antibody-secreting cells; LLPC, long-lived plasma cells; ER, endoplasmic reticulum; SLO, secondary lymphoid organs; MSC, mesenchymal stromal cells; FRC, fibroblastic reticular cells; OC, osteoclasts; NALT, nasal-associated lymphoid tissues; MALT, mucosa-associated lymphoid tissues.
As ASCs differentiate and mature, they alter their surface protein expression profile. These proteins can be used as the phenotypic markers for identifying subsets within the ASC population. Notably, there is no universally accepted or “standardized” phenotype to identify ASCs—in humans or in mice (which make it difficult to compare across studies). Nevertheless, human ASCs in general lose their surface immunoglobulins and can typically be identified based on the expression of one or more of the following cell surface markers: CD19, CD27, CD38, CD138, and BCMA (40–46) (Figure 2). In mice, blood ASCs typically reduce or lose the expression of B220, MHC-II, and BCR. Additional markers such as Sca-1, TACI, and CD98, have also recently been used for identifying ASCs in mice—on the basis of CD138 expression (43, 45–49); however, it is unclear how this combination translates to humans or other animal models.
In humans, blood ASCs are typically identified as CD19intCD27hiCD38hi (gated on CD3−CD14−IgD− cells) (41). However, our recent studies along with others have demonstrated that human blood ASCs are heterogeneous and can be further subgrouped on the basis of the additional expression of CD19 and CD138 (40, 50) (Figures 1B, 2, Table 1). Of the five different ASC subsets we identified, three were CD19+ whereas two were CD19− subsets. Among them, populations 2 (pop 2; CD19+IgD−CD27+CD38hiCD138−) and 3 (pop 3; CD19+IgD−CD27+CD38hiCD138+) are the most abundant, and population 5 (pop 5; CD19−IgD−CD27+CD38hiCD138+) resembles LLPCs by surface markers (Table 1). The other ASC subsets contained a mixed population of B cells and other cell types and thus, the study focused on pops 2, 3 (CD19+), and 5 (CD19−). Based on RNAseq analysis, pops 2 and 3 are very similar transcriptionally whereas pop 5 is quite distinct (Figure 1B). More interestingly, all pops 2, 3, and 5 have similar potential for survival in an in vitro BM mimetic system, suggesting the importance of extrinsic factors in their survival.
The exit of ASCs from SLOs into the blood is guided by the expression of their S1P receptor 1 (S1P1) (51, 52). Subsequently, in concert with changes in surface markers, blood ASCs modulate the expression of chemokine receptors that direct them to local tissues (Figure 1C). For example, CXCR3 directs ASCs to inflamed tissues, CCR9 guides them to the small intestine, CCR10 directs the cells to a variety of tissues, including the colon, lung, trachea, or mammary glands (53), and, importantly, CXCR4 leads them to the BM rich in CXCL12 (54–56). It is also thought that ASCs are retained in the BM by CXCR4 because they fail to accumulate in the BM microenvironment in CXCR4−/− mice (54).
It still remains unclear how the expression of chemokine receptors on ASCs is regulated, although transcription factor c-Myb may be involved (57). In the absence of c-Myb, no ASCs are detected in the BM. Whether this phenomenon is due to inability of c-Myb deficient ASCs to migrate along a CXCL12 gradient or if c-Myb is involved in ASC survival will need further elucidation. Nonetheless, the migratory capability of ASCs appears to be decreased as they mature (55, 58), along with upregulation of Blimp-1 (59–61). This phenomenon may serve as another means of retaining ASCs in the BM or other tissues. Clearly, migration to specific sites rich in survival factors enhances ASC survival; but whether fundamental intrinsic differences are imprinted during ASC differentiation or whether maintenance is solely dependent on homing markers alone is not well-established. Thus, the nature (intrinsic) or nurture (extrinsic) is a critical debate for LLPC generation. To further its complexity, ASC maturation to a LLPC may actually require a combination of both.
As previously mentioned, LLPCs are non-dividing, long-lasting terminally differentiated ASCs. In mice and humans, most LLPCs reside in the BM (12, 62, 63) (Figure 1D). These specialized ASCs persist and are thought to autonomously and continuously produce Abs for decades or beyond, independently of antigen stimulation (13–15). BM ASCs are also thought to be unresponsive to further antigenic stimulation (64). However, unlike IgG ASCs, a recent report identified a population of IgM BCR expressing ASCs that may sense antigen (65). In mice, ASCs in the spleen and the BM are identified based on the expression of B220, CD138, and Sca-1 subsets, with implications of B220− ASCs as mature subsets (46–48). Human BM ASCs are heterogeneous, with few overlapping markers with mice, and can be grouped on the basis of expression of CD19, CD27, CD38, and CD138 (66) (Figure 1D, Table 1). To identify the human BM LLPC subset, we interrogated each compartment for microbial specificity against viral infection (measles and mumps) that occurred decades ago and found that only the CD19−CD38hiCD138+ subset contained these long-lived viral specificities. We assigned the CD19+ BM populations as short-lived plasma cells (SLPCs), which include pops A and B that did not comprise of the long-lived specificities (66) (Table 1). Others have alluded to these results but did not show long-lived antigen specificities (67). Thus, it appears that the ASC fraction that downregulates CD19 expression and escapes apoptotic cell death eventually becomes LLPCs.
The B cell to ASC differentiation involves an extensive reorganization of transcriptional networks (68, 69) (Figure 2). In order for ASCs to synthesize and secrete large amounts of Abs, these regulatory networks massively upregulate transcription of the heavy and light chain mRNA transcripts (up to 100–1,000-folds). ASCs have unique transcriptomes where >70% transcripts encode the IgH and IgL chains for Ab synthesis (49). The differentiation of ASCs is regulated at the transcriptional, post-translational, and epigenetic levels. In this section, we discuss the intrinsic cellular programs—including signaling pathways and metabolic alterations as well as transcriptional, post-transcriptional, and epigenetic networks—involved in regulation of ASC differentiation, maturation, and maintenance.
Aspects of ASC biology, metabolism, homeostasis, migration, and survival are controlled by their distinct gene expression programs (or transcriptional regulatory networks) (49, 70, 71). These regulatory programs promote features to produce large quantities of Abs and to shed B cell characteristics. They function through two groups of antagonistic transcription factors, in which acquisition of plasma cell-specific transcription results in termination of B cell-specific transcriptional program (39, 72–74).
Transcription factors maintaining the B cell program are mainly BCL6 and PAX5 (75), although Bach2 and IRF8 are also important (39, 69). BCL6 is highly expressed in GC B cells and promotes cell proliferation and survival. Transcription factors facilitating ASC differentiation include Blimp1 (encoded by Prdm1) (76), XBP1, and IRF4 (39, 60, 61, 69, 77–79). These ASC-“specific” transcription factors are uniquely upregulated in plasma cell transcriptome (49, 70), with Blimp1 and XBP1 required for Ab secretion whereas IRF4 is likely essential for survival (61, 80–82). Importantly, Blimp1 and XBP1 play a multifunctional role in both instructing differentiation and maintaining homeostasis. Blimp1 inactivation downregulates the unfolded protein response (UPR), thus negatively affecting the cell secretory capabilities (60, 61). Also, Blimp1 positively regulates mTORC1 activity (61) as loss of Raptor negatively affects ASC maturation and Ab secretion (but not survival) (48). Finally, XBP1 has been shown to be an important regulator of the UPR (83).
In addition to these two reciprocally-regulated groups of transcription factors, a number of other factors have been found differently expressed by ASCs in distinct compartments (i.e., SLOs, blood, and BM). Examples include anti-apoptotic factors, such as Mcl-1, Bcl-2, BclxL, Bim, and BCLw, which are increased, and pro-apoptotic factors, such as Bax and BID, which are decreased, to promote ASC maintenance (84–87). Importantly, expression of Mcl-1 appears to be required for ASC survival in the BM and is mediated by BCMA, a TNF receptor (85), potentially under the control of IRF4 (71) or Zbtb20 (61, 88–90).
During differentiation, ASCs adapt and utilize various signaling pathways to accommodate the need for high-rate Ab production and maintenance of homeostasis (Figure 2). Thus, activation of the UPR, the cellular response to ER stress due to accumulation of misfolded (damaged) proteins, is essential for increased Ab secretion during ASC differentiation from B cells (61, 81, 91, 92). In most cells, upregulation of the UPR indicates the inability of the cells to recycle proteins via ubiquitination or signify the hijacking of this pathway (i.e., by virus). In these situations, the UPR typically results in apoptosis as a means to eliminate a virally infected or dysfunctional cell. In the case of ASCs, the UPR is upregulated due to stress from the massive protein secretion in order to maintain the cell's survival since this imbalance could quickly lead to apoptosis. This pathway strongly involves XBP1 through ATF6 and IRE-1 during differentiation (61, 91, 92). Nevertheless, little is known about the regulation of this pathway in human ASCs after differentiation or how it may be modulated to ensure the maintenance of LLPCs. Thus, identifying the molecular mechanisms that regulate this pathway could lead to modulating LLPC formation and maintenance.
Antibody secreting cells (ASCs) must also adapt their metabolism to produce Abs during circadian nutrient fluctuations while maintaining cellular homeostasis. As terminally-differentiated cells, ASCs do not actively remodel their cytoplasm through cell division but rely on autophagy to recycle their protein aggregates and organelles in order to optimize energy for survival. Multiple autophagy genes, such as Atg5, Atg9, and Atg13, are upregulated in ASCs and likely play a role as LLPCs mature (49, 66, 93). This increased autophagic activity enables LLPCs to regulate the production of Abs when nutrients are limited (93). Interestingly, hypoxia is a known inducer of autophagocytic pathways, and we recently showed that hypoxic conditions enhance the survival of human ASCs in vitro (94). Whether autophagy programs are upregulated prior to BM localization or in response to the hypoxic BM microenvironment is still not clear.
Another important signaling pathway for ASCs is the mTOR. The mTOR kinase is a major regulator of many cellular processes, including survival and proliferation. mTOR signaling, mainly through the mTORC1 signaling complex (mTORC1), regulates the biosynthesis of cellular macromolecules, including proteins, nucleic acids, and fatty acids, as well as glycolysis and organelle biosynthesis (95, 96). Blimp1 positively regulates mTOR signaling as B cells differentiate into an ASC (48, 61, 97). However, in humans, as ASCs mature into BM LLPCs, mTORC1 activity is downregulated (94) and autophagy is increased (66). Additionally, the survival of human BM LLPCs, unlike that of BM SLPCs or early minted blood ASCs, is resistant to mTOR inhibitors, illustrating downregulation of mTOR pathways for LLPC maturation (94). Similar findings were corroborated in mouse studies (48).
As ASCs evolve into LLPCs, major metabolic changes occur. In addition to differences in mTOR signaling that functionally distinguishes LLPCs from SLPCs, a sundry of metabolic mechanisms differs between these two cell types. LLPCs have higher glucose uptake than SLPCs, and utilize glucose for Ab glycosylation (98). Moreover, LLPCs have increased expression of the glucose transporter, GLUT1 (98, 99). Recently, the ATP-degrading enzyme ENPP1, a regulator of glucose metabolism, was shown to be required for the development and survival of LLPCs in mice (100). In both mice and human, higher maximal respiratory capacity was shown in LLPCs compared to SLPCs (98), suggesting differences in respiratory capacity that may be linked to survival advantages. Nonetheless, more studies are warranted to understand whether versatility of energy utilization may determine survival advantages in unique nutrient-deprived environments.
Post-transcriptional regulation of mRNA expression is a major mechanism to rewire the transcriptome and proteome. This type of regulation defines the fate of mRNAs, including immunoglobulin transcripts, through multiple steps of mRNA processing, including alternative splicing of pre-mRNAs, mRNA stability and turnover, 3' UTR regulation, and translational control (101–103). The two key players in post-transcriptional regulation of protein expression are non-coding RNAs (mainly miRNAs) and associated RNA-binding proteins (RBPs) (101, 103, 104). In committed ASCs, post-transcriptional regulation is employed in several processes and can play a role in the magnitude of immunoglobulin expression (Figure 2). The processing of IgH mRNA is modulated post-transcriptionally (which requires Blimp-1) (60, 61). Expression of the glucose transporter GLUT1 is also regulated at the post-transcriptional level (98, 99). However, whether post-transcriptional regulation of mRNA and non-coding RNAs plays a role in LLPC maturation needs further evaluation.
RNA-binding proteins (RBPs) are involved in regulating the B cell programs and thus, can also influence ASC differentiation (101, 102). An example is HuR, an RBP splicing regulator ubiquitously found in the nucleus and capable of nucleo-cytoplasmic shuttling (105). Depletion of HuR results in unbalanced mitochondrial metabolism and impaired B cell proliferation and differentiation, which has a negative impact on ASC differentiation (106). Another major RBP involved in ASC differentiation is the splicing factor hnRNPLL, a member of the hnRNP (heterogeneous nuclear ribonucleoprotein) family. hnRNPLL is specifically induced in ASCs and, through regulating mRNA alternative splicing and stability, facilitates B cell differentiation and ASC Ab secretion (107). Also, hnRNPLL and the transcription elongation factor ELL2 (elongation factor, RNA polymerase II, 2), a regulator of pre-mRNA processing in plasma cells, modulate the ratio of secreted vs. membrane-encoding IgH transcripts. Most interesting, ELL2 was responsible for differentially processed transcripts such as BCMA (108).
Our recent integrated transcriptomic and proteomic analysis distinguishing early minted ASCs and BM LLPCs identified a novel RBP, hnRNP A1, as a potential post-transcriptional modulator (94). Blocking hnRNP A1 led to reduced survival of both human blood ASCs and BM LLPCs (109). Thus, unlike HuR or hnRNPLL, which are involved in regulating the differentiation of B cells to ASCs, hnRNP A1 may be unique and important in the regulation of the maturation of early blood ASCs and BM LLPCs. Altogether, these data highlight the important role of post-transcriptional regulation in the differentiation as well as maturation of ASCs, which can also influence the survival of LLPCs.
Ezh2, a histone methyltransferase that catalyzes trimethylation of histone H3 (H3K27me3), has been shown to epigenetically regulate gene expression during ASC differentiation. Ezh2 expression is upregulated during ASC differentiation, and Ezh2-deficient ASCs exert reduced levels of the UPR, glucose metabolism, and mitochondrial respiration, resulting in decreased Ab secretion (110, 111) (Figure 2). Thus, Ezh2 plays a role in ASC differentiation by promoting metabolic changes that are important for Ab secretion. Interestingly, this enzyme can interact directly with Blimp1 (60) and represses the B cell program (110). It will be important to understand if Ezh2 expression is modified as ASCs mature into a LLPC.
The notion that some ASC subsets exist for only a few days whereas others live for decades is thought to be chiefly determined extrinsically, i.e., largely dependent on where ASCs reside. In both mice and humans, most LLPCs reside in a specialized BM microenvironment, known as the survival niche (12, 62, 63) (Figure 1D). Removal of ASCs from this specialized residence leads to rapid cell death. Thus, these niches enable ASC maintenance and longevity—via intracellular crosstalk or direct ASC interactions with neighboring cells and/or through soluble factors produced locally by neighboring cells (12, 112, 113). It is believed that these niches support only a limited ASC number due to their physical confines (112). Thus, the BM environment must be stable to protect their resident cells yet dynamic enough to adapt to new arrivals.
Although LLPCs are highly enriched in the BM (63), recent studies have described several tissue-specific niches (TSNs) that afford LLPC survival advantages. LLPCs can also be found, albeit at low frequencies, in other tissues throughout the body, such as the nasal-associated lymphoid tissues (NALT) (114), human tonsillar lymphoid tissues (115), human mucosa or mucosa-associated lymphoid tissues (MALT) (116, 117), human intestine (118–120), spleen (67, 121), and human salivary gland microenvironment (in primary Sjögren's syndrome) (122) (Figure 2). In mice, the spleen appears to provide extrinsic signals to determine the cell survival (71, 123). Thus, outside the BM, TSNs may also be capable of maintaining LLPCs, but may be unique for specific isotypes or specificities (114–116, 118, 120, 122).
The BM niche is the primary “home” for LLPCs. This ASC niche represents a highly-complex microenvironment which includes multiple cell types such as mesenchymal stromal/stem cells (MSCs) (113, 124), eosinophils (125, 126), megakaryocytes (127), basophils (128), monocytes (129), macrophages (130), dendritic cells (131), T cells (132), osteoclasts (133), and fat cells (134) (Figure 1D). Notably, the BM is physiologically hypoxic—with oxygen tensions range ~1–6%, depending upon the vicinity of blood vessels (135–138). Despite the rich cellular BM composition, it still remains a mystery whether LLPCs preferentially home to the BM or pro-actively adapt upon arrival to the extreme hypoxic BM environment.
Mesenchymal stromal/stem cells (MSCs) are the principal organizer of the BM microniche involved in supporting LLPC survival. Evidence shows that this support is mediated through cell-cell contacts and/or soluble signals (62, 94–145). BM ASCs are in direct contact with MSCs (126, 146). This interaction is thought to be mediated mainly via ASC adhesion receptors, VLA-4 and LFA-1 (140, 145, 147, 148) and their natural ligands, VCAM-1 and ICAM-1, respectively, on MSCs, and are thought to anchor them to the extracellular matrix (ECM) (149). MSCs also assist ASC survival through the secretion of soluble molecules. For example, MSCs secrete CXCL12 that interacts with CXCR4 on ASCs (54, 55, 62, 150), which direct them to close proximity of MSCs. In addition to chemokines, MSCs secrete IL-6 that is critical for ASC differentiation and survival (62). Moreover, interaction of VLA-4 and fibronectin was shown to be essential for MSC-mediated survival of ASCs (148). MSCs also secrete TGF-β1 and TGF-β2, which are involved in B cell homeostasis and IgA responses in mice (139, 151–154). Finally, MSCs produce ECM-modifying molecules, such as heparan sulfate (HS) (130, 155, 156), which is an unconventional “receptor” that induces APRIL oligomerization and may trigger BCMA-mediated survival signaling of ASCs (157, 158).
The interaction between MSCs and ASCs via MSC-produced CXCL12 and ASC membrane-bound CXCR4 is well-established (159). However, it was unclear whether cell-cell contact between MSCs and ASCs was essential (126, 139, 144, 159) or dispensable (94, 150). Possible explanations for this discrepancy may be due to distinct MSC subsets for each study. Our recent work confirms that human BM-derived MSCs provided sufficient soluble factors for ASC survival without the need for cell-cell contact (94). While the persistence of BM ASCs is supported in general by MSCs, a unique subset of CXCL12+ MSCs are obligatory to form and maintain the survival niche (126, 146). Alternatively, the requirements for the survival of ASCs as they mature into LLPCs may vary, supporting a model that different MSC subsets are needed for distinct ASC populations (specific to blood or BM).
Recently, a unique subset of lymph node stromal cells found in the medullary cords provided a major source of ASC survival factors, including CXCL12, IL-6, APRIL, and BAFF (160). Distinct from T zone fibroblastic reticular cells (FRCs), the medullary FRC subset (MedRCs) together with macrophages promoted ASC survival by soluble signals similar to our observations with BM stromal cells. Additionally, FRCs at the GC-T cell zone interface may also have similar characteristics (160, 161). In summary, particular stromal cells in specific locations (i.e., lymph nodes, BM, or other tissue sites) may ultimately orchestrate ASC survival.
Extracellular vesicles (EVs) from BM MSCs provide additional evidence that MSC-ASC cell-cell contacts are non-essential (162). Both MSCs and ASCs are rare cell types in the BM microniche and EVs may provide survival factors over greater distances compared to the local paracrine factors which are limited by proximity. MSC-derived EVs have been shown to be involved in multiple myeloma (163), but our group recently showed that human BM MSC-derived EVs can support healthy ASC survival in vitro (162). Although the exact mechanism was not determined, EVs probably mediate the delivery and transfer of diverse biologically-active molecules over greater distances.
In addition to MSCs, ASCs are also surrounded by other cell types in the BM microenvironment. Among them, eosinophils (125) and osteoclasts (OCs) (133) (Figure 1D) have been shown to promote ASC survival. Eosinophils have high-turnover, are located in the ASC vicinity, and produce survival factors, including IL-6 and APRIL (and BAFF) (125, 126). Although Chu et al. showed that eosinophils are needed for ASC survival (125), other investigators found that they are not essential (164–166). This discrepancy may be attributed to redundancy of ASC survival factors among a variety of accessory cells. Monocyte-derived OCs, which are able to support myeloma cell growth (167), also appeared to support the survival of ASCs in vitro (133). Interestingly, OCs produce BAFF and APRIL (168), but their ASC survival benefits may actually be independent of either APRIL or BAFF (133).
Other accessory cell types that are capable of promoting ASC maintenance include megakaryocytes (125, 127), basophils (125, 128), dendritic cells (131), Tfh (161, 169), and Treg (125, 170). Again, some of these cell types in BM niche may have redundant functions or secrete overlapping factors with other BM cell types and thus, may play overlapping roles for ASC survival. For example, IL-6 is paramount for ASC survival and is produced by both MSCs and eosinophils. Similarly, APRIL, another important ASC survival factor, can be secreted by neutrophils, eosinophils, megakaryocytes, osteoclasts, as well as BM MSCs (125, 127, 171). At this point, redundant functions from many BM cell types facilitate ASC persistence; however, the BM MSCs appear to be essential beyond IL-6 production for ASC survival (39, 94, 125, 126, 172). The precise soluble factors and molecular mechanisms are not known, but specific clues suggest the MSC surrounding ECM.
In addition to via cell-cell interactions, BM niches also support the process of ASC maturation by soluble components that ultimately alter the cell phenotype into a LLPC.
The APRIL/BAFF:BCMA signaling axis is a well-characterized pathway in ASC survival (161, 173, 174). APRIL or BAFF, two members of the TNF family of ligands, promotes ASC survival and longevity in vitro (125, 175, 176), in mice (123, 129, 177), and ex vivo (in humans) (94). These signals are primarily triggered and delivered through the interactions of BAFF/APRIL with one of their shared receptors, BCMA (176). BCMA is predominantly expressed on terminally differentiated B cells with upregulation on LLPCs (178) (Figure 2). BCMA is required for maintenance of ASCs residing within BM niches, which likely acts through APRIL (or BAFF) in combination with IL-6 (176) or CD4+ T cells (123). Moreover, APRIL also promotes IgA PC survival in human mucosa (116). Molecularly, APRIL mediates cell survival primarily by activation of Akt, Erk1/2, JNK, and NF-κB signaling pathways, which leads to upregulation of antiapoptotic genes. APRIL:BCMA has shown to sustain expression of Mcl-1, an important Bcl2-family antiapoptotic factor that is expressed in ASCs (85, 176).
Syndecan-1 (SDC-1), otherwise known as CD138, a heparan sulfate proteoglycan (HSPG), has been shown to be a pro-survival factor for ASCs (179) (Figure 2). While BAFF predominately binds BAFF receptor (BAFF-R; which is expressed on immature B cells), APRIL can bind through HSPGs (129, 180, 181) and thereby through SDC-1, APRIL oligomerizes and triggers BCMA mediating ASC survival. Alternatively, binding to APRIL may enable syndecans to deliver survival signals through intracellular tails (182). In humans, SDC-1 expression had been used as a LLPC surrogate (3, 42, 180); however, our group recently showed that SDC-1 can be found in early blood ASCs even though SDC-1neg ASCs could eventually upregulate SDC-1 in our in vitro BM mimetic cultures (40). Not surprisingly, both SDC-1+ and SDC-1neg ASCs survived similarly, concluding that upregulation of SDC-1 plays a role in ASC maintenance.
Although there was some debate whether BAFF and APRIL equally support ASC survival (177), BAFF provides no survival advantage in our in vitro BM mimetic cultures (Figure 3). Only APRIL enhanced ex vivo survival of human early minted blood ASCs (94). Also, BAFF and APRIL do not appear to be involved in the pro-survival support to ASCs of OCs, which is reported to be fully cell-cell dependent (133). To our surprise, APRIL provided no additional enhancement over the BM MSC secretome for BM LLPCs (183) (Figure 4). Together, these data suggest that APRIL may only be needed transiently in the early blood ASC phase, permanently altering the phenotype into the LLPC maturation program.
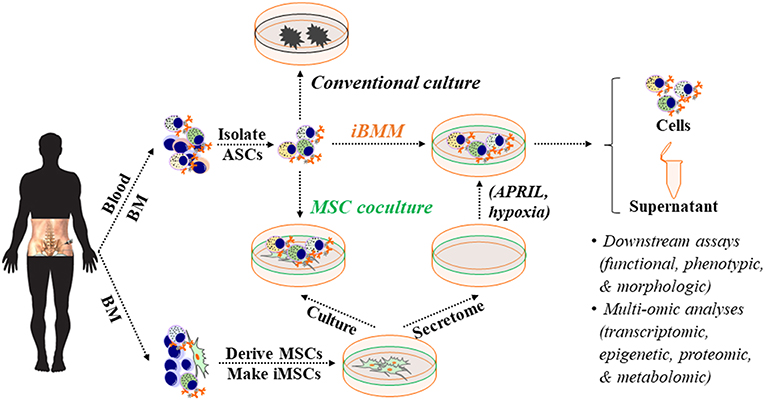
Figure 3. An in vitro BM microniche mimic (iBMM) for studying human ASCs ex vivo. Primary MSCs are derived from human autologous or allogeneic BM samples, expanded, and irradiated. Populations or single blood or BM ASCs are isolated and co-cultured with BM-derived iMSCs or in the cell-free secretome that is harvested from BM-derived MSCs. Typically, blood or BM ASCs quickly die off ex vivo or in conventional culture but survive for weeks under these conditions. The cellular materials and supernatants obtained from the ex vivo ASC cultures can be collected for downstream assays and multi-omic analyses for studying human ASC biology. BM, bone marrow; ASC, antibody-secreting cells; MSC, mesenchymal stromal/stem cells; iMSC, irradiated MSC; APRIL, a proliferation-inducing ligand.
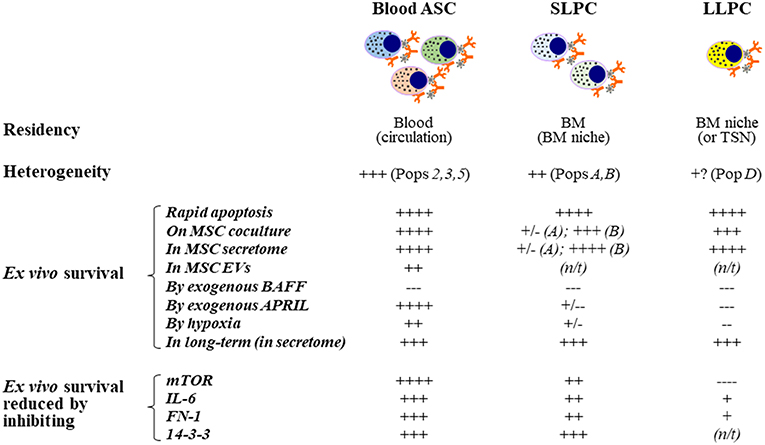
Figure 4. Extrinsic cues for human ex vivo ASC maintenance: Findings from using the in vitro human BM mimic. Blood ASCs circulate transiently while short- and long-lived PCs reside in tissue-specific niches like the BM. Both circulating ASC and local PC populations are heterogeneous in humans and can be classified into distinct subsets based on their expression of CD19, CD38, and CD138. These populations are named Pop2, Pop3, or Pop5 in blood and PopA, PopB, or PopD in the BM. Typically, each of these ASC populations undergo rapid apoptosis ex vivo; however, they can survive for several weeks to months when cultured on BM MSCs, cultured in the cell-free MSC secretome, or cultured in BM MSC-derived extracellular vesicles, albeit at a lower potency. Using this approach, we have evaluated the effect of molecules on ex vivo human ASC survival by adding exogenous molecules, altering the environment where the cells are cultured, and by inhibiting pathways. APRIL and hypoxia offer survival advantages to human blood ASCs but appear to have little or no effects on human BM SLPCs and LLPCs. In contrast, BAFF does not increase survival of ASCs ex vivo even though it is essential for B cell survival. Interestingly, inhibition of mTOR has also elucidated a dependence on this pathway for human blood ASCs but less reliance on this pathway for BM LLPCs. Other pathways have been evaluated similarly as indicated. ASC, antibody-secreting cells; PC, plasma cells; SLPC, short-lived PCs; LLPC, long-lived PCs; MSC, mesenchymal stromal/stem cells; BM, bone marrow; SLO, secondary lymphoid organs; TSN, tissue-specific niches; ex-, exogenous; EVs, extracellular vesicles; N/T, not tested or not available; long-term survival, >8 weeks.
IL-6 signaling has been known to play a critical role in ASC differentiation and long-term survival (Figure 2). IL-6 is important for the differentiation of B cells into ASCs and acts as a pro-survival factor during ASC migration (112). Inhibiting IL-6 signaling leads to a profound reduction of human ASCs (94, 150). Early minted blood ASC subsets have a relatively high expression of the IL-6R (~32–77%), as shown in our recent study (40). Thus, during the acute phase of an infection when B cells differentiate to ASCs, when there is an abundance of free IL-6, ASCs signal through IL-6 through the cell surface IL-6 receptor (IL-6R). However, after resolution of the infection during steady state, free IL-6 levels decrease dramatically with most of it bound to the soluble IL-6 receptor (sIL-6R). The fact that, unlike blood ASCs, BM ASC have low expression of the surface IL-6R (66), might have implicated distinct mechanisms for IL-6 signaling in early minted ASCs and LLPCs.
Fortunately, IL-6 can act through two distinct signaling pathways: classic and trans-signaling. In the classic pathway, IL-6 first binds to the membrane-bound IL-6R (also known as gp80 or CD126); this interaction leads to dimerization and activation of the signal transducing protein gp130 (also known as IL-6ST or CD130), a transmembrane protein important for signal transduction following cytokine engagement through activation of the Jak-1/STAT and Akt/MAPK/ERK-1/2 signal transduction pathways, to eventually upregulate STAT3 (184–186). In the absence of IL-6R, IL-6 has no binding affinity for IL-6ST; therefore, only cells that express IL-6R can be stimulated by IL-6 (187). Thus, the effect of IL-6 through classic signaling is rather limited because it is restricted to only cell types that express the IL-6R such as early minted blood ASC (40, 188, 189). The trans-signaling activates the IL-6/STAT3 through extracellular IL-6 initially binding to its soluble IL-6R (sIL-6R), then this IL-6/sIL-6R complex subsequently binds to IL-6ST (190). Through trans-signaling, IL-6 (and the sIL-6R) stimulates cells that lack the membrane-bound IL-6R. Interestingly, IL-6ST is virtually expressed on all cells in the body (191), including BM LLPCs. It will be interesting to corroborate evidence from the transcriptomes of early minted ASCs and BM LLPCs for IL-6ST expression and investigate whether IL-6 trans-signaling plays a role in LLPC survival.
As aforementioned, CXCR4 (CD184) and its ligand CXCL12 (SDF-1) play an essential role in directing blood ASCs to BM (42, 54, 55). Whether CXCL12 can directly also act as an ASC survival factor in vivo (192), or merely functions as a retention factor to maintain ASCs in survival niches is not known (193). One study showed that CXCL12:CXCR4 signaling promotes ASC survival in vitro (62). Nonetheless, further studies are needed to discriminate survival vs. homing mechanisms of CXCL12.
Additional survival factors reported to date include TGF-β1 (as mentioned previously) (139, 153, 154), IGF-1, TNF-a, HGF, VEGF, IL-21 (125, 130, 159, 161, 173), and fibronectin-1 (FN-1) and YWHAZ (14-3-3 zeta/delta) (94, 148) (Figure 2). Among some of the cell surface molecules involved in ASC survival are CD28 (131, 194, 195), CD93 (196), CD44 (62), CD37 (197), FcgRIIb (198), LFA-1, CD49d (VLA-4), CD49e (VLA-5) (130, 142, 148), CD73 (199), CD13 (YWHAZ receptor) (200, 201), and CD123 (IL-3R) (202). Although some of the factors, such as CD28 (131, 195) and CD93 (196), have been shown to be important for the Ab secretion and survival of BM ASCs, the other factors will probably need further evaluation to understand if they are essential or redundant factors for healthy LLPC maturation and maintenance.
Mouse and human ASCs undergo rapid apoptosis ex vivo (62, 94, 142, 162). Therefore, studies to understand human ASC survival in vitro have not been possible. To overcome this issue, we developed a novel culture system that mimics the human BM microniche to support ASC survival ex vivo to interrogate mechanisms of LLPC maturation (94). To establish this system, we derived and expanded primary human BM MSCs, which are spindle-shaped, plastic-adherent, and phenotyped as CD90+CD73+CD45−CD19− (94) (Figure 3). We initially utilized irradiated BM MSCs as feeders for in vitro cultures of ASCs but quickly found their pro-survival support was entirely driven through paracrine effects, thus defining cell-free BM-derived MSC secretome (94). In contrast, we did not find prolonged ASC survival from cell types found in the blood (94).
We also found that the BM MSC secretome alone was not sufficient for long-lived ASC survival and exogenous APRIL and BM hypoxic conditions provided enhanced longevity (Figures 3, 4). We refer to the BM MSC secretome, exogenous APRIL, and hypoxic conditions as the human cell-free in vitro BM mimetic (iBMM). Previous systems supported human ASCs with co-cultures of supportive cells for a few days or from in vitro generated ASCs (139, 175, 203). Our unique system maintains freshly isolated human ASCs ex vivo for 8–12 weeks and beyond (94).
The development of iBMM has been critical for studying human ASCs in vitro. It provides powerful tools to follow the human ASC maturation process in the BM microniche to characterize the survival factors, signaling pathways, or metabolic programs important for LLPC generation. Moreover, by coupling recent advances in single cell molecular analysis, epigenetics, proteomics, and metabolomics together with the in vitro iBMM, we will be able to dissect the mechanisms of LLPC generation and provide novel insights to their survival and modulation of Ig secretion.
Additionally, the iBMM or cell-free ASC survival system provides a rapid novel system for monoclonal Ab discovery. Currently, monoclonal antibody discovery is limited by cost and time, limiting the ability to identify rare clones. With this system, a priori selection of rare but targeted ASC clones can be identified prior to monoclonal Ab generation. For example, Abs for only neutralizing epitopes can be selected for monoclonal Ab generation, thereby decreasing cost and time.
The ability to support human blood and BM ASC survival ex vivo for months may also provide novel assays to measure vaccine durability. Most vaccine candidates go through extensive clinical testing to assess immunogenicity, safety, and efficacy, and at this time, assessment of vaccine longevity requires the tincture of time. By using the BM mimetic, antigen specificity and neutralization capacity (i.e., protectiveness), of the Ab secreted from circulating ASCs can easily be assessed prior to serum level. Additionally, durability of the vaccines may also be assessed in vitro using the iBMM.
In summary, the generation of LLPCs is complex and involves both intrinsic and extrinsic factors. B cell differentiation to an ASC is immensely intricate involving GC and non-GC reactions together with upregulating the ASC programs. Although ASC differentiation is necessary it is not sufficient for LLPC generation, which involves BM survival factors to maintain ASC Ab secretion as well as other extrinsic elements in the microniche that appear to fundamentally transform early minted ASCs into a LLPC phenotypes. The novel iBMM culture system provides tools for studying LLPC generation and maintenance. Coupling with the recent advances in transcriptomics, at both a high-throughput scale and single-cell resolution, this system has the potential to answer questions about the intrinsic and extrinsic factors that regulate differentiation, maturation, and longevity of human LLPCs. Despite the progress in understanding the extrinsic cues that support ASC survival and Ab secretion, more studies are needed to characterize the complex maturation and maintenance of LLPCs.
DN, CJ, and FL wrote the manuscript. IS provided important editorial input.
This work was supported by the following grants: NIH/NIAID 1R01AI121252, R21AI109601, 1P01AI125180, P01A1078907, R37AI049660, U19AI1109962, U19AI110483, R01CA238471, U01AI141993, and the Bill & Melinda Gates Foundation Grant INV-002351.
FL founded MicroB-plex, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Jacob J, Kassir R, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. I The architecture and dynamics of responding cell populations. J Exp Med. (1991) 173:1165–75. doi: 10.1084/jem.173.5.1165
2. MacLennan IC, Toellner KM, Cunningham AF, Serre K, Sze DM, Zuniga E, et al. Extrafollicular antibody responses. Immunol Rev. (2003) 194:8–18. doi: 10.1034/j.1600-065X.2003.00058.x
3. Smith KG, Hewitson TD, Nossal GJ, Tarlinton DM. The phenotype and fate of the antibody-forming cells of the splenic foci. Eur J Immunol. (1996) 26:444–8. doi: 10.1002/eji.1830260226
4. Amanna IJ, Carlson NE, Slifka MK. Duration of humoral immunity to common viral and vaccine antigens. N Engl J Med. (2007) 357:1903–15. doi: 10.1056/NEJMoa066092
5. Blanchard-Rohner G, Pulickal AS, Jol-van der Zijde CM, Snape MD, Pollard AJ. Appearance of peripheral blood plasma cells and memory B cells in a primary and secondary immune response in humans. Blood. (2009) 114:4998–5002. doi: 10.1182/blood-2009-03-211052
6. Halliley JL, Kyu S, Kobie JJ, Walsh EE, Falsey AR, Randall TD, et al. Peak frequencies of circulating human influenza-specific antibody secreting cells correlate with serum antibody response after immunization. Vaccine. (2010) 28:3582–7. doi: 10.1016/j.vaccine.2010.02.088
7. Ho F, Lortan JE, MacLennan IC, Khan M. Distinct short-lived and long-lived antibody-producing cell populations. Eur J Immunol. (1986) 16:1297–301. doi: 10.1002/eji.1830161018
8. Kyu SY, Kobie J, Yang H, Zand MS, Topham DJ, Quataert SA, et al. Frequencies of human influenza-specific antibody secreting cells or plasmablasts post vaccination from fresh and frozen peripheral blood mononuclear cells. J Immunol Methods. (2009) 340:42–7. doi: 10.1016/j.jim.2008.09.025
9. Nossal GJ, Makela O. Autoradiographic studies on the immune response. I. The kinetics of plasma cell proliferation. J Exp Med. (1962) 115:209–230. doi: 10.1084/jem.115.1.209
10. Wrammert J, Smith K, Miller J, Langley WA, Kokko K, Larsen C, et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature. (2008) 453:667–71. doi: 10.1038/nature06890
11. Hammarlund E, Thomas A, Amanna IJ, Holden LA, Slayden OD, Park B, et al. Plasma cell survival in the absence of B cell memory. Nat Commun. (2017) 8:1781. doi: 10.1038/s41467-017-01901-w
12. Radbruch A, Muehlinghaus G, Luger EO, Inamine A, Smith KG, Dorner T, et al. Competence and competition: the challenge of becoming a long-lived plasma cell. Nat Rev Immunol. (2006) 6:741–50. doi: 10.1038/nri1886
13. Manz RA, Lohning M, Cassese G, Thiel A, Radbruch A. Survival of long-lived plasma cells is independent of antigen. Int Immunol. (1998) 10:1703–11. doi: 10.1093/intimm/10.11.1703
14. Manz RA, Thiel A, Radbruch A. Lifetime of plasma cells in the bone marrow. Nature. (1997) 388:133–4. doi: 10.1038/40540
15. Slifka MK, Antia R, Whitmire JK, Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity. (1998) 8:363–72. doi: 10.1016/S1074-7613(00)80541-5
16. Amanna IJ, Slifka MK. Contributions of humoral and cellular immunity to vaccine-induced protection in humans. Virology. (2011) 411:206–15. doi: 10.1016/j.virol.2010.12.016
17. Herrera D, Rojas OL, Duarte-Rey C, Mantilla RD, Angel J, Franco MA. Simultaneous assessment of rotavirus-specific memory B cells and serological memory after B cell depletion therapy with rituximab. PLoS ONE. (2014) 9:e97087. doi: 10.1371/journal.pone.0097087
18. Allen CD, Okada T, Tang HL, Cyster JG. Imaging of germinal center selection events during affinity maturation. Science. (2007) 315:528–31. doi: 10.1126/science.1136736
19. Amanna IJ, Slifka MK. Mechanisms that determine plasma cell lifespan and the duration of humoral immunity. Immunol Rev. (2010) 236:125–38. doi: 10.1111/j.1600-065X.2010.00912.x
20. Bannard O, Cyster JG. Germinal centers: programmed for affinity maturation and antibody diversification. Curr Opin Immunol. (2017) 45:21–30. doi: 10.1016/j.coi.2016.12.004
21. Smith KG, Light A, Nossal GJ, Tarlinton DM. The extent of affinity maturation differs between the memory and antibody-forming cell compartments in the primary immune response. EMBO J. (1997) 16:2996–3006. doi: 10.1093/emboj/16.11.2996
22. He J, Tsai LM, Leong YA, Hu X, Ma CS, Chevalier N, et al. Circulating precursor CCR7(lo)PD-1(hi) CXCR5(+) CD4(+) T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity. (2013) 39:770–81. doi: 10.1016/j.immuni.2013.09.007
23. Linterman MA, Beaton L, Yu D, Ramiscal RR, Srivastava M, Hogan JJ, et al. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J Exp Med. (2010) 207:353–63. doi: 10.1084/jem.20091738
24. Ozaki K, Spolski R, Feng CG, Qi CF, Cheng J, Sher A, et al. A critical role for IL-21 in regulating immunoglobulin production. Science. (2002) 298:1630–4. doi: 10.1126/science.1077002
25. Rasheed MA, Latner DR, Aubert RD, Gourley T, Spolski R, Davis CW, et al. Interleukin-21 is a critical cytokine for the generation of virus-specific long-lived plasma cells. J Virol. (2013) 87:7737–46. doi: 10.1128/JVI.00063-13
26. Song S, Matthias PD. The transcriptional regulation of germinal center formation. Front Immunol. (2018) 9:2026. doi: 10.3389/fimmu.2018.02026
27. Tangye SG, Ma CS, Brink R, Deenick EK. The good, the bad and the ugly–TFH cells in human health and disease. Nat Rev Immunol. (2013) 13:412–26. doi: 10.1038/nri3447
28. Weinstein JS, Bertino SA, Hernandez SG, Poholek AC, Teplitzky TB, Nowyhed HN, et al. B cells in T follicular helper cell development and function: separable roles in delivery of ICOS ligand and antigen. J Immunol. (2014) 192:3166–79. doi: 10.4049/jimmunol.1302617
29. Stone SL, Peel JN, Scharer CD, Risley CA, Chisolm DA, Schultz MD, et al. T-bet transcription factor promotes antibody-secreting cell differentiation by limiting the inflammatory effects of IFN-gamma on B cells. Immunity. (2019) 50:1172–87.e1177. doi: 10.1016/j.immuni.2019.04.004
30. Bortnick A, Chernova I, Quinn WJ 3rd, Mugnier M, Cancro MP, Allman D. Long-lived bone marrow plasma cells are induced early in response to T cell-independent or T cell-dependent antigens. J Immunol. (2012) 188:5389–96. doi: 10.4049/jimmunol.1102808
31. Foote JB, Mahmoud TI, Vale AM, Kearney JF. Long-term maintenance of polysaccharide-specific antibodies by IgM-secreting cells. J Immunol. (2012) 188:57–67. doi: 10.4049/jimmunol.1100783
32. Taillardet M, Haffar G, Mondiere P, Asensio MJ, Gheit H, Burdin N, et al. The thymus-independent immunity conferred by a pneumococcal polysaccharide is mediated by long-lived plasma cells. Blood. (2009) 114:4432–40. doi: 10.1182/blood-2009-01-200014
33. Corti D, Lanzavecchia A. Efficient methods to isolate human monoclonal antibodies from memory B cells and plasma cells. Microbiol Spectr. (2014) 2:129–39. doi: 10.1128/microbiolspec.AID-0018-2014
34. Corti D, Voss J, Gamblin SJ, Codoni G, Macagno A, Jarrossay D, et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science. (2011) 333:850–6. doi: 10.1126/science.1205669
35. Eyer K, Doineau RCL, Castrillon CE, Briseno-Roa L, Menrath V, Mottet G, et al. Single-cell deep phenotyping of IgG-secreting cells for high-resolution immune monitoring. Nat Biotechnol. (2017) 35:977–82. doi: 10.1038/nbt.3964
36. Hibi T, Dosch HM. Limiting dilution analysis of the B cell compartment in human bone marrow. Eur J Immunol. (1986) 16:139–45. doi: 10.1002/eji.1830160206
37. Kometani K, Nakagawa R, Shinnakasu R, Kaji T, Rybouchkin A, Moriyama S, et al. Repression of the transcription factor Bach2 contributes to predisposition of IgG1 memory B cells toward plasma cell differentiation. Immunity. (2013) 39:136–47. doi: 10.1016/j.immuni.2013.06.011
38. Lanzavecchia A. Dissecting human antibody responses: useful, basic and surprising findings. EMBO Mol Med. (2018) 10:e8879. doi: 10.15252/emmm.201808879
39. Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol. (2015) 15:160–71. doi: 10.1038/nri3795
40. Garimalla S, Nguyen DC, Halliley JL, Tipton C, Rosenberg AF, Fucile CF, et al. Differential transcriptome and development of human peripheral plasma cell subsets. JCI Insight. (2019) 4:126732. doi: 10.1172/jci.insight.126732
41. Kaminski DA, Wei C, Qian Y, Rosenberg AF, Sanz I. Advances in human B cell phenotypic profiling. Front Immunol. (2012) 3:302. doi: 10.3389/fimmu.2012.00302
42. Medina F, Segundo C, Campos-Caro A, Gonzalez-Garcia I, Brieva JA. The heterogeneity shown by human plasma cells from tonsil, blood, and bone marrow reveals graded stages of increasing maturity, but local profiles of adhesion molecule expression. Blood. (2002) 99:2154–61. doi: 10.1182/blood.V99.6.2154
43. Pracht K, Meinzinger J, Daum P, Schulz SR, Reimer D, Hauke M, et al. A new staining protocol for detection of murine antibody-secreting plasma cell subsets by flow cytometry. Eur J Immunol. (2017) 47:1389–92. doi: 10.1002/eji.201747019
44. Ribatti D. The discovery of plasma cells: an historical note. Immunol Lett. (2017) 188:64–7. doi: 10.1016/j.imlet.2017.06.006
45. Tellier J, Nutt SL. Standing out from the crowd: how to identify plasma cells. Eur J Immunol. (2017) 47:1276–9. doi: 10.1002/eji.201747168
46. Wilmore JR, Jones DD, Allman D. Protocol for improved resolution of plasma cell subpopulations by flow cytometry. Eur J Immunol. (2017) 47:1386–8. doi: 10.1002/eji.201746944
47. Chernova I, Jones DD, Wilmore JR, Bortnick A, Yucel M, Hershberg U, et al. Lasting antibody responses are mediated by a combination of newly formed and established bone marrow plasma cells drawn from clonally distinct precursors. J Immunol. (2014) 193:4971–9. doi: 10.4049/jimmunol.1401264
48. Jones DD, Gaudette BT, Wilmore JR, Chernova I, Bortnick A, Weiss BM, et al. mTOR has distinct functions in generating versus sustaining humoral immunity. J Clin Invest. (2016) 126:4250–61. doi: 10.1172/JCI86504
49. Shi W, Liao Y, Willis SN, Taubenheim N, Inouye M, Tarlinton DM, et al. Transcriptional profiling of mouse B cell terminal differentiation defines a signature for antibody-secreting plasma cells. Nat Immunol. (2015) 16:663–73. doi: 10.1038/ni.3154
50. Arumugakani G, Stephenson SJ, Newton DJ, Rawstron A, Emery P, Doody GM, et al. Early emergence of CD19-negative human antibody-secreting cells at the plasmablast to plasma cell transition. J Immunol. (2017) 198:4618–28. doi: 10.4049/jimmunol.1501761
51. Kabashima K, Haynes NM, Xu Y, Nutt SL, Allende ML, Proia RL, et al. Plasma cell S1P1 expression determines secondary lymphoid organ retention versus bone marrow tropism. J Exp Med. (2006) 203:2683–90. doi: 10.1084/jem.20061289
52. Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. (2004) 427:355–60. doi: 10.1038/nature02284
53. Kunkel EJ, Kim CH, Lazarus NH, Vierra MA, Soler D, Bowman EP, et al. CCR10 expression is a common feature of circulating and mucosal epithelial tissue IgA Ab-secreting cells. J Clin Invest. (2003) 111:1001–10. doi: 10.1172/JCI200317244
54. Hargreaves DC, Hyman PL, Lu TT, Ngo VN, Bidgol A, Suzuki G, et al. A coordinated change in chemokine responsiveness guides plasma cell movements. J Exp Med. (2001) 194:45–56. doi: 10.1084/jem.194.1.45
55. Hauser AE, Debes GF, Arce S, Cassese G, Hamann A, Radbruch A, et al. Chemotactic responsiveness toward ligands for CXCR3 and CXCR4 is regulated on plasma blasts during the time course of a memory immune response. J Immunol. (2002) 169:1277–82. doi: 10.4049/jimmunol.169.3.1277
56. Nie Y, Waite J, Brewer F, Sunshine MJ, Littman DR, Zou YR. The role of CXCR4 in maintaining peripheral B cell compartments and humoral immunity. J Exp Med. (2004) 200:1145–56. doi: 10.1084/jem.20041185
57. Good-Jacobson KL, O'Donnell K, Belz GT, Nutt SL, Tarlinton DM. c-Myb is required for plasma cell migration to bone marrow after immunization or infection. J Exp Med. (2015) 212:1001–9. doi: 10.1084/jem.20150191
58. Bowman EP, Kuklin NA, Youngman KR, Lazarus NH, Kunkel EJ, Pan J, et al. The intestinal chemokine thymus-expressed chemokine (CCL25) attracts IgA antibody-secreting cells. J Exp Med. (2002) 195:269–75. doi: 10.1084/jem.20010670
59. Kallies A, Hasbold J, Tarlinton DM, Dietrich W, Corcoran LM, Hodgkin PD, et al. Plasma cell ontogeny defined by quantitative changes in blimp-1 expression. J Exp Med. (2004) 200:967–77. doi: 10.1084/jem.20040973
60. Minnich M, Tagoh H, Bonelt P, Axelsson E, Fischer M, Cebolla B, et al. Multifunctional role of the transcription factor Blimp-1 in coordinating plasma cell differentiation. Nat Immunol. (2016) 17:331–43. doi: 10.1038/ni.3349
61. Tellier J, Shi W, Minnich M, Liao Y, Crawford S, Smyth GK, et al. Blimp-1 controls plasma cell function through the regulation of immunoglobulin secretion and the unfolded protein response. Nat Immunol. (2016) 17:323–30. doi: 10.1038/ni.3348
62. Cassese G, Arce S, Hauser AE, Lehnert K, Moewes B, Mostarac M, et al. Plasma cell survival is mediated by synergistic effects of cytokines and adhesion-dependent signals. J Immunol. (2003) 171:1684–90. doi: 10.4049/jimmunol.171.4.1684
63. Chu VT, Berek C. The establishment of the plasma cell survival niche in the bone marrow. Immunol Rev. (2013) 251:177–88. doi: 10.1111/imr.12011
64. McMillan R, Longmire RL, Yelenosky R, Lang JE, Heath V, Craddock CG. Immunoglobulin synthesis by human lymphoid tissues: normal bone marrow as a major site of IgG production. J Immunol. (1972) 109:1386–94.
65. Blanc P, Moro-Sibilot L, Barthly L, Jagot F, This S, de Bernard S, et al. Mature IgM-expressing plasma cells sense antigen and develop competence for cytokine production upon antigenic challenge. Nat Commun. (2016) 7:13600. doi: 10.1038/ncomms13600
66. Halliley JL, Tipton CM, Liesveld J, Rosenberg AF, Darce J, Gregoretti IV, et al. Long-lived plasma cells are contained within the CD19(-)CD38(hi)CD138(+) subset in human bone marrow. Immunity. (2015) 43:132–45. doi: 10.1016/j.immuni.2015.06.016
67. Mei HE, Wirries I, Frolich D, Brisslert M, Giesecke C, Grun JR, et al. A unique population of IgG-expressing plasma cells lacking CD19 is enriched in human bone marrow. Blood. (2015) 125:1739–48. doi: 10.1182/blood-2014-02-555169
68. Alinikula J, Lassila O. Gene interaction network regulates plasma cell differentiation. Scand J Immunol. (2011) 73:512–9. doi: 10.1111/j.1365-3083.2011.02556.x
69. Nutt SL, Taubenheim N, Hasbold J, Corcoran LM, Hodgkin PD. The genetic network controlling plasma cell differentiation. Semin Immunol. (2011) 23:341–9. doi: 10.1016/j.smim.2011.08.010
70. Kassambara A, Reme T, Jourdan M, Fest T, Hose D, Tarte K, et al. GenomicScape: an easy-to-use web tool for gene expression data analysis. Application to investigate the molecular events in the differentiation of B cells into plasma cells. PLoS Comput Biol. (2015) 11:e1004077. doi: 10.1371/journal.pcbi.1004077
71. Tellier J, Nutt SL. Plasma cells: the programming of an antibody-secreting machine. Eur J Immunol. (2019) 49:30–7. doi: 10.1002/eji.201847517
72. Calame KL, Lin KI, Tunyaplin C. Regulatory mechanisms that determine the development and function of plasma cells. Annu Rev Immunol. (2003) 21:205–30. doi: 10.1146/annurev.immunol.21.120601.141138
73. Igarashi K, Ochiai K, Itoh-Nakadai A, Muto A. Orchestration of plasma cell differentiation by Bach2 and its gene regulatory network. Immunol Rev. (2014) 261:116–25. doi: 10.1111/imr.12201
74. Shapiro-Shelef M, Calame K. Regulation of plasma-cell development. Nat Rev Immunol. (2005) 5:230–42. doi: 10.1038/nri1572
75. Cobaleda C, Schebesta A, Delogu A, Busslinger M. Pax5: the guardian of B cell identity and function. Nat Immunol. (2007) 8:463–70. doi: 10.1038/ni1454
76. Shapiro-Shelef M, Lin KI, McHeyzer-Williams LJ, Liao J, McHeyzer-Williams MG, Calame K. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity. (2003) 19:607–20. doi: 10.1016/S1074-7613(03)00267-X
77. Klein U, Casola S, Cattoretti G, Shen Q, Lia M, Mo T, et al. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat Immunol. (2006) 7:773–82. doi: 10.1038/ni1357
78. Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature. (2001) 412:300–7. doi: 10.1038/35085509
79. Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, Singh H. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity. (2006) 25:225–36. doi: 10.1016/j.immuni.2006.07.009
80. Hu CC, Dougan SK, McGehee AM, Love JC, Ploegh HL. XBP-1 regulates signal transduction, transcription factors and bone marrow colonization in B cells. EMBO J. (2009) 28:1624–36. doi: 10.1038/emboj.2009.117
81. Taubenheim N, Tarlinton DM, Crawford S, Corcoran LM, Hodgkin PD, Nutt SL. High rate of antibody secretion is not integral to plasma cell differentiation as revealed by XBP-1 deficiency. J Immunol. (2012) 189:3328–38. doi: 10.4049/jimmunol.1201042
82. Todd DJ, McHeyzer-Williams LJ, Kowal C, Lee AH, Volpe BT, Diamond B, et al. XBP1 governs late events in plasma cell differentiation and is not required for antigen-specific memory B cell development. J Exp Med. (2009) 206:2151–9. doi: 10.1084/jem.20090738
83. Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. (2001) 107:881–91. doi: 10.1016/S0092-8674(01)00611-0
84. Carrington EM, Vikstrom IB, Light A, Sutherland RM, Londrigan SL, Mason KD, et al. BH3 mimetics antagonizing restricted prosurvival Bcl-2 proteins represent another class of selective immune modulatory drugs. Proc Natl Acad Sci USA. (2010) 107:10967–71. doi: 10.1073/pnas.1005256107
85. Peperzak V, Vikstrom I, Walker J, Glaser SP, LePage M, Coquery CM, et al. Mcl-1 is essential for the survival of plasma cells. Nat Immunol. (2013) 14:290–7. doi: 10.1038/ni.2527
86. Strasser A, Whittingham S, Vaux DL, Bath ML, Adams JM, Cory S, et al. Enforced BCL2 expression in B-lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proc Natl Acad Sci USA. (1991) 88:8661–5. doi: 10.1073/pnas.88.19.8661
87. Yoshida T, Mei H, Dorner T, Hiepe F, Radbruch A, Fillatreau S, et al. Memory B and memory plasma cells. Immunol Rev. (2010) 237:117–39. doi: 10.1111/j.1600-065X.2010.00938.x
88. Chevrier S, Emslie D, Shi W, Kratina T, Wellard C, Karnowski A, et al. The BTB-ZF transcription factor Zbtb20 is driven by Irf4 to promote plasma cell differentiation and longevity. J Exp Med. (2014) 211:827–40. doi: 10.1084/jem.20131831
89. Shaffer AL, Emre NC, Lamy L, Ngo VN, Wright G, Xiao W, et al. IRF4 addiction in multiple myeloma. Nature. (2008) 454:226–31. doi: 10.1038/nature07064
90. Wang Y, Bhattacharya D. Adjuvant-specific regulation of long-term antibody responses by ZBTB20. J Exp Med. (2014) 211:841–56. doi: 10.1084/jem.20131821
91. Gass JN, Gifford NM, Brewer JW. Activation of an unfolded protein response during differentiation of antibody-secreting B cells. J Biol Chem. (2002) 277:49047–54. doi: 10.1074/jbc.M205011200
92. Ma Y, Shimizu Y, Mann MJ, Jin Y, Hendershot LM. Plasma cell differentiation initiates a limited ER stress response by specifically suppressing the PERK-dependent branch of the unfolded protein response. Cell Stress Chaperones. (2010) 15:281–93. doi: 10.1007/s12192-009-0142-9
93. Pengo N, Scolari M, Oliva L, Milan E, Mainoldi F, Raimondi A, et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol. (2013) 14:298–305. doi: 10.1038/ni.2524
94. Nguyen DC, Garimalla S, Xiao H, Kyu S, Albizua I, Galipeau J, et al. Factors of the bone marrow microniche that support human plasma cell survival and immunoglobulin secretion. Nat Commun. (2018) 9:3698. doi: 10.1038/s41467-018-05853-7
95. Ben-Sahra I, Manning BD. mTORC1 signaling and the metabolic control of cell growth. Curr Opin Cell Biol. (2017) 45:72–82. doi: 10.1016/j.ceb.2017.02.012
96. Dibble CC, Manning BD. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol. (2013) 15:555–64. doi: 10.1038/ncb2763
97. Setz CS, Hug E, Khadour A, Abdelrasoul H, Bilal M, Hobeika E, et al. PI3K-mediated Blimp-1 activation controls B cell selection and homeostasis. Cell Rep. (2018) 24:391–405. doi: 10.1016/j.celrep.2018.06.035
98. Lam WY, Becker AM, Kennerly KM, Wong R, Curtis JD, Llufrio EM, et al. Mitochondrial pyruvate import promotes long-term survival of antibody-secreting plasma cells. Immunity. (2016) 45:60–73. doi: 10.1016/j.immuni.2016.06.011
99. Lam WY, Jash A, Yao CH, D'Souza L, Wong R, Nunley RM, et al. Metabolic and transcriptional modules independently diversify plasma cell lifespan and function. Cell Rep. (2018) 24:2479–92.e2476. doi: 10.1016/j.celrep.2018.07.084
100. Wang H, Gonzalez-Garcia I, Traba J, Jain S, Conteh S, Shin DM, et al. ATP-degrading ENPP1 is required for survival (or persistence) of long-lived plasma cells. Sci Rep. (2017) 7:17867. doi: 10.1038/s41598-017-18028-z
101. Ivanov P, Anderson P. Post-transcriptional regulatory networks in immunity. Immunol Rev. (2013) 253:253–72. doi: 10.1111/imr.12051
102. Martinez NM, Lynch KW. Control of alternative splicing in immune responses: many regulators, many predictions, much still to learn. Immunol Rev. (2013) 253:216–36. doi: 10.1111/imr.12047
103. Turner M, Galloway A, Vigorito E. Noncoding RNA and its associated proteins as regulatory elements of the immune system. Nat Immunol. (2014) 15:484–91. doi: 10.1038/ni.2887
104. Shukla S, Oberdoerffer S. Co-transcriptional regulation of alternative pre-mRNA splicing. Biochim Biophys Acta. (2012) 1819:673–83. doi: 10.1016/j.bbagrm.2012.01.014
105. Fan XC, Steitz JA. Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J. (1998) 17:3448–60. doi: 10.1093/emboj/17.12.3448
106. Diaz-Munoz MD, Bell SE, Fairfax K, Monzon-Casanova E, Cunningham AF, Gonzalez-Porta M, et al. The RNA-binding protein HuR is essential for the B cell antibody response. Nat Immunol. (2015) 16:415–25. doi: 10.1038/ni.3115
107. Chang X, Li B, Rao A. RNA-binding protein hnRNPLL regulates mRNA splicing and stability during B-cell to plasma-cell differentiation. Proc Natl Acad Sci USA. (2015) 112:E1888–97. doi: 10.1073/pnas.1422490112
108. Benson MJ, Aijo T, Chang X, Gagnon J, Pape UJ, Anantharaman V, et al. Heterogeneous nuclear ribonucleoprotein L-like (hnRNPLL) and elongation factor, RNA polymerase II, 2 (ELL2) are regulators of mRNA processing in plasma cells. Proc Natl Acad Sci USA. (2012) 109:16252–7. doi: 10.1073/pnas.1214414109
109. Nguyen DC, Saney C, Ley A, Sanz I, Lee FE. Human plasma cells switch on and turn off antibody secretion. J Immunol. (2019) 202(Suppl 1):123.20.
110. Guo M, Price MJ, Patterson DG, Barwick BG, Haines RR, Kania AK, et al. EZH2 represses the B cell transcriptional program and regulates antibody-secreting cell metabolism and antibody production. J Immunol. (2018) 200:1039–52. doi: 10.4049/jimmunol.1701470
111. Scharer CD, Barwick BG, Guo M, Bally APR, Boss JM. Plasma cell differentiation is controlled by multiple cell division-coupled epigenetic programs. Nat Commun. (2018) 9:1698. doi: 10.1038/s41467-018-04125-8
112. Tangye SG. Staying alive: regulation of plasma cell survival. Trends Immunol. (2011) 32:595–602. doi: 10.1016/j.it.2011.09.001
113. Tokoyoda K, Hauser AE, Nakayama T, Radbruch A. Organization of immunological memory by bone marrow stroma. Nat Rev Immunol. (2010) 10:193–200. doi: 10.1038/nri2727
114. Liang B, Hyland L, Hou S. Nasal-associated lymphoid tissue is a site of long-term virus-specific antibody production following respiratory virus infection of mice. J Virol. (2001) 75:5416–20. doi: 10.1128/JVI.75.11.5416-5420.2001
115. van Laar JM, Melchers M, Teng YK, van der Zouwen B, Mohammadi R, Fischer R, et al. Sustained secretion of immunoglobulin by long-lived human tonsil plasma cells. Am J Pathol. (2007) 171:917–27. doi: 10.2353/ajpath.2007.070005
116. Huard B, McKee T, Bosshard C, Durual S, Matthes T, Myit S, et al. APRIL secreted by neutrophils binds to heparan sulfate proteoglycans to create plasma cell niches in human mucosa. J Clin Invest. (2008) 118:2887–95. doi: 10.1172/JCI33760
117. Lemke A, Kraft M, Roth K, Riedel R, Lammerding D, Hauser AE. Long-lived plasma cells are generated in mucosal immune responses and contribute to the bone marrow plasma cell pool in mice. Mucosal Immunol. (2016) 9:83–97. doi: 10.1038/mi.2015.38
118. Di Niro R, Mesin L, Raki M, Zheng NY, Lund-Johansen F, Lundin KE, et al. Rapid generation of rotavirus-specific human monoclonal antibodies from small-intestinal mucosa. J Immunol. (2010) 185:5377–83. doi: 10.4049/jimmunol.1001587
119. Landsverk OJ, Snir O, Casado RB, Richter L, Mold JE, Reu P, et al. Antibody-secreting plasma cells persist for decades in human intestine. J Exp Med. (2017) 214:309–17. doi: 10.1084/jem.20161590
120. Mesin L, Di Niro R, Thompson KM, Lundin KE, Sollid LM. Long-lived plasma cells from human small intestine biopsies secrete immunoglobulins for many weeks in vitro. J Immunol. (2011) 187:2867–74. doi: 10.4049/jimmunol.1003181
121. Mahevas M, atin P, Huetz F, Descatoire M, Cagnard N, Bole-Feysot C, et al. B cell depletion in immune thrombocytopenia reveals splenic long-lived plasma cells. J Clin Invest. (2013) 123:432–42. doi: 10.1172/JCI65689
122. Szyszko EA, Brokstad KA, Oijordsbakken G, Jonsson MV, Jonsson R, Skarstein K. Salivary glands of primary Sjogren's syndrome patients express factors vital for plasma cell survival. Arthritis Res Ther. (2011) 13:R2. doi: 10.1186/ar3220
123. Thai LH, Le Gallou S, Robbins A, Crickx E, Fadeev T, Zhou Z, et al. BAFF and CD4(+) T cells are major survival factors for long-lived splenic plasma cells in a B-cell-depletion context. Blood. (2018) 131:1545–55. doi: 10.1182/blood-2017-06-789578
124. Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. (2014) 505:327–34. doi: 10.1038/nature12984
125. Chu VT, Frohlich A, Steinhauser G, Scheel T, Roch T, Fillatreau S, et al. Eosinophils are required for the maintenance of plasma cells in the bone marrow. Nat Immunol. (2011) 12:151–9. doi: 10.1038/ni.1981
126. Zehentmeier S, Roth K, Cseresnyes Z, Sercan O, Horn K, Niesner RA, et al. Static and dynamic components synergize to form a stable survival niche for bone marrow plasma cells. Eur J Immunol. (2014) 44:2306–17. doi: 10.1002/eji.201344313
127. Winter O, Moser K, Mohr E, Zotos D, Kaminski H, Szyska M, et al. Megakaryocytes constitute a functional component of a plasma cell niche in the bone marrow. Blood. (2010) 116:1867–75. doi: 10.1182/blood-2009-12-259457
128. Rodriguez Gomez M, Talke Y, Goebel N, Hermann F, Reich B, Mack M. Basophils support the survival of plasma cells in mice. J Immunol. (2010) 185:7180–5. doi: 10.4049/jimmunol.1002319
129. Belnoue E, Pihlgren M, McGaha TL, Tougne C, Rochat AF, Bossen C, et al. APRIL is critical for plasmablast survival in the bone marrow and poorly expressed by early-life bone marrow stromal cells. Blood. (2008) 111:2755–64. doi: 10.1182/blood-2007-09-110858
130. Belnoue E, Tougne C, Rochat AF, Lambert PH, Pinschewer DD, Siegrist CA. Homing and adhesion patterns determine the cellular composition of the bone marrow plasma cell niche. J Immunol. (2012) 188:1283–91. doi: 10.4049/jimmunol.1103169
131. Rozanski CH, Arens R, Carlson LM, Nair J, Boise LH, Chanan-Khan AA, et al. Sustained antibody responses depend on CD28 function in bone marrow-resident plasma cells. J Exp Med. (2011) 208:1435–46. doi: 10.1084/jem.20110040
132. Tokoyoda K, Zehentmeier S, Hegazy AN, Albrecht I, Grun JR, Lohning M, et al. Professional memory CD4+ T lymphocytes preferentially reside and rest in the bone marrow. Immunity. (2009) 30:721–30. doi: 10.1016/j.immuni.2009.03.015
133. Geffroy-Luseau A, Jego G, Bataille R, Campion L, Pellat-Deceunynck C. Osteoclasts support the survival of human plasma cells in vitro. Int Immunol. (2008) 20:775–82. doi: 10.1093/intimm/dxn035
134. Rosen CJ, Ackert-Bicknell C, Rodriguez JP, Pino AM. Marrow fat and the bone microenvironment: developmental, functional, and pathological implications. Crit Rev Eukaryot Gene Expr. (2009) 19:109–24. doi: 10.1615/CritRevEukarGeneExpr.v19.i2.20
135. Eliasson P, Jonsson JI. The hematopoietic stem cell niche: low in oxygen but a nice place to be. J Cell Physiol. (2010) 222:17–22. doi: 10.1002/jcp.21908
136. Harrison JS, Rameshwar P, Chang V, Bandari P. Oxygen saturation in the bone marrow of healthy volunteers. Blood. (2002) 99:394. doi: 10.1182/blood.V99.1.394
137. Parmar K, Mauch P, Vergilio JA, Sackstein R, Down JD. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc Natl Acad Sci USA. (2007) 104:5431–6. doi: 10.1073/pnas.0701152104
138. Winkler IG, Barbier V, Wadley R, Zannettino AC, Williams S, Levesque JP. Positioning of bone marrow hematopoietic and stromal cells relative to blood flow in vivo: serially reconstituting hematopoietic stem cells reside in distinct nonperfused niches. Blood. (2010) 116:375–85. doi: 10.1182/blood-2009-07-233437
139. Bonnaure G, Gervais-St-Amour C, Neron S. Bone marrow mesenchymal stem cells enhance the differentiation of human switched memory B lymphocytes into plasma cells in serum-free medium. J Immunol Res. (2016) 2016:7801781. doi: 10.1155/2016/7801781
140. Calame KL. Plasma cells: finding new light at the end of B cell development. Nat Immunol. (2001) 2:1103–8. doi: 10.1038/ni1201-1103
141. Kayaba A, Itoh-Nakadai A, Niibe K, Shirota M, Funayama R, Sugahara-Tobinai A, et al. Bone marrow PDGFRalpha+Sca-1+-enriched mesenchymal stem cells support survival of and antibody production by plasma cells in vitro through IL-6. Int Immunol. (2018) 30:241–53. doi: 10.1093/intimm/dxy018
142. Minges Wols HA, Underhill GH, Kansas GS, Witte PL. The role of bone marrow-derived stromal cells in the maintenance of plasma cell longevity. J Immunol. (2002) 169:4213–21. doi: 10.4049/jimmunol.169.8.4213
143. Rasmusson I, Le Blanc K, Sundberg B, Ringden O. Mesenchymal stem cells stimulate antibody secretion in human B cells. Scand J Immunol. (2007) 65:336–43. doi: 10.1111/j.1365-3083.2007.01905.x
144. Traggiai E, Volpi S, Schena F, Gattorno M, Ferlito F, Moretta L, et al. Bone marrow-derived mesenchymal stem cells induce both polyclonal expansion and differentiation of B cells isolated from healthy donors and systemic lupus erythematosus patients. Stem Cells. (2008) 26:562–9. doi: 10.1634/stemcells.2007-0528
145. Underhill GH, Minges Wols HA, Fornek JL, Witte PL, Kansas GS. IgG plasma cells display a unique spectrum of leukocyte adhesion and homing molecules. Blood. (2002) 99:2905–12. doi: 10.1182/blood.V99.8.2905
146. Zehentmeier S, Cseresnyes Z, Escribano Navarro J, Niesner RA, Hauser AE. Automated quantification of hematopoietic cell - stromal cell interactions in histological images of undecalcified bone. J Vis Exp. (2015) e52544. doi: 10.3791/52544
147. DiLillo DJ, Hamaguchi Y, Ueda Y, Yang K, Uchida J, Haas KM, et al. Maintenance of long-lived plasma cells and serological memory despite mature and memory B cell depletion during CD20 immunotherapy in mice. J Immunol. (2008) 180:361–71. doi: 10.4049/jimmunol.180.1.361
148. Roldan E, Garcia-Pardo A, Brieva JA. VLA-4-fibronectin interaction is required for the terminal differentiation of human bone marrow cells capable of spontaneous and high rate immunoglobulin secretion. J Exp Med. (1992) 175:1739–47. doi: 10.1084/jem.175.6.1739
149. De Grandis M, Lhoumeau AC, Mancini SJ, Aurrand-Lions M. Adhesion receptors involved in HSC and early-B cell interactions with bone marrow microenvironment. Cell Mol Life Sci. (2016) 73:687–703. doi: 10.1007/s00018-015-2064-2
150. Ellyard JI, Avery DT, Mackay CR, Tangye SG. Contribution of stromal cells to the migration, function and retention of plasma cells in human spleen: potential roles of CXCL12, IL-6 and CD5(2019) 4. Eur J Immunol. (2005) 35:699–708. doi: 10.1002/eji.200425442
151. Cazac BB, Roes J. TGF-beta receptor controls B cell responsiveness and induction of IgA in vivo. Immunity. (2000) 13:443–51. doi: 10.1016/S1074-7613(00)00044-3
152. Gros MJ, Naquet P, Guinamard RR. Cell intrinsic TGF-beta 1 regulation of B cells. J Immunol. (2008) 180:8153–8. doi: 10.4049/jimmunol.180.12.8153
153. Kyurkchiev D, Bochev I, Ivanova-Todorova E, Mourdjeva M, Oreshkova T, Belemezova K, et al. Secretion of immunoregulatory cytokines by mesenchymal stem cells. World J Stem Cells. (2014) 6:552–70. doi: 10.4252/wjsc.v6.i5.552
154. Yamazaki S, Iwama A, Takayanagi S, Eto K, Ema H, Nakauchi H. TGF-beta as a candidate bone marrow niche signal to induce hematopoietic stem cell hibernation. Blood. (2009) 113:1250–6. doi: 10.1182/blood-2008-04-146480
155. Amable PR, Teixeira MV, Carias RB, Granjeiro JM, Borojevic R. Protein synthesis and secretion in human mesenchymal cells derived from bone marrow, adipose tissue and Wharton's jelly. Stem Cell Res Ther. (2014) 5:53. doi: 10.1186/scrt442
156. Harvey A, Yen TY, Aizman I, Tate C, Case C. Proteomic analysis of the extracellular matrix produced by mesenchymal stromal cells: implications for cell therapy mechanism. PLoS ONE. (2013) 8:e79283. doi: 10.1371/journal.pone.0079283
157. Kimberley FC, van Bostelen L, Cameron K, Hardenberg G, Marquart JA, Hahne M, et al. The proteoglycan (heparan sulfate proteoglycan) binding domain of APRIL serves as a platform for ligand multimerization and cross-linking. FASEB J. (2009) 23:1584–95. doi: 10.1096/fj.08-124669
158. Reijmers RM, Groen RW, Kuil A, Weijer K, Kimberley FC, Medema JP, et al. Disruption of heparan sulfate proteoglycan conformation perturbs B-cell maturation and APRIL-mediated plasma cell survival. Blood. (2011) 117:6162–71. doi: 10.1182/blood-2010-12-325522
159. Healy ME, Bergin R, Mahon BP, English K. Mesenchymal stromal cells protect against caspase 3-mediated apoptosis of CD19(+) peripheral B cells through contact-dependent upregulation of VEGF. Stem Cells Dev. (2015) 24:2391–402. doi: 10.1089/scd.2015.0089
160. Huang HY, Rivas-Caicedo A, Renevey F, Cannelle H, Peranzoni E, Scarpellino L, et al. Identification of a new subset of lymph node stromal cells involved in regulating plasma cell homeostasis. Proc Natl Acad Sci USA. (2018) 115:E6826–35. doi: 10.1073/pnas.1712628115
161. Zhang Y, Tech L, George LA, Acs A, Durrett RE, Hess H, et al. Plasma cell output from germinal centers is regulated by signals from Tfh and stromal cells. J Exp Med. (2018) 215:1227–43. doi: 10.1084/jem.20160832
162. Nguyen DC, Lewis HC, Joyner C, Warren V, Xiao H, Kissick HT, et al. Extracellular vesicles from bone marrow-derived mesenchymal stromal cells support ex vivo survival of human antibody secreting cells. J Extracell Vesicles. (2018) 7:1463778. doi: 10.1080/20013078.2018.1463778
163. Roccaro AM, Sacco A, Maiso P, Azab AK, Tai YT, Reagan M, et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J Clin Invest. (2013) 123:1542–55. doi: 10.1172/JCI66517
164. Bortnick A, Chernova I, Spencer SP, Allman D. No strict requirement for eosinophils for bone marrow plasma cell survival. Eur J Immunol. (2018) 48:815–21. doi: 10.1002/eji.201747229
165. Cravedi P, Lessman DA, Heeger PS. Eosinophils are not required for the induction and maintenance of an alloantibody response. Am J Transplant. (2013) 13:2696–702. doi: 10.1111/ajt.12404
166. Haberland K, Ackermann JA, Ipseiz N, Culemann S, Pracht K, Englbrecht M, et al. Eosinophils are not essential for maintenance of murine plasma cells in the bone marrow. Eur J Immunol. (2018) 48:822–8. doi: 10.1002/eji.201747227
167. Abe M, Hiura K, Wilde J, Shioyasono A, Moriyama K, Hashimoto T, et al. Osteoclasts enhance myeloma cell growth and survival via cell-cell contact: a vicious cycle between bone destruction and myeloma expansion. Blood. (2004) 104:2484–91. doi: 10.1182/blood-2003-11-3839
168. Moreaux J, Cremer FW, Reme T, Raab M, Mahtouk K, Kaukel P, et al. The level of TACI gene expression in myeloma cells is associated with a signature of microenvironment dependence versus a plasmablastic signature. Blood. (2005) 106:1021–30. doi: 10.1182/blood-2004-11-4512
169. Krautler NJ, Suan D, Butt D, Bourne K, Hermes JR, Chan TD, et al. Differentiation of germinal center B cells into plasma cells is initiated by high-affinity antigen and completed by Tfh cells. J Exp Med. (2017) 214:1259–67. doi: 10.1084/jem.20161533
170. Glatman Zaretsky A, Konradt C, Depis F, Wing JB, Goenka R, Atria DG, et al. T regulatory cells support plasma cell populations in the bone marrow. Cell Rep. (2017) 18:1906–16. doi: 10.1016/j.celrep.2017.01.067
171. Abe M, Kido S, Hiasa M, Nakano A, Oda A, Amou H, et al. BAFF and APRIL as osteoclast-derived survival factors for myeloma cells: a rationale for TACI-Fc treatment in patients with multiple myeloma. Leukemia. (2006) 20:1313–5. doi: 10.1038/sj.leu.2404228
172. Tokoyoda K, Egawa T, Sugiyama T, Choi BI, Nagasawa T. Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity. (2004) 20:707–18. doi: 10.1016/j.immuni.2004.05.001
173. Klein B, Tarte K, Jourdan M, Mathouk K, Moreaux J, Jourdan E, et al. Survival and proliferation factors of normal and malignant plasma cells. Int J Hematol. (2003) 78:106–13. doi: 10.1007/BF02983377
174. Matthes T, Dunand-Sauthier I, Santiago-Raber ML, Krause KH, Donze O, Passweg J, et al. Production of the plasma-cell survival factor a proliferation-inducing ligand (APRIL) peaks in myeloid precursor cells from human bone marrow. Blood. (2011) 118:1838–44. doi: 10.1182/blood-2011-01-332940
175. Jourdan M, Cren M, Robert N, Bollore K, Fest T, Duperray C, et al. IL-6 supports the generation of human long-lived plasma cells in combination with either APRIL or stromal cell-soluble factors. Leukemia. (2014) 28:1647–56. doi: 10.1038/leu.2014.61
176. O'Connor BP, Raman VS, Erickson LD, Cook WJ, Weaver LK, Ahonen C, et al. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med. (2004) 199:91–8. doi: 10.1084/jem.20031330
177. Benson MJ, Dillon SR, Castigli E, Geha RS, Xu S, Lam KP, et al. Cutting edge: the dependence of plasma cells and independence of memory B cells on BAFF and APRIL. J Immunol. (2008) 180:3655–9. doi: 10.4049/jimmunol.180.6.3655
178. Coquery CM, Erickson LD. Regulatory roles of the tumor necrosis factor receptor BCMA. Crit Rev Immunol. (2012) 32:287–305. doi: 10.1615/CritRevImmunol.v32.i4.10
179. McCarron MJ, Park PW, Fooksman DR. CD138 mediates selection of mature plasma cells by regulating their survival. Blood. (2017) 129:2749–59. doi: 10.1182/blood-2017-01-761643
180. Ingold K, Zumsteg A, Tardivel A, Huard B, Steiner QG, Cachero TG, et al. Identification of proteoglycans as the APRIL-specific binding partners. J Exp Med. (2005) 201:1375–83. doi: 10.1084/jem.20042309
181. Mackay F, Schneider P, Rennert P, Browning J. BAFF AND APRIL: a tutorial on B cell survival. Annu Rev Immunol. (2003) 21:231–64. doi: 10.1146/annurev.immunol.21.120601.141152
182. Couchman JR. Syndecans: proteoglycan regulators of cell-surface microdomains? Nat Rev Mol Cell Biol. (2003) 4:926–37. doi: 10.1038/nrm1257
183. Nguyen DC, Saney C, Sanz I, Lee FE. Human bone marrow plasma cell survival is independent of APRIL. J Immunol. (2019) 202(Suppl 1):181.16.
184. Rodriguez-Bayona B, Ramos-Amaya A, Lopez-Blanco R, Campos-Caro A, Brieva JA. STAT-3 activation by differential cytokines is critical for human in vivo-generated plasma cell survival and Ig secretion. J Immunol. (2013) 191:4996–5004. doi: 10.4049/jimmunol.1301559
185. Rose-John S. Coordination of interleukin-6 biology by membrane bound and soluble receptors. Adv Exp Med Biol. (2001) 495:145–51. doi: 10.1007/978-1-4615-0685-0_19
186. Taga T, Kishimoto T. Gp130 and the interleukin-6 family of cytokines. Annu Rev Immunol. (1997) 15:797–819. doi: 10.1146/annurev.immunol.15.1.797
187. Taga T, Hibi M, Hirata Y, Yamasaki K, Yasukawa K, Matsuda T, et al. Interleukin-6 triggers the association of its receptor with a possible signal transducer, gp130. Cell. (1989) 58:573–81. doi: 10.1016/0092-8674(89)90438-8
188. Oberg HH, Wesch D, Grussel S, Rose-John S, Kabelitz D. Differential expression of CD126 and CD130 mediates different STAT-3 phosphorylation in CD4+CD25- and CD25high regulatory T cells. Int Immunol. (2006) 18:555–63. doi: 10.1093/intimm/dxh396
189. Schaper F, Rose-John S. Interleukin-6: biology, signaling and strategies of blockade. Cytokine Growth Factor Rev. (2015) 26:475–87. doi: 10.1016/j.cytogfr.2015.07.004
190. Rose-John S, Heinrich PC. Soluble receptors for cytokines and growth factors: generation and biological function. Biochem J. (1994) 300:281–90. doi: 10.1042/bj3000281
191. Hibi M, Murakami M, Saito M, Hirano T, Taga T, Kishimoto T. Molecular cloning and expression of an IL-6 signal transducer, gp130. Cell. (1990) 63:1149–57. doi: 10.1016/0092-8674(90)90411-7
192. Minges Wols HA, Ippolito JA, Yu Z, Palmer JL, White FA, Le PT, et al. The effects of microenvironment and internal programming on plasma cell survival. Int Immunol. (2007) 19:837–46. doi: 10.1093/intimm/dxm051
193. Beck TC, Gomes AC, Cyster JG, Pereira JP. CXCR4 and a cell-extrinsic mechanism control immature B lymphocyte egress from bone marrow. J Exp Med. (2014) 211:2567–81. doi: 10.1084/jem.20140457
194. Nair JR, Carlson LM, Koorella C, Rozanski CH, Byrne GE, Bergsagel PL, et al. CD28 expressed on malignant plasma cells induces a prosurvival and immunosuppressive microenvironment. J Immunol. (2011) 187:1243–53. doi: 10.4049/jimmunol.1100016
195. Rozanski CH, Utley A, Carlson LM, Farren MR, Murray M, Russell LM, et al. CD28 promotes plasma cell survival, sustained antibody responses, and BLIMP-1 upregulation through its distal PYAP proline motif. J Immunol. (2015) 194:4717–28. doi: 10.4049/jimmunol.1402260
196. Chevrier S, Genton C, Kallies A, Karnowski A, Otten LA, Malissen B, et al. CD93 is required for maintenance of antibody secretion and persistence of plasma cells in the bone marrow niche. Proc Natl Acad Sci USA. (2009) 106:3895–900. doi: 10.1073/pnas.0809736106
197. van Spriel AB, de Keijzer S, van der Schaaf A, Gartlan KH, Sofi M, Light A, et al. The tetraspanin CD37 orchestrates the alpha(4)beta(1) integrin-Akt signaling axis and supports long-lived plasma cell survival. Sci Signal. (2012) 5:ra82. doi: 10.1126/scisignal.2003113
198. Xiang Z, Cutler AJ, Brownlie RJ, Fairfax K, Lawlor KE, Severinson E, et al. FcgammaRIIb controls bone marrow plasma cell persistence and apoptosis. Nat Immunol. (2007) 8:419–29. doi: 10.1038/ni1440
199. Conter LJ, Song E, Shlomchik MJ, Tomayko MM. CD73 expression is dynamically regulated in the germinal center and bone marrow plasma cells are diminished in its absence. PLoS ONE. (2014) 9:e92009. doi: 10.1371/journal.pone.0092009
200. Sliva D, Gu M, Zhu YX, Chen J, Tsai S, Du X, et al. 14-3-3zeta interacts with the alpha-chain of human interleukin 9 receptor. Biochem J. (2000) 345(Pt 3):741–7. doi: 10.1042/bj3450741
201. Takatsuka S, Yamada H, Haniuda K, Saruwatari H, Ichihashi M, Renauld JC, et al. IL-9 receptor signaling in memory B cells regulates humoral recall responses. Nat Immunol. (2018) 19:1025–34. doi: 10.1038/s41590-018-0177-0
202. Merville P, Dechanet J, Grouard G, Durand I, Banchereau J. T cell-induced B cell blasts differentiate into plasma cells when cultured on bone marrow stroma with IL-3 and IL-(2018) 10. Int Immunol. (1995) 7:635–43. doi: 10.1093/intimm/7.4.635
Keywords: immunoglobulin, B cell, antibody-secreting cell, long-lived plasma cell, differentiation, maturation, maintenance
Citation: Nguyen DC, Joyner CJ, Sanz I and Lee FE-H (2019) Factors Affecting Early Antibody Secreting Cell Maturation Into Long-Lived Plasma Cells. Front. Immunol. 10:2138. doi: 10.3389/fimmu.2019.02138
Received: 14 July 2019; Accepted: 27 August 2019;
Published: 11 September 2019.
Edited by:
Michael R. Gold, University of British Columbia, CanadaReviewed by:
Stephen Nutt, Walter and Eliza Hall Institute of Medical Research, AustraliaCopyright © 2019 Nguyen, Joyner, Sanz and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: F. Eun-Hyung Lee, Zi5lLmxlZUBlbW9yeS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.